

Abstract
Despite the ongoing vaccination against the life-threatening COVID-19, there is need for viable therapeutic interventions. The S-adenosyl-l-Methionine (SAM) dependent 2-O’-ribose methyltransferase (2′-O-MTase) of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) presents a therapeutic target against COVID-19 infection. In a bid to profile bioactive principles from natural sources, a custom-made library of 226 phytochemicals from African medicinal plants with especially anti-malarial activity was screened for direct interactions with SARS-CoV-2 2′-O-MTase (S2RMT) using molecular docking and molecular dynamics (MD) simulations as well as binding free energies methods. Based on minimal binding energy lower than sinefungin (a reference methyl-transferase inhibitor) and binding mode analysis at the catalytic site of S2RMT, a list of 26 hit phytocompounds was defined. The interaction of these phytocompounds was compared with the 2′-O-MTase of SARS-CoV and MERS-CoV. Among these compounds, the lead phytocompounds (LPs) viz: mulberrofuran F, 24-methylene cycloartenol, ferulate, 3-benzoylhosloppone and 10-hydroxyusambarensine interacted strongly with the conserved KDKE tetrad within the substrate binding pocket of the 2′-O-MTase of the coronavirus strains which is critical for substrate binding. The thermodynamic parameters analyzed from the MD simulation trajectories of the LPs-S2RMT complexes presented an eminent structural stability and compactness. These LPs demonstrated favorable druggability and in silico ADMET properties over a diverse array of molecular computing descriptors. The LPs show promising prospects in the disruption of S2RMT capping machinery in silico. However, these LPs should be validated via in vitro and in vivo experimental models.
Keywords: Coronavirus, SARS-CoV-2, 2-O’-ribosemethyltransferase, Phytochemicals, Molecular docking, Molecular dynamics, Mulberrofuran F
Graphical abstract

1. Introduction
The coronavirus disease-19 (COVID-19), was classified a worrisome global pandemic by the World Health Organization (WHO), following the virulent infection rate of Severe Acute Respiratory Syndrome Coronavirus 2 – (SARS-CoV-2) in humans [1]. Recent epidemiological findings present a cumulative total of about over 246 million confirmed cases and 5 million deaths have been reported since the start of the outbreak [2]. The SARS-CoV-2 belongs to one of the two zoonotic coronaviruses, the other ones being the Middle East Respiratory Syndrome Coronavirus (MERS-CoV). MERS-CoV and SARS-CoV have engendered severe respiratory disorder in mankind since the 21st century commenced [3]. SARS-CoV-2 has been described to be part of the most virulent viruses of this century, with the most fatalities till date [4]. Coronaviruses are described as rapidly evolving viruses, with a high rate of genomic mutation [5]. Recently, several variants of SARS-CoV-2 have been identified: the United Kingdom (UK), South Africa and Brazil variants are cited in the several literatures as (B.1.1.7 for UK, 501Y.V2 or 20C/501Y.V2B.1.351 for South Africa and P.1 for Brazil variants) [6]. This, along with a high infection rate has made the development of drugs quite elusive. Like the earlier coronaviruses, SARS-CoV-2 makes use of its cell environment for its replication and survival [7]. The viral RNA maintains its integrity through the “cap”, a unique organization towards the 5‘ end of the RNA molecule which comprise of a C-2′-O-methyl-ribosyladenine and N-methylated guanosine triphosphate; an arrangement similar to the host cell's RNA [8,9]. The “cap” structure plays significant function in pre-mRNA splicing, mRNA export, RNA stability and escaping the cellular innate immune system [10]. However, in humans, the cap is established in the nucleus of the cell, on the newly transcribed RNA to which the virus has no access. Consequently, they have to own their specified cap-synthesizing enzymes [9]. The last methylation step that cumulates into the RNA cap requires two enzymes, non-structural proteins (nsp) 14 and 16. The nsp 14 for GTP nucleobase, N-7 methylation while the nsp 16 for C-2′-O methylation of the following nucleotide. Both enzymes are methyltransferases (MTases) that depend on S-adenosylmethionine (SAM) [11,12]. Nsp14 when complexed to nsp10 has been reported to reduce cases of mismatched nucleotides via its exoribonuclease domain (ExoN) [13]. The 2′-O-ribose methyltransferase (2′-O-MTase) activity of nsp16 is also influenced by the enzyme's association with nsp10 [13], The activity of nsp16 functioning as a 2′-O-ribose methyltransferase (2′-O-MTase) is also influenced by its association with nsp10 [14,15]. These properties indicate nsp14 and nsp16 as promising therapeutic targets for SARS-CoV-2, especially nsp16 being a very promising molecular target for structural drug design. The 2′-O methyltransferase (MTase) is also essential for coronaviruses replication (in cell cultures) [16,17]. Identifying bioactive compounds with therapeutic activities against these targets is a necessary step to designing potent antiviral agents. Initial large-scale screening of bio-active agents capable of inhibiting target proteins, using bioinformatics tools have been variously reported [18], [19], [20], [21], [22], [23]. The use of plants and their parts, ‘herbal remedies’, in traditional medicine has been well documented. These plants are used as concoctions, decoctions, infusions etc. Indeed, the efficacy of many of these remedies has been attributed to their bioactive phyotcompounds [24]. Compounds derived from plants have been known to possess enormous structural diversity that has served as good starting points for investigating new drug [25].
There are several reports that focuses on the use of computation methods to screen different databases and libraries of natural compounds for potential inhibitors of several targets of SARS-CoV-2, this information have been compiled in some reviews [26,27]. Though there are few reports that targets 2′-O-MTase as a viable therapeutic target [28,29], there is no report on the repurposing of antimalarial compounds against SARS-CoV-2 2′-O-MTase
The inhibitory potential of phytocompounds against viral methyltransferases have been well documented [30,31]. This approach can be exploited in the quest for inhibitors of important targets against the novel SARS-CoV-2. In the wake of the ravaging (and still evolving) effect of the COVID-19 pandemic, the dearth of effective anti-viral drugs, and the relatively long process of drug discovery, computational simulation techniques has been a viable tool employed to study the evolving mutations [32,33], and for screening possible novel drug candidates [34], [35], [36], [37], [38]. In this study, we employ computational techniques to predict the interactions of a list of bioactive phytocompounds (BP) that were compiled from literature search and known to be derived from African medicinal plants against SARS-CoV-2 and other coronaviruses 2′-O-MTase.
2. Methods
2.1. Retrieval and preparation of protein structure for molecular docking
The 3D structure of nsp-16/10 of SARS-CoV-2 complexed with its native substrate (PDBID: 6WRZ), and previous viruses SARS-CoV (PDB ID: 3R24) and MERS-CoV (PDB ID: 5YNB), were retrieved from the Protein Data Bank (http://www.rcsb.org). Existing ligands and water molecules associated with the protein structures were removed and missing hydrogen atoms were added. Using MGL-AutoDockTools (ADT, v1.5.6), the Kollamn charges were added as the partial atomic charge [39]. The non-polar hydrogens were merged while the polar hydrogens were added to the proteins. This procedure was applied to all proteins and then saved into a dockable pdbqt format for docking calculations.
2.2. Ligand preparation for molecular docking
The structure data format (SDF) of the reference inhibitors (sinefungin and S-adenosyl-L-homocysteine (SAH)) and 226 phytocompounds were downloaded from the PubChem database (www.pubchem.ncbi.nlm.nih.gov). These ligands were converted to mol2 using Open babel [40]. Compounds that were unavailable on the database were drawn using ChemDraw version 19, the same was converted to mol2 chemical format.
2.3. Virtual screening and active site targeted molecular docking of phytocompounds
The screening of the 226 bioactive compounds against SARS-CoV-2 2′-O-ribose methyltransferase (S2RMT) was performed using AutoDock Vina [41].]. Based on the docking scores, interaction in the catalytic site and binding poses, 26 hit phytocompounds were selected. These hit phytocompounds were docked for interaction with the active pockets of the S2RMT of other strains (SARS-CoV and MERS-CoV). For all the docking
In OpenBabel that is incorporated into PyRx 0.8. the Universal Force Field (UFF) was used as the energy minimization parameter and conjugate gradient descent as the optimization algorithm. The energy of all the ligands were minimized using conjugate gradient descent as the optimization algorithm in OpenBabel that is incorporated into PyRx 0.8. The active sites of the three enzymes were defined by the grid boxes and presented in Table 1 . All other parameters were kept as default.
Table 1.
Binding site coordinates of nsp16 protease of coronaviruses.
| Dimensions | SARS-CoV-2 (Å) | SARS-CoV (Å) | MERS-CoV (Å) |
|---|---|---|---|
| Center_x | 89.26 | 54.25 | 89.26 |
| Center_y | 16.92 | 60.82 | 16.92 |
| Center_z | 26.44 | 65.17 | 26.44 |
| Size x | 31.00 | 26.34 | 31.00 |
| Size y | 29.63 | 25.41 | 29.63 |
| Size z | 31.34 | 20.81 | 31.34 |
2.4. Molecular dynamics simulation
Desmond module of Schrodinger 2019–4 was employed for the MD simulation of the LPs-S2RMT. Water boxes were added to the proteins subsequent to addition of the missing hydrogen atoms and removal of any ligand in the TIP3P molecules solvent system [42] under orthorhombic periodic boundary conditions for 10 Å, buffer region with OPLS3 force field. An isothermal–isobaric ensemble (constant number of particles N, constant pressure P and constant temperature T) which is an ensemble of Nose-Hoover thermostat [43] and barostat was applied to maintain the constant temperature (310 K) and pressure (1 bar) of the systems, respectively. An energy minimization of 1000 steps with steepest descent followed by conjugate gradient algorithms was utilized. The Parameters such as temperature, salt concentration, and pH were set at the physiological values (310 K, 0.154 M NaCl and 7.0, respectively) during the simulation period. Multiple time step RESPA integration (Reference System Propagator Algorithms) was used in the dynamics study for bonded, near and far-bonded interactions with 2, 2 and 6 fs, respectively. The data were collected for every 100 ps, and the obtained trajectory was analyzed with Maestro graphical interphase (Schrödinger Release 2021–1: Maestro, Schrödinger, LLC, New York, NY, 2021). Various structural parameters, like Root Mean Square Deviation (RMSD), Root Mean Square Fluctuations (RMSF), Radius of Gyration (rGyr), Intramolecular Hydrogen Bonds (intraHB), Molecular Surface Area (MolSA), Solvent Accessible Surface Area (SASA) and Polar Surface Area (PSA) were calculated as a function of time to explore the structural behavior of the proteins, ligands and protein-ligand complexes. In order to estimate the free energy change that describes the binding of these LPs through the MD trajectories, MM-GBSA calculations were carried out, and free energy estimations were computed for 11 snapshots (one every 10 ns as shown in Fig. S19: supplementary data).
2.5. PCA and FEL analysis and covariance matrix generation
PCA and FEL analysis and covariance matrix generation were performed through covar and anaeig GROMACS modules with Desmond MD trajectories and represented by matplotlib Python library.
2.6. Physicochemical and pharmacokinetic study
The LPs for S2RMT were submitted for drug-likeness and ADMET filtering analysis. The SwissADME webserver (http://www.swissadme.ch/index.php) was used to analysis the drug-likeness using the Lipinski and Veber filtering tools [44]. Several molecular descriptors on the SuperPred webserver (http://lmmd.ecust.edu.cn/admetsar1/predict/) was used to analysis the predicted Absorption, Distribution, Metabolism, Excretion and toxicity (ADME/tox [45]. The canonical SMILES of the LPs were used for the analysis.
3. Results
3.1. Molecular docking of phytocompounds with the target protein
The virtual screening of 226 bioactive phytocompounds from African medicinal plants against S2RMT demonstrated varying degrees of estimated binding energies as presented in Table S2: supplementary data. From the results obtained, a hit list of 26 BP with binding affinities higher than the reference inhibitors, Sinefungin (−7.7 Kcal/mol) and SAH (−8.2 Kcal/mol), with notable interaction with the catalytic residues. The interactions of the top 26 ranked BP with S2RMT were further compared with those of SARS-CoV and MERS-CoV2’-O-MTase. From these analyzes, the four lead phytocompounds (LPs) with the highest binding affinity to the S2RMT were further selected viz: mulberrofuran F, a flavonoid; 24-methylene cycloartenol ferulate, a pentacyclic triterpenes; 10′-hydroxyusambarensine, an indole alkaloid; and 3-benzoylhosloppone, an abietane diterpenes (Table 2 ) with quantified free binding energy of (−10.7, −10.1, −9.4 and −9.2 Kcal/mol, respectively). The LPs interacted with SARS-CoV and MERS-CoV 2′-O-MTase with binding affinities of (−9.4, −8.9, −10.5 and −9.6 Kcal/mol) and (−8.7, −8.9, −10.1, and −9.9 Kcal/mol), respectively (Fig. 1 ). It was observed the Mulberrofuran F (−10.7 Kcal/mol) the topmost ranked phytocompound to the S2RMT displayed a lower binding affinity of −9.2 and 8.7 Kcal/mol to SAR CoV and MERS-CoV2’-O-MTase. On the other hand, 10-Hydroxyusambarensine demonstrated the highest binding affinity to SARS-CoV and MERS-CoV 2′-O-MTase (−10.5 and −10.1 Kcal/mol, respectively). Thus, the compounds displayed selectivity for different strain of the coronaviruses base on their affinity. The structural stability of the S2RMT complexed with the LPs was analyzed through MD simulations.
Table 2.
Structure of reference inhibitors (sinefungin and SAH) and the top docked BP with the active site residues of SAR CoV-2 2′-O-MTase.
| S/No | Bioactive Compounds | Class of compound | Plant species (Family) | |
|---|---|---|---|---|
| S1 | Sinefungin | 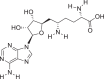 |
Nucleoside | |
| S2 | S-adenosyl-l-homocysteine(SAH) | 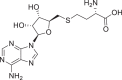 |
Nucleoside | |
| 1 | Mulberrofuran F | 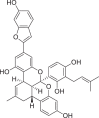 |
Isoprenylated flavonoids | Morusmesozygia (Moraceae) |
| 2 | 24-Methylene cycloartenol ferulate | 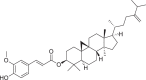 |
Pentacyclic triterpenes | Entandrophrag maangolense (Meliaceae) |
| 3 | 10 -Hydroxyusambarensine | 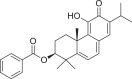 |
Indole alkaloids | Strychnosus ambarensis (Loganiaceae) |
| 4 | 3- Benzoylhosloppone | 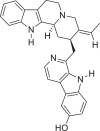 |
Abietane diterpenes | Hoslundiaopposita (Lamiaceae) |
Fig. 1.

Binding energies of the ten lead phytocompounds from the docking analysis of 226 phytocompounds and reference compounds docked to the active site of coronaviruses 2-O-methyltransferase. The red dotted line shows the top 4 docked compounds. 168 = 2, 3, 19 -trihydroxy-urs-12–20-en-28-oic acid.
3.2. Molecular interactions between the lead phytocompounds and coronaviruses 2′-O-MTase
The interactions of the LPs with the amino acid residues of coronaviruses 2′-O-MTase is given in Table 3 . The active site directed docking of sinefungin and SAH to SARS-CoV-2, SARS-CoV and MERS-CoV 2′-O-MTase revealed that it actively interacted with the catalytic site residues majorly through conventional hydrogen-bond, in most cases the reference compounds served as H-Donor from its hydrozyl or amino group. In the three coronaviruses 2′-O-MTase, sinefungin and SAH were docked into the substrate binding cavity as SAM (Fig. 2 ). In the same manner mulberrofuran F, 24-methylene cycloartenol ferulate, 10-hydroxyusambarensine and 3-benzoylhosloppone interacted with the catalytic residues of the substrate binding pocket (SBP) that is localized in a canyon of the three coronaviruses [46]. Mulberrofuran F interacted via a hydrogen bond to ASP6897 which is part of the amino acid involved in methionine binding in the pocket of the second subdivision of the SBP [46]. The benzofuran-2-yl ring of Mulberrofuran F was responsible for the hydrophobic interaction with the SBP via Pi-Pi, T-shaped, Pi-alkyl and alkyl interactions (Table 3 and Fig. 2). 24-Methylene cycloartenol ferulate interacted with both the nucleoside and amino acid (methionine binding) pocket sub unit of SAM binding cleft of S2RMT. The same binding pattern was observed with SARS-CoV and MERS-CoV 2′-O-MTase. The 3-O-feruloyl moiety was directed into the cleft of S2RMT, interacting via H-bonds, while the cycloartenol moiety interacted with the nucleoside binding residue via Pi-alkyl and alkyl hydrophobic interaction (Fig. 2). The hydroxyl and amino group of the 9H-pyrido[3,4-b]indol-6-ol moiety of 10-Hydroxyusambarensine interacted with catalytic residues of S2RMT such as ASP6897 and ASP6912 via hydrogen bonds while the 1H-indolo[2,3-a]quinolizine moiety formed most of the hydrophobic (P-alkyl and alkyl) interactions. A Pi-Sigma interaction with PRO6932 alongside carbon hydrogen bonds were formed between the indolopyridocoline moiety and the SAM binding residues (ASN6899 and GLY6871) of S2RMT (Fig. 2 and Table 3). For 3- Benzoylhosloppone the carboyl and hydroxyl group were responsible for the conventional and carbon hydrogen bonds to the catalytic residues S2RMT. The benzoyl ring interacted via Pi-alkyl and alkyl interactions to MET6929 and LEU6898 residues belonging to the amino acid (methionine binding) pocket a subunit of the SAM binding cleft of S2RMT. In a similar binding mode as the reference compounds, it was further observed that the LPs interacted with SARS-CoV and MERS-CoV2’-O-MTase in similar binding pattern as with S2RMT. They were docked into the ligand binding cleft of the two proteins and interacted with the catalytic and substrate binding residues (Fig. 2, Fig. 3, Fig. 4 ). The interacting amino acids are represented in Table 3.
Table 3.
Interactions of top docked compounds and reference inhibitors with active site residues of coronaviruses 2′-O-MTase.
| Compounds | Coronavirus | Hydrogen bonds (Bond distance) | Other interactions |
|---|---|---|---|
| Sinefungin | SARS-Cov-2 | ||
| TYR6930(3.36) ASP6912(3.00, 2.97) ASP6897(2.29) SER6872 MET6929(3.37) TYR6930 (2.01) ASP6928(2.71, 2.83, 2.01) GLY6869(2.78) ASP6897(2.61, 2.70) CYS6913(2.91) | CYS6913MET6929 | ||
| S-Adenosyl-l-Homocysteine | TYR6930(3.29) ASP6912(3.00, 2.79) ASP6897(2.29) ASP6928(2.65, 2.83, 2.01) GLY6869(2.36) ASP6897(1.98) | SER6872 MET6929 ASP6897 TYR6930 | |
| Mulberrofuran F | SER6872(2.41) GLY6869 (2.13) ASP6897(2.80) | ASP6897 ASP6899 PHE6947 PRO6932 LEU6898 MET6929 CYS6913 | |
| 24-Methylene cycloartenol ferulate | SER6999(3.31) GLU6971(2.81) HIS6972(3.37) TYR6930(3.56) THR6934(3.56) LYS6935(3.72) SER7000 (3.50) | LYS6935 LEU6898 MET6929TYR6930 HIS6972 | |
| 10 -Hydroxyusambarensine | ASP6897(2.27) TYR6930(2.16) ASP6912(2.54) ASN6899(3.60) | PRO6932 MET6929 PHE6947 LEU6898 | |
| 3- Benzoylhosloppone | LYS6968(3.05) ASP6928(2.79) GLY6869(3.61) ASP6897(3.76) | TYR6930 PRO6932 LEY6898 MET6929 | |
| Sinefungin | SARS-COV | GLY75(2.77) GLY71(2.39) ASP73(2.54) ASP130 (2.18, 1.87) SER74(3.25) ASP99(3.32) TYR132(3.23) | LEU100 (2.95, 3.52,4.80) |
| S-Adenosyl-l-Homocysteine | TYR132(3.22) ASP114(2.65) ASP130(2.13) ASN43(2.70) ASP99(2.11) | SER74 PRO80 ASP99 TYR132 | |
| Mulberrofuran F | SER201(2.73) SER202(3.21) GLY71(2.40) | TYR132 PRO134 MET42 | |
| 24-Methylene cycloartenol ferulate | ASP133(2.04) | GLY71 ASP114 PRO134 TYR132 LEU100 | |
| 10 -Hydroxyusambarensine | ASP99(2.41) GLY71(2.35) | ASP75 ASP99 ASP130 LEU100 PRO134 TYR132 MET131 CYS115 | |
| 3- Benzoylhosloppone | LYS170(2.81) ASP130(2.60) | GLY71 PRO134 LEU100MET131 | |
| Sinefungin | MERS-CoV | ASN101(2.07) ASN98 (2.51) ASP75(3.02) ASP99(1.92, 3.36) ASP130(3.72) GLY71 (2.35) GLY73 (3.72, 3.133) | LYS170 ASN43 |
| S-Adenosyl-l-Homocysteine | ASN43(2.48) TYR47(2.78) GLY81(2.99) CYS111(2.96) GLY71(2.81) ASP130(2.85) GLY73(2.98) ASP99(2.75) | MET131(3.60) LEU100 (3.60) | |
| Mulberrofuran F | TYR132(2.74) ASN101(3.72) ASP99(3.72) | PRO134MET131ASP75 PHE149LEU100 | |
| 24-Methylene cycloartenol ferulate | ASP114(3.44) TYR132(2.74) LEU100 (3.25) | PRO134 PHE149 LEU100 LYS76 HIS41 CYS115 | |
| 10 -Hydroxyusambarensine | TYR132(2.54) ASP114(3.07) ASP99(2.37) LEU100 (3.75) | PRO134 MET131 PHE149 | |
| 3- Benzoylhosloppone | TYR132(2.74) GLY71(3.25) ASP99(3.72) ASP130(3.72) LEU130(3.72) LYS170 | MET131 PHE149 | |
Fig. 2.

Amino acid interactions of top lead phytocompounds from the docking analysis and reference inhibitors in substrate binding cavity SARS-CoV-2 2′-O-MTase. (S) solvent-accessible surface view. The top four ranked phytocompounds in sticks representation are represented by colors: (a) cyan: sinefungin (b) orange: SAM (c) gold: mulberrofuran F (d) red: 24-methylene cycloartenol ferulate (e) blue: 10–hydroxyusambarensine (f) Green: 3-benzoylhosloppone. Types of interactions are represented by light purple-dotted line: Green-dotted lines: H-bonds; hydrophobic interactions (Pi-Alkyl, Alkyl and pi-stacking); yellow-dotted lines: purple-dotted line: Pi-Pi T Shaped; Pi-sulfur interactions, pi-stacking interactions, with three-letter abbreviations of amino acids.
Fig. 3.

Amino acid interactions of phytocompounds and reference inhibitors in substrate binding cavity SARS-CoV 2′-O-MTase. (S) solvent-accessible surface view. The top four ranked phytocompounds in sticks representation are represented by colors: (a) cyan: sinefungin (b) orange: SAM (III) gold: mulberrofuran F (d) red: 24-methylene cycloartenol ferulate (e) blue: 10–Hydroxyusambarensine (f) Green: 3-benzoylhosloppone.
Fig. 4.

Amino acid interactions of phytocompounds lead phytocompounds from the docking analysis and reference inhibitors in substrate binding cavity MERS-CoV2’-O-MTase. (S) solvent-accessible surface view. The top four ranked phytocompounds in sticks representation are represented by colors: (a) cyan: sinefungin (b) orange: SAM (c) gold: mulberrofuran F (d) red: 24-methylene cycloartenol ferulate (e) blue: 10–hydroxyusambarensine (f) green: 3-benzoylhosloppone.
3.3. Result from molecular dynamic analysis
An in-depth 100 ns MD simulation was performed on the LPs complexed with S2RMT. In other to access the stability of the bound system and the structural integrity upon the binding of the phytochemicals, the MD simulation trajectories of the complex systems were compared to that of the unbound systems. The following thermodynamic parameters (RMSD, RMSF SASA, RoG, and number of H-bonds) protein secondary structure, ligand properties and protein-ligand contacts were computed from the trajectories, the plots were presented as a function of time frame.
3.3.1. Protein secondary structure
Protein secondary structure elements (SSE) of the S2RMT such as the alpha-helices and beta-strands were monitored throughout the simulation. Fig. 8a shows the SSE distribution by residue index. Fig. 8b summarizes the SSE composition, while Fig. 8c monitors each residue and its SSE assignment over time. The result of the analysis showed that 19.75% was Helix, 15.28% was strands, while 35.05% was Total SSE (Fig. 5 ).
Fig. 8.

The Radius of gyration (RoG) plots of molecular dynamics (MD) simulation of SARS-Cov-2 2′-O-MTase complexed to the four lead phytochemicals from the docking analysis.
Fig. 5.

Secondary structural analysis of SARS-Cov-2 2′-O-MTase during 100 ns MD simulation (a) SSE distribution by residue (b) summary of the SSE composition for each trajectory frame (c) residue and its SSE assignment over time.
3.3.2. Thermodynamic parameters
Root mean square deviation analysis
The RMSD plots for the five systems show that they were equilibrated before 10 ns. The systems exhibited the same progression of RMSD with minimal fluctuation with average RMSD values of 6.83043, 5.674218, 6.124726, 6.369042 and 5.989651 Å for the unbound enzyme, and S2RMT complexed to 10-Hydroxyusambarensine, Mulberrofuran F, 3-Benzoylhosloppone, 24-Methyono cycloartenol, respectively. The binding of the lead phytochemicals reduced the fluctuation in the phytochemical-enzyme complex system; this indicates a more compacted structure upon the binding of the phytochemicals (Fig. 6 ). The LP- S2RMT systems were further analyzed in Figs. S1–S4 (suplemantery data) The Cα shows the RMSD evolution of a protein (left Y-axis). The ligand RMSD (right Y-axis) indicates how stable the LPs are with respect to the S2RMTP and its binding pocket.
Fig. 6.

The Backbone-Root Mean Square Deviation (RMSD) plots of molecular dynamics (MD) simulation of SARS-Cov-2 2′-O-MTase complexed to the four lead phytochemicals from the docking analysis.
Root mean square fluctuation analysis
The RMSF plots reveal the flexibility of the amino acid residues of the protein. Higher fluctuations are observed at the N and C terminal ends of the proteins due to terminal motions. The mean RMSF values for the systems are 2.37097, 2.642513, 3.20722, 2.304798 and 2.51605 Å for the unbound enzyme and S2RMT complexed to 10-Hydroxyusambarensine, Mulberrofuran F, 3-Benzoylhosloppone, 24-Methyono cycloartenol, respectively. The phytochemicals bound SARS-Cov-2 2′-O-MTase complexes displayed higher RMSF values when compared to the unbound enzyme (Fig. 7 ).
Fig. 7.

Per residue Root Mean Square Fluctuations (RMSF) plots of molecular dynamics (MD) simulation of SARS-Cov-2 2′-O-MTase complexed to the four lead phytochemicals from the docking analysis.
The RMSF plots of the LP- S2RMT systems were analyzed to reveal the secondary structure elements (alpha-helical and beta-strand) regions that interacted with the LP. For the 4 LP- S2RMT systems, the highest fluctuation was observed with the amino acid residues close to residue no. 300 and after residue no. 350. These residues weren't involved in interaction with the ligand. A minimal fluctuation was observed with the interacting amino acid residues before amino acid residue no. 150. The catalytic and substrate binding residue were stable throughout the simulation period. (Fig. S5: supplementary data)
The RMSF of the LP with respect to the S2RMT complexes was further analyzed. The atomic breakdown of the LP that corresponds to the 2D structure in the top panel (Fit Ligand on Protein) line shows the ligand fluctuations, with respect to the protein. A large degree of fluctuation was observed during the simulation period in Mulberrofuran F atoms especially around atom no. 33 with respect to the protein, though the internal atoms of the ligands experienced fewer fluctuations (Fig. S6a). For 24-Methylene cycloartenol ferulate-protein complex the highest fluctuation was around atom no. 34 and 44 (Fig. S6b). Atoms around the later interacted via hydrophobic contacts with the binding site residue. In the case of 10–Hydroxyusambarensine-protein complex the atoms were stable with a lesser degree of fluctuations at atom no. 34 a hydroxyl moiety (Fig. S6c). Atoms of the benzoyl ring moiety of 3- Benzoylhosloppone were the most stable, while the alkyl, carbonyl and hydroxyl group on hosloppone moiety caused some level of fluctuations (Fig. S6d).
The radius of gyration (RoG) analysis
The extent of the compactness of the enzyme upon binding of the ligands is measured from the RoG plots and values. A stably folded protein structure presents a steady RoG plot. Fig. 8 shows the RoG plots of the five systems. The plots for the systems show a steady progression with minimal fluctuations. The mean RoG values calculated for the S2RMT systems are 21.94529, 22.34246, 22.72907, 22.32701 and 22.66686 for the unbound enzyme and the enzyme complexed to 10-Hydroxyusambarensine, Mulberrofuran F, 3-Benzoylhosloppone, 24-Methyono cycloartenol, respectively. The unbound and the enzyme complexed to the lead phytochemicals displayed very close mean RoG values indicating compacted systems (Fig. 8).
The surface accessible surface area analysis
The measure of solvent accessible by the surface of the enzymes was computed from the generated SASA values for the systems. Both RoG and SASA plots indicates the level of structural unfolding of proteins with reference to its original structure. Fig. 9 show the SASA plots for the enzymes systems. The average SASA values for the S2RMT systems are 20,326.16, 21,156.28, 21,112.91, 20,899.48 and 20,900.15 for the unbound enzyme and the enzyme complexed to 10-Hydroxyusambarensine, Mulberrofuran F, 3-Benzoylhosloppone, 24-Methyono cycloartenol, respectively (Fig. 9).
Fig. 9.

The Surface Accessible Surface Area (SASA) plots of molecular dynamics (MD) simulation of SARS-Cov-2 2′-O-MTase complexed to the four lead phytochemicals from the docking analysis.
The changes in the number of H-bonds
The average number of hydrogen bonds for the unbound enzyme, 10-Hydroxyusambarensine, Mulberrofuran F, 3-Benzoylhosloppone, 24-Methyono cycloartenol complexes are 53.72927, 46.89011, 48.31968, 50.53147 and 48.3956. In the AChE systems, a slight reduction in average number of hydrogen bond was observed in the complexes when compared to the unbound protein (Fig. 10 ).
Fig. 10.

The changes in the number of H-bonds during the MDS trajectory of SARS-Cov-2 2′-O-MTase complexed to the four lead phytochemicals from the docking analysis.
3.3.3. Protein-ligand contacts
The S2RMT interactions or contacts with the LPs were monitored throughout the simulation. The 2D-trajectory interaction diagram (Fig. 11, Fig. 12, Fig. 13, Fig. 14 ) and the stacked bar interactions plots were categorized by type (Hydrogen Bonds, Hydrophobic, Ionic and Water Bridges) and summarized in Fig. 11, Fig. 12, Fig. 13, Fig. 14. From the total of 21 amino acids contact made, CYS6913 and ASP6873 maintained contact for about 50% and 20% of the simulation time, respectively, via H-Bonds to mulberrofuran F. TYR6930 maintained the highest contact via hydrophobic interaction mulberrofuran F. Almost all the residue maintained some level of contact during the simulation time via water bridges (Fig. 11b). 24-Methylene cycloartenol ferulate made contact with 25 amino acid residues. H-bond contacts were sparsely maintained, while residue like TYR6930, VAL6937 and LEU6898 interacting via hydrophobic interaction-maintained contact for ∼40% 18% and 15% of the simulation time, respectively. Most residues interacting with water bridges maintained some level of prolonged contact (Fig. 12 b). From the total of 18 contacts made, LYS6933, TYR6930 and ASP6912 interacting via H-bond maintained contact for at least 30% of the simulation time with 10–Hydroxyusambarensine. PHE6947 maintained the highest hydrophobic contact time (>50%) with 10–Hydroxyusambarensine Unlike the first 2 compounds 10–Hydroxyusambarensine maintained short ionic contact with ASP6879 and ASP6912. The 2D-trajectory interaction diagram (Fig. 13 a,b) depicts that 3- Benzoylhosloppone maintained a H-bonding with TYR6930 for 66% of the simulation time, while it maintained a hydrophobic contact for about 33% of during the period of simulation with PHE6947 (Fig. 14 a,b).
Fig. 11.

(a) A schematic details of binding groups of mulberrofuran F interacting with the amino acid residues of SARS-Cov-2 2′-O-MTase (S2RMT) during the period of 100 ns MD simulation analysis. Interactions that occured more than 30.0% of the simulation time in the selected trajectory (0.00 through 100.00 ns), are shown (b) simulation interactions plot showing categorized S2RMT- mulberrofuran F interactions.
Fig. 12.

(a)A schematic details of binding groups of 24-Methylene cycloartenol ferulate interacting with the amino acid residues of SARS-Cov-2 2′-O-MTase (S2RMT) during the period of 100 ns MD simulation analysis. Interactions that occured more than 30.0% of the simulation time in the selected trajectory (0.00 through 100.00 ns) are shown (b) simulation interactions plot showing categorized S2RMT- 24-Methylene cycloartenol ferulate interactions.
Fig. 13.

(a) A schematic details of binding groups of 10 -Hydroxyusambarensine interacting with the amino acid residues of SARS-Cov-2 2′-O-MTase (S2RMT) during the period of 100 ns MD simulation analysis. Interactions that occurred more than 30.0% of the simulation time in the selected trajectory (0.00 through 100.00 ns) are shown (b) simulation interactions plot showing categorized S2RMT-10 -Hydroxyusambarensine interactions.
Fig. 14.

(a) A schematic details of binding groups of 3-Benzoylhosloppone interacting with the amino acid residues of SARS-Cov-2 2′-O-MTase (S2RMT) during the period of 100 ns MD simulation analysis. Interactions that occurred more than 30.0% of the simulation time in the selected trajectory (0.00 through 100.00 ns) are shown (b) simulation interactions plot showing categorized S2RMT-3-Benzoylhosloppone interactions.
A timeline representation of the interactions and contacts (H-bonds, Hydrophobic, Ionic, Water bridges) summarized in Figs. S11–S14 is presented in the supplementary data.
3.3.4. Ligand properties
The LPs properties analyzed on its reference conformation. From the plots (Figs. S7a–10a), we observed stable RMSD fluctuations (<3.0 Å) for most cases indicating no huge dynamical alterations during the course of simulations. All the compounds in ligand-protein systems showed a stable rGyr profile, suggesting no conformational alterations (expansion or compression) (Fig. S7b–S10b). Except for mulberrofuran F the other three displayed no intramolecular hydrogen bond during the simulation run (Figs. S7c–S10c). The MolSA, SASA and PSA plots for all the four compounds during the simulation run showed minimal fluctuations, indicating an impressively stable complex upon the binding of compounds to the active sites of the protein. Other ligand properties such as the Ligand Torsion Profile (LTP) was analyzed, the results are presented in the Figs. S15–S18 (supplementary data). The ligand torsions plot summarizes the conformational evolution of every rotatable bond (RB) in the ligand throughout the simulation trajectory (0.00 through 100.00 ns). The 2D schematic of a ligand, rotatable bonds, conformation and torsion are represented in the Figs. S15–S18 (supplementary Data).
3.3.5. MM-GBSA method for estimating phytocompound binding free energy
The computed free energy estimations for 11 snapshots (one every 10 ns) are summarized in average values and their standard deviation in Table 4 . All the LPs yielded dG values that collaborates the docking analysis, with Mulberrofuran F possessing the highest binding free energy (dG). The evolution of the binding free energy for the four systems is graphically presented as a function of the time during the simulation (Fig. S19: supplementary data).
Table 4.
MMGBSA obtained dG average values and their standard deviation for the four studied compounds.
| Compound | RMSD value at 100 ns (Å) | dG Average (kcal/mol) | dG Standard deviation |
|---|---|---|---|
| 10-Hydroxyusambarensine | 9.618 | −112.4300034 | 21.67643475 |
| Mulberrofuran F | 6.735 | −140.1412904 | 18.02256363 |
| 3-Benzoylhosloppone | 4.481 | −132.1901051 | 12.52935498 |
| 24-Methyono cycloartenol | 6.782 | −139.3845749 | 26.35954092 |
4. Principal component and free energy landscapes analysis
The free energy landscape representations generated by the two first principal components (PC1 and PC2) of the complexes with each one of the inhibitors show similar PCA distribution in the SARS-Cov-2 2′-O-MTase-ligand complexes. Additionally, all of them have differences with the PCA distribution for free protein system. It was observed more different metastable conformations with low-energy states, represented as free energy basins in the blue regions, for those complexes with inhibitor respect to the observed in the free protein. Besides, only one region near to the minimum energy was detected, while rest of complex show more of one metastable region with the minimum value (Fig. 15 ).
Fig. 15.

Free energy landscape (FEL) between first and second principal components (PC1, PC2) graph representation for SARS-Cov-2 2′-O-MTase complexed with (a) mulberrofuran F (b) 24-methylene cycloartenol ferulate (c) 10 –hydroxyusambarensine (d) 3- benzoylhosloppone and (e) without any compound systems.
Regarding the traces of covariance matrix, the most relevant evidence is the difference of trace between free protein (10.7905 nm2) and protein binds with Mulberrofuran F (8.35602 nm2). Thus, these results suggest SARS-Cov-2 2′-O-MTase structure obtains a greater compaction when is binding of Mulberrofuran F due to trace decrease in the complex. The rest of ligands don't show a considerable increase in complex compaction respect protein (Table 5 ).
Table 5.
Trace of the covariance matrix for each SARS-Cov-2 2′-O-MTase-compound complex.
| Compound | Trace of covariance matrix (nm2) |
|---|---|
| No compound (Free protein) | 10.7905 |
| 10-Hydroxyusambarensine (16) | 10.9227 |
| Mulberrofuran F (113) | 8.35602 |
| 3-Benzoylhosloppone (119) | 10.9818 |
| 24-Methyono cycloartenol (164) | 18.2716 |
4.1. Drug-likeness and pharmacokinetic properties of selected compounds
The result for the predictive druglikeness and ADMET filtering analyzes for the LPs presented in Table 6 . For ADMET analyzes, the molecular descriptors used for the filtering included blood brain barrier (BBB) penetration, this who compounds could cross the blood brain barrier, the aqueous solubility (AS), predicts the solubility of each LPs in water at 25 °C. The various cytochrome P450 descriptors were used to assess the cytochrome P450 inhibitory activities of the LPs. The human intestinal absorption (HIA), predicts the intestinal absorption of the LPs after oral administration. The drug likeness and ADMET analysis of 10–hydroxyusambarensine have been reported in our previous paper [18] while the 3 reported herein (mulberrofuran F, 24-methylene cycloartenol ferulate, and 3- benzoylhosloppone) fulfilled the all the requirement for Lipinski analysis with corresponding favorable predicted ADMET parameters. The in silico druglikeness and ADMET properties suggested mulberrofuran F to have low GI absorption, while 24-methylene cycloartenol ferulate, and 3- benzoylhosloppone have high GI absorption. The three compounds had high probability of absorption, subcellular distribution, and low toxicity [47]. The ADMET analysis shows that the LPs have the ability to be absorbed in the human intestine, high aqueous solubility, low acute oral toxicity with a good bioavailability score (Table 6).
Table 6.
Physicochemical properties of the top-binding phytocompounds from African plants to SARS-CoV-2 2′-O-MTase.
| a) Physiochemical properties | Mulberrofuran F | 24-Methylene cycloartenol ferulate | 3- Benzoylhosloppone |
|---|---|---|---|
| Molecular weight (g/mol) | 630.68 | 630.68 | 418.52 |
| Num. heavy atoms | 47 | 44 | 31 |
| Num. arom. heavy atoms | 27 | 6 | 6 |
| Num. rotatable bonds | 4 | 9 | 4 |
| Num. H-bond acceptors | 8 | 4 | 4 |
| Hydrogen bond donor | 5 | 1 | 1 |
| cLogP | 4.55 | 4.55 | 2.61 |
| Molar Refractivity | 179 | 179 | 101.11 |
| TPSA (Ų) | 132.75 | 55.76 | |
| Lipinski violation | 1 | 1 | 0 |
| Drug likeness | |||
| Lipinski | Yes | Yes | Yes |
| Veber | Yes | Yes | Yes |
| Bioavailability Score | 0.55 | 0.17 | 0.55 |
| (b) ADMET SAR | Absorption (Probability) | ||
| Blood-Brain Barrier | BBB+ (0.565) | BBB+ (0.649) | BBB+ (0.835) |
| Human Intestinal Absorption | HIA+ (0.984) | HIA+ (0.973) | HIA+ (0.974) |
| Caco-2 Permeability | Caco2+ (0.577) | Caco2+ (0.745) | Caco2+ (0.678 |
| P-glycoprotein Substrate | Non-substrate (0.727) | Substrate (0.795) | Substrate (0.752) |
| P-glycoprotein Inhibitor | Non-inhibitor (0.656) | Non-inhibitor (0.606) | Non-inhibitor (0.806) |
| Renal Organic Cation Transporter | Non-inhibitor (0.910) | Non-inhibitor (0.797) | Non-inhibitor (0.814) |
| Distribution (Probability) | |||
| Subcellular localization | Mitochondria (0.786) | Mitochondria (0.802) | Mitochondria (0.838) |
| Metabolism | Metabolism | Metabolism | |
| CYP450 2C9 Substrate | Non-substrate (0.780) | Non-substrate (0.758) | Non-substrate (0.777) |
| CYP450 2D6 Substrate | Non-substrate (0.852) | Non-substrate (0.812) | Non-substrate (0.912) |
| CYP450 3A4 Substrate | Non-substrate (0.567) | Non-substrate (0.822) | Non-substrate (0.813 |
| CYP450 1A2 Inhibitor | Non-inhibitor (0.5154) | Non-inhibitor (0.5814) | Non-inhibitor (0.5814) |
| CYP450 2C9 Inhibitor | Non-inhibitor (0.8197) | Non-inhibitor (0.500) | Non-inhibitor (0.539) |
| CYP Inhibitory Promiscuity | Low CYP Inhibitory Promiscuity (0.8818) | Low CYP Inhibitory Promiscuity (0.729) | Low CYP Inhibitory Promiscuity (0.815) |
| Toxicity | |||
| AMES Toxicity | Non-AMES toxic (0.506) | Non-AMES toxic (0.50) | Non-AMES toxic (0.882) |
| Carcinogens | Non-carcinogens (0.934) | Non-carcinogens (0.9712) | Non-carcinogens (0.912) |
| Acute Oral Toxicity | III (0.429) | IV (0.607) | III (0.749) |
| Rat Acute Toxicity LD50, mol/kg | 3.3280 | 1.4139 | 1.9882 |
| Aqueous solubility (LogS) | −4.3480 | −5.8146 | −4.7646 |
| Pharmacokinetics | |||
| GI absorption | low | High | High |
| Log Kp (skin permeation) cm/s | −4.53 | −1.42 | −1.42 |
5. Discussion
SARS-CoV-2 is a virulent and highly evolving virus, whereas the drug discovery process has not matched the increasing therapeutic need of this viral infection [48]. Naturally existing phytocompounds from plants are potential bioactive repositories, including antiviral activity, which, if adequately explored, could provide affordable, accessible and available use as therapeutic agents against coronavirus infections [49]. Like other coronavirus, the SARS-CoV-2 evades host immune detection and reduces the chance of immune response in the incubation period of 2 to 14 days. This evasion of immune detection is projected to be achieved through the modification of viral mRNA by 2′ O-methyltransferase activity of nsp16/nsp10 which enables the virus to escape detection by the host's innate immune mechanism [50]. Compounds that block viral immune evasion through the suppression of viral RNA 2′-O-methylation, will encourage early expression of interferon-stimulated genes which in turn will serve to impede SARS-CoV-2 replication [51]. The 2′-O-Methyl transferase activity of nsp-16 is S-adenosyl-methionine (SAM)-dependent and regulated by nsp10 binding. The binding of SAM induces essential conformational changes, in the enzyme, that favors RNA affinity and methylation [52]. Hence it is expected that compounds that interacts with the SAM binding site may elicit host response against the virus. Structure based drug design has employed molecular docking to predict the binding-conformation of ligands in the binding site of target receptor and the strength of association (binding affinity) [53]. In the present study, we screened 226 bioactive phytocompounds from various African plants against nsp16 of SARS-CoV 2. The docking, interactive and binding free energy analysis identified the LPs (Mulberrofuran F, 24-Methylene cycloartenol ferulate, 10-Hydroxyusambarensine and 3-Benzoylhosloppone) with high potential and selective inhibition of the coronaviruses nsp16 protein. Hydroxyusambarensine is an alkaloid from the roots of Strychnosusambarensis, previously reported as an antimalarial [54]. Mulberrofuran F isolated from Morus alba, has been used to treat hypotension [55,56]. 24-Methylene cycloartenol ferulate, also called γ-Oryzanol (OZ) has been identified in various cereals, including barley, rice bran and corn [57]. It has been reported to exhibit antioxidant, anti-lipidemic, anti-diabetic and neuro-modulatory properties [57,58]. These LPs interacted with the surface residues (Lys-46, Asp-130, Lys-170 and Glu-203) at the bottom of the central groove, thatcatalysis the transfer SAM methyl group within the substrate binding pocket [46]. In all strains of CoV, the catalytic tetrad (Lys46, Asp130, Lys170) and Glu203 are conserved [59], this may have been responsible for the high binding potential to the three CoV understudied. Though the LPs interacted with the catalytic residues in a similar binding pattern as the SAH (the product of methylation of SAM) and sinefugine (a known inhibitor), they interacted with a stronger binding affinity than these compounds. Thus, these compounds may be able to bind to the S2RMT tightly and hence compromise the RNA methylation function of the enzyme, this will in turn, disrupt the capping machinery, prevent evasion of recognition by the host innate immune system [60], [61], [62] and preclude the viruses from resisting the IFN-mediated antiviral response [10,16]. To further understand the dynamic behavior of the LPs at the binding site of S2RMT, MD simulation was employed [63]. The binding patterns and per-residue amino acid interactions of the LPs-S2RMT complexes in the dynamic state collaborated with those done from the static docking analysis. The various thermodynamics parameters that were analyzed from the 100 ns atomistic MDS trajectory files of the LPs-S2RMT complexes revealed stable complexes that can be adapted into other forms of experiments. The comparison of the RMSD plots for the complex systems shows that the binding of the LPs to S2RMT did not cause any structure deformation in the protein [64]. From the RMSF plots analysis of the four systems, the higher fluctuation that was observed with the interacting residues is consistent with previous reports, where higher structural fluctuations occurred in ligand binding sites of catalytic loop regions [65]. The RoG and SASA plots of all the systems did not show fluctuation above the optimum of >2 Å further indicating that the structural integrity of the proteins was preserved [66]. The binding free energy that is measured from the simulation trajectories provides more accurate computation of ligand binding affinities than the static docking analysis [67]. These results were calculated based on the total binding free energy of the complex. In these calculations, the binding free energy (∆Gbind) measures the affinity of a ligand to its target protein. Thus, the ∆Gbind calculations are important to gain in-depth knowledge about the binding modes of the hits in drug design [68]. The results from the binding free energy calculation (MM-GBSA) agreed with that from the docking analysis; further establishing Mulberrofuran F as the most potent phytocompound. Also, from the predictive drug-likeness, pharmacokinetic and ADMET filtering analyzes, the top docked phytocompounds were predicted to be druggable and nontoxic. The result from the filtering analyzes showed descriptors that suggests a favorable ADMET and pharmacokinetic properties. This further indicates the druggable potential of the LPs [69,70]. The LPs displayed properties that suggest their ability to cross the BBB, hence their potential to ensure overall viral clearance in the brain cells [47]. Also, the LPs expressed high possibility of human intestinal absorption and not susceptible to the permeability-glycoprotein (P-gp, a drug efflux pump). Therefore, it is suggested to be well absorbed into the blood stream, subverting the restraining effect of the P-gp to pump compounds back into the intestinal lumen [71].
6. Conclusion
Herein, we have virtually screened a list of 226 bioactive phytocompounds compiled from a literature search of compounds from African medicinal flora with reported bioactivity against infectious diseases (including viral infection). Altogether the top four docked compounds demonstrated higher binding affinity than the reference inhibitors to the coronaviruses 2′-O-MTase, nevertheless, they displayed similar binding pattern as the reference inhibitors. These phytocompounds were identified to interact with important catalytic residue in the substrate binding site of SARS-CoV-2, SARS-CoV and MERS-CoV 2′-O-MTase as the reference inhibitors, hence, they may disrupt the RNA capping machinery, the replication and survival of the viruses. These potential inhibitors of SARS-CoV-2 2′-O-MTase were stable in a simulated dynamic condition and exhibited positive drug-likeness in the ADMET studies, thus, they are well adaptable for a recommended in vitro and in vivo experimental studies as anti-COVID-19 agents.
Funding
This work has been funded by the Fundación Séneca de la Región de Murcia under Project 20,988/PI/18. This research was partially supported by the omputer resources and the technical support provided by Barcelona Supercomputing Center (BCV-2021–1–0010), Poznan Supercomputing Center, the e-infrastructure program of the Research Council of Norway via the supercomputer center of UiT−the Arctic University of Norway, and by the supercomputing infrastructure of the NLHPC (ECM-02), Powered@NLHPC
Institutional review board statement
Not applicable.
Informed consent statement
Not applicable.
Data availability
All data supporting the findings of this study are available within the article and its supplementary materials.
Ethical approval
Not required.
CRediT authorship contribution statement
Gideon A. Gyebi: Conceptualization, Visualization, Writing – original draft, Methodology. Oludare M. Ogunyemi: Methodology, Writing – review & editing. Adedotun A. Adefolalu: Writing – review & editing. Alejandro Rodríguez-Martínez: Methodology. Juan F. López-Pastor: Methodology. Antonio J. Banegas-Luna: Methodology. Horacio Pérez-Sánchez: Supervision. Adegbenro P. Adegunloye: Writing – review & editing. Olalekan B. Ogunro: Writing – review & editing. Saheed O. Afolabi: Writing – review & editing.
Declaration of Competing Interest
The authors declare that they have no competing interests.
Acknowledgements
This work is funded by grants from the Spanish Ministry of Economy and Competitiveness (CTQ2017-87974-R), and by the Fundación Séneca de la Región de Murcia under Project 20988/PI/18. This research was partially supported by the supercomputing infrastructure of Poznan Supercomputing Center, the e-infrastructure program of the Research Council of Norway via the supercomputer center of UiT−the Arctic University of Norway, and by the supercomputing infrastructure of the NLHPC (ECM-02), Powered@NLHPC. The authors appreciate the members of the BioNet-AP: Bioinformatics Network for African Phytomedicine.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.molstruc.2022.133019.
Appendix. Supplementary materials
References
- 1.Li X., Wang W., Zhao X., Zai J., Zhao Q., Li Y., Chaillon A. Transmission dynamics and evolutionary history of 2019-nCoV. J. Med. Virol. 2020;92(5):501–511. doi: 10.1002/jmv.25701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization . World Health Organization; 2021. Coronavirus Disease (COVID-19) Weekly Epidemiological. https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19—19-october-2021. [Google Scholar]
- 3.da Costa V.G., Moreli M.L., Saivish M.V. The emergence of SARS, MERS and novel SARS-2 coronaviruses in the 21st century. Arch. Virol. 2020:1–10. doi: 10.1007/s00705-020-04628-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou P., YANG X., wANG X., HU B., w Z.H.A.N.G., Hr Si, y zHu, li B., cl HuanG, cHen H.D., cHen J., luo y, Guo H., rD JianG, liu M.Q., y cHen, Xr SHen, WanG X., zHenG X.S., zHao K., cHen Q.J., DenG F., ll liu, yan B., zHan F.X., yy WanG, Xiao G.F., zl SHi. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen Y.W., Yiu C.-P.B., Wong K.-Y. Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CL pro) structure: virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000Research. 2020;9(129) doi: 10.12688/f1000research.22457.1. [DOI] [PMC free article] [PubMed] [Google Scholar]; eCollection.
- 6.Horby P., Huntley C., Davies N., Edmunds J., Ferguson N., Medley G., Semple C. NERVTAG; 2021. NERVTAG Note On B. 1.1. 7 Severity. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/955239/NERVTAG_paper_on_variant_of_concern__VOC__B 1(7) [Google Scholar]
- 7.Ziebuhr J. The Nidoviruses. Springer; 2006. The coronavirus replicase: insights into a sophisticated enzyme machinery; pp. 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bradrick S.S. Causes and consequences of flavivirus RNA methylation. Front. Microbiol. 2017;8:2374. doi: 10.3389/fmicb.2017.02374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramanathan A., Robb G.B., Chan S.-H. mRNA capping: biological functions and applications. Nucleic Acids Res. 2016;44(16):7511–7526. doi: 10.1093/nar/gkw551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nallagatla S.R., Toroney R., Bevilacqua P.C. A brilliant disguise for self RNA: 5’-end and internal modifications of primary transcripts suppress elements of innate immunity. RNA Biol. 2008;5(3):140–144. doi: 10.4161/rna.5.3.6839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Y., Cai H., Xiang N., Tien P., Ahola T., Guo D. Functional screen reveals SARS coronavirus nonstructural protein nsp14 as a novel cap N7 methyltransferase. Proc. Natl. Acad. Sci. 2009;106(9):3484–3489. doi: 10.1073/pnas.0808790106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bouvet M., Imbert I., Subissi L., Gluais L., Canard B., Decroly E. RNA 3′-end mismatch excision by the severe acute respiratory syndrome coronavirus nonstructural protein nsp10/nsp14 exoribonuclease complex. Proc. Natl. Acad. Sci. 2012;109(24):9372–9377. doi: 10.1073/pnas.1201130109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eckerle L.D., Becker M.M., Halpin R.A., Li K., Venter E., Lu X., Scherbakova S., Graham R.L., Baric R.S., Stockwell T.B. Infidelity of SARS-CoV Nsp14-exonuclease mutant virus replication is revealed by complete genome sequencing. PLoS Pathog. 2010;6(5) doi: 10.1371/journal.ppat.1000896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Y., Su C., Ke M., Jin X., Xu L., Zhang Z., Wu A., Sun Y., Yang Z., Tien P. Biochemical and structural insights into the mechanisms of SARS coronavirus RNA ribose 2′-O-methylation by nsp16/nsp10 protein complex. PLoS Pathog. 2011;7(10) doi: 10.1371/journal.ppat.1002294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Decroly E., Debarnot C., Ferron F., Bouvet M., Coutard B., Imbert I., Gluais L., Papageorgiou N., Sharff A., Bricogne G. Crystal structure and functional analysis of the SARS-coronavirus RNA cap 2′-O-methyltransferase nsp10/nsp16 complex. PLoS Pathog. 2011;7(5) doi: 10.1371/journal.ppat.1002059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daffis S., Szretter K.J., Schriewer J., Li J., Youn S., Errett J., Lin T.-Y., Schneller S., Zust R., Dong H. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature. 2010;468(7322):452–456. doi: 10.1038/nature09489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Decroly E., Imbert I., Coutard B., Bouvet M., Selisko B., Alvarez K., Gorbalenya A.E., Snijder E.J., Canard B. Coronavirus nonstructural protein 16 is a cap-0 binding enzyme possessing (nucleoside-2′ O)-methyltransferase activity. J. Virol. 2008;82(16):8071–8084. doi: 10.1128/JVI.00407-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gyebi G.A., Ogunro O.B., Adegunloye A.P., Ogunyemi O.M., Afolabi S.O. Potential inhibitors of coronavirus 3-chymotrypsin-like protease (3CLpro): an in silico screening of alkaloids and terpenoids from African medicinal plants. J. Biomol. Struct. Dyn. 2020;39(9):3396–3408. doi: 10.1080/07391102.2020.1764868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boopathi S., Poma A.B., Kolandaivel P. Novel 2019 coronavirus structure, mechanism of action, antiviral drug promises and rule out against its treatment. J. Biomol. Struct. Dyn. 2020:1–10. doi: 10.1080/07391102.2020.1758788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muralidharan N., Sakthivel R., Velmurugan D., Gromiha M.M. Computational studies of drug repurposing and synergism of lopinavir, oseltamivir and ritonavir binding with SARS-CoV-2 protease against COVID-19. J. Biomol. Struct. Dyn. 2020;39(7):2673–2678. doi: 10.1080/07391102.2020.1752802. [DOI] [PubMed] [Google Scholar]
- 21.Elfiky A.A., Azzam E.B. Novel guanosine derivatives against MERS CoV polymerase: an in silico perspective. J. Biomol. Struct. Dyn. 2020;39(8):2923–2931. doi: 10.1080/07391102.2020.1758789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fox T., Kollman P.A. The application of different solvation and electrostatic models in molecular dynamics simulations of ubiquitin: how well is the X-ray structure “maintained”? Proteins Struct. Funct. Bioinf. 1996;25(3):315–334. doi: 10.1002/(SICI)1097-0134(199607)25:3<315::AID-PROT4>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 23.Gyebi G.A., Adegunloye A.P., Ibrahim I.M., Ogunyemi O.M., Afolabi S.O., Ogunro O.B. Prevention of SARS-CoV-2 cell entry: insight from in silico interaction of drug-like alkaloids with spike glycoprotein, human ACE2, and TMPRSS2. J. Biomol. Struct. Dyn. 2020:1–25. doi: 10.1080/07391102.2020.1835726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petrovska B.B. Historical review of medicinal plants’ usage. Pharmacogn. Rev. 2012;6(11):1. doi: 10.4103/0973-7847.95849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shen B. A new golden age of natural products drug discovery. Cell. 2015;163(6):1297–1300. doi: 10.1016/j.cell.2015.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khan N., Chen X., Geiger J.D. Possible therapeutic use of natural compounds against COVID-19. J. Cell. Signal. 2021;2(1):63–79. [PMC free article] [PubMed] [Google Scholar]
- 27.Xian Y., Zhang J., Bian Z., Zhou H., Zhang Z., Lin Z., Xu H. Bioactive natural compounds against human coronaviruses: a review and perspective. Acta Pharm. Sin. B. 2020;10(7):1163–1174. doi: 10.1016/j.apsb.2020.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sharma K., Morla S., Goyal A., Kumar S. Computational guided drug repurposing for targeting 2′-O-ribose methyltransferase of SARS-CoV-2. Life Sci. 2020;259 doi: 10.1016/j.lfs.2020.118169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang Y., Liu L., Manning M., Bonahoom M., Lotvola A., Yang Z., Yang Z.Q. Structural analysis, virtual screening and molecular simulation to identify potential inhibitors targeting 2′-O-ribose methyltransferase of SARS-CoV-2 coronavirus. J. Biomol. Struct. Dyn. 2020;40(3):1331–1346. doi: 10.1080/07391102.2020.1828172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Naik B., Gupta N., Ojha R., Singh S., Prajapati V.K., Prusty D. High throughput virtual screening reveals SARS-CoV-2 multi-target binding natural compounds to lead instant therapy for COVID-19 treatment. Int. J. Biol. Macromol. 2020;1(160):1–17. doi: 10.1016/j.ijbiomac.2020.05.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saldívar-González F.I., Gómez-García A., Chavez-Ponce de Leon D.E., Sánchez-Cruz N., Ruiz-Rios J., Pilón-Jiménez B.A., Medina-Franco J.L. Inhibitors of DNA methyltransferases from natural sources: a computational perspective. Front. Pharmacol. 2018;9:1144. doi: 10.3389/fphar.2018.01144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen A.T., Altschuler K., Zhan S.H., Chan Y.A., Deverman B.E. COVID-19 CG enables SARS-CoV-2 mutation and lineage tracking by locations and dates of interest. Elife. 2021;10:e63409. doi: 10.7554/eLife.63409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Desai S., Rashmi S., Rane A., Dharavath B., Sawant A., Dutt A. An integrated approach to determine the abundance, mutation rate and phylogeny of the SARS-CoV-2 genome. Brief. Bioinf. 2021;22(2):1065–1075. doi: 10.1093/bib/bbaa437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Padhi A.K., Shukla R., Saudagar P., Tripathi T. High-throughput rational design of the remdesivir binding site in the RdRp of SARS-CoV-2: implications for potential resistance. Iscience. 2021;24(1) doi: 10.1016/j.isci.2020.101992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Padhi A.K., Seal A., Khan J.M., Ahamed M., Tripathi T. Unraveling the mechanism of arbidol binding and inhibition of SARS-CoV-2: insights from atomistic simulations. Eur. J. Pharmacol. 2021;894 doi: 10.1016/j.ejphar.2020.173836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Padhi A.K., Dandapat J., Saudagar P., Uversky V.N., Tripathi T. Interface-based design of the favipiravir-binding site in SARS-CoV-2 RNA-dependent RNA polymerase reveals mutations conferring resistance to chain termination. FEBS Lett. 2021 doi: 10.1002/1873-3468.14182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ogunyemi O.M., Gyebi G.A., Ibrahim I.M., Olaiya C.O., Ocheje J.O., Fabusiwa M.M., Adebayo J.O. Dietary stigmastane-type saponins as promising dual-target directed inhibitors of SARS-CoV-2 proteases: a structure-based screening. RSC Adv. 2021;11(53):33380–33398. doi: 10.1039/d1ra05976a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gyebi G.A., Ogunyemi O.M., Ibrahim I.M., Afolabi S.O., Adebayo J.O. Dual targeting of cytokine storm and viral replication in COVID-19 by plant-derived steroidal pregnanes: an in silico perspective. Comput. Biol. Med. 2021;134 doi: 10.1016/j.compbiomed.2021.104406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30(16):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O'Boyle N.M., Banck M., James C.A., Morley C., Vandermeersch T., Hutchison G.R. Open babel: an open chemical toolbox. J. Cheminform. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trott O., Olson A.J. AutoDock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31(2):455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jorgensen W. Optimized intermolecular potential functions for liquid hydrocarbons. I. Am. Chem. Sot. 1984;106:6638–6646. [Google Scholar]
- 43.Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79(2):926–935. 0. [Google Scholar]
- 44.Martyna G.J., Klein M.L., Tuckerman M. Nosé–Hoover chains: the canonical ensemble via continuous dynamics. J. Chem. Phys. 1992;97(4):2635–2643. [Google Scholar]
- 45.Daina A., Michielin O., Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheng F., Li W., Zhou Y., Shen J., Wu Z., Liu G., Lee P.W., Tang Y. admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model. 2012;52(11):3099–3105. doi: 10.1021/ci300367a. [DOI] [PubMed] [Google Scholar]
- 47.Krafcikova P., Silhan J., Nencka R., Boura E. Structural analysis of the SARS-CoV-2 methyltransferase complex involved in RNA cap creation bound to sinefungin. Nat. Commun. 2020;11(1):1–7. doi: 10.1038/s41467-020-17495-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zanin L., Saraceno G., Panciani P.P., Renisi G., Signorini L., Migliorati K., Fontanella M.M. SARS-CoV-2 can induce brain and spine demyelinating lesions. Acta Neurochir. 2020:1–4. doi: 10.1007/s00701-020-04374-x. (Wien) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu C., Liu Y., Yang Y., Zhang P., Zhong W., Wang Y., Wang Q., Xu Y., Li M., Li X. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B. 2020;10(5):766–788. doi: 10.1016/j.apsb.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mani J.S., Johnson J.B., Steel J.C., Broszczak D.A., Neilsen P.M., Walsh K.B., Naiker M. Natural product-derived phytochemicals as potential agents against coronaviruses: a review. Virus Res. 2020 doi: 10.1016/j.virusres.2020.197989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hyde J.L., Diamond M.S. Innate immune restriction and antagonism of viral RNA lacking 2׳-O methylation. Virology. 2015;479:66–74. doi: 10.1016/j.virol.2015.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Encinar J.A., Menendez J.A. Potential drugs targeting early innate immune evasion of sars-coronavirus 2 via 2’-O-methylation of viral RNA. Viruses. 2020;12(5):525. doi: 10.3390/v12050525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Aouadi W., Blanjoie A., Vasseur J.-J., Debart F., Canard B., Decroly E. Binding of the methyl donor S-adenosyl-l-methionine to middle east respiratory syndrome coronavirus 2′-O-methyltransferase nsp16 promotes recruitment of the allosteric activator nsp10. J. Virol. 2017;91(5) doi: 10.1128/JVI.02217-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meng X.-Y., Zhang H.-X., Mezei M., Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011;7(2):146–157. doi: 10.2174/157340911795677602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Frédérich M., Tits M., Hayette M.-P., Brandt V., Penelle J., DeMol P., Llabres G., Angenot L. 10 ‘-Hydroxyusambarensine, a new antimalarial bisindole alkaloid from the roots of strychnos u sambarensis. J. Nat. Prod. 1999;62(4):619–621. doi: 10.1021/np980375m. [DOI] [PubMed] [Google Scholar]
- 56.Fukai T., Hano Y., HIRAKURA K., NOMURA T., Uzawa J., FUKUSHIMA K. Structures of two natural hypotensive diels-alder type adducts, mulberrofurans f and g, from the cultivated mulberry tree (morus lhou koidz.) Chem. Pharm. Bull. 1985;33(8):3195–3204. doi: 10.1248/cpb.33.3195. [DOI] [PubMed] [Google Scholar]
- 57.Tang W., Eisenbrand G. Springer; 1992. Panax Ginseng CA Mey, Chinese drugs of Plant Origin; pp. 711–737. [Google Scholar]
- 58.Ito J., Sawada K., Ogura Y., Xinyi F., Rahmania H., Mohri T., Kohyama N., Kwon E., Eitsuka T., Hashimoto H. Definitive evidence of the presence of 24-methylenecycloartanyl ferulate and 24-methylenecycloartanyl caffeate in barley. Sci. Rep. 2019;9(1):1–11. doi: 10.1038/s41598-019-48985-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ishihara M., Ito Y., Nakakita T., Maehama T., Hieda S., Yamamoto K., Ueno N. Clinical effect of gamma-oryzanol on climacteric disturbance-on serum lipid peroxides (author's transl) Nihon Sanka Fujinka Gakkai Zasshi. 1982;34(2):243–251. [PubMed] [Google Scholar]
- 60.Chen Y., Guo D. Molecular mechanisms of coronavirus RNA capping and methylation. Virol. Sin. 2016;31(1):3–11. doi: 10.1007/s12250-016-3726-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hornung V., Ellegast J., Kim S., Brzózka K., Jung A., Kato H., Poeck H., Akira S., Conzelmann K.-K., Schlee M. 5′-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314(5801):994–997. doi: 10.1126/science.1132505. (New York, N.Y.) [DOI] [PubMed] [Google Scholar]
- 62.Züst R., Cervantes-Barragan L., Habjan M., Maier R., Neuman B.W., Ziebuhr J., Szretter K.J., Baker S.C., Barchet W., Diamond M.S. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol. 2011;12(2):137–143. doi: 10.1038/ni.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Abbas Y.M., Pichlmair A., Górna M.W., Superti-Furga G., Nagar B. Structural basis for viral 5′-PPP-RNA recognition by human IFIT proteins. Nature. 2013;494(7435):60–64. doi: 10.1038/nature11783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhao H., Caflisch A. Molecular dynamics in drug design. Eur. J. Med. Chem. 2015;91:4–14. doi: 10.1016/j.ejmech.2014.08.004. [DOI] [PubMed] [Google Scholar]
- 65.Cheng X., Ivanov I. Molecular dynamics, Computational toxicology. 2012;929:243–285. doi: 10.1007/978-1-62703-050-2_11. [DOI] [PubMed] [Google Scholar]
- 66.Dong Y.W., Liao M.L., Meng X.L., Somero G.N. Structural flexibility and protein adaptation to temperature: molecular dynamics analysis of malate dehydrogenases of marine molluscs. Proc. Natl. Acad. Sci. 2018;115(6):1274–1279. doi: 10.1073/pnas.1718910115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sinha S., Wang S.M. Classification of VUS and unclassified variants in BRCA1 BRCT repeats by molecular dynamics simulation. Comput. Struct. Biotechnol. J. 2020;18:723–736. doi: 10.1016/j.csbj.2020.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Perez A., Morrone J.A., Simmerling C., Dill K.A. Advances in free-energy-based simulations of protein folding and ligand binding. Curr. Opin. Struct. Biol. 2016;36:25–31. doi: 10.1016/j.sbi.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kollman P.A., Massova I., Reyes C., Kuhn B., Huo S., Chong L., Lee M., Lee T., Duan Y., Wang W. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res. 2000;33(12):889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 70.Hughes J.D., Blagg J., Price D.A., Bailey S., DeCrescenzo G.A., Devraj R.V., Ellsworth E., Fobian Y.M., Gibbs M.E., Gilles R.W. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008;18(17):4872–4875. doi: 10.1016/j.bmcl.2008.07.071. [DOI] [PubMed] [Google Scholar]
- 71.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug. Deliv. Rev. 1997;23(1–3):3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the findings of this study are available within the article and its supplementary materials.