

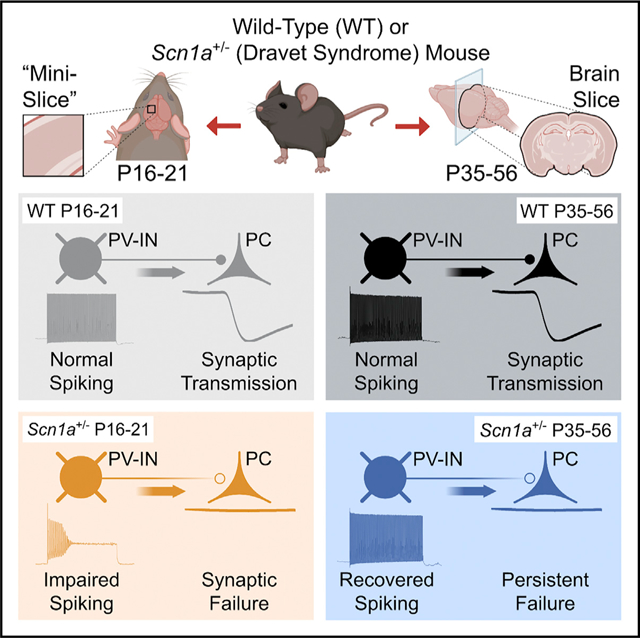
SUMMARY
Dravet syndrome is a neurodevelopmental disorder characterized by epilepsy, intellectual disability, and sudden death due to pathogenic variants in SCN1A with loss of function of the sodium channel subunit Nav1.1. Nav1.1-expressing parvalbumin GABAergic interneurons (PV-INs) from young Scn1a+/− mice show impaired action potential generation. An approach assessing PV-IN function in the same mice at two time points shows impaired spike generation in all Scn1a+/− mice at postnatal days (P) 16–21, whether deceased prior or surviving to P35, with normalization by P35 in surviving mice. However, PV-IN synaptic transmission is dysfunctional in young Scn1a+/− mice that did not survive and in Scn1a+/− mice ≥ P35. Modeling confirms that PV-IN axonal propagation is more sensitive to decreased sodium conductance than spike generation. These results demonstrate dynamic dysfunction in Dravet syndrome: combined abnormalities of PV-IN spike generation and propagation drives early disease severity, while ongoing dysfunction of synaptic transmission contributes to chronic pathology.
In brief
Dravet syndrome is caused by variants in SCN1A with loss of function of Nav1.1 sodium channels. Kaneko et al. use the “mini-slice” to record at two developmental time points. Impaired spike generation of Nav1.1-expressing PV interneurons in Scn1a+/− mice is transient, while abnormalities of PV interneuron synaptic transmission persist.
Graphical Abstract
INTRODUCTION
Dravet syndrome (DS) is a neurodevelopmental disorder characterized by temperature-sensitive seizures, treatment-resistant epilepsy, developmental delay, and intellectual disability with features of autism spectrum disorder (ASD), and increased incidence of sudden unexpected death in epilepsy (SUDEP). DS is due to heterozygous pathogenic loss-of-function variants in the gene SCN1A encoding the voltage-gated sodium (Na+) channel α subunit Nav1.1 (Dravet, 2011; Scheffer, 2012). While new pharmacological therapies for DS have emerged, including cannabidiol (CBD) (Kaplan et al., 2017; Khan et al., 2018) and fenfluramine (Ceulemans et al., 2016; Schoonjans et al., 2017), and antisense oligonucleotide-based approaches may be on the horizon (Han et al., 2020), treatment remains essentially palliative, and there is no cure. Previous studies in experimental mouse models of DS (Scn1a+/− mice) show prominent dysfunction of cerebral cortex GABAergic interneurons (INs) at early time points (second and third postnatal weeks), particularly the parvalbumin (PV)-expressing subclass (PV-INs) (Yu et al., 2006; Ogiwara et al., 2007; Tai et al., 2014; De Stasi et al., 2016). However, recent data suggest that impairments of spike generation may be transient (Favero et al., 2018), which raises the question as to the mechanism of chronic epilepsy, intellectual disability, ASD, and ongoing sudden death in Scn1a+/− mice and DS patients. This finding also has important implications for emerging efforts to increase expression of SCN1A as a potential therapeutic strategy (Colasante et al., 2019; Han et al., 2020; Yamagata et al., 2020).
Accurate assessment of PV-IN function across development is confounded by the fact that most data on PV-IN function and dysfunction in DS derive from recordings from acutely dissociated neurons or brain slices from Scn1a+/− mice before or at onset of epilepsy (prior to postnatal day 21 [P21]); hence, a proportion of Scn1a+/− mice included in prior studies conducted at or before P21 would have been expected to die from status epilepticus or a SUDEP-like phenomenon if not used for experiments. It is possible—and perhaps likely—that mice remaining alive represent a nonrandom subset of the initial population. There is a spectrum of disease severity in Scn1a+/− mice (Hawkins et al., 2016, 2017), similar to that observed in humans (Harkin et al., 2007; Mei et al., 2019), the basis of which is unclear but may involve genetic modifiers (Hawkins and Kearney, 2016; Hawkins et al., 2016; Calhoun et al., 2017). Restated, if we could retrospectively record PV-INs at early time points (P16–P21) in those Scn1a+/− mice where PV-IN function was later determined to be normal (e.g., P35–P56), would these cells be normal or abnormal? Conversely, would PV-INs in those Scn1a+/− mice deceased at early time points (<P35) have exhibited functional recovery if these mice had survived to and been assessed at or beyond P35? Here, we address this critical question using an experimental paradigm to assess PV-IN function in the same mice at both time points (≤P21 and ≥P35) in “mini-slices” from young (P16–P21) Scn1a.PV-Cre.tdTomato mice and then again in acute brain slices at later time points (P35–P56) from those mice that remained alive. We find impaired action potential generation at early time points (P16–P21) in PV-INs from Scn1a+/− mice that did and did not survive until at least P35 (i.e., all mice).
We also reconcile the apparent normalization of PV-IN spike generation with the ongoing pathology in Scn1a+/− mice during the chronic phase of the disorder. We examined unitary synaptic transmission at PV-IN synapses in Scn1a+/− versus age-matched wild-type control mice across development by performing multiple whole-cell patch-clamp recordings of genetically identified PV-INs and target principal cells (PCs). We found severe impairments in synaptic transmission at PV-IN:PC synapses selectively in those P16–P21 Scn1a+/− mice that were subsequently deceased prior to P35. Spike generation of PV-INs from Scn1a+/− mice surviving beyond P35 was found to have normalized by P35–P56, as described previously (Favero et al., 2018); yet we again identified dysfunction of synaptic transmission at PV-IN:PC synapses at this later time point.
We conclude that PV-IN axonal dysfunction is a core pathomechanism of chronic DS and suggest that the safety factor for axonal conduction is less than for spike generation. Results highlight a potential marker of SUDEP risk and illuminate differential mechanisms of epilepsy onset versus chronic pathology in DS, shifting the locus of dysfunction from that of PV-IN spike generation to that of synaptic transmission.
RESULTS
Targeting PV interneurons in wild-type versus Scn1a+/− mice across development
We used an established mouse model of DS (Scn1a+/− mice) in which a loxP-flanked neo cassette replaces exon 1 of the Scn1a gene, creating a null allele (Mistry et al., 2014; STAR Methods), which recapitulates the core features of DS in human patients, including temperature-sensitive seizures, chronic epilepsy (with onset of seizures at or around P18), approximately 30% death from seizure-related causes (compared with 20% lifetime risk in humans; Cooper et al., 2016), and behavioral deficits consistent with features of ASD (Berkvens et al., 2015; He et al., 2018; Xiong et al., 2016). Results of a cross to double-transgenic PV-Cre.tdTomato mice yielded triple-transgenic Scn1a.PV-Cre.tdT mice and age-matched wild-type PV-Cre.tdT littermate controls (Favero et al., 2018) and facilitated comparison of PV-INs between genotypes and across development.
To facilitate recordings from the same mice at two time points (pre-weaning and young adult), we developed a technique referred to here as “mini-slices” (see STAR Methods) generated from microbiopsies of sensorimotor neocortices of young (P16–P21) mice (Figures 1B and S1), followed by recordings via standard acute brain slice preparation from the hemispheres contralateral to the mini-slice biopsies at later time points (P35–P56; i.e., in those mice that survived to young adulthood; Figure 1). Scn1a+/− mice recorded at P16–P21 were retrospectively separated into two groups based on whether they were deceased prior to P35 (Scn1a+/− P16–P21 SUDEP) or survived to P35–P56 (Scn1a+/− P16–P21 SURVIVOR). Scn1a+/− mice surviving to and recorded at P35–P56 via conventional brain slice physiology were defined as a third group (Scn1a+/− P35–P56; Figure 1). Rate of spontaneous death was 26.2% (22 of 84) in Scn1a+/− mice [Scn1a+/− P16-P21 SUDEP, n = 22; Scn1a+/− P16–P21 SURVIVOR, n = 62), similar to 25.1% in our prior study (Favero et al., 2018) and in the published literature (e.g., Hawkins et al., 2017; Han et al., 2020). The same mini-slice procedure in wild-type mice (pre-weaning, wild type [WT] P16–P21; young adult, WT P35–P56) was associated with 0% mortality (0 of 37).
Figure 1. Analysis of PV interneurons across development.
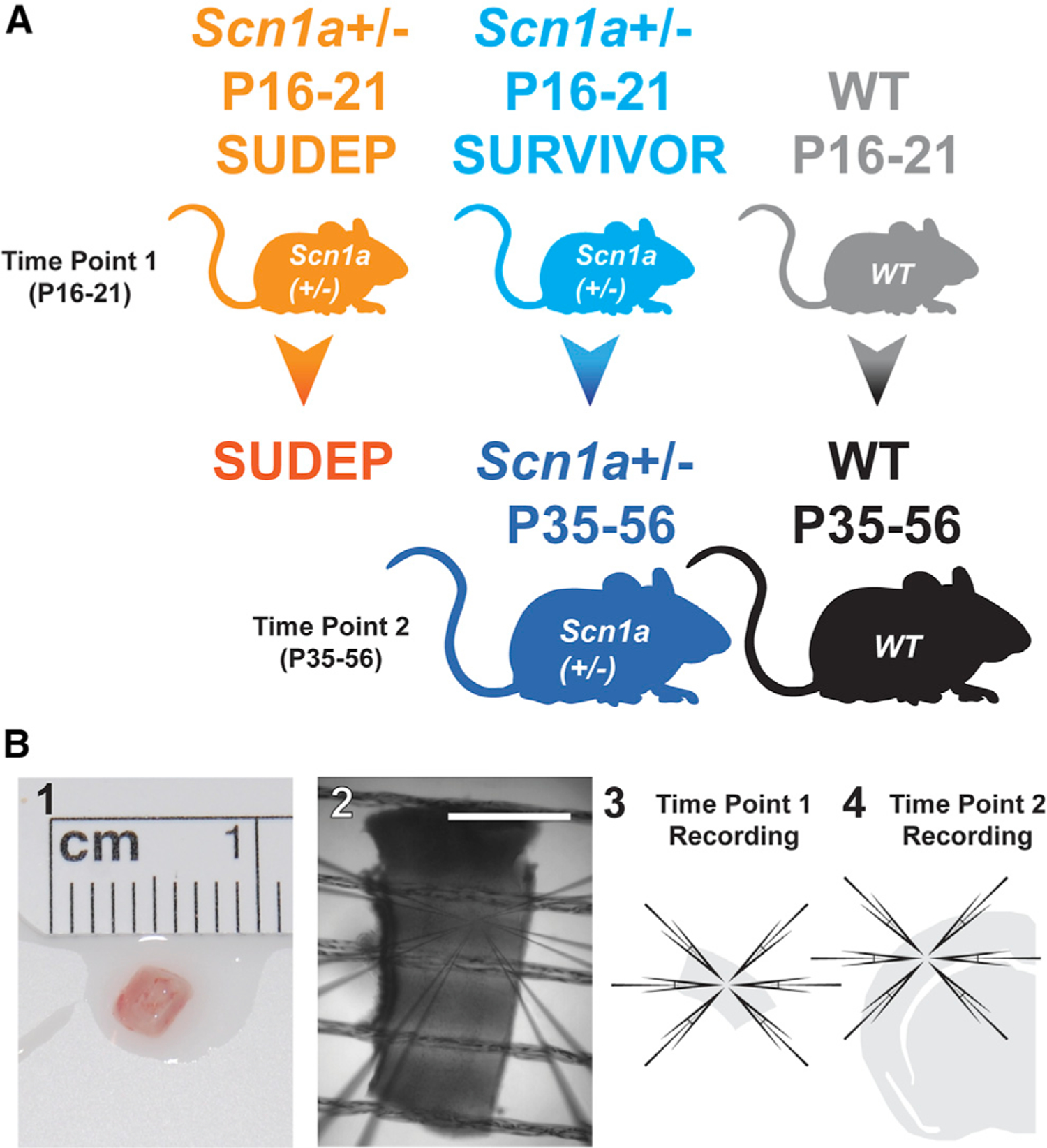
(A) Five experimental groups included (1) pre-weaning Scn1a+/− mice recorded at P16–P21 via mini-slice that were deceased by P35 (Scn1a+/− P16–P21 SUDEP; orange), (2) Scn1a+/− mice recorded at P16–P21 via mini-slice that survive to P35–P56 (Scn1a+/− P16–P21 SURVIVOR; light blue), (3) surviving Scn1a+/− mice recorded at P35–P56 via conventional acute brain-slice techniques (Scn1a+/− P35–P56; dark blue; i.e., the same mice as in Scn1a+/− P16–P21 SURVIVOR mice but recorded again at P35–P56 to generate a separate dataset), (4) pre-weaning wild-type (WT) littermates at P16–P21 (WT P16–P21; gray), and (5) young adult WT recorded at P35–P56 via acute brain slice (WT P35–P56; black; i.e., the same mice as in WT P16–P21).
(B) Mini-slices (1–3) were generated from biopsies of somatosensory neocortex of P16–P21 mice. Acute brain slices (4) were generated using standard techniques from surviving Scn1a+/− P35–P56 mice and from age-matched WT P35–P56 littermates. Scale bar in (2), 1 mm.
Hence, we defined five experimental datasets and focused on six key comparisons: Scn1a+/− P16–P21 SUDEP versus Scn1a+/− P16–P21 SURVIVOR (to assess for any difference between Scn1a+/− mice recorded at P16–P21 that die prior to P35 versus those that survive to young adulthood), Scn1a+/− P16–P21 SUDEP versus WT P16–P21 and Scn1a+/− P16–P21 SURVIVOR versus WT P16–P21 (to determine the difference between the two groups of pre-weaning Scn1a+/− mice versus age-matched WT littermate controls), Scn1a+/− P16-P21 SURVIVOR versus Scn1a+/− P35–P56 (to assess the developmental trajectory of PV-IN excitability in surviving Scn1a+/− mice), Scn1a+/− P35–P56 versus WT P35–P56 (to compare surviving young-adult Scn1a+/− mice with age-matched WT controls), and WT P16–P21 versus WT P35–P56 (to assess the developmental trajectory of PV-IN excitability in WT mice).
PV-INs from all Scn1a+/− mice show impairments in action potential generation at early time points
We then performed whole-cell patch-clamp recordings from identified tdTomato-positive PV-INs in layer 4 primary somatosensory neocortex. Recordings from Scn1a+/− P16–P21 SUDEP versus Scn1a+/− P16–P21 SURVIVOR mice addressed a key unresolved question: whether PV-INs recorded in surviving Scn1a+/− P35–P56 mice were ever abnormal (i.e., would such cells have been abnormal if recorded at earlier time points).
We found that PV-INs recorded in mini-slices from Scn1a+/− P16–21 SUDEP mice (Figure 2A1) and PV-INs recorded in mini-slices from Scn1a+/− P16–P21 SURVIVOR mice (Figure 2A2) all exhibited markedly abnormal electrical excitability relative to PV-INs recorded in mini-slices from age-matched littermate control WT P16–P21 mice (Figure 2A4) and to PV-INs from young-adult Scn1a+/− P35–P56 mice (Figure 2A3), across a range of measures that depend on Na+ channel function, including indicators of action potential generation and repetitive firing (Figures 2B and 2C; Table S1). PV-Ins from WT mice exhibited higher firing frequency at the later time point (WT P35–P56 relative to WT P16–P21), consistent with normal maturation of PV-IN excitability (Goldberg et al., 2011). Identical results were obtained after normalizing for minor differences in input resistance (Table S1; see STAR Methods). We constructed a linear mixed-effects model to account for variability between mice as well as the necessarily imbalanced sizes of the experimental groups; the model was fit to the data with mouse as a random factor.
Figure 2. Transient impairments in action potential generation in PV interneurons in all Scn1a+/− mice.
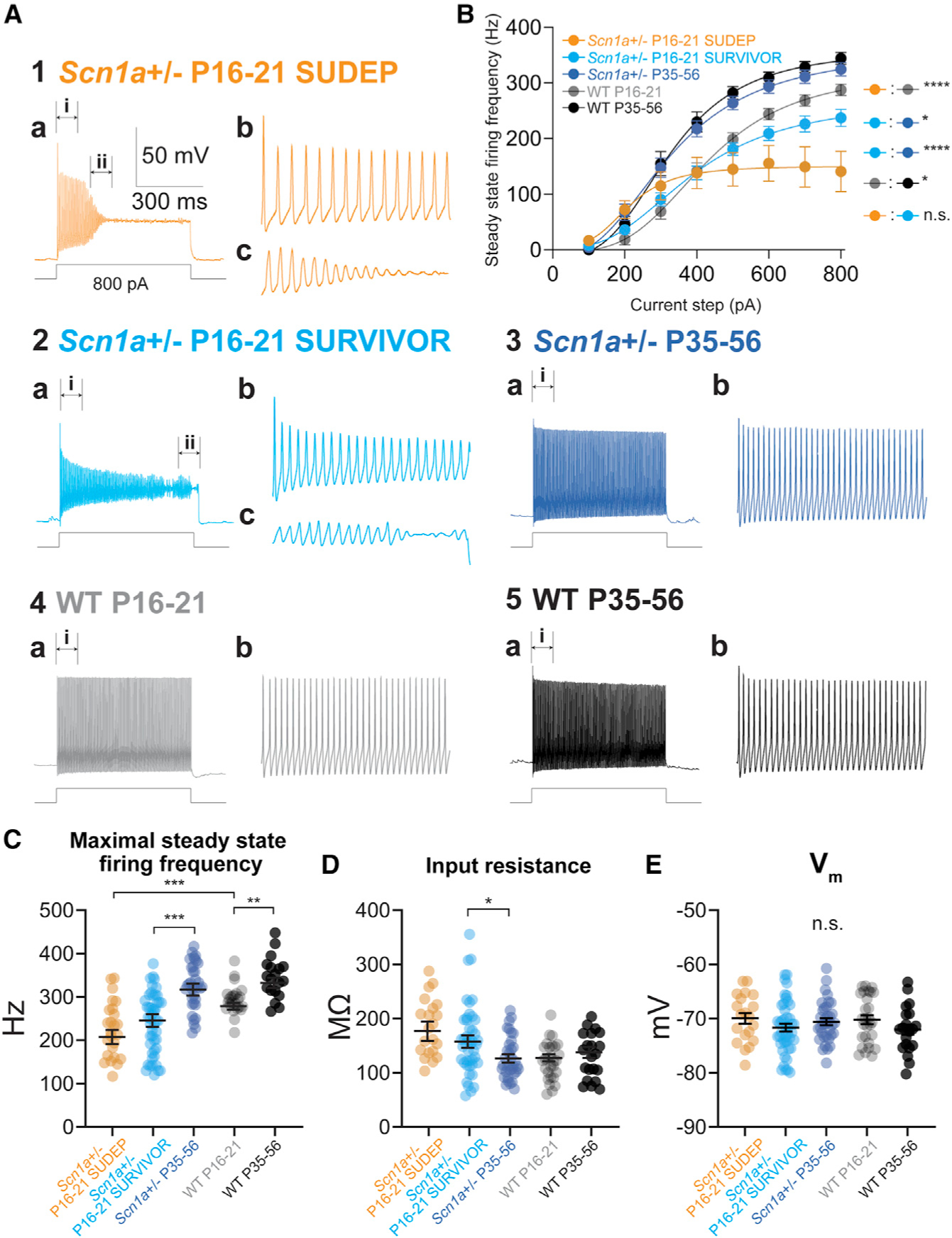
(A) Representative current-clamp recordings from PV-Ins from all groups (1–5). Note that PV-Ins from all Scn1a+/− mice recorded at P16–P21, including those that did or did not survive to P35–P56, exhibited markedly abnormal excitability with action potential failure (shown in detail in 1a–c and 2a–c), while Scn1a+/− P35–P56 mice exhibit normal action potential generation and repetitive firing similar to WT P35–P56 mice.
(B) Input-output curves show steady-state firing frequency for PV-Ins from each group. Excitability of P16–P21 Scn1a+/− mice that did (SURVIVOR) and did not (SUDEP) survive to young adulthood was decreased relative to age-matched WT P16–P21 and to PV-INs from surviving Scn1a+/− P35–P56 mice. See Table S1.
(C–E) Summary data for maximal steady-state firing frequency (C), input resistance (D), and resting membrane potential (E) in PV-Ins from each group.
*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 via mixed-effects modeling (see STAR Methods). n.s., not significant.
In contrast, we found no differences in various passive membrane properties (Figures 2E and S2; Table S1), although we did find small genotype differences in input resistance (Figure 2D; Table S1) and measures related to this (rheobase current injection; membrane time constant; Figure S2; Table S1).
Data obtained in WT P16–P21 mice via the mini-slice procedure were qualitatively similar to prior recordings from PV-INs from mouse neocortex obtained via standard acute brain slice techniques (Goldberg et al., 2011). However, to determine whether the mini-slice alters physiology of PV-INs, we directly compared PV-INs in mini-slice versus acute brain slice from P16–P21 Scn1a+/− mice and found no differences across a broad range of measures (n = 11 cells from four mice each; Figure S4; Table S2). The mini-slice procedure had no effect on body weight or on temperature threshold for seizure induction measured at P35 for Scn1a+/− mice that did versus did not undergo the mini-slice procedure (see Figure S4).
Hence, action potential generation is impaired at early time points in all PV-INs from Scn1a+/− mice at ≤P21, including those that do and do not survive to P35, while confirming our prior results (Favero et al., 2018) that action potential generation of PV-INs from Scn1a+/− mice has normalized by P35–P56.
Group-specific impairments in PV-IN synaptic transmission in Scnla+/− mice
The findings reported above represent somewhat of a paradox and raise two critical questions relevant to pathomechanisms of DS. Action potential generation and repetitive and high-frequency firing of PV-INs was impaired in all Scn1a+/− mice at P16–P21, yet some mice were deceased by P35 while others survived to P35–P56. The basis for this profound difference is unclear. Second, PV-INs from young-adult Scn1a+/− P35–P56 mice had recovered high-frequency discharge characteristic of mature fast-spiking PV-INs (Jonas et al., 2004; Hu et al., 2014; Favero et al., 2018). However, Scn1a+/− mice that survive to P35 continue to exhibit chronic epilepsy, features of ASD, and ongoing SUDEP. Hence, the chronic pathology in DS cannot be attributed to abnormal PV-IN spike generation. We hypothesized that persistent deficits might exist in synaptic transmission — perhaps due to impaired spike propagation — given the dependence of PV-IN axonal function on a high Na+ channel density at the axon initial segment (AIS) (Hu and Jonas, 2014).
To assay the fidelity of GABAergic transmission at PV-IN synapses, we performed multiple whole-cell patch-clamp recordings and measured the properties of evoked inhibitory post-synaptic currents (IPSCs) (in voltage clamp) recorded in PCs in response to action potentials in presynaptic PV-INs (generated in current clamp; Figure 3A). PCs were patched using an internal solution containing high chloride (Cl) such that ECl− = −17 mV and inhibitory postsynaptic potentials (IPSPs) were recorded as depolarizations, while IPSCs were recorded as large inward currents at a holding potential of −80 mV (STAR Methods). This approach facilitated detection of small IPSCs versus synaptic failures.
Figure 3. Persistent impairments in AP propagation in PV interneurons.
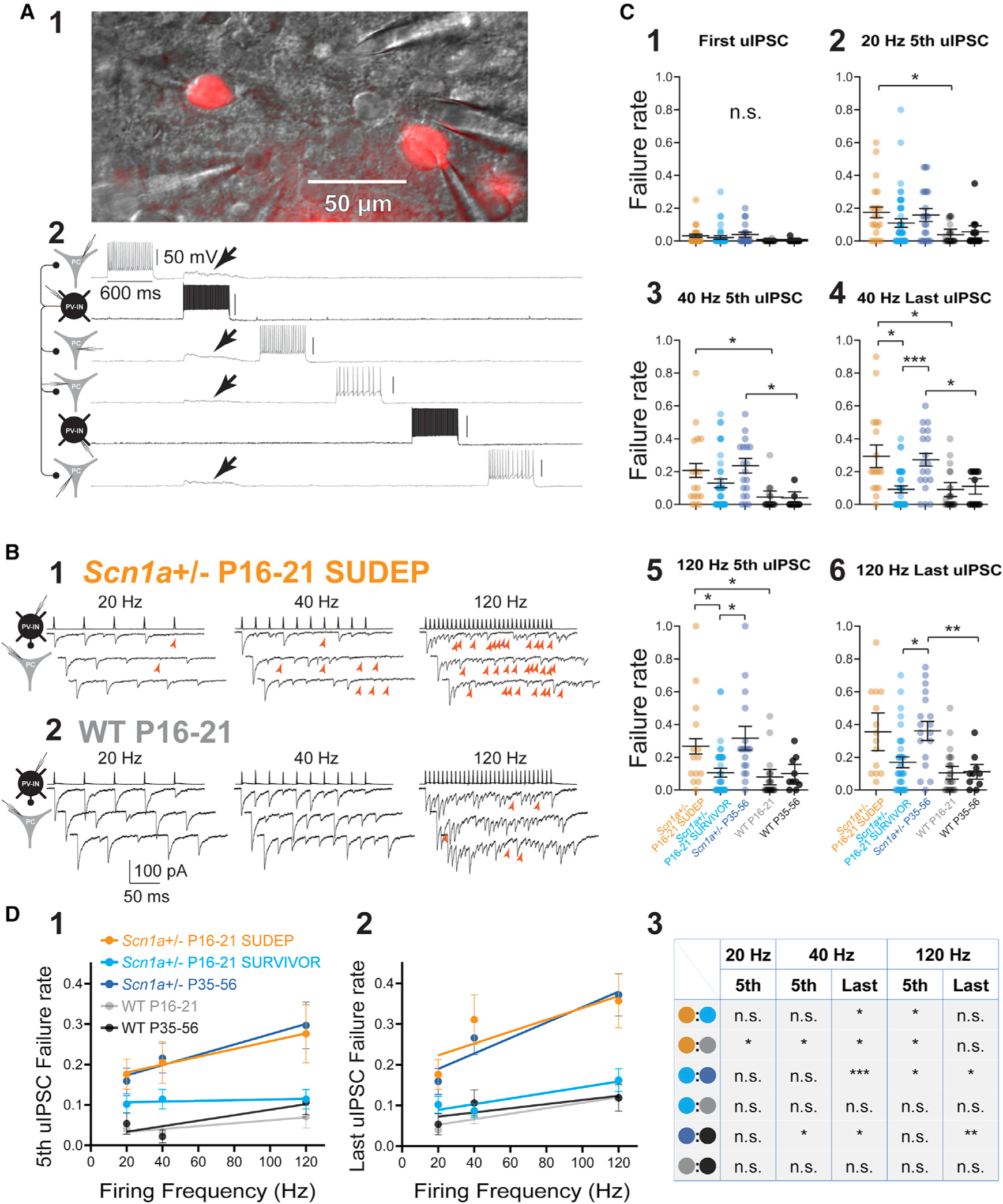
(A) Multiple whole-cell recording from tdTomato-positive PV-INs as well as principal cells. Shown is a representative image of two PV-INs (cells no. 2 and no. 5) and four PCs (cells no. 1, no. 3, no. 4, and no. 6) in the mini-slice preparation under infrared differential interference contrast (IR-DIC) and corresponding epifluorescence (1;showing tdTomato-positive PV-INs). Four synaptic connections were identified from cell no. 2 (in black),which produce depolarization in cells no. 1, no. 2, no. 3, and no. 4 (arrows; in gray; 2).
(B) Representative examples of evoked unitary inhibitory postsynaptic currents (uIPSCs) in an Scn1a+/− P16–P21 SUDEP mouse (1)and age-matched WT P16–P21 littermate (2). Presynaptic PV-INs were recorded in current clamp, and IPSCs were recorded in PCs in voltage clamp as inward currents. Red arrowheads, synaptic failures.
(C) Summary data for failure rate (range, 0 to 1), calculated as the number of failures divided by total stimuli (failures plus number of success). See Table S1. Note that failures occurred at frequencies that PV-INs could clearly generate at the soma and were more pronounced with successive stimulation (4 and 6) yet were still present early in trains (3 and 5).
(D) Relationship between failure rate and presynaptic firing frequency across groups. *p < 0.05; **p < 0.01; ***p < 0.001; n.s., not significant; via mixed-effects modeling. See Table S1.
The PV-IN:PC synapse is known to be a reliable connection with a low (near-zero) failure rate due to high release probability from multiple boutons per connection (Galarreta and Hestrin, 1998; Kraushaar and Jonas, 2000; Ali et al., 2001; Beierlein et al., 2003; Goldberg et al., 2005; Hu et al., 2014). We stimulated PV-INs with single action potentials and found a low rate of synaptic failures in WT P16–P21 (0.00 ± 0.01; n = 23 connections from 10 mice) and P35–P56 mice (0.00 ± 0.01; n = 13 connections from 10 mice). However, we detected failures in Scn1a+/− mice, including Scn1a+/− P16–P21 SUDEP (0.03 ± 0.01; n = 21 connections from seven mice; p = 0.2412 versus WT P16–P21) and Scn1a+/− P35–P56 (0.04 ± 0.02; n = 23 connections from 13 mice; p = 0.2344 versus WT P35–P56), with a lower but still non-zero failure rate in Scn1a+/− P16–P21 SURVIVOR mice (0.02 ± 0.01; n = 55 connections from 22 mice; p = 0.7725 versus Scn1a+/− P16–P21 SUDEP and 0.6161 versus WT P16–P21).
We then stimulated presynaptic PV-INs to discharge trains of repetitive action potentials and identified a marked increase in frequency- or activity-dependent synaptic failure specifically in Scn1a+/− P16–P21 SUDEP and Scn1a+/− P35–P56 mice, but not Scn1a+/− P16–P21 SURVIVOR (Figures 3B and 3C; Table S1). For example, in approximately one-third of trials, the final stimulus in a 10-pulse train at 40 Hz resulted in failure at PV-IN:PC connections in Scn1a+/− P16–P21 SUDEP mice (0.29 ± 0.07; n = 21 connections from seven mice), a rate that was 3-fold higher than for Scn1a+/− P16–P21 no SUDEP mice (0.09 ± 0.02; n = 55 connections from 22 mice; p = 0.0378 versus Scn1a+/− P16–P21 SUDEP via mixed-effects modeling with post hoc correction for multiple comparisons via Holm-Sidak test) or for age-matched littermate control WT P16–P21 mice (0.09 ± 0.04; n = 23 connections from 10 mice; p = 0.0432 versus Scn1a+/− P16–P21 SUDEP). Failure rate was also 3-fold higher in young-adult Scn1a+/− P35–P56 mice (0.27 ± 0.04; n = 23 connections from 13 mice) than for age-matched littermate control WT P35–P56 mice (0.11 ± 0.05; n = 13 connections from 10 mice; p = 0.0378 versus Scn1a+/− P35–P56; Figure 3C4). For the last event in a 30-pulse train at 120 Hz, failures occurred in approximately one-third of trials at PV-IN:PC synapses from surviving Scn1a+/− P35–P56 mice (Figure 3C6).
Failures may be due to impaired action potential propagation for the following reasons. First, the presence of a presynaptic action potential was directly confirmed in the multiple whole-cell recording configuration, ruling out failure of spike generation (only apparent in PV-INs from Scn1a+/− mice during trains of repetitive firing in response to prolonged, large-amplitude current injections). Second, the PV-IN:PC connection is characterized by large-amplitude IPSCs that were further augmented by our experimental conditions, and this—combined with <2 pA peak-to-peak noise of our recording system — makes it unlikely that we were incorrectly labeling small IPSCs as failures. We performed an analysis of the contingency of the amplitude of the IPSC in response to the second presynaptic action potential (IPSC2) based on the success or failure of the first event (IPSC1) in a paired-pulse paradigm (Figure S6A). Action potential invasion of the presynaptic terminal leads to spike-evoked calcium influx that engages short-term synaptic plasticity mechanisms (synaptic depression in the case of the PV-IN synapse; Zucker and Regehr, 2002; Xiang et al., 2002; Beierlein et al., 2003); hence, presence or absence of synaptic depression of IPSC2 can be used as an indicator of whether calcium influx occurred and hence whether a somatic action potential arrived successfully at the presynapse. At neocortical PV-IN:PC connections in Scn1a+/− mice, the amplitude of IPSC2 was smaller if IPSC1 was a success versus a failure (31.9 ± 7.0 pA for success versus 48.8 ± 11.0 pA for failure; n = 20; p = 0.0023; Figure S6B1), consistent with synaptic depression (paired pulse ratio [PPR] = 0.65). However, if the response to the first presynaptic action potential resulted in failure, the amplitude of IPSC2 was the same as the mean amplitude of IPSC1 when IPSC1 was a success (48.9 ± 11.0 pA for IPSC2 if IPSC1 was a failure versus 54.7 ± 10.8 pA for IPSC1 if IPSC1 was a success; n = 20; p = 0.17; Figure S6B2). Such data suggest that presynaptic calcium channels did not open in trials in which a failure was observed, consistent with failure of propagation of the somatic action potential to the synapse.
In addition, we found that synaptic latency (measured from the peak of the action potential to onset of the unitary IPSC) was prolonged in Scn1a+/− P16–P21 SUDEP mice relative to age-matched littermate control and in young-adult Scn1a+/− P35–P56 mice versus WT P35–P56 control (Figures 4A and 4B; Table S1), being 0.87 ± 0.08 ms in Scn1a+/− P16–P21 SUDEP (n = 21 connections from seven mice), 0.78 ± 0.05 in Scn1a+/− P16–P21 SURVIVOR (n = 55 connections from 22 mice; p = 0.6923 versus Scn1a+/− P16–P21 SUDEP), 0.87 ± 0.08 ms in Scn1a+/− P35–P56 (n = 23 connections from 13 mice), 0.61 ± 0.04 in WT P16–P21 (n = 23 connections from 10 mice; p = 0.0229 versus Scn1a+/− P16–P21 SUDEP; p = 0.0366 versus Scn1a+/− P16–P21 SURVIVOR), and 0.62 ± 0.05 ms in WT P35–P56 (p = 0.0469 versus Scn1a+/− P35–P56 via mixed-effects modeling with post hoc correction via Holm-Sidak test). While we did find that the presynaptic action potential (AP) half-width was slightly longer in Scn1a+/− P16–P21 SUDEP and Scn1a+/− P16–P21 SURVIVOR than in WT P16–P21 mice (Table S1), this difference was much smaller than and hence cannot account for the observed difference in synaptic latency and is more likely due to the prolonged rising phase of the AP (with lower dV/dt) in Scn1a+/− P16–P21 mice relative to WT.
Figure 4. Prolonged synaptic latency at PV interneuron connections.
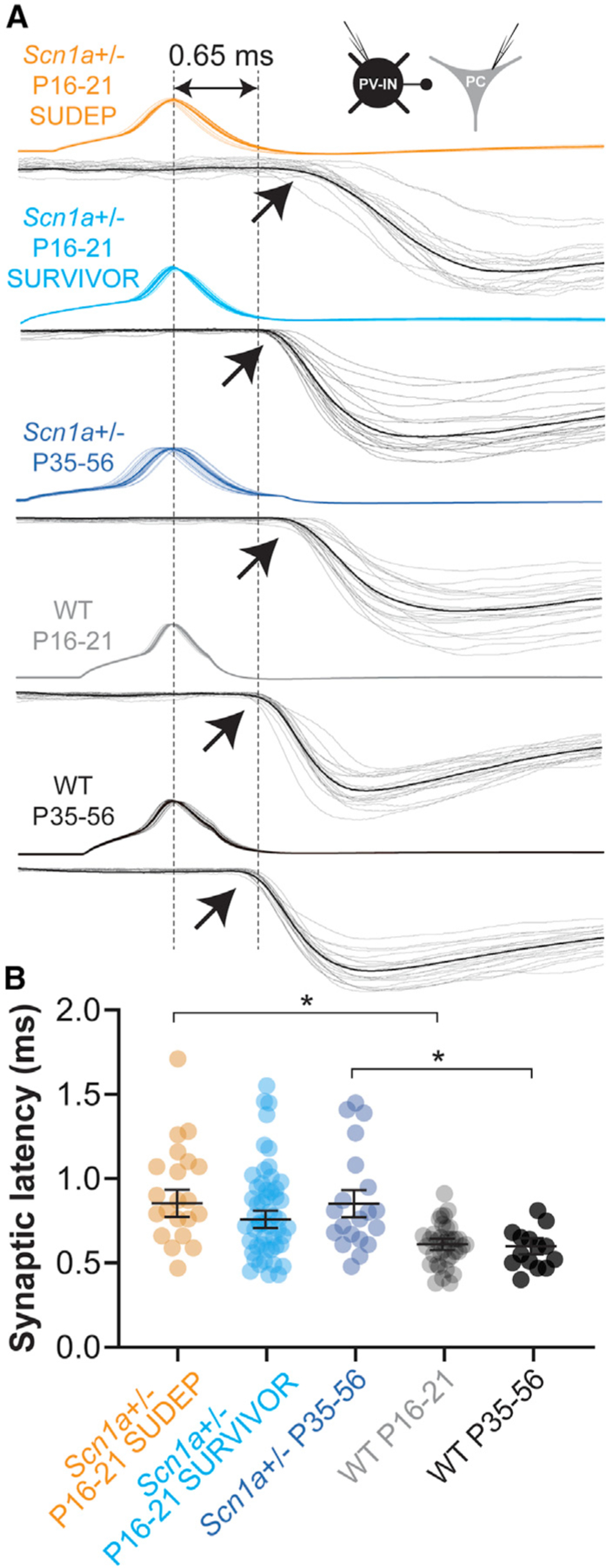
(A) Representative examples from each group illustrating synaptic latency, measured from peak of the presynaptic AP (dotted line at left) to onset (arrows) of the postsynaptic uIPSC (mean, black; individual traces, gray). The dotted line at right corresponds to a latency of 0.65 ms, to approximate the mean value for WT mice (at P16–P21 and P35–P56).
(B) Summary data. *p < 0.05 via mixed-effects modeling.
See Table S1.
Synaptic latency is determined by axon length, conduction velocity, and synaptic delay (Sabatini and Regehr, 1999), as well as release probability (Boudkkazi et al., 2007, 2011), and prolonged latency could reflect slowed conduction velocity along PV-IN axons. This interpretation is supported by prior work showing increased synaptic latency at PV-IN:PC synapses in rat dentate gyrus with low concentrations of tetrodotoxin (TTX) (Hu and Jonas, 2014). We found that bath application of low concentrations of TTX (10 nM; estimated to block approximately 50% of Nav1.1-containing voltage-gated Na+ channels; Smith and Goldin, 1998) had minimal effect on the PV-IN firing pattern in WT P16–P21 mice (Figure 5A) but led to a 2-fold increase in failure rate in a frequency-dependent manner similar to what was observed in age-matched pre-weaning Scn1a+/− P16–P21 SUDEP mice. Failure rate for the fifth pulse in the train was 0.08 ± 0.03 for control and 0.15 ± 0.05 after application of 10 nM TTX (n = 11 connections from four mice; p = 0.020 versus control via two-tailed Student’s t test); failure rate for the last pulse in a 120-Hz train was 0.12 ± 0.04 in control and 0.23 ± 0.07 after 10 nM TTX (p = 0.047 versus control; Figures 5B, 5D, and 5E). Synaptic latency also increased from 0.49 ± 0.02 to 0.64 ± 0.06 ms after application of TTX (p = 0.016 versus control; Figures 5C and 5F). Hence, loss of Nav1.1 in PV-INs may preferentially affect synaptic transmission over spike generation.
Figure 5. Low concentrations of TTX reproduce the effect of heterozygous loss of Nav1.1 on PV-IN synaptic transmission.
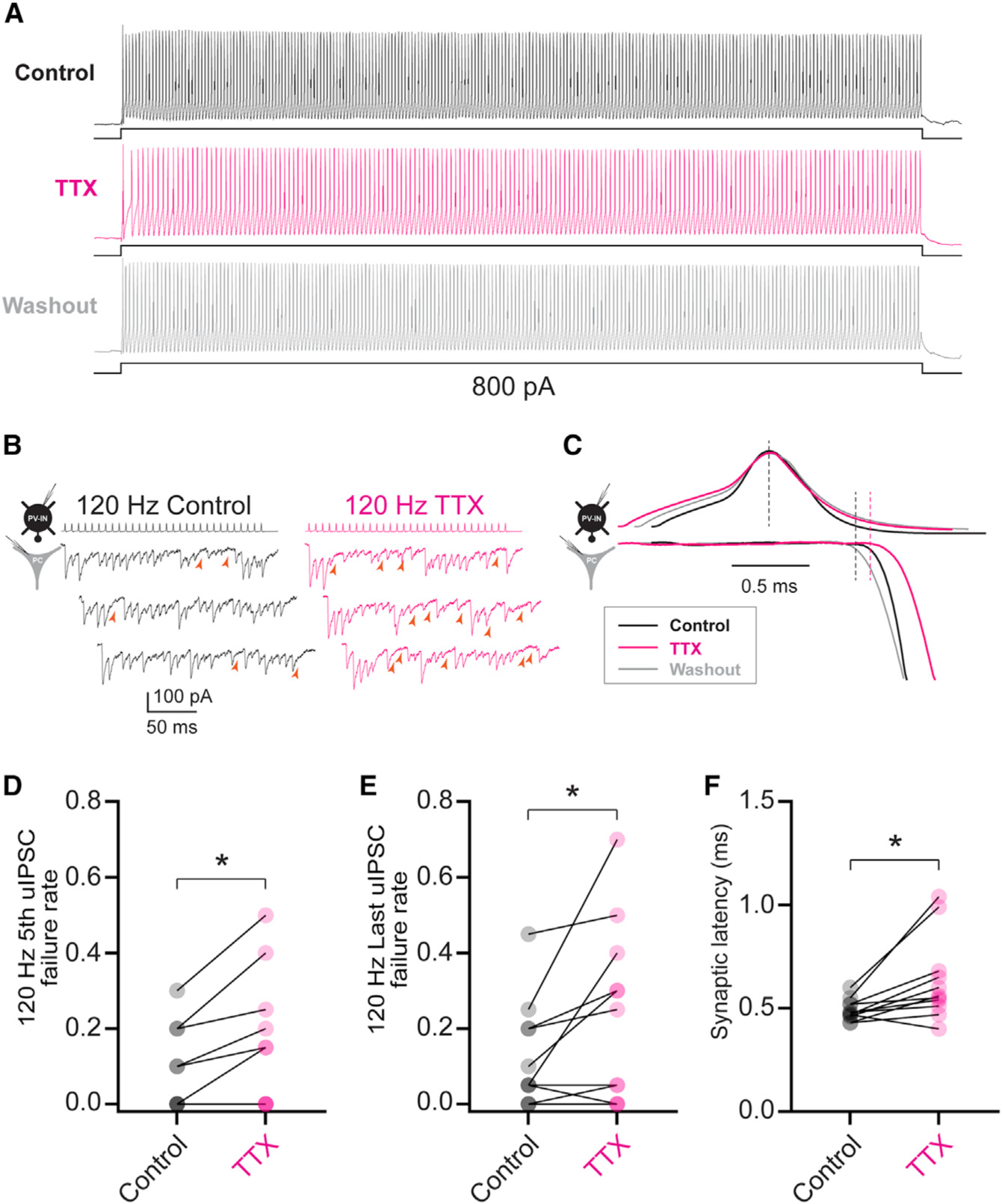
(A) Electrophysiological discharge pattern of a PV-IN from a P18 WT.PV-Cre.tdT mouse in response to a 600-ms, 800-pA current injection before (top, black) and after (middle; magenta) bath application of 10 nM TTX, with washout (bottom; gray).
(B) Repetitive stimulation of presynaptic PV-INs elicited IPSCs in postsynaptic principal cells (left) with a higher failure rate after bath application of 10 nM TTX (right). Note that PV-INs can follow 120-Hz stimulation in the presence of TTX.
(C) Prolonged synaptic latency after TTX application. Shown is a presynaptic action potential aligned to peak (top), with the onset of the postsynaptic IPSC response (bottom).
(D–F) Summary data show increased failure rate for the fifth (D) and last (E) IPSC in a train at 120 Hz and for synaptic latency (F). *p < 0.05.
Expression of Nav1.1 at PV interneuron axons
Direct whole-cell voltage-clamp recordings from PV-INs in dentate gyrus indicate the presence of Na+ conductance along the axon (Hu and Jonas, 2014; Hu et al., 2018). PV-IN axons in cerebral cortex of rodent and human are extensively myelinated along internodal regions and between axonal branch points (Micheva et al., 2016; Stedehouder et al., 2017; Benamer et al., 2020). Using immunohistochemistry and confocal microscopy, we confirmed that Nav1.1 is expressed at the AIS of PV-INs (Figure S7A), consistent with previous results (Ogiwara et al., 2007; Li et al., 2014; Goff and Goldberg, 2019). We also found Nav1.1 expression along axons, at branch points (Figure S7A1), and at presumptive pre-terminal axons (Figure S7A3). We used automated filtering, segmentation, and extraction from reconstructed z stacks (Figures S7B–S7D) and found that Nav1.1 more prominently labeled branching structures (mean, 3.60 ± 0.24 branches per labeled structure; n = 6,718 puncta from n = 10 z stacks from n = 3 mice from WT P35–P56 mice) relative to pan-Nav (1.74 ± 0.37 branches per labeled structure; n = 1,409 puncta from n = 7 z stacks; p < 0.0001 via mixed-effects model; Figure S7E; see STAR Methods). Nav1.1-positive structures were shorter in length (Nav1.1, 33.8 ± 0.92 mm; pan-Nav, 48.5 ± 4.4 mm; p = 0.0009) and less linear (ratio index for Nav1.1, 2.74 ± 0.03; pan-Nav, 4.98 ± 0.17; p < 0.00001; Figures S7F and S7G). We compared Nav1.1 immunolabeling in young-adult WT P35–P56 and Scn1a+/− P35–P56 mice and found a marked reduction in overall normalized intensity of Nav1.1 immunoreactivity in Scn1a+/− P35–P56 relative to WT (WT, 96.3 ± 10.6; Scn1a+/−, 72.7 ± 6.0; p = 0.025), with a reduction in labeling of branching structures (WT, 3.60 ± 0.24 branches/puncta; Scn1a+/−, 2.56 ± 0.10; p = 0.00009) and decreased length of labeled puncta (WT, 33.8 ± 0.92 μm; Scn1a+/−, 29.3 ± 0.3 μm; p < 0.00001) but no change in linearity of puncta (WT, 2.74 ± 0.03; Scn1a+/−, 2.84 ± 0.06; p = 0.013; Figures S7I–S7L), supporting the possibility of a preferential decrease in Nav1.1 at distal axons and branching structures relative to the AIS.
Alterations in PV interneuron:principal cell connection probability
We next assessed for differences in connection rate between PV-INs and PCs in Scn1a+/− versus WT mice, as evidence of decreased connectivity—perhaps due to fewer connections between and/or fewer synaptic boutons per synapse at PV-IN:PC connections—could potentially explain the decreased unitary IPSC (uIPSC) amplitude observed in pre-weaning Scn1a+/− mice. The connection rate of neocortical PV-INs to PCs obtained via paired recordings in brain slice varies widely, ranging from 25% to 50% in the published literature (Packer and Yuste, 2011; Espinoza et al., 2018; Vickers et al., 2018), and depends on slice preparation technique, distance between recorded cells, and other considerations.
We found that PV-IN:PC connection rate was 21.9% (21 connections out of a maximum possible of 96) in Scn1a+/− P16–P21 SUDEP mice yet was 32.6% (105/322) in Scn1a+/− P16–P21 SURVIVOR, similar to the connection rate of 35.0% (77/220) observed in age-matched littermate WT P16–P21 mice (p = 0.023 versus Scn1a+/− P16–P21 SUDEP via chi square test). Connection rate was 18.8% (46/245) in surviving young-adult Scn1a+/− P35–P56 mice and 33.8% (52/154) in age-matched WT P35–P56 (p < 0.0011 versus Scn1a+/− P35–P56 via chi square test; Figure S8B). Hence, we found lower connection probability selectively in Scn1a+/− P16–P21 SUDEP and in surviving young-adult Scn1a+/− P35–P56 mice.
We performed immunohistochemical labeling (see STAR Methods) for synaptotagmin-2 (Syt2), a specific marker of PV-IN synapses (Sommeijer and Levelt, 2012; Bouhours et al., 2017; van Lier et al., 2020), and confirmed that Syt2-positive boutons were perisomatic and PV positive (Figure S8A). We then quantified the number of PV-IN boutons surrounding the cell bodies of PCs in all experimental groups (Figure S8B) and found a small but statistically significant decrease in the number of Syt2-positive puncta around PCs in Scn1a+/− P16–P21 SUDEP relative to Scn1a+/− P16–P21 SURVIVOR and WT P16–P21 and in Scn1a+/− P35–P56 relative to WT P35–P56 (Figure S8C). The number of puncta around PCs was 10.4 ± 0.3 in Scn1a+/− P16–P21 SUDEP mice (n = 142 cells from six slices from two mice), 12.9 ± 0.3 in Scn1a+/− P16–P21 SURVIVOR (n = 147 cells from eight slices from six mice; ANOVA (4, 706) = 1.615; p < 0.0001 versus Scn1a+/− P16–P21 SUDEP with post hoc correction for multiple comparisons via Holm-Sidak test), 12.7 ± 0.3 in Scn1a+/− P35–P56 (n = 163 cells from eight slices from four mice; p = 0.65 versus Scn1a+/− P16–P21 SURVIVOR), 13.3 ± 0.3 in WT P16–P21 (n = 122 cells from six slices from three mice; p < 0.0001 versus Scn1a+/− P16–P21 SUDEP; p = 0.65 versus Scn1a+/− P16–P21 SURVIVOR), and 14.2 ± 0.4 in WT P35–P56 (n = 134 cells from seven slices from three mice; p = 0.001 versus Scn1a+/− P35–P56; p = 0.12 versus WT P16–P21). Such findings are consistent with a decreased number of synaptic terminals at PV-IN:PC synapses in Scn1a+/− P16–P21 SUDEP and in Scn1a+/− P35–P56 mice and compatible with the observed differences in connection probability. This may contribute to the observed differences in uIPSC amplitude at PV-IN:PC synapses in Scn1a+/− mice but likely cannot account for the observed increases in synaptic failure rates.
A multi-compartment interneuron model supports a preferential impact of reduction in Na+ conductance on action potential propagation
Because we could not directly attribute synaptic transmission failure to failure of AP propagation, we constructed a multi-compartment model of a PV-IN to assess the differential effects of reduction of Na+ current density at the soma and AIS versus axon on features of AP initiation and propagation (see STAR Methods).
Selectively reducing Nav1.1 by 50% (to model heterozygous loss of Nav1.1) in specific neuronal compartments (“deletion paradigm”) caused AP failure (Figure 6). Failure could be of initiation or of propagation (Figure 6D). When Nav1.1 was partially deleted in the soma, AIS, and nodes (“–soma –AIS –nodes” in Figures 6B–6D, modeling Scn1a+/− P16–P21 SUDEP mice), spike generation fails at the soma and AIS, as seen experimentally (Figure 2A1). This was also true for the “–soma –AIS” deletion paradigm. However, when Nav1.1 was impaired specifically at the nodes (“–nodes” deletion paradigm, modeling Scn1a+/− P35–P56), APs continued to be generated at the AIS but did not propagate to the terminal.
Figure 6. A multi-compartment model supports the selective effect of Na+ reduction at the axon on the fidelity of synaptic transmission at an interneuron synapse.
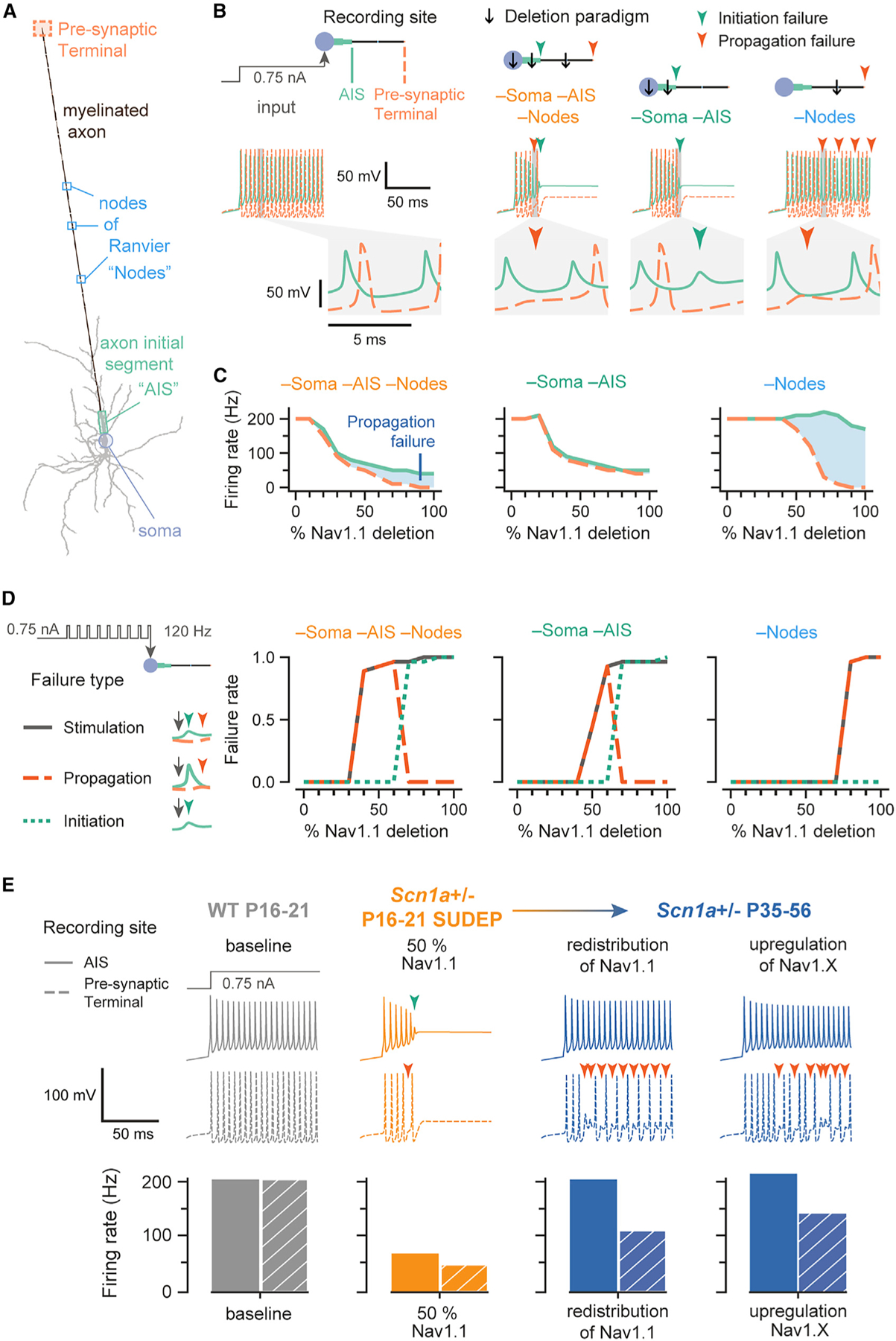
(A) A morphologically realistic PV-IN with a single myelinated axon, nodes of Ranvier (“nodes”), and pre-synaptic terminal.
(B) Shown are membrane potential traces recorded at the AIS (solid light green line) and terminal (dashed light orange line), following injection of 0.75 nA into the soma at t = 20 ms. The enlarged trace shows the action potential generated at the AIS and propagated along the axon to the terminal. The compartment(s) of Nav1.1 deletion is indicated by a black arrow, the combination of which is a “deletion paradigm.” Colored arrows represent the time of a failure (initiation, dark green; propagation, dark orange).
(C) Average firing rate of a neuron with progressive Nav1.1 deletion. Differences in firing rate between the AIS (solid light green line) and pre-synaptic terminal (dashed light orange line) represent propagation failures (shaded blue area), which increase with further deletion.
(D) Failure of stimulation corresponds to a pulse being delivered (at 120 Hz) but without an action potential detected at the pre-synaptic terminal within 2 ms. Failure of propagation refers to an action potential generated at the AIS that did not reach the terminal. Failure of initiation was considered to occur under conditions when a pulse was delivered, but an action potential was not generated at the AIS. For example, propagation failure in the –soma –AIS –nodes deletion paradigm first appears at 30% Nav1.1 deletion but returns to zero at 70% Nav1.1 deletion because there is no longer any spike initiation.
(E) The model generates potential explanations for the experimental observations. Traces were from the AIS (solid lines) and pre-synaptic terminal (dashed lines) with failures represented as in (B). Bar plots show the firing rate of the AIS (solid) and pre-synaptic terminal (hatched pattern). Baseline Nav1.1 in the model represents WT P16–P21 in the experimental setup (gray), while 50% Nav1.1 (in the soma, AIS, and nodes) represents Scn1a+/− P16–P21 SUDEP (orange) or Scn1a+/− P16–P21 SURVIVOR. The model can simulate different ways to recover the properties of the Scn1a+/− P35–P56 group, where action potential generation has recovered but action potential propagation is still impaired. The first is a redistribution of Nav1.1 from nodes to soma and AIS such that Nav1.1 conductance in soma and AIS was 120% of baseline (20% Nav1.1 addition) and nodal Nav1.1 conductance was 40% of its baseline (60% Nav1.1 deletion). The Scn1a+/− P35–P56 group was also modeled by upregulation of another transient sodium current by 120% in the soma and 130% in the AIS.
See STAR Methods and Figure S9 for details.
The firing rate for both AIS and presynaptic terminal recording sites dramatically decreased with further Nav1.1 deletion in both the –soma –AIS –nodes and –soma –AIS paradigms. Progressive deletion at the nodes had no effect on the firing rate at the AIS.
We then stimulated the model with brief repetitive current injections (Figure 6D). Failures could be that of stimulation (a pulse was delivered but an AP was not recorded at the presynaptic terminal), initiation (an AP was not recorded at the AIS), or propagation (an AP was recorded at the AIS, but not at the terminal). Progressive loss of Nav1.1 in the –soma –AIS –nodes deletion paradigm yielded substantial propagation failures first apparent at 40% Nav1.1 deletion and initiation failures at 30% Nav1.1 deletion. Similarly, the –soma –AIS deletion paradigm exhibited propagation failures from 50% Nav1.1 deletion and initiation failures from 70% Nav1.1 deletion. As expected, the –nodes deletion paradigm showed only propagation failures, which were apparent from 80% Nav1.1 deletion.
The model was also used to explore possible mechanisms for the functional recovery seen in Scn1a+/− P35–P56 mice (Figure 6E). One option explored was the possibility that Nav1.1 protein could be redistributed from the axon to the AIS, leading to recovery of normal spike generation but worsening of spike propagation. The second option involved upregulation of Na+ conductance, modeling a condition of compensatory upregulation of, for example, Nav1.3 and/or Nav1.6 subunits, as has been proposed (Favero et al., 2018; Figure S9).
The results of the model provide proof of principle that Nav1.1 deletion in specific compartments leads to differential effects on AP initiation and propagation and supports the mechanism of transition from Scn1a+/− P16–P21 to P35–P56 as potentially occurring via redistribution of Nav1.1 or the upregulation of other Nav1.X Na+ channel subunits.
A dual optogenetic approach supports existence of frequency-dependent spike propagation failure in Scn1a+/− mice
To further confirm the above inferences, we used a dual optogenetic approach in which an excitatory opsin was expressed at the PV-IN soma while a second spectrally distinct opsin was expressed preferentially at synaptic terminals (Figures 7A and S10), facilitating selective generation of somatic versus axo-synaptic spikes. We used Cre-dependent AAVs to express the excitatory opsin hChR2 (AAV9.Ef1a.DIO.hChR2(E123A).EYFP), which is preferentially targeted to the axon and synapse (Baker et al., 2016; Grubb and Burrone, 2010; Mattis et al., 2012), as well as a soma-targeted, red-shifted opsin (AAV9.hSyn.DIO.ChrimsonR.mRuby2.ST; Pégard et al., 2017), in Scn1a.PV-Cre mice and WT PV-Cre littermate controls. We then performed whole-cell voltage-clamp recording of IPSCs in PCs in response to blue (490 nm; ChR2) and red-light (660 nm; ChrimsonR) photostimulation (optogenetically evoked IPSCs [oIPSCs]; Figures 7C and 7D). We confirmed that ChrimsonR was selectively expressed at the soma of PV-INs, and hChR2 was expressed preferentially along the axon and at synaptic terminals (Figure 7B).
Figure 7. A dual optogenetics strategy demonstrates impaired PV interneuron spike propagation in Scn1a+/− mice.
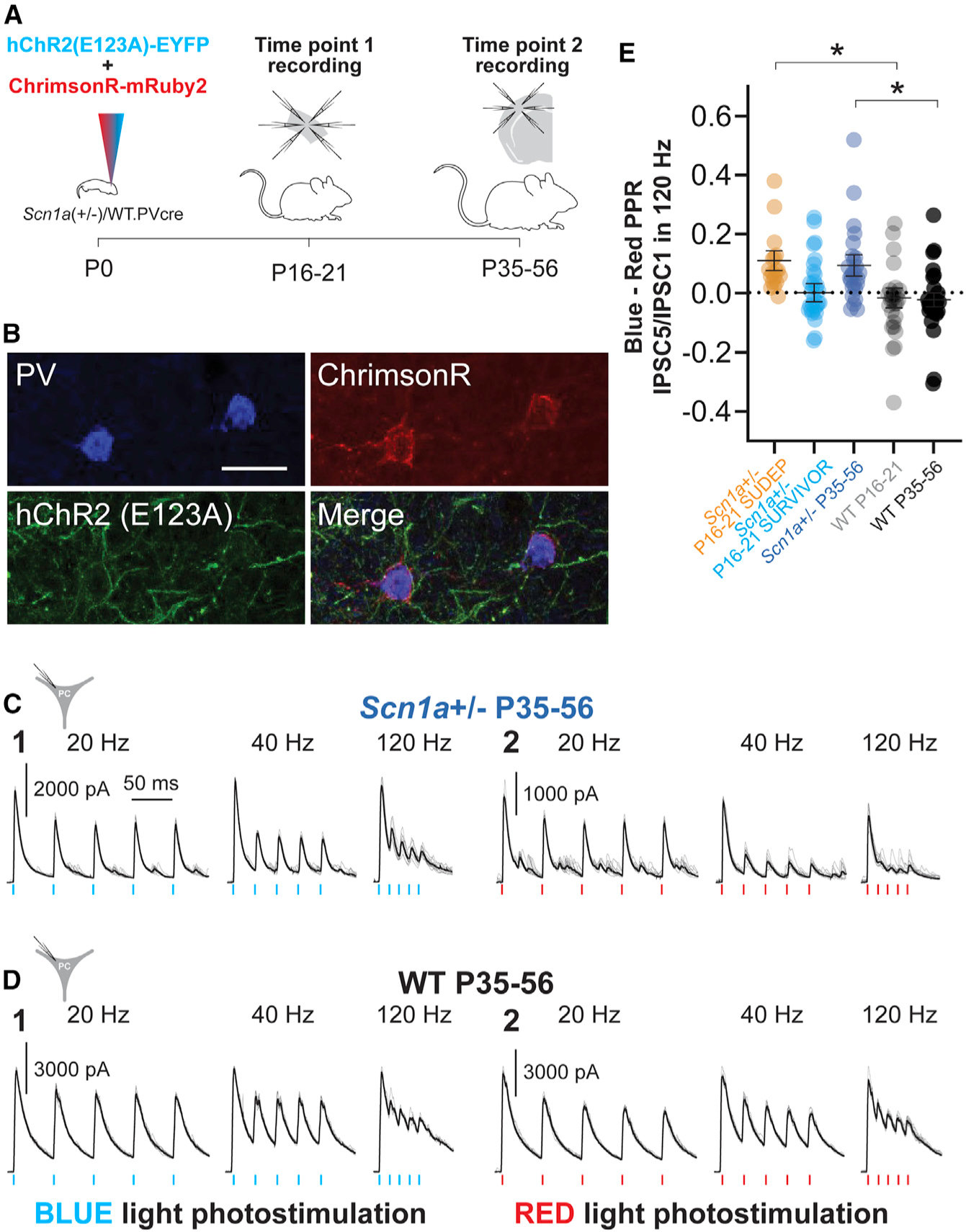
(A) Experimental timeline showing viral injection of both AAV9.DIO.ChrimsonR.mRuby2 and AAV.DIO.hChR2.eYFP into PV-Cre mice at P0 (left) and time points 1 (mini-slice; center) and 2 (acute brain slice; right) recordings.
(B) Representative confocal images showing expression of PV (Alexa Fluor 405; blue), hChR2.eYFP (green), and ChrimsonR.mRuby2 (red). Scale bar, 20 μm.
(C and D) Representative data for evoked oIPSCs recorded in principal cells in response to repetitive photostimulation with ChR2 (C1 and D1) or ChrimsonR (C2 and D2) in Scn1a+/− P35–P56 mice
(C) and WT P35–P56 mice (D).
(E) Summary data for the index of oIPSCn/oIPSC1 in response to blue-light photostimulation minus oIPSCn/oIPSC1 in response to red-light photostimulation (blue-red index). *p<0.05 via mixed-effects modeling.
See Table S3.
oIPSCs were recorded as large outward currents at a holding potential of +10 mV (ECl = −110 mV; see STAR Methods). Recording of PCs combined with photostimulation of surrounding PV-INs revealed a progressive decrease in the amplitude of successive oIPSCs with repeated red-light (somatic) stimulation that was far greater than the decrease observed in response to blue-light (axon and terminal) stimulation (Figures 7C–7E). We quantified the ratio of oIPSC2/oIPSC1 and oIPSC5/oIPSC1 at 20, 40, and 120 Hz for blue-light (hChR2) and red-light (ChrimsonR) stimulation (Figures 7C, 7D, and S10; Table S3). To compare responses between groups, we calculated a blue:red index, defined as the arithmetic difference between the blue oIPSC2/oIPSC1 or oIPSC5/oIPSC1 ratio and the red oIPSC2/ oIPSC1 or oIPSC5/oIPSC1 ratio. A value of zero reflects no difference in the dynamics of the response to red and blue light and suggests and the somatically generated response is equally efficacious to the axo-synaptic response during repetitive stimulation; this index yields a positive value if there is a greater decrease in oIPSC amplitude with successive stimulation in response to red (somatic) relative to blue light and reflects enhanced efficacy of the axo-synaptic response during repetitive stimulation (Figure 7E; Table S3).
There was no significant difference between genotypes or ages in the axon- and synaptic-terminal-evoked oIPSC2/oIPSC1 and oIPSC5/oIPSC1 ratio. However, we found marked differences in soma-evoked oIPSC5/oIPSC1 and oIPSC2/oIPSC1 ratio between groups (Figure S10; Table S3). This yielded a blue:red index value near zero for Scn1a+/− P16–P21 SURVIVOR as well as WT P16–P21 and P35–P56, indicating no difference in the frequency-dependent dynamics of the oIPSC between somatic and axo-synaptic photostimulation in these groups. However, we found that recordings in Scn1a+/− P16–P21 SUDEP and Scn1a+/− P35–P56 mice exhibited significantly larger values for this blue:red index (Figure 7E; Table S3), indicating a relative reduction in the somatically evoked oIPSC response. For example, the oIPSC5/oIPSC1 index at 120 Hz was 0.11 ± 0.03 for Scn1a+/− P16–P21 SUDEP (n = 18 PCs from eight mice), −0.01 ± 0.04 for Scn1a+/− P16–P21 SURVIVOR (n = 30 PCs from 10 mice; p = 0.0424 versus Scn1a+/− P16–P21 SUDEP via mixed-effects model), 0.10 ± 0.04 for Scn1a+/− P35–P56 (n = 26 PCs from six mice; p = 0.1563 versus Scn1a+/− P16–P21 SURVIVOR), −0.01 ± 0.03 for WT P16–P21 (n = 26 PCs from eight mice; p = 0.0424 versus Scn1a+/− P16–P21 SUDEP; p = 0.9042 versus Scn1a+/− P16–P21 SURVIVOR), and −0.02 ± 0.02 for WT P35–P56 (n = 28 PCs from eight mice; p = 0.0431 versus Scn1a+/− P35–P56 and 0.8723 versus WT P16–P21). We confirmed that PV-INs at all age groups could reliably follow brief (1-ms) pulses of red-light photostimulation at 120 Hz (Figure S10).
This result is consistent with a frequency- and activity-dependent failure of synaptic transmission with somatic stimulation that can be “bypassed” via direct axo-synaptic stimulation. Such findings support the results of the multiple whole-cell electrophysiology experiments and modeling, indicating that failures of synaptic transmission reflect frequency-dependent failure of the somatic AP to efficiently drive neurotransmitter release at PV-IN synapses in Scn1a+/− mice.
DISCUSSION
Advances in genetic testing in the neurodevelopmental disorders have led to the identification of hundreds of gene associations (Parenti et al., 2020). Understanding how such genetic variation leads to disease has proven challenging (Sahin and Sur, 2015; del Pino et al., 2018). This remains the case for DS, despite being the most common of the epileptic encephalopathies, affecting 1 in 15,000 individuals in the United States (Wu et al., 2015).
Our prior work demonstrated that abnormalities in PV-IN spike generation in Scn1a+/− mice were in fact transient and restricted to early developmental time points (Yu et al., 2006; Ogiwara et al., 2007; Tai et al., 2014; Tsai et al., 2015; De Stasi et al., 2016); PV-INs in young-adult Scn1a+/− P35–P56 mice had recovered the ability to discharge sustained trains of APs at high frequency (Favero et al., 2018). The AIS—defined by ankyrin-G immunoreactivity—was longer in PV-INs from Scn1a+/− mice as compared with WT controls (perhaps suggesting a prior history of decreased excitability; Grubb et al., 2011; Bender and Trussell, 2012; Wefelmeyer et al., 2016; Yamada and Kuba, 2016). In addition, we had found a clear “hump” in the phase plot in PV-INs from Scn1a+/− P35–P56 mice that was not seen in WT P16–P21 or P35–P56 mice (Figure S3A), suggesting spatiotemporal separation of the AIS and somatic spikes, hypothesized to be due to an extension of or distal shift in the location and/or extent of the AIS, perhaps secondary to upregulation of non-Nav1.1 Na+ channel subunits and/or to slowing of spike propagation.
However, Scn1a+/− mice in the chronic phase of the disorder continue to exhibit seizures (albeit at decreased frequency), ASD-related phenotypes, and SUDEP-like phenomena—as is the case with human patients and which cannot be explained by impaired AP generation by PV-INs. Hence, the primary goal of the present study was to investigate PV-IN synaptic transmission as an indirect measure of spike propagation. We hypothesized that PV-INs are able to reconstitute high-frequency firing at the soma via still unidentified mechanisms yet cannot recover axonal excitability resulting from loss of Nav1.1. We confirmed that PV-INs in young-adult Scn1a+/− P35–P56 mice indeed recover fast spiking yet exhibit impaired synaptic transmission, as evidenced by markedly increased failure rates (Figure 3) and prolonged synaptic delay (Figure 4). This establishes PV-IN axonal dysfunction is an important locus of pathology in the chronic phase of DS. Such a conclusion makes logical sense, as prior work showed that low concentrations of TTX estimated to block ~50% of Na+ channels produce only mild effects on AP generation in PV-INs, consistent with the presence of a “supercritical” density of Na+ channels at the AIS (Hu and Jonas, 2014). However, high Na+ channel density is required for spike reliability and propagation speed. While PV-INs in Scn1a+/− P16–P21 mice do exhibit impairments in AP generation, these cells are still able to discharge sustained trains of APs at >200 Hz (Figures 2B and 2C), just as PV-INs in WT mice retain fast-spiking capabilities in the presence of low concentrations of TTX (Figure 5).
The mini-slice technique to evaluate electrophysiological properties across development
Our prior work (Favero et al., 2018; Tran et al., 2020; Mattis et al., 2022) and much of the work in the field is limited by the obvious fact that we could only record from young-adult Scn1a+/− mice that had actually survived. Hence, it could be the case that PV-INs found to exhibit normal high-frequency firing at ≥P35 had always been normal, with PV-IN dysfunction being present only in those mice deceased prior to P35. Hence, a secondary aim of the study was to resolve this issue, which required development of a method to perform targeted recordings from PV-INs from the same animal at two different time points and within-animal comparison. This approach confirmed that PV-IN dysfunction is preset at early time points, even in those animals that survive to young adulthood and allowed us to show that recovery of PV-IN AP generation occurs in the same animals in which PV-IN excitability was directly shown to be dysfunctional at earlier time points. Furthermore, identification of combined abnormalities in spike generation and synaptic transmission in Scn1a+/− mice that exhibit early mortality may represent a marker predictive of SUDEP. This mini-slice technique could prove widely applicable to the study of risk factors for mortality in various preclinical models of severe neurological disease.
We validated the mini-slice approach extensively: in a dedicated cohort of age-matched littermates, we demonstrated that recordings from PV-INs obtained via mini-slice did not differ from those obtained via standard acute brain slice preparation (Figure S4; Table S2). Furthermore, mini-slice performed at P16–P21 had no effect on growth or mortality and did not alter temperature threshold for seizure induction at P35 (Figure S4). This confirms recordings obtained in mini-slice are similar to what would have been obtained via standard acute brain slice and that no obvious long-term impairments were caused by the procedure. It should be noted that inflammation with microglial activation likely occurs at the biopsy site with surrounding neuronal cell death; hence, all experiments in acute slice performed at the second time point were performed in the contralateral hemisphere. This approach allowed us to record from PV-INs in the same layer and brain area at both time points.
Combined early impairments in spike generation and spike propagation as a risk factor for early mortality
PV-INs from all Scn1a+/− mice showed impairments in spike generation at early time points, regardless of survival to or death prior to P35, while PV-INs had recovered high-frequency firing in Scn1a+/− mice surviving to adulthood. However, the fidelity of synaptic transmission at PV-IN:PC synapses was decreased selectively at P16–P21 in those Scn1a+/− mice that subsequently died of SUDEP relative to mice that survived (Figures 3C and 3D). This was confirmed by a dual optogenetics approach that allowed us to separate somatic and axo-synaptic stimulation of PV-INs (Figures 7 and S10) and revealed a relative impairment of somatically generated, but not axo-synaptic oIPSCs (Figure 7E), suggesting that direct stimulation of PV-IN terminals can bypass PV-IN axonal dysfunction. And while PV-INs from Scn1a+/− P35–P56 mice did not show impairments in spike generation (Figures 2, S2C, S2F, and S3, consistent with the results of our previous study; Favero et al., 2018), we found frequency-dependent impairment in synaptic transmission at PV-IN:PC synapses at this young-adult time point, similar to that observed in Scn1a+/− P16–P21 SUDEP mice (Figures 3, 4, and 6). These results suggest that (1) combined early impairments in spike generation and synaptic transmission may be a risk factor for early mortality in Scn1a+/− mice, (2) severe impairments in synaptic transmission are involved in the pathophysiology of continued symptoms of DS in the chronic phase, and (3) targeted modulation of axonal or presynaptic function may represent a therapeutic approach to this disorder.
Implications for development of novel therapies for Dravet syndrome
We provide evidence that PV-INs in Scn1a+/− mice exhibit impaired synaptic transmission, that early impairments in PV-IN synaptic transmission may predict SUDEP risk, and that such deficits persist across development and may contribute to the defining pathology of DS in the chronic phase of the disorder. This has important implications for efforts to increase expression of SCN1A as a therapeutic strategy (Colasante et al., 2019; Han et al., 2020; Yamagata et al., 2020). Upregulation of SCN1A may be ineffective if administered after disease onset if chronic dysfunction is driven by a mechanism other than haploinsufficiency of Nav1.1. Of note, AAV-mediated upregulation of the voltage-gated Na+ channel b1 accessory sub-unit—which regulates α subunit expression, plasma membrane localization, and subcellular targeting, among other functions (O’Malley and Isom, 2015)—led to decreased seizure frequency, improved performance on cognitive tests, and reduced mortality in Scn1a+/− mice (Niibori et al., 2020). Our findings suggest that increasing fidelity of PV-IN synaptic transmission, perhaps via upregulation of Nav1.1 specifically in the axo-synaptic compartment, such as at nodes and/or axonal branch points (Micheva et al., 2016; Stedehouder et al., 2017; Benamer et al., 2020) or by otherwise bypassing axonal dysfunction, may be important for the treatment of DS.
Limitations of the study
A limitation of the present study is the lack of detailed anatomical reconstruction of individual PV-INs. However, our conclusions are supported by the results of the computational modeling and the dual optogenetics experiment. Yet some questions remain unanswered, such as the mechanistic basis of the observed differences in synaptic transmission between the Scn1a+/− P16–P21 SUDEP and SURVIVOR conditions.
We consider it unlikely that the main findings could be due exclusively to changes in presynaptic calcium channels at PV-IN synapses in Scn1a+/− mice, such as in functional properties, density, and/or subtype identity. Were this the case, one would expect a change in short-term synaptic plasticity (summarized in Table S1). It is not clear how such an alternative explanation could account for the stochastic yet frequency-dependent nature of the observed failures of synaptic transmission.
There is unlikely to be a singular cellular or circuit mechanism of DS or of any complex neurodevelopmental disorder. It is known that excitability of somatostatin (SST) and vasoactive intestinal peptide (VIP)-INs are also impaired in DS (Tai et al., 2014; Rubinstein et al., 2015; Almog et al., 2021) and that VIP-IN dysfunction persists in young-adult Scn1a+/− mice (Goff and Goldberg, 2019). The present work focused on neocortical PV-INs, yet it is clear that the hippocampus, and, in particular, the dentate gyrus, is a prominent locus of pathology in DS models (Liautard et al., 2013; Dutton et al., 2017; Stein et al., 2019; Mattis et al., 2022) and in human patients (Van Poppel et al., 2012; Gaily et al., 2013).
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Ethan M. Goldberg (goldberge@chop.edu).
Materials availability
This study did not generate any new reagents.
Data and code availability
Source data is available at https://doi.gin.g-node.org/10.12751/g-node.4adqko and from the corresponding author upon request.
All original code has been deposited at GitHub (https://github.com/GoldbergNeuroLab/Kaneko-et-al.−2022) and at https://doi.org/10.5281/zenodo.6298722 and is publicly available as of the date of publication.
Any additional information required to reanalyse the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All procedures and experiments were approved by the Institutional Animal Care and Use Committee at The Children’s Hospital of Philadelphia and were conducted in accordance with the ethical guidelines of the National Institutes of Health. Both male and female mice were used in equal proportions; the key genotype- and age-dependent group differences were observed for both male and female mice (not shown). After weaning at P21, mice were group-housed with up to five mice per cage and maintained on a 12-h light/dark cycle with ad libitum access to food and water.
Mouse strains used in this study included: Scn1a+/− mice on a 129S6.SvEvTac background (RRID: MMRRC_037107-JAX) generated by a targeted deletion of exon 1 of the Scn1a gene, PV-Cre mice (B6; 129P2-Pvalbtm1(cre)Arbr/J; RRID:IMSR_JAX:017320); tdTomato reporter/Ai14 mice (Rosa-CAG-LSL-tdTomato; RRID: IMSR_JAX:007914; on a C57BL/6J background); wild-type 129S6.SvEvTac (Taconic Biosciences model #129SVE; RRID:IMSR_TAC: 129sve); and wild-type C57BL/6J (RRID:IMSR_JAX:000664).
Homozygous PV-Cre mice were crossed to homozygous Ai14 mice to generate PV-Cre.tdT double heterozygotes on a C57BL/6J background. Female PV-Cre.tdT double heterozygotes were then crossed to male 129S6.Scn1a+/− mice to generate Scn1a.PV-Cre.tdT mice and WT.PV-Cre.tdT littermate controls. The genotype of all mice was determined via PCR of tail snips obtained at P7 and was re-confirmed for each mouse after they were sacrificed for slice preparation. All mice used for experiments were on a strict 50:50 129S6:C57BL/6J genetic background, and Scn1a+/− mice on this background have been shown to replicate the salient core phenotypic features of Dravet Syndrome observed in human patients (Miller et al., 2014; Mistry et al., 2014). We observed similar rates of spontaneous death (22/84, or 26.2% by P56) for Scn1a.PV-Cre.tdT mice in a prior study (Favero et al., 2018).
METHOD DETAILS
Viral injections
Adeno-associated viruses (AAV) encoding the red-shifted channelrhodopsin variant ChrimsonR (Klapoetke et al., 2014) tagged with mRuby2 (AAV9.hSyn.DIO.ChrimsonR.mRuby2.ST; Addgene Catalog No. 105448; titer, 2.13 × 1013 GC/mL; made available for distribution by Edward Boyden and Hillel Adesnik) and eYFP-tagged hChR2 tagged (AAV9.EF1a.DIO.hChR2(E123A).EYFP; Addgene Catalog No. 35507; titer, 0.73 × 1013 GC/mL) were injected into S1 through the skull of mice age P0 using a NanoFil syringe (10 μL; WPI) under ice anesthesia. Mice recovered on a heating pad within 10 min and were placed in their home cage.
Acute slice preparation
Mice were deeply anesthetized with 5% isoflurane and transcardially perfused with ice-cold aCSF containing (in mM): KCl, 2.5; NaH2PO4, 1.25; HEPES, 20; N-Methyl-D-glucamine (NMDG), 93; L-ascorbic acid (sodium salt), 5; thiourea, 2; sodium pyruvate, 3; CaCl2, 0.5; MgSO4, 10; D-glucose, 25; N-acetyl-L-cysteine, 12; NaHCO3, 30; with pH adjusted to 7.30 with HCl and osmolarity adjusted to 310 mOsm, and equilibrated with 95% O2 and 5% CO2. Slices recovered in modified aCSF for 30 min at 32°C. Slices were transferred to standard (recording) aCSF containing (in mM): NaCl, 125; KCl, 2.5; MgSO4, 2; NaH2PO4, 1.25; CaCl2, 2; D-glucose, 20; NaHCO3, 26. Slices were maintained at room temperature for up to 3 h before recording. Slices were then transferred to a recording chamber on the stage of an upright microscope (Olympus) and continuously perfused using a diaphragm pump (Smoothflow Pump Q Series, Model #Q-100-TT-ULP-ES, TACMINA CORPORATION, Japan) and suction (PC-21, NARISHIGE, Japan) with recording aCSF at a rate of 3 mL/min at 30 ± 1°C.
Mini-slice preparation
Mice were placed in a stereotaxic apparatus (Kopf Model #963) and deeply anesthetized via inhalation of 3–5% isoflurane in oxygen for induction and maintained at 1.5–2% during the surgery. Under sterile conditions, a 3 mm diameter craniotomy was made overlying primary somatosensory cortex using a hand-held drill (Osada EXL-M40) equipped a 1.0 mm diameter drill bit (NeoBurr #EF4). Then, four shallow cuts were made using a #10 scalpel blade in a 1–2 mm square, avoiding surface arteries/arterioles and veins/venules. The rectangular approximately 2 × 2 × 0.8–1.0 mm depth biopsy specimen was then removed using the tip of the scalpel blade (Figure 1B1). The collected tissue was immediately placed into ice-cold, oxygenated, modified aCSF (see Acute slice preparation) containing 3% low-melting point agarose (Lonza; Catalog Number 50101). The tissue block was allowed to harden in a plastic mold on ice, oriented so as to obtain coronal sections to facilitate subsequent visualization of neocortical layers, and then mounted immediately on the specimen holder of a Leica VT-1200S vibratome using cyanoacrylate glue and sliced at 300 μm. Hemostasis was achieved with Gelfoam® sterile sponges (Pfizer) soaked in aCSF. The craniotomy bone flap was replaced and the scalp was sutured. Mini-slices were then subject to the same post-sectioning and recording procedures as acute brain slices (see above).
Recordings of electrophysiological membrane/firing properties and unitary IPSCs
PV-INs were identified by tdTomato expression visualized with epifluorescence and displayed non-accommodating trains of relatively brief action potentials. Spiny stellate cells (PCs; principal cells) were identified by morphology under infrared differential interference contrast (IR-DIC) and the presence of a regular-spiking firing pattern. Whole-cell recordings were obtained from layer 4 primary somatosensory cortex (S1; “barrel”). Patch pipettes were pulled from borosilicate glass using a P-97 puller (Sutter Instruments) and filled with intracellular solution containing (in mM): K-gluconate, 65; KCl, 65; MgCl2, 2; HEPES, 10; EGTA, 0.5; Phosphocreatine-Tris2, 10; ATP (magnesium salt), 4; GTP (sodium salt), 0.3; pH was adjusted to 7.30 with KOH, and osmolarity adjusted to 290 mOsm. Pipettes had a resistance of 3–4 MΩ when filled and placed in recording solution. Unitary IPSCs were obtained via 2–6 simultaneous patch clamp recordings from neurons located within 100 μm of one another in a mini-slice in pre-weaning mice (Figure 1B) or a thalamocortical slice (Agmon and Connors, 1991) in young adult mice (Figure 3A1 and 3A2). Recordings in young adult mice were performed in sensorimotor cortex of the hemisphere contralateral to that of prior mini-slice biopsy.
Voltage was sampled at 100 kHz with a MultiClamp 700B amplifier (Molecular Devices), filtered at 2 kHz by Lowpass and Bessel filters, digitized using a DigiData 1550B, and acquired using pClamp11 software. Recordings were discarded if the cell had an unstable resting membrane potential and/or a membrane potential of PV-INs greater (less negative) than −60 mV, or if access resistance increased by > 20% during the recording. We did not correct for liquid junction potential.
Recordings of optogenetic evoked IPSCs
PV-INs were identified by mRuby2 expression visualized with epifluorescence and PCs were identified as above. To record optogenetically-evoked spikes in PV-INs (Figure S10), we used a regular intracellular solution containing (in mM): K-gluconate, 130; KCl, 6.3; MgCl2, 1; HEPES, 10; EGTA, 0.5; Phosphocreatine-Tris2, 10; ATP (magnesium salt), 4; GTP (sodium salt), 0.3; pH was adjusted to 7.30 with KOH, and osmolarity adjusted to 290 mOsm. To record optogenetic evoked IPSCs in PCs (Figure 7), we used a cesium-based intracellular solution containing, in mM: cesium methanesulfonate, 125; HEPES, 15; EGTA, 0.5; QX-314, 2; Phosphocreatine-Tris2, 10; tetraethylammonium chloride, 2; ATP (magnesium salt), 4; GTP (sodium salt), 0.3; pH was adjusted to 7.30 with KOH, and osmolarity adjusted to 290 mOsm.
Immunohistochemistry
For visualization of PV-IN axonal boutons, microbiopsies collected from P16–21 mice were drop-fixed in 4% PFA. Mice from P35–56 mice were deeply anesthetized with isoflurane, then transcardially perfused with 10–15 mL of 4% PFA in PBS and postfixed for 24 h. Brain tissue was then transferred to 30% sucrose solution for 24–48 h and then 40 μm coronal sections through S1 were made using a sliding microtome (American Optical). After washing in PBS, the slices were blocked (3% normal goat serum, 2% BSA, 0.3% Triton X-100 in PBS) for 1 h at room temperature to decrease nonspecific staining, and then washed in PBS. The sections were then transferred into solution containing mouse-α-synaptotagmin-2 primary antibody (1:5000; ZFIN ZDB-ATB-081002–25) with 1% normal goat serum, 0.2% BSA, and 0.3% Triton X-100 in PBS at 4°C overnight in the dark. After washing in PBS, the incubated sections were transferred into a solution containing the secondary antibody (Alexa Fluor 488-conjugated goat anti-mouse IgG2a; 1:500, Molecular Probes A21131) with 2% BSA and 0.3% Triton X-100 in PBS at 4°C overnight in the dark. Sections were then mounted on glass slides and imaged using a confocal microscope (Leica SP8) equipped with a 40X objective (Leica HC PL APO 40X NA 1.30 OIL CS2). Bouton density was counted surrounding putative principle cell somata in a randomly selected 100 × 100 μm area in layer 4 primary somatosensory neocortex.
For validation of optogenetic experiments, Scn1a.PV-Cre.tdT and WT.PV-Cre.tdT mice injected in the subdural space with AAV9.hSyn.DIO.ChrimsonR.mRuby2.ST and AAV9.EF1a.DIO.hChR2(E123A).EYFP at P0 and processed for immunohistochemistry at P35–56 as described above. Tissue sections were transferred to a solution containing rabbit anti-PV antibody (1:1000; Swant PV27), 1% normal goat serum, 0.2% BSA, and 0.3% Triton X-100 in PBS, and incubated at 4°C overnight in the dark. After washing in PBS, the incubated sections were transferred into solution containing secondary antibody (Alexa Fluor 405; 1:500, Thermo Fisher A31556) with 2% BSA and 0.3% Triton X-100 in PBS at 4°C overnight in the dark. Sections were mounted on glass slides and confocal microscopy was performed to visualize cells immunopositive for PV and co-expressing hChR2-eYFP and ChrimsonR-mRuby2.
Na + channel immunohistochemistry was performed on free-floating brain tissue sections following light fixation and permeabilization as described previously (Goff and Goldberg, 2019). Briefly, isoflurane-anesthetized mice were transcardially perfused with 1% paraformaldehyde and 0.5% MeOH in PBS; then, the brain were removed and post-fixed in perfusate at room temperature for 1 h 50 μm thick sections were cut on a Leica VT-1200S vibratome, and then blocked and permeabilized with 0.2% Triton X-100 (Sigma) and 10% normal goat serum in PBS for one hour at room temperature. Slices were stained overnight at 4°C with a primary antibody directed against Nav1.1 (NeuroMab K74/71) or Pan-Nav (NeuroMab N419/40) in PBS with 3% BSA (BSA, Sigma) and 0.2% Triton X-100. The following day, the slices were washed with PBS and stained with a secondary antibody, Alexa Fluor 488-conjugated goat anti-mouse IgG1 or IgG2 respectively (Molecular Probes), in PBS, with 3% BSA and 0.2% Triton X-100. Slices were mounted, cover-slipped, and sealed before imaging on a Leica TCS SP8 confocal microscope. All imagining was done at 80X using identical laser power, exposure, and averaging parameters. Several Z-stacks of Layer IV barrel cortex and spanning the thickness of each section were taken using 1 μm steps for each sample. The dimensions of each z stack were 150 μm × 150 μm × 20–40 μm thickness.
For automated analysis of confocal images, Z-stacks were first imported into ImageJ, and the green channel (Nav1.1 or Pan-Nav) was exported as a.tif image sequence excluding any sections near the surface of the slice which tended to show brighter non-specific labeling. These sequences were imported into MATLAB, where each image was segmented in the following sequence: each plane in the sequence was binarized using Otsu’s method to determine a global threshold; then, the activecontour function was used to refine the generated mask for 100 iterations using the edge detection method; and finally, the image mask was refined using imopen and imfill. The utilized functions are all available through the MATLAB Image Processing and computer vision toolbox, and accessed through the image segmenter app. A fraction of the segmented images were checked against a blind scorer and were found to be in good agreement with well-defined Nav1.1 and Pan-Nav puncta. Puncta with a total length smaller than 2 μm or with an area greater than 160 μm2 were filtered out of analysis, as these were not usually reported as staining by a blind scorer but rather seemed to be non-specific labeling of large structures like blood vessels/nuclei and visual noise. The individual images were then reconstructed into a 3D volume, and segmentation analysis was done in 3D using the regionprops function. From this, we extracted the major and minor axis length, number of branch points, and a 2D flattened representation of each puncta for calculation of linearity as the ratio of the major to the minor axis. Mean intensity values for each puncta were normalized by subtracting the median background value of each slice of the z stack. “Branches” were defined by the built-in MATLAB function as the number of branch points for an individual puncta, with a “branchpoint” corresponding to a point on the extracted 3D volume where a pixel splits into two or more structures. Linearity was defined as the unit-less ratio of major to minor axis.
Loss of Nav1.1 in Scn1a+/− mice results in lower fluorescence intensity (Figures S7H and S7I). Because of this, we did not to use a simple threshold level for puncta detection; instead, the multistep algorithm described above achieves greater consistency across experiments and tissue sections with different overall fluorescence intensity levels and maintained performance in conditions with relatively lower fluorescence intensity and corrected for near-threshold “dropout” of low(er) intensity puncta in Scn1a+/− mice.
Electrophysiology data analysis
All analysis was performed manually in Clampfit (pCLAMP) or using routines written in MATLAB. Resting membrane potential (Vm) was calculated using the average value of a 1 s sweep with no direct current injection. Input resistance (Rm) was calculated using the response to hyperpolarizing current injections near rest using Rm = V/I. AP threshold was calculated as the value at which the derivative of the voltage (dV/dt) first reached 10 mV/ms. Spike height refers to the absolute maximum voltage value of an individual AP, while spike amplitude was calculated as the difference between spike height and AP threshold for a given AP. AP rise time is the time from AP threshold to the peak of the AP. AP half-width (AP 1/2-width) is defined as the width of the AP (in ms) at half-maximal amplitude. AP afterhyperpolarization (AHP) amplitude is calculated as the depth of the afterhyperpolarization (in mV) relative to AP threshold. Unless indicated, all quantification of single spike properties was done using the first AP elicited at rheobase.
Rheobase was determined as the minimum current injection that elicited APs using a 600 ms sweep at 25 pA intervals. Maximal instantaneous firing was calculated using the smallest interspike interval (ISI) elicited at near-maximal current injection. Maximal steady-state firing was defined as the maximal mean firing frequency during the last 300 ms of a suprathreshold 600 ms current injection, with a minimum requirement for a spike being an amplitude of 40 mV with a clear AP threshold of dV/dt > 10 mV/ms and height overshooting at least 0 mV. All I-f plots were created using the steady-state firing calculated for each current step, counting failures as 0 for subsequent current steps.
The amplitude of the unitary IPSC was calculated as the difference between baseline and peak from the average of 10–20 consecutive sweeps obtained with a 15 s inter-sweep interval to facilitate recovery from short-term synaptic depression. The paired pulse ratio (PPR) was calculated from this data as the ratio of the second (IPSC2) to the first IPSC. At 40 and 120 Hz, the amplitude of the subsequent IPSCs in the train was calculated as the difference between the peak amplitude and the extrapolation of the single exponential fit to the decay of the preceding IPSC. uIPSC latency was measured from the peak of the presynaptic AP to the onset of the IPSC. Failure was defined as the absence of a transient current greater than 5 pA occurring within 5 ms after the presynaptic AP.
For the contingency analysis, all unitary connections from Scn1a+/− mice exhibiting IPSC1 failure were pooled. We defined Type 1 IPSC2 as the IPSC2 following success of IPSC1, and Type 2 IPSC2 as IPSC2 following a failure of IPSC1.
NEURON model
A detailed interneuron model was derived from that specified in (Berecki et al., 2019). Data on neuronal morphology and ion channel biophysical properties were from the Blue Brain Project neocortical microcircuit portal (https://portal.bluebrain.epfl.ch/resources/models; Ramaswamy et al., 2015), and formulated using Hodgkin-Huxley kinetics. Previously, values were fitted using BluePyOpt to fit the electrophysical characteristics of continuous non-accommodating fast-spiking PV-INs. Here, the model was adapted to explore the effects of altered Nav1.1 conductances in the axon initial segment (AIS) and the nodes of Ranvier (“Nodes”) by spacing a 1 μm node every 33 μm (Stedehouder et al., 2017). In accordance with existing data, Nodes contained Nav1.1 and Kv3 channels with soma-to-axon and Na + -to-K+ conductance ratios per (Hu et al., 2018). Simulations were performed using NEURON and Python. Temp was 34°C. Current was injected at the soma and action potentials recorded at 26.5 μm (the length of the AIS) and 1056.5 μm (pre-synaptic terminal and last node). The current was either continuous (i.e., a rectangular pulse delivered in current-clamp) or pulsed at specific frequencies. An action potential was measured if the membrane potential crossed −20 mV. Nav1.1 was selectively deleted at specific compartments, proportional to the baseline conductance of each compartment.
As in Figure 6, failure rate was the number of failures divided by total stimuli. A failure could be that of stimulation (a pulse was sent but an action potential was not recorded, for any reason, at the pre-synaptic terminal within 5 ms), initiation (a pulse was sent but an action potential was not recorded at the AIS), or propagation (an action potential was recorded at the AIS but not at the pre-synaptic terminal).
To assess potentials mechanisms that could exhibit functional recovery as seen in Scn1a+/− P35–56 mice (action potential generation recovery but still exhibiting impaired propagation), Nav1.1 conductances were either redistributed from the nodes to the AIS, or another transient sodium conductance, imitating a possible source of Nav1.3 or Nav1.6 channels, was upregulated. The necessary amount redistribution and upregulation for the model was determined quantitatively (Figure S9).
QUANTIFICATION AND STATISTICAL ANALYSIS
Linear mixed-effects modeling of the data was performed to account for the fact that more than one cell or synaptic connection was recorded from some mice. The model was fitted to the data with mouse as a random factor. The model fitting was implemented in MATLAB using the fitlme function, and normality of residues was confirmed using qqplot. Post-hoc Holm Sidak correction was applied for the deduced p values to correct for multiple comparison performed between experimental groups. Statistical significance was defined as p < 0.05 after post-hoc correction with p values reported exactly. All values are reported as mean ± SEM. Numbers for mice, cells, and synapses, are reported in the text, Figure Legends, and/or Supplemental Tables.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit anti-parvalbumin | Swant | Cat# PV27; RRID:AB_2631173 |
| ZNP-1 (Mouse anti-synaptotagmin 2) | ZFIN | Cat# ZDB-ATB-081002-25; RRID:AB_10013783 |
| Mouse anti-Nav1.1 (K74/71) | NeuroMab | Cat# 75–023; RRID:AB_2877321 |
| Mouse anti-Pan-Nav1 (N419/40) | NeuroMab | Cat# 75–405; RRID:AB_2877589 |
| Alexa Fluor 405 goat anti-rabbit | Molecular Probes | Cat# A31556 |
| Alexa Fluor 488 goat anti-mouse (IgG2a) | Molecular Probes | Cat# A21131 |
| Alexa Fluor 488 goat anti-mouse (IgG1) | Molecular Probes | Cat# A21121 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| pAAV-hSyn-DIO-ChrimsonR-mRuby2-ST | Addgene | Cat# 105448-AAV9 |
| pAAV-Ef1a-DIO-hChR2(E123A)-EYFP | Addgene | Cat# 35507-AAV9 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Tetrodotoxin citrate | abcam | Cat# ab120055 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Wild-type 129S6.SvEvTac Mice | Taconic Biosciences | RRID:IMSR_TAC:129sve |
| Wild-type C57BL/6J Mice | Jackson Laboratory | RRID:IMSR_JAX:000664 |
| Scn1a+/− Mice | Laboratory of Jennifer Kearney, Northwestern University | RRID:MMRRC_037107-JAX |
| Pvalb-Cre Mice | Jackson Laboratory | RRID:IMSR_JAX:017320 |
| Ai14 Mice | Jackson Laboratory | RRID:IMSR_JAX:007914 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Primer: Jax007914/Ai14D wt-f: AAG GGA GCT GCA GTG GAG TA | Integrated DNA Technologies | N/A |
| Primer: Jax007914/Ai14D wt-r: CCG AAA ATC TGT GGG AAG TC | Integrated DNA Technologies | N/A |
| Primer: Jax007914/Ai14D mt-f: GGC ATT AAA GCA GCG TAT CC | Integrated DNA Technologies | N/A |
| Primer: Jax007914/Ai14D mt-r: CTG TTC CTG TAC GGC ATG G | Integrated DNA Technologies | N/A |
| Primer: Jax017320/PvCre mt-f: AAA TGC TTC TGT CCG TTT GC | Integrated DNA Technologies | N/A |
| Primer: Jax017320/PvCre mt-r: ATG TTT AGC TGG CCC AAA TG | Integrated DNA Technologies | N/A |
| Primer: Jax017320/PvCre wt-f: CAG AGC AGG CAT GGT GAC TA | Integrated DNA Technologies | N/A |
| Primer: Jax017320/PvCre wt-r: AGT ACC AAG CAG GCA GGA GA | Integrated DNA Technologies | N/A |
| Primer: MMRRC_037107-JAX/Scn1atm1Kea, common-f: AGT CTG TAC CAG GCA GAA CTT G | Integrated DNA Technologies | N/A |
| Primer: MMRRC_037107-JAX/Scn1atm1Kea, wt-r: CCC TGA GAT GTG GGT GAA TAG | Integrated DNA Technologies | N/A |
| Primer: MMRRC_037107-JAX/Scn1atm1Kea, mt-r: AGA CTG CCT TGG GAA AAG CG | Integrated DNA Technologies | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| pClamp 11.0 | Molecular Devices | N/A |
| MATLAB | MathWorks | N/A |
| GraphPad Prism 9.0.1 | GraphPad Software Inc. | RRID: SCR_002798 https://www.graphpad.com/scientific-software/prism/ |
| Adobe Illustrator 2020 | Adobe | N/A |
| Fiji:ImageJ | Open Source at https://fiji.sc | RRID:SCR_002285 |
| BioRender | BioRender | N/A |
| Source data | This paper | https://doi.gin.g-node.org/10.12751/g-node.4adqko |
| Resource code | This paper | https://doi.org/10.5281/zenodo.6298722 |
Highlights.
“Mini-slices” allow recording of neurons from the same mouse at two time points
PV interneurons show transient impairment of spike generation in Dravet syndrome mice
Dysfunction of PV-IN synaptic transmission is long lasting
ACKNOWLEDGMENTS
We would like to thank Bernardo Rudy, Joanna Mattis, and Laura Mcgarry for comments on a previous version of the manuscript; Xiaohong Zhang for expert technical support and mouse colony maintenance; Melody Cheng for assistance with generation of the graphical abstract; and Jennifer Kearney for the gift of Scn1a+/− mice. This work was supported by the National Institute of Neurological Disorders and Stroke of the National Institutes of Health under F31NS111803 (to K.M.G.) and K08NS097633 and R01NS110869 (to E.M.G.), the Dravet Syndrome Foundation (to A.S.), an ERC Consolidator Grant (SYNAPSEEK) (to T.P.V.), and the NOMIS Foundation through the NOMIS Fellowships program at IST Austria (to C.C.). The graphical abstract was prepared using BioRender software (BioRender.com).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
We worked to ensure sex balance in the selection of non-human subjects. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110580.
REFERENCES
- Agmon A, and Connors BW (1991). Thalamocortical responses of mouse somatosensory (barrel) cortex in vitro. Neuroscience 41, 365–379. [DOI] [PubMed] [Google Scholar]
- Ali AB, Rossier J, Staiger JF, and Audinat E (2001). Kainate receptors regulate unitary IPSCs elicited in pyramidal cells by fast-spiking interneurons in the neocortex. J. Neurosci. 21, 2992–2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almog Y, Fadila S, Brusel M, Mavashov A, Anderson K, and Rubinstein M (2021). Developmental alterations in firing properties of hippocampal CA1 inhibitory and excitatory neurons in a mouse model of Dravet syndrome. Neurobiol. Dis. 148, 105209. [DOI] [PubMed] [Google Scholar]
- Baker CA, Elyada YM, Parra A, and Bolton MML (2016). Cellular resolution circuit mapping with temporal-focused excitation of soma-targeted channelrhodopsin. Elife 5, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beierlein M, Gibson JR, and Connors BW (2003). Two dynamically distinct inhibitory networks in layer 4 of the neocortex. J. Neurophysiol. 90, 2987–3000. [DOI] [PubMed] [Google Scholar]
- Benamer N, Vidal M, Balia M, and Angulo MC (2020). Myelination of parvalbumin interneurons shapes the function of cortical sensory inhibitory circuits. Nat. Commun. 11, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender KJ, and Trussell LO (2012). The physiology of the axon initial segment. Annu. Rev. Neurosci. 35, 249–265. [DOI] [PubMed] [Google Scholar]
- Berecki G, Bryson A, Terhag J, Maljevic S, Gazina EV, Hill SL, and Petrou S (2019). SCN1A gain of function in early infantile encephalopathy. Ann. Neurol. 85, 514–525. [DOI] [PubMed] [Google Scholar]
- Berkvens JJL, Veugen I, Veendrick-Meekes MJBM, Snoeijen-Schouwenaars FM, Schelhaas HJ, Willemsen MH, Tan IY, and Aldenkamp AP (2015). Autism and behavior in adult patients with Dravet syndrome (DS). Epilepsy Behav. 47, 11–16. [DOI] [PubMed] [Google Scholar]
- Boudkkazi S, Carlier E, Ankri N, Caillard O, Giraud P, Fronzaroli-Molinieres L, and Debanne D (2007). Release-dependent variations in synaptic latency: a putative code for short- and long-term synaptic dynamics. Neuron 56, 1048–1060. [DOI] [PubMed] [Google Scholar]
- Boudkkazi S, Fronzaroli-Molinieres L, and Debanne D (2011). Presynaptic action potential waveform determines cortical synaptic latency. J. Physiol. 589, 1117–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouhours B, Gjoni E, Kochubey O, and Schneggenburger R (2017). Synaptotagmin2 (syt2) drives fast release redundantly with syt1 at the output synapses of parvalbumin-expressing inhibitory neurons. J. Neurosci. 37, 4604–4617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun JD, Hawkins NA, Zachwieja NJ, and Kearney JA (2017). Cacna1g is a genetic modifier of epilepsy in a mouse model of Dravet syndrome. Epilepsia 58, e111–e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceulemans B, Schoonjans AS, Marchau F, Paelinck BP, and Lagae L (2016). Five-year extended follow-up status of 10 patients with Dravet syndrome treated with fenfluramine. Epilepsia 57, e129–e134. [DOI] [PubMed] [Google Scholar]
- Colasante G, Lignani G, Brusco S, Di Berardino C, Carpenter J, Giannelli S, Valassina N, Bido S, Ricci R, Castoldi V, et al. (2019). dCas9-based Scn1a gene activation restores inhibitory interneuron excitability and attenuates seizures in Dravet syndrome mice. Mol. Ther. 28, 235–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper MS, Mcintosh A, Crompton DE, McMahon JM, Schneider A, Farrell K, Ganesan V, Gill D, Kivity S, Lerman-Sagie T, et al. (2016). Mortality in Dravet syndrome. Epilepsy Res. 128, 43–47. [DOI] [PubMed] [Google Scholar]
- De Stasi AM, Farisello P, Marcon I, Cavallari S, Forli A, Vecchia D, Losi G, Mantegazza M, Panzeri S, Carmignoto G, et al. (2016). Unaltered network activity and interneuronal firing during spontaneous cortical dynamics in vivo in a mouse model of severe myoclonic epilepsy of infancy. Cereb. Cortex 26, 1778–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Pino I, Rico B, and Marin O (2018). Neural circuit dysfunction in mouse models of neurodevelopmental disorders. Curr. Opin. Neurobiol. 48,174–182. [DOI] [PubMed] [Google Scholar]
- Dravet C (2011). The core Dravet syndrome phenotype. Epilepsia 52, 3–9. [DOI] [PubMed] [Google Scholar]
- Dutton SBB, Dutt K, Papale LA, Helmers S, Goldin AL, and Escayg A (2017). Early-life febrile seizures worsen adult phenotypes in Scn1a mutants. Exp. Neurol. 293, 159–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinoza C, Guzman SJ, Zhang X, and Jonas P (2018). Parvalbumin+ interneurons obey unique connectivity rules and establish a powerful lateral-inhibition microcircuit in dentate gyrus. Nat. Commun. 9, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favero M, Sotuyo NP, Lopez E, Kearney JA, and Goldberg EM (2018). Atransient developmental window offast-spiking interneuron dysfunction in a mouse model of dravet syndrome. J. Neurosci. 38, 7912–7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaily E, Anttonen AK, Valanne L, Liukkonen E, Traskelin AL, Polvi A, Lommi M, Muona M, Eriksson K, and Lehesjoki AE (2013). Dravet syndrome: new potential genetic modifiers, imaging abnormalities, and ictal findings. Epilepsia 54, 1577–1585. [DOI] [PubMed] [Google Scholar]
- Galarreta M, and Hestrin S (1998). Frequency-dependent synaptic depression and the balance of excitation and inhibition in the neocortex. Nat. Neurosci. 1, 587–594. [DOI] [PubMed] [Google Scholar]
- Goff KM, and Goldberg EM (2019). Vasoactive intestinal peptide-expressing interneurons are impaired in a mouse model of Dravet syndrome. Elife 8, 1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg EM, Watanabe S, Chang SY, Joho RH, Huang ZJ, Leonard CS, and Rudy B (2005). Specific functions of synaptically localized potassium channels in synaptic transmission at the neocortical GABAergic fast- spiking cell synapse. J. Neurosci. 25, 5230–5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg EM, Jeong H-Y, Kruglikov I, Tremblay R, Lazarenko RM, and Rudy B (2011). Rapid developmental maturation of neocortical FS cell intrinsic excitability. Cereb. Cortex 21, 666–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb MS, and Burrone J (2010). Channelrhodopsin-2 localised tothe axon initial segment. PLoS One 5, e13761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb MS, Shu Y, Kuba H, Rasband MN, Wimmer VC, and Bender KJ (2011). Short- and long-term plasticity at the axon initial segment. J. Neurosci. 31, 16049–16055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Z, Chen C, Christiansen A, Ji S, Lin Q, Anumonwo C, Liu C, Leiser SC, Meena, Aznarez I, et al. (2020). Antisense oligonucleotides increase Scn1a expression and reduce seizures and SUDEP incidence in a mouse model of Dravet syndrome. Sci. Transl. Med. 12, eaaz6100. [DOI] [PubMed] [Google Scholar]
- Harkin LA, McMahon JM, Iona X, Dibbens L, Pelekanos JT, Zuberi SM, Sadleir LG, Andermann E, Gill D, Farrell K, et al. (2007). The spectrum of SCN1A-related infantile epileptic encephalopathies. Brain 130, 843–852. [DOI] [PubMed] [Google Scholar]
- Hawkins NA, and Kearney JA (2016). Hlf is a genetic modifier of epilepsy caused by voltage-gated sodium channel mutations. Epilepsy Res. 119, 20–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins NA, Zachwieja NJ, Miller AR, Anderson LL, and Kearney JA (2016). Fine mapping of a Dravet syndrome modifier locus on mouse chromosome 5 and candidate gene analysis by RNA-seq. PLoS Genet. 12, e1006398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins NA, Anderson LL, Gertler TS, Laux L, George AL, and Kearney JA (2017). Screening of conventional anticonvulsants in a genetic mouse model of epilepsy. Ann. Clin. Transl. Neurol. 4, 326–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He N, Li BM, Li ZX, Wang J, Liu XR, Meng H,Tang B, Bian WJ, Shi YW, and Liao WP (2018). Few individuals with Lennox-Gastaut syndrome have autism spectrum disorder: a comparison with Dravet syndrome. J. Neurodev. Disord. 10, 4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, and Jonas P (2014). A supercritical density of Na+ channels ensures fast signaling in GABAergic interneuron axons. Nat. Neurosci. 17, 686–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Gan J, and Jonas P (2014). Interneurons. Fast-spiking, parvalbumin+ GABAergic interneurons:from cellulardesignto microcircuit function. Science 345, 1255263. [DOI] [PubMed] [Google Scholar]
- Hu H, Roth FC, Vandael D, and Jonas P (2018). Complementarytuning of Na+ and K+ channel gating underlies fast and energy-efficient action potentials in GABAergic interneuron axons. Neuron 98, 156–165.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas P, Bischofberger J, Fricker D, and Miles R (2004). Interneuron diversity series: fast in, fast out-temporal and spatial signal processing in hippocampal interneurons. Trends Neurosci. 27, 30–40. [DOI] [PubMed] [Google Scholar]
- Kaplan JS, Stella N, Catterall WA, and Westenbroek RE (2017). Canna- bidiol attenuates seizures and social deficits in a mouse model of Dravet syndrome. Proc. Natl. Acad. Sci. U. S. A. 774,11229–11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan AA, Shekh-Ahmad T, Khalil A, Walker MC, and Ali AB (2018). Cannabidiol exerts antiepileptic effects by restoring hippocampal interneuron functions in a temporal lobe epilepsy model. Br. J. Pharmacol. 175, 20972115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klapoetke NC, Murata Y, Kim SS, Pulver SR, Birdsey-Benson A, Cho YK, Morimoto TK, Chuong AS, Carpenter EJ, Tian Z, et al. (2014). Independent optical excitation of distinct neural populations (Nature Methods (2014) 11,(338–346. Nat. Methods 11, 972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraushaar U, and Jonas P (2000). Efficacy and stability of quantal GABA release at a hippocampal interneuron-principal neuron synapse. J. Neurosci. 20, 5594–5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T,Tian C, Scalmani P, Frassoni C, Mantegazza M, Wang Y, Yang M, Wu S, and Shu Y (2014). Action potential initiation in neocortical inhibitory interneurons. PLoS Biol. 12, e1001944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liautard C, Scalmani P, Carriero G, De Curtis M, Franceschetti S, and Mantegazza M (2013). Hippocampal hyperexcitability and specific epileptiform activity in a mouse model of Dravet syndrome. Epilepsia 54,1251–1261. [DOI] [PubMed] [Google Scholar]
- Mattis J, Somarowthu A, Goff KM, Yom J, Sotuyo NP, McGarry LM, Feng H, Kaneko K, and Goldberg EM (2022). Corticohippocampal circuit dysfunction in a mouse model of Dravet syndrome. eLife 11:e69293. 10.7554/eLife.69293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattis J, Tye KM, Ferenczi EA, Ramakrishnan C, O’Shea DJ, Prakash R, Gunaydin LA, Hyun M, Fenno LE, Gradinaru V, et al. (2012). Principles for applying optogenetic tools derived from direct comparative analysis of microbial opsins. Nat. Methods 9, 159–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei D, Cetica V, Marini C, and Guerrini R (2019). Dravet syndrome as part ofthe clinical and genetic spectrum ofsodium channel epilepsies and encephalopathies. Epilepsia 60, S2–S7. [DOI] [PubMed] [Google Scholar]
- Micheva KD, Wolman D, Mensh BD, Pax E, Buchanan J, Smith SJ, and Bock DD (2016).Alargefraction ofneocortical myelin ensheathesaxons of local inhibitory neurons. Elife 5, 1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AR, Hawkins NA, Mccollom CE, and Kearney JA (2014). Mapping genetic modifiers of survival in a mouse model of Dravet syndrome. Genes Brain Behav. 13, 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry AM, Thompson CH, Miller AR, Vanoye CG, George AL, and Kearney JA (2014). Strain- and age-dependent hippocampal neuron sodium currents correlate with epilepsy severity in Dravet syndrome mice. Neurobiol. Dis. 65, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niibori Y, Lee SJ, Minassian BA, and Hampson DR (2020). Sexually divergent mortality and partial phenotypic rescue after gene therapy in a mouse model of Dravet syndrome. Hum. Gene Ther. 31, 339–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Malley HA, and Isom LL (2015). Sodium channel b subunits: emerging targets in channelopathies. Annu. Rev. Physiol. 77, 481–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, Takeuchi T, Itohara S, Yanagawa Y, Obata K, et al. (2007). Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J. Neurosci. 27, 5903–5914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packer AM, and Yuste R (2011). Dense, unspecific connectivity of neocortical parvalbumin-positive interneurons: a canonical microcircuit for inhibition? J. Neurosci. 31, 13260–13271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parenti I, Rabaneda LG, Schoen H, and Novarino G (2020). Neurodevelopmental disorders: from genetics to functional pathways. Trends Neurosci. 43, 608–621. [DOI] [PubMed] [Google Scholar]
- Pégard NC, Mardinly AR, Oldenburg IA, Sridharan S, Waller L, and Adesnik H (2017). Three-dimensional scanless holographic optogenetics with temporal focusing (3D-SHOT). Nat. Commun. 8, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy S, Courcol JD, Abdellah M, Adaszewski SR, Antille N, Arsever S, Atenekeng G, Bilgili A, Brukau Y, Chalimourda A, et al. (2015). The neocortical microcircuit collaboration portal: a resource for rat somatosensory cortex. Front. Neural Circuits 9, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein M, Han S, Tai C, Westenbroek RE, Hunker A, Scheuer T, and Catterall WA (2015). Dissecting the phenotypes of Dravet syndrome by gene deletion. Brain 138, 2219–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, and Regehr WG (1999). Timing of synaptic transmission. Annu. Rev. Physiol. 61, 521–542. [DOI] [PubMed] [Google Scholar]
- Sahin M, and Sur M (2015). Genes, circuits, and precision therapies for autism and related neurodevelopmental disorders. Science 350, aab3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffer IE (2012). Diagnosis and long-term course of Dravet syndrome. Eur. J. Paediatr. Neurol. 16, S5. [DOI] [PubMed] [Google Scholar]
- Schoonjans A, Paelinck BP, Marchau F, Gunning B, Gammaitoni A, Galer BS, Lagae L, and Ceulemans B (2017). Low-dose fenfluramine significantly reduces seizure frequency in Dravet syndrome: a prospective study of a new cohort of patients. Eur. J. Neurol. 24, 309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RD, and Goldin AL (1998). Functional analysis of the rat I sodium channel in Xenopus oocytes. J. Neurosci. 18, 811–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommeijer JP, and Levelt CN (2012). Synaptotagmin-2 is a reliable marker for parvalbumin positive inhibitory boutons in the mouse visual cortex. PLoS One 7, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stedehouder J, Couey JJ, Brizee D, Hosseini B, Slotman JA, Dirven CMF, Shpak G, Houtsmuller AB, and Kushner SA (2017). Fast-spiking parvalbumin interneurons are frequently myelinated in the cerebral cortex of mice and humans. Cereb. Cortex 27, 5001–5013. [DOI] [PubMed] [Google Scholar]
- Stein RE, Kaplan JS, Li J, and Catterall WA (2019). Hippocampal deletion of Nav1.1 channels in mice causes thermal seizures and cognitive deficit characteristic of Dravet syndrome. Proc. Natl. Acad. Sci. U S A. 116, 16571–16576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai C, Abe Y, Westenbroek RE, Scheuer T, and Catterall WA (2014). Impaired excitability of somatostatin- and parvalbumin-expressing cortical interneurons in a mouse model of Dravet syndrome. Proc. Natl. Acad. Sci. U S A. 111, e3139–e3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran CH, Vaiana M, Nakuci J, Somarowthu A, Goff KM, Goldstein N, Murthy P, Muldoon SF, and Goldberg EM (2020). Interneuron desynchronization precedes seizures in a mouse model of Dravet syndrome. J. Neurosci. 40, 2764–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MS, Lee ML, Chang CY, Fan HH, Yu IS, Chen YT, You JY, Chen CY, Chang FC, Hsiao JH, et al. (2015). Functional and structural deficits of the dentate gyrus network coincide with emerging spontaneous seizures in an Scn1a mutant dravet syndrome model during development. Neurobiol. Dis. 77, 35–48. [DOI] [PubMed] [Google Scholar]
- van Lier M, Hadi Saiepour M, Kole K, Cheyne JE, Zabouri N, Blok T, Qin Y, Ruimschotel E, Alexander Heimel J, Lohmann C, et al. (2020). Disruption of critical period plasticity in a mouse model of neurofibromatosis type 1. J. Neurosci. 40, 5495–5509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Poppel K, Patay Z, Roberts D, Clarke DF, McGregor A, Perkins FF, and Wheless JW (2012). Mesial temporal sclerosis in a cohort of children with SCN1A gene mutation. J. Child Neurol. 27, 893–897. [DOI] [PubMed] [Google Scholar]
- Vickers ED, Clark C, Osypenko D, Fratzl A, Kochubey O, Bettler B, and Schneggenburger R (2018). Parvalbumin-interneuron output synapses show spike-timing-dependent plasticity that contributes to auditory map remodeling. Neuron 99, 720–735.e6. [DOI] [PubMed] [Google Scholar]
- Wefelmeyer W, Puhl CJ, and Burrone J (2016). Homeostatic plasticity of subcellular neuronal structures: from inputs to outputs. Trends Neurosci. 39, 656–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YW, Sullivan J, McDaniel SS, Meisler MH, Walsh EM, Li SX, and Kuzniewicz MW (2015). Incidence of dravet syndrome in a US population. Pediatrics 136, e1310–e1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Z, Huguenard JR, and Prince DA (2002). Synaptic inhibition of pyramidal cells evoked by different interneuronal subtypes in layer V of rat visual cortex. J. Neurophysiol. 88, 740–750. [DOI] [PubMed] [Google Scholar]
- Xiong Z, Yi L, Cao D, He W, Chen J, Gao S, and Sun X (2016). Dravet syndrome with autism inherited from a paternal mosaic heterozygous mutation on SCN1A. J. Neurol. Sci. 369, 53–56. [DOI] [PubMed] [Google Scholar]
- Yamada R, and Kuba H (2016). Structural and functional plasticity at the axon initial segment. Front. Cell. Neurosci. 10, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata T, Raveau M, Kobayashi K, Miyamoto H, Tatsukawa T, Ogiwara I, Itohara S, Hensch TK, and Yamakawa K (2020). CRISPR/dCas9-based Scn1a gene activation in inhibitory neurons ameliorates epileptic and behavioral phenotypes of Dravet syndrome model mice. Neurobiol. Dis. 141, 104954. [DOI] [PubMed] [Google Scholar]
- Yu FH, Mantegazza M, Westenbroek RE, Robbins C.a., Kalume F, Burton K.a., Spain WJ, McKnight GS, Scheuer T, and Catterall WA (2006). Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 9, 1142–1149. [DOI] [PubMed] [Google Scholar]
- Zucker RS, and Regehr WG (2002). Short-term synaptic plasticity. Annu. Rev. Physiol. 64, 355–405. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data is available at https://doi.gin.g-node.org/10.12751/g-node.4adqko and from the corresponding author upon request.
All original code has been deposited at GitHub (https://github.com/GoldbergNeuroLab/Kaneko-et-al.−2022) and at https://doi.org/10.5281/zenodo.6298722 and is publicly available as of the date of publication.
Any additional information required to reanalyse the data reported in this paper is available from the lead contact upon request.