

Abstract
Combination therapies have emerged to mitigate Plasmodium drug resistance, which has hampered the fight against malaria. M5717 is a potent multistage antiplasmodial drug under clinical development, which inhibits parasite protein synthesis. The combination of M5717 with pyronaridine, an inhibitor of hemozoin formation, displays potent activity against blood stage Plasmodium infection. However, the impact of this therapy on liver infection by Plasmodium remains unknown. Here, we employed a recently described 3D culture-based hepatic infection platform to evaluate the activity of the M5717-pyronaridine combination against hepatic infection by P. berghei. This effect was further confirmed in vivo by employing the C57BL/6J rodent Plasmodium infection model. Collectively, our data demonstrate that pyronaridine potentiates the activity of M5717 against P. berghei hepatic development. These preclinical results contribute to the validation of pyronaridine as a suitable partner drug for M5717, supporting the clinical evaluation of this novel antiplasmodial combination therapy.
Keywords: pyronaridine, M5717, combination therapy, liver stage infection, malaria, drug discovery
In 2020 alone, malaria was responsible for more than 600 000 deaths worldwide, resulting from over 200 million infections by Plasmodium parasites, the causative agents of disease. The burden of malaria is mostly felt in the African region and primarily affects children under the age of 5.1
Plasmodium parasites, of which P. falciparum is the deadliest to humans, cycle between a mammalian host and an insect vector. An obligatory and clinically silent pre-erythrocytic stage of Plasmodium infection takes place in the host’s liver cells,2,3 where the parasite develops within a parasitophorous vacuole, giving rise to thousands of red blood cell-infectious merozoites,2,3 leading to the erythrocytic stage of infection. The latter is responsible for the clinical manifestations of malaria4 and for the transmission to the mosquito vector upon ingestion of gametocytes during a blood meal.5
Most pharmacological approaches against malaria focus on the clinically relevant erythrocytic stage of infection. However, the obligatorily nature of the pre-erythrocytic stage of the parasite’s life cycle provides an appealing opportunity for prophylaxis.3 Only few drugs target the hepatic stage of infection, a scarcity that can be explained by the constraints in experimental access to sporozoites and the lack of drug screening models that faithfully recapitulate the liver microenvironment. We have recently established a 3D infection platform, employing spheroids of human hepatic cell lines that maintain a stable hepatic phenotype over the course of 4 weeks and enable the evaluation of drugs against hepatic infection by the rodent P. berghei parasite, in either static or dynamic culture conditions.6
M5717, or DDD107498, a drug candidate under clinical development, was shown to inhibit protein synthesis by targeting P. falciparum’s translation elongation factor 2 (eEF2).7 In preclinical studies, M5717 displayed nanomolar-ranged activity across the life cycle of Plasmodium parasites, including blocking of transmission. The pre-erythrocytic activity of M5717 against P. yoelii and P. berghei infection was also demonstrated in vitro.6,7 Employing our in vitro platform, we estimated a 10 nM minimum inhibitory concentration of M5717, when administered during the hepatic development of Plasmodium.6 This observation correlated with the plasma concentration that cleared liver-stage P. berghei infection in vivo when M5717 was administered as a radical cure6 or as a prophylactic treatment, prior to infection.7 M5717’s novel mechanism of action, potent multistage antiplasmodial activity, and long plasma half-life make it a promising candidate for the treatment and prevention of malaria.8 Since generation of resistance to M5717 was identified in preclinical studies,7 suitable partner drugs have been sought. Pyronaridine (Pyro), a hemozoin formation inhibitor,9−11 was identified as a suitable combination partner for M5717 in a mouse model of human malaria.12 No detrimental pharmacodynamic interactions were observed between the two drugs, and M5717 did not impact the killing rate of Pyro. Moreover, the latter cleared M5717-related resistance and delayed parasite recrudescence.12 The effectiveness of the combination of M5717 and Pyro against pre-erythrocytic stage parasites remains unknown.
In this work, we assessed the impact of M5717 combination with Pyro on the pre-erythrocytic stage of the malaria parasite’s life cycle, employing preclinical models of P. berghei infection. We show that Pyro is not detrimental to the pre-erythrocytic activity of M5717, rather potentiating its activity against P. berghei hepatic infection in vitro and in vivo. Altogether, our results support the selection of Pyro as a suitable partner drug for M5717.
Pre-erythrocytic Activity of the M5717-Pyro Combination
As a hemozoin inhibitor, pyronaridine is not expected to possess liver-stage antiplasmodial activity. However, drug–drug interactions are an important aspect in the development of new combination therapies. Therefore, the effect of the M5717-Pyro combination against the pre-erythrocytic stage of Plasmodium infection was compared to that of M5717 in monotherapy, employing luciferase-expressing P. berghei (Pb-Luc) parasites.
In vitro drug activity was assessed in HepG2 spheroids, as previously established and validated for drug evaluation purposes (Figure 1A).6
Figure 1.
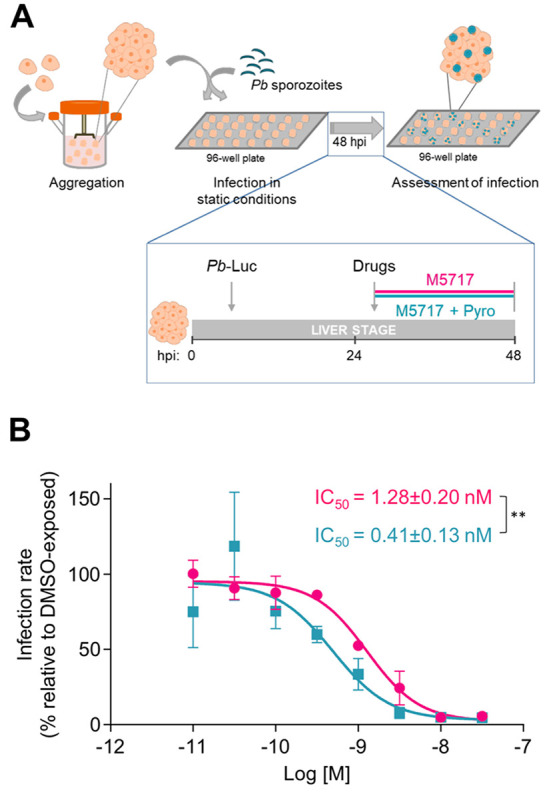
In vitro assessment of the pre-erythrocytic activity of M5717-Pyro combination in Pb-Luc-infected HepG2 spheroids. (A) Schematic representation of the static infection platform employing HepG2 spheroids and Pb-Luc sporozoites (not at scale) and the drug exposure mode used for IC50 determination. (B) Dose–response curves and IC50 determination of M5717 (pink) and M5717-Pyro combination (M5717 + Pyro, blue). Cells were plated at a density of 2.5 × 104 cell/well and infected at a cell:spz ratio of 1:2. Infection rate was normalized to that of DMSO-exposed spheroids and results are represented as the mean ± SD of at least three independent experiments. Statistical analysis was performed using an unpaired t test. ** P ≤ 0.01.
A range of eight concentrations of M5717, employed in monotherapy or combined with a fixed dose of 1 μM of Pyro, was added to Pb-Luc-infected HepG2 spheroids at 24 h post infection (hpi) (Figure 1A). Twenty-four hours later, at 48 hpi, the impact of each drug concentration on the infection rate was assessed by bioluminescence, and the half-maximal inhibitory concentration (IC50) of M5717 alone or in combination with Pyro was determined (Figure 1B). The IC50 of M5717 (1.3 ± 0.2 nM) was in agreement to what had been previously determined in this platform.6 The addition of 1 μM of Pyro resulted in a statistically significant 3-fold reduction of the IC50 of M5717 (0.4 ± 0.1 nM). Importantly, no dose-dependent toxicity toward the host cell was noted, as observed by the percentage of cell viability, which was relatively constant across the experimental conditions tested (Figure S1).
Next, the difference between the impact of the administration of M5717 alone or in combination with Pyro observed in vitro was confirmed in vivo, in a rodent model of P. berghei infection (Figure 2). Mice were injected with Pb-Luc sporozoites, and the success of the liver infection was confirmed by live bioluminescence after 24 h. Drugs were then administered by oral gavage, and their impact on the liver load was determined by real time quantitative PCR (RT-qPCR) 24 h later, at 48 hpi (Figure 2A). A dose of 0.3 mg/kg of M5717 per mouse body weight was selected, as it has previously been shown to not abolish infection completely.6 Pyro was employed at 12 mg/kg of mouse body weight, a dose previously assessed in vivo in combination with M5717, in the context of erythrocytic activity.12 Mice that received the drug vehicle alone were used as controls. Our results show that mice from all experimental groups were similarly infected prior to drug administration (Figure 2B and Figure S2A). Pharmacokinetic analysis of the blood of the treated mice by liquid chromatography (LC)-mass spectrometry (MS)/MS showed that the plasma concentrations of Pyro in monotherapy and in combination with M5717 remained stable throughout the duration of parasite exposure to the drugs (i.e., from 28 to 48 hpi; Table S1). Unfortunately, we were unable to quantify M5717 at the selected concentration using this methodology (Table S1). Nevertheless, 0.3 mg/kg M5717 drastically reduced infection at 48 hpi to 22% of that of vehicle-treated mice, while Pyro administered in monotherapy had no impact on the P. berghei liver load (Figure 2C). Importantly, the combination of Pyro with M5717 further reduced infection to 9%, a 2.3-fold reduction relative to that observed upon administration of M5717 alone (Figure 2C). A similar decrease was observed when the same dose of Pyro was combined with a slightly higher dose of 0.4 mg/kg of M5717, resulting in a 2.2-fold reduction of infection upon administration of the combination, when compared with administration of M5717 alone (Figure S2B).
Figure 2.
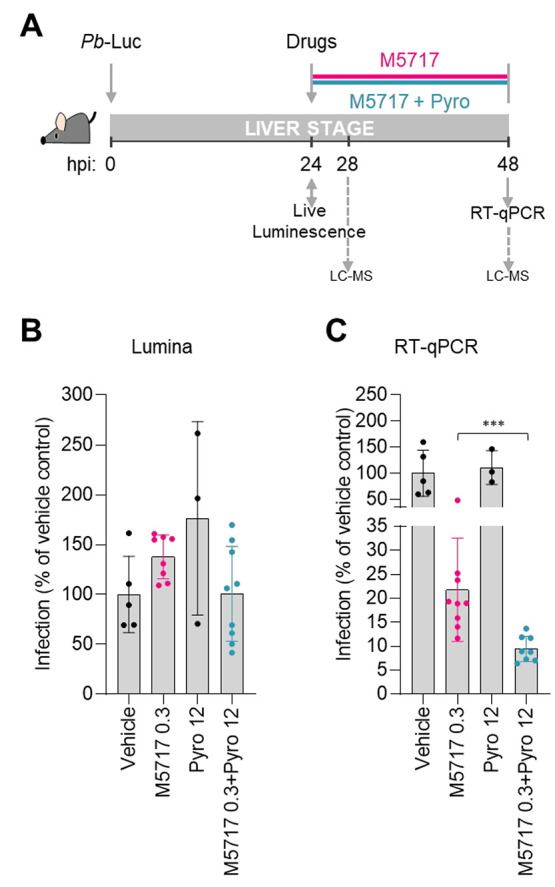
In vivo assessment of the pre-erythrocytic activity of M5717-Pyro combination. (A) Schematic representation of in vivo drug administration and analysis of liver infection. C57BL/6J mice were infected with 3 × 104Pb-Luc sporozoites. At 24 h postinfection (hpi), the liver load was assessed by live bioluminescence and the drugs of interest were administered. At 48 hpi the impact of M5717 (pink) or M5717-Pyro combination (blue) on the liver load of the same mice was evaluated by RT-qPCR analysis. At 28 and 48 hpi, a blood sample was collected for LC-MS analysis. (B) Assessment of the liver load at 24 hpi by live bioluminescence. (C) Assessment of the infection rate at 48 hpi by gene expression analysis (RT-qPCR). Infection (B,C) was normalized to the vehicle-treated control group, and results are represented as mean ± SD of three to seven mice from one independent experiment. Statistical analysis of luminescence data was performed by one-way ANOVA, whereas analysis of RT-qPCR data was performed using a Mann–Whitney test. *** P ≤ 0.001.
Collectively, the in vitro and in vivo assessment of Pyro as a possible drug partner of M5717 against the hepatic stage of infection by P. berghei shows not only that the former is not detrimental to the latter but also that addition of Pyro consistently potentiates M5717’s antiplasmodial effect during this phase of the parasite’s life cycle.
Between 2010 and 2018, the global incidence rate and the number of malaria deaths have declined. However, despite the long lasting struggle to end this infectious disease, this progress is coming to a halt.1 New antiplasmodial strategies are, therefore, required to accelerate the progress toward the goal of malaria eradication. In a context where a highly desirable effective vaccine against malaria remains unavailable, vector control measures and therapeutics persist as the cornerstone of disease control.
As for other infectious diseases, such as tuberculosis, the treatment of malaria resorts to combination therapies as a means of decreasing the risk of emergence of drug resistance.13 In the search for a suitable combination partner for a given drug, several aspects need to be taken into consideration, including each drug’s mode of action, pharmacokinetic properties, and toxicity.13 Crucially, combinations should be assessed for drug–drug interactions that may compromise the efficacy of either drug. M5717-resistant Plasmodium parasites have been identified in vitro during the preclinical characterization of this drug, highlighting the need to combine it with a suitable partner drug.7 A second study identified Pyro as an appropriate candidate for combination with M5717, given its different target, faster action, matching half-life, and ability to suppress the selection of M5717-resistant mutants when tested against the asexual erythrocytic stage of the Plasmodium life cycle.12 Importantly, in vitro isobologram studies and in vivo testing revealed a non detrimental interaction between both drugs.12
Besides its efficacy against asexual blood-stage Plasmodium parasites, M5717 is also active against exoerythrocytic parasite forms. Thus, it was important to ensure that its combination with Pyro would not hamper these relevant prophylactic and transmission-blocking activities. Our in vitro and in vivo observations demonstrate that the combination of M5717 with Pyro does not impair its pre-erythrocytic activity. Interestingly, our results point to a potentiation of this activity, both in vitro and in vivo. As an inhibitor of hemozoin formation, Pyro is not expected to possess liver stage activity. This has been formally demonstrated in an in vitro model of P. yoelii infection of mouse primary hepatocytes, where infected cells were exposed to Pyro throughout the 48 h of hepatic infection.14 At nanomolar-range doses (100, 10, and 1 nM), Pyro did not display activity against the parasite’s hepatic stages. At higher doses, which included the 1 μM dose employed in this study, Pyro was hepatotoxic, which did not allow the assessment of its impact on the infection rate.19 In our in vitro 3D model, 1 μM Pyro did not show hepatotoxicity, as confirmed by different analytical methods (Figure S3). Rather, the viability of cell spheroids that were exposed to the M5717-Pyro combination was comparable to that of spheroids that were exposed solely to M5717. The absence of pre-erythrocytic activity of Pyro was further supported by our in vivo observations, which show that the administration of this drug to Pb-Luc-infected mice has no impact on the ongoing liver infection. Thus, the enhanced pre-erythrocytic activity of the combination is not justified by the addition of the activities of the individual drugs. This type of interaction is not unprecedented, as chloroquine, an antiplasmodial drug structurally and target related to Pyro, has been described to potentiate primaquine’s activity against the pre-erythrocytic stage of infection, even though chloroquine has no inhibitory activity of its own.15 This effect was shown to be specific to this combination, as it was not observed when chloroquine was combined with atovaquone.15 Interestingly, this potentiation effect was only noted in primary hepatocytes but not in HepG2-A16 cells, which have reduced cytochrome P450 activities.15 The fact that we observed a potentiation of M5717’s activity in vitro using the HepG2 cell line may be related to the enhanced physiological relevance of the 3D hepatic system employed.16−18 Similar to the enhancement of primaquine’s activity by chloroquine,15 the identification of the molecular mechanism underlying the potentiation of M5717’s activity by Pyro described in this study requires further experimental work.
An assessment of the effect of a drug combination against the pre-erythrocytic stage of Plasmodium infection was first carried out in 2016 for Malarone.19 This combination of atovaquone and proguanil, employed both for the chemoprevention and the treatment of malaria, was already known to display a synergistic effect against the erythrocytic stage of infection20 and was next demonstrated to also be synergistic against developing hepatic P. yoelii parasites, providing a pharmacological basis for its success in chemoprevention.19 Although this study highlighted the importance of widening the evaluation of antiplasmodial drug combinations against the pre-erythrocytic stage of the parasite’s life cycle, the literature on this subject remains rather limited. Other drug combinations have since been evaluated in the pre-erythrocytic context, such as the combination of several antiretroviral drugs21 or of metformin with primaquine22 against rodent Plasmodium infection, and of ivermectin and chloroquine against P. cynomolgi in rhesus macaques.23 Our study now adds to the scarce information available on the pre-erythrocytic activity of antiplasmodial drug combinations and further validates the physiologically relevant 3D in vitro hepatic platform to provide data that correlates with the outcome of drug evaluation in vivo.
This study identifies Pyro not only as a suitable partner drug for M5717 but also as an enhancer of this drug’s activity against the pre-erythrocytic stage of Plasmodium infection. Our results demonstrate the added value of this drug combination for malaria chemoprevention, informing the clinical development of this compound and paving the way for its deployment in the field.
Methods
Ethics Statement
Animal experiments carried out at Instituto de Medicina Molecular João Lobo Antunes (iMM JLA, Lisbon, Portugal) were performed in strict compliance with the guidelines of the institute’s animal ethics committee (ORBEA), which also approved the study, and the Federation of European Laboratory Animal Science Associations (FELASA). Animal experiments carried out at the Swiss Tropical and Public Health Institute (Swiss TPH, Basel, Switzerland) were in agreement with local and national regulations of laboratory animal welfare in Switzerland (awarded permission no. 2693). Protocols were regularly reviewed and revised following approval by the local authority (Veterinäramt Basel Stadt).
Mice, Cell Sources, and Parasites
Male C57BL/6J mice (Charles River Laboratories), 7 to 12 weeks old, were housed in specific pathogen-free (SPF) conditions at iMM JLA’s rodent facility. Female NMRI mice (Charles River laboratories), with 20–22 g of body weight were housed and manipulated in Swiss TPH’s animal facility. The in vitro studies were performed by employing the HepG2 cell line purchased from ATCC (CRL/10741). Infection procedures were performed employing sporozoites recovered from infected Anopheles stephensi mosquitoes, reared and maintained in the insectarium facilities of iMM JLA and of Swiss TPH. Uninfected mosquitoes were allowed to feed on mice infected with luciferase-expressing P. berghei ANKA (Pb-Luc) parasites. Sporozoites were freshly isolated from mosquito salivary glands into RPMI 1640 medium (Thermo Fisher Scientific) or phosphate-buffered saline (PBS), macerated and filtered through a 40 μm cell strainer prior to infection experiments.
Two-Dimensional (2D) Cell Culture
HepG2 2D cultures were maintained in T-flasks in static conditions, as previously described (Arez et al., 2019). Briefly, HepG2 cells were cultured in low glucose DMEM (Thermo Fisher Scientific) supplemented with 1% (v/v) pen/strep and 10% (v/v) FBS. Cells were passaged twice every week at a cell inoculum of 5 × 104 cell/cm2. Cells were maintained in an incubator with humidified environment, at 37 °C and 5% CO2.
Three-Dimensional (3D) Cell Culture (Spheroids)
Hepatic cell spheroids were generated and maintained in dynamic suspension cultures under magnetic stirring (2mag AG), in an incubator with humidified environment at 37 °C and 5% CO2. HepG2 cells were inoculated as single cell suspensions (3 × 105 cell/mL) in 125 mL spinners (Corning, Merck KGaA, Darmstadt, Germany), in the culture medium employed for the 2D cell culture. HepG2 3D cultures started at an agitation rate of 40 rpm, and reached up to 120 rpm by the end of culture time. Medium replacement was performed in order to attain 100% medium renovation per week. Therefore, 50% of the culture medium was replaced every 3 days. To this end, spheroids were sedimented by centrifugation, followed by pellet resuspension in the adequate proportion of pre-existent and fresh culture medium supplemented with 5% (v/v) FBS.
Determination of Cell Viability and Concentration
Cell viability of spheroid cultures was assessed as previously described (Arez et al. 2019). Briefly, spheroids were incubated with a cell-permeant dye (fluorescein diacetate, Sigma-Aldrich, Merck KGaA, Darmstadt, Germany) at 10 μg/mL and a DNA dye (TO-PRO-3 Iodide, Thermo Fisher Scientific) at 1 μM for detection of viable and dead cells, respectively. Visual inspection of spheroids was performed using an inverted fluorescence microscope (Leica DMI6000). The cell density of 2D and 3D cultures was determined by the Trypan blue exclusion method, as previously described (Rebelo et al., 2014).
In Vitro Plasmodium Infection
Spheroids were inoculated at 5 × 105 cell/mL in infection medium (basal culture medium supplemented with 5% (v/v) FBS, 1:300 Amphotericin B (250 μg/mL) and 1:1000 Gentamycin 50 mg/mL, all from Thermo Fisher Scientific), on the day of infection, in ultralow attachment flat-bottom 96-well plates (Corning, Merck KGaA, Darmstadt, Germany). Sporozoites were added to 3D cultures at a 1:2 cell:spz ratio. Sporozoite addition in microplates was followed by a centrifugation step at 1800g for 5 min, after which plates were maintained in static conditions at 37 °C and 5% CO2 for 48 h. The infection rates were assessed by bioluminescence, as a measurement of luciferase activity.
In Vitro Assessment of Plasmodium Infection by Bioluminescence
The infection load of Pb-Luc-infected cells was assessed by employing a commercially available Firefly Luciferase Assay Kit from Biotium, following the manufacturer’s instructions. Briefly, cells and spheroids were washed twice with PBS and later incubated in lysis buffer diluted 1:4 in Milli-Q water. Samples underwent several freeze–thaw cycles, alternated with agitation steps at 500 rpm, until complete cell lysis was confirmed by visual inspection. Bioluminescence was measured using a microplate reader (Infinite 200 PRO, Tecan Trading AG), and the light reaction of each well was measured for 100 ms.
Drug Assays
M5717 and Pyro were synthesized and supplied by Merck KGaA, Darmstadt, Germany.
In Vitro Drug Assays
Stock solutions of 10 mM were prepared in DMSO. Working solutions of compounds employed in drug assays were prepared freshly at concentrations ranging from 0.01 to 30 nM for M5717 or at a fixed concentration of 1 μM for Pyro. Pb-infected cells were exposed to the aforementioned drug dilutions at 24 hpi and cultured for an additional 24 h. For the dose–response analysis, cell viability and infection rate were assessed at the end of the drug incubation period (48 hpi), as previously described (Arez et al., 2019). Briefly, cell metabolic activity was determined by incubation of adherent cells and spheroids for 50 min at 37 °C, protected from light, with 1× PrestoBlue Cell Viability Reagent (A-13262, Thermo Fisher Scientific). The fluorescence of the culture supernatant was measured at 560 and 590 nm excitation and emission wavelengths, respectively. Cell metabolic activity was used as a measurement of cell viability and employed in the normalization of the infection rates. Samples were further analyzed for bioluminescence as described above.
In Vivo Drug Assays
Male C57BL/6J mice were infected by intravenous injection (i.v.) of 3 × 104 firefly luciferase-expressing P. berghei sporozoites. Hepatic infection was confirmed at 24 hpi by live bioluminescence, prior to compound administration, as previously described.24 Briefly, mice were anesthetized with isofluorane and subcutaneously injected with 200 μL of D-Luciferin (PerkinElmer) dissolved in PBS (10 mg/mL). After 5 min, mice were anesthetized by an intraperitoneal injection of a ketamine/xylazine solution. Image acquisition took place at approximately 10 min after D-Luciferin injection, employing the in vivo IVIS Lumina Imaging System (Caliper LifeSciences, Waltham, MA, U.S.A.). P. berghei bioluminescence was measured as total flux (photons/s) and analyzed with the Living Image software (version 3.0, PerkinElmer, Waltham, MA, U.S.A.). Compounds were solubilized in a solution of 70% Tween-80 and 30% ethanol, followed by a 10-fold dilution in water, and administered by oral gavage at 24 hpi. M5717 was administered at 0.4 or 0.3 mg/kg of mouse weight, whereas Pyro was administered at 12 mg/kg. An equivalent amount of drug vehicle was administered as a control. At 48 hpi, mice were sacrificed and livers were collected to denaturing solution (4 M guanidine thiocyanate, 25 mM sodium citrate pH 7.0, 0.5% (w/v) sarcosyl and 0.7% (v/v) b-mercaptoethanol in DEPC-treated water) for parasite load quantification by quantitative real-time PCR (qRT-PCR). Liver samples were then mechanically homogenized, and RNA was extracted using the TripleXtractor directRNA Kit (GRiSP), according to the manufacturer’s recommendations. Complementary DNA (cDNA) was synthesized from 1 μg of RNA, using the NZY First-strand cDNA Synthesis Kit (NZYTech). P. berghei load was quantified by qRT-PCR, employing primers specific for Pb 18S RNA (5′-AAGCATTAAATAAAGCGAATACATCCTTAC-3′ and 5′-GGAGATTGGTTTTGACGTTTATGTG-3′). Gene expression levels were normalized to the endogenous mouse housekeeping gene hypoxanthine-guanine phosphoribosyltransferase (Hprt) (primers 5′-TTTGCTGACCTGCTGGATTAC-3′ and 5′- CAAGACATTCTTTCCAGTTAAAGTTG-3′). The RT-qPCR reaction was performed in a total volume of 10 μL using the NZYSpeedy qPCR Green, ROX (NZYtech), employing the ViiA 7 System (Applied Biosystems). The comparative ΔΔCT method was used for analysis of RT-qPCR results.
Data Analysis and Statistics
Nonlinear regression analysis of the normalized results for the determination of IC50 values and statistical analysis were performed employing GraphPad Prism version 6 for Windows (GraphPad software, La Jolla California U.S.A.). Outliers were identified by the ROUT method. Normality was assessed by the Shapiro-Wilk normality test. Significant differences were determined using a parametric or nonparametric t test, considering paired conditions when subjected to the same batch of spz, or using one-way ANOVA. P values are presented for statistically significant results (*, P < 0.05, **, P < 0.01, *** P < 0.001, ****P < 0.0001), as indicated in each figure legend.
Acknowledgments
We acknowledge Ana Filipa Teixeira and Ana Parreira for mosquito production and infection. This work was funded by the healthcare business of Merck KGaA, Darmstadt, Germany (CrossRef Funder ID: 10.13039/100009945). M.P. is a recipient of a “Concurso de Estímulo ao Emprego Científico” Principal Investigator award of Fundação para a Ciência e Tecnologia, Portugal (FCT), with ref. N. CEECIND/03539/2017. D.F. is funded by FCT project CRCNA/BRB/0281/2019. F.A. is recipient of a PhD fellowship PD/BD/128371/2017, funded by FCT.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.1c00640.
Supplementary Methods; Supplementary Figures 1, 2, and 3; Supplementary Table 1 (PDF)
Author Contributions
¶ D.F. and F.A. contributed equally to the work. Conceptualization: D.F., F.A., T.S., C.B., M.P., P.M.A. Methodology: D.F., F.A., I.R.G., G.N., D.M., T.H.H.B. Formal analysis: D.F., F.A., I.R.G., T.H.H.B., C.B., M.P .Writing–original draft: D.F., F.A. Writing–review and editing: T.S., C.B., M.P., P.M.A. Funding acquisition: T.S., M.P., P.M.A. Supervision: T.S., C.B., M.P., P.M.A.
The authors declare the following competing financial interest(s): D.F., F.A., T.S., C.B., M.P., and P.M.A. are inventors on a patent WO 2018162623. T.H.H.B and T.S. are employees of the healthcare business of Merck KGaA, Darmstadt, Germany, which funded the work and provided novel compounds to be tested in the platforms developed.
Supplementary Material
References
- WHO . World Malaria Report 2021; World Health Organization: Geneva, Switzerland; 2021.
- Lindner S. E.; Miller J. L.; Kappe S. H. Malaria parasite pre-erythrocytic infection: preparation meets opportunity. Cellular microbiology. 2012, 14 (3), 316–24. 10.1111/j.1462-5822.2011.01734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudencio M.; Rodriguez A.; Mota M. M. The silent path to thousands of merozoites: the Plasmodium liver stage. Nature reviews Microbiology. 2006, 4 (11), 849–56. 10.1038/nrmicro1529. [DOI] [PubMed] [Google Scholar]
- White N. J.; Pukrittayakamee S.; Hien T. T.; Faiz M. A.; Mokuolu O. A.; Dondorp A. M. Malaria. Lancet. 2014, 383 (9918), 723–35. 10.1016/S0140-6736(13)60024-0. [DOI] [PubMed] [Google Scholar]
- Meibalan E.; Marti M. Biology of malaria transmission. Cold Spring Harb Perspect Med. 2017, 7 (3), a025452 10.1101/cshperspect.a025452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arez F.; Rebelo S. P.; Fontinha D.; Simao D.; Martins T. R.; Machado M.; Fischli C.; Oeuvray C.; Badolo L.; Carrondo M. J. T.; Rottmann M.; Spangenberg T.; Brito C.; Greco B.; Prudencio M.; Alves P. M. Flexible 3D Cell-Based Platforms for the Discovery and Profiling of Novel Drugs Targeting Plasmodium Hepatic Infection. ACS Infect Dis. 2019, 5 (11), 1831–42. 10.1021/acsinfecdis.9b00144. [DOI] [PubMed] [Google Scholar]
- Baragana B.; Hallyburton I.; Lee M. C. S.; Norcross N. R.; Grimaldi R.; Otto T. D.; Proto W. R.; Blagborough A. M.; Meister S.; Wirjanata G.; Ruecker A.; Upton L. M.; Abraham T. S.; Almeida M. J.; Pradhan A.; Porzelle A.; Martinez M. S.; Bolscher J. M.; Woodland A.; Luksch T.; Norval S.; Zuccotto F.; Thomas J.; Simeons F.; Stojanovski L.; Osuna-Cabello M.; Brock P. M.; Churcher T. S.; Sala K. A.; Zakutansky S. E.; Jimenez-Diaz M. B.; Sanz L. M.; Riley J.; Basak R.; Campbell M.; Avery V. M.; Sauerwein R. W.; Dechering K. J.; Noviyanti R.; Campo B.; Frearson J. A.; Angulo-Barturen I.; Ferrer-Bazaga S.; Gamo F. J.; Wyatt P. G.; Leroy D.; Siegl P.; Delves M. J.; Kyle D. E.; Wittlin S.; Marfurt J.; Price R. N.; Sinden R. E.; Winzeler E. A.; Charman S. A.; Bebrevska L.; Gray D. W.; Campbell S.; Fairlamb A. H.; Willis P. A.; Rayner J. C.; Fidock D. A.; Read K. D.; Gilbert I. H. A novel multiple-stage antimalarial agent that inhibits protein synthesis. Nature. 2015, 522 (7556), 315–320. 10.1038/nature14451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy J. S.; Yalkinoglu O.; Odedra A.; Webster R.; Oeuvray C.; Tappert A.; Bezuidenhout D.; Giddins M. J.; Dhingra S. K.; Fidock D. A.; Marquart L.; Webb L.; Yin X.; Khandelwal A.; Bagchus W. M. Safety, pharmacokinetics, and antimalarial activity of the novel plasmodium eukaryotic translation elongation factor 2 inhibitor M5717: a first-in-human, randomised, placebo-controlled, double-blind, single ascending dose study and volunteer infection study. Lancet Infect Dis. 2021, 21 (12), 1713–24. 10.1016/S1473-3099(21)00252-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft S. L.; Duparc S.; Arbe-Barnes S. J.; Craft J. C.; Shin C.-S.; Fleckenstein L.; Borghini-Fuhrer I.; Rim H.-J. Review of pyronaridine anti-malarial properties and product characteristics. Malar J. 2012, 11, 270. 10.1186/1475-2875-11-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X. Y.; Chen C.; Gao F. H.; Zhu P. E.; Guo H. Z. [Synthesis of new antimalarial drug pyronaridine and its analogues (author’s transl)]. Yao Xue Xue Bao. 1982, 17 (2), 118–125. [PubMed] [Google Scholar]
- Zheng X. Y.; Xia Y.; Gao F. H.; Chen C. [Synthesis of 7351, a new antimalarial drug (author’s transl)]. Yao Xue Xue Bao. 1979, 14 (12), 736. [PubMed] [Google Scholar]
- Rottmann M.; Jonat B.; Gumpp C.; Dhingra S. K.; Giddins M. J.; Yin X.; Badolo L.; Greco B.; Fidock D. A.; Oeuvray C.; Spangenberg T. Preclinical Antimalarial Combination Study of M5717, a Plasmodium falciparum Elongation Factor 2 Inhibitor, and Pyronaridine, a Hemozoin Formation Inhibitor. Antimicrob. Agents Chemother. 2020, 64 (4), e02181-19 10.1128/AAC.02181-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosten F.; Brasseur P. Combination therapy for malaria: the way forward?. Drugs 2002, 62 (9), 1315–1329. 10.2165/00003495-200262090-00003. [DOI] [PubMed] [Google Scholar]
- Basco L. K.; Ringwald P.; Franetich J. F.; Mazier D. Assessment of pyronaridine activity in vivo and in vitro against the hepatic stages of malaria in laboratory mice. Trans R Soc. Trop Med. Hyg. 1999, 93 (6), 651–2. 10.1016/S0035-9203(99)90085-8. [DOI] [PubMed] [Google Scholar]
- Dembélé L.; Franetich J. F.; Soulard V.; Amanzougaghene N.; Tajeri S.; Bousema T.; van Gemert G. J.; Le Grand R.; Dereuddre-Bosquet N.; Baird J. K.; Mazier D.; Snounou G. Chloroquine Potentiates Primaquine Activity against Active and Latent Hepatic Plasmodia Ex Vivo: Potentials and Pitfalls. Antimicrob. Agents Chemother. 2020, 65 (1), e01416-20. 10.1128/AAC.01416-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebelo S. P.; Costa R.; Estrada M.; Shevchenko V.; Brito C.; Alves P. M. HepaRG microencapsulated spheroids in DMSO-free culture: novel culturing approaches for enhanced xenobiotic and biosynthetic metabolism. Arch. Toxicol. 2015, 89 (8), 1347–58. 10.1007/s00204-014-1320-9. [DOI] [PubMed] [Google Scholar]
- Gaskell H.; Sharma P.; Colley H. E.; Murdoch C.; Williams D. P.; Webb S. D. Characterization of a functional C3A liver spheroid model. Toxicol Res. (Camb). 2016, 5 (4), 1053–65. 10.1039/C6TX00101G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leite S. B.; Wilk-Zasadna I.; Zaldivar J. M.; Airola E.; Reis-Fernandes M. A.; Mennecozzi M.; Guguen-Guillouzo C.; Chesne C.; Guillou C.; Alves P. M.; Coecke S. Three-dimensional HepaRG model as an attractive tool for toxicity testing. Toxicol. Sci. 2012, 130 (1), 106–16. 10.1093/toxsci/kfs232. [DOI] [PubMed] [Google Scholar]
- Barata L.; Houzé P.; Boutbibe K.; Zanghi G.; Franetich J. F.; Mazier D.; Clain J. In Vitro Analysis of the Interaction between Atovaquone and Proguanil against Liver Stage Malaria Parasites. Antimicrob. Agents Chemother. 2016, 60 (7), 4333–5. 10.1128/AAC.01685-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canfield C. J.; Pudney M.; Gutteridge W. E. Interactions of Atovaquone with Other Antimalarial Drugs against Plasmodium falciparum in Vitro. Experimental Parasitology. 1995, 80 (3), 373–81. 10.1006/expr.1995.1049. [DOI] [PubMed] [Google Scholar]
- Machado M.; Sanches-Vaz M.; Cruz J. P.; Mendes A. M.; Prudêncio M. Inhibition of Plasmodium Hepatic Infection by Antiretroviral Compounds. Front Cell Infect Microbiol. 2017, 7, 329. 10.3389/fcimb.2017.00329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vera I. M.; Grilo Ruivo M. T.; Lemos Rocha L. F.; Marques S.; Bhatia S. N.; Mota M. M.; Mancio-Silva L. Targeting liver stage malaria with metformin. JCI Insight. 2019, 4 (24), e127441 10.1172/jci.insight.127441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanachayangkul P.; Im-Erbsin R.; Tungtaeng A.; Kodchakorn C.; Roth A.; Adams J.; Chaisatit C.; Saingam P.; Sciotti R. J.; Reichard G. A.; Nolan C. K.; Pybus B. S.; Black C. C.; Lugo-Roman L. A.; Wegner M. D.; Smith P. L.; Wojnarski M.; Vesely B. A.; Kobylinski K. C. Safety, Pharmacokinetics, and Activity of High-Dose Ivermectin and Chloroquine against the Liver Stage of Plasmodium cynomolgi Infection in Rhesus Macaques. Antimicrob. Agents Chemother. 2020, 64 (9), e00741-20. 10.1128/AAC.00741-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploemen I. H.; Prudêncio M.; Douradinha B. G.; Ramesar J.; Fonager J.; van Gemert G. J.; Luty A. J.; Hermsen C. C.; Sauerwein R. W.; Baptista F. G.; Mota M. M.; Waters A. P.; Que I.; Lowik C. W.; Khan S. M.; Janse C. J.; Franke-Fayard B. M. Visualisation and quantitative analysis of the rodent malaria liver stage by real time imaging. PLoS One. 2009, 4 (11), e7881 10.1371/journal.pone.0007881. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.