

Summary:
The formation of three-dimensional kidney tissue (organoids) from human pluripotent stem cell lines provides a valuable tool to examine kidney function in an in vitro model and could be used for regenerative medicine approaches. Kidney organoids have the potential to model kidney diseases and congenital defects, be used for drug development, and to further our understanding of acute kidney injury, fibrosis, and chronic kidney disease. In this review, we examine the current stage of pluripotent stem cell–derived kidney organoid technology, challenges, shortcomings, and regenerative potential of kidney organoids in the future.
Keywords: Kidney organoids, pluripotent stem cells, kidney disease
The etiology of kidney disease is multifactorial, but the outcomes are usually a progressive decline in kidney function. It can arise from genetic abnormalities such as polycystic kidney disease (PKD), can be acquired as a result of ingestion of nephrotoxic drugs causing acute kidney injury (AKI), or can be a consequence of other conditions such as diabetes and hypertension, which are the leading causes of chronic kidney disease (CKD).1 Irrespective of the foundations for kidney disease, it can become chronic and eventually progress toward end-stage renal disease (ESRD), requiring life-long renal replacement therapy. The prevalence of kidney disease is increasing worldwide, with more than 10% of the population estimated to suffer from CKD.1 Prognosis for patients with CKD and ESRD is poor because kidneys have a very limited capacity to regenerate. The only treatments available for patients who progress from CKD to ESRD are dialysis and transplantation, but there are an insufficient number of kidneys to meet the current and growing demand.2 As such, there is an urgent need to develop new therapies and treatments. This can be achieved only through understanding the diverse mechanisms by which kidney disease arises at both the cellular and molecular levels to develop targeted therapies.
Cellular reprogramming technology developed by Takahashi and Yamanaka in 2006 has revolutionized the field of stem cell research.3 This technology reprograms somatic cells such as fibroblasts into an embryonic stem cell–like state using ectopic expression of specific pluripotency genes (eg, SOX2, KLF4, c-MYC, NANOG, and OCT4). The resulting reprogrammed cells, called induced pluripotent stem cells (iPSCs), have the potential to differentiate into any mature cell type, as well as self-renew. When iPSCs are made from individuals with genetic diseases, the resulting lines provide an unlimited source of diseased human tissue. Although these disease iPSCs can be compared directly with healthy control iPSCs to better understand the pathogenesis of the disease, this often is problematic because of the wide phenotypic variation seen between genetically unrelated but nevertheless healthy iPSC lines. However, the recent development of gene editing technologies such as the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9 system has largely overcome this issue, as in many cases the patient-specific iPSC lines can be corrected genetically to create isogenic control lines to compare against (Tables 1 and 2).
Table 1.
List of Human iPSC, Embryonic Stem Cell Healthy, and Patient/Disease Cell Lines Made to Study Kidney Organoids
| M/F | Pathology | References | |||
|---|---|---|---|---|---|
| Embryonic stem cell lines | |||||
| H1 | M | Normal | 29,73 | ||
| H9 | F | Normal | 14,19,24,29,45,49,62,65,68,69,73,83 | ||
| UM77-2 | F | Normal | 54 | ||
| SEEC3 | M | Normal | 84 | ||
| Envy (hES3) | F | Normal | 44,69 | ||
| Man11 | F | Normal | 74 | ||
| Man13 | M | Normal | 74 | ||
| HUES1 | F | Normal | 74 | ||
| iPSC lines | M/F | Age | Reprogramming | Source | |
| WTC-11 | M | Adult | Episomal | Skin fibroblast | 19,49,85 |
| CRL1502-c32 | F | <1 y | Episomal | Skin fibroblast | 15,44,47,81,86 |
| CRL1502.3 | F | <1 y | Episomal | Skin fibroblast | 69 |
| IMR90-1 | F | Fetal | Lentivirus | Lung fibroblast | 83 |
| IMR90-4 | F | Fetal | Lentivirus | Lung fibroblast | 87 |
| BCRTi004-A | F | Adult | Sendai virus | Urinary | 87 |
| BCRTi005-A | F | Adult | Sendai virus | Urinary | 87 |
| 201B7 | F | Adult | Lentivirus | Skin fibroblast | 18,53 |
| RN7 | N/A | Adult | Sendai virus | Peripheral blood | 53 |
| BJFF.6 | M | <1 y | Sendai virus | Fibroblast | 68 |
| MANZ-2-2 | F | Adult | Sendai virus | Fibroblast | 15 |
| BJ RiPS | M | mRNA | Fibroblast | 15 | |
| LUMC0099iCTRL04 | F | Adult | mRNA | Skin fibroblast | 44 |
| Patient/disease cell lines | Reprogramming | Source | Pathology | Citation | |
| Nephrin | Sendai virus | Skin fibroblasts | E725D nephrin | 52 | |
| IFT140 | Episomal | Fibroblast | c.634G>A and c.2176C>G | 60 | |
| IFT140 corrected | CRISPR/Cas9 | 60 | |||
| ARPKD | Episomal | Fibroblast | c.11630delT | 29 | |
| ARPKD corrected | CRISPR/Cas9 | 29 | |||
| PKD1−/− | CRISPR/Cas9 | H9 | PKD1 deletion | 24,88 | |
| PKD2−/− | CRISPR/Cas9 | H9 | PKD2 deletion | 24,88 | |
| PODXL−/− | CRISPR/Cas9 | H9 | Podocalyxin deficient | 49,88 |
Abbreviations: M/F, male/female; mRNA, messenger RNA.
Table 2.
List of Human iPSC Embryonic Stem Cell Reporter Lines Made Using CRISPR/Cas9 to Study Kidney Organoids
| Line Name | Target Gene | Parental Line | Reporter Expression | References |
|---|---|---|---|---|
| Lineage-tracing lines | ||||
| SIX2Cre/Cre | SIX2 | CRL-2429 | SIX2 lineage | 89 |
| SIX2CreERT2 | SIX2 | CRL-2429 | Inducible SIX2 lineage tracing | 89 |
| SIX2Cre/ Cre:GAPDH dual | SIX2/GAPDH | SIX2Cre/Cre | SIX2 lineage tracing | 89 |
| SIX2CreERT2/ CreERT2: GAPDH dual | SIX2/GAPDH | SIX2CreERT2/mCreERT2 | Inducible SIX2 lineage tracing | 89 |
| Pax2−/−* | PAX2 | 201b7 | Early development, nephron progenitor | 18 |
| NPHS1-GFP | NPHS1 | 201b7 | Mature podocyte | 49,53 |
| SIX2-GFP | SIX2 | N/A | Nephron progenitor | 90 |
| NPHS1-GFP | NPHS1 | N/A | Mature podocyte | 90 |
| SIX2-GFP/ NPHS1-mKate | SIX2/NPHS1 | Six2-GFP | Nephron progenitor/mature podocyte | 90 |
| Single reporter lines | ||||
| HNF1A-inducible | HNF1A | SEEC3 | Up-regulates proximal tubule-related markers | 84 |
| HNF1B−/− | HNF1B | MANZ-2-2 | Failure of proximal tubule and thick ascending limb segment formation | 15 |
| GAPTrapLuc2 | GAPDH | H9 | Ubiquitous | 69 |
| SOX17mCherry | SOX7 | hES3 | Ubiquitous | 69 |
| GAPTrap Td* Tomato | GAPDH | RM3.5 | Ubiquitous | 69 |
| SIX2EGFP | SIX2 | CRL-2429 | Early nephron, stromal populations | 89 |
| CITED1mCherry | CITED1 | CRL-2429 | Early intermediate mesoderm, NPs, early nephron | 89 |
| GATA3mCherry | GATA3 | CRL-2429 | SLC12A1-, collecting duct, GATA3+ interstitial | 89 |
| MAFBmTagBFP2 | MAFB | CRL-2429 | Podocytes | 47,89 |
| LRP2mTagBFP2 | LRP2 | CRL-2429 | Proximal tubule | 89 |
| HNF4αYFP | HNF4α | PCS-201-010 | Proximal tubule | 89 |
| Dual reporter lines | ||||
| SIX2EGFP: CITED1mCherry | SIX2/CITED1 | SIX2EGFP | NPs, early nephron, stromal populations | 89 |
| MAFBBFP: GATA3mCherry | MAFB/GATA3 | MAFBm TagBFP2 | Podocytes (MAFB+), collecting duct (GATA3+), interstitial population (GATA3+) | 47,89 |
| MAFB-P2A-eGFP | MAFB | H9 hESC | Podocyte (MaFB+) | 43 |
Abbreviations: GAPDH, glyceraldehyde-3-phosphate dehydrogenase; hESC, human embryonic stem cell; NP, nephron progenitor.
Reporter/transgenic lines made using Transcription Activator-Like Effector Nucleases (TALEN).
The ability of iPSCs to differentiate into all cell types in the body has been exploited recently to generate organoids—small three-dimensional masses of multicellular tissue that approximate mini-organs.4 Organoid protocols have been developed for many organ systems by modulating growth factors and cell culture conditions including brain,5 intestine,6 heart,7 pancreas,8 lung,9 and liver.10 Development of organoid models has bridged the gap between two-dimensional cell lines and in vivo models allowing researchers to study more complex biological functions, providing a basis for translational research. For instance, intestinal organoids have been used to study microbial interactions,11 while brain organoids have been used to study Zika virus infections12 to understand the pathogenic mechanisms and facilitate development of targeted therapies.
In recent years, a number of groups have developed protocols to coax iPSCs into kidney organoids (Fig. 1).13–18 These methods generate fetal-stage nephrons with primitive glomeruli, tubules segmented into proximal and distal segments, and stromal populations (Fig. 2). In the case of the Taguchi and Nishinakamura18 protocol, a ureteric bud/collecting duct also is generated; however, the presence of a collecting system in the other methods is less clear. Together, these protocols have provided a way to study human kidney tissue in a dish, thus opening up a host of new research avenues to explore. For instance, kidney organoids can be used to understand how different kidney diseases manifest and consequently dissect specific disease pathways from the very early stages of disease development. They also can provide opportunities for the discovery of novel disease-specific biomarkers, potentially allowing early detection of at-risk patients or with early signs of kidney disease. In contrast to mouse and cell line models, iPSCs and organoids provide a biologically relevant model to study drug toxicity, allowing for high-throughput, high-content analysis in a human context.19
Figure 1.
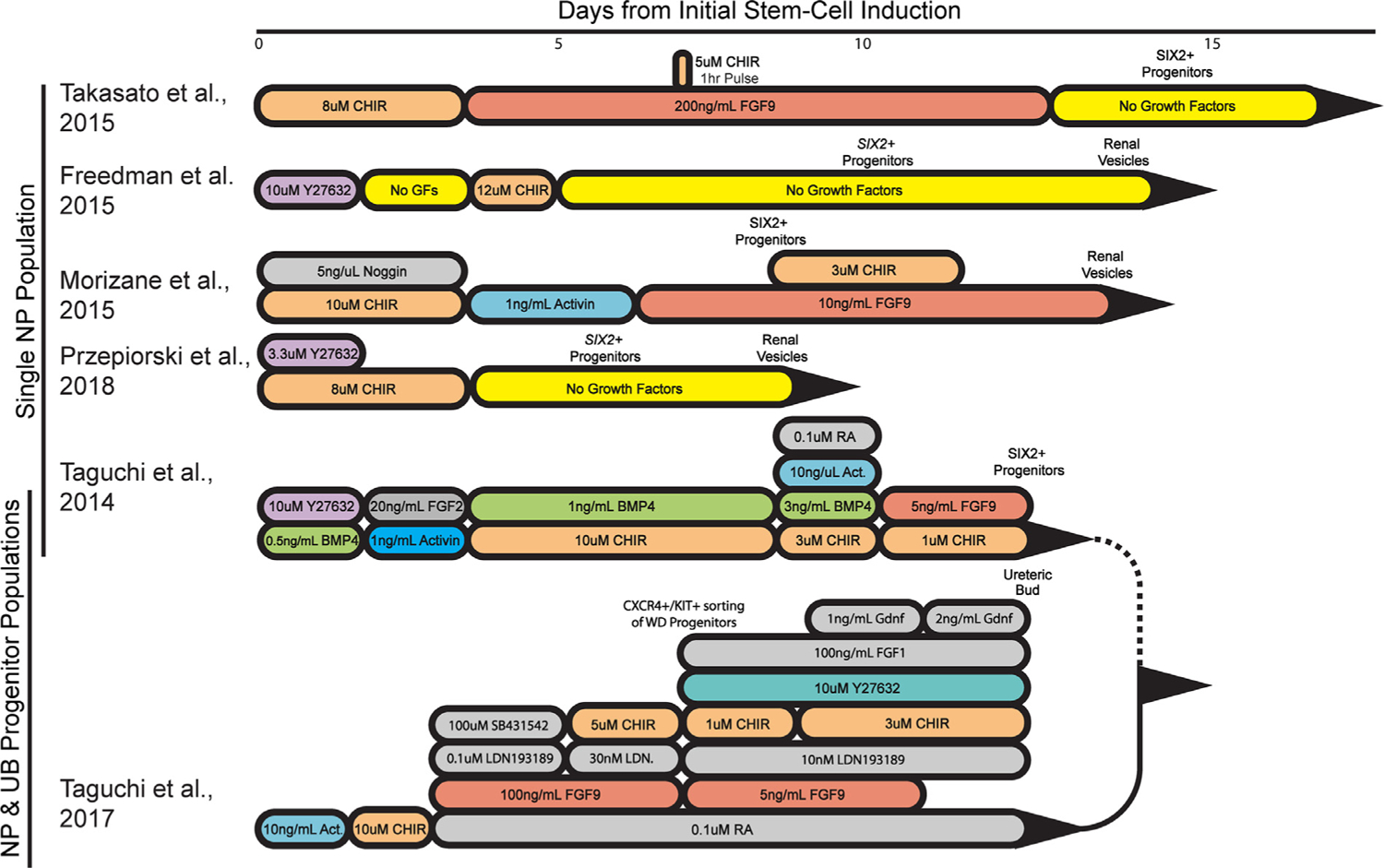
Comparison of human Pluripotent Stem Cells-derived kidney organoid protocols. An illustration highlighting the compounds used to differentiate PSCs toward a nephron progenitor state, including their concentrations and window of use. Compounds used across protocols are color coordinated. The cell types induced by each step of differentiation are noted above the protocol timeline. Single NP population refers to protocols generating organoids derived from nephron progenitors. NP and UB progenitor populations refers to organoids generated by combining nephron and ureteric bud progenitors. Abbreviations: Act, activin; BMP4, bone morphogenetic protein 4; CHIR, CHIR99021; CXCR4, C-X-C chemokine receptor type 4; FGF, fibroblast growth factor; GDNF, glial cell line–derived neurotrophic factor; GF, growth factor; KIT, mast/stem cell growth factor receptor Kit; LDN, LDN193189 is a bone morphogenetic protein pathway inhibitor; NP, nephron progenitor; RA, retinoic acid; SB431542, an activin/bone morphogenetic protein/transforming growth factor-β pathway inhibitor; SIX2, SIX homeobox 2 italicized for transcripts, nonitalicized for protein; WD, Wolffian duct; Y27632, a p160 ROCK inhibitor.
Figure 2.
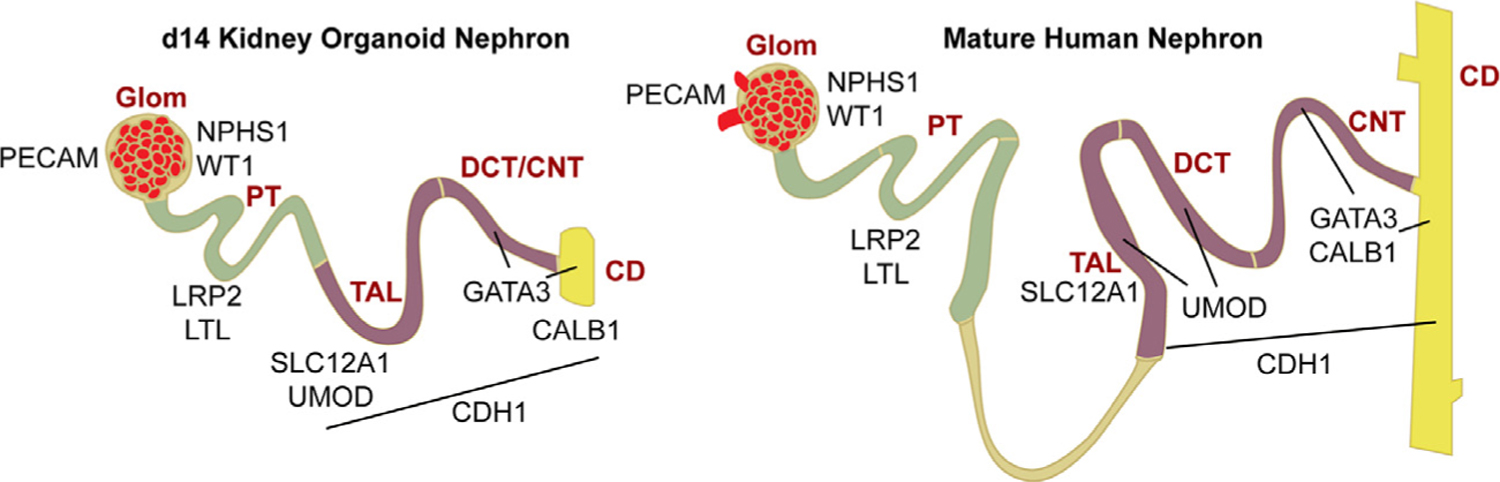
Schematic of day 14 (D14) kidney (left) organoid nephrons expressing segment-specific markers based on findings from Przepiorski et al15 in comparison with the mature human kidney (right). The glomerulus, expresses NPHS1 and WT1 in the podocytes, and PECAM1 in the endothelial cells. Segments 1 to 3 of the proximal tubule (S1, S2, S3) expressed LRP2 and Lotus tetragonologus lectin. The thick ascending limb expressed SLC12A1 and UMOD. The loop of Henley was not present in the kidney organoids. The distal convoluted tubule expressed UMOD, GATA3, and CALB1. The connecting tubule expressed GATA3 and CALB1. The CD expressed CALB1 and GATA3. CDH1 expressed TAL to CD. Abbreviations: CALB1, calbindin 1; CNT, connecting tubule; DCT; distal convoluted tubule; Glom, glomerulus; LoH, loop of Henley; LRP2, LDL receptor related protein 2; LTL, Lotus tetragonologus lectin; PECAM1, platelet and endothelial cell adhesion molecule-1; PT, proximal tubule; TAL, thick ascending limb; UMOD, uromodulin; WT1, Wilms’ tumor-1.
Here, we review how iPSC technology and the use of kidney organoids has advanced our understanding of kidney diseases and how it can be used in the future for potential treatments.
MODELING PKD
PKD is a slow-developing disease characterized by accumulation of fluid-filled cysts in the kidney.20 Defects in the primary cilia (a cellular mechanosensory organelle) of the tubular epithelial cells lead to fluid build-up and a progressive decline in kidney function, eventually leading to renal failure.21 In the case of PKD, a number of disease-causing mutations have been identified. Heterozygous mutations in PKD1 or PKD2 encoding for polycystin proteins (PC1 and PC2) are responsible for the autosomal-dominant form of PKD. Autosomal-recessive PKD is characterized by the presence of biallelic mutations in PKHD1, encoding a receptor-like protein called fibrocystin/polyductin. All three PKD proteins form a receptor complex on the cilium where they are thought to play a role in calcium and mechanistic target of rapamycin signaling and maintenance of cellular proliferation.21–23 Use of mice and in vitro cell-line models to study PKD has helped to elucidate how specific pathways are perturbed, leading to cyst formation. However, these models do not fully recapitulate the development and progression of cysts in human beings.23 For instance, mice heterozygous for Pkd1 or Pkd2 show a milder form of cyst formation in comparison with a double-heterozygote Pkd1+/−/Pkd2+/−, which is inconsistent with the dominant phenotype seen for human PKD1 or PKD2.23 Use of iPSCs derived from human PKD patients would provide a complementary model to animal models and cell lines.
The first disease-specific iPSC line to model kidney disease was developed by Freedman et al21 in 2013 and came from individuals with PKD1 mutations associated with autosomal-dominant PKD. The reprogrammed patient iPSCs showed a significant reduction in PC2 protein on ciliary shafts. Differentiation of the PKD-iPSCs into hepatoblasts and epithelial cells showed that PC2 was mislocalized to the plasma membrane instead of the cilium, but this could be corrected by overexpression of PC1. This study showed that PC1 plays a role in localizing PC2 to the cilium and highlighted the utility of using iPSCs to further elucidate specific disease mechanisms.
This group went on to develop a protocol to generate kidney organoids from iPSCs and used CRISPR/Cas9 gene editing to generate PKD1 and PKD2 knockout lines. Kidney organoids from the knockout PKD-iPSCs developed fluid-filled cysts after extended culture, in comparison with the unedited isogenic controls. Although the frequency of cyst formation was low and a disease mechanism still needs to be clarified, this study showed that kidney organoids can be used as a functional model to study genetic kidney diseases.13 A follow-up study by Cruz et al24 in 2017 extended this work by defining the modulators of cyst formation in the kidney organoids. They described a model in which cyst formation is driven by hyperproliferation of tubule cells and showed that PC1 regulates tubule architecture by interacting with the extracellular matrix. Furthermore, knocking down PC2 with small interfering RNA showed that it was necessary for correct localization of PC1 in human cells. Taken together, these studies confirmed prior observations that PC1 and PC2 interact and are dependent on each other for correct expression, function, and localization to the cilium.25–28
In a recent report from 2019, Low et al29 used autosomal-recessive PKD (ARPKD) patient-derived iPSCs containing compound heterozygous mutations to model cystogenesis in kidney organoids. They corrected the mutations using CRISPR/Cas9 and performed a comparative analysis with the isogenic control. As shown in previous studies, cyst formation was present in the ARPKD organoids and dilation of the proximal tubule preceded the distal tubule, which is consistent with the initiation of cyst formation in vivo. As proof of principle, they used this model to test previously described therapeutic compounds, which blocked cyst formation in the ARPKD organoids, proving that kidney organoids are a viable model to screen potential PKD therapeutics.
CONGENITAL KIDNEY DISEASE MODELS
Kidney organoids are developmentally equivalent to a first trimester kidney based on their RNA profiles derived from RNA sequencing analysis and immunohistologic comparison with fetal human tissue.15,16 The current findings support kidney organoids as a fitting model for congenital kidney defects and could be used to study early human kidney development in vitro. Two recent studies have generated knockout iPSC lines for the genes PAX2 and HNF1B, which are associated with congenital anomalies of the kidney and urinary tract. In mice, Pax2 is expressed during embryonic kidney development in the intermediate mesoderm, ureteric bud, metanephric mesenchyme, and primitive nephron.30,31 Nishinakamura’s32 laboratory generated PAX2-deficient iPSCs to determine how loss of PAX2 contributes to autosomal-dominant renal coloboma syndrome, which leads to underdeveloped kidneys and eventually ESRD. Despite many studies confirming the role PAX2 plays in kidney development and disease manifestation in mice, little is known about its function in human beings. To understand how PAX2 expression affects renal development at the earliest stages, PAX2-deficient iPSCs were differentiated into metanephric mesenchyme (nephron progenitors) using their previously established protocol (Fig. 1).17,32 Coculturing PAX2-deficient metanephric mesenchyme with the spinal cord to induce nephrogenesis in vitro showed that deficiency of PAX2 did not affect nephron formation in contrast with what was observed when culturing metanephric mesenchyme isolated from Pax2-deficient mice. This discrepancy may be the result of the protocol not faithfully recapitulating in vivo nephrogenesis, such as lacking ureteric bud induction as suggested by Kaku et al.32 Alternatively, loss of PAX2 may be compensated by PAX8, which is closely related to PAX2 and is known from rodent and zebrafish studies to be functionally redundant in certain contexts.33,34 Despite no overt nephrogenic phenotype when PAX2-deficient iPSCs were converted into nephron progenitors, a role for PAX2 in directing the differentiation of the ureteric bud was found with a disruption in mesenchymal-to-epithelial transition and reduced expression of the PAX2 target gene E-cadherin (CDH-1).18 It would be interesting to determine the impact PAX2 mutations have on interactions between ureteric bud and metanephric mesenchyme in this new culture model because previous studies have shown that even heterozygous mutations resulted in renal hypoplasia owing to a decrease in branching and consequently nephron induction.35
Expression of HNF1B is important for maintenance of the ureteric bud and patterning of the nephron.36,37 Heterozygous mutations in human beings lead to the development of renal cysts and diabetes syndrome, characterized by various kidney deformities such as hypoplastic cystic kidneys.38 The Davidson15 laboratory has generated knockout HNF1B iPSCs and found that kidney organoids made from these iPSCs lack proximal tubules and the thick ascending limb segment, but retain a GATA binding protein 3 (GATA3+) segment that may represent a connecting segment or possibly a collecting duct-like population. Podocyte clusters appear attached to this GATA3+ segment, highly reminiscent of the phenotype described in the mouse knockout model, and providing good evidence for conserved functionality in HNF1B between human beings and mouse.15,36,39 Another defect described in HNF1B mutant mice is a deficiency in branching of the ureteric tips.40 Because a bona fide ureteric bud population has not been confirmed in the Przepiorski et al15 protocol, assessment of the role of HNF1B in the human ureteric bud may be best pursued by producing kidney organoids using the method described by Taguchi et al17 in 2017 (Fig. 1). Based on their approach, early stage organoid ureteric bud progenitors can be isolated and cultured separately in a Matrigel (Corning, Corning, NY) culture system. Alternatively, HNF1B−/− iPSCs could be differentiated directly into ureteric epithelium and cultured in Matrigel.18,41
GLOMERULAR-RELATED DISEASES
Glomeruli within the organoids are relatively immature and require culture in flow chambers or transplantation in vivo to develop glomerular capillaries and more elaborate foot processes.42–46 Nevertheless, aspects of glomerular-related diseases still can be studied because the podocytes in the organoids form rudimentary foot processes and slit diaphragms,42,46,47 in contrast to immortalized and primary podocytes.48 Hence, kidney organoid-derived podocytes can provide valuable information about disease biology as shown by recent studies. As proof of this principle, in 2015 Freedman et al13 generated podocalyxin (PODXL) knockout iPSC lines using CRISPR/Cas9 and made the discovery that PODXL−/− organoids form podocytes with abnormal cell junctions with decreased distance between adjacent podocytes and a reduction in microvilli.,49 Ultrastructural analysis of Podxl−/− mice confirmed the presence of a similar phenotype49 and this new role for PODXL in cell-to-cell adhesion is supported further by recent findings that mutations in PODXL are associated with autosomal-dominant focal segmental glomerulosclerosis50 and congenital nephrotic syndrome in human beings.51
NEPHRIN (encoded by NPHS1) is a major component of the slit-diaphragm and causes congenital nephrotic syndrome when mutated. Taguchi et al17 made an iPSC line from an individual carrying a point mutation in the NPHS1 gene and generated kidney organoids using their protocol (Fig. 1).17 They found that, although foot processes still form in NPHS1-deficient kidney organoids, NEPHRIN did not localize to the cell surface normally and the formation of the slit-diaphragm was reduced significantly compared with corrected organoids. In addition, they discovered that the mutation altered the phosphorylation status of NEPHRIN and this was associated with a failure to recruit other slit-diaphragm proteins.52 In a follow-up study, Yoshimura et al53 refined their nephron progenitor protocol in 201918 to generate a near-pure population of podocytes in vitro (induced podocytes). In a global transcriptional analysis using RNA sequencing, comparison of adult human podocytes and induced podocytes showed a high similarity in gene signatures associated with slit diaphragm genes and podocyte transcription factors. Treatment of the induced podocytes with puromycin aminonucleoside, which is used to induce podocyte injury in vivo, compromised expression of slit diaphragm proteins and reduced levels of phosphorylated NEPHRIN similar to observations in animals.53 In 2018, Hale et al47 also generated iPSCs from patients with congenital nephrotic syndrome carrying different NPHS1 mutations and generated kidney organoids. In this study, it was found that the patient-derived podocytes were enlarged and had reduced protein levels of NEPHRIN and PODOCIN in comparison with the control podocytes. Taken together, these studies nicely show how podocyte-specific disease pathways and physiology can be modeled using organoids.
In 2018, Harder et al54 generated a single-cell RNA sequencing data set from human kidney organoids and compared it with a published data set from the developing human kidney.55 This analysis showed the presence of two podocyte clusters in the organoid: an immature cluster and a more mature cluster with higher NPHS1 expression. They then compared their organoid data with microarray data from dissected glomeruli from individuals with CKD of various etiologies and found a subset of genes common to the immature organoid podocyte cluster and diseased glomeruli. They concluded that diseased podocytes showed an increase in immature podocyte genes, suggesting that podocyte dedifferentiation may be a feature of diseased glomeruli in CKD. These results show how human kidney organoids can be used to shed new insights into the molecular basis of glomerular disease.
CHARACTERIZING AND VALIDATING NOVEL PATHOGENIC MUTATIONS
Kidney cells derived from iPSCs also can be used to test the pathogenicity of previously uncharacterized DNA variants. For instance, in the study by Huang et al,56 iPSC lines were made from individuals from a family with autosomal-dominant PKD but no mutations in the PKD1 and PKD2 genes. Genetic analysis showed a deletion in the 5’ untranslated region of the SAMSN1 gene (a tumor-suppressor gene previously unlinked to PKD57–59) that reduced expression of SAMSN1 by 35%. This hypomorphic SAMSN1 mutation compromised the differentiation of iPSCs into kidney-like cells, raising the possibility that it contributes to cystogenesis in PKD, although more work is needed to explore this.
In another study from the Little60,61 laboratory, cells from an individual with nephronophthisis-related ciliopathy that carried a previously uncharacterized mutation in IFT140 were reprogrammed and corrected using CRISPR/Cas9 in a one-step protocol. Kidney organoids generated from the proband iPSCs had abnormally shortened, club-shaped cilia in the distal CDH1+ tubule in comparison with corrected organoids. Transcriptome profiling of the tubules isolated from these organoids showed disease mechanisms beyond ciliary defects, such as changes in apicobasal cell polarity, proliferation, and cellular junctions, similar to that observed in other types of nephronophthisis.60 The one-step reprogramming/correction approach in this study is an interesting approach because it may reduce variation between control and disease cell lines as a result of a common history in handling and passaging. Taken together, disease-specific iPSCs and kidney organoids have utility in functionally assessing DNA variants of unknown significance and may uncover new pathogenic pathways as well as identify commonalities between disease subtypes.
MODELING NEPHROTOXIC INJURY IN ORGANOIDS
Another potential application for kidney organoids is the modeling of AKI in response to nephrotoxins. This was shown independently by the Bonventre13,14 and Little labs16,62 by exposing kidney organoids to cisplatin and gentamicin (two drugs that cause AKI in a clinical setting) and reporting an up-regulation of the proximal tubule injury marker Hepatitis A virus cellular receptor 1 aka kidney injury molecule-113,14 as well as an apoptotic response16 in a dose-dependent manner. The specificity of this injury and whether these nephrotoxins are the most optimal injury inducers remains to be explored in detail. This system has the potential to discover new kidney injury biomarkers, an area of high interest given that the current predictors of kidney injury suffer from a lack of specificity and sensitivity.63 The recent observation that kidney organoids maintained in culture for extended periods of time show signs of activated myofibroblasts and collagen deposition15 raises the possibility that the kidney organoid system also can be used as a model to study mechanisms and biomarkers of fibrosis. This development would be important because both CKD and repeated AKI lead to renal fibrosis with a progressive loss of kidney function.
In addition to tubular injury, a model of toxin-induced podocyte damage also has been reported.47 In 2018, Hale et al47 used a reporter line, MAFB-mTagBFP2, (Table 2) in which podocytes become fluorescently labeled in kidney organoids. By using this reporter, they show that doxorubicin (a chemotherapeutic drug that is toxic to podocytes) dose-dependently reduces glomeruli fluorescence and diameter. At lower concentrations of drug treatment, they noted the presence of apoptotic podocytes, providing a proof of principle that kidney organoids can be used for toxicity screening.
Kidney organoids also lend themselves to drug development approaches. Although rodent and cell line models have been successful at recapitulating and advancing our understanding of kidney injury, they have not been as successful at predicting effectiveness in human beings.64 Human kidney organoids thus could serve as a new screening tool for new preclinical therapeutic candidates. For instance, Lemos et al65 in 2018 identified MYC proto-oncogene (MYC) as a potential therapeutic target of fibrosis in vivo and, as proof of concept, treated kidney organoids with interleukin 1β to model fibrotic events. Treatment of kidney organoids with a MYC inhibitor (+)-JQ1 attenuated proliferation of profibrotic cells and reduced MYC accumulation, validating use of kidney organoids to study fibrosis and screening for compounds to alleviate injury. Coupled with automated high-throughput screening discussed by Czerniecki et al,19 testing of hundreds of compounds for toxicity or to mitigate injury could help expedite the screening process.
CHALLENGES IN CURRENT KIDNEY ORGANOID METHODS
Organoid Maturity and Vascularization
RNA sequencing analysis of kidney organoids has shown that kidney organoids are similar to fetal human kidneys and follow similar developmental pathways.43,49,54,66,67 Although this makes the organoid system suited for the study of kidney development and pediatric renal disorders, the lack of maturation makes it more challenging to model adult human diseases. At present, extended time in culture does not greatly improve organoid maturation. In fact, a comparison of the developmental trajectory of podocytes in human kidney organoids generated by the Morizane et al14 protocol to podocytes in human beings showed that organoid podocytes down-regulate their mature markers at later stages of culture. A similar loss of podocyte markers, as well as markers of the proximal and distal tubule segments, was observed in aged organoids generated by the Przepiorski et al15 protocol in 2018. In addition, this group also found that CDH1+ distal tubules in older organoids were stained with lotus tetragonolobus lectin, traditionally used as a proximal tubule marker. Colocalization of the two markers has not been observed in nephrons from mature human kidney samples. This suggests that the distal tubule of kidney organoids may change its identity over time and adds further evidence that long-term culture causes undesirable changes in kidney organoid viability.68,69
One likely contributor to poor organoid maturation and the decline in viability over time is a lack of adequate vasculature and perfusion. Although varying degrees of endothelial cell numbers are seen in each protocol, there is in general a lack of a well-developed peritubular capillary bed and glomerular capillary tufts. This overall lack of vasculature may lead to limited oxygen and nutrient availability in the interior of the organoid once they reach a certain size. Studies using cultured embryoid bodies of various sizes found that a radius of 400 μm was associated with a more than 50% reduction in oxygen and growth factors in the core compared with embryoid bodies with radii of 200 μm or less.70–72 Different groups have tackled this challenge in different ways. Several laboratories have shown that transplantation of kidney organoids into immunocompromised mice leads to a recruitment of an elaborate host-derived endothelial network with greater maturation of podocytes and tubules.29,43,44,46,73,74 Replicating these effects in vitro remains a difficult task and simply enhancing the endogenous organoid endothelial cell population using growth factors19 or small molecules29 does not lead to improved integration into the podocyte clusters. Instead, it appears that fluid flow is an important factor and, when this is provided in specialized fluidic chambers, the endogenous vascular network becomes more elaborate and endothelial cells are stimulated to invade the podocyte clusters.45 Establishing flow in kidney organoid cultures is rather laborious but, hopefully, with improvements in culturing technologies, this can be incorporated without limiting the current ease of using this system.
Ureteric Epithelium
With the exception of the Taguchi and Nishinakamura18 protocol, kidney organoid protocols that are derived from one progenitor pool do not convincingly generate a ureteric bud (UB) or the subsequent collecting duct (CD) epithelium.66,68,75 Some of the confusion as to whether a collecting duct is formed in these organoids may lie with the fact that there are very few markers of the UB and immature CD that are not expressed by the connecting segment.76,77 Thus, epithelial structures that have been considered CD based on markers such as GATA3 may in fact be connecting segments (Fig. 2). Alternatively, the UB/CD epithelium may be forming in organoids, but are not mature enough to express definitive markers such as WNT9B. By contrast, Nishinakamura and colleagues18 solved the UB/CD issue by modifying their protocol to induce a separate UB population from iPSCs based on mouse development studies. By combining induced UB and induced nephron progenitors, Taguchi and Nishinakamura18 were able to reconstruct the CD branching process, UB-nephron progenitor niches, and, consequently, generate fetal kidney structures that more faithfully replicate what is seen in vivo.
Another cell population that has proven to be important in guiding the organization of kidney development is the interstitium. Based on developmental and RNA sequencing analysis, there are multiple stromal compartments in the kidney.55,78,79 Even though kidney organoids do contain stromal cells,15,65,80 most recent RNA sequencing data have suggested that these populations are not fully representative of the native kidney.66 Taguchi and Nishinakamura18 solved this problem as well by incorporating endogenous mouse embryonic kidney interstitium with their induced mouse UB and nephron progenitors. They found that the stromal cells were essential for enhancing branching morphogenesis of the ureteric tree. Based on these observations it is likely that the other protocols, although easier to use, will need to be optimized further to improve the functionality of their stromal cell populations.
Variability and Off-Target Cell Populations
One of the major challenges in the kidney organoid field is the variability that arises not only between different protocols but also between assays. Variation between the Takasato et al80 (2015) and Morizane et al62 (2015) protocols has been examined by single-cell RNA sequencing. It was found that the Morizane protocol was more enriched in podocytes whereas, conversely, the Takasato et al80 protocol tended to generate more tubular cells.62,68 This variation in podocyte versus tubule enrichment may lie in the difference in timing and concentration of CHIR99021 (Wnt agonist) and fibroblast growth factor 9 in the two protocols (Fig. 1).16 Both protocols formed off-target populations, such as neuronal cells, with these nonrenal cells comprising up to 20% of the total organoid cell population, which expanded with extended culturing time. The neuronal cells could be suppressed by the inclusion of a brain-derived neurotrophic factor-neurotrophin receptor tyrosine kinase 2 inhibitor, providing an additional way to optimize the protocols in which these off-target cells are problematic.68
Another single-cell RNA sequencing study by Subramanian et al67 in 2019 examined the reproducibility of kidney organoids generated by the Takasato80 and Morizane62 protocols using four different iPSC lines at different passages and differentiation time points. Their analysis showed that the major source of cellular variation in the organoids came from using different iPSC lines, rather than between different clones of the same line. For example, one of the iPSC lines tended to generate kidney organoids that were enriched in distal tubular segments. There also were differences in the proportion of off-target cells between organoids generated from different iPSC lines, with some containing less than 2% and others upwards of 43% off-target cells, primarily neuronal.67 This suggests that different iPSC lines may have propensities for certain cellular compartments. In 2019, Phipson et al81 found that the major source of variation was between experimental batches rather than cell lines, with main differences in nephron maturation, patterning, and proportions of on-target versus off-target cells. Interestingly, implantation into immunocompromised mice reduced the off-target population, suggesting that their growth is suppressed or that they cannot survive when placed in an in vivo setting.81
PERSPECTIVES
In this review, we have provided an overview of the current kidney organoid field and how these organoids are being used in various ways to model diseases and injuries in vitro. Although the kidney organoid field has made advances toward creating functional nephron units, there are still many developmental steps that need to be improved before obtaining a well-patterned mini-kidney. The use of single cell RNA sequencing data may help to identify various missing factors that will be necessary for the cellular programming of the progenitor populations, including stromal cells to recapitulate the normal kidney environment.
Even with modifying protocols based on specific functional read-outs, there still remains the challenge of incorporating vasculature. Creating a vascular network has been a significant obstacle for the organoid field including liver, intestine, lung, and retina. In addition to the methods outlined earlier, bioengineering fields continue to develop methods to improve vascularization including scaffolds, fluidics, and polymers, which could be adapted to a kidney organoid system. It is vital that the field continues to move forward with developing new protocols to improve function and to develop new and innovative ways to generate a reproducible microenvironment that reduces the heterogeneity of kidney organoids from experiment to experiment.
In the short term, the utility of organoids may be most suited to functional readouts for cellular, developmental, and genetic mechanisms, and screening for nephrotoxins (Fig. 3). If the main goal is to use kidney organoids in cell replacement therapies and clinical trials, then stringent quality controls need to be established and adopted by the community to reduce variability and misinterpretation of data. This includes unifying protocols and developing a more representative in vitro human kidney model. Toward this goal, the American Society for Cell Biology has established a group to develop guidelines for researchers performing organoid research to facilitate reproducible, transparent data and how to communicate the findings more effectively to the public.82
Figure 3.
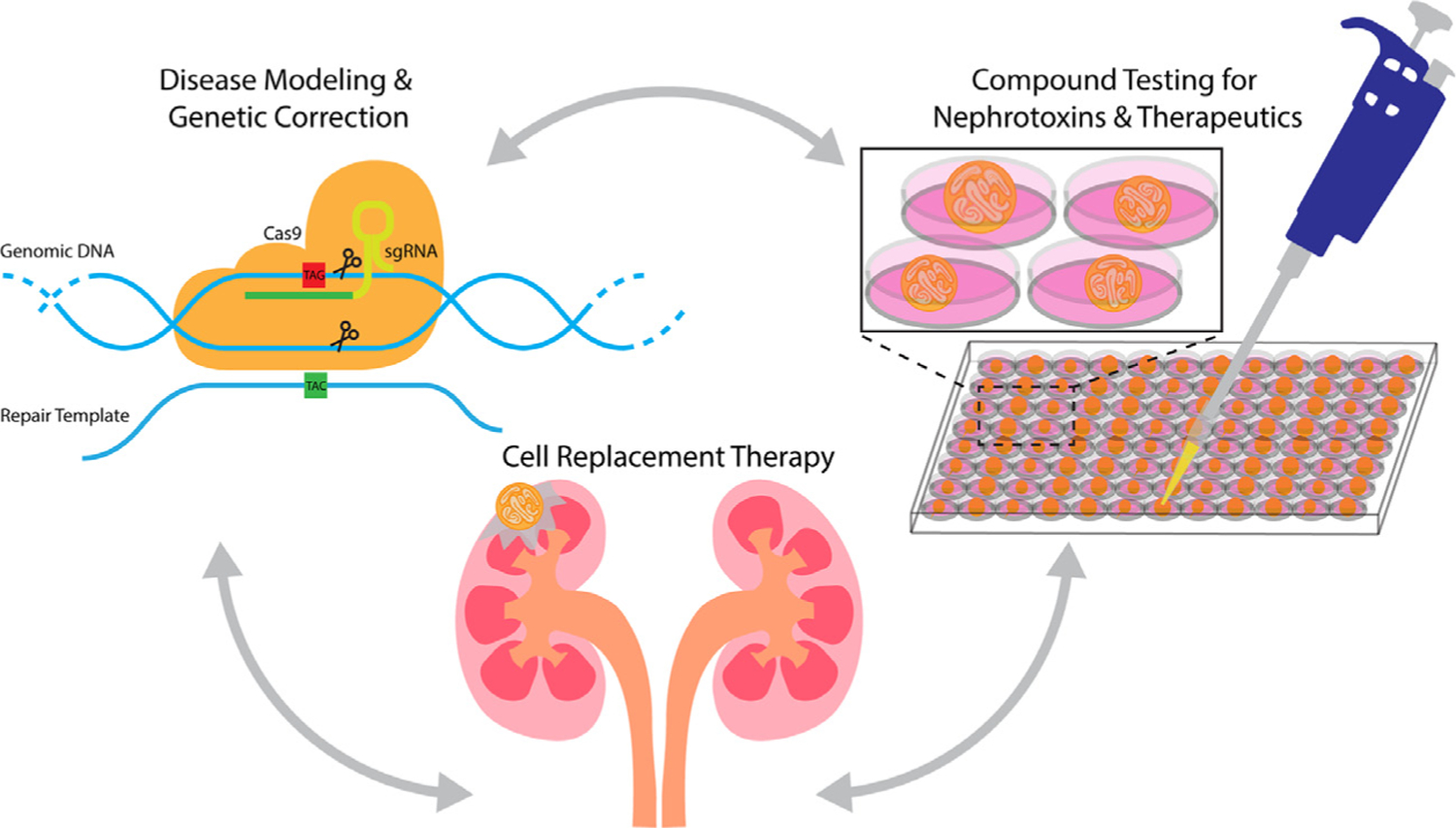
Kidney organoids in the short term can be used to study genetic disease modeling, compound, and nephro-toxicity testing. Future prospective studies for kidney organoids should transition into regenerative research. sgRNA, single guide RNA.
ACKNOWLEDGMENTS
We would like to thank Yvette Aban for professional assistance with the design of our figure illustrations.
Financial support:
Supported by the National Institutes of Health grants 2R01DK069403, 1R01DK112652, 1P30DK079307 (N.A.H.), and T32DK061296 (E.B.E.); the US Department of Defense grant W81XWH-17-1-0610 (N.A.H.); ASN Foundation for Kidney Research Ben J. Lipps Research Fellowship Program (A.P.); and the Health Research Council of New Zealand grants 15/057 and 17/425 (A.J.D.).
Footnotes
Conflict of interest statement: none.
REFERENCES
- 1.Mills KT, Xu Y, Zhang W, Bundy JD, Chen C-S, Kelly TN, et al. A systematic analysis of worldwide population-based data on the global burden of chronic kidney disease in 2010. Kidney Int. 2015;88:950–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robinson BM, Akizawa T, Jager KJ, Kerr PG, Saran R, Pisoni RL. Factors affecting outcomes in patients reaching end-stage kidney disease worldwide: differences in access to renal replacement therapy, modality use, and haemodialysis practices. Lancet. 2016;388:294–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–76. [DOI] [PubMed] [Google Scholar]
- 4.Wiegand C, Banerjee I. Recent advances in the applications of iPSC technology. Curr Opin Biotechnol. 2019;60:250–8. [DOI] [PubMed] [Google Scholar]
- 5.Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, et al. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501:373–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spence JR, Mayhew CN, Rankin SA, Kuhar MF, Vallance JE, Tolle K, et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature. 2011;470:105–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Giacomelli E, Bellin M, Sala L, van Meer BJ, Tertoolen LG, Orlova VV, et al. Three-dimensional cardiac microtissues composed of cardiomyocytes and endothelial cells co-differentiated from human pluripotent stem cells. Development. 2017;144: 1008–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pagliuca FW, Millman JR, Gürtler M, Segel M, Dervort A, Ryu JH, et al. Generation of functional human pancreatic β cells in vitro. Cell. 2014;159:428–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dye BR, Hill DR, Ferguson MA, Tsai YH, Nagy MS, Dyal R, et al. In vitro generation of human pluripotent stem cell derived lung organoids. Elife. 2015;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takebe T, Sekine K, Enomura M, Koike H, Kimura M, Ogaeri T, et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature. 2013;499:481–4. [DOI] [PubMed] [Google Scholar]
- 11.Leslie JL, Huang S, Opp JS, Nagy MS, Kobayashi M, Young VB, et al. Persistence and toxin production by Clostridium difficile within human intestinal organoids result in disruption of epithelial paracellular barrier function. Infect Immun. 2015;83: 138–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garcez PP, Loiola EC, da Costa R, Higa LM, Trindade P, Delvecchio R, et al. Zika virus impairs growth in human neurospheres and brain organoids. Science. 2016;352:816–8. [DOI] [PubMed] [Google Scholar]
- 13.Freedman BS, Brooks CR, Lam AQ, Fu H, Morizane R, Agrawal V, et al. Modelling kidney disease with CRISPR-mutant kidney organoids derived from human pluripotent epiblast spheroids. Nat Commun. 2015;6:8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morizane R, Lam AQ, Freedman BS, Kishi S, Valerius MT, Bonventre JV. Nephron organoids derived from human pluripotent stem cells model kidney development and injury. Nat Biotechnol. 2015;33:1193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Przepiorski A, Sander V, Tran T, Hollywood JA, Sorrenson B, Shih J-HH, et al. A simple bioreactor-based method to generate kidney organoids from pluripotent stem cells. Stem Cell Rep. 2018;11:470–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takasato M, Er PX, Chiu HS, Maier B, Baillie GJ, Ferguson C, et al. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature. 2015; 526:564–8. [DOI] [PubMed] [Google Scholar]
- 17.Taguchi A, Kaku Y, Ohmori T, Sharmin S, Ogawa M, Sasaki H, et al. Redefining the in vivo origin of metanephric nephron progenitors enables generation of complex kidney structures from pluripotent stem cells. Cell Stem Cell. 2013;14:53–67. [DOI] [PubMed] [Google Scholar]
- 18.Taguchi A, Nishinakamura R. Higher-order kidney organogenesis from pluripotent stem cells. Cell Stem Cell. 2017;21:730–46. e736. [DOI] [PubMed] [Google Scholar]
- 19.Czerniecki SM, Cruz NM, Harder JL, Menon R, Annis J, Otto EA, et al. High-throughput screening enhances kidney organoid differentiation from human pluripotent stem cells and enables automated multidimensional phenotyping. Cell Stem Cell. 2018;22: 929–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chapin HC, Caplan MJ. The cell biology of polycystic kidney disease. J Cell Biol. 2010;191:701–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Freedman BS, Lam AQ, Sundsbak JL, Iatrino R, Su X, Koon SJ, et al. Reduced ciliary polycystin-2 in induced pluripotent stem cells from polycystic kidney disease patients with PKD1 mutations. J Am Soc Nephrol. 2013;24:1571–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghata J, Cowley BD Jr. Polycystic kidney disease. Compr Physiol. 2017;7:945–75. [DOI] [PubMed] [Google Scholar]
- 23.Wu G, Tian X, Nishimura S, Markowitz GS, D’Agati V, Park JH, et al. Trans-heterozygous Pkd1 and Pkd2 mutations modify expression of polycystic kidney disease. Hum Mol Genet. 2002;11:1845–54. [DOI] [PubMed] [Google Scholar]
- 24.Cruz NM, Song X, Czerniecki SM, Gulieva RE, Churchill AJ, Kim Y, et al. Organoid cystogenesis reveals a critical role of microenvironment in human polycystic kidney disease. Nat Mater. 2017;16:1112–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gainullin VG, Hopp K, Ward CJ, Hommerding CJ, Harris PC. Polycystin-1 maturation requires polycystin-2 in a dose-dependent manner. J Clin Invest. 2015;125:607–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Su X, Wu M, Yao G, El-Jouni W, Luo C, Tabari A, et al. Regulation of polycystin-1 ciliary trafficking by motifs at its C-terminus and polycystin-2 but not by cleavage at the GPS site. J Cell Sci. 2015;128:4063–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Su Q, Hu F, Ge X, Lei J, Yu S, Wang T, et al. Structure of the human PKD1/PKD2 complex. Science. 2018;361. [DOI] [PubMed] [Google Scholar]
- 28.Parnell SC, Magenheimer BS, Maser RL, Pavlov TS, Havens MA, Hastings ML, et al. A mutation affecting polycystin-1 mediated heterotrimeric G-protein signaling causes PKD. Hum Mol Genet. 2018;27:3313–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Low JH, Li P, Chew EGY, Zhou B, Suzuki K, Zhang T, et al. Generation of human PSC-derived kidney organoids with patterned nephron segments and a de novo vascular network. Cell Stem Cell. 2019;25:373–87. e379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harshman LA, Brophy PD. PAX2 in human kidney malformations and disease. Pediatr Nephrol. 2012;27:1265–75. [DOI] [PubMed] [Google Scholar]
- 31.Dressler GR, Woolf AS. Pax2 in development and renal disease. Int J Dev Biol. 1999;43:463–8. [PubMed] [Google Scholar]
- 32.Kaku Y, Taguchi A, Tanigawa S, Haque F, Sakuma T, Yamamoto T, et al. PAX2 is dispensable for in vitro nephron formation from human induced pluripotent stem cells. Sci Rep. 2017;7:4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bouchard M, Souabni A, Mandler M, Neubüser A, Busslinger M. Nephric lineage specification by Pax2 and Pax8. Genes Dev. 2002;16:2958–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharma R, Sanchez-Ferras O, Bouchard M. Pax genes in renal development, disease and regeneration. Semin Cell Dev Biol. 2015;44:97–106. [DOI] [PubMed] [Google Scholar]
- 35.Porteous S, Torban E, Cho NP, Cunliffe H, Chua L, McNoe L, et al. Primary renal hypoplasia in humans and mice with PAX2 mutations: evidence of increased apoptosis in fetal kidneys of Pax2(1Neu) +/− mutant mice. Hum Mol Genet. 2000;9:1–11. [DOI] [PubMed] [Google Scholar]
- 36.Heliot C, Desgrange A, Buisson I, Prunskaite-Hyyryläinen R, Shan J, Vainio S, et al. HNF1B controls proximal-intermediate nephron segment identity in vertebrates by regulating Notch signalling components and Irx1/2. Development. 2013; 140:873–85. [DOI] [PubMed] [Google Scholar]
- 37.Lokmane L, Heliot C, Garcia-Villalba P, Fabre M, Cereghini S. vHNF1 functions in distinct regulatory circuits to control ureteric bud branching and early nephrogenesis. Development. 2010;137: 347–57. [DOI] [PubMed] [Google Scholar]
- 38.Bockenhauer D, Jaureguiberry G. HNF1B-associated clinical phenotypes: the kidney and beyond. Pediatr Nephrol. 2016;31:707–14. [DOI] [PubMed] [Google Scholar]
- 39.Massa F, Garbay S, Bouvier R, Sugitani Y, Noda T, Gubler M-CC, et al. Hepatocyte nuclear factor 1β controls nephron tubular development. Development.2013;140:886–96. [DOI] [PubMed] [Google Scholar]
- 40.Desgrange A, Heliot C, Skovorodkin I, Akram SU, Heikkilä J, Ronkainen V-P, et al. HNF1B controls epithelial organization and cell polarity during ureteric bud branching and collecting duct morphogenesis. Development. 2017;144:4704–19. [DOI] [PubMed] [Google Scholar]
- 41.Mae S II, Ryosaka M, Toyoda T, Matsuse K, Oshima Y, Tsujimoto H, et al. Generation of branching ureteric bud tissues from human pluripotent stem cells. Biochem Biophys Res Commun. 2018;495:954–61. [DOI] [PubMed] [Google Scholar]
- 42.Nishinakamura R, Sharmin S, Taguchi A. Induction of nephron progenitors and glomeruli from human pluripotent stem cells. Pediatr Nephrol. 2017;32:195–200. [DOI] [PubMed] [Google Scholar]
- 43.Tran T, Lindstrom NO, Ransick A, De Sena Brandine G, Guo Q, Kim AD, et al. In vivo developmental trajectories of human podocyte inform in vitro differentiation of pluripotent stem cell-derived podocytes. Dev Cell. 2019;50:102–16. e106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van den Berg CW, Ritsma L, Avramut MC, Wiersma LE, van den Berg BM, Leuning DGG, et al. Renal subcapsular transplantation of PSC-derived kidney organoids induces neo-vasculogenesis and significant glomerular and tubular maturation in vivo. Stem Cell Rep. 2018;10:751–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Homan KA, Gupta N, Kroll KT, Kolesky DB, Skylar-Scott M, Miyoshi T, et al. Flow-enhanced vascularization and maturation of kidney organoids in vitro. Nat Methods. 2019;16:255–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sharmin S, Taguchi A, Kaku Y, Yoshimura Y, Ohmori T, Sakuma T, et al. Human induced pluripotent stem cell-derived podocytes mature into vascularized glomeruli upon experimental transplantation. J Am Soc Nephrol. 2016;27:1778–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hale LJ, Howden SE, Phipson B, Lonsdale A, Er PX, Ghobrial I, et al. 3D organoid-derived human glomeruli for personalised podocyte disease modelling and drug screening. Nat Commun. 2018; 9:5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chittiprol S, Chen P, Petrovic-Djergovic D, Eichler T, Ransom RF. Marker expression, behaviors, and responses vary in different lines of conditionally immortalized cultured podocytes. Am J Physiol Renal Physiol. 2011;301:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim YK, Refaeli I, Brooks CR, Jing P, Gulieva RE, Hughes MR, et al. Gene-edited human kidney organoids reveal mechanisms of disease in podocyte development. Stem Cells. 2017;35:2366–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin FJ, Yao L, Hu XQ, Bian F, Ji G, Jiang GR, et al. First identification of PODXL nonsense mutations in autosomal dominant focal segmental glomerulosclerosis. Clin Sci (Lond). 2019;133:9–21. [DOI] [PubMed] [Google Scholar]
- 51.Kang HG, Lee M, Lee KB, Hughes M, Kwon BS, Lee S, et al. Loss of podocalyxin causes a novel syndromic type of congenital nephrotic syndrome. Exp Mol Med. 2017;49:e414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tanigawa S, Islam M, Sharmin S, Naganuma H, Yoshimura Y, Haque F, et al. Organoids from nephrotic disease-derived iPSCs identify impaired NEPHRIN localization and slit diaphragm formation in kidney podocytes. Stem Cell Rep. 2018;11:727–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yoshimura Y, Taguchi A, Tanigawa S, Yatsuda J, Kamba T, Takahashi S, et al. Manipulation of nephron-patterning signals enables selective induction of podocytes from human pluripotent stem cells. J Am Soc Nephrol. 2019;30:304–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Harder JL, Menon R, Otto EA, Zhou J, Eddy S, Wys NL, et al. Organoid single cell profiling identifies a transcriptional signature of glomerular disease. JCI Insight. 2019;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Menon R, Otto EA, Kokoruda A, Zhou J, Zhang Z, Yoon E, et al. Single-cell analysis of progenitor cell dynamics and lineage specification in the human fetal kidney. Development. 2018; 145:258798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang CY, Ho MC, Lee JJ, et al. Generation of induced pluripotent stem cells derived from an autosomal dominant polycystic kidney disease patient with a p.Ser1457fs mutation in PKD1. Stem Cell Res. 2017;24:139–43. [DOI] [PubMed] [Google Scholar]
- 57.Sueoka S, Kanda M, Sugimoto H, Shimizu D, Nomoto S, Oya H, et al. Suppression of SAMSN1 expression is associated with the malignant phenotype of hepatocellular carcinoma. Ann Surg Oncol. 2015;22(Suppl 3):60. [DOI] [PubMed] [Google Scholar]
- 58.Noll JE, Hewett DR, Williams SA, Vandyke K, Kok C, To LB, et al. SAMSN1 is a tumor suppressor gene in multiple myeloma. Neoplasia. 2014;16:572–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yan Y, Zhang L, Xu T, Zhou J, Qin R, Chen C, et al. SAMSN1 is highly expressed and associated with a poor survival in glioblastoma multiforme. PLoS One. 2013;8:e81905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Forbes TA, Howden SE, Lawlor K, Phipson B, Maksimovic J, Hale L, et al. Patient-iPSC-derived kidney organoids show functional validation of a ciliopathic renal phenotype and reveal underlying pathogenetic mechanisms. Am J Hum Genet. 2018;102:816–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Howden SE, Thomson JA, Little MH. Simultaneous reprogramming and gene editing of human fibroblasts. Nat Protoc. 2018;13:875–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Morizane R, Bonventre JV. Generation of nephron progenitor cells and kidney organoids from human pluripotent stem cells. Nat Protoc. 2017;12:195–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nassirpour R, Ramaiah SK, Whiteley LO. Nephron segment specific microRNA biomarkers of pre-clinical drug-induced renal toxicity: opportunities and challenges. Toxicol Appl Pharmacol. 2016;312:34–41. [DOI] [PubMed] [Google Scholar]
- 64.Zuk A, Palevsky PM, Fried L, Harrell FE, Khan S, McKay DB, et al. Overcoming translational barriers in acute kidney injury: a report from an NIDDK workshop. Clin J Am Soc Nephrol. 2018;13:1113–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lemos DR, McMurdo M, Karaca G, Wilflingseder J, Leaf IA, Gupta N, et al. Interleukin-1beta activates a MYC-dependent metabolic switch in kidney stromal cells necessary for progressive tubulointerstitial fibrosis. J Am Soc Nephrol. 2018;29:1690–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Combes AN, Zappia L, Er PX, Oshlack A, Little MH. Single-cell analysis reveals congruence between kidney organoids and human fetal kidney. Genome Med. 2019;11:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Subramanian A, Sidhom EH, Emani M, et al. Single cell census of human kidney organoids shows reproducibility and diminished off-target cells after transplantation. Nat Commun. 2019;10:5462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu H, Uchimura K, Donnelly EL, Kirita Y, Morris SA, Humphreys BD. Comparative analysis and refinement of human PSC-derived kidney organoid differentiation with single-cell transcriptomics. Cell Stem Cell. 2018;23:869–81. e868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kumar SV, Er PX, Lawlor KT, Motazedian A, Scurr M, Ghobrial I, et al. Kidney micro-organoids in suspension culture as a scalable source of human pluripotent stem cell-derived kidney cells. Development. 2019;146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Winkle AP, Gates ID, Kallos MS. Mass transfer limitations in embryoid bodies during human embryonic stem cell differentiation. Cells Tissues Organs. 2012;196:34–47. [DOI] [PubMed] [Google Scholar]
- 71.Zhao F, Pathi P, Grayson W, Xing Q, Locke BR, Ma T. Effects of oxygen transport on 3–D human mesenchymal stem cell metabolic activity in perfusion and static cultures: experiments and mathematical model. Biotechnol Prog. 2005;21:1269–80. [DOI] [PubMed] [Google Scholar]
- 72.Wu J, Rostami MR, Olaya DP, Tzanakakis ES. Oxygen transport and stem cell aggregation in stirred-suspension bioreactor cultures. PLoS One. 2014;9:e102486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Garreta E, Prado P, Tarantino C, Oria R, Fanlo L, Marti E, et al. Fine tuning the extracellular environment accelerates the derivation of kidney organoids from human pluripotent stem cells. Nat Mater. 2019;18:397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bantounas I, Ranjzad P, Tengku F, Silajdžić E, Forster D, Asselin M-C, et al. Generation of functioning nephrons by implanting human pluripotent stem cell-derived kidney progenitors. Stem Cell Rep. 2018;10:766–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Islam M, Nishinakamura R. How to rebuild the kidney: recent advances in kidney organoids. J Biochem. 2019;166:7–12. [DOI] [PubMed] [Google Scholar]
- 76.Combes AN, Phipson B, Lawlor KT, Dorison A, Patrick R, Zappia L, et al. Correction: Single cell analysis of the developing mouse kidney provides deeper insight into marker gene expression and ligand-receptor crosstalk. Development. 2019;146. [DOI] [PubMed] [Google Scholar]
- 77.Lindstrom NO, McMahon JA, Guo J, Tran T, Guo Q, Rutledge E, et al. Conserved and divergent features of human and mouse kidney organogenesis. J Am Soc Nephrol. 2018;29:785–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Boivin FJ, Sarin S, Lim J, Javidan A, Svajger B, Khalili H, et al. Stromally expressed β-catenin modulates Wnt9b signaling in the ureteric epithelium. PLOS One. 2015;10:e0120347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lindstrom NO, Guo J, Kim AD, Tran T, Guo Q, De Sena Brandine G, et al. Conserved and divergent features of mesenchymal progenitor cell types within the cortical nephrogenic niche of the human and mouse kidney. J Am Soc Nephrol. 2018;29: 806–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Takasato M, Er PX, Chiu HS, Little MH. Generation of kidney organoids from human pluripotent stem cells. Nat Protoc. 2016;11:1681–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Phipson B, Er PX, Combes AN, Forbes TA, Howden SE, Zappia L, et al. Evaluation of variability in human kidney organoids. Nat Methods. 2019;16:79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lehmann R, Lee CM, Shugart EC, Benedetti M, Charo RA, Gartner Z, et al. Human organoids: a new dimension in cell biology. Mol Biol Cell. 2019;30:1129–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Musah S, Dimitrakakis N, Camacho DM, Church GM, Ingber DE. Directed differentiation of human induced pluripotent stem cells into mature kidney podocytes and establishment of a Glomerulus Chip. Nat Protoc. 2018;13:1662–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hiratsuka K, Monkawa T, Akiyama T, et al. Induction of human pluripotent stem cells into kidney tissues by synthetic mRNAs encoding transcription factors. Sci Rep. 2019;9:913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Francipane MG, Han B, Oxburgh L, Sims-Lucas S, Li Z, Lagasse E. Kidney-in-a-lymph node: a novel organogenesis assay to model human renal development and test nephron progenitor cell fates. J Tissue Eng Regen Med. 2019;13:1724–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Combes AN, Phipson B, Lawlor KT, et al. Single cell analysis of the developing mouse kidney provides deeper insight into marker gene expression and ligand-receptor crosstalk. Development. 2019;146:dev178673. [DOI] [PubMed] [Google Scholar]
- 87.Hariharan K, Stachelscheid H, Rossbach B, et al. Parallel generation of easily selectable multiple nephronal cell types from human pluripotent stem cells. Cell Mol Life Sci. 2019;76:179–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Freedman BS. Modeling kidney disease with iPS cells. Biomark Insights. 2015;10(Suppl 1):153–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Vanslambrouck JM, Wilson SB, Tan KS, et al. A toolbox to characterize human induced pluripotent stem cell-derived kidney cell types and organoids. J Am Soc Nephrol. 2019;30:1811–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Borestrom C, Jonebring A, Guo J, et al. A CRISP(e)R view on kidney organoids allows generation of an induced pluripotent stem cell-derived kidney model for drug discovery. Kidney Int. 2018;94:1099–110. [DOI] [PubMed] [Google Scholar]