

Abstract
A variety of targeted anticancer agents have been successfully introduced into clinical practice, largely reflecting their ability to inhibit specific molecular alterations that are required for disease progression. However, not all malignant cells rely on such alterations to survive, proliferate, disseminate and/or evade anticancer immunity, implying that many tumours are intrinsically resistant to targeted therapies. Radiotherapy is well known for its ability to activate cytotoxic signalling pathways that ultimately promote the death of cancer cells, as well as numerous cytoprotective mechanisms that are elicited by cellular damage. Importantly, many cytoprotective mechanisms elicited by radiotherapy can be abrogated by targeted anticancer agents, suggesting that radiotherapy could be harnessed to enhance the clinical efficacy of these drugs. In this Review, we discuss preclinical and clinical data that introduce radiotherapy as a tool to elicit or amplify clinically actionable signalling pathways in patients with cancer.
Over the past two decades, targeted anticancer agents have revolutionized the clinical management of a wide range of malignancies, largely reflecting the selective inhibition of aberrantly activated signalling pathways that are required for the survival, proliferation, dissemination and/or immunoevasion of cancer cells1. However, various tumours can be intrinsically resistant to targeted anticancer agents, because not all malignancies harbour genetic alterations that promote aberrant signal transduction (such as KRAS mutations), or because such signal transduction pathways emerge from epigenetic alterations or stress-responsive transcriptional programmes that are either not present or inactive at baseline2, two situations that equally result in a lack of targetable alterations. In the former scenario, personalized, in-depth genetic characterization of the tumour might enable the identification of patients who are likely to benefit from targeted anticancer agents, such as those with non-small-cell lung carcinoma (NSCLC) driven by KRASG12C, who are now eligible to receive the KRASG12C-specific agent sotorasib (as second-line or later-line of therapy)3. A similar approach has also been successfully used in the latter scenario, for example by identifying patients with PD-L1+ tumours of various histologies, who are likely to benefit from immune-checkpoint inhibitors (ICIs)4. Moreover, at least in principle, cancer cells that lack a targetable alteration at baseline might become sensitized to certain targeted anticancer agents by harnessing the principle of ‘non-oncogene addition’, which involves rendering malignant cells dependent on otherwise non-oncogenic (and therapeutically actionable) signalling pathways5.
More than 50% of patients with cancer receive radiotherapy as part of the clinical management of their disease either with curative intent (especially, but not exclusively in the context of early-stage disease6,7) or in palliative settings in order to contain the symptoms of metastatic disease, such as pain8. Radiotherapy is often used as a preoperative debulking intervention to facilitate surgical excision, as well as postoperatively (and less so intra-operatively) to control residual microscopic disease9-11. In all of these applications, radiotherapy has been demonstrated to reduce the incidence of local recurrence9-11. From a molecular standpoint, radiotherapy causes direct and reactive oxygen species (ROS)-dependent damage to DNA (and other molecules), potentially culminating in the permanent inactivation of cell division (cellular senescence) or the initiation of cell death programmes12,13 (BOX 1). Intriguingly, cancer cells that succumb to radiotherapy also release abundant antigenic material as they emit immunostimulatory signals that support tumour-targeting immune responses14. Furthermore, accumulating preclinical and clinical findings suggest that the ultimate efficacy of radiotherapy might depend, at least in certain settings, on engagement of the patient’s immune system15,16 (BOX 2).
Box 1 ∣. Cytotoxic pathways elicited by radiotherapy.
Radiotherapy mediates cytotoxic effects that originate either from direct damage or reactive oxygen species (ROS)-dependent damage to macromolecules. According to a commonly accepted model, DNA is the macromolecule most affected by radiotherapy, resulting in a variety of lesions with a predominance of double-stand breaks. These lesions rapidly activate the so-called DNA damage response (DDR), which initially operates in an adaptive, cytoprotective mode, involving a temporary arrest of cellular proliferation (generally at the G2–M transition) that enables DNA repair and the recovery of cellular homeostasis. However, if DNA damage is excessive and ultimately remains unrepaired, the DDR can switch to a cytotoxic mode, in which it can initiate the active demise of cells with DNA damage deemed to be beyond repair335. Such a cytotoxic DDR most often involves activation of p53, which upon phosphorylation by ATM or the ATM substrate CHEK2 is stabilized and coordinates the expression of various proteins involved in mitochondrial apoptosis, including BAX and its activators BBC3 (also known as PUMA) and PMAIP1 (also known as NOXA). PUMA, NOXA and other so-called BH3 only proteins favour the oligomerization of BAX and BAK1 at the outer mitochondrial membrane, culminating in its permeabilization, and (1) irreversible mitochondrial inactivation coupled with oxidative macromolecular damage (the actual cause of cell death) and (2) the activation of proteolytic enzymes including caspases (which regulate the kinetic and immunological manifestations of cell death)13,266. Importantly, especially in the context of p53 defects, cancer cells arrested by radiotherapy at the G2–M transition can illicitly slip into defective mitosis characterized by multinucleation or micronucleation336. This process, which is commonly referred to as mitotic catastrophe, ultimately leads to cell death (either during mitosis, or during interphase in daughter cells) or permanent proliferative inactivation (so-called cellular senescence) resulting in mitosis-incompetent cells336. Of note, data published in 2020 point to extranuclear damage, especially oxidative damage to lipid layers, as another contributor to the anticancer effects of radiotherapy337,338. In this setting, cytotoxicity emerges from a non-apoptotic variant of regulated cell death commonly known as ferroptosis337,338. Intriguingly, ROS participate in both DDR-dependent apoptosis and ferroptosis, which is in line with an abundant literature linking hypoxia with radioresistance24.
Box 2 ∣. Immunomodulatory pathways elicited by radiotherapy.
Besides promoting senescence and the death of malignant cells, radiotherapy mediates a panel of immunostimulatory effects that (at least in certain settings) are expected to contribute to clinical efficacy. These effects largely reflect the ability of radiotherapy to promote the antigenicity and adjuvanticity of cancer cells downstream of: (1) transcriptional upregulation of genes that encode antigenic neoepitopes that are otherwise silenced, resulting in the engagement of adaptive immunity339; (2) increased exposure of MHC class I molecules on the cell surface, thus facilitating the recognition of irradiated cells by CD8+ T cells340; (3) upregulation of natural killer cell activating ligands, thus favouring the activation of antigen-independent effector responses340; (4) accumulation of mitochondrial DNA in the cytosol of irradiated cells, culminating in type I interferon secretion upon activation of cyclic GMP–AMP synthase58,341; and (5) activation of immunogenic cell death and the consequent release of multiple immunostimulatory molecules that ultimately support T cell activation, including a panel of so-called damage-associated molecular patterns342. However, radiotherapy can also mediate immunosuppressive effects, including the upregulation of PD-L1340 and the accumulation of CD4+CD25+FOXP3+ regulatory T cells279. Thus, at least in immunologically competent tumours (those that are susceptible to anticancer immunity), the efficacy of radiotherapy is influenced by the balance between activation of immunostimulatory and immunosuppressive signalling pathways. Importantly, many of these pathways can be harnessed using immunotherapeutic approaches designed to enhance the efficacy of radiotherapy, as currently investigated in a large number of clinical trials343.
Of note, the macromolecular damage imposed by radiotherapy affects both malignant and non-malignant cells, although the latter are relatively more radioresistant than the former as they can usually harness efficient repair mechanisms and are generally less proliferative12. Consistent with this notion, the acute adverse effects of radiotherapy tend to be more pronounced in non-malignant tissues with rapid cellular turnover, such as the gastrointestinal epithelium17. Thus, the terminal fate of irradiated cancer cells depends on their ability to successfully cope with the damage inflicted by radiotherapy by activating cytoprotective pathways that might enable avoidance of cellular senescence or regulated cell death, including (but not limited to): (1) proficient DNA damage resolution via the DNA damage response (DDR)12; (2) mitogenic signalling via surface-exposed receptors, such as HER218 and MET19, or signal transducers thereof, such as PI3K20 and MTOR21; (3) cellular stress management upon activation of macroautophagy (herein referred to as autophagy), which is an evolutionary conserved pathway for the preservation of cellular and organismal homeostasis22; and (4) microenvironmental reconfiguration and immunoevasion, as driven by TGFβ23 and PD-L1 signalling24 (FIG. 1). Consistent with this notion, numerous targeted anticancer agents and ICIs have been tested for their ability to enhance the efficacy of radiotherapy25,26. Such a conceptual approach, however, has thus far achieved limited clinical success, potentially reflecting the notion that many tumours do not display non-oncogene addiction at baseline, but rather acquire such dependency during therapy through the emergence and/or expansion of treatment-resistant clones via positive selection5,27. This limitation suggests a crucial role for administration schedules in the efficacy of radiotherapy-containing therapeutic combinations28.
Fig. 1 ∣. Cytoprotective pathways elicited by radiotherapy.

Ionizing radiation damages a variety of macromolecules including nuclear DNA, either directly or upon generation of reactive oxygen species. Such damage is often detected by a molecular complex encompassing meiotic recombination 11 (MRE11), DNA repair protein Rad50 (RAD50) and Nijmegen breakage syndrome 1 (NSB1) in co-operation with members of the poly(ADP-ribose) polymerase (PARP) protein family. Formation of this complex results in the sequential activation of ATM, CHEK2 and p53. Alternatively or concomitantly, the DNA damage induced by radiotherapy drives the activation of ATR and consequently CHEK1, DNA-dependent protein kinase (DNA-PK) or WEE1 signalling. Ultimately, these pathways converge on the inhibition of cyclin-dependent kinases (CDKs) resulting in arrested cell-cycle progression at specific checkpoints, which enables DNA repair and hence supports radioresistance (part a). The DNA damage response elicited by radiotherapy also promotes (directly or indirectly) the hyperactivation of PI3K signalling, resulting in the delivery of cytoprotective signals via AKT1 and MTOR (part b), the activation of autophagy (which is generally under negative regulation by MTOR) (part c), as well as the synthesis, secretion and activation of transforming growth factor-β1 (TGFβ1) (part d). GPCR, G-protein-coupled receptor; LAP, latency associated peptide; LTBP1, latent transforming growth factor-β binding protein 1; PTEN, phosphatase and tensin homologue; RTK, receptor tyrosine kinase.
Combining radiotherapy with targeted anticancer agents might be challenging in clinical settings, given that non-malignant tissues resist the detrimental effects of radiotherapy by harnessing the same cytoprotective mechanisms that support radioresistance (such as a proficient DDR)12. These overlapping mechanisms of resistance might explain the limited clinical success of such approaches when implemented according to standard-of-care (SOC) protocols that have been developed for each agent employed as standalone therapeutic interventions. This issue highlights the importance of a close collaboration between radiation oncologists and medical oncologists during trial design, with the aim of identifying a good compromise between efficacy and toxicity. For example, combining radiotherapy with an anti-PD-L1 antibody in patients with NSCLC creates a high risk of pneumonitis from each modality. Thus, investigators in a phase II trial with results published in June 2021 selected sub-ablative doses of preoperative stereotactic body radiotherapy (SBRT), which successfully avoided such pulmonary toxicities in patients with resectable NSCLC who were also receiving the anti-PD-L1 antibody durvalumab as a neoadjuvant therapy29.
In this Review, we build on the pioneering work of Coleman and colleagues to further develop the innovative concept (originally introduced in 2013) of harnessing radiotherapy early in the course of treatment as a method for sensitizing malignant cells to targeted anticancer agents, thus expanding the range of oncological indications in which these drugs are effective30. As we summarize preclinical data supporting this novel use of radiotherapy, we discuss key aspects — including, but not limited to, administration schedules and potential for toxicities — for such a strategy to be successfully implemented in the clinic. We surmise that (at least for certain indications) this approach might improve the extent of local, and possibly systemic, disease control by enabling the early eradication of radiosensitive cancer cells, as well as the suppression of cancer cells that survive radiotherapy in the context of acquired radioresistance mediated by non-oncogene addiction.
DDR signalling
The best studied cellular effect of radiotherapy involves the direct and ROS-dependent formation of DNA single-strand and double-strand breaks (DSBs), which rapidly initiate the DDR as a cytoprotective response31. The primary goal of the DDR is to establish a reversible cell-cycle arrest that enables DNA repair and restoration of genomic integrity in cells harbouring damaged DNA31. In line with this notion, a proficient DDR is associated with reduced sensitivity to radiotherapy12 and various DNA-damaging chemotherapeutic agents32. However, genomic instability generally supports malignant transformation and tumour progression33, and indeed tumours of various histologies are known to harbour deletions or loss-of-function mutations in various genes encoding components of the DDR, such as BRCA1 and ATM34. These defects often elicit dependence on complementary non-oncogenic DDR pathways or accrued anti-apoptotic signalling, which has driven the development of various targeted therapies designed to harness the principle of synthetic lethality31. As a standalone example, poly(ADP-ribose) polymerase (PARP) inhibitors are now clinically approved drugs for use in patients with breast or ovarian cancers harbouring BRCA1 or BRCA2 mutations35. Along similar lines, acquired resistance to the DNA-damaging agent cisplatin has been shown to emerge alongside PARP1 hyperactivation and non-oncogene addiction to PARP1 (REF.36). Thus, DDR-targeting agents might also be effective in targeting malignant cells that acquire resistance to radiotherapy (Supplementary Table 1). Importantly, non-malignant cells are generally less sensitive to DNA-damaging agents than their malignant counterparts (reflecting, at least in part, their reduced proliferative rate)12, which offers a therapeutic opportunity to combine radiotherapy with DDR-targeting agents in the context of acceptable toxicity (at least a priori).
ATM, ATR and DNA-PK inhibitors.
The serine/threonine kinase ATM has a major role in DSB repair upon irradiation, in part owing to the capacity to drive (at least initially) cytoprotective transcriptional programmes transduced by checkpoint kinase 2 (CHEK2) and orchestrated by p53 (REF.37). Consistent with this notion, germline ATM alterations, which lead to the human autosomal recessive disorder ataxia–telangiectasia, explain the extraordinary sensitivity of patients with ataxia–telangiectasia to ionizing radiation38, as well as the inability of their lymphocytes to properly repair RT-induced DNA damage39. Moreover, basal ATM activation has been linked with radioresistance in stem-like cells isolated from patients with glioblastoma40, and pharmacological ATM inhibition can boost the cytostatic and/or cytotoxic effects of radiotherapy on both human and mouse cancer cells in vitro40-44 and in vivo45-47, especially in the context of TP53 mutations45-47.
ATR is also involved in the repair of radiotherapy-induced DNA damage, although cytoprotective signals associated with ATR activation are mostly transduced via CHEK1, as opposed to CHEK2. ATR inhibition exacerbates the cytostatic and/or cytotoxic effects of radiotherapy in multiple preclinical tumour models44,48-52, including patient-derived xenografts established from patients with triple-negative breast cancer (TNBC) recurring after chemotherapy53. Similar effects have also been documented with DNA-dependent protein kinase (DNA-PK) inhibitors, which target yet another component of the early DDR that can be activated by radiotherapy54-56. Of note, DNA-PK inhibitors appear to completely spare non-malignant tissues (at least in mouse models)57 and therefore stand out as promising combinatorial partners for radiotherapy.
Importantly, radiotherapy can also synergize with ATM and ATR inhibitors via immunostimulatory mechanisms. In particular, both ATM inhibitors (such as KU60019) and ATR-targeting agents (such as ceralasertib) have been shown to: (1) stimulate type I interferon (IFN) release upon cyclic GMP–AMP synthase (CGAS) activation driven by the cytosolic accumulation of nuclear (as opposed to mostly mitochondrial, as in the case of radiotherapy)58 DNA fragments in preclinical models of breast, lung and pancreatic cancer59-61; and (2) abrogate the immunosuppressive effects of radiotherapy including the accumulation of CD4+CD25+FOXP3+ regulatory T cells and PD-L1 expression by cancer cells in preclinical models of hepatocellular carcinoma (HCC) and colorectal cancer (CRC)62,63. These findings are in line with the notion that ATM drives immunosuppressive NF-κB signalling in radioresistant cancer cells64, potentially linked to accrued genomic instability and indolent CGAS activation by micronuclei65. However, the observation that ATM silencing exacerbates PD-L1 overexpression driven by radiation in mouse models of pancreatic cancer, resulting in increased sensitivity to PD-L1 blockade, provides evidence to the contrary61. Whether this apparent discrepancy originates from an off-target effect of ceralasertib or the activation of compensatory pathways emerging from stable ATM knockdown remains to be elucidated.
Inhibitors of the MRN complex.
DSBs elicited by irradiation activate ATM via the so-called MRN complex, which encompasses meiotic recombination 11 (MRE11), DNA repair protein Rad50 (RAD50) and Nijmegen breakage syndrome 1 (NSB1)66. MRE11 dysfunction has been linked to enhanced tumorigenesis (independent of p53 and ATM) in a mouse model of oncogene-driven mammary carcinoma and to hypersensitivity to DNA-damaging agents as well as ATR, CHEK1 and PARP1 inhibitors in breast cancer cell lines and cancer stem cells derived from patients with CRC67-69. Consistent with this notion, women with TNBC expressing limited levels of MRN complex components (defined as <10% of nuclei staining for MRE11 or NBS1) have superior disease-specific survival than patients with abundant MRE11 or NBS1 expression67, while overexpression of the MRN complex is correlated with a poor response to neoadjuvant radiotherapy in patients with rectal cancer70. Moreover, the transgene-driven expression of a RAD50 variant that weakens the interactions between MRN components has been shown to enhance the sensitivity of nasopharyngeal cancer cells to radiation, both in vitro and in vivo71. However, high MRE11 levels (defined as >25th percentile) are also predictive of improved overall survival (OS) in patients with muscle-invasive bladder cancer receiving radiotherapy72. Whether this latter observation reflects the ability of MRE11 to drive type I IFN signalling in response to cytosolic DNA73 remains to be clarified. Irrespective, MRE11 can be cleaved into an inactive variant that lacks nuclease and DNA-binding activities but still assembles with the MRN complex in response to histone deacetylase (HDAC) inhibitors74. Consistent with a key role for MRE11 in DNA repair following radiotherapy, various HDAC inhibitors, including the clinically approved agents panobinostat and romidepsin, have been shown to disrupt the DDR and synergize with radiotherapy without notable increases in systemic toxicity in various xenograft models of urothelial carcinoma75,76.
PARP inhibitors.
PARPs are a superfamily of DNA repair enzymes with functions that are essential for the survival of cancer cells bearing homologous recombination defects, such as those imposed by loss-of-function BRCA1 and BRCA2 mutations77. Various PARPs, including PARP1, are recruited to radiotherapy-induced DSBs and activate the DDR by promoting DNA end resection via the MRN complex78, and rapidly decondensing chromatin at sites of DNA damage79, pointing to a potential synergy between PARP inhibitors and radiotherapy in the control of malignant cells. Consistent with this notion, various FDA-approved and experimental PARP inhibitors, including olaparib80-83, talazoparib84, veliparib82,85 and fluzoparib86, are able to interfere with the cytoprotective DDR elicited by radiation in cancer cells, resulting in increased DSB accumulation and apoptotic cell death. Similarly, combined PARP and ATR inhibition has profound radiosensitizing effects in patient-derived glioblastoma stem-like cells (which have highly proficient DDR) by abrogating the G2–M arrest promoted by radiotherapy, ultimately driving malignant cells towards mitotic catastrophe87. While PARP inhibitors are currently approved as monotherapies for patients with BRCA1-mutant and BRCA2-mutant cancers, preclinical evidence supports the possibility that radiotherapy can be harnessed to either elicit or aggravate non-oncogene addiction to PARP in both the presence and absence of functional BRCA1/2 proteins81-83,86, as well as in malignant cells lacking polybromo 1 (PBRM1), which encodes a chromatin remodelling enzyme affected by loss-of-function mutations in 40% of clear cell renal cell carcinomas88. Of note, while combining radiotherapy with PARP inhibitors is expected to be effective in homologous-recombination-incompetent tumours, reliable tools to detect such a ‘BRCAness’ phenotype in malignancies bearing functional BRCA1 and BRCA2 are missing89. Conversely, identifying tumours that utilize alternative non-homologous end-joining for DSB repair in the absence of homologous recombination stands out as a promising tool to predict sensitivity to radiotherapy plus PARP inhibitors90.
Intriguingly, multiple FDA-approved PARP inhibitors (such as olaparib, rucaparib and niraparib) have been shown to promote robust CGAS-dependent type I IFN secretion by malignant cells following the cytosolic accumulation of nuclear DNA or micronuclei, thus promoting anticancer immunity91. Radiotherapy largely drives type I IFN secretion via mitochondrial (rather than nuclear) DNA58, suggesting that PARP inhibitors might robustly boost the tumour-targeting immune response driven by radiotherapy (BOX 2). Accordingly, niraparib has been shown to improve radiation-induced CD8+ T cell activation in a mouse model of EGFR-mutant NSCLC, correlating with superior CGAS signalling in malignant cells92. Both PARP inhibitors and radiotherapy have been linked with PD-L1 upregulation in certain settings16,93, implying that the addition of ICIs stands out as a potential strategy to further improve the therapeutic efficacy of this combination.
Inhibitors of cell-cycle checkpoint kinases.
CHEK1, CHEK2 and WEE1 G2 checkpoint kinase (WEE1) are also involved in the DDR. However, while WEE1 phosphorylates cyclin-dependent kinase 1 (CDK1) to prevent mitotic entry, CHEK1 and CHEK2 mainly act as downstream effectors of ATR and ATM, respectively, to block the transition from the G1 to the S phase of the cell cycle12. WEE1 and CHEK1 are upregulated by radiation, not only in vitro94-96, but also in patients with disease recurrence following radiotherapy, as demonstrated in patients with radioresistant human papillomavirus-positive (HPV+) head and neck squamous cell carcinoma (HNSCC)97. Consistent with this notion, experimental inhibitors of WEE1 (REFS98-100) and CHEK1/2 (REFS101-104) have been shown to compromise cellular adaptation to radiotherapy by abrogating activation of the irradiation-induced G2 checkpoint, thus promoting mitotic catastrophe, an effect that is further aggravated by PARP inhibition105-107. Of note, CHEK1 inhibitors might also synergize with radiotherapy by compromising the formation of RAD51 nuclear foci, which is critical for homologous recombination108,109.
The orally available pan-CDK inhibitor AZD5438 has been shown to mediate considerable radiosensitizing effects on radioresistant NSCLC cell lines, both in vitro and in vivo110. Nonetheless, more attention has been focused on specific CDK4/6 inhibitors (that inhibit cell-cycle progression at the G1–S transition), especially following the approval of palbociclib, ribociclib and later abemaciclib (all in combination with an aromatase inhibitor) as first-line therapies for patients with advanced-stage and/or metastatic oestrogen-receptor-positive (ER+), HER2− breast cancer111,112. A growing body of preclinical literature demonstrates that these agents can be successfully combined with radiotherapy, resulting in delayed repair of radiation-induced DNA damage113-115, prolonged cell-cycle blockade116, and enhanced apoptosis117, especially (but not exclusively) in models that retain p53 function114,116,118. Importantly, research involving xenograft models of glioblastoma and atypical teratoid rhabdoid tumour119 as well as immunocompetent models of ER+ breast cancer and TNBC116, has demonstrated improved tumour control when palbociclib is administered after completion of (rather than before or concomitant with) hypofractionated radiation. Similar findings have been obtained with abemaciclib in a study involving several human NSCLC cell lines that were either maintained in vitro or xenografted into immunodeficient mice114. These data support the notion that cancer cells that can escape the G2–M arrest are selected for by radiotherapy and that further disease progression in this malignant cell population can then be inhibited using CDK4/6 inhibitors, thus highlighting the critical importance of treatment schedule for optimal therapeutic effects.
Intriguingly, multiple agents that interfere with cell-cycle progression can also mediate immunomodulatory effects that can be maximized by radiotherapy. For example, adavosertib (a first-in-class, orally available WEE1 kinase inhibitor) reportedly enhances the CD8+ T cell responses driven by single-fraction radiation against a variety of tumours in immunocompetent mouse models120,121. However, although this combination seems to promote PD-L1 expression (suggesting benefit from the addition of ICIs)120, the extent of tumour shrinkage was found to correlate with PD-L1 downregulation in immunocompetent models of breast cancer121. Whether this apparent discrepancy reflects variations in radiation dose (8 Gy versus 12 Gy) or intrinsic tumour features currently remains unclear. A similar radiation-enhanced antitumour immune response has been obtained with the experimental CHEK1/2 inhibitor AZD7762 followed by single-dose (17 Gy) irradiation in an immunocompetent mouse model of melanoma122. In this setting, AZD7762 followed by irradiation (but not either intervention alone) resulted in micronucleation and abundant secretion of type I IFN by cultured malignant cells, eliciting robust systemic anticancer immune responses when tested in vivo122. Whether delivering AZD7762 after radiotherapy would further enhance the immunotherapeutic effects of the combination therapy remains to be investigated. Along similar lines, despite an abundant preclinical literature suggesting that radiotherapy and CDK4/6 inhibitors stand out as promising combination partners owing to their ability to activate poorly overlapping immunostimulatory pathways123, mechanistic evidence supporting this possibility is currently lacking.
Clinical considerations.
PARP inhibitors provide one of the first validations of the clinical utility of synthetic lethality124. Specifically, PARP inhibitors are not only approved as monotherapies for use in patients with breast, ovarian or prostate cancer harbouring loss-of-function BRCA1, BRCA2 or ATM mutations125,126, but have been and are being extensively tested in combination with various treatment strategies including chemotherapy, radiotherapy and immunotherapy. Monotherapy with PARP inhibitors is generally well tolerated, with common toxicities including myelosuppression, gastrointestinal symptoms and fatigue, albeit with a low risk (<1%) of secondary malignancies owing to DNA12,127. Similarly, standard-dose radiotherapy administered in combination with olaparib has demonstrated an acceptable safety profile in patients with locally advanced or metastatic HNSCC or TNBC128,129. Conversely, olaparib combined with higher-dose radiotherapy and concurrent cisplatin was poorly tolerated by patients with locally advanced NSCLC, resulting in severe oesophageal and haematological toxicities, as well as pulmonary adverse events130. While conformal radiotherapy schedules and techniques enabling improved pulmonary and oesophageal sparing should be implemented in order to further explore therapeutic combinations involving PARP inhibitors, other strategies can also enhance efficacy. For example, preliminary clinical evidence demonstrates that olaparib combined with alpelisib (an FDA-approved α-specific PI3K inhibitor) or buparlisib (an orally available experimental broad-spectrum PI3K inhibitor) is well tolerated and has synergistic effects in patients with advanced-stage and/or recurrent ovarian or breast tumours131,132. The safety and efficacy of adding radiotherapy to this combination, however, remains to be clinically investigated.
Unlike PARP inhibitors, ATM, ATR, DNA-PK, WEE1 and CHEK1 inhibitors are still in early-phase clinical development and have largely not been investigated in combination with radiotherapy. Berzosertib, a first-in-class ATR inhibitor previously assessed for safety and preliminary efficacy as monotherapy or combined with various chemotherapies in patients with advanced-stage solid tumours133,134, is currently being investigated in combination with radiotherapy in patients with brain metastases from NSCLC (NCT02589522), in those with chemotherapy-resistant breast cancer (NCT04052555), and (combined with cisplatin) in those with locally advanced HNSCC (NCT02567422). Similarly, the ATR inhibitor ceralasertib135-136 (NCT02223923) is currently being tested in combination with radiotherapy in patients with advanced-stage solid tumours, while the DNA-PK inhibitor peposertib (formerly known as nedisertib)137 is currently being investigated in combination with radiotherapy and SOC chemotherapy in at least nine phase I/II basket trials (NCT02516813, NCT03724890, NCT03770689, NCT04068194, NCT04071236, NCT04172532, NCT04533750, NCT04555577 and NCT04750954). Preliminary evidence suggests that both the WEE1 inhibitor adavosertib and the CHEK1 inhibitor prexasertib are well tolerated when combined with radiotherapy or chemoradiotherapy in patients with pancreatic cancer138 and in those with advanced-stage HNSCC139, resulting in several phase I/II clinical trials testing similar regimens (NCT02585973, NCT02555644, NCT03028766 and NCT04460937). The final results of these studies, however, have thus far not been reported.
The clinical development of AZD5438 and AZD7762 has been discontinued owing to exposure and/or tolerability issues140,141. Conversely, data from several cohorts of patients with metastatic ER+ breast cancer who received CDK4/6 inhibitors suggest that concomitant administration of CDK4/6 inhibitors with radiotherapy does not significantly increase the incidence of neutropenia relative to that observed with CDK4/6 inhibitors alone142-146. Other notable toxicities include sporadic episodes of high-grade but reversible intestinal toxicities when the bowel is located within the radiation field147. Similarly, radiotherapy combined with ribociclib is well tolerated with preliminary signs of clinical activity in children with newly diagnosed diffuse intrinsic pontine glioma148. Importantly, preclinical findings from our research group and others demonstrate that treatment schedule is a major determinant of efficacy when radiation is combined with CDK4/6 inhibitors116,119. These findings inspired the design of a randomized phase II trial comparing the efficacy of palbociclib plus letrozole versus the same regimen preceded by SBRT in patients with oligometastatic (five or fewer metastases) ER+ HER2− breast cancer (NCT04563507). Various other phase I/II trials assessing the role of radiotherapy in combination with palbociclib, ribociclib or abemaciclib in patients with breast cancer (NCT03691493, NCT04334330, NCT04605562 and NCT04923542) or in those with other solid tumours, including HNSCC (NCT03024489), glioma (NCT03355794) and prostate cancer (NCT04298983), are ongoing.
PI3K signalling
The PI3K signal transduction cascade is the most frequently dysregulated pathway in human cancer149. PI3K, which exists in several isoforms, is generally activated by receptor tyrosine kinases (RTKs) or G-protein-coupled receptors and promotes the phosphorylation-dependent activation of AKT serine/threonine kinase 1 (AKT1). In turn, active AKT1 can phosphorylate a variety of substrates that promote cellular survival and anabolism including MTOR, thus supporting cellular proliferation20. Multiple mechanisms can lead to aberrant PI3K signalling in malignant cells: (1) genetic alterations affecting specific PI3K-coding genes such as phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit-α (PIK3CA) as well as AKT1, MTOR, and phosphatase and tensin homologue (PTEN), which encodes a prominent PI3K antagonist149-151; (2) genetic or epigenetic defects that culminate in the overexpression or hyperactivation of oncogenic RTKs, including EGFR, HER2, MET and KIT149,152; and (3) additional features such as the composition of the local microbiome, which has been linked with PI3K activation in patients with lung cancer153 and dysregulated insulin signalling, pointing to a role for insulinaemia-controlling strategies including metformin (an FDA-approved drug for type 2 diabetes mellitus) and ketogenic diets as promising partners for combination with PI3K inhibitors154,155. Considerable efforts have been dedicated to the development of PI3K inhibitors for clinical use, culminating in the approval of alpelisib in combination with fulvestrant for use in patients with advanced-stage and/or metastatic ER+ HER2− breast cancer harbouring PIK3CA mutations156.
Extensive evidence links PI3K hyperactivation to the emergence of radioresistance. For example, activating PIK3CA mutations have been associated with an increased risk of local treatment failure in patients undergoing SBRT for primary or metastatic lung lesions157 and in those receiving whole-brain radiotherapy for brain metastases158. Similarly, hyperactivation of AKT1 downstream of PTEN loss has been associated with an elevated risk of relapse after radiotherapy in patients with prostate cancer harbouring copy number increases in MYC159. Moreover, patients with HNSCC or nasopharyngeal carcinoma with high levels of EGFR expression160,161, women with HER2+ breast cancer162, those with locally invasive prostate cancer staining positively for vascular endothelial growth factor A (VEGFA)163, and patients with uterine cervical cancer harbouring alterations in FGFR1, FGFR2, FGFR3 and FGFR4 (REFS164,165) have high rates of disease recurrence following radiotherapy. Finally, radiotherapy can drive cytoprotective PI3K signalling by promoting the upregulation or activation of various upstream activators of PI3K, including (but not limited to) EGFR166-169, HER2 (REF.170), MET171-174, VEGFR2 (REFS175-178) and FGFR2 (REF.179), which has been consistently associated with radioresistance coupled with the acquisition of mesenchymal and stem-like features by malignant cells180-183. Thus, multiple nodes of the PI3K signal transduction cascade stand out as promising targets for interventions designed to overcome acquired resistance to radiotherapy184,185 (Supplementary Table 2).
PI3K, AKT and MTOR inhibitors.
An abundant body of preclinical literature demonstrates that pharmacological inhibition of PI3K synergizes with radiation in a variety of tumour models. For example, the orally available PI3K/MTOR inhibitor dactolisib has been shown to substantially increase the radiosensitivity of human prostate cancer cells in vitro, correlating with increased biomarkers of an epithelial (over mesenchymal) phenotype182. Similar findings have been obtained with the poorly selective PI3K inhibitor LY294002 (REFS167,186,187) and the orally available PI3Kα/δ inhibitor pictilisib188 in models of human high-grade glioma, especially when combined with radiation and temozolomide (which mimics the SOC for patients with glioblastoma)189. Moreover, irradiation has been shown to confer improved tumour control in immunocompromised mice bearing human pancreatic tumours190 or HNSCCs harbouring PIK3CA mutations191 when combined with the PI3K inhibitors HS-173 or taselisib, respectively. Conversely, neither the pan-PI3K inhibitor buparlisib nor the dual PI3Kα/β/δ/γ and MTOR inhibitor apitolisib mediates radiosensitizing effects in cultured HNSCC cells, despite effective PI3K inhibition192. These findings suggest that, at least in certain cell types, compensatory mechanisms might preserve the radioresistant phenotype despite adequate PI3K inhibition. Supporting this possibility, the experimental AKT1 inhibitor MK-2206 has been shown to block AKT1 signalling in PTEN−/− glioblastoma cells but failed to improve the therapeutic activity of radiotherapy owing to aberrant downstream activation of MTOR193. Consistent with this notion, dactolisib demonstrated excellent synergy with radiation in preclinical models of endometrial cancer194, an effect that was linked with reduced PI3K, MTOR and VEGFA activity194,195. Moreover, pharmacological inhibition of AKT1 or MTOR after (but not before) fractionated irradiation has been shown to enhance the loss of clonogenicity of cultured radioresistant human prostate cancer cells196. Finally, co-administration of the MTOR inhibitor vistusertib with buparlisib or alpelisib appears to sensitize radioresistant human oral squamous cell carcinoma cells to radiation197. Whether similar effects can be achieved in immunocompetent preclinical models remains largely uninvestigated. Irrespective of these and other unknown aspects, PI3K inhibition synergizes with PARP inhibitors in preclinical models of BRCA1-defective and p53-defective breast cancer198, PTEN-deficient and p53-deficient prostate cancer199, and PTEN-deficient endometrial cancer200,201, suggesting that concurrent inhibition of PARP and PI3K in combination with radiotherapy is a promising therapeutic approach.
RTK inhibitors.
Pharmacological agents targeting EGFR (such as AG1478)167 as well as expression of a dominant-negative form of the type III EGFR variant (EGFRvIII)202 reportedly increase the radiosensitivity of glioblastoma cells, correlating with abrogated AKT1 phosphorylation. Similar data have been obtained with clinically available anti-EGFR monoclonal antibodies (nimotuzumab and cetuximab) in human NSCLC cells growing in vitro or xenografted into immunodeficient mice203,204. Moreover, treatment with the dual EGFR/HER2 tyrosine-kinase inhibitors lapatinib or pyrotinib reduces HER2 phosphorylation and downstream AKT1 activation, ultimately sensitizing HER2+ breast and gastric carcinoma cells to radiation205,206. HER2 has also been shown to mediate AKT1-dependent immunosuppressive effects including the suppression of cytosolic DNA sensing and consequent abrogation of CGAS signal transducer stimulator of IFN response cGAMP interactor 1 (STING1) signalling207, as well as upregulation of the phagocytosis inhibitor CD47 (REF.208). Accordingly, dual blockade of CD47 and HER2 has been demonstrated to maximize macrophage-dependent phagocytosis and promote the eradication of radioresistant breast cancer cells208.
Cediranib, an orally available VEGFR inhibitor, limits radiation-induced VEGFR phosphorylation in endothelial cells, thus sensitizing lung cancer and CRC xenografts to fractionated radiation, at least partly owing to aggravated vascular disruption209,210. Similarly, apatinib (a selective VEGFR2 inhibitor also known as rivoceranib) promotes radiation-induced cell death in HCC xenografts, largely via suppression of radiation-induced PI3K signalling211. Moreover, genetic inhibition of VEGFA exacerbates the extent of DNA damage in irradiated nasopharyngeal carcinoma cells via a mechanism that involves compensatory activation of MTOR signalling and inhibition of autophagy, a cytoprotective pathway that (among other functions) supports DNA repair212. The pan-FGFR inhibitors LY2874455 and AZD4547 have been shown to sensitize multiple cancer cell lines and two human NSCLC xenografts to carbon ion and conventional radiation, respectively165,213, correlating with decreased AKT1 phosphorylation in vitro213. Finally, inhibition of MET with the FDA-approved agents crizotinib173 and tepotinib174, as well as with the experimental ATP-competitive inhibitor JNJ38877605 (REF.214), has been shown to improve the efficacy of radiation in mouse models of HNSCC and glioblastoma, an effect that was linked to the ability of MET inhibitors to overcome the enrichment of radioresistant stem-like cells that tend to occur in the context of sub-ablative doses of radiation.
In vivo data obtained from mouse fibrosarcoma cells growing in immunocompetent hosts demonstrate that optimal tumour control is achieved when anti-VEGFR2 antibodies are delivered shortly before (but not shortly after) irradiation, owing to interception of very rapid-onset VEGFA signalling driven by irradiation215. Along similar lines, AZD4547 ameliorates the radiosensitivity of glioma neurospheres orthotopically implanted into immunodeficient mice, albeit only when FGFR2-driven DNA repair, involving nuclear PTEN phosphorylation, is successfully inhibited179. These data lend further support to the critical importance of treatment schedule for the therapeutic efficacy of combination regimens involving radiotherapy.
Clinical considerations.
Over the past three decades, a large number of drugs targeting PI3K, its activators or its effectors have been developed, including dozens of agents that have ultimately received regulatory approval for use in patients1. These agents (which comprise small molecules and monoclonal antibodies), as well as hitherto experimental drugs targeting PI3K signalling at one of its nodes, are being (or already have been) extensively tested in combination with radiotherapy in hundreds of clinical trials.
The EGFR-targeting antibody cetuximab has been shown to improve the extent of locoregional tumour control and lead to extended OS in patients with HNSCC receiving definitive radiotherapy216,217. However, this approach is rarely used in clinical practice, as subsequent studies demonstrated the superiority of cisplatin-based chemoradiotherapy218-220. Moreover, the combination of cetuximab and radiotherapy has been associated with severe dermatitis (grade 3–4 in 32.5% of patients)221, reflecting the convergence of toxicities separately associated with each approach222. The EGFR inhibitors gefitinib and erlotinib also improve response rates in patients with brain-metastatic NSCLC receiving whole-brain radiotherapy223, as do bevacizumab and cediranib in patients with recurrent high-grade glioma224 and newly diagnosed glioblastoma225,226, respectively. Moreover, data from a pilot study suggest that <250-mg daily doses of apatinib can be safely combined with palliative radiotherapy in men with metastatic prostate cancer, resulting in synergistic effects on pain management227.
Promising findings on safety and clinical activity have been obtained by combining radiotherapy with the experimental EGFR-targeting antibody nimotuzumab in patients with various solid tumours including NHSCC228, glioblastoma229 and diffuse intrinsic pontine glioma230. In a small pilot trial involving 11 patients with stage III–IVB HNSCC, alpelisib combined with cetuximab and intensity-modulated radiotherapy was well tolerated and was associated with a radiological complete response in all patients231. Similarly, daily alpelisib (200 mg) appears to be a safe combination partner for concurrent cisplatin-based chemoradiation in patients with locoregionally advanced HNSCC, with dose-limiting toxicities emerging only at the 250-mg dose232. However, data on the clinical efficacy of this combination are currently not available. Buparlisib has been tested in combination with palliative thoracic radiotherapy (20 Gy in five fractions) in a cohort of 22 patients with NSCLC, demonstrating an acceptable safety profile, target engagement and a reduction in tumour hypoxia233. Conversely, the unacceptable toxicities of buparlisib and radiotherapy combined with temozolomide documented in patients with newly diagnosed glioblastoma led to discontinuation of this approach234. Similarly, both pictilisib and dactolisib have been discontinued owing to limited therapeutic efficacy and a high risk of gastrointestinal toxicities235-239.
Daily everolimus (an MTOR inhibitor) appears to be safe and tolerable in combination with fractionated radiotherapy following prostatectomy in men with prostate cancer240, as well as combined with chemoradiation in patients with locally advanced rectal cancer241. Again, results in patients with newly diagnosed glioblastoma were disappointing. Specifically, adding everolimus to radiotherapy plus concurrent and adjuvant temozolomide failed to improve progression-free survival (PFS) despite an increase in toxicity in a phase II study242. On the contrary, several randomized controlled trials have demonstrated that trastuzumab following adjuvant radiotherapy improves locoregional control and prolongs both PFS and OS in patients with HER2+ breast cancer243-247, albeit with a risk of sporadic acute cardiotoxicities248,249. Similarly, combining radiotherapy with lapatinib in patients with HER2+ breast cancer250 or HNSCC251,252 appears to be safe, with at least some clinical activity. Conversely, the clinical development of the MET inhibitor JNJ38877605 has been abandoned owing to excessive renal toxicities253. Nonetheless, approaches combining radiotherapy with JNJ38877605, dactolisib or other agents that are poorly tolerated as monotherapies might be feasible owing to the potential for lower (and hence less toxic) doses of these agents to be used, potentially opening avenues for further clinical investigation.
Official sources list hundreds of ongoing clinical studies investigating the use of radiotherapy in combination with FDA-approved (and less so investigational) RTK and/or MTOR inhibitors, generally involving concurrent administration schedules and enrolling patients for whom these drugs are approved as monotherapies. Conversely, only a few studies assessing the therapeutic profile of radiotherapy combined with PI3K inhibitors are currently ongoing and these largely focus on patients with primary or metastatic brain lesions (NCT03696355, NCT04192981) and HNSCC (NCT02113878), two indications in which patients usually receive radiotherapy as part of the current SOC.
TGFβ signalling
TGFβ antagonizes the cytotoxic effects of radiotherapy by promoting DNA repair and hence favouring cancer cell survival (BOX 3). Accordingly, TGFβ has attracted attention as a promising target of novel anticancer therapeutics, leading to the development of various agents targeting canonical TGFβ signalling, including the TGFβ-targeting antibody fresolizumab as well as inhibitors of transforming growth factor-β receptor 1 (TGFβR1) or the TGFβ effectors smooth muscle and MAD-related protein 2 (SMAD2) and SMAD3, some of which are currently being tested in patients as monotherapies or in combination with radiotherapy254 (Supplementary Table 3).
Box 3 ∣. TGFβ signalling.
TGFβ is a pleiotropic cytokine with a wide range of biological effects on a number of targets, including (but not limited to) malignant, stromal and immune cells254. Although TGFβ is generally regarded as an tumour-suppressive agent, it can paradoxically also support tumour progression via several cancer cell-intrinsic mechanisms, including the epithelial-to-mesenchymal transition and the acquisition of stem-like features254. Moreover, TGFβ production by malignant cells and cancer-associated fibroblasts (CAFs) promotes a variety of immunosuppressive effects, including repressed antigen presentation on MHC class I and the upregulation of PD-L1 by cancer cells, as well as the stimulation of regulatory T cell differentiation and tumour infiltration by myeloid-derived suppressor cells (MDSCs)254,344. TGFβ is also involved in stromal remodelling driven by CAFs, which favours the establishment of a fibrotic tumour microenvironment that arrests infiltration by tumour-targeting T cells and thus limits the efficacy of several therapies345-347. Activation of TGFβ in the irradiated tumour microenvironment depends on a series of highly regulated events beyond transcriptional upregulation: (1) assembly of a supramolecular complex containing a long TGFβ precursor and latent TGFβ binding protein 1 (LTBP1), which occurs in the endoplasmic reticulum; (2) proteolytic and oxidative processing to obtain mature TGFβ complexed with the so-called latency-associated peptide (LAP) and LTBP1, which occurs in the Golgi apparatus; (3) secretion of the TGFβ–LAP–LTBP1 complex into the microenvironment; and (4) release of bioactive TGFβ348,349. On release from LAP, TGFβ binds to a heterodimeric receptor composed of TGFβ receptor 1 (TGFβR1) and TGFβR2, generally culminating in the activation of cytoprotective and immunosuppressive transcriptional programmes orchestrated by smooth muscle and MAD-related protein (SMAD) 2 (REF.349). Moreover, TGFβ signalling has also been linked to radioresistance as a consequence of robust DNA repair via ATM and p53 activation350.
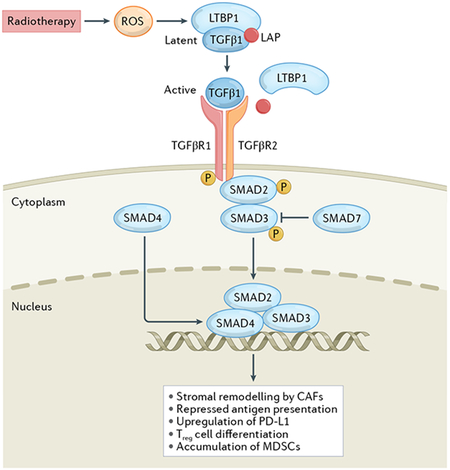
Genetic and pharmacological inhibition of TGFβ1 signalling in mammary epithelial cells leads to compromised ATM phosphorylation and ultimately to improved radiosensitivity255. Moreover, loss of TGFβ competence in HPV+ HNSCC cells results in a defective DDR and increased sensitivity to DNA-damaging agents, including radiation256. Similarly, pretreatment with the TGFβ-neutralizing antibodies 13C4 and 1D11, as well as the TGFβR1 inhibitor LY364947, aggravates the loss of clonogenicity imposed by irradiation on cultured human breast cancer cells, and enhances the control of mouse 4T1 mammary tumours established in syngeneic immunocompetent mice achieved by single-fraction (8 Gy) and fractionated (12 Gy in three fractions) radiotherapy257. Comparable findings have been obtained in preclinical models of glioblastoma258-260 and NSCLC261. TGFβ also appears to promote the maintenance of the stem cell pool (which is enriched in the setting of acquired radioresistance) in multiple cancers262. Accordingly, treatment with the dual TGFβR1/TGFβR2 inhibitor LY2109761 has been associated with attenuation of radiation-driven epithelial-to-mesenchymal transition in a preclinical model of glioblastoma involving the orthotopic implantation of stem-like glioblastoma precursor cells in immunodeficient mice259.
Notably, radiotherapy-elicited TGFβ signalling also promotes systemic effects outside the radiation field, including accrued tumour dissemination as a consequence of metastatic niche formation and inhibition of antitumour immunity. For example, thoracic radiation (10 Gy) elevates circulating TGFβ1 levels in mice bearing MMTV/PyVmT-driven mammary carcinomas, which correlate with increased numbers of blood-borne malignant cells and aggravated metastatic seeding263. In line with this notion, inhibition of autocrine or paracrine TGFβ signalling with the neutralizing antibody 2G7, as well as conditional deletion of TGFBR2, reduces the extent of radiation-initiated metastatic dissemination in the MMTV/PyVmT model263. TGFβ also has robust immunosuppressive effects that antagonize the ability of fractionated irradiation to elicit anticancer immunity264. Thus, administration of 1D11 enables the regression of syngeneic mammary and colorectal tumours exposed to fractionated radiation as it elicits systemic antitumour immunity against non-irradiated lung metastases or synchronous tumours (the so-called abscopal effect)264,265. This effect correlates with reduced phosphorylation of SMAD2 and SMAD3, as well as a genetic signature of IFNγ signalling with compensatory upregulation of PD-L1 and PD-L2 (REFS264,265). Accordingly, the addition of an anti-PD-1 antibody has been shown to extend the survival benefits enabled by radiation plus TGFβ blockade in several preclinical tumour models264-266. Moreover, dual inhibition of TGFβR2 and PD-L1 using the bifunctional antibody bintrafusp-α has been shown to strongly enhance the extent of radiotherapy-induced antitumour immunity compared with interventions targeting either pathway alone267,268.
Importantly, TGFβ also mediates fibrotic reactions and promotes injury to non-malignant tissues, hence contributing to the adverse effects of radiotherapy on certain organs. In a cohort of patients with NSCLC receiving definitive radiotherapy, the development of radiotherapy-induced lung injury and poor clinical responses were associated with high circulating TGFβ levels269. Mice exposed to focal radiation can develop pulmonary fibrosis270, oral mucositis271 and skin irritation272 correlating with accrued TGFβ signalling and SMAD activation. Moreover, pharmacological induction of TGFβ1 exacerbates radiation-induced heart and intestinal injuries in rats273. Consistent with this notion, LY2109761 has been shown to limit the extent of SMAD1 and SMAD2 phosphorylation, as well as the radiation-induced expression of genes associated with inflammation or angiogenesis, such as Il7 in the irradiated mouse lung, ultimately having an antifibrotic effect274. Similarly, topical administration of recombinant human SMAD7 (which represses the activation of SMAD proteins involved in TGFβ signalling)271 fused with a cell-penetrating peptide, alleviated radiotherapy-induced oral mucositis in an orthotopic xenograft model of oral cancer275, while genetic inhibition of SMAD3 via transcutaneous delivery of a specific small-interfering RNA ameliorated skin irritation after high-dose irradiation (45 Gy) in mice272. Altogether, these findings strongly support the therapeutic targeting of TGFβ signalling in order to inhibit the cytoprotective, immunosuppressive and profibrotic pathways elicited by radiotherapy.
Clinical considerations.
No specific TGFβ-targeted therapies are currently approved for use in patients with cancer276. The combination of fresolimumab (a fully human antibody directed against human TGFβ1, TGFβ2 and TGFβ3) plus radiotherapy (7.5 Gy in three fractions) has been evaluated in a randomized phase II trial involving women with previously treated metastatic breast cancer277. This trial was designed to compare two different doses of fresolimumab (1 mg/kg versus 10 mg/kg) with targeted radiotherapy delivered to a single metastatic lesion. Toxicities were deemed acceptable (only two of 23 treated patients developed keratoacanthoma) although clinical responses were limited to stable disease in three patients with no abscopal responses demonstrated277, potentially owing to immune dysfunction at baseline278. Nonetheless, patients receiving 10 mg/kg fresolimumab had significantly longer median OS durations than those receiving the 1-mg/kg dose (16.0 months versus 7.6 months; HR 2.73, 95% CI 1.02–7.30; P = 0.039)277. Investigations of immune parameters demonstrated better activation of anticancer immunity in patients receiving the 10-mg/kg dose, manifesting as an increase in peripheral blood mononuclear cell counts and an enhanced CD8+ T cell central memory pool277. Potentially explaining the limited objective response rate observed in this trial, radiation combined with inhibition of TGFβ signalling has been shown to drive the secretion of inhibin subunit-β A (INHBA) homodimers from mouse and human breast cancer cells, which are known to support the recruitment of immunosuppressive regulatory T cells in vivo and might explain the lack of responsiveness among patients receiving fresolimumab279. A randomized phase I/II study investigating the benefit of adding galunisertib (a selective TGFβR1 inhibitor) to temozolomide-based chemoradiotherapy in patients with newly diagnosed glioma revealed no differences in efficacy, safety or pharmacokinetic variables between the two treatment arms280. Thus, although safety appears to be acceptable, regimens combining radiotherapy with inhibition of TGFβ signalling might require the addition of other immunomodulatory agents to achieve clinical efficacy281. A number of ongoing clinical trials are currently investigating this approach in a variety of indications including early-stage NSCLC (NCT02581787), metastatic breast cancer (NCT03524170, NCT04756505), intrahepatic cholangiocarcinoma (NCT04708067), oesophageal squamous cell carcinoma (NCT04481256) and nasopharyngeal carcinoma (NCT04605562), in some of these settings as part of a multimodal treatment regimen also including ICIs or ICI-like strategies. The results of these studies are eagerly awaited.
Autophagy
Reflecting its potent cytoprotective effects, autophagy (BOX 4) has attracted considerable attention as a target of compounds that can be utilized clinically as chemosensitizers or radiosensitizers282. However, a variety of hitherto unresolved challenges have prevented the identification of clinically viable inhibitors of autophagy other than chloroquine and hydroxychloroquine, two non-specific lysosomal fusion inhibitors that are mainly used for malaria prevention283 (Supplementary Table 3).
Box 4 ∣ Autophagy.
Autophagy is a catabolic process through which cytoplasmic material, including damaged or dispensable organelles and/or portions thereof, are sequestered within newly formed double-membraned vacuoles (known as autophagosomes) and delivered to lysosomes for degradation351. As such, autophagy generally mediates robust cytoprotective functions by supporting the preservation of cellular homeostasis in response to stress351. Indeed, the autophagic flux of material can be finely tuned in response to cellular demands22. Specifically, autophagy is mediated by members of the ATG family of evolutionarily conserved proteins, the coordinated activity of which is under tonic inhibition by MTOR22. Importantly, although proficient autophagy in non-malignant cells generally antagonizes malignant transformation by favouring the preservation of genetic, oxidative and metabolic homeostasis, established cancer cells can also harness autophagy in support of disease progression and resistance to therapy352. Moreover, autophagy has a context-dependent, dual role in the initiation of anticancer immunity, being necessary for the emission of danger signals by malignant cells responding to immunogenic chemotherapy, but also inhibiting both antigen presentation and radiotherapy-induced secretion of type I interferon (IFN)353. DDR, DNA damage response.
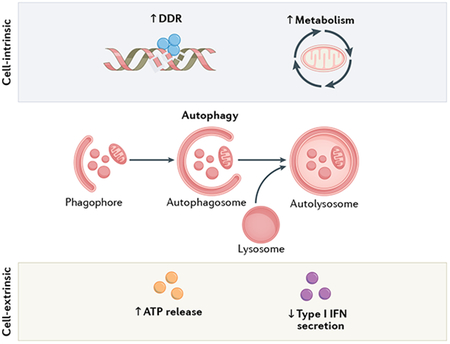
Radiotherapy drives dose-dependent autophagic responses driven by DNA damage and ROS production coupled with endoplasmic reticulum (ER) stress284,285, and several reports have proposed a link between increased autophagic flux and the cytotoxicity of radiotherapy. Most of these studies, however, harnessed non-specific intervention strategies such as MTOR inhibitors (which are known to have radiosensitizing effects owing to inhibition of PI3K signalling, as discussed above)286-290 or calorie restriction (which operates at the whole-body level)291,292 to promote autophagy. Moreover, these studies have often drawn conclusions based on knockdown of a single component of the apparatus, despite virtually all of them having a plethora of concurrent autophagy-independent functions293,294. Conversely, proficient autophagic responses support the survival of irradiated cells, at least in part by antagonizing the generation of ROS295 and promoting DNA repair via homologous recombination296. Indeed, autophagy defects typically result in the downregulation of proteins involved in homologous recombination (such as CHEK1) and other DNA repair pathways296,297, as well as inhibition of DDR-related histone ubiquitination upon accumulation of the autophagic substrate sequestosome 1 (SQSTM1, best known as p62)298, culminating in exacerbated accumulation of radiotherapy-elicited DSBs and global genomic instability299. Moreover, preclinical data suggest that hypoxia-associated radioresistance might, at least partially, involve activation of autophagy300,301.
High levels of autophagy have been linked with radioresistance in HNSCC cell lines302 and in patients with CRC303. Moreover, chloroquine has been shown to enhance the ER-stress-linked death of cultured mouse sarcoma cells driven by radiation304 as well as the radiosensitivity of mouse breast cancer cells growing in immunocompetent syngeneic mice58. Similar effects have been observed with 3-methyladenine, a non-specific inhibitor of the autophagy-related PI3K catalytic subunit type 3, in preclinical models of HCC and in oesophageal cancer xenografts305,306. These findings are supported by the radiosensitizing effects of genetic methods of autophagy inhibition, including the miR-214-dependent silencing of ATG12 (REF.303) in cultured CRC cells, as well as the deletion of Atg5, Atg7 or beclin 1 (Becn1) in mouse models of CRC and breast cancer, both in vitro and in vivo58,307. Consistent with this notion, proficient autophagic responses protect the non-malignant bone marrow following irradiation308, largely by compensating for radiation-induced genotoxic stress via BRCA1-dependent DDR activation309. Of note, depletion of BECN1 results in a compromised radiation-induced DDR independent of autophagy310, lending further support to the notion that signalling pathways not directly involved in autophagy that are nonetheless controlled by components of the autophagic machinery might also have a role in the acquisition of radioresistance294.
Importantly, autophagy is also involved in the regulation of both natural and radiotherapy-driven anticancer immunity in a highly context-dependent manner311-313. Indeed, stable depletion of ATG5 or BECN1 compromises the sensitivity of syngeneic immunocompetent mouse models of CRC exposed to single-dose irradiation (8 Gy)307. Conversely, the deletion of Atg5 or Atg7 ameliorates the efficacy of fractionated irradiation (8 Gy in three fractions) in mouse mammary carcinomas growing in syngeneic immunocompetent hosts58. Data from mechanistic experiments demonstrate that this apparent discrepancy reflects the fact that anticancer immunity driven by these models preferentially involves autophagy-dependent ATP release versus autophagy-inhibited type I IFN secretion, respectively58,307. Whether such a different requirement for ATP release versus type I IFN secretion depends on radiation dose, cell type or other unknown variables remains to be determined.
Clinical considerations.
Chloroquine and hydroxychloroquine are both available as potential combination partners for radiotherapy (and potentially other treatments including chemotherapy) in clinical settings314, although the safety profile of these agents is far from optimal, probably reflecting their broad lysosomotropism315. Consistent with this notion, a phase I–II clinical trial testing hydroxychloroquine plus radiotherapy and temozolomide in patients with newly diagnosed glioblastoma identified a maximum tolerated dose of 600 mg/kg per day, a dose that was insufficient to achieve consistent inhibition of autophagy in patients and failed to improve OS316. Along similar lines, a single-centre, open-label, dose-finding phase I trial enrolling patients with newly diagnosed glioblastoma identified 200 mg/day as the maximum tolerated dose for chloroquine when administered in combination with radiotherapy (59.4 Gy in 33 fractions), resulting in severe toxicities (six chloroquine-related clinically serious events and one death) and a median OS duration of 16 months (which is comparable to that achieved with SOC approaches)317. Conversely, 150 mg/day chloroquine combined with whole-brain irradiation (30 Gy in ten fractions over 2 weeks) did not affect quality of life outcomes or increase the incidence or severity of adverse events in patients with brain metastases enrolled in a randomized double-blind, placebo-controlled phase II study318. However, despite a mild amelioration of brain metastases-specific PFS (relative risk 0.31, 95% CI 0.1–0.9; P = 0.046), no significant difference in OS could be documented relative to radiotherapy alone318. Similar findings emerged from a pilot study involving 20 patients with newly diagnosed brain metastases from primary lung, breast or ovarian cancers who received daily chloroquine (250 mg) and whole-brain radiotherapy (37.5 Gy in 2.5-Gy daily fractions)319. Accordingly, very few clinical trials testing chloroquine or hydroxychloroquine remain active. Trials continuing to investigate this approach include a phase II study testing radiotherapy plus hydroxychloroquine and capecitabine in patients with resectable pancreatic cancer (NCT01494155), a phase I trial investigating partial brain radiotherapy plus temozolomide, chloroquine and tumour-treating fields in patients with newly diagnosed glioblastoma (NCT04397679), and a phase II study assessing the effects of radiotherapy combined with chloroquine in the same indication (NCT02432417). Thus, although an abundant preclinical literature suggests that inhibition of autophagy stands out as a promising strategy to improve the efficacy of radiotherapy, the clinical translation of this concept is hindered by the lack of safe and specific inhibitors283. Further complicating this issue, strategies involving the specific delivery of autophagy inhibitors to cancer cells might have to be developed for these agents to achieve acceptable tolerability and good efficacy (alone as well as combined with radiotherapy), thus reflecting the key role of autophagy in the initiation of tumour-targeting immunity320.
Conclusions
Taken together, the data discussed herein lend strong support to the notion that radiotherapy can be used to activate cytoprotective signalling pathways associated with radioresistance and thus create a state of non-oncogene addiction that renders tumour cells vulnerable to targeted therapies (FIG. 2). With only a few exceptions, however, this concept has yet to be implemented in the clinic, reflecting the existence of several obstacles in translating results from preclinical settings. Nonetheless, a few key points emerge from the literature discussed above.
Fig. 2 ∣. Targeting the pro-survival pathways induced by radiotherapy in cancer.

At least in part owing to intratumoural heterogeneity, radiotherapy alone is often unable to kill all malignant cells and thus does not mediate complete tumour eradication. In this setting, cancer cells generally resist the cytostatic and/or cytotoxic effects of radiotherapy along with the activation of various cytoprotective signalling pathways. Importantly, such cytoprotective pathways establish a state of non-oncogene addition that can be targeted using specific agents in order to achieve superior disease control.
Firstly, administration schedule clearly has a critical role in the efficacy of therapeutic regimens involving radiotherapy116,196,215. However, only a few preclinical studies have thus far compared the efficacy of different treatment schedules with the aim of identifying an optimal approach prior to focusing on mechanistic aspects. Thus, different treatment schedules might have resulted in superior preclinical and possibly clinical efficacy in at least some of the studies that have suggested only limited synergy between radiotherapy and targeted therapies.
Secondly, clinical studies investigating the role of radiotherapy in combination with agents targeting the mechanisms discussed herein have typically used conventional fractionation schedules. However, an expanding body of literature demonstrates that both dose and fractionation schedule have clinically relevant effects on the signal transduction cascades elicited by radiotherapy. For example, radiation delivered as three fractions of 8 Gy each has robust immunostimulatory effects in preclinical models of breast cancer, while a single radiation dose of 20 Gy fails to do so as a consequence of the upregulation of a cytosolic exonuclease (TREX1) that shuts down type I IFN secretion by irradiated cells321. This observation implies that combining radiotherapy with hitherto experimental TREX1 inhibitors would result in limited synergy if the radiation dose and fractionation schedules employed failed to elicit meaningful TREX1 expression. Similar considerations apply to each of the pathways discussed herein, for which limited preclinical investigation of optimal dose and fractionation approaches has been undertaken prior to clinical testing. As an added layer of complexity, recapitulating the standard radiotherapy regimens used clinically (such as 2 Gy in 30 fractions, which is commonly used for the clinical management of patients with NSCLC)322 is not always feasible in preclinical models. At least in part, this lack of accurate modelling reflects the practical, ethical and experimental constraints associated with delivering anaesthesia to rodents on a daily basis over several weeks323.
Thirdly, most of the preclinical studies testing radiation in combination with agents targeting radiotherapy-driven non-oncogene addiction have involved human cancer cell lines maintained in vitro or xenografted into immunodeficient hosts. While cell lines offer a number of advantages, novel experimental platforms including patient-derived organoids and patient-derived xenografts might be superior in terms of recapitulating neoplasm-specific features and enabling the identification of personalized combination regimens that might be highly effective (although difficult to test in large patient cohorts)324,325. That said, both patient-derived organoid and patient-derived xenograft models also fall short in assessing the potential effects of treatment on the immune system (be it positive, and hence supporting efficacy, or detrimental, and hence limiting efficacy). Indeed, both radiotherapy16,326 and targeted anticancer agents93 are now known to mediate a number of clinically relevant immunomodulatory effects that can no longer be overlooked in an era in which translational research is increasingly being used to guide the design of clinical studies.
Finally, clinically actionable biomarkers enabling the identification of tumours that are likely to respond to radiotherapy in combination with drugs that block the cytoprotective signalling pathways associated with radioresistance are generally missing327. In specific settings (such as radiotherapy combined with PARP inhibitors), baseline features that are associated with treatment sensitivity might exist (such as homologous recombination defects), but remain difficult to assess in a reliable manner89. In other scenarios, for example regimens combining radiotherapy with autophagy inhibitors, activation of autophagy might emerge only after irradiation, which renders assessment even more complex (at least within the tumour microenvironment). Whether circulating biomarkers can be helpful in this setting remains completely unexplored.
Of note, growing levels of expectation exist regarding the efficacy of regimens that combine radiotherapy with anti-PD-1 or anti-PD-L1 antibodies in patients with various cancer types, reflecting encouraging data from clinical trials involving patients with advanced-stage NSCLC29,328,329. Intriguingly, PD-L1 expression at baseline has been associated with improved responsiveness to ICIs in multiple clinical settings330, although patients with NSCLC appear to obtain benefit from the addition of durvalumab to chemoradiotherapy irrespective of baseline PD-L1 status331. Despite a lack of mechanistic evidence, it is tempting to speculate that such an observation reflects the ability of radiotherapy to promote PD-L1 expression via several different mechanisms, as discussed in this Review. Further supporting this possibility, patients with PD-L1− NSCLC at baseline appear to obtain greater levels of benefit from the addition of SBRT to pembrolizumab than those with PD-L1+ NSCLC (in whom pembrolizumab is more likely to be active as monotherapy)332. Yet another promising strategy involves combining radiotherapy with agents that target inhibitor of apoptosis proteins (IAPs), such as xevinapant333,334. Whether radiotherapy is capable of driving the upregulation or activation of IAPs, however, remains unclear.
In conclusion, while additional research is needed, we surmise that well-designed preclinical studies conceived to comparatively assess various dose and fractionation schedules in immunocompetent preclinical models could unlock the clinical potential of radiotherapy as a means to elicit cytoprotective pathways that can then be inhibited using targeted anticancer therapies. Future studies will reveal which patient subgroups can derive superior clinical benefits from this innovative use of radiotherapy.
Supplementary Material
Key points.
Targeted anticancer agents are commonly used in the treatment of various solid and haematological malignancies.
Not all tumours are sensitive to these agents, largely reflecting the lack of or inactivity of the targetable alteration.
Radiotherapy is also frequently used for the treatment of cancer, owing to its prominent cytostatic and cytotoxic effects on malignant cells.
A wide panel of cytoprotective pathways can be activated by radiotherapy, thus limiting therapeutic efficacy.
However, these signal transduction cascades can be effectively inhibited with targeted anticancer agents, potentially supporting superior treatment efficacy.
Radiotherapy stands out as a promising tool to elicit clinically actionable signalling pathways in cancer.
Acknowledgements
The work of L.C.C. is supported by US National Institutes of Health (NIH) P01 (#P01CA120964) and R35 (#R35CA197588) grants. The work of S.C.F. is supported by a Breakthrough Level 2 grant from the US Department of Defense (DoD), Breast Cancer Research Program (BRCP) (#BC180476) and by the 2019 Laura Ziskin Prize in Translational Research (#ZP-6177) from Stand Up to Cancer (SU2C). The work of L.G. is supported by a Breakthrough Level 2 grant from the US DoD BRCP (#BC180476P1), by the 2019 Laura Ziskin Prize in Translational Research (#ZP-6177, PI: Formenti) from SU2C, by a Mantle Cell Lymphoma Research Initiative (MCL-RI, PI: Chen-Kiang) grant from the Leukaemia and Lymphoma Society (LLS), by a startup grant from the Department of Radiation Oncology at Weill Cornell Medicine (New York, USA), by a Rapid Response Grant from the Functional Genomics Initiative (New York, USA), by industrial collaborations with Lytix (Oslo, Norway) and Phosplatin (New York, USA), and by donations from Phosplatin (New York, USA), the Luke Heller TECPR2 Foundation (Boston, USA), Onxeo (Paris, France), Ricerchiamo (Brescia, Italy) and Sotio a.s. (Prague, Czech Republic).
Footnotes
Competing interests
L.S. has received research funding from Puretech. L.C.C. has acted as a consultant and/or advisor to Agios Pharmaceuticals, Faeth Therapeutics, Larkspur Therapeutics and Volastra Therapeutics, has received research funding from Petra Pharmaceuticals and is a co-founder of, and holds equity in, Agios Pharmaceuticals, Faeth Therapeutics, Larkspur Therapeutics and Volastra Therapeutics. S.C.F. has acted as a consultant and/or advisor to AstraZeneca, Bayer, Bristol Myers Squibb, Eisai, Elekta, EMD Serono, GlaxoSmithKline, Janssen, MedImmune, Merck US, Regeneron, Varian and ViewRay, and has received research funding from Bristol Myers Squibb, Eli-Lilly, Merck, Regeneron and Varian; and other support from Pfizer. L.G. has acted as a consultant and/or advisor to AstraZeneca, Boehringer Ingelheim, Inzen, The Longevity Labs, the Luke Heller TECPR2 Foundation, OmniSEQ and Onxeo, and has received research funding from Lytix, and Phosplatin. G.P. declares no competing interests.
Supplementary information
The online version contains supplementary material available at https://doi.org/10.1038/s41571-021-00579-w.
References
- 1.Bedard PL, Hyman DM, Davids MS & Siu LL Small molecules, big impact: 20 years of targeted therapy in oncology. Lancet 395, 1078–1088 (2020). [DOI] [PubMed] [Google Scholar]
- 2.Boumahdi S & de Sauvage FJ The great escape: tumour cell plasticity in resistance to targeted therapy. Nat. Rev. Drug Discov 19, 39–56 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Sotorasib edges closer to approval. Cancer Discov. 11, OF2 (2021). [DOI] [PubMed] [Google Scholar]
- 4.Doroshow DB et al. PD-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat. Rev. Clin. Oncol 18, 345–362 (2021). [DOI] [PubMed] [Google Scholar]
- 5.Luo J, Solimini NL & Elledge SJ Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 136, 823–837 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harmenberg U, Hamdy FC, Widmark A, Lennernäs B & Nilsson S Curative radiation therapy in prostate cancer. Acta Oncol. 50, 98–103 (2011). [DOI] [PubMed] [Google Scholar]
- 7.Nakano T, Ohno T, Ishikawa H, Suzuki Y & Takahashi T Current advancement in radiation therapy for uterine cervical cancer. J. Radiat. Res 51, 1–8 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Spencer K, Parrish R, Barton R & Henry A Palliative radiotherapy. BMJ 360, k821 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Riet FG et al. Preoperative radiotherapy in breast cancer patients: 32 years of follow-up. Eur. J. Cancer 76, 45–51 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Calvo FA et al. ESTRO/ACROP IORT recommendations for intraoperative radiation therapy in primary locally advanced rectal cancer. Clin. Transl. Radiat. Oncol 25, 29–36 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zaorsky NG et al. The evolution of brachytherapy for prostate cancer. Nat. Rev. Urol 14, 415–439 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pilié PG, Tang C, Mills GB & Yap TA State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol 16, 81–104 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galluzzi L et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 25, 486–541 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galluzzi L, Buqué A, Kepp O, Zitvogel L & Kroemer G Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol 17, 97–111 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Formenti SC et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat. Med 24, 1845–1851 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rodriguez-Ruiz ME, Vitale I, Harrington KJ, Melero I & Galluzzi L Immunological impact of cell death signaling driven by radiation on the tumor microenvironment. Nat. Immunol 21, 120–134 (2020). [DOI] [PubMed] [Google Scholar]
- 17.Dalwadi SM, Herman JM, Das P & Holliday EB Novel radiotherapy technologies in the treatment of gastrointestinal malignancies. Hematol. Oncol. Clin. North. Am 34, 29–43 (2020). [DOI] [PubMed] [Google Scholar]
- 18.Oh DY & Bang YJ HER2-targeted therapies–a role beyond breast cancer. Nat. Rev. Clin. Oncol 17, 33–48 (2020). [DOI] [PubMed] [Google Scholar]
- 19.Guo R et al. MET-dependent solid tumours–molecular diagnosis and targeted therapy. Nat. Rev. Clin. Oncol 17, 569–587 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fruman DA et al. The PI3K pathway in human disease. Cell 170, 605–635 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu GY & Sabatini DM mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol 21, 183–203 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rybstein MD, Bravo-San Pedro JM, Kroemer G & Galluzzi L The autophagic network and cancer. Nat. Cell Biol 20, 243–251 (2018). [DOI] [PubMed] [Google Scholar]
- 23.McLaughlin M et al. Inflammatory microenvironment remodelling by tumour cells after radiotherapy. Nat. Rev. Cancer 20, 203–217 (2020). [DOI] [PubMed] [Google Scholar]
- 24.Barker HE, Paget JT, Khan AA & Harrington KJ The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat. Rev. Cancer 15, 409–425 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vanneste BGL et al. Immunotherapy as sensitizer for local radiotherapy. Oncoimmunology 9, 1832760 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moding EJ, Kastan MB & Kirsch DG Strategies for optimizing the response of cancer and normal tissues to radiation. Nat. Rev. Drug Discov 12, 526–542 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vitale I, Shema E, Loi S & Galluzzi L Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat. Med 27, 212–224 (2021). [DOI] [PubMed] [Google Scholar]
- 28.Petroni G & Galluzzi L Impact of treatment schedule on the efficacy of cytostatic and immunostimulatory agents. Oncoimmunology 10, 1889101 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Altorki NK et al. Neoadjuvant durvalumab with or without stereotactic body radiotherapy in patients with early-stage non-small-cell lung cancer: a single-centre, randomised phase 2 trial. Lancet Oncol. 22, 824–835 (2021). [DOI] [PubMed] [Google Scholar]
- 30.Coleman CN et al. Radiation-induced adaptive response: new potential for cancer treatment. Clin. Cancer Res 26, 5781–5790 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang RX & Zhou PK DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal. Transduct. Target. Ther 5, 60 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Galluzzi L et al. Molecular mechanisms of cisplatin resistance. Oncogene 31, 1869–1883 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Sansregret L, Vanhaesebroeck B & Swanton C Determinants and clinical implications of chromosomal instability in cancer. Nat. Rev. Clin. Oncol 15, 139–150 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Tubbs A & Nussenzweig A Endogenous DNA damage as a source of genomic instability in cancer. Cell 168, 644–656 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ashworth A & Lord CJ Synthetic lethal therapies for cancer: what’s next after PARP inhibitors? Nat. Rev. Clin. Oncol 15, 564–576 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Michels J et al. Cisplatin resistance associated with PARP hyperactivation. Cancer Res. 73, 2271–2280 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Shiloh Y & Ziv Y The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol 14, 197–210 (2013). [PubMed] [Google Scholar]
- 38.Taylor AM et al. Ataxia telangiectasia: a human mutation with abnormal radiation sensitivity. Nature 258, 427–429 (1975). [DOI] [PubMed] [Google Scholar]
- 39.Imray FP & Kidson C Perturbations of cell-cycle progression in ɣ-irradiated ataxia telangiectasia and Huntington’s disease cells detected by DNA flow cytometric analysis. Mutat. Res 112, 369–382 (1983). [DOI] [PubMed] [Google Scholar]
- 40.Carruthers R et al. Abrogation of radioresistance in glioblastoma stem-like cells by inhibition of ATM kinase. Mol. Oncol 9, 192–203 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Golding SE et al. Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol. Cancer Ther 8, 2894–2902 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vecchio D et al. Predictability, efficacy and safety of radiosensitization of glioblastoma-initiating cells by the ATM inhibitor KU-60019. Int. J. Cancer 135, 479–491 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Tang S, Li Z, Yang L, Shen L & Wang Y A potential new role of ATM inhibitor in radiotherapy: suppressing ionizing radiation-activated EGFR. Int. J. Radiat. Biol 96, 461–468 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takeuchi M et al. Anti-tumor effect of inhibition of DNA damage response proteins, ATM and ATR, in endometrial cancer cells. Cancers 11, 1913 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Durant ST et al. The brain-penetrant clinical ATM inhibitor AZD1390 radiosensitizes and improves survival of preclinical brain tumor models. Sci. Adv 4, eaat1719 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karlin J et al. Orally bioavailable and blood-brain barrier-penetrating ATM inhibitor (AZ32) radiosensitizes intracranial gliomas in mice. Mol. Cancer Ther 17, 1637–1647 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Biddlestone-Thorpe L et al. ATM kinase inhibition preferentially sensitizes p53-mutant glioma to ionizing radiation. Clin. Cancer Res 19, 3189–3200 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fokas E et al. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis. 3, e441 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Foote KM et al. Discovery of 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-(methylsulfonyl)cyclopropyl]pyrimidin-2-yl}-1H-indole (AZ20): a potent and selective inhibitor of ATR protein kinase with monotherapy in vivo antitumor activity. J. Med. Chem 56, 2125–2138 (2013). [DOI] [PubMed] [Google Scholar]
- 50.Dunne V et al. Inhibition of ataxia telangiectasia related-3 (ATR) improves therapeutic index in preclinical models of non-small cell lung cancer (NSCLC) radiotherapy. Radiother. Oncol 124, 475–481 (2017). [DOI] [PubMed] [Google Scholar]
- 51.Wengner AM et al. The novel ATR inhibitor BAY 1895344 is efficacious as monotherapy and combined with DNA damage-inducing or repair-compromising therapies in preclinical cancer models. Mol. Cancer Ther 19, 26–38 (2020). [DOI] [PubMed] [Google Scholar]
- 52.Pires IM et al. Targeting radiation-resistant hypoxic tumour cells through ATR inhibition. Br. J. Cancer 107, 291–299 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tu X et al. ATR inhibition is a promising radiosensitizing strategy for triple-negative breast cancer. Mol. Cancer Ther 17, 2462–2472 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zenke FT et al. Pharmacologic inhibitor of DNA-PK, M3814, potentiates radiotherapy and regresses human tumors in mouse models. Mol. Cancer Ther 19, 1091–1101 (2020). [DOI] [PubMed] [Google Scholar]
- 55.Fok JHL et al. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat. Commun 10, 5065 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Timme CR, Rath BH, O’Neill JW, Camphausen K & Tofilon PJ The DNA-PK inhibitor VX-984 enhances the radiosensitivity of glioblastoma cells grown in vitro and as orthotopic xenografts. Mol. Cancer Ther 17, 1207–1216 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Willoughby CE et al. Selective DNA-PKcs inhibition extends the therapeutic index of localized radiotherapy and chemotherapy. J. Clin. Invest 130, 258–271 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yamazaki T et al. Mitochondrial DNA drives abscopal responses to radiation that are inhibited by autophagy. Nat. Immunol 21, 1160–1171 (2020). [DOI] [PubMed] [Google Scholar]
- 59.Feng X et al. ATR inhibition potentiates ionizing radiation-induced interferon response via cytosolic nucleic acid-sensing pathways. EMBO J. 39, e104036 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dillon MT et al. ATR inhibition potentiates the radiation-induced inflammatory tumor microenvironment. Clin. Cancer Res 25, 3392–3403 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang Q et al. Inhibition of ATM increases interferon signaling and sensitizes pancreatic cancer to immune checkpoint blockade therapy. Cancer Res. 79, 3940–3951 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sheng H et al. ATR inhibitor AZD6738 enhances the antitumor activity of radiotherapy and immune checkpoint inhibitors by potentiating the tumor immune microenvironment in hepatocellular carcinoma. J. Immunother. Cancer 8, e000340 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vendetti FP et al. ATR kinase inhibitor AZD6738 potentiates CD8+ T cell-dependent antitumor activity following radiation. J. Clin. Invest 128, 3926–3940 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.He H, Chang R, Zhang T, Yang C & Kong Z ATM mediates DAB2IP-deficient bladder cancer cell resistance to ionizing radiation through the p38MAPK and NF-κB signaling pathway. Mol. Med. Rep 16, 1216–1222 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bakhoum SF et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bian L, Meng Y, Zhang M & Li D MRE11-RAD50-NBS1 complex alterations and DNA damage response: implications for cancer treatment. Mol. Cancer 18, 169 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fagan-Solis KD et al. A P53-independent dna damage response suppresses oncogenic proliferation and genome instability. Cell Rep. 30, 1385–1399 e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mattiello L et al. The targeting of MRE11 or RAD51 sensitizes colorectal cancer stem cells to CHK1 inhibition. Cancers (Basel) 13, 1957 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Manic G et al. Control of replication stress and mitosis in colorectal cancer stem cells through the interplay of PARP1, MRE11 and RAD51. Cell Death Differ. 28, 2060–2082 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ho V et al. Overexpression of the MRE11-RAD50-NBS1 (MRN) complex in rectal cancer correlates with poor response to neoadjuvant radiotherapy and prognosis. BMC Cancer 18, 869 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chang L et al. Targeting Rad50 sensitizes human nasopharyngeal carcinoma cells to radiotherapy. BMC Cancer 16, 190 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Choudhury A et al. MRE11 expression is predictive of cause-specific survival following radical radiotherapy for muscle-invasive bladder cancer. Cancer Res. 70, 7017–7026 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kondo T et al. DNA damage sensor MRE11 recognizes cytosolic double-stranded DNA and induces type I interferon by regulating STING trafficking. Proc. Natl Acad. Sci. USA 110, 2969–2974 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nicholson J et al. E3 ligase cIAP2 mediates downregulation of MRE11 and radiosensitization in response to HDAC inhibition in bladder cancer. Cancer Res. 77, 3027–3039 (2017). [DOI] [PubMed] [Google Scholar]
- 75.Groselj B et al. Radiosensitization in vivo by histone deacetylase inhibition with no increase in early normal tissue radiation toxicity. Mol. Cancer Ther 17, 381–392 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Paillas S et al. The histone deacetylase inhibitor romidepsin spares normal tissues while acting as an effective radiosensitizer in bladder tumors in vivo. Int. J. Radiat. Oncol. Biol. Phys 107, 212–221 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bryant HE et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917 (2005). [DOI] [PubMed] [Google Scholar]
- 78.Caron MC et al. Poly(ADP-ribose) polymerase-1 antagonizes DNA resection at double-strand breaks. Nat. Commun 10, 2954 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Strickfaden H et al. Poly(ADP-ribosyl)ation-dependent transient chromatin decondensation and histone displacement following laser microirradiation. J. Biol. Chem 291, 1789–1802 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu C et al. PARP inhibitor olaparib increases the sensitization to radiotherapy in FaDu cells. J. Cell Mol. Med 24, 2444–2450 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bi Y et al. Radiosensitization by the PARP inhibitor olaparib in BRCA1-proficient and deficient high-grade serous ovarian carcinomas. Gynecol. Oncol 150, 534–544 (2018). [DOI] [PubMed] [Google Scholar]
- 82.Michmerhuizen AR et al. PARP1 inhibition radiosensitizes models of inflammatory breast cancer to ionizing radiation. Mol. Cancer Ther 18, 2063–2073 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cho EJ et al. Preclinical evaluation of radiation therapy of BRCA1-associated mammary tumors using a mouse model. Int. J. Biol. Sci 17, 689–701 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Soni A et al. Inhibition of Parp1 by BMN673 effectively sensitizes cells to radiotherapy by upsetting the balance of repair pathways processing DNA double-strand breaks. Mol. Cancer Ther 17, 2206–2216 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tuli R et al. Radiosensitization of pancreatic cancer cells in vitro and in vivo through poly (ADP-ribose) polymerase inhibition with ABT-888. Transl. Oncol 7, 439–445 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Luo J et al. Fluzoparib increases radiation sensitivity of non-small cell lung cancer (NSCLC) cells without BRCA1/2 mutation, a novel PARP1 inhibitor undergoing clinical trials. J. Cancer Res. Clin. Oncol 146, 721–737 (2020). [DOI] [PubMed] [Google Scholar]
- 87.Ahmed SU et al. Selective inhibition of parallel DNA damage response pathways optimizes radiosensitization of glioblastoma stem-like cells. Cancer Res. 75, 4416–4428 (2015). [DOI] [PubMed] [Google Scholar]
- 88.Chabanon RM et al. PBRM1 deficiency confers synthetic lethality to DNA repair inhibitors in cancer. Cancer Res. 81, 2888–2902 (2021). [DOI] [PubMed] [Google Scholar]
- 89.Lord CJ & Ashworth A BRCAness revisited. Nat. Rev. Cancer 16, 110–120 (2016). [DOI] [PubMed] [Google Scholar]
- 90.Zhao B, Rothenberg E, Ramsden DA & Lieber MR The molecular basis and disease relevance of non-homologous DNA end joining. Nat. Rev. Mol. Cell Biol 21, 765–781 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chabanon RM et al. PARP inhibition enhances tumor cell-intrinsic immunity in ERCC1-deficient non-small cell lung cancer. J. Clin. Invest 129, 1211–1228 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang N et al. PARP inhibitor niraparib as a radiosensitizer promotes antitumor immunity of radiotherapy in EGFR-mutated non-small cell lung cancer. Clin. Transl. Oncol 23, 1827–1837 (2021). [DOI] [PubMed] [Google Scholar]
- 93.Petroni G, Buqué A, Zitvogel L, Kroemer G & Galluzzi L Immunomodulation by targeted anticancer agents. Cancer Cell 39, 310–345 (2021). [DOI] [PubMed] [Google Scholar]
- 94.Wang WJ et al. MYC regulation of CHK1 and CHK2 promotes radioresistance in a stem cell-like population of nasopharyngeal carcinoma cells. Cancer Res. 73, 1219–1231 (2013). [DOI] [PubMed] [Google Scholar]
- 95.Zhang P et al. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat. Cell Biol 16, 864–875 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yan Y, Black CP & Cowan KH Irradiation-induced G2/M checkpoint response requires ERK1/2 activation. Oncogene 26, 4689–4698 (2007). [DOI] [PubMed] [Google Scholar]
- 97.Barker HE et al. CHK1 inhibition radiosensitizes head and neck cancers to paclitaxel-based chemoradiotherapy. Mol. Cancer Ther 15, 2042–2054 (2016). [DOI] [PubMed] [Google Scholar]
- 98.Richer AL et al. WEE1 kinase inhibitor AZD1775 has preclinical efficacy in LKB1-deficient non-small cell lung cancer. Cancer Res. 77, 4663–4672 (2017). [DOI] [PubMed] [Google Scholar]
- 99.Lee YY et al. Anti-tumor effects of Wee1 kinase inhibitor with radiotherapy in human cervical cancer. Sci. Rep 9, 15394 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yang L et al. Wee1 kinase inhibitor AZD1775 effectively sensitizes esophageal cancer to radiotherapy. Clin. Cancer Res 26, 3740–3750 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mitchell JB et al. In vitro and in vivo radiation sensitization of human tumor cells by a novel checkpoint kinase inhibitor, AZD7762. Clin. Cancer Res 16, 2076–2084 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Morgan MA et al. Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res. 70, 4972–4981 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Patties I et al. The Chk1 inhibitor SAR-020106 sensitizes human glioblastoma cells to irradiation, to temozolomide, and to decitabine treatment. J. Exp. Clin. Cancer Res 38, 420 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zeng L, Nikolaev A, Xing C, Della Manna DL & Yang ES CHK1/2 inhibitor prexasertib suppresses NOTCH signaling and enhances cytotoxicity of cisplatin and radiation in head and neck squamous cell carcinoma. Mol. Cancer Ther 19, 1279–1288 (2020). [DOI] [PubMed] [Google Scholar]
- 105.Karnak D et al. Combined inhibition of Wee1 and PARP1/2 for radiosensitization in pancreatic cancer. Clin. Cancer Res 20, 5085–5096 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Parsels LA et al. PARP1 trapping and DNA replication stress enhance radiosensitization with combined WEE1 and PARP inhibitors. Mol. Cancer Res 16, 222–232 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Vance S et al. Selective radiosensitization of p53 mutant pancreatic cancer cells by combined inhibition of Chk1 and PARP1. Cell Cycle 10, 4321–4329 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Parmar K et al. The CHK1 inhibitor prexasertib exhibits monotherapy activity in high-grade serous ovarian cancer models and sensitizes to PARP inhibition. Clin. Cancer Res 25, 6127–6140 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Choi C et al. Checkpoint kinase 1 (CHK1) inhibition enhances the sensitivity of triple-negative breast cancer cells to proton irradiation via Rad51 downregulation. Int. J. Mol. Sci 21, 2691 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Raghavan P et al. AZD5438, an inhibitor of Cdk1, 2, and 9, enhances the radiosensitivity of non-small cell lung carcinoma cells. Int. J. Radiat. Oncol. Biol. Phys 84, e507–e514 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.O’Leary B, Finn RS & Turner NC Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol 13, 417–430 (2016). [DOI] [PubMed] [Google Scholar]
- 112.Bosacki C et al. CDK 4/6 inhibitors combined with radiotherapy: a review of literature. Clin. Transl. Radiat. Oncol 26, 79–85 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Göttgens EL et al. Inhibition of CDK4/CDK6 enhances radiosensitivity of HPV negative head and neck squamous cell carcinomas. Int. J. Radiat. Oncol. Biol. Phys 105, 548–558 (2019). [DOI] [PubMed] [Google Scholar]
- 114.Naz S et al. Abemaciclib, a selective CDK4/6 inhibitor, enhances the radiosensitivity of non-small cell lung cancer in vitro and in vivo. Clin. Cancer Res 24, 3994–4005 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Huang CY et al. Palbociclib enhances radiosensitivity of hepatocellular carcinoma and cholangiocarcinoma via inhibiting ataxia telangiectasia-mutated kinase-mediated DNA damage response. Eur. J. Cancer 102, 10–22 (2018). [DOI] [PubMed] [Google Scholar]
- 116.Petroni G et al. Radiotherapy delivered before CDK4/6 inhibitors mediates superior therapeutic effects in ER(+) breast cancer. Clin. Cancer Res 27, 1855–1863 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Xie X et al. CDK4/6 inhibitor palbociclib amplifies the radiosensitivity to nasopharyngeal carcinoma cells via mediating apoptosis and suppressing dna damage repair. Onco Targets Ther. 12, 11107–11117 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fernández-Aroca DM et al. P53 pathway is a major determinant in the radiosensitizing effect of palbociclib: implication in cancer therapy. Cancer Lett. 451, 23–33 (2019). [DOI] [PubMed] [Google Scholar]
- 119.Hashizume R et al. Inhibition of DNA damage repair by the CDK4/6 inhibitor palbociclib delays irradiated intracranial atypical teratoid rhabdoid tumor and glioblastoma xenograft regrowth. Neuro Oncol. 18, 1519–1528 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Patel P et al. Enhancing direct cytotoxicity and response to immune checkpoint blockade following ionizing radiation with Wee1 kinase inhibition. Oncoimmunology 8, e1638207 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang B, Sun L, Yuan Z & Tao Z Wee1 kinase inhibitor AZD1775 potentiates CD8+ T cell-dependent antitumour activity via dendritic cell activation following a single high dose of irradiation. Med. Oncol 37, 66 (2020). [DOI] [PubMed] [Google Scholar]
- 122.Chao HH et al. Combination of CHEK1/2 inhibition and ionizing radiation results in abscopal tumor response through increased micronuclei formation. Oncogene 39, 4344–4357 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Petroni G, Formenti SC, Chen-Kiang S & Galluzzi L Immunomodulation by anticancer cell cycle inhibitors. Nat. Rev. Immunol 20, 669–679 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Huang A, Garraway LA, Ashworth A & Weber B Synthetic lethality as an engine for cancer drug target discovery. Nat. Rev. Drug Discov 19, 23–38 (2020). [DOI] [PubMed] [Google Scholar]
- 125.Paluch-Shimon S & Cardoso F PARP inhibitors coming of age. Nat. Rev. Clin. Oncol 18, 69–70 (2021). [DOI] [PubMed] [Google Scholar]
- 126.Li N et al. An open-label, multicenter, single-arm, phase II study of fluzoparib in patients with germline BRCA1/2 mutation and platinum-sensitive recurrent ovarian cancer. Clin. Cancer Res 27, 2452–2458 (2021). [DOI] [PubMed] [Google Scholar]
- 127.Sonnenblick A, de Azambuja E, Azim HA Jr. & Piccart M An update on PARP inhibitors–moving to the adjuvant setting. Nat. Rev. Clin. Oncol 12, 27–41 (2015). [DOI] [PubMed] [Google Scholar]
- 128.Karam SD et al. Final report of a phase I trial of olaparib with cetuximab and radiation for heavy smoker patients with locally advanced head and neck cancer. Clin. Cancer Res 24, 4949–4959 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Loap P et al. Combination of olaparib and radiation therapy for triple negative breast cancer: preliminary results of the RADIOPARP phase 1 trial. Int. J. Radiat. Oncol. Biol. Phys 109, 436–440 (2021). [DOI] [PubMed] [Google Scholar]
- 130.de Haan R et al. Phase I and pharmacologic study of olaparib in combination with high-dose radiotherapy with and without concurrent cisplatin for non-small cell lung cancer. Clin. Cancer Res 27, 1256–1266 (2021). [DOI] [PubMed] [Google Scholar]
- 131.Konstantinopoulos PA et al. Olaparib and α-specific PI3K inhibitor alpelisib for patients with epithelial ovarian cancer: a dose-escalation and dose-expansion phase 1b trial. Lancet Oncol. 20, 570–580 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Matulonis UA et al. Phase I dose escalation study of the PI3kinase pathway inhibitor BKM120 and the oral poly (ADP ribose) polymerase (PARP) inhibitor olaparib for the treatment of high-grade serous ovarian and breast cancer. Ann. Oncol 28, 512–518 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yap TA et al. Phase I trial of first-in-class ATR inhibitor M6620 (VX-970) as monotherapy or in combination with carboplatin in patients with advanced solid tumors. J. Clin. Oncol 38, 3195–3204 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Thomas A et al. Phase I study of ATR inhibitor M6620 in combination with topotecan in patients with advanced solid tumors. J. Clin. Oncol 36, 1594–1602 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Dillon MT et al. PATRIOT: a phase I study to assess the tolerability, safety and biological effects of a specific ataxia telangiectasia and Rad3-related (ATR) inhibitor (AZD6738) as a single agent and in combination with palliative radiation therapy in patients with solid tumours. Clin. Transl. Radiat. Oncol 12, 16–20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kim ST et al. Phase I study of ceralasertib (AZD6738), a novel DNA damage repair agent, in combination with weekly paclitaxel in refractory cancer. Clin. Cancer Res 27, 4700–4709 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.van Bussel MTJ et al. A first-in-man phase 1 study of the DNA-dependent protein kinase inhibitor peposertib (formerly M3814) in patients with advanced solid tumours. Br. J. Cancer 124, 728–735 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Cuneo KC et al. Dose escalation trial of the Wee1 inhibitor adavosertib (AZD1775) in combination with gemcitabine and radiation for patients with locally advanced pancreatic cancer. J. Clin. Oncol 37, 2643–2650 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yang ES et al. A phase 1b trial of prexasertib in combination with chemoradiation in patients with locally advanced head and neck squamous cell carcinoma. Radiother. Oncol 157, 203–209 (2021). [DOI] [PubMed] [Google Scholar]
- 140.Boss DS et al. Safety, tolerability, pharmacokinetics and pharmacodynamics of the oral cyclin-dependent kinase inhibitor AZD5438 when administered at intermittent and continuous dosing schedules in patients with advanced solid tumours. Ann. Oncol 21, 884–894 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sausville E et al. Phase I dose-escalation study of AZD7762, a checkpoint kinase inhibitor, in combination with gemcitabine in US patients with advanced solid tumors. Cancer Chemother. Pharmacol 73, 539–549 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Beddok A et al. Concurrent use of palbociclib and radiation therapy: single-centre experience and review of the literature. Br. J. Cancer 123, 905–908 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ratosa I et al. Cyclin-dependent kinase 4/6 inhibitors combined with radiotherapy for patients with metastatic breast cancer. Clin. Breast Cancer 20, 495–502 (2020). [DOI] [PubMed] [Google Scholar]
- 144.Ippolito E et al. Concurrent radiotherapy with palbociclib or ribociclib for metastatic breast cancer patients: preliminary assessment of toxicity. Breast 46, 70–74 (2019). [DOI] [PubMed] [Google Scholar]
- 145.Meattini I, Desideri I, Scotti V, Simontacchi G & Livi L Ribociclib plus letrozole and concomitant palliative radiotherapy for metastatic breast cancer. Breast 42, 1–2 (2018). [DOI] [PubMed] [Google Scholar]
- 146.Chowdhary M et al. Safety and efficacy of palbociclib and radiation therapy in patients with metastatic breast cancer: initial results of a novel combination. Adv. Radiat. Oncol 4, 453–457 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Guerini AE et al. A single-center retrospective safety analysis of cyclin-dependent kinase 4/6 inhibitors concurrent with radiation therapy in metastatic breast cancer patients. Sci. Rep 10, 13589 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.DeWire M et al. A phase I/II study of ribociclib following radiation therapy in children with newly diagnosed diffuse intrinsic pontine glioma (DIPG). J. Neurooncol 149, 511–522 (2020). [DOI] [PubMed] [Google Scholar]
- 149.Hoxhaj G & Manning BD The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 20, 74–88 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Thorpe LM, Yuzugullu H & Zhao JJ PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 15, 7–24 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Vasan N et al. Double PIK3CA mutations in cis increase oncogenicity and sensitivity to PI3Kα inhibitors. Science 366, 714–723 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ruiz-Saenz A et al. HER2 amplification in tumors activates PI3K/Akt signaling independent of HER3. Cancer Res. 78, 3645–3658 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Tsay JJ et al. Airway microbiota is associated with upregulation of the PI3K pathway in lung cancer. Am. J. Respir. Crit. Care Med 198, 1188–1198 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hopkins BD et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 560, 499–503 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hopkins BD, Goncalves MD & Cantley LC Insulin-PI3K signalling: an evolutionarily insulated metabolic driver of cancer. Nat. Rev. Endocrinol 16, 276–283 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.André F et al. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N. Engl. J. Med 380, 1929–1940 (2019). [DOI] [PubMed] [Google Scholar]
- 157.Lockney NA et al. PIK3CA mutation is associated with increased local failure in lung stereotactic body radiation therapy (SBRT). Clin. Transl. Radiat. Oncol 7, 91–93 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lockney NA et al. Phosphatidylinositol-3-kinase mutations are associated with increased local failure in brain metastases treated with radiation. Int. J. Radiat. Oncol. Biol. Phys 101, 833–844 (2018). [DOI] [PubMed] [Google Scholar]
- 159.Zafarana G et al. Copy number alterations of c-MYC and PTEN are prognostic factors for relapse after prostate cancer radiotherapy. Cancer 118, 4053–4062 (2012). [DOI] [PubMed] [Google Scholar]
- 160.Ang KK et al. Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res. 62, 7350–7356 (2002). [PubMed] [Google Scholar]
- 161.Chua DT, Nicholls JM, Sham JS & Au GK Prognostic value of epidermal growth factor receptor expression in patients with advanced stage nasopharyngeal carcinoma treated with induction chemotherapy and radiotherapy. Int. J. Radiat. Oncol. Biol. Phys 59, 11–20 (2004). [DOI] [PubMed] [Google Scholar]
- 162.Brollo J et al. Locoregional recurrence in patients with HER2 positive breast cancer. Breast 22, 856–862 (2013). [DOI] [PubMed] [Google Scholar]
- 163.Green MM et al. Expression of vascular endothelial growth factor (VEGF) in locally invasive prostate cancer is prognostic for radiotherapy outcome. Int. J. Radiat. Oncol. Biol. Phys 67, 84–90 (2007). [DOI] [PubMed] [Google Scholar]
- 164.Yoshimoto Y et al. Mutation profiling of uterine cervical cancer patients treated with definitive radiotherapy. Gynecol. Oncol 159, 546–553 (2020). [DOI] [PubMed] [Google Scholar]
- 165.Darwis NDM et al. FGFR signaling as a candidate therapeutic target for cancers resistant to carbon ion radiotherapy. Int. J. Mol. Sci 20, 4563 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Chen DJ & Nirodi CS The epidermal growth factor receptor: a role in repair of radiation-induced DNA damage. Clin. Cancer Res 13, 6555–6560 (2007). [DOI] [PubMed] [Google Scholar]
- 167.Li HF, Kim JS & Waldman T Radiation-induced Akt activation modulates radioresistance in human glioblastoma cells. Radiat. Oncol 4, 43 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Chinnaiyan P et al. Mechanisms of enhanced radiation response following epidermal growth factor receptor signaling inhibition by erlotinib (Tarceva). Cancer Res. 65, 3328–3335 (2005). [DOI] [PubMed] [Google Scholar]
- 169.Park CM et al. Ionizing radiation enhances matrix metalloproteinase-2 secretion and invasion of glioma cells through Src/epidermal growth factor receptor-mediated p38/Akt and phosphatidylinositol 3-kinase/Akt signaling pathways. Cancer Res. 66, 8511–8519 (2006). [DOI] [PubMed] [Google Scholar]
- 170.Cao N et al. NF-kappaB-mediated HER2 overexpression in radiation-adaptive resistance. Radiat. Res 171, 9–21 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Rose Li,Y. et al. Mutational signatures in tumours induced by high and low energy radiation in Trp53 deficient mice. Nat. Commun 11, 394 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.De Bacco F et al. Induction of MET by ionizing radiation and its role in radioresistance and invasive growth of cancer. J. Natl Cancer Inst 103, 645–661 (2011). [DOI] [PubMed] [Google Scholar]
- 173.Luttich L et al. Tyrosine kinase c-MET as therapeutic target for radiosensitization of head and neck squamous cell carcinomas. Cancers 13, 1865 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Nisa L et al. Targeting the MET receptor tyrosine kinase as a strategy for radiosensitization in locoregionally advanced head and neck squamous cell carcinoma. Mol. Cancer Ther 19, 614–626 (2020). [DOI] [PubMed] [Google Scholar]
- 175.Sofia Vala I et al. Low doses of ionizing radiation promote tumor growth and metastasis by enhancing angiogenesis. PLoS ONE 5, e11222 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Gorski DH et al. Blockage of the vascular endothelial growth factor stress response increases the antitumor effects of ionizing radiation. Cancer Res. 59, 3374–3378 (1999). [PubMed] [Google Scholar]
- 177.Knizetova P et al. Autocrine regulation of glioblastoma cell cycle progression, viability and radioresistance through the VEGF-VEGFR2 (KDR) interplay. Cell Cycle 7, 2553–2561 (2008). [DOI] [PubMed] [Google Scholar]
- 178.Gomez-Roman N et al. Radiation responses of 2D and 3D glioblastoma cells: a novel, 3D-specific radioprotective role of VEGF/Akt signaling through functional activation of NHEJ. Mol. Cancer Ther 19, 575–589 (2020). [DOI] [PubMed] [Google Scholar]
- 179.Ma J et al. Inhibition of nuclear PTEN tyrosine phosphorylation enhances glioma radiation sensitivity through attenuated DNA repair. Cancer Cell 35, 504–518.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lammering G, Valerie K, Lin PS, Hewit TH & Schmidt-Ullrich RK Radiation-induced activation of a common variant of EGFR confers enhanced radioresistance. Radiother. Oncol 72, 267–273 (2004). [DOI] [PubMed] [Google Scholar]
- 181.Lammering G et al. EGFRvIII-mediated radioresistance through a strong cytoprotective response. Oncogene 22, 5545–5553 (2003). [DOI] [PubMed] [Google Scholar]
- 182.Chang L et al. Acquisition of epithelial-mesenchymal transition and cancer stem cell phenotypes is associated with activation of the PI3K/Akt/mTOR pathway in prostate cancer radioresistance. Cell Death Dis. 4, e875 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Ni J et al. Epithelial cell adhesion molecule (EpCAM) is associated with prostate cancer metastasis and chemo/radioresistance via the PI3K/Akt/mTOR signaling pathway. Int. J. Biochem. Cell Biol 45, 2736–2748 (2013). [DOI] [PubMed] [Google Scholar]
- 184.Fruman DA & Rommel C PI3K and cancer: lessons, challenges and opportunities. Nat. Rev. Drug Discov 13, 140–156 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Janku F, Yap TA & Meric-Bernstam F Targeting the PI3K pathway in cancer: are we making headway? Nat. Rev. Clin. Oncol 15, 273–291 (2018). [DOI] [PubMed] [Google Scholar]
- 186.Kim IA et al. Selective inhibition of Ras, phosphoinositide 3 kinase, and Akt isoforms increases the radiosensitivity of human carcinoma cell lines. Cancer Res. 65, 7902–7910 (2005). [DOI] [PubMed] [Google Scholar]
- 187.Brognard J, Clark AS, Ni Y & Dennis PA Akt/protein kinase B is constitutively active in non-small cell lung cancer cells and promotes cellular survival and resistance to chemotherapy and radiation. Cancer Res. 61, 3986–3997 (2001). [PubMed] [Google Scholar]
- 188.Hasslacher S et al. Inhibition of PI3K signalling increases the efficiency of radiotherapy in glioblastoma cells. Int. J. Oncol 53, 1881–1896 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Shi F et al. The PI3K inhibitor GDC-0941 enhances radiosensitization and reduces chemoresistance to temozolomide in GBM cell lines. Neuroscience 346, 298–308 (2017). [DOI] [PubMed] [Google Scholar]
- 190.Park JH et al. Radiosensitization of the PI3K inhibitor HS-173 through reduction of DNA damage repair in pancreatic cancer. Oncotarget 8, 112893–112906 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Zumsteg ZS et al. Taselisib (GDC-0032), a potent β-sparing small molecule inhibitor of PI3K, radiosensitizes head and neck squamous carcinomas containing activating PIK3CA alterations. Clin. Cancer Res 22, 2009–2019 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Glorieux M, Dok R & Nuyts S The influence of PI3K inhibition on the radiotherapy response of head and neck cancer cells. Sci. Rep 10, 16208 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Djuzenova CS et al. Differential effects of the Akt inhibitor MK-2206 on migration and radiation sensitivity of glioblastoma cells. BMC Cancer 19, 299 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Miyasaka A et al. PI3K/mTOR pathway inhibition overcomes radioresistance via suppression of the HIF1-α/VEGF pathway in endometrial cancer. Gynecol. Oncol 138, 174–180 (2015). [DOI] [PubMed] [Google Scholar]
- 195.Yu CC et al. Targeting the PI3K/AKT/mTOR signaling pathway as an effectively radiosensitizing strategy for treating human oral squamous cell carcinoma in vitro and in vivo. Oncotarget 8, 68641–68653 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Eke I et al. Exploiting radiation-induced signaling to increase the susceptibility of resistant cancer cells to targeted drugs: AKT and mTOR inhibitors as an example. Mol. Cancer Ther 17, 355–367 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Chuang FC et al. PI3k inhibitors (BKM120 and BYL719) as radiosensitizers for head and neck squamous cell carcinoma during radiotherapy. PLoS ONE 16, e0245715 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Juvekar A et al. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov. 2, 1048–1063 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Gonzalez-Billalabeitia E et al. Vulnerabilities of PTEN-TP53-deficient prostate cancers to compound PARP-PI3K inhibition. Cancer Discov. 4, 896–904 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Bian X et al. PTEN deficiency sensitizes endometrioid endometrial cancer to compound PARP-PI3K inhibition but not PARP inhibition as monotherapy. Oncogene 37, 341–351 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Philip CA et al. Inhibition of PI3K-AKT-mTOR pathway sensitizes endometrial cancer cell lines to PARP inhibitors. BMC Cancer 17, 638 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Lammering G et al. Inhibition of the type III epidermal growth factor receptor variant mutant receptor by dominant-negative EGFR-CD533 enhances malignant glioma cell radiosensitivity. Clin. Cancer Res 10, 6732–6743 (2004). [DOI] [PubMed] [Google Scholar]
- 203.Akashi Y et al. Enhancement of the antitumor activity of ionising radiation by nimotuzumab, a humanised monoclonal antibody to the epidermal growth factor receptor, in non-small cell lung cancer cell lines of differing epidermal growth factor receptor status. Br. J. Cancer 98, 749–755 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Raben D et al. The effects of cetuximab alone and in combination with radiation and/or chemotherapy in lung cancer. Clin. Cancer Res 11, 795–805 (2005). [PubMed] [Google Scholar]
- 205.Yu T et al. Radiosensitizing effect of lapatinib in human epidermal growth factor receptor 2-positive breast cancer cells. Oncotarget 7, 79089–79100 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Huang T et al. Pyrotinib enhances the radiosensitivity of HER2-overexpressing gastric and breast cancer cells. Oncol. Rep 44, 2634–2644 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Wu S et al. HER2 recruits AKT1 to disrupt STING signalling and suppress antiviral defence and antitumour immunity. Nat. Cell Biol 21, 1027–1040 (2019). [DOI] [PubMed] [Google Scholar]
- 208.Candas-Green D et al. Dual blockade of CD47 and HER2 eliminates radioresistant breast cancer cells. Nat. Commun 11, 4591 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Cao C et al. Vascular endothelial growth factor tyrosine kinase inhibitor AZD2171 and fractionated radiotherapy in mouse models of lung cancer. Cancer Res. 66, 11409–11415 (2006). [DOI] [PubMed] [Google Scholar]
- 210.Melsens E et al. The VEGFR inhibitor cediranib improves the efficacy of fractionated radiotherapy in a colorectal cancer xenograft model. Eur. Surg. Res 58, 95–108 (2017). [DOI] [PubMed] [Google Scholar]
- 211.Liao J et al. Apatinib potentiates irradiation effect via suppressing PI3K/AKT signaling pathway in hepatocellular carcinoma. J. Exp. Clin. Cancer Res 38, 454 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Chen L et al. VEGF knockdown enhances radiosensitivity of nasopharyngeal carcinoma by inhibiting autophagy through the activation of mTOR pathway. Sci. Rep 10, 16328 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.SenthilKumar G et al. FGFR inhibition enhances sensitivity to radiation in non-small cell lung cancer. Mol. Cancer Ther 19, 1255–1265 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.De Bacco F et al. MET inhibition overcomes radiation resistance of glioblastoma stem-like cells. EMBO Mol. Med 8, 550–568 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Truman JP et al. Endothelial membrane remodeling is obligate for anti-angiogenic radiosensitization during tumor radiosurgery. PLoS ONE 5, e12310 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Bonner JA et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N. Engl. J. Med 354, 567–578 (2006). [DOI] [PubMed] [Google Scholar]
- 217.Bonner JA et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol. 11, 21–28 (2010). [DOI] [PubMed] [Google Scholar]
- 218.Vermorken JB et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N. Engl. J. Med 359, 1116–1127 (2008). [DOI] [PubMed] [Google Scholar]
- 219.Mesia R et al. Chemoradiotherapy with or without panitumumab in patients with unresected, locally advanced squamous-cell carcinoma of the head and neck (CONCERT-1): a randomised, controlled, open-label phase 2 trial. Lancet Oncol. 16, 208–220 (2015). [DOI] [PubMed] [Google Scholar]
- 220.Giralt J et al. Panitumumab plus radiotherapy versus chemoradiotherapy in patients with unresected, locally advanced squamous-cell carcinoma of the head and neck (CONCERT-2): a randomised, controlled, open-label phase 2 trial. Lancet Oncol. 16, 221–232 (2015). [DOI] [PubMed] [Google Scholar]
- 221.Bonomo P et al. Incidence of skin toxicity in squamous cell carcinoma of the head and neck treated with radiotherapy and cetuximab: a systematic review. Crit. Rev. Oncol. Hematol 120, 98–110 (2017). [DOI] [PubMed] [Google Scholar]
- 222.Tougeron D et al. Skin inflammatory response and efficacy of anti-epidermal growth factor receptor therapy in metastatic colorectal cancer (CUTACETUX). Oncoimmunology 9, 1848058 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Zheng MH et al. Combining whole-brain radiotherapy with gefitinib/erlotinib for brain metastases from non-small-cell lung cancer: a meta-analysis. Biomed. Res. Int 2016, 5807346 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Kulinich DP et al. Radiotherapy versus combination radiotherapy-bevacizumab for the treatment of recurrent high-grade glioma: a systematic review. Acta Neurochir. 163, 1921–1934 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Andronesi OC et al. Early changes in glioblastoma metabolism measured by MR spectroscopic imaging during combination of anti-angiogenic cediranib and chemoradiation therapy are associated with survival. NPJ Precis. Oncol 1, 20 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Batchelor TT et al. Improved tumor oxygenation and survival in glioblastoma patients who show increased blood perfusion after cediranib and chemoradiation. Proc. Natl Acad. Sci. USA 110, 19059–19064 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Zhao F et al. Apatinib alone or combined with radiotherapy in metastatic prostate cancer: results from a pilot, multicenter study. Oncotarget 8, 110774–110784 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Yamamoto N et al. Phase 2 study of nimotuzumab in combination with concurrent chemoradiotherapy in patients with locally advanced non-small-cell lung cancer. Clin. Lung Cancer 22, 134–141 (2021). [DOI] [PubMed] [Google Scholar]
- 229.Du XJ et al. Efficacy and safety of nimotuzumab in addition to radiotherapy and temozolomide for cerebral glioblastoma: a phase II multicenter clinical trial. J. Cancer 10, 3214–3223 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Fleischhack G et al. Nimotuzumab and radiotherapy for treatment of newly diagnosed diffuse intrinsic pontine glioma (DIPG): a phase III clinical study. J. Neurooncol 143, 107–113 (2019). [DOI] [PubMed] [Google Scholar]
- 231.Dunn LA et al. A phase 1b study of cetuximab and BYL719 (Alpelisib) concurrent with intensity modulated radiation therapy in stage III-IVB head and neck squamous cell carcinoma. Int. J. Radiat. Oncol. Biol. Phys 106, 564–570 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Day D et al. Phase I trial of alpelisib in combination with concurrent cisplatin-based chemoradiotherapy in patients with locoregionally advanced squamous cell carcinoma of the head and neck. Oral. Oncol 108, 104753 (2020). [DOI] [PubMed] [Google Scholar]
- 233.McGowan DR et al. Buparlisib with thoracic radiotherapy and its effect on tumour hypoxia: a phase I study in patients with advanced non-small cell lung carcinoma. Eur. J. Cancer 113, 87–95 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Wen PY et al. Phase I, open-label, multicentre study of buparlisib in combination with temozolomide or with concomitant radiation therapy and temozolomide in patients with newly diagnosed glioblastoma. ESMO Open 5, e000673 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Vanacker H, Cassier PA & Bachelot T The complex balance of PI3K inhibition. Ann. Oncol 32, 127–128 (2021). [DOI] [PubMed] [Google Scholar]
- 236.Wise-Draper TM et al. A phase Ib study of the dual PI3K/mTOR inhibitor dactolisib (BEZ235) combined with everolimus in patients with advanced solid malignancies. Target. Oncol 12, 323–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Rodon J et al. Phase 1/1b dose escalation and expansion study of BEZ235, a dual PI3K/mTOR inhibitor, in patients with advanced solid tumors including patients with advanced breast cancer. Cancer Chemother. Pharmacol 82, 285–298 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Salazar R et al. Phase II study of BEZ235 versus everolimus in patients with mammalian target of rapamycin inhibitor-naive advanced pancreatic neuroendocrine tumors. Oncologist 23, 766–e90 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Carlo MI et al. A phase Ib study of BEZ235, a dual inhibitor of phosphatidylinositol 3-Kinase (PI3K) and mammalian target of rapamycin (mTOR), in patients with advanced renal cell carcinoma. Oncologist 21, 787–788 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Narayan V et al. Phase 1 trial of everolimus and radiation therapy for salvage treatment of biochemical recurrence in prostate cancer patients following prostatectomy. Int. J. Radiat. Oncol. Biol. Phys 97, 355–361 (2017). [DOI] [PubMed] [Google Scholar]
- 241.Gelsomino F et al. A dose-finding and biomarker evaluation phase Ib study of everolimus in association with 5-fluorouracil and pelvic radiotherapy as neoadjuvant treatment of locally advanced rectal cancer (E-LARC study). Clin. Colorectal Cancer 16, 410–415.e1 (2017). [DOI] [PubMed] [Google Scholar]
- 242.Chinnaiyan P et al. A randomized phase II study of everolimus in combination with chemoradiation in newly diagnosed glioblastoma: results of NRG Oncology RTOG 0913. Neuro Oncol. 20, 666–673 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Cao L et al. Trastuzumab improves locoregional control in HER2-positive breast cancer patients following adjuvant radiotherapy. Medicine 95, e4230 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Jeon SH et al. Effects of trastuzumab on locoregional recurrence in human epidermal growth factor receptor 2-overexpressing breast cancer patients treated with chemotherapy and radiotherapy. Breast Cancer Res. Treat 172, 619–626 (2018). [DOI] [PubMed] [Google Scholar]
- 245.Sun GY et al. Trastuzumab provides a comparable prognosis in patients with HER2-positive breast cancer to those with HER2-negative breast cancer: post hoc analyses of a randomized controlled trial of post-mastectomy hypofractionated radiotherapy. Front. Oncol 10, 605750 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Abi Jaoude J et al. De-intensifying radiation therapy in HER-2 positive breast cancer: to boost or not to boost? Int. J. Radiat. Oncol. Biol. Phys 108, 1040–1046 (2020). [DOI] [PubMed] [Google Scholar]
- 247.Chumsri S et al. Incidence of late relapses in patients with HER2-positive breast cancer receiving adjuvant trastuzumab: combined analysis of NCCTG N9831 (Alliance) and NRG Oncology/NSABP B-31. J. Clin. Oncol 37, 3425–3435 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Bonzano E, Guenzi M & Corvò R Cardiotoxicity assessment after different adjuvant hypofractionated radiotherapy concurrently associated with trastuzumab in early breast cancer. In Vivo 32, 879–882 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Sayan M et al. Acute cardiotoxicity with concurrent trastuzumab and hypofractionated radiation therapy in breast cancer patients. Front. Oncol 9, 970 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Khan M, Zhao Z, Arooj S, Zheng T & Liao G Lapatinib plus local radiation therapy for brain metastases from HER-2 positive breast cancer patients and role of trastuzumab: a systematic review and meta-analysis. Front. Oncol 10, 576926 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Harrington K et al. Randomised phase II study of oral lapatinib combined with chemoradiotherapy in patients with advanced squamous cell carcinoma of the head and neck: rationale for future randomised trials in human papilloma virus-negative disease. Eur. J. Cancer 49, 1609–1618 (2013). [DOI] [PubMed] [Google Scholar]
- 252.Harrington K et al. Postoperative adjuvant lapatinib and concurrent chemoradiotherapy followed by maintenance lapatinib monotherapy in high-risk patients with resected squamous cell carcinoma of the head and neck: a phase III, randomized, double-blind, placebo-controlled study. J. Clin. Oncol 33, 4202–4209 (2015). [DOI] [PubMed] [Google Scholar]
- 253.Lolkema MP et al. The c-Met tyrosine kinase inhibitor JNJ-38877605 causes renal toxicity through species-specific insoluble metabolite formation. Clin. Cancer Res 21, 2297–2304 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Derynck R, Turley SJ & Akhurst RJ TGFβ biology in cancer progression and immunotherapy. Nat. Rev. Clin. Oncol 18, 9–34 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Kirshner J et al. Inhibition of transforming growth factor-β1 signaling attenuates ataxia telangiectasia mutated activity in response to genotoxic stress. Cancer Res. 66, 10861–10869 (2006). [DOI] [PubMed] [Google Scholar]
- 256.Liu Q et al. Subjugation of TGFβ signaling by human papilloma virus in head and neck squamous cell carcinoma shifts DNA repair from homologous recombination to alternative end joining. Clin. Cancer Res 24, 6001–6014 (2018). [DOI] [PubMed] [Google Scholar]
- 257.Bouquet F et al. TGFβ1 inhibition increases the radiosensitivity of breast cancer cells in vitro and promotes tumor control by radiation in vivo. Clin. Cancer Res 17, 6754–6765 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Hardee ME et al. Resistance of glioblastoma-initiating cells to radiation mediated by the tumor microenvironment can be abolished by inhibiting transforming growth factor-β. Cancer Res. 72, 4119–4129 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Zhang M et al. Blockade of TGF-β signaling by the TGFβR-I kinase inhibitor LY2109761 enhances radiation response and prolongs survival in glioblastoma. Cancer Res. 71, 7155–7167 (2011). [DOI] [PubMed] [Google Scholar]
- 260.Gonzalez-Junca A et al. Positron emission tomography imaging of functional transforming growth factor β (TGFβ) activity and benefit of TGFβ inhibition in irradiated intracranial tumors. Int. J. Radiat. Oncol. Biol. Phys 109, 527–539 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Du S et al. Attenuation of the DNA damage response by transforming growth factor-beta inhibitors enhances radiation sensitivity of non-small-cell lung cancer cells in vitro and in vivo. Int. J. Radiat. Oncol. Biol. Phys 91, 91–99 (2015). [DOI] [PubMed] [Google Scholar]
- 262.Bellomo C, Caja L & Moustakas A Transforming growth factor β as regulator of cancer stemness and metastasis. Br. J. Cancer 115, 761–769 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Biswas S et al. Inhibition of TGF-β with neutralizing antibodies prevents radiation-induced acceleration of metastatic cancer progression. J. Clin. Invest 117, 1305–1313 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Vanpouille-Box C et al. TGFβ is a master regulator of radiation therapy-induced antitumor immunity. Cancer Res. 75, 2232–2242 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Rodríguez-Ruiz ME et al. TGFβ blockade enhances radiotherapy abscopal efficacy effects in combination with anti-PD1 and anti-CD137 immunostimulatory monoclonal antibodies. Mol. Cancer Ther 18, 621–631 (2019). [DOI] [PubMed] [Google Scholar]
- 266.Rodriguez-Ruiz ME et al. Apoptotic caspases inhibit abscopal responses to radiation and identify a new prognostic biomarker for breast cancer patients. Oncoimmunology 8, e1655964 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Lind H et al. Dual targeting of TGF-β and PD-L1 via a bifunctional anti-PD-L1/TGF-βRII agent: status of preclinical and clinical advances. J. Immunother. Cancer 8, e000433 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Lan Y et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-β. Sci. Transl. Med 10, eaan5488 (2018). [DOI] [PubMed] [Google Scholar]
- 269.Wang S et al. Plasma Levels of IL-8 and TGF-β1 predict radiation-induced lung toxicity in non-small cell lung cancer: a validation study. Int. J. Radiat. Oncol. Biol. Phys 98, 615–621 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Kim H et al. LXA(4)-FPR2 signaling regulates radiation-induced pulmonary fibrosis via crosstalk with TGF-β/Smad signaling. Cell Death Dis. 11, 653 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Han G et al. Preventive and therapeutic effects of Smad7 on radiation-induced oral mucositis. Nat. Med 19, 421–428 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Lee JW et al. Inhibition of Smad3 expression in radiation-induced fibrosis using a novel method for topical transcutaneous gene therapy. Arch. Otolaryngol. Head. Neck Surg 136, 714–719 (2010). [DOI] [PubMed] [Google Scholar]
- 273.Boerma M, Wang J, Sridharan V, Herbert JM & Hauer-Jensen M Pharmacological induction of transforming growth factor-beta1 in rat models enhances radiation injury in the intestine and the heart. PLoS ONE 8, e70479 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Flechsig P et al. LY2109761 attenuates radiation-induced pulmonary murine fibrosis via reversal of TGF-β and BMP-associated proinflammatory and proangiogenic signals. Clin. Cancer Res 18, 3616–3627 (2012). [DOI] [PubMed] [Google Scholar]
- 275.Luo J et al. Smad7 promotes healing of radiotherapy-induced oral mucositis without compromising oral cancer therapy in a xenograft mouse model. Clin. Cancer Res 25, 808–818 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Ciardiello D, Elez E, Tabernero J & Seoane J Clinical development of therapies targeting TGFβ: current knowledge and future perspectives. Ann. Oncol 31, 1336–1349 (2020). [DOI] [PubMed] [Google Scholar]
- 277.Formenti SC et al. Focal irradiation and systemic TGFβ blockade in metastatic breast cancer. Clin. Cancer Res 24, 2493–2504 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Formenti SC et al. Baseline T cell dysfunction by single cell network profiling in metastatic breast cancer patients. J. Immunother. Cancer 7, 177 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.De Martino M et al. Activin a promotes regulatory T-cell-mediated immunosuppression in irradiated breast cancer. Cancer Immunol. Res 9, 89–102 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Wick A et al. Phase 1b/2a study of galunisertib, a small molecule inhibitor of transforming growth factor-beta receptor I, in combination with standard temozolomide-based radiochemotherapy in patients with newly diagnosed malignant glioma. Invest. New Drugs 38, 1570–1579 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Vanpouille-Box C & Formenti SC Dual transforming growth factor-β and programmed death-1 blockade: a strategy for immune-excluded tumors? Trends Immunol. 39, 435–437 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Levy JMM, Towers CG & Thorburn A Targeting autophagy in cancer. Nat. Rev. Cancer 17, 528–542 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Galluzzi L, Bravo-San JMP, Levine B, Green DR & Kroemer G Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov 16, 487–511 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Jin X et al. Role of autophagy in high linear energy transfer radiation-induced cytotoxicity to tumor cells. Cancer Sci. 105, 770–778 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Galati S, Boni C, Gerra MC, Lazzaretti M & Buschini A Autophagy: a player in response to oxidative stress and DNA damage. Oxid. Med. Cell Longev 2019, 5692958 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Wang WJ et al. NVP-BEZ235, a novel dual PI3K/mTOR inhibitor, enhances the radiosensitivity of human glioma stem cells in vitro. Acta Pharmacol. Sin 34, 681–690 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Cao C et al. Inhibition of mammalian target of rapamycin or apoptotic pathway induces autophagy and radiosensitizes PTEN null prostate cancer cells. Cancer Res. 66, 10040–10047 (2006). [DOI] [PubMed] [Google Scholar]
- 288.Albert JM, Kim KW, Cao C & Lu B Targeting the Akt/mammalian target of rapamycin pathway for radiosensitization of breast cancer. Mol. Cancer Ther 5, 1183–1189 (2006). [DOI] [PubMed] [Google Scholar]
- 289.Woo Y et al. Rapamycin promotes ROS-mediated cell death via functional inhibition of xCT expression in melanoma under γ-irradiation. Front. Oncol 11, 665420 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Zhuang W et al. Induction of autophagy promotes differentiation of glioma-initiating cells and their radiosensitivity. Int. J. Cancer 129, 2720–2731 (2011). [DOI] [PubMed] [Google Scholar]
- 291.Saleh AD et al. Caloric restriction augments radiation efficacy in breast cancer. Cell Cycle 12, 1955–1963 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Simone BA et al. Caloric restriction coupled with radiation decreases metastatic burden in triple negative breast cancer. Cell Cycle 15, 2265–2274 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Classen F et al. Autophagy induced by ionizing radiation promotes cell death over survival in human colorectal cancer cells. Exp. Cell Res 374, 29–37 (2019). [DOI] [PubMed] [Google Scholar]
- 294.Galluzzi L & Green DR Autophagy-independent functions of the autophagy machinery. Cell 177, 1682–1699 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Kimmelman AC & White E Autophagy and tumor metabolism. Cell Metab. 25, 1037–1043 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Liu EY et al. Loss of autophagy causes a synthetic lethal deficiency in DNA repair. Proc. Natl Acad. Sci. USA 112, 773–778 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Hewitt G & Korolchuk VI Repair, reuse, recycle: the expanding role of autophagy in genome maintenance. Trends Cell Biol. 27, 340–351 (2017). [DOI] [PubMed] [Google Scholar]
- 298.Wang Y et al. Autophagy regulates chromatin ubiquitination in DNA damage response through elimination of SQSTM1/p62. Mol. Cell 63, 34–48 (2016). [DOI] [PubMed] [Google Scholar]
- 299.Park JM, Tougeron D, Huang S, Okamoto K & Sinicrope FA Beclin 1 and UVRAG confer protection from radiation-induced DNA damage and maintain centrosome stability in colorectal cancer cells. PLoS One 9, e100819 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Chen X et al. Autophagy enhanced the radioresistance of non-small cell lung cancer by regulating ROS level under hypoxia condition. Int. J. Radiat. Biol 93, 764–770 (2017). [DOI] [PubMed] [Google Scholar]
- 301.Chaachouay H et al. AMPK-independent autophagy promotes radioresistance of human tumor cells under clinical relevant hypoxia in vitro. Radiother. Oncol 116, 409–416 (2015). [DOI] [PubMed] [Google Scholar]
- 302.Jing Q et al. Wnt3a promotes radioresistance via autophagy in squamous cell carcinoma of the head and neck. J. Cell Mol. Med 23, 4711–4722 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Hu JL et al. Inhibition of ATG12-mediated autophagy by miR-214 enhances radiosensitivity in colorectal cancer. Oncogenesis 7, 16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Zheng X et al. Inhibiting autophagy with chloroquine enhances the anti-tumor effect of high-LET carbon ions via ER stress-related apoptosis. Med. Oncol 34, 25 (2017). [DOI] [PubMed] [Google Scholar]
- 305.Tseng HC et al. Sensitizing effect of 3-methyladenine on radiation-induced cytotoxicity in radio-resistant HepG2 cells in vitro and in tumor xenografts. Chem. Biol. Interact 192, 201–208 (2011). [DOI] [PubMed] [Google Scholar]
- 306.Chen Y et al. Combining radiation with autophagy inhibition enhances suppression of tumor growth and angiogenesis in esophageal cancer. Mol. Med. Rep 12, 1645–1652 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Ko A et al. Autophagy inhibition radiosensitizes in vitro, yet reduces radioresponses in vivo due to deficient immunogenic signalling. Cell Death Differ. 21, 92–99 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Lin W et al. Autophagy confers DNA damage repair pathways to protect the hematopoietic system from nuclear radiation injury. Sci. Rep 5, 12362 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Xu F et al. Autophagy promotes the repair of radiation-induced DNA damage in bone marrow hematopoietic cells via enhanced STAT3 signaling. Radiat. Res 187, 382–396 (2017). [DOI] [PubMed] [Google Scholar]
- 310.Xu F et al. Nuclear localization of Beclin 1 promotes radiation-induced DNA damage repair independent of autophagy. Sci. Rep 7, 45385 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Janji B, Hasmim M, Parpal S, Berchem G & Noman MZ Firing up the cold tumors by targeting Vps34. Oncoimmunology 9, 1809936 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Arensman MD et al. Anti-tumor immunity influences cancer cell reliance upon ATG7. Oncoimmunology 9, 1800162 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Pietrocola F, Bravo-San Pedro JM, Galluzzi L & Kroemer G Autophagy in natural and therapy-driven anticancer immunosurveillance. Autophagy 13, 2163–2170 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Manic G, Obrist F, Kroemer G, Vitale I & Galluzzi L Chloroquine and hydroxychloroquine for cancer therapy. Mol. Cell Oncol 1, e29911 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Muller R Systemic toxicity of chloroquine and hydroxychloroquine: prevalence, mechanisms, risk factors, prognostic and screening possibilities. Rheumatol. Int 41, 1189–1202 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Rosenfeld MR et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 10, 1359–1368 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Compter I et al. Chloroquine combined with concurrent radiotherapy and temozolomide for newly diagnosed glioblastoma: a phase IB trial. Autophagy 17, 2604–2612 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Rojas-Puentes LL et al. Phase II randomized, double-blind, placebo-controlled study of whole-brain irradiation with concomitant chloroquine for brain metastases. Radiat. Oncol 8, 209 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Eldredge HB et al. Concurrent whole brain radiotherapy and short-course chloroquine in patients with brain metastases: a pilot trial. J. Radiat. Oncol 2, 315–321 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Clarke AJ & Simon AK Autophagy in the renewal, differentiation and homeostasis of immune cells. Nat. Rev. Immunol 19, 170–183 (2019). [DOI] [PubMed] [Google Scholar]
- 321.Vanpouille-Box C et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun 8, 15618 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Roach MC, Bradley JD & Robinson CG Optimizing radiation dose and fractionation for the definitive treatment of locally advanced non-small cell lung cancer. J. Thorac. Dis 10, S2465–S2473 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Stollings LM et al. Immune modulation by volatile anesthetics. Anesthesiology 125, 399–411 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Byrne AT et al. Interrogating open issues in cancer precision medicine with patient-derived xenografts. Nat. Rev. Cancer 17, 254–268 (2017). [DOI] [PubMed] [Google Scholar]
- 325.Kim J, Koo BK & Knoblich JA Human organoids: model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol 21, 571–584 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Demaria S et al. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int. J. Radiat. Oncol. Biol. Phys 58, 862–870 (2004). [DOI] [PubMed] [Google Scholar]
- 327.Lhuillier C, Vanpouille-Box C, Galluzzi L, Formenti SC & Demaria S Emerging biomarkers for the combination of radiotherapy and immune checkpoint blockers. Semin. Cancer Biol 52, 125–134 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Landman Y et al. Durvalumab after concurrent chemotherapy and high-dose radiotherapy for locally advanced non-small cell lung cancer. Oncoimmunology 10, 1959979 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Antonia SJ et al. Durvalumab after chemoradiotherapy in stage III non-small-cell lung cancer. N. Engl. J. Med 377, 1919–1929 (2017). [DOI] [PubMed] [Google Scholar]
- 330.Nishino M, Ramaiya NH, Hatabu H & Hodi FS Monitoring immune-checkpoint blockade: response evaluation and biomarker development. Nat. Rev. Clin. Oncol 14, 655–668 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Paz-Ares L et al. Outcomes with durvalumab by tumour PD-L1 expression in unresectable, stage III non-small-cell lung cancer in the PACIFIC trial. Ann. Oncol 31, 798–806 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Theelen W et al. Effect of pembrolizumab after stereotactic body radiotherapy vs pembrolizumab alone on tumor response in patients with advanced non-small cell lung cancer: results of the PEMBRO-RT phase 2 randomized clinical trial. JAMA Oncol. 5, 1276–1282 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Sun XS et al. Debio 1143 and high-dose cisplatin chemoradiotherapy in high-risk locoregionally advanced squamous cell carcinoma of the head and neck: a double-blind, multicentre, randomised, phase 2 study. Lancet Oncol. 21, 1173–1187 (2020). [DOI] [PubMed] [Google Scholar]
- 334.Le Tourneau C et al. Phase I trial of debio 1143, an antagonist of inhibitor of apoptosis proteins, combined with cisplatin chemoradiotherapy in patients with locally advanced squamous cell carcinoma of the head and neck. Clin. Cancer Res 26, 6429–6436 (2020). [DOI] [PubMed] [Google Scholar]
- 335.Galluzzi L, Yamazaki T & Kroemer G Linking cellular stress responses to systemic homeostasis. Nat. Rev. Mol. Cell Biol 19, 731–745 (2018). [DOI] [PubMed] [Google Scholar]
- 336.Vitale I, Galluzzi L, Castedo M & Kroemer G Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat. Rev. Mol. Cell Biol 12, 385–392 (2011). [DOI] [PubMed] [Google Scholar]
- 337.Lei G et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 30, 146–162 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Ye LF et al. Radiation-induced lipid peroxidation triggers ferroptosis and synergizes with ferroptosis inducers. ACS Chem. Biol 15, 469–484 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Lhuillier C et al. Radiotherapy-exposed CD8+ and CD4+ neoantigens enhance tumor control. J. Clin. Invest 131, e138740 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Wennerberg E et al. Immune recognition of irradiated cancer cells. Immunol. Rev 280, 220–230 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Yamazaki T & Galluzzi L Mitochondrial control of innate immune signaling by irradiated cancer cells. Oncoimmunology 9, 1797292 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Golden EB et al. Radiation fosters dose-dependent and chemotherapy-induced immunogenic cell death. Oncoimmunology 3, e28518 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Turchan WT, Pitroda SP & Weichselbaum RR Radiotherapy and immunotherapy combinations in the treatment of patients with metastatic disease: current status and future focus. Clin. Cancer Res 27, 5188 (2021). [DOI] [PubMed] [Google Scholar]
- 344.Batlle E & Massagué J Transforming growth factor-β signaling in immunity and cancer. Immunity 50, 924–940 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Mariathasan S et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Chakravarthy A, Khan L, Bensler NP, Bose P & De Carvalho DD TGF-β-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat. Commun 9, 4692 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Galluzzi L, Chan TA, Kroemer G, Wolchok JD & Lopez-Soto A The hallmarks of successful anticancer immunotherapy. Sci. Transl. Med 10, eaat7807 (2018). [DOI] [PubMed] [Google Scholar]
- 348.Jobling MF et al. Isoform-specific activation of latent transforming growth factor β (LTGF-β) by reactive oxygen species. Radiat. Res 166, 839–848 (2006). [DOI] [PubMed] [Google Scholar]
- 349.Barcellos-Hoff MH & Cucinotta FA New tricks for an old fox: impact of TGFβ on the DNA damage response and genomic stability. Sci. Signal 7, re5 (2014). [DOI] [PubMed] [Google Scholar]
- 350.Wiegman EM, Blaese MA, Loeffler H, Coppes RP & Rodemann HP TGFβ-1 dependent fast stimulation of ATM and p53 phosphorylation following exposure to ionizing radiation does not involve TGFβ-receptor I signalling. Radiother. Oncol 83, 289–295 (2007). [DOI] [PubMed] [Google Scholar]
- 351.Galluzzi L et al. Molecular definitions of autophagy and related processes. EMBO J. 36, 1811–1836 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Galluzzi L et al. Autophagy in malignant transformation and cancer progression. EMBO J. 34, 856–880 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.White E, Lattime EC & Guo JY Autophagy regulates stress responses, metabolism, and anticancer immunity. Trends Cancer 7, 778–789 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.