

Abstract
Hemophilia A and B are hereditary bleeding disorders, characterized by factor VIII or IX deficiencies, respectively. For many decades, prophylaxis with coagulation factor concentrates (replacement therapy) was the standard‐of‐care approach in hemophilia. Since the 1950s, when prophylaxis started, factor concentrates have been improved with virus inactivation and molecule modification to extend its half‐life. The past years have brought an intense revolution in hemophilia care, with the development of nonfactor therapy and gene therapy. Emicizumab is the first and only nonreplacement agent to be licensed for prophylaxis in people with hemophilia A, and real‐world data show similar efficacy and safety from the pivotal studies. Other nonreplacement agents and gene therapy have ongoing studies with promising results. Innovative approaches, like subcutaneous factor VIII and lipid nanoparticles, are in the preclinical phase. These novel agents, such as extended half‐life concentrates and emicizumab, have been available in resource‐constrained countries through the constant efforts of the World Federation of Haemophilia Humanitarian Aid Program. Despite the wide range of new approaches and therapies, the main challenge remains the same: to guarantee treatment for all. In this article, we discuss the evolution of hemophilia care, global access to hemophilia treatment, and the current and future strategies that are now under development. Finally, we summarize relevant new data on this topic presented at the ISTH 2021 virtual congress.
Keywords: blood coagulation factors, emicizumab, factor IX, factor VIII, genetic therapy, hemophilia
Essentials.
Replacement therapy has been the standard of care for hemophilia since the late 1950s.
Emicizumab, the first nonfactor therapy for hemophilia A, changed the hemophilia care scenario.
Rebalancing agents and gene therapy are new options with ongoing studies and promising results.
The main challenge remains the same: guarantee treatment for all.
1. INTRODUCTION
Hemophilia A and B are congenital X‐linked bleeding disorders caused by factor VIII (FVIII) or factor IX (FIX) deficiency, respectively. Clinical manifestation correlates with the residual endogenous clotting factor activity. People with FVIII or FIX plasma levels of <1 IU/dL are classified as having severe hemophilia and may have spontaneous bleeding events. In moderate (1‐5 IU/dL) and mild (5 to ≤40 IU/dL) hemophilia, 1 bleeding symptoms are usually associated with trauma or surgical procedures. In hemophilia, the most frequent bleeds occur in joints and muscles, resulting in chronic and progressive arthropathy with significant crippling morbidity. In addition, there is a risk of life‐threatening bleeding, such as intracranial hemorrhage, particularly in people with severe phenotype, in the absence of adequate treatment. 2
Since the 1950s, several significant achievements have happened for hemophilia A and B, resulting in considerable improvements in hemophilia care and patient quality of life (Figure 1). Among these, the main improvement is the availability of safe options to replace the missing clotting factor and restore hemostasis. The so‐called replacement therapy, using plasma‐derived or recombinant products, has been considered the cornerstone for hemophilia treatment and ensures the adoption of prophylactic therapy. 2 , 3 However, even with the progress achieved with bioengineered clotting factors, including extended half‐life (EHL) FVIII or FIX products, replacement therapy is costly, requires burdensome frequent intravenous injections, and has the risk for development of inhibitors. 2 These factors compromise adherence and access to adequate treatment for people with hemophilia worldwide.
FIGURE 1.
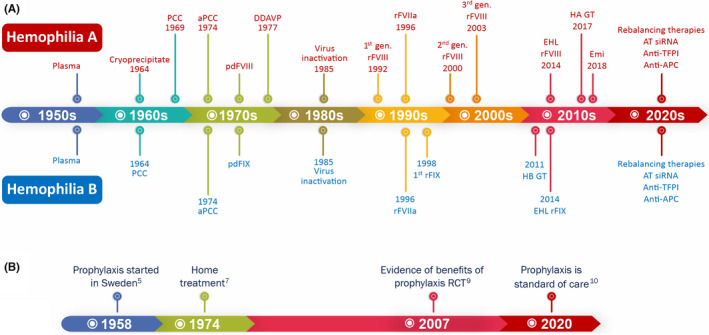
Hemophilia care evolution. (A) Evolution of hemostatic agents. (B) Improvement in hemophilia care. Abbreviations: anti‐APC, anti‐activated protein C; anti‐TFPI, anti‐tissue factor pathway inhibitor; aPCC, activated prothrombin complex concentrate; AT siRNA, small interfering RNA targeting antithrombin; DDAVP, desmopressin; EHL rFVIII, extended half‐life recombinant factor VIII concentrate; EHL rFIX, extended half‐life recombinant factor IX concentrate; Emi, emicizumab; HA GT, hemophilia A gene therapy; HB GT, hemophilia B gene therapy; PCC, prothrombin complex concentrate; pdFIX, plasma‐derived factor IX concentrate; pdFVIII, plasma‐derived factor VIII concentrate; RCT, randomized controlled trial; rFIX, recombinant factor IX concentrate; rFVIII, recombinant factor VIII concentrate
New products have recently been developed. Nonreplacement therapies, including emicizumab and rebalancing products, are transforming the approach for hemophilia treatment. These products are administered subcutaneously and are effective prophylactic options, regardless of the presence of inhibitors. More recently, adeno‐associated virus (AAV) vector‐mediated gene therapy trials for hemophilia A and B have presented promising results. 2 , 3
Despite the several new options and strategies for hemophilia care, access to treatment still represents a critical challenge for most people with hemophilia worldwide 4 and more needs to be done to guarantee adequate treatment for all people with hemophilia. In this article, we discuss the evolution of hemophilia care, the access to hemophilia treatment worldwide, and the current and future strategies that are now under development. Finally, we summarize relevant new data on this topic presented at the ISTH 2021 virtual congress.
2. IMPROVING HEMOPHILIA CARE
Until the early 1960s, the only available treatment for hemophilia was based on whole blood or fresh plasma transfusion, which was insufficient to avoid most bleeding complications. Consequently, most people with severe hemophilia died in childhood and early adulthood. 3 In 1958, Inga Nilsson and colleagues in Malmo, Sweden, were pioneers to use the regular prophylactic infusion of a local factor human fraction I‐0 (antihemophilic factor concentrate) containing FVIII to convert the bleeding phenotype temporarily from severe to moderate, resulting in a significant decrease in bleeding episodes and reduction of the impact of arthropathy, particularly when started at an early age. 5
Nevertheless, only after the advent of cryoprecipitate 6 and, later, lyophilized plasma‐derived FVIII and FIX concentrates, did home treatment become possible, a crucial step to guarantee the early treatment of the bleeding episodes and regular prophylaxis. 7 However, the first plasma‐derived clotting factor concentrates had no viral inactivation methods applied to their manufacturing process. It was only in 1985 that these methods were incorporated, reducing the risk of blood‐borne infections drastically. 3 Meanwhile, the cloning of FVIII and FIX genes (in 1982 and 1984, respectively) enabled the development of virus‐free recombinant FVIII and FIX concentrates. 8
The availability of safe FVIII and FIX products contributed to the evolution of prophylaxis as a feasible treatment modality. The benefits of prophylaxis have been recognized since the first publications. However, primary prophylaxis became the evidence‐based standard of care for hemophilia after results from a randomized clinical trial conducted by Manco‐Johnson et al. 9 In fact, prophylaxis is preferable to episodic treatment, even when using a lower dose of factor concentrates. Long‐term prophylaxis is recognized as the standard of care for all people with hemophilia with severe clinical phenotype 10 and has been proven to be effective in preventing life‐threatening bleeds and joint damage 9 (Figure 1).
3. NEW THERAPIES FOR HEMOPHILIA
3.1. Replacement therapy: Extended half‐life products
For many decades, hemophilia treatment was based on replacement therapy using plasma‐derived clotting factors or recombinant products. However, advancing technology made it possible to develop unmodified recombinant products with a standard half‐life (SHL), and clotting factor concentrates with EHL. 3
EHL clotting factor concentrates are bioengineered molecules with increased half‐life by at least 1.3 times over that of SHL FVIII or FIX concentrates. 11 Different technologies were used for the development of EHL products. These include the conjugation with polyethylene glycol and fusion with other proteins, such as albumin and or the fragment crystallizable (Fc) of IgG1. Table 1 summarizes the characteristics of the currently licensed EHL products.
TABLE 1.
Currently licensed extended half‐life recombinant factor VIII and factor IX products
| Product | Method to extend half‐life | Dosing scheme | Half‐life, h | Immunogenicity PTP | Immunogenicity PUP | References |
|---|---|---|---|---|---|---|
| Factor VIII | ||||||
| Efmoroctocog alfa (Elocta, Eloctate) | IgG1‐Fc‐fusion |
25–65 IU/kg every 3‐5 days |
19 (OSA) 20.9 (CSA) |
No inhibitor No anaphylaxis |
31.1% inhibitor 15.6% high‐titer inhibitor No anaphylaxis |
17, 21, 22, 101 |
| Rurioctocog alfa pegol (Adynovi, Adynovate) | Random PEGylation |
45 ± 5 IU/kg twice per week |
14.3–16 (OSA) |
No inhibitor No anaphylaxis |
– | 18 |
| Damoctocog alfa pegol (BAY 94‐9027, JIVI) | Site‐specific PEGylation |
30–60 IU/kg every 3‐7 days a |
19 (OSA) |
No inhibitor 1.5% hypersensibility (n = 2) 3.7% anti‐PEG Ab |
– | 20 |
| Turoctocog alfa pegol (N8‐GP, Esperoct) | Site‐specific glycoPEGylation |
50 IU/kg every 4 days 75 IU/kg per week |
19.9 (OSA) |
0.6% inhibitor 12.3% anti‐PEG Ab (>12yo) 29.4% anti‐PEG Ab (<12 yo) |
– | 19, 23, 24 |
| Factor IX | ||||||
| Eftrenonacog alfa (Alprolix) | IgG1‐Fc‐fusion |
50 IU/kg per week 100 IU/kg every 10 days |
82.1 |
No inhibitor No anaphylaxis |
3% inhibitor | 12, 13 |
| Nonacog beta pegol (N9‐GP, Refixia, Rebinyn) | Site‐specific glycoPEGylation | 40 IU/kg per week |
111 |
No inhibitor | 6.1% inhibitor | 15, 102, 103 |
| Albutrepenonacog alfa (Idelvon) | Albumin fusion |
35–50 IU/kg/wk 75 IU/kg every 10‐14 days |
101.7 |
No inhibitor 1.6% hypersensibility (n = 1) |
… | 16 |
Abbreviations: Ab, antibody; CSA, chromogenic substrate assay; OSA, one‐stage clotting assay; PTP, previously treated patients; PUP, previously untreated patients.
Dosing scheme may vary between different countries.
EHL products were developed to lead to higher factor peaks and trough levels, decreasing the frequency of intravenous injections and reducing the burden of prophylaxis. These strategies to improve pharmacokinetic (PK) parameters resulted in a significant extension of FIX concentrates half‐life, usually 3 to 5 times longer than SHL‐FIX products. 12 , 13 , 14 , 15 , 16 However, EHL–recombinant FVIII (rFVIII) products achieved only 1.5 to 1.8 times longer half‐life than SHL‐FVIII products 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 This minor improvement in PK parameters is probably due to the function of von Willebrand factor (VWF). In the circulation, FVIII needs to be bound to VWF for stabilization. Therefore, the maximum half‐life achieved by EHL‐rFVIII products is the same as VWF’s half‐life. 25
More recently, a new EHL‐rFVIII product has been under development to overcome this effect. BIVV001 (rFVIIIFc‐VWF‐XTEN) is a novel fusion protein with two different technologies. A single recombinant B‐domain deleted (BDD) FVIII protein is fused to the FVIII‐binding D′D3 domain of VWF via IgG1 dimeric Fc domain and two XTEN polypeptides. The covalent link to the VWF D′D3 domain prevents binding between the rFVIII and endogenous VWF. This strategy confers to BIVV001 a fourfold longer half‐life than SHL‐FVIII products, a benefit similar to those from the EHL‐rFIX products. 26 Until later 2021, BIVV001 was on phase 3 clinical trial and not yet commercially available.
3.2. Nonreplacement therapy
Although replacement therapy has been the standard therapeutic option to repair the hemostatic defect in hemophilia for several decades, it has limitations and challenges. That includes the burden from recurrent intravenous infusions, which compromises the adherence and, as a result, the efficacy of prophylactic treatments, even using EHL products. In this context, nonreplacement therapies could fulfill these unmet needs in hemophilia care. 2
Nonreplacement therapy is a class of products developed using strategies beyond the concept of replacing the deficient clotting factor. These novel agents aim to either restore the hemostasis using mimetic products or establish the rebalance of the hemostasis, inhibiting the anticoagulant pathways. In addition, these nonreplacement products are administered subcutaneously, overcoming the burden associated with frequent intravenous administration. The subcutaneous route can be particularly exciting when caring for young children or patients with poor venous access. These agents also address the challenging scenario of managing patients with neutralizing anti‐FVIII or anti‐FIX antibodies (inhibitors), whose bleeding episodes are more frequent and difficult to control (Table 2).
TABLE 2.
Nonreplacement therapies
| Emicizumab | Concizumab | Marstacimab | Fitusiran | SerpinPC | |
|---|---|---|---|---|---|
| Manufacturer | Roche | NovoNordisk | Pfizer | Sanofi | Centessa / ApcinteX |
| Molecule | Humanized bispecific mAB | Humanized mAB | Humanized mAB | siRNA | Modified serpin |
| Mechanism of action | Factor VIII mimetics | Anti‐TFPI | Anti‐TFPI | Reduces AT production | APC inhibitor |
| Administration route and frequency |
SC weekly, every 2 or every 4 wks |
SC daily |
SC weekly |
SC every other month |
SC every 4 wks |
| Development status | Approved | Phase 3 (recruiting) | Phase 3 (recruiting) | Multiple phase 3 trials (completed, recruiting) | Active phase 1/2 |
| Indication | HA with or without inhibitors | HA and HB, with or without inhibitors | HA and HB, with or without inhibitors | HA and HB, with or without inhibitors | HA and HB, with or without inhibitors |
| References | 27, 29, 30, 31 | 42, 43 | 41 | 44, 45 | 47 |
Abbreviations: APC, activated protein C; AT, antithrombin; HA, hemophilia A; HB, hemophilia B; mAb, monoclonal antibody; SC, subcutaneous; TFPI, tissue factor pathway inhibitor.
3.2.1. Factor VIII mimetics
In this setting, the approach to substitute rather than replace FVIII was highly successful. Emicizumab is the first nonreplacement therapy approved for prophylaxis in patients with hemophilia A with and without inhibitors. It is a humanized bispecific monoclonal antibody with binding sites to activated factor IX and factor X, mimicking FVIII in its cofactorial activity. 27 It increases thrombin generation in patients with hemophilia A, regardless of their inhibitor status. Other advantages are its long half‐life and good bioavailability, making it possible to achieve a stable hemostatic effect with subcutaneous dosing each 1, 2, or 4 weeks. 28
A series of emicizumab pivotal clinical trials (HAVEN 1‐4) have confirmed efficacy and safety for children and adults with hemophilia A, with and without inhibitors. 27 , 28 , 29 , 30 , 31 These trials have also reported a significant impact on the quality of life for both children and adults. Efficacy and safety in the pediatric population were also confirmed in HOHOEMI trial, which included children as young as 4 months old. 32 Reports from different hemophilia centers have confirmed the safety reported in the pivotal studies but observed a higher risk of breakthrough bleeds in older people. 33
Prophylaxis with emicizumab in previously untreated people is still under debate, while the incidence and consequences of anti‐FVIII inhibitors are unknown in this scenario. Many trials are on the way to try to address this open issue. HAVEN 7 is now recruiting and will evaluate early prophylaxis with emicizumab in children under 12 months of age with hemophilia A without inhibitors. The Hemophilia Inhibitor Prevention Trial is a phase 3 randomized, open‐label study that will compare inhibitor data from previously untreated people under prophylaxis with an EHL‐rFVIII concentrate or emicizumab (NCT04303559). Another trial will also assess inhibitor data from primary prophylaxis with emicizumab and a concomitant low dose of simoctocog alfa, a recombinant FVIII concentrate (NCT04030052). Both trials are now recruiting.
Emicizumab also had a positive impact on the challenging scenario of periprocedural management. In the past few years, many reports with real‐world data have shown the safety of managing minor and major procedures in people with and without inhibitors. 33 , 34
Currently, we observe a learning curve for both patients and clinicians has advanced since emicizumab was licensed for people with hemophilia A. This disruptive approach to hemophilia also changed the way bleeds are identified and treated. Therefore, data on real‐world use, including bleeding and periprocedural management, is essential to build knowledge outside clinical trials. Societies and groups of experts have also issued guidelines on bleeding treatment, perioperative management, and laboratory surveillance for emicizumab. 35 , 36 , 37 , 38
Other unanswered questions come from scenarios where emicizumab probably does not fully substitute FVIII. One of them is bone metabolism and health. The EmiMK study will evaluate bone health in patients under emicizumab (NCT04131036). Prophylaxis for sports is another intriguing scenario to be addressed in the STEP study, where emicizumab and FVIII concentrates will be compared for prophylaxis for provoked bleeding (NCT05022459).
Emicizumab use in other populations is also currently under evaluation in clinical trials. HAVEN 6 is designed to evaluate prophylaxis with emicizumab in people with mild and moderate hemophilia A without inhibitors (NCT04158648). Another ongoing trial will address the efficacy of prophylaxis with emicizumab in acquired hemophilia A (NCT04188639).
Since emicizumab was approved, new activated FVIII mimetic bispecific antibodies have been developed, such as BS‐027125 (Bioverativ, Waltham, MA, USA) 39 and Mim8 (Novo Nordisk, Bagsvaerd, Denmark). Preclinical analysis of Mim8, including in vitro assays with FVIII deficient human plasma and in vivo assay using a hemophilia A mouse model, suggest that this new bispecific antibody could be more potent than emicizumab. 40
3.2.2. Rebalancing therapies
This new class of products restores the hemostatic capacity in the blood, mainly inhibiting different natural anticoagulant pathways, establishing the hemostatic balance even in the absence of FVIII or FIX and the presence of inhibitors. At present, no rebalancing agents are licensed, and their use is limited to clinical trials (Table 2).
Tissue factor pathway inhibitor (TFPI) is an anticoagulant protein that can reversibly inhibit activated factor X, either by direct inhibition or by complexing with activated factor X and then inhibiting tissue factor and activated factor VII. Two monoclonal antibodies with anti‐TFPI activity are now in phase 3 clinical studies: concizumab and marstacimab. The first agent may be administered subcutaneously, at 0.15 or 0.25 mg/kg, once daily. Marstacimab is evaluated with a single loading dose of 300 mg and then 150 mg or 300 mg weekly, depending on the person’s bleeding phenotype. Updates for both molecules were presented at ISTH 2021 congress (see ISTH 2021 Virtual Congress below). 41 , 42 , 43
Fitusiran, an antithrombin small interfering RNA, is now an ongoing phase 3 trial. In the randomized, open‐label ATLAS‐INH study (NCT03417102), 38 people with inhibitor (29 with hemophilia A and 9 with hemophilia B) with a mean age of 26.8 years (± 9.8) received once‐monthly fixed doses of fitusiran 80 mg subcutaneously. A significant reduction of bleeding episodes was observed when compared to 19 people with inhibitor (16 with hemophilia A and 3 with hemophilia B) with a mean age of 28.4 years (± 11.1) receiving on‐demand bypassing agents (BPAs), with 65.8% of patients with zero bleeding events in the fitusiran arm, compared to 5.3% in the on‐demand BPA group. In addition, two people (5.3%) receiving fitusiran were reported with venous thrombosis events, and fitusiran was discontinued for one person. 44 In the phase 3 ATLAS‐A/B study (NCT03417245), people with severe hemophilia A or B without inhibitor, previously treated with on‐demand factor concentrate, were randomly assigned to receive a fixed dose of fitusiran 80 mg once monthly or continuous with on‐demand treatment. As expected, a significant reduction was observed in the fitusiran arm, but only 50.6% (40 of 79 people who completed 9 months of fitusiran prophylaxis) had zero treated bleeds during the study period. 45
SerpinPC is a highly specific activated protein C inhibitor, currently on phase 1/2a trial. 46 In a press release in September 2021, Centessa Pharmaceuticals and its subsidiary ApcinteX Limited announced some results from its phase 2a proof‐of‐concept trial (AP‐0101). Twenty‐three people with severe hemophilia and on‐demand therapy were enrolled in this trial (19 with hemophilia A and 4 with hemophilia B, both without inhibitors). Three doses were evaluated (0.3, 0.6, and 1.2 mg/kg) with subcutaneous administration every 4 weeks, and the primary outcome was assessed after 24 weeks of follow‐up. Results presented a median 88% reduction in all bleeds (from 36.0 to 4.4) and a median 94% decrease in spontaneous joint bleeds (from 21.1 to 2.2) for the highest dose. No venous thromboembolism or other concerning adverse effects were observed. Two people developed anti‐drug antibodies (ADAs) but displayed no apparent impacts on their bleeding phenotype. 47
3.3. Gene therapy for hemophilia
Hemophilia has always been an attractive candidate for gene therapy. For decades, hemophilia gene therapy research groups have been dedicated to finding the ideal strategy for achieving lasting plasma factor levels with a one‐time treatment. Current gene therapy clinical trials for hemophilia are using the same strategy. They are based on intravenous administration of the liver‐directed delivery of FVIII or FIX transgene using recombinant nonintegrating AAV vectors (Figure 2A).
FIGURE 2.
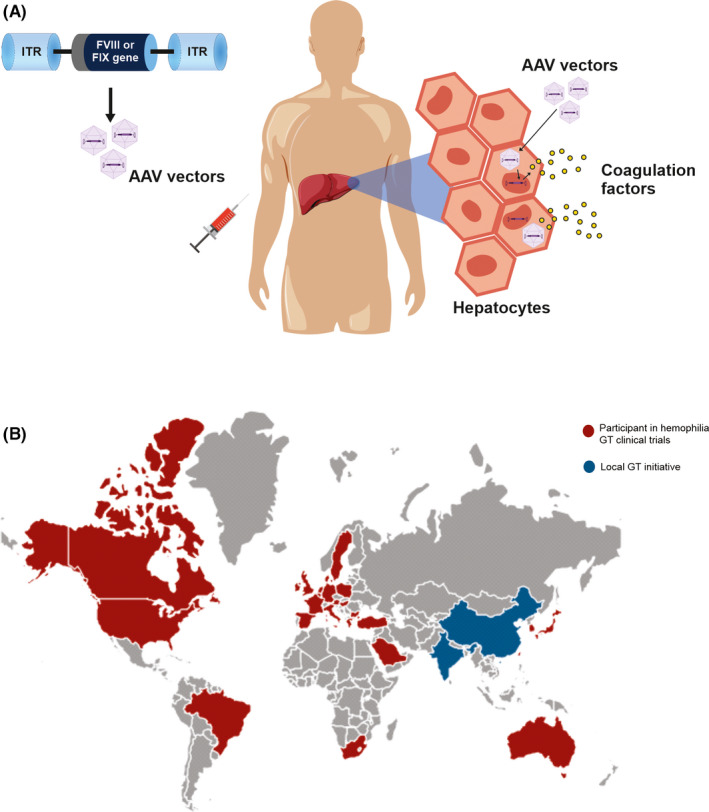
Gene therapy for hemophilia. (A) Adeno‐associated virus (AAV) vector‐mediated gene therapy: factor VIII (FVIII) or factor IX (FIX) transgene is delivered to hepatocytes by an AAV vector. After efficient transduction, liver cells will express factor VIII or IX. (B) Worldwide distribution of participants in gene therapy trials. Abbreviations: GT, gene therapy; ITR, inverted terminal repeats
The first AAV liver‐directed gene therapy clinical trial for hemophilia B was critical to establish the current successful strategies. 48 This study used an AAV2 vector administered into the hepatic artery and revealed essential issues related to AAV vector immunogenicity. One was associated with the preexisting immunity due to the presence of neutralizing antibodies (NAbs) to the AAV vector capsid. NAbs are AAV serotype specific and may impair the vector transduction efficacy. A plausible strategy to avoid this negative effect is to exclude people with anti‐AAV vector antibodies. Another critical issue is related to the AAV capsid‐mediated cellular immune response. Transduced cells may have a transient expression of AAV vector capsids peptides in its surface, which can induce a CD8 T‐cell response and the destruction of transduced hepatocytes, clinically recognized by an increase in liver transaminase (alanine aminotransferase [ALT]) and/or decreasing the transgene expression.
In 2011, the first successful liver‐directed AAV vector‐based gene therapy for hemophilia B was reported, with two critical strategies for AAV immunogenicity management. 49 , 50 The first was the exclusion of patients with anti‐AAV NAbs. In addition, immunosuppression with corticosteroids was used in response to ALT elevation to control AAV capsid‐mediated cellular immune response.
AAV vector‐mediated gene therapy trials for hemophilia A and B clinical have shown promising results. Some participants have presented meaningful expression of FVIII 51 , 52 , 53 , 54 , 55 , 56 or FIX, 57 , 58 , 59 , 60 , 61 , 62 , 63 shifting their phenotype from severe to mild or even achieving normal factor levels after a single vector injection. Table 3 shows the ongoing hemophilia A and B gene therapy clinical trials.
TABLE 3.
Ongoing AAV‐base gene therapy clinical trials for hemophilia A and B
| Program (Sponsor) | Product | Dose (vg/kg) | Status | Reference |
|---|---|---|---|---|
| Hemophilia A trials | ||||
| BMN 270, GENEr8‐1 (Biomarin) |
valoctocogene roxaparvovec rAAV5‐BDDFVIII |
4 × 1013 6 × 1013 |
Phase 1/2: active (n = 15) Phase 3: active (n = 134) |
51, 52, 53, 54, 67 |
| SB‐525, ALTA and AFFINE (Sangamo, Pfizer) |
PF−07055480 giroctocogene fitelparvovec rAAV2/6‐hFVIII |
9 × 1011 2 × 1012 1 × 1013 3 × 1013 |
Phase 1/2: active (n = 11) Phase 3: recruiting |
63, 104 |
| GO8 (UCL) | AAV2/8‐HLP‐FVIII‐V3 |
6 × 1011 2 × 1012 6 × 1012 |
Phase 1: recruiting | ‐ |
| SPK8011 (Spark) | rAAV‐SPK200‐BDDFVIIIco |
5 × 1011 1 × 1012 1.5 × 1012 2 × 1012 |
Phase 1/2: active (n = 18) |
55 |
| BAY 2599023, DTX201 (Bayer, Ultragenyx) |
BAY 2599023 rAAVhu37‐hFVIIIco |
5 × 1012 1 × 1013 2 × 1013 |
Phase 1/2: recruiting (n = 8) | 59 |
| GS001 (Institute of Hematology & Blood Diseases Hospital, China) | GS001 |
2 × 1012 6 × 1012 2 × 1013 |
Phase 1/2: recruiting | |
| Hemophilia B trials | ||||
| BENEGENE‐2 (Spark, Pfizer) |
PF−06838435 fidanacogene elaparvovec rAAV‐SPK100‐hFIX‐Padua |
5 × 1011 |
Phase 2: active (n = 15) Phase 3: recruiting |
|
|
AMT 061, HOPE‐B (uniQure) |
etranacogene dezaparvovec AAV5‐Padua hFIX |
2 × 1013 |
Phase 2b: active (n = 3) Phase 3: active (n = 54) |
61, 62 |
| FLT‐180a, B‐AMAZE (UCL and Freeline) |
verbrinacogene setparvovec AAV2/S3‐FRE1‐Ti‐FIXco1 |
3.84 × 1011 6.4 × 1011 8.32 × 1011 1.28 × 1012 7.7 × 1011 a |
Phase 1/2: active (n = 10) | 105 |
For trials with more than one dosing scheme, doses considered therapeutic are in bold.
Abbreviations: AAV, adeno‐associated virus; UCL, University College London; vg/kg, vector genomes per kilogram of body weight.
B‐AMAZE study results suggested that the dose of 7.7 × 1011 vg/kg was optimal, after evaluation of the four doses as mentioned.
Despite the encouraging results of the recent hemophilia gene therapy clinical trials, several issues remain unclear and unresolved. The variability in FVIII and FIX expression levels among the clinical trial participants and the unpredictable responses are observed in both hemophilia A 52 , 53 , 54 , 55 , 64 and B gene therapy trials. 59 , 60 , 61 , 62 , 63 , 65 , 66 Furthermore, the cellular immune response is AAV vector dose dependent, and the ideal immunosuppressive therapy still needs to be determined. In addition, safety concerns due to integration and potential malignancy risk will need long‐term follow‐up. 57 However, long‐term durability seems to be another bold challenge, particularly for hemophilia A gene therapy. 52 , 67
After initial phase 1 and 2 trials confirming safety and exploring efficacy outcomes, more extensive phase 3 clinical trials for hemophilia A and B have been conducted in many centers, even outside North America and Europe. Centers in Australia, Japan, Brazil, South Africa, Turkey, Taiwan, Saudi Arabia, and other countries also include participants in hemophilia gene therapy clinical trials (Figure 2B). This widespread participation of hemophilia centers worldwide may help achieve the target patient number faster, despite the difficulties associated with AAV‐seroprevalence limitations. In addition, although it is early to predict, the involvement of several centers around the world in prelicensed clinical trials may help in the future to establish and improve infrastructure and knowledge for other potential market sites, increasing access to this possible therapeutic alternative.
Also, local gene therapy programs for hemophilia have been carried out in Japan, China, and India. 68 Another engaging initiative has been organized by St. Jude Children’s Research Hospital, a phase 2 feasibility trial of AAV‐mediated hemophilia B gene therapy in low‐ and middle‐income countries. This program has two stages. In stage 1, vector infusion will happen at St. Jude, in Memphis, Tennessee, and patients will be monitored at their local sites. In stage 2, infusion and monitoring will happen at the low and middle‐income countries’ sites. This initiative expects to prove the principle of the feasibility to perform and give access to gene therapy for patients in resource‐constrained settings. 68 , 69
4. GLOBAL DISTRIBUTION OF PROCOAGULANT PRODUCTS
Despite the significant advances in hemophilia care in the last decades, access to adequate diagnosis and treatment is still considerably unbalanced.
FVIII usage per capita is a critical parameter that has been used to estimate access to clotting factor products for years. Although the consumption of new nonfactor products will require consideration when assessing this parameter, it remains a helpful tool to evaluate the local availability of hemophilia treatment. It is suggested that 1 IU of FVIII per capita is the minimum amount of FVIII concentrates to guarantee the long‐term survival of people with hemophilia. 70 On the other hand, according to the European consensus, the minimum to provide the standard‐of‐care treatment is 4 IU of FVIII, and 0.5 IU of FIX concentrates per capita. 71 According to the World Federation of Haemophilia (WFH) annual global survey, in 2020, 78% of FVIII products were consumed in countries from the Americas and Europe, which encompass 31% of the world's hemophilia population. This disproportion is even more pronounced if we consider the gross national income, based upon World Bank economic ratings. Only 12.3% of the FVIII consumed in 2020 was destined to treat 63% of the world’s hemophilia population living in low‐ and lower‐middle‐income countries. 72
4.1. Novel therapies around the globe
Nowadays, the use of EHL products is increasing. In Ireland, this is the only FVIII and FIX concentrates used. 72 The latest WFH Annual Global Survey reported the current scenario for hemophilia therapies, including EHL factor concentrates. In 2020, 23 countries purchased EHL‐rFVIII concentrates for their patients, using 1 604 990 663 units of this product. EHL‐rFIX products are available in 18 countries, with a total consumption of 565 695 806 units in 2020. The Humanitarian Aid Program has distributed 40 673 500 units of EHL FVIII and FIX products to 37 and 18 countries, respectively. 72 Recently, Krumb et al 73 published data on the adoption of emicizumab in Europe. Of the 144 contacted hemophilia treatment centers, 46 responded to the survey, representing 21 countries. Emicizumab data were available on 43 centers. This agent was available for people with inhibitors in all of them, but approval for people without inhibitors was more restricted (in 37 centers, 88.1%).
In the WFH 2020 Annual Global Survey, 62 countries reported that emicizumab was available for people with or without inhibitors. 72 According to the manufacturer, emicizumab is now licensed for prophylaxis in people with hemophilia A with inhibitors in >100 countries and for people without inhibitors in >80 countries. As a result, >10 000 people worldwide are on prophylaxis with emicizumab. 74
In countries where emicizumab is approved for people both with and without inhibitors, it has been widely prescribed and is now one of the leading agents in use. Few agents have been incorporated into clinical practice at such a fast pace. According to Hermans and Makris, 75 25% to 35% of people with hemophilia A without inhibitors are under prophylaxis with emicizumab in Israel, the United Kingdom, and Belgium. In a real‐world cost estimate in the United States, emicizumab seems to be economically favorable compared to other agents for prophylaxis in people with or without inhibitors. 76
Few data are available regarding the use of new therapies in Latin America. In Brazil, until 2021, there were no EHL products distributed through the Inherited Coagulopathies Program from the Ministry of Health. Prophylaxis with emicizumab has just become available for people with hemophilia A with inhibitors who failed the immune tolerance induction (ITI) protocol. 77 Chile has a similar scenario, with no EHL products and emicizumab provided for people with inhibitors who failed ITI (Dr Verónica Soto, personal communication, June 2021). Both Argentina and Colombia have a few people on EHL products, and emicizumab is available for people with inhibitors and selected people without inhibitors (Drs Daniela Neme and Adriana Linares, personal communication, June 2021). In Mexico, EHL factor concentrates are expected to be available by 2022, and emicizumab is provided for people with inhibitors, particularly those who failed ITI (Dr Jaime Garcia Chavez, personal communication, June 2021). According to the 2020 WFH Annual Global Survey, Ecuador has some people on EHL products; Bolivia and Venezuela are participants of the Humanitarian Aid Program, and Uruguay has only SHL factor concentrates. 72
4.2. WFH Humanitarian Aid Program
In 1996, WFH started the Humanitarian Aid Program, an important initiative to provide access to treatment for people with inherited bleeding disorders in resource‐constrained countries. Nevertheless, until 2014, the donations of procoagulant products were limited and sporadic, and its use was restricted to emergencies since there were not enough factor concentrates to provide sustained on‐demand treatment. 4
However, in 2014, pharmaceutical companies Sanofi (formerly Biogen and Bioverativ) and Sobi announced the donation of 1 billion IU of EHL FVIII and FIX concentrates over 10 years. 4 As a result of this expressive increase in donated products through the WFH Humanitarian Aid Program, it was possible to expand the goals and number of people and countries that benefit from this initiative. Since 2016, donated EHL products have provided low‐dose prophylaxis for people from resource‐constrained countries. In 2016, the number of people on prophylaxis with donated products rose from 0 to 852, with 458 people <10 years old. 4 In 2020, the cumulative number of people on prophylaxis with donated products was 1804 (including 1145 people <10 years old). 78 The Humanitarian Aid Program helped expand the use of EHL products worldwide for prophylaxis 79 and surgical procedures. 4 , 80
Several pharmaceutical companies continue to help increase access to treatment through the Humanitarian Aid Program. Despite the challenges and logistical restrictions with the COVID‐19 pandemic, the program was not interrupted and was crucial to guarantee access to treatment for people in 69 countries in 2020. 78
In 2019, Roche also became a contributor to the program and, in 2020, started to donate emicizumab. 78 According to Dr Assad Haffar, the director of the Humanitarian Aid Program, in June 2021, 633 people with hemophilia A from 26 countries were on prophylaxis with donated emicizumab, including 356 (56.2%) people <12 years old. Among these patients on emicizumab, 227 (35.9%) are people with hemophilia A with inhibitors. The remaining 406 (64.1%) people with hemophilia A who do not have inhibitors but met the criteria to receive prophylaxis with emicizumab were frequent bleeders (annualized bleeding rate [ABR] ≥6) or history of life‐threatening bleeding episodes (Dr Assad Haffar, personal communication, June 2021).
5. ISTH 2021 VIRTUAL CONGRESS
5.1. Nonreplacement therapy
Almost 4 years after its first approval, real‐world data elucidated emicizumab’s performance outside controlled clinical trials. The ISTH 2021 virtual congress had reports from multiple cohorts, including people with and without inhibitors, with efficacy and safety results comparable to those from the original trials. 81 , 82 , 83 , 84 , 85 , 86 , 87 , 88 , 89
Alternative dosing regimens have also been proposed. Fischer et al 90 reported the experience of prescribing only entire emicizumab vials for maintenance in people with severe hemophilia A, with adjusted intervals between doses. They reported that 79% of people had zero bleeds during follow‐up.
Also, in the Netherlands, Bukkems et al 91 reported an alternative simulated dosing regimen, with a target median emicizumab plasma concentration between 40 and 60 µg/mL at steady state. The authors reported that costs were saved in up to 60% of the virtual population and could reach a median of almost 60 000 euros.
Another interesting report of a small cohort (n = 3) with a reduced‐dose regimen for emicizumab from Malaysia has shown satisfactory clinical outcomes. With a mean dose of 1.8 mg/kg every 4 weeks, only one bleeding event was reported after up to 132 weeks of follow‐up. Patients reported improved quality of life and could dismiss walking aids. 92
Regarding the rebalancing agents, updates on efficacy and safety for concizumab and marstacimab were presented at the ISTH 2021 virtual congress.
Astermark et al 42 presented data from concizumab in people with hemophilia A/B with inhibitors (explorer 4) and hemophilia A without inhibitors (explorer 5). Subjects in the explorer4 trial were randomly assigned to either on‐demand recombinant activated factor VII followed by concizumab or straight to concizumab throughout the whole study period. A total of 61 subjects were enrolled, and 51 had completed the extension period when the data were presented. As for efficacy, the ABR was 6.4 for hemophilia A without inhibitors, 3.8 for hemophilia A with inhibitors, and 6.2 for hemophilia B with inhibitors. Joint ABR was 5.2, 2.7, and 3.8 for each group, respectively. Interestingly, 15 of the 61 subjects enrolled developed concizumab ADAs. ADAs were transient and presented with a low titer in most cases, with no clinical impact.
An open‐label study with marstacimab prophylaxis was presented by Mahlangu et al. 41 Twenty subjects with hemophilia A or B, with or without inhibitors, were enrolled. No ADAs or serious adverse events related to marstacimab were reported. As for efficacy, when comparing data from before and after prophylaxis with marstacimab, the ABR decreased from 20.2 to 1.5 in the higher‐dose (300 mg weekly) arm, and 17.4 to 2.7 in the lower‐dose (150 mg weekly) group.
5.2. Gene therapy
5.2.1. Gene therapy for hemophilia A
Results from the phase 1/2 trial with valoctocogene roxaparvovec (AAV5‐hFVIII‐SQ) were updated after participants completed a 5‐year follow‐up period. There was a sustained reduction in ABR for both dose cohorts (4 × 1013 vector genomes per kilogram of body weight [vg/kg] and 6 × 1013 vg/kg). All participants remained off FVIII prophylaxis 5 years after infusion. Mean FVIII activities measured by chromogenic assay were 5.6 and 11.6 IU/dL for the lower‐ and higher‐vector dose, respectively. 51
The GENEr8‐1 phase 3 trial reported results after 134 people with severe hemophilia A were dosed with valoctocogene roxaparvovec and followed up for at least 52 weeks. All participants received a single infusion of 6 × 1013 vg/kg. At weeks 49 to 52, the mean FVIII activity for 132 participants was 42.9 IU/dL (chromogenic assay). Among 17 participants with a minimum follow‐up of 2 years, the mean FVIII activity was 24.4 IU/dL at week 104. Regarding clinical outcomes, 80% of the participants reported zero treated bleeds 4 weeks after infusion. The ABR decreased by 83.8% from baseline after week 4, with statistical superiority over FVIII concentrate prophylaxis. ALT elevation was reported in 115 participants (85.8%), and 106 (79.1%) received corticosteroids due to this ALT elevation. The average duration of corticosteroid use was 33 months, and three serious adverse events related to corticosteroid use were reported. 53
George et al., 55 shared updated results on a phase 1/2 trial for SPK‐8011, after 18 participants with hemophilia A were treated with four different dosing protocols (5 × 1011 vg/kg, 1 × 1012 vg/kg, 2 × 1012 vg/kg, and 1.5 × 1012 vg/kg). Only 17 participants had completed 1 year of follow‐up. Of this group, two participants completely lost the FVIII expression. The remaining 15 participants had a mean FVIII activity of 11 ± 6.8% of the normal value (one‐stage FVIII assay) and had a 91.2% reduction in ABR. Transaminitis was also a significant adverse event in this trial. Corticosteroids were used prophylactically by 5 participants and on‐demand by 10 participants.
Results from the phase 1/2 trial with BAY 2599023 (AAVhu37.hFVIIIco) were also presented. So far, eight people were infused with three different dosing schemes (0.5 × 1013 vg/kg, 1 × 1013 vg/kg, and 2 × 1013 vg/kg). Investigators have stated that BAY 2599023 delivered protective FVIII levels sustained over time. Follow‐up duration ranged from 12 to 100 weeks. ALT elevation was observed in four people, all managed with corticosteroids. Two additional patients from the higher‐dose cohort were prescribed prophylactic corticosteroids. 59
5.2.2. Gene therapy for hemophilia B
Gene therapy for hemophilia B was also addressed at the ISTH 2021 virtual congress. UniQure reported results for three clinical trials. The first was a phase 1/2 trial for AMT‐060 (AAV5‐hFIX). Ten patients were equally divided between two different dosing cohorts (5 × 1012 vg/kg and 2 × 1013 vg/kg) and were followed for 5 years. Mean FIX activity was 5.2% in the lower‐dose cohort and 7.2% in the second group. ABR and FIX consumption declined compared to baseline, and all participants remained prophylaxis free. No major safety concerns were observed, with no sustained liver enzyme elevation. An enhanced construct, with FIX Padua as the therapeutic transgene, was later developed and is now the product used in the ongoing phase 3 trial. 60
Following AMT‐060, the updates for a phase 2b trial with etranacogene dezaparvovec (AAV5‐Padua hFIX, AMT‐061) were presented, following completion of a 30‐month evaluation period. Unlike other gene therapy trials, preexisting anti‐AAV5 neutralizing antibodies were assessed but did not represent an exclusion criterion for this study. Mean FIX activity was 44.2% (range, 36%‐52%) 2 years after infusion, and no relationship between response and anti‐AAV5 neutralizing antibody status was observed. 61
HOPE‐B is a phase 3 study that evaluates the efficacy and safety of etranacogene dezaparvovec (AAV5‐Padua hFIX, AMT‐061). So far, 54 patients have been dosed and completed a minimum follow‐up of 6 months. Mean FIX activity was 41.3 IU/dL at 26 weeks in participants without anti‐AAV5 neutralizing antibodies (n = 31) and 32.7 IU/dL in those with positive anti‐AAV5 neutralizing antibodies (n = 23). No significant correlation was found between anti‐AAV5 neutralizing antibody titer and FIX activity, bleeding phenotype, or safety. 62 Investigators also shared some data on the HOPE‐B cohort participant diagnosed with hepatocellular carcinoma over a year after AMT‐061 infusion. Tumor sample analysis showed that AAV integration was very infrequent, and after an extensive analysis, hepatocellular carcinoma occurrence was deemed unrelated to gene therapy. 93
6. FUTURE DIRECTIONS
The following years promise to continue the revolution in the hemophilia care scenario, as many new therapies are under development or ongoing preclinical and early‐phase clinical trials.
SIG‐001 consists of 1.5 mm alginate spheres encapsulating genetically modified allogeneic cells engineered to express human FVIII. The capsules shield the cells from the host’s immune system, and the conjugation to alginate biomaterial avoids pericapsular fibrotic overgrowth. In preclinical trials, SIG‐001 could produce functional FVIII in a dose‐dependent manner and correct the bleeding phenotype of hemophilia A mice. After no concerns regarding safety or toxicology were observed in mice and nonhuman primates, a phase 1/2 clinical trial was announced. SIG‐001 would be administered in the peritoneal cavity through laparoscopy in 18 patients. 94 However, on July 9, 2021, the company announced that the study was put on clinical hold by the US Food and Drug Administration after a participant developed inhibitors to FVIII. Three participants had been dosed so far.
Currently, there are preclinical studies on subcutaneous agents for hemophilia care. The first is FVIII‐ABD, a subcutaneous FVIII that has shown good availability (ranging from 15.3% to almost 50%, depending on the animal model studied). 95 Another strategy is the coadministration of recombinant FVIII and recombinant VWF fragments containing the D3 domain (VWF‐12 and VWF‐13). This strategy resulted in a good availability (up to 18.5%) for subcutaneous human recombinant FVIII, with slow absorption and prolonged half‐life. 96
As previously discussed, AAV vectors are widely used in gene transfer clinical trials for hemophilia A and B. Among its many benefits, AAV vectors are replication defective and target many different cells and tissues. Nevertheless, the unpredictable postinfusion response and the slow but progressive loss of expression over the years are still issues to be addressed. In this scenario, lentiviral vectors emerge as interesting candidates for gene transfer with long‐term expression. Since lentivirus integrates into the host’s cell chromatin, its genome is maintained after each cellular duplication. Preclinical studies have shown multiyear transgene liver expression in mice and dogs with hemophilia A. 97 , 98 More recently, some improvements in lentiviral vectors have been studied. Modifications in the vector surface to decrease T‐cell–mediated immunogenicity and increased resistance to phagocytosis have led to higher FIX expression, reaching values up to 300% of normal. 99
Beyond viral vectors, lipid nanoparticles are also being studied for gene transfer. Chen et al 100 have described the use of FVIII‐encoding mRNA, packaged into liver‐directed lipid nanoparticles. After a single intravenous injection, hemophilia A mice have presented with a variable range of FVIII activity and maintained therapeutic FVIII levels up to 5 to 7 days after infusion.
7. SUMMARY
Remarkable improvement in hemophilia care was achieved during the past decades. The availability of safe and effective clotting factor concentrates was crucial for prophylaxis feasibility, starting at a young age for people with severe hemophilia. New bioengineered clotting factors, such as EHL products, helped ameliorate the burden of frequent intravenous administration. However, the risk for inhibitors continues to be the major complication of hemophilia replacement therapy.
More recently, the development of nonreplacement therapies represents a unique alternative in the effective prevention of bleeding, regardless of the presence of inhibitors. Currently, emicizumab has become the preferable option for prophylaxis for people with hemophilia A with inhibitors, with several advantages for people with noninhibitor hemophilia A. These new therapies have also been used in resource‐constrained countries through the WFH Humanitarian Aid Program and contributed to increasing the chance of elective surgeries and the number of people on prophylaxis in low‐income countries.
In addition, gene therapy clinical trials for hemophilia have shown promising results, and this modality of treatment may become an attractive alternative for hemophilia management, even in resource‐constrained countries.
Thus, in the near future, one of the recurrent challenges for hemophilia management will be to define the most appropriate treatment according to the needs of each person and local treatment availability.
RELATIONSHIP DISCLOSURE
MCO received research support from BioMarin, Novo Nordisk, Pfizer, Sanofi, Roche, and Takeda; participated as speaker and/or in advisory boards sponsored by Bayer, BioMarin, Novo Nordisk, Pfizer, Roche, Sanofi, and Takeda; and participated as a council member of Novo Nordisk Haemophilia Foundation and as a grant reviewer supported by Grifols. GGY participated as a speaker sponsored by BioMarin, Novo Nordisk, Pfizer, and Roche.
AUTHOR CONTRIBUTIONS
The authors co‐wrote the manuscript and approved the final version.
ACKNOWLEDGMENTS
We thank Michel Antonio Bueno Moraes for the illustrations support and Dr Assad Haffar from the World Federation of Haemophilia for contributing data regarding the WFH Humanitarian Aid Program.
Ozelo MC, Yamaguti‐Hayakawa GG. Impact of novel hemophilia therapies around the world. Res Pract Thromb Haemost. 2022;6:e12695. doi: 10.1002/rth2.12695
Handling Editor: Dr Johnny Mahlangu
This manuscript was developed based on an invited State of the Art Session presented by Prof. Margareth Ozelo at the XXIX Congress of the International Society on Thrombosis and Haemostasis, July 17–21, 2021, Philadelphia, PA, USA.
Contributor Information
Margareth C. Ozelo, Email: margaret@unicamp.br.
Gabriela G. Yamaguti‐Hayakawa, @gabiyamaguti.
REFERENCES
- 1. Blanchette VS, Key NS, Ljung LR, et al. Definitions in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost. 2014;12(11):1935‐1939. [DOI] [PubMed] [Google Scholar]
- 2. Mancuso ME, Mahlangu JN, Pipe SW. The changing treatment landscape in haemophilia: from standard half‐life clotting factor concentrates to gene editing. Lancet. 2021;397(10274):630‐640. [DOI] [PubMed] [Google Scholar]
- 3. Mannucci PM. Hemophilia therapy: the future has begun. Haematologica. 2020;105(3):545‐553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pierce GF, Haffar A, Ampartzidis G, et al. First‐year results of an expanded humanitarian aid programme for haemophilia in resource‐constrained countries. Haemophilia. 2018;24(2):229‐235. [DOI] [PubMed] [Google Scholar]
- 5. Nilsson IM, Berntorp E, Lofqvist T, Pettersson H. Twenty‐five years’ experience of prophylactic treatment in severe haemophilia A and B. J Intern Med. 1992;232(1):25‐32. [DOI] [PubMed] [Google Scholar]
- 6. Pool JG, Shannon AE. Production of high‐potency concentrates of antihemophilic globulin in a closed‐bag system. N Engl J Med. 1965;273(27):1443‐1447. [DOI] [PubMed] [Google Scholar]
- 7. Le Quesne B, Britten MI, Maragaki C, Dormandy KM. Home treatment for patients with haemophilia. Lancet. 1974;2(7879):507‐509. [DOI] [PubMed] [Google Scholar]
- 8. Pipe SW. Recombinant clotting factors. Thromb Haemost. 2008;99(5):840‐850. [DOI] [PubMed] [Google Scholar]
- 9. Manco‐Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357(6):535‐544. [DOI] [PubMed] [Google Scholar]
- 10. Srivastava A, Santagostino E, Dougall A, et al. WFH Guidelines for the Management of Hemophilia. Haemophilia. 2020;26(Suppl 6):1‐158. [DOI] [PubMed] [Google Scholar]
- 11. Mahlangu J, Young G, Hermans C, Blanchette V, Berntorp E, Santagostino E. Defining extended half‐life rFVIII‐A critical review of the evidence. Haemophilia. 2018;24(3):348‐358. [DOI] [PubMed] [Google Scholar]
- 12. Powell JS, Pasi KJ, Ragni MV, et al. Phase 3 study of recombinant factor IX Fc fusion protein in hemophilia B. N Engl J Med. 2013;369(24):2313‐2323. [DOI] [PubMed] [Google Scholar]
- 13. Nolan B, Klukowska A, Shapiro A, et al. Final results of the PUPs B‐LONG study: evaluating safety and efficacy of rFIXFc in previously untreated patients with hemophilia B. Blood Adv. 2021;5(13):2732‐2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Collins PW, Young G, Knobe K, et al. Recombinant long‐acting glycoPEGylated factor IX in hemophilia B: a multinational randomized phase 3 trial. Blood. 2014;124(26):3880‐3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chan AK, Alamelu J, Barnes C, et al. Nonacog beta pegol (N9‐GP) in hemophilia B: First report on safety and efficacy in previously untreated and minimally treated patients. Res Pract Thromb Haemost. 2020;4(7):1101‐1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Santagostino E, Martinowitz U, Lissitchkov T, et al. Long‐acting recombinant coagulation factor IX albumin fusion protein (rIX‐FP) in hemophilia B: results of a phase 3 trial. Blood. 2016;127(14):1761‐1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mahlangu J, Powell JS, Ragni MV, et al. Phase 3 study of recombinant factor VIII Fc fusion protein in severe hemophilia A. Blood. 2014;123(3):317‐325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Konkle BA, Stasyshyn O, Chowdary P, et al. Pegylated, full‐length, recombinant factor VIII for prophylactic and on‐demand treatment of severe hemophilia A. Blood. 2015;126(9):1078‐1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Giangrande P, Andreeva T, Chowdary P, et al. Clinical evaluation of glycoPEGylated recombinant FVIII: Efficacy and safety in severe haemophilia A. Thromb Haemost. 2017;117(2):252‐261. [DOI] [PubMed] [Google Scholar]
- 20. Reding MT, Ng HJ, Poulsen LH, et al. Safety and efficacy of BAY 94–9027, a prolonged‐half‐life factor VIII. J Thromb Haemost. 2017;15(3):411‐419. [DOI] [PubMed] [Google Scholar]
- 21. Nolan B, Mahlangu J, Pabinger I, et al. Recombinant factor VIII Fc fusion protein for the treatment of severe haemophilia A: Final results from the ASPIRE extension study. Haemophilia. 2020;26(3):494‐502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Konigs C, Ozelo MC, Dunn A, et al. Final results of PUPs A‐LONG study: evaluating safety and efficacy of rFVIIIFc in previously untreated patients with haemophilia A. Res Pract Thromb Haemost. 2020;4(Suppl1):8. [Google Scholar]
- 23. Chowdary P, Carcao M, Holme PA, et al. Fixed doses of N8‐GP prophylaxis maintain moderate‐to‐mild factor VIII levels in the majority of patients with severe hemophilia A. Res Pract Thromb Haemost. 2019;3(3):542‐554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Meunier S, Alamelu J, Ehrenforth S, et al. Safety and efficacy of a glycoPEGylated rFVIII (turoctocog alpha pegol, N8‐GP) in paediatric patients with severe haemophilia A. Thromb Haemost. 2017;117(9):1705‐1713. [DOI] [PubMed] [Google Scholar]
- 25. Pipe SW, Montgomery RR, Pratt KP, Lenting PJ, Lillicrap D. Life in the shadow of a dominant partner: the FVIII‐VWF association and its clinical implications for hemophilia A. Blood. 2016;128(16):2007‐2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Konkle BA, Shapiro AD, Quon DV, et al. BIVV001 Fusion Protein as Factor VIII Replacement Therapy for Hemophilia A. N Engl J Med. 2020;383(11):1018‐1027. [DOI] [PubMed] [Google Scholar]
- 27. Oldenburg J, Mahlangu JN, Kim B, et al. Emicizumab Prophylaxis in Hemophilia A with Inhibitors. N Engl J Med. 2017;377(9):809‐818. [DOI] [PubMed] [Google Scholar]
- 28. Callaghan MU, Negrier C, Paz‐Priel I, et al. Long‐term outcomes with emicizumab prophylaxis for hemophilia A with or without FVIII inhibitors from the HAVEN 1–4 studies. Blood. 2021;137(16):2231‐2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mahlangu J, Oldenburg J, Paz‐Priel I, et al. Emicizumab Prophylaxis in Patients Who Have Hemophilia A without Inhibitors. N Engl J Med. 2018;379(9):811‐822. [DOI] [PubMed] [Google Scholar]
- 30. Pipe SW, Shima M, Lehle M, et al. Efficacy, safety, and pharmacokinetics of emicizumab prophylaxis given every 4 weeks in people with haemophilia A (HAVEN 4): a multicentre, open‐label, non‐randomised phase 3 study. Lancet Haematol. 2019;6(6):e295‐e305. [DOI] [PubMed] [Google Scholar]
- 31. Young G, Liesner R, Chang T, et al. A multicenter, open‐label phase 3 study of emicizumab prophylaxis in children with hemophilia A with inhibitors. Blood. 2019;134(24):2127‐2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shima M, Nogami K, Nagami S, et al. A multicentre, open‐label study of emicizumab given every 2 or 4 weeks in children with severe haemophilia A without inhibitors. Haemophilia. 2019;25(6):979‐987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McCary I, Guelcher C, Kuhn J, et al. Real‐world use of emicizumab in patients with haemophilia A: Bleeding outcomes and surgical procedures. Haemophilia. 2020;26(4):631‐636. [DOI] [PubMed] [Google Scholar]
- 34. Jimenez‐Yuste V, Rodriguez‐Merchan EC, Matsushita T, Holme PA. Concomitant use of bypassing agents with emicizumab for people with haemophilia A and inhibitors undergoing surgery. Haemophilia. 2021;27(4):519‐530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Castaman G, Santoro C, Coppola A, et al. Emergency management in patients with haemophilia A and inhibitors on prophylaxis with emicizumab: AICE practical guidance in collaboration with SIBioC, SIMEU, SIMEUP, SIPMeL and SISET. Blood Transfus. 2020;18(2):143‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Collins PW, Liesner R, Makris M, et al. Treatment of bleeding episodes in haemophilia A complicated by a factor VIII inhibitor in patients receiving Emicizumab. Interim guidance from UKHCDO Inhibitor Working Party and Executive Committee. Haemophilia. 2018;24(3):344‐347. [DOI] [PubMed] [Google Scholar]
- 37. Coppola A, Castaman G, Santoro RC, et al. Management of patients with severe haemophilia a without inhibitors on prophylaxis with emicizumab: AICE recommendations with focus on emergency in collaboration with SIBioC, SIMEU, SIMEUP, SIPMeL and SISET. Haemophilia. 2020;26(6):937‐945. [DOI] [PubMed] [Google Scholar]
- 38. Escuriola‐Ettingshausen C, Auerswald G, Konigs C, et al. Optimizing the management of patients with haemophilia A and inhibitors in the era of emicizumab: Recommendations from a German expert panel. Haemophilia. 2021;27(3):e305‐e313. [DOI] [PubMed] [Google Scholar]
- 39. Leksa NC, Aleman MM, Goodman AG, Rabinovich D, Peters R, Salas J. Intrinsic differences between FVIIIa mimetic bispecific antibodies and FVIII prevent assignment of FVIII‐equivalence. J Thromb Haemost. 2019;17(7):1044‐1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ostergaard H, Lund J, Greisen PJ, et al. A factor VIIIa‐mimetic bispecific antibody, Mim8, ameliorates bleeding upon severe vascular challenge in hemophilia A mice. Blood. 2021;138(14):1258‐1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mahlangu J, Lamas JL, Morales JC, et al. Long‐term Safety and Efficacy of the Anti‐TFPI Monoclonal Antibody Marstacimab in Patients with Severe Hemophilia A or B: Results from a Phase 2 Long‐term Treatment Study. Res Pract Thromb Haemost. 2021;5(Suppl 2):82. [Google Scholar]
- 42. Astermark J, Angchaisuksiri P, Benson G, et al. Longer‐term Efficacy and Safety of Concizumab Prophylaxis in Hemophilia A and Hemophilia A/B with Inhibitors: Results from the Main and Extension Parts of Concizumab Phase 2 Trials. Res Pract Thromb Haemost. 2021;5(Suppl 2):390.33870024 [Google Scholar]
- 43. Shapiro A, Cepo K, Tønder SM, Young G, Jímenez‐Yuste V. Safety and Efficacy of Concizumab Prophylaxis Following a Switch from rFVIIa on‐demand Treatment: Sub‐analysis Results from the Phase 2 Explorer4 Trial in Patients with Hemophilia A or B with Inhibitors. Res Pract Thromb Haemost. 2021;5(Suppl 2):381. [Google Scholar]
- 44. Young G, Srivastava A, Kavakli K, et al. Efficacy and Safety of Fitusiran Prophylaxis, an siRNA Therapeutic, in a Multicenter Phase 3 Study (ATLAS‐INH) in People with Hemophilia A or B, with Inhibitors (PwHI). Blood. 2021;138(Suppl 1):4.34236429 [Google Scholar]
- 45. Srivastava A, Rangarajan S, Kavakli K, et al. Fitusiran, an investigational siRNA therapeutic targeting antithrombin for the treatment of hemophilia: first results from a phase 3 study to evaluate efficacy and safety in people with hemophilia A or B without inhibitors (ATLAS‐A/B). Blood. 2021;138(Suppl 2):LBA‐3. [Google Scholar]
- 46. Polderdijk SG, Adams TE, Ivanciu L, Camire RM, Baglin TP, Huntington JA. Design and characterization of an APC‐specific serpin for the treatment of hemophilia. Blood. 2017;129(1):105‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Centessa Pharmaceuticals announces positive topline data from proof‐of‐concept study of SerpinPC in severe hemophilia A and B patients not on prophylaxis 2021. https://investors.centessa.com/news‐releases/news‐release‐details/centessa‐pharmaceuticals‐announces‐positive‐topline‐data‐proof/. Accessed November 27, 2021.
- 48. Manno CS, Pierce GF, Arruda VR, et al. Successful transduction of liver in hemophilia by AAV‐Factor IX and limitations imposed by the host immune response. Nat Med. 2006;12(3):342‐347. [DOI] [PubMed] [Google Scholar]
- 49. Nathwani AC, Reiss UM, Tuddenham EG, et al. Long‐term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med. 2014;371(21):1994‐2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nathwani AC, Tuddenham EG, Rangarajan S, et al. Adenovirus‐associated virus vector‐mediated gene transfer in hemophilia B. N Engl J Med. 2011;365(25):2357‐2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pasi K, Rangarajan S, Robinson TM, et al. Hemostatic response is maintained for up to 5 years following treatment with valoctocogene roxaparvovec, an AAV5‐hFVIII‐SQ gene therapy for severe hemophilia A. Res Pract Thromb Haemost. 2021;5(Suppl 2):90. [Google Scholar]
- 52. Pasi KJ, Laffan M, Rangarajan S, et al. Persistence of haemostatic response following gene therapy with valoctocogene roxaparvovec in severe haemophilia A. Haemophilia. 2021;27(6):947‐956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ozelo MC, Mahlangu J, Pasi K, et al. Efficacy and Safety of Valoctocogene Roxaparvovec Adeno‐associated Virus Gene Transfer for Severe Hemophilia A: Results from the Phase 3 GENEr8‐1 Trial. Res Pract Thromb Haemost. 2021;5(Suppl 2):89. [Google Scholar]
- 54. Ozelo MC, Mahlangu J, Pasi KJ, et al. Valoctocogene Roxaparvovec Gene Therapy for Hemophilia A. N Engl J Med. 2022;386(11):1013–1025. [DOI] [PubMed] [Google Scholar]
- 55. George LA, Monahan PE, Eyster ME, et al. Multiyear Factor VIII Expression after AAV Gene Transfer for Hemophilia A. N Engl J Med. 2021;385(21):1961‐1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Visweshwar N, Harrington TJ, Leavitt AD, et al. Updated Results of the Alta Study, a Phase 1/2 Study of Giroctocogene Fitelparvovec (PF‐07055480/SB‐525) Gene Therapy in Adults with Severe Hemophilia a. Blood. 2021;138(Suppl 1):564. [Google Scholar]
- 57. George LA, Ragni MV, Rasko JEJ, et al. Long‐Term Follow‐Up of the First in Human Intravascular Delivery of AAV for Gene Transfer: AAV2‐hFIX16 for Severe Hemophilia B. Mol Ther. 2020;28(9):2073‐2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Samelson‐Jones BJ, Sullivan SK, Rasko JEJ, et al. Follow‐up of More Than 5 Years in a Cohort of Patients with Hemophilia B Treated with Fidanacogene Elaparvovec Adeno‐Associated Virus Gene Therapy. Blood. 2021;138(Suppl 1):3975. [Google Scholar]
- 59. Pipe SW, Hay C, Sheehan J, et al. Evolution of AAV vector gene therapy is ongoing in hemophilia. Will the unique features of BAY 2599023 address the outstanding needs? Res Pract Thromb Haemost. 2021;5(Suppl 2):490. [Google Scholar]
- 60. Miesbach W, Leebek FWG. Five year Data Confirms Stable FIX Expression and Sustained Reductions in Bleeding and factor IX use Following AMT‐ 060 Gene Therapy in Adults with Severe or Moderate‐severe Hemophilia B. Res Pract Thromb Haemost. 2021;5(Suppl 2):90. [Google Scholar]
- 61. Gomez E, Giermasz A, Castaman G, et al. Etranacogene dezaparvovec (AAV5‐Padua hFIX variant, AMT‐061), an enhanced vector for gene transfer in adults with severe or moderate‐severe hemophilia B: 2.5 year data from a phase 2b trial. Res Pract Thromb Haemost. 2021;5(Suppl 2):487. [Google Scholar]
- 62. Leebeek FWG, Miesbach W, Recht M, et al. Clinical Outcomes in Adults with Hemophilia B with and without Pre‐existing Neutralizing Antibodies to AAV5: 6 Month Data from the Phase 3 Etranacogene Dezaparvovec HOPE‐B Gene Therapy Trial. Res Pract Thromb Haemost. 2021;5(Suppl 2):92. [Google Scholar]
- 63. Chowdary P, Shapiro S, Makris M, et al. Factor IX Expression within the Normal Range Prevents Spontaneous Bleeds Requiring Treatment Following FLT180a Gene Therapy in Patients with Severe Hemophilia B: Long‐Term Follow‐up Study of the B‐Amaze Program. Blood. 2021;138(Suppl 1):3967. [Google Scholar]
- 64. Rangarajan S, Walsh L, Lester W, et al. AAV5‐Factor VIII Gene Transfer in Severe Hemophilia A. N Engl J Med. 2017;377(26):2519‐2530. [DOI] [PubMed] [Google Scholar]
- 65. George LA, Sullivan SK, Giermasz A, et al. Hemophilia B Gene Therapy with a High‐Specific‐Activity Factor IX Variant. N Engl J Med. 2017;377(23):2215‐2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Konkle BA, Walsh CE, Escobar MA, et al. BAX 335 hemophilia B gene therapy clinical trial results: potential impact of CpG sequences on gene expression. Blood. 2021;137(6):763‐774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pasi KJ, Rangarajan S, Mitchell N, et al. Multiyear Follow‐up of AAV5‐hFVIII‐SQ Gene Therapy for Hemophilia A. N Engl J Med. 2020;382(1):29‐40. [DOI] [PubMed] [Google Scholar]
- 68. Reiss UM, Zhang L, Ohmori T. Hemophilia gene therapy‐New country initiatives. Haemophilia. 2021;27(Suppl 3):132‐141. [DOI] [PubMed] [Google Scholar]
- 69. Pierce GF. Uncertainty in an era of transformative therapy for haemophilia: Addressing the unknowns. Haemophilia. 2021;27(Suppl 3):103‐113. [DOI] [PubMed] [Google Scholar]
- 70. Evatt BL. Observations from Global Survey 2001: an emerging database for progress. Haemophilia. 2002;8(2):153‐156. [DOI] [PubMed] [Google Scholar]
- 71. Giangrande PLF, Peyvandi F, O'Mahony B, et al. Kreuth IV: European consensus proposals for treatment of haemophilia with coagulation factor concentrates. Haemophilia. 2017;23(3):370‐375. [DOI] [PubMed] [Google Scholar]
- 72.Annual Global Survey 2020. World Federation of Hemophilia. https://www1.wfh.org/publications/files/pdf‐2045.pd. Accessed November 25, 2021.
- 73. Krumb E, Fijnvandraat K, Makris M, et al. Adoption of emicizumab (Hemlibra(R)) for hemophilia A in Europe: Data from the 2020 European Association for Haemophilia and Allied Disorders survey. Haemophilia. 2021;27(5):736‐743. [DOI] [PubMed] [Google Scholar]
- 74.roche.com [Internet]. Roche ‐ Hemlibra (emicizumab). Available from: https://www.roche.com/products/product‐details.htm?productId=4889d5d4‐c688‐4db5‐8805‐12a57b1c95a1. Accessed November 25, 2021.
- 75. Hermans C, Makris M. Disruptive technology and hemophilia care: The multiple impacts of emicizumab. Res Pract Thromb Haemost. 2021;5(4):e12508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Samelson‐Jones BJ, Guelcher C, Kuhn J, et al. Real‐world cost estimates of initiating emicizumab in US patients with haemophilia A. Haemophilia. 2021;27(4):591‐598. [DOI] [PubMed] [Google Scholar]
- 77. Coordenação Geral de Sangue e Hemoderivados ‐ CGSH/DAET/SAES/MS & Coordenação de gestão de protocolos clínicos e diretrizes terapêuticas ‐ CPCDT/CGGTS/DGITIS/SCTIE/MS. Protocolo de uso do emicizumabe para tratamento de indivíduos com hemofilia A e inibidores do fator VIII refratários ao tratamento de imunotolerância [Internet]. Brasília, 2021. Available from: http://conitec.gov.br/images/Consultas/Relatorios/2021/20210708_Protocolo_de_uso_emicizumabe_CP_64.pdf. Accessed December 2, 2021.
- 78. World Federation of Hemophilia . WFH Humanitarian Aid Program ‐ Impact Report 2020 [Internet]. World Federation of Hemophilia, 2021 [cited 2021 Nov 25]. Available from: https://www1.wfh.org/publications/files/pdf‐1889.pdf. Accessed November 25, 2021.
- 79. Lambert C, Meite N, Sanogo I, Lobet S, Hermans C. Feasibility and outcomes of low‐dose and low‐frequency prophylaxis with recombinant extended half‐life products (Fc‐rFVIII and Fc‐rFIX) in Ivorian children with hemophilia: two‐year experience in the setting of World Federation of Haemophilia Humanitarian Aid Programme. Haemophilia. 2021;27(1):33‐40. [DOI] [PubMed] [Google Scholar]
- 80. El Ekiaby M, Haffar A. Low‐dose surgical prophylaxis: optimization of use of World Federation of Hemophilia Humanitarian Aid donated clotting factor concentrates to developing countries. Haemophilia. 2020;26(Suppl 3):11‐15. [DOI] [PubMed] [Google Scholar]
- 81. Barg AA, Budnik I, Avishai E, et al. Emicizumab prophylaxis and monitoring: real world data. Res Pract Thromb Haemost. 2021;5(Suppl 2):81.33537532 [Google Scholar]
- 82. Campaniço S, Rodrigues F, Pereira A, Rocha E, Cevadinha G, Catarino C. Is zero bleeds in pwh an achievable goal? 3 years of emicizumab in persons with hemophilia A and inhibitors in a Portuguese hemophilia treatment centers. Res Pract Thromb Haemost. 2021;5(Suppl 2):409. [Google Scholar]
- 83. Fujii T, Yamasaki N, Inoue T, Fujii T. A bleed suppression efficacy of emicizumab in patients with hemophilia A whose therapies switched from conventional prophylaxis. Res Pract Thromb Haemost. 2021;5(Suppl 2):388. [Google Scholar]
- 84. Garza MTP, Aranda AA, Madrazo RM, Duron NP, González CSG. Real world experience with emicizumab with hemophilia A treated in private practice in Mexico. Res Pract Thromb Haemost. 2021;5(Suppl 2):476. [Google Scholar]
- 85. Heine SI, Graf N. Real life use of emicizumab in pediatric patients without inhibitors. Res Pract Thromb Haemost. 2021;4(Suppl 1):429. [Google Scholar]
- 86. Larkin N, Singleton E, Benson J, et al. Real‐world experience in introducing emicizumab prophylaxis for adults with haemophilia A without inhibitors. Res Pract Thromb Haemost. 2021;5(Suppl 2):408. [Google Scholar]
- 87. Toscano MN, Lopez‐Santiago N, Pedroza MDLG, et al. Decreased annual bleeding rate in paediatric patients with hemophilia a and inhibitors treated with emicizumab vs bypassing agents experience at the National Institute of Pediatrics, Mexico City, Mexico. Res Pract Thromb Haemost. 2021;5(Suppl 2):504. [Google Scholar]
- 88. Wall C, Xiang H, Palmer BP, et al. Efficacy and safety of emicizumab prophylaxis in severe haemophilia A without Inhibitors: a report from the UK Haemophilia Centre Doctors’ Organisation (UKHCDO). Res Pract Thromb Haemost. 2021;5(Suppl 2):388. [Google Scholar]
- 89. Windyga J, Zdziarska J, Stefanska‐Windyga E, et al. Emicizumab in the treatment of adult haemophilia patients real‐world data. Res Pract Thromb Haemost. 2021;5(Suppl 2):457. [Google Scholar]
- 90. Fischer K, Donners AA, Urbanus RT, et al. Real‐world experience of emicizumab treatment using entire vials only. Res Pract Thromb Haemost. 2021;5(Suppl 2):508. [Google Scholar]
- 91. Bukkems LH, Fischer K, Kremer‐Hovinga I, et al. Emicizumab dosing in children and adults with hemophilia A: simulating a user‐friendly and cost‐efficient regimen. Res Pract Thromb Haemost. 2021;5(Suppl 2):398. [DOI] [PubMed] [Google Scholar]
- 92. Tang ASO, Leong TS, Ko CT, Chew LP. Efficacy of reduced‐dose emicizumab in haemophilia A with inhibitors: real world experience in East Malaysia. Res Pract Thromb Haemost. 2021;5(Suppl 2):428. [Google Scholar]
- 93. Schmidt M, Foster GR, Coppens M, et al. Liver safety case report from the phase 3 HOPE‐B Gene Therapy Trial in adults with hemophilia B. Res Pract Thromb Haemost. 2021;5(Suppl 2):93. [Google Scholar]
- 94. Shapiro AD, Konkle BA, Croteau SE, et al. First‐in‐human phase 1/2 clinical trial of SIG‐001, an innovative shielded cell therapy platform, for hemophilia Α. Blood. 2020;136(Suppl 1):8.32614959 [Google Scholar]
- 95. Herbener P, Schüttrumpf J, Daufenbach J, Kistner S. A next generation recombinant factor viii for subcutaneous hemophilia A prophylaxis. Res Pract Thromb Haemost. 2020;4(Suppl 1):579. [Google Scholar]
- 96. Vollack‐Hesse N, Oleshko O, Werwitzke S, Solecka‐Witulska B, Kannicht C, Tiede A. Recombinant VWF fragments improve bioavailability of subcutaneous factor VIII in hemophilia A mice. Blood. 2021;137(8):1072‐1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Cantore A, Nair N, Della Valle P, et al. Hyperfunctional coagulation factor IX improves the efficacy of gene therapy in hemophilic mice. Blood. 2012;120(23):4517‐4520. [DOI] [PubMed] [Google Scholar]
- 98. Cantore A, Ranzani M, Bartholomae CC, et al. Liver‐directed lentiviral gene therapy in a dog model of hemophilia B. Sci Transl Med. 2015;7(277):277‐328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Cantore A, Naldini L. WFH State‐of‐the‐art paper 2020: in vivo lentiviral vector gene therapy for haemophilia. Haemophilia. 2021;27(Suppl 3):122‐125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Chen CY, Tran DM, Cavedon A, et al. Treatment of hemophilia a using factor VIII messenger RNA lipid nanoparticles. Mol Ther Nucleic Acids. 2020;20:534‐544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Young G, Mahlangu J, Kulkarni R, et al. Recombinant factor VIII Fc fusion protein for the prevention and treatment of bleeding in children with severe hemophilia A. J Thromb Haemost. 2015;13(6):967‐977. [DOI] [PubMed] [Google Scholar]
- 102. Young G, Collins PW, Colberg T, et al. Nonacog beta pegol (N9‐GP) in haemophilia B: a multinational phase III safety and efficacy extension trial (paradigm4). Thromb Res. 2016;141:69‐76. [DOI] [PubMed] [Google Scholar]
- 103. Tiede A, Abdul‐Karim F, Carcao M, et al. Pharmacokinetics of a novel extended half‐life glycoPEGylated factor IX, nonacog beta pegol (N9‐GP) in previously treated patients with haemophilia B: results from two phase 3 clinical trials. Haemophilia. 2017;23(4):547‐555. [DOI] [PubMed] [Google Scholar]
- 104. Konkle BA, Stine K, Visweshwar N, et al. Updated follow‐up of the Alta Study, a phase 1/2, open label, adaptive, dose‐ranging study to assess the safety and tolerability of SB‐525 gene therapy in adult patients with severe hemophilia A. Blood. 2019;134(Suppl 1):2060. [Google Scholar]
- 105. Chowdary P, Shapiro S, Makris M, et al. A novel adeno associated virus (AAV) gene therapy (FLT180a) achieves normal FIX activity levels in severe hemophilia B (HB) patients (B‐AMAZE Study). Res Pract Thromb Haemost. 2020;4(Suppl 2):1. [Google Scholar]