

Abstract
Objectives:
Cigarette smoking is an established risk factor for pancreatic ductal adenocarcinoma (PDAC). In this project we investigated the effect of smoking and the role of histone deacetylase 4 (HDAC4) in PDAC invasion and metastasis.
Methods:
Cells were treated with 4-(methylnitro-s-amino)-1-(3-pyridyl)-1-butanone (NNK) and cigarette smoke extract (CSE) and the mRNA levels of HDACs were measured by real-time polymerase chain reaction. Invasion was measured using the Matrigel Invasion Assay. Syngeneic PDAC mice were treated with NNK and metastasis measured. Human PDAC primary and metastatic tissues were analyzed by immunohistochemistry.
Results:
Levels of HDAC4 mRNA were increased by smoking. Smoking compounds significantly promoted invasion of cancer cells and promoted metastasis of PDAC cells to different organs including the liver and the lung, whereas inhibition of HDAC4 prevented this effect. The effect of HDAC4 inhibition on preventing smoking-induced metastasis was greater in the liver compared to the lung. We found that HDAC4 is highly expressed in primary and metastatic PDAC tumors.
Conclusions:
We found that HDAC4 is the only HDAC induced by smoking among all HDACs analyzed. We found that smoking promotes invasion and metastasis of PDAC cells through a mechanism that involves HDAC4 and that HDAC4 is a promising target for preventing PDAC metastasis.
Keywords: pancreatic cancer, HDAC4, metastasis
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is an aggressive disease with limited effective therapies.1,2 Approximately 55,440 people were diagnosed with PDAC in 2018 and about 44,330 patients died from the disease in the US alone.3
The five-year survival rate for PDAC patients is estimated at 7%, making it the third most common cancer-related cause of death in the US even though it is twelfth in incidence.2,4 A major reason for this devastating outcome is the high metastatic rate of the disease combined with a lack of effective strategies for early detection and treatment of metastasis. Nearly 80% of the diagnosed patients have regional or distal metastasis. At autopsy, it was found that over 80% of PDAC patients have liver metastasis and nearly 45% have lung metastasis.5
Smoking, alcoholism, diabetes and obesity play major roles in the promotion of PDAC.6–8 However, little is known about how these risk factors promote the disease, especially metastasis. Cigarette smoking is an established major risk factor for PDAC.9,10 It increases the risk of PDAC up to 6-fold dependent on the intensity and the duration of smoking.10–12 Smoking promotes metastasis in multiple cancers13,14 and speeds up pancreatic cancer development and progression,15,16 including liver and lung metastases. The reason for certain tumor types to metastasize to the liver and the lung at the early stage of the disease is poorly understood.
Post-translational and epigenetic changes were shown to be involved in cancer development.17 Histone deacetylases (HDACs) are critical regulators of fundamental cellular events such as cell cycle, differentiation, and apoptosis.18 It was shown that HDAC3 promotes early PDAC in mice and that HDAC1 regulates epithelial to mesenchymal transition (EMT) in pancreatic cancer cells by directly interacting and stabilizing Zeb1 transcription factor.19,20 Epithelial to mesenchymal transition is an early and critical process of cell transformation during cancer cell metastasis. Many members of the HDAC family play a role in cancer promotion and metastasis; however, the mechanisms involved by each HDAC protein seem to be different. Previous reports showed a significant role played by inflammatory cells in the promotion of PDAC in both humans and animal models and there is increasing evidence that pro-cancer M2-like macrophages play a major role in the promotion of various cancers including pancreatic cancer.21,22 Our data indicate that inhibition of HDACs decreases the level of pro-cancer M2-like macrophages and PDAC progression.23
Accumulating evidence show a role of HDAC4 in cancer promotion. Published data indicate that HDAC4 is highly expressed in many cancers and that it promotes tumor growth and is associated with worse outcome in esophageal, colon, glioblastoma, ovarian, and gastric cancers.24–28 Even though the role of HDAC4 in pancreatic cancer invasion is not known, HDAC4 was shown to promote invasion in osteosarcoma.29
Thus far, the role of HDAC4 in the pathogenesis of pancreatic cancer is still unclear, and the expression of HDAC4 in pancreatic cancer metastatic tissues has not been reported. Hence, understanding the role of HDAC4 in pancreatic cancer cell invasion and metastasis is of great significance to curing the disease.
We have shown before that the HDAC class I and II inhibition20 decreased pancreatic lesion formation in a PDAC mouse model using the Pdx-Cre; LSL-Kras (KC) mice and that a dual inhibitor for HDAC (class I and II) and GSK-3β prevents cancer development and inhibits metastasis in KPC mice.23 Histone deacetylases of the classes I and II has 10 members. Therefore, it is important to determine which member of the HDAC family is involved in PDAC progression, especially in the process of metastasis. In this study, we investigated the role of HDACs in the context of smoking induced metastasis. We investigated the effect of smoking on the pancreatic cancer cells’ ability to metastasize to the liver and the lung, the two major PDAC metastasis destinations. We used metastatic syngeneic PDAC mice. We also investigated the role of HDAC4 in mediating the smoking induced PDAC invasion and metastasis.
MATERIAL AND METHODS
Reagents
Smoking compound NNK was purchased from ChemSyn Science Laboratories (Lenexa, Kan) and cigarette smoke extract (CSE) from Murty Pharmaceuticals Inc (Lexington, Ky). Suberoylanilide Hydroxamic Acid (Saha) was purchased from Cayman Chemical Company (Ann Arbor, Mich), Lmk-235 from Santa Cruz Biotechnology Inc (Santa Cruz, Calif), and HDAC4 antibody from Cell Signaling Technology (Danvers, Mass). All other chemicals were from Sigma Aldrich (St. Louis, Mo).
Cell Culture Experiments
The poorly differentiated human pancreatic cancer MIA PaCa-2 cell line and the moderately differentiated BxPC-3 human cell line were obtained from the American Type Culture Collection (Manassas, Va). MIA PaCa-2 cells were cultured in 1/1 D-MEM/F-12 medium (GIBCO Invitrogen Corporation, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS), L-glutamine (final concentration 4mM), and 1% antibiotics/antimycotics solution (Omega Scientific, Tarzana, Calif). BxPC-3 cells were cultured in RPMI-1640 supplemented with 10% FBS and 1% of antibiotic/antimycotics solution. Cells were maintained at 37°C in a humidified atmosphere containing 5% CO2 and were used between passages 2 and 10.
Real-time Quantitative Polymerase Chain Reaction
Total RNA was extracted using Trizol from Thermo Fisher (Rockford, Ill). Reverse transcription reaction was carried out using High-Capacity Reverse Transcription Kit from Thermo Fisher. We used real-time quantitative polymerase chain reaction (RT-PCR) for quantifying mRNA levels using the iTaq Universal SYBR Green Supermix (Bio-Rad, Hercules, Calif) and BioRad cfx96 platform according to the manufacturer’s protocol. Gene expression levels were normalized to that of GAPDH. Primers were purchased from Integrated DNA Technologies (IDT, Coralville, Iowa). The sequences of primers used for RT-PCR were as follow: h-HDAC1-F; CCAAGTACCACAGCGATGAC, h-HDAC1-R; TGGACAGTCCTCACCAACG, h-HDAC2-F; TGAAGGAGAAGGAGGTCGAA, h-HDAC2-R; GGATTTATCTTCTTCCTTAACGTCTG, h-HDAC3-F; CACCATGCCAAGAAGTTTGA, h-HDAC3-R; CCCGAGGGTGGTACTTGAG, h-HDAC4-F; GGCCCACCGGAATCTGAAC, h-HDAC4-R; GAACTCTGGTCAAGGGAACTG, h-HDAC5-F; TCTTGTCGAAGTCAAAGGAGC, h-HDAC5-R; GAGGGGAACTCTGGTCCAAAG, h-HDAC6-F; AAGAAGACCTAATCGTGGGACT, h-HDAC6-R; GCTGTGAACCAACATCAGCTC, h-HDAC7-F; GGCGGCCCTAGAAAGAACAG, h-HDAC7-R; CTTGGGCTTATAGCGCAGCTT, h-HDAC8-F; TCGCTGGTCCCGGTTTATATC, h-HDAC8-R; TACTGGCCCGTTTGGGGAT, h-HDAC9-F; AGTAGAGAGGCATCGCAGAGA, h-HDAC9-R; GGAGTGTCTTTCGTTGCTGAT, h-HDAC10-F; CAGTTCGACGCCATCTACTTC, h-HDAC10-R; CAAGCCCATTTTGCACAGCTC, h-GAPDH-F; CCAGGTGGTCTCCTCTGACTTCAACA, h-GAPDH-R; AGGGTCTCTCTCTTCCTCTTGTGCTC.
Invasion Measurements
Invasion was assessed by using the Matrigel Invasion Chamber Assay (Corning, Bedford, Mass).
Immunohistochemistry
Tissues were fixed with formalin and paraffin embedded. Staining was performed as shown before.23 Human tissue samples were received from Nebraska University through the Rapid autopsy program and were used at the Cedars-Sinai Medical Center (CSMC) with IRB protocol number 50715.
Syngeneic Metastatic PDAC Mice
The 2 months old B6.129J mice were injected with NNK (25 mg/kg) or saline intra-peritoneally (i.p.) once a week for 4 weeks. 1×106 UN-KPC961-Luc cells23,30 were then injected into the spleen of mice followed by a treatment for 6 weeks with i.p. injections of pan-HDAC I/II inhibitor Saha (50 mg/kg), HDAC4 inhibitor Lmk-235 (3 mg/Kg), or saline 3 times per week. Mice were then killed and organs collected for analysis. All animal studies were performed according to the guidence of IACUC and after the approval of IACUC Protocol # 5970 at the Cedars-Sinai Medical Center. All mice were housed in Association for Assessment and Accreditation of Laboratory Animal Care-accredited facilities and used in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals.
Statistics
Statistical analysis of the studies were performed by using Student’s t test, one-way ANOVA, or Fisher’s exact test with GraphPad Prism 6 (GraphPad, San Diego, Calif). Statistical significance was achieved when P value was smaller than 0.05. The data showed is the result section is from at least 3 different experiments.
RESULTS
Smoking Promotes Transcription of HDAC4
Our previously published data showed that pan-inhibition of HDACs 1 to 10 prevented PDAC progression induced by smoking.20 Therefore, we first, determined the effect of smoking compounds NNK and CSE on mRNA level of HDACs 1 to 10. We found that NNK and CSE had no significant effect on HDACs 1 to 3 and induced a significant decrease in HDACs 5 to 10 in both pancreatic cancer cell lines MIA PaCa-2 and BxPC-3 (Figs. 1A, B). However, the only HDAC mRNA increased by smoking compounds in both cell lines was HDAC4 (Figs. 1A, B). In addition to HDACs, we measured the levels of sirtuins and found that sirtuins 1, 2, 3, and 4 were significantly decreased in the presence of NNK and CSE; however, none of the sirtuins mRNA level was increased by both smoking compounds in both cell lines (Fig. 1A, B). Sirtuin 7 mRNA level was increased by CSE but not with NNK (Fig. 1A, B).
FIGURE 1.
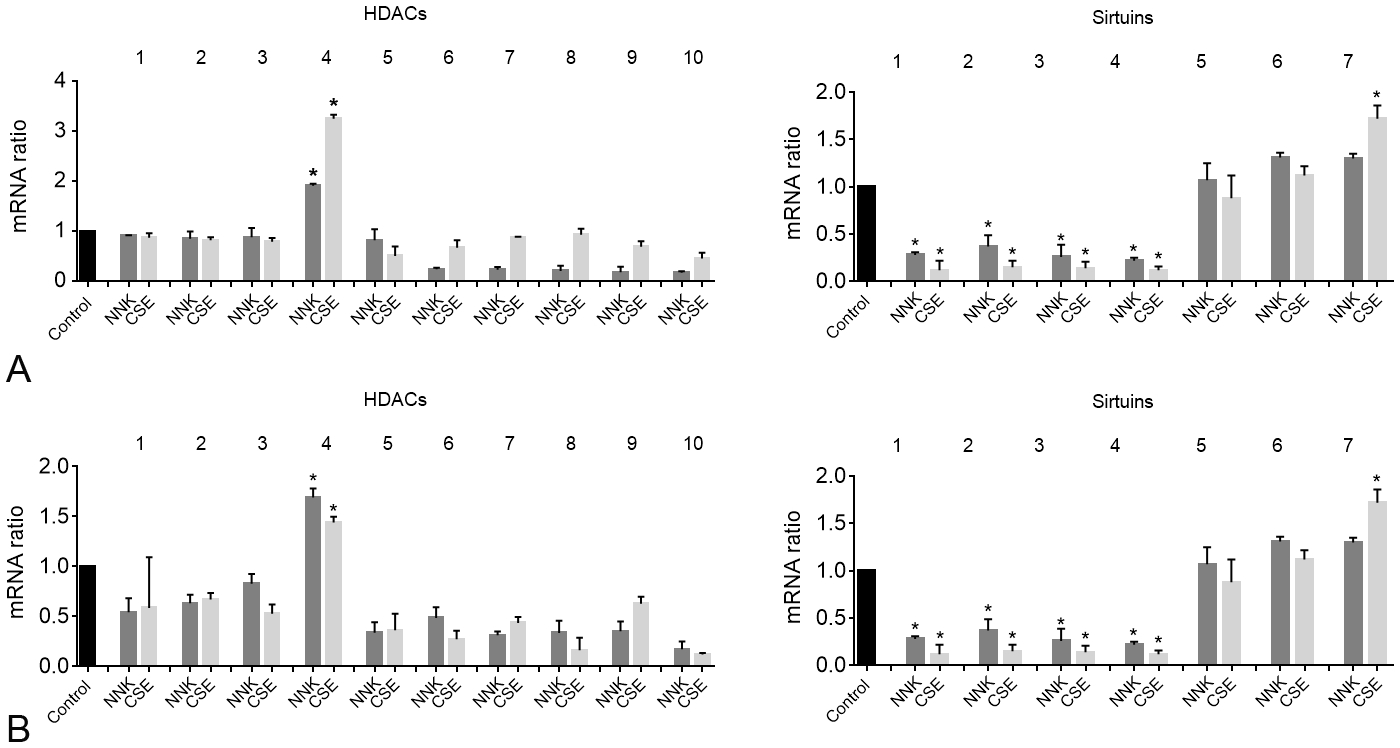
Smoking compounds NNK and CSE increase HDAC4 mRNA levels in pancreatic cancer cells. mRNA level was measured by RT-PCR in MIA PaCa-2 (A) and BxPC-3 (B) pancreatic cancer cells. *P < 0.05 vs control.
Next, we performed staining of the human primary pancreatic cancer and metastatic tumors in the liver and the lung tissues collected through the Nebraska rapid autopsy program. We found a high expression level of HDAC4 in the primary PDAC as well as in the PDAC metastatic tumors in the liver and the lung of PDAC patients compared to morphologically normal-looking tissues (Fig. 2).
FIGURE 2.
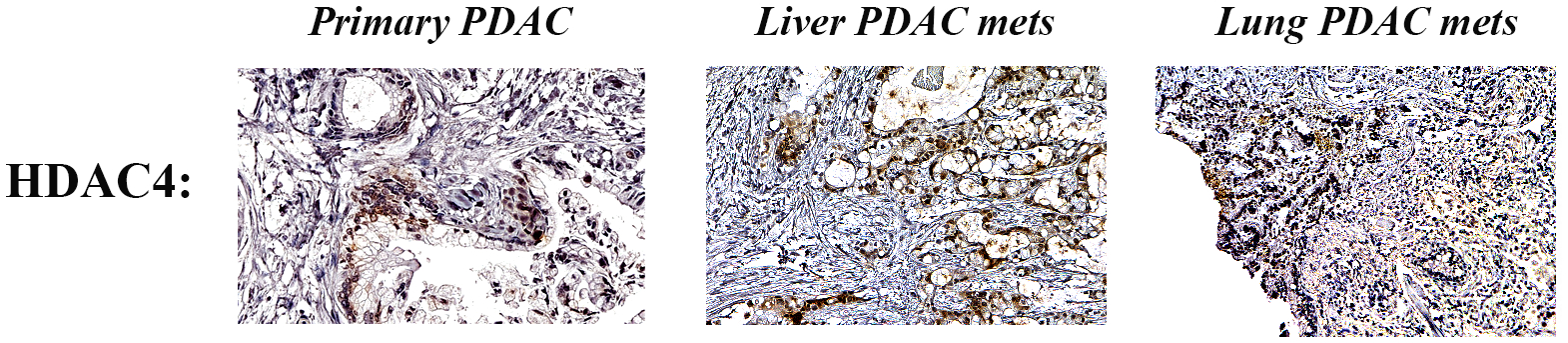
Expression of HDAC4 in primary PDAC and in liver and lung metastases (mets). Immunohistochemistry staining of HDAC4 in human PDAC tissues from primary, liver, and lung mets.
Smoking-induced Cancer Cell Invasion is Mediated by HDAC4
To determine the effect of specific inhibition of HDAC4 on PDAC cell invasion we treated cells with NNK or CSE with and without the specific HDAC4 inhibitor Lmk-235 and measured cancer cells invasion. The Matrigel Invasion Assay data showed that smoking compounds NNK and CSE significantly increased invasion of MIA PaCa-2 (Figs. 3A, B) and BxPC-3 (Fig. 3C) cancer cells by 50% to 80%. This effect was abolished when adding HDAC4 inhibitor Lmk-235, which decreased basal invasion ability as well as invasion induced by NNK and CSE in both cell lines by up to 70% (Figs. 3B, C).
FIGURE 3.
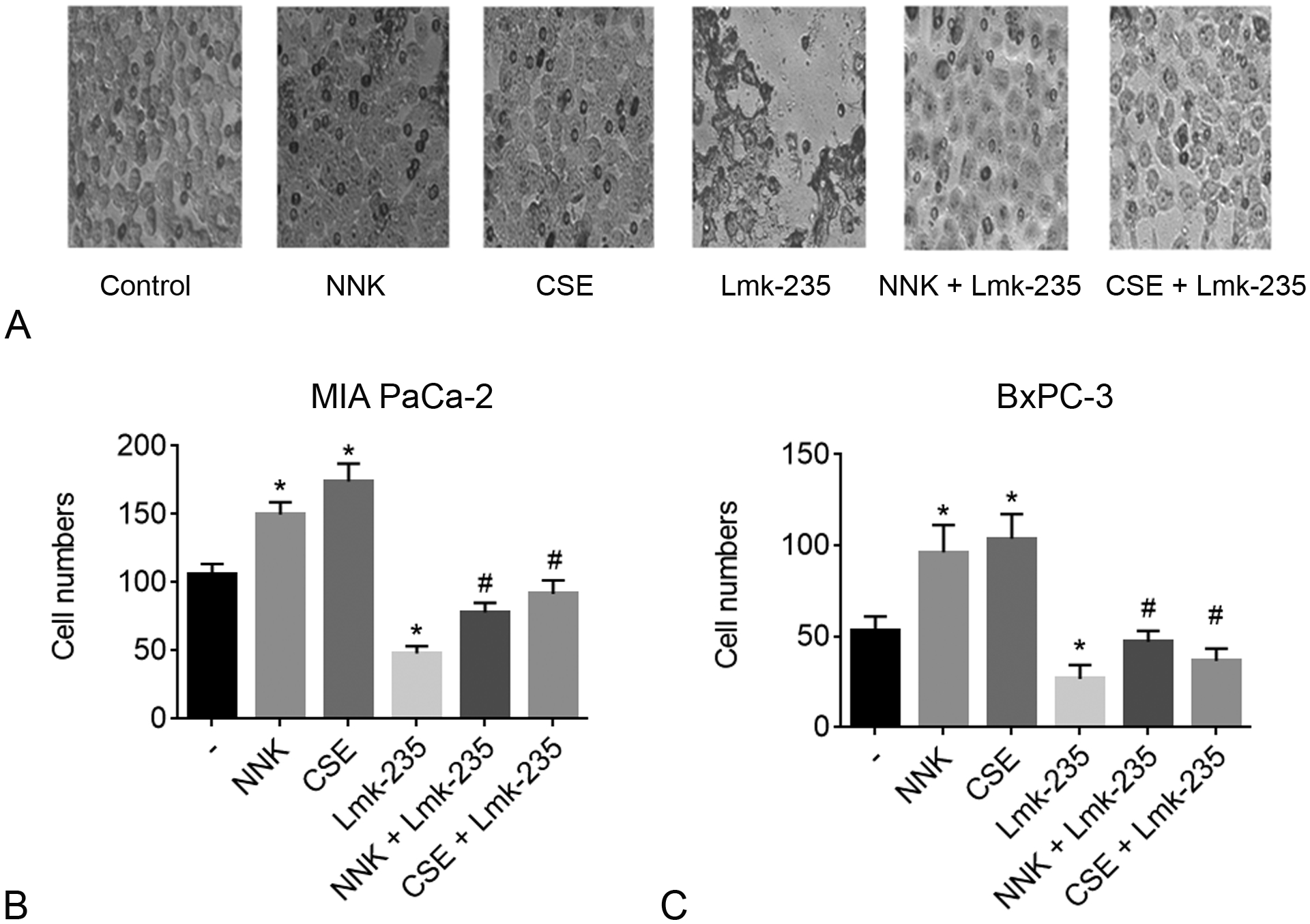
Smoking promotes invasion, whereas HDAC4 inhibitor reverses this effect in pancreatic cancer cells. MIA PaCa-2 and BxPC-3 cells were cultured with NNK (1 μM), CSE (0.4 μg/ml) with and without HDAC4 inhibitor Lmk-235 (5 μM) for 24h and cell invasion was measured using the Matrigel Invasion Assay. A, Images of MIA PaCa-2 invading cells. B and C, Quantification of cells invading the lower level of the plate in MIA PaCa-2 (B) and BxPC-3 (C) cells. *P < 0.05 vs control. #P < 0.05 vs NNK or CSE alone.
Smoking Promotes Metastasis and HDAC4 Inhibition Reverses This Effect in Syngeneic PDAC Mice
To study the role of HDACs, of the class I and II, and HDAC4 in promoting pancreatic cancer metastasis we developed a syngeneic mouse model of metastatic PDAC by injecting 1 × 106 UN-KPC961-Luc cells30 into the spleen of C57BL.129J mice. Four weeks before surgery mice were exposed to NNK (one injection at the dose of 100 mg/kg per week for 4 weeks) or mock treatment. Two days after cancer cell inoculation, the mice were treated with i.p. injections of pan-HDAC inhibitor Saha (50 mg/kg, 3 times per week), HDAC4 inhibitor Lmk-235 (3 mg/kg, 3 times per week) or vehicle control for 6 weeks.
To determine the effect of NNK and HDAC inhibition on metastasis we quantified the number of organs showing metastatic lesions. We found that 7 organs, in addition to the peritoneum, are mostly affected by metastasis. The organs are liver, lung, stomach, intestines, colon, kidney, and pancreas. Analysis of the number of organs affected by metastatic lesions (Fig. 4A) showed that NNK increased the number of affected organs from an average of 4.2 to 6.8 organs (Fig. 4B). Saha and Lmk-235 decreased the basal level of metastasis to 3.1 and 3.8, respectively, and decreased the NNK-induced level of metastasis from 6.8 to 3.6 and 3.8, respectively. The decrease in basal level of metastasis by Saha was significant (−26%) but the decrease by Lmk-235 was not significant (−10%). However, the decrease in metastasis, in the presence of NNK, by both Saha (−47%) and Lmk-235 (−44%) was significant (Fig. 4B).
FIGURE 4.
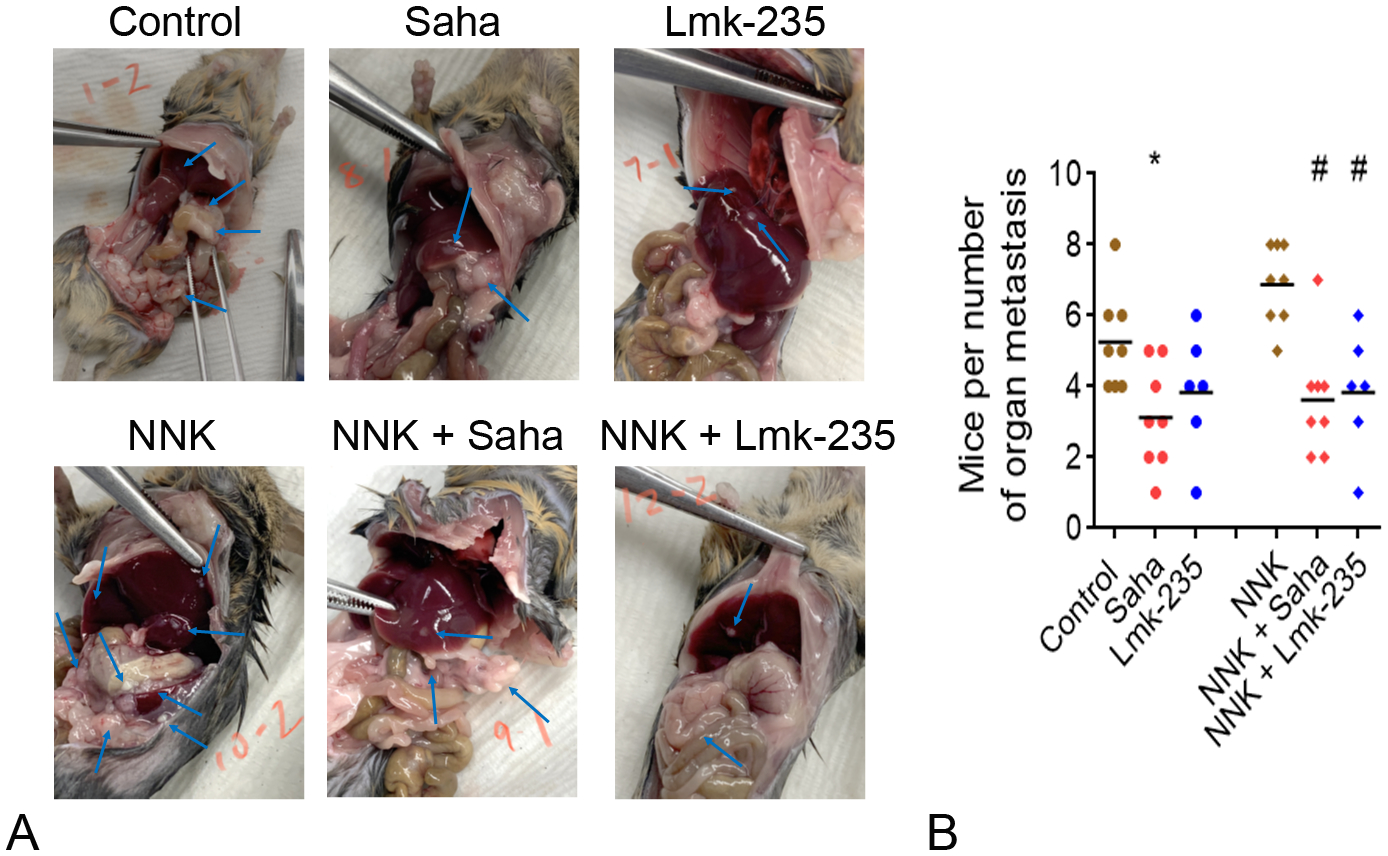
Smoking promotes metastasis in a mouse model of pancreatic cancer and this effect is prevented by HDAC4 inhibition. B6/129 mice were pre-treated with i.p. injections of NNK (25 mg/kg) once a week for 4 weeks. Mice underwent a surgery to inject UNKPC-961 pancreatic cancer cells (1 × 106 cells) in the spleen and treatment with Saha (50 mg/kg) or Lmk-235 (3 mg/kg) started 2 days after surgery. The i.p. injections of Saha, Lmk-235 or saline were performed 3 times per week until mice were killed after 6 weeks. A, Images of PDAC metastases in mice abdomen. B, Quantification of the number of organs in mice abdomen affected by PDAC metastatic lesions. N = 6 to 8 mice per group. *P < 0.05 vs control. #P < 0.05 vs NNK.
Liver and lung are the main metastatic sites for PDAC in humans. Therefore, we specifically quantified the level of PDAC metastatic lesions in the liver and the lung of the syngeneic mice. We found that NNK treatment promoted metastasis to the liver and the lung (Figs. 5A, 6A). Metastasis quantification by counting the number of tumors in the liver and the lung shows a significant increase in mice treated with NNK compared to control mice. The increase was 80% for liver and 60% for lung metastasis (Figs. 5B, 6B). This effect was reversed in mice treated with HDAC inhibitor Saha and HDAC4 specific inhibitor Lmk-235. The decrease in liver and lung metastasis by Saha was significant in NNK-treated mice (Figs. 5B, 6B). To confirm the data observed by quantifying the number of metastases we macroscopically quantified the area covered by metastases in the liver and the lung. We found a significant increase in metastases’ areas in the liver and the lung by NNK. The increase was prevented by Saha and Lmk-235 (Figs. 5C, 6C). It is worth noting that the basal level of liver and lung metastasis was slightly but not significantly affected by Saha and Lmk-235, except the significant decrease in the number of liver metastatic lesions by Lmk-235 (Fig. 5B).
FIGURE 5.
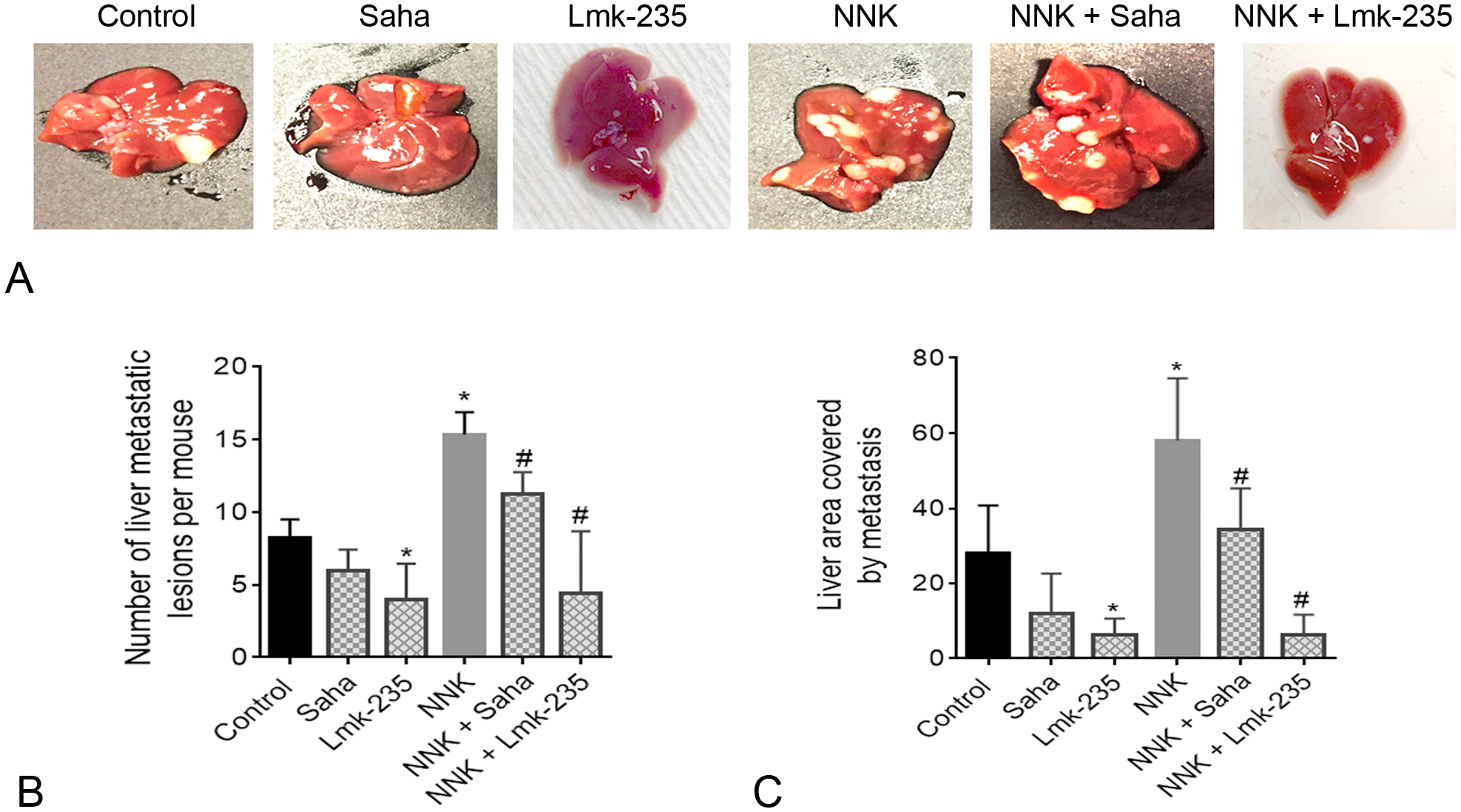
Smoking promotes metastasis to the liver and this effect is prevented by HDAC4 inhibition. A, Images of liver metastatic lesions. B, Quantification of the liver number of metastases. C, Quantification of the ratio of liver area occupied by metastases. *P < 0.05 vs control. #P < 0.05 vs NNK alone.
FIGURE 6.
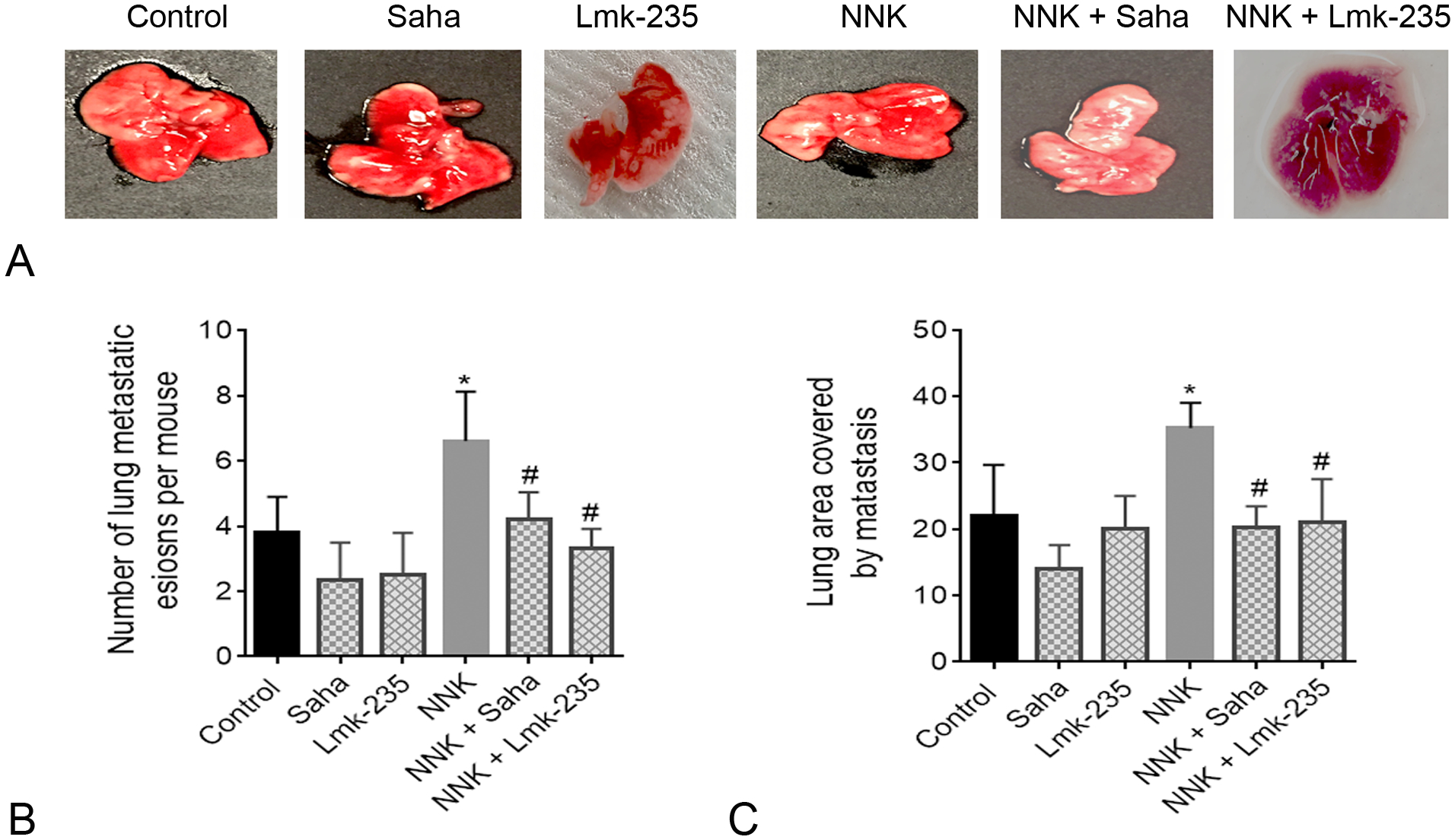
Smoking promotes metastasis to the lung and this effect is prevented by HDAC4 inhibition. A, Images of lung metastatic lesions. B, Quantification of the lung number of metastases. C, Quantification of the ratio of lung area occupied by metastases. *P < 0.05 vs control. #P < 0.05 vs NNK alone.
It is also worth noting that Saha and Lmk-235 treatments did not induce any apparent toxicity or any weight change in mice.
The data of this study indicate that exposure to the smoking compound NNK significantly increased tumor metastasis in general, and specifically in the liver and the lung, and that HDAC4 inhibition prevented this effect. The effect of HDAC4 inhibition on preventing metastasis to the liver was much stronger than its effect on preventing metastasis to the lung suggesting that the mechanisms of metastasis to these two organs are partially different.
DISCUSSION
Although HDACs have been shown to play a critical role in promoting pancreatic cancer, their effect on regulating pancreatic cancer invasion and metastasis is not very well understood. Moreover, very little is known about the effect of smoking on HDACs and how this interaction mediates PDAC metastasis.
Our data indicate that HDAC4 is the only HDAC, of the class I and II family of HDACs, to be increased when cells are treated with smoking compounds. Therefore, we investigated the role of HDAC4 in the PDAC metastasis with and without smoking compounds. We found that HDAC4 plays a major role in mediating smoking induced invasion and metastasis of pancreatic cancer.
We have developed a syngeneic metastatic mouse model of pancreatic cancer using the UN-KPC961 cells injected in the spleen of mice.23,30 We found that treatment by the smoking compound NNK stimulated cancer cell metastasis by 60% in general and by 80% and 60% to the liver and the lung, respectively. Published data showed that 80% of PDAC patients at autopsy have liver metastasis and 45% have lung metastasis.5 More importantly, we found that HDAC (class I and II) inhibitor Saha and HDAC4 specific inhibitor Lmk-235 prevented the increase in metastasis induced by the smoking compound NNK. Inhibitors Saha and Lmk-235 decreased the NNK-induced metastasis to the eight organs analyzed by 47% and 44%, respectively. The number of metastatic lesions in the liver in the presence of NNK was decreased by Saha and Lmk-235 by 30% and 70%, respectively, and to the lung by 40% and 50%, respectively. In general, the data indicate that HDAC4 inhibition shows a better effect on preventing NNK-induced metastasis to the liver compared to Saha, whereas the effect of pan-HDAC and HDAC4 inhibitions on preventing lung metastasis are not significantly different.
Inhibition of HDAC4 decreased the basal level of metastasis in the liver by 70% but slightly affected the basal level of metastasis to the lung suggesting that HDAC4 May be more critical for liver metastasis compared to lung metastasis. The data suggest HDAC4 as a major mediator of the basal and NNK induced metastasis to the liver with less involvement in the metastasis to the lung. More investigation is needed to understand why some patients have liver PDAC metastasis, some have more of the PDAC lung metastasis, and others have both sites involved. However, our data suggest that HDAC4 is a major player in metastasis to the liver compared to the lung.
The data indicate that HDAC4 fully mediates the invasiveness of PDAC cells induced by smoking and partially regulates the smoking-independent invasive level. This is the first data to show regulation of invasiveness by HDAC4 in pancreatic cancer cells. The data confirm the observation of a similar effect in osteosarcoma.29
We have shown previously that HDAC3 plays an important role in PDAC growth in animal models of the disease.20 Here, we show that HDAC4 is playing a role in promoting invasion of the cancer cells and metastasis. Our data indicate that HDAC4 is highly induced in human PDAC cancer cells when treated with smoking compounds NNK or CSE. None of the other HDACs of the class I and II was affected by smoking compounds.
Our data give new insights about the mechanism of metastasis induced by smoking. The data confirm that HDAC4 is regulated by smoking compounds and that it regulates PDAC invasion and metastasis.
Acknowledgments
This work was supported by the SOCCI Luke Wu-Jei Chang Discovery Fund, the Cedars-Sinai Cancer at Cedars-Sinai Medical Center through the 2021 Cedars-Sinai Cancer Project Acceleration, and NCI P01 award CA233452.
Footnotes
Disclosure: The authors have no conflicts of interest to disclose.
REFERENCES
- 1.DeSantis CE, Lin CC, Mariotto AB, et al. Cancer treatment and survivorship statistics, 2014. CA Cancer J Clin. 2014;64:252–271. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. [DOI] [PubMed] [Google Scholar]
- 3.Idachaba S, Dada O, Abimbola O, et al. A Review of Pancreatic Cancer: Epidemiology, Genetics, Screening, and Management. Open Access Maced J Med Sci 2019;7:663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rahib L, Smith BD, Aizenberg R, et al. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913–2921. [DOI] [PubMed] [Google Scholar]
- 5.Yachida S, Iacobuzio-Donahue CA. The pathology and genetics of metastatic pancreatic cancer. Arch Pathol Lab Med. 2009;133:413–422. [DOI] [PubMed] [Google Scholar]
- 6.Edderkaoui M, Thrower E. Smoking and pancreatic disease. J Cancer Ther. 2013;4:34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lucenteforte E, La Vecchia C, Silverman D, et al. Alcohol consumption and pancreatic cancer: a pooled analysis in the International Pancreatic Cancer Case-Control Consortium (PanC4). Ann Oncol. 2012;23:374–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yadav D, Lowenfels AB. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology. 2013;144:1252–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lowenfels AB, Maisonneuve P. Environmental factors and risk of pancreatic cancer. Pancreatology. 2003;3:1–7. [DOI] [PubMed] [Google Scholar]
- 10.Raimondi S, Lowenfels AB, Morselli-Labate AM, et al. Pancreatic cancer in chronic pancreatitis; aetiology, incidence, and early detection. Best Pract Res Clin Gastroenterol. 2010;24:349–358. [DOI] [PubMed] [Google Scholar]
- 11.Iodice S, Gandini S, Maisonneuve P, et al. Tobacco and the risk of pancreatic cancer: a review and meta-analysis. Langenbecks Arch Surg. 2008;393:535–545. [DOI] [PubMed] [Google Scholar]
- 12.Whittemore AS, Paffenbarger RS Jr, Anderson K, et al. Early precursors of pancreatic cancer in college men. J Chronic Dis. 1983;36:251–256. [DOI] [PubMed] [Google Scholar]
- 13.Abrams JA, Lee PC, Port JL, et al. Cigarette smoking and risk of lung metastasis from esophageal cancer. Cancer Epidemiol Biomarkers Prev. 2008;17:2707–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murin S, Inciardi J. Cigarette smoking and the risk of pulmonary metastasis from breast cancer. Chest. 2001;119:1635–1640. [DOI] [PubMed] [Google Scholar]
- 15.Anderson MA, Zolotarevsky E, Cooper KL, et al. Alcohol and tobacco lower the age of presentation in sporadic pancreatic cancer in a dose-dependent manner: a multicenter study. Am J Gastroenterol. 2012;107:1730–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maisonneuve P, Lowenfels AB. Epidemiology of pancreatic cancer: an update. Dig Dis. 2010;28:645–656. [DOI] [PubMed] [Google Scholar]
- 17.Kanwal R, Gupta S. Epigenetic modifications in cancer. Clin Genet. 2012;81:303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marks P, Rifkind RA, Richon VM, et al. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2001;1:194–202. [DOI] [PubMed] [Google Scholar]
- 19.Aghdassi A, Sendler M, Guenther A, et al. Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut. 2012;61:439–448. [DOI] [PubMed] [Google Scholar]
- 20.Edderkaoui M, Xu S, Chheda C, et al. HDAC3 mediates smoking-induced pancreatic cancer. Oncotarget. 2016;7:7747–7760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cho HJ, Jung JI, Lim do Y, et al. Bone marrow-derived, alternatively activated macrophages enhance solid tumor growth and lung metastasis of mammary carcinoma cells in a Balb/C mouse orthotopic model. Breast Cancer Res. 2012;14:R81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu CY, Xu JY, Shi XY, et al. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab Invest. 2013;93:844–854. [DOI] [PubMed] [Google Scholar]
- 23.Edderkaoui M, Chheda C, Soufi B, et al. An inhibitor of GSK3B and HDACs kills pancreatic cancer cells and slows pancreatic tumor growth and metastasis in mice. Gastroenterology. 2018;155:1985–1998.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zeng LS, Yang XZ, Wen YF, et al. Overexpressed HDAC4 is associated with poor survival and promotes tumor progression in esophageal carcinoma. Aging. 2016;8:1236–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilson AJ, Byun DS, Nasser S, et al. HDAC4 promotes growth of colon cancer cells via repression of p21. Mol Biol Cell. 2008;19:4062–4075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mottet D, Pirotte S, Lamour V, et al. HDAC4 represses p21(WAF1/Cip1) expression in human cancer cells through a Sp1-dependent, p53-independent mechanism. Oncogene. 2009;28:243–256. [DOI] [PubMed] [Google Scholar]
- 27.Shen YF, Wei AM, Kou Q, et al. Histone deacetylase 4 increases progressive epithelial ovarian cancer cells via repression of p21 on fibrillar collagen matrices. Oncol Rep. 2016;35:948–954. [DOI] [PubMed] [Google Scholar]
- 28.Kang ZH, Wang CY, Zhang WL, et al. Histone deacetylase HDAC4 promotes gastric cancer SGC-7901 cells progression via p21 repression. PloS One. 2014;9:e98894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cao K, Wang H, Fang Y, et al. Histone deacetylase 4 promotes osteosarcoma cell proliferation and invasion by regulating expression of proliferating cell nuclear antigen. Front Oncol. 2019;9:870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Torres MP, Rachagani S, Souchek JJ, et al. Novel pancreatic cancer cell lines derived from genetically engineered mouse models of spontaneous pancreatic adenocarcinoma: applications in diagnosis and therapy. PloS One. 2013;8:e80580. [DOI] [PMC free article] [PubMed] [Google Scholar]