

SUMMARY
Melanoma is one of the deadliest cancers, yet the cells of origin and mechanisms of tumor initiation remain unclear. The majority of melanomas emerge from clear skin without a precursor lesion, but it is unknown whether these melanomas can arise from melanocyte stem cells (MCSCs). Here, we employ mouse models to define the role of MCSCs as melanoma cells of origin, demonstrate that MCSC quiescence acts as a tumor suppressor, and identify the extrinsic environmental and molecular factors required for the critical early steps of melanoma initiation. Specifically, melanomas originate from melanoma-competent MCSCs upon stimulation by ultraviolet-B (UVB) which induces MCSC activation and translocation via an inflammation-dependent process. Moreover, the chromatin remodeling factor Hmga2 in the skin plays a critical role in UVB-mediated melanomagenesis. These findings delineate melanoma formation from melanoma-competent MCSCs following extrinsic stimuli, and suggest that abrogation of Hmga2 function in the microenvironment can suppress MCSC-originating cutaneous melanomas.
INTRODUCTION
Adult stem cells are compelling candidates of cancer cells of origin as they are long lived, uni/multi-potent and have significant self-renewal potential (White and Lowry, 2015). Recent mathematical models have been used to suggest that the number of stem cell divisions is significantly correlated with cancer risk (Tomasetti and Vogelstein, 2015), however, cell extrinsic factors may also significantly contribute to the initiation of tumor formation from its cellular origin (Wu et al., 2016). Furthermore, recent studies have shown that oncogenic combinations sufficient to induce cutaneous squamous cell carcinomas (SCCs) from their cancer cells of origin (Lapouge et al., 2011; White et al., 2011) were unable to induce tumor formation while in a stem cell quiescent period (White et al., 2014). Therefore, along with the identification of cancer cells of origin in each tissue type, elucidating how a tumor initiates from its cellular origin is indispensable to develop effective preventative strategies for each type of cancer.
This is particularly crucial in cutaneous melanoma, one of the deadliest forms of cancer that continues to have a gradually increasing annual incidence and mortality rate (Siegel et al., 2015). While many studies have identified the genetic etiologies of this cancer (Dankort et al., 2009; Miller and Mihm, 2006), little is known regarding the environmental causes and cellular conditions responsible for melanoma initiation from adult stem cells. The data presented here, using lineage-tracing of melanocyte stem cells (MCSCs) in melanoma mouse models, identifies adult MCSCs as melanoma cells of origin and shows that ultraviolet-B (UVB) can act as an extrinsic factor which promotes melanoma initiation through MCSC activation and translocation, in a dose- and inflammation-dependent manner. We further demonstrate the mechanistic requirement for the chromatin remodeling factor Hmga2 in the cutaneous microenvironment for UVB-mediated MCSC activation and translocation, as well as melanomagenesis from tumor-competent adult stem cells.
RESULTS
The Tyr-CreER transgene can efficiently label quiescent melanocyte stem cells
To determine the role of adult melanocyte stem cells (MCSCs) in cutaneous melanoma initiation, our experimental scheme required mouse models capable of reliable spatial and temporal control of inducible oncogenic expression with lineage tracing markers in adult MCSCs. Hence, we first determined whether expression of the Tyr-CreER transgenic allele could efficiently target quiescent MCSCs in the adult mouse skin.
As murine MCSCs are located in the hair follicles of the dorsal and ventral skin, both MCSCs and hair follicle stem cells undergo cyclical periods of rest (telogen) and activation (anagen) that define the hair cycle (Lang et al., 2013). Murine follicular MCSCs during telogen are found within the hair germ of quiescent hair follicles of the dorsal skin (Figure 1A). L-dopachrome tautomerase (Dct, or Tyrosinase Related Protein 2, Trp2) is expressed in both MCSCs and differentiated melanocytes of the anagen hair bulb, whereas Tyrosinase Related Protein 1 (Tyrp1, Trp1) is found only in differentiated melanocytes (Figures S1A–B) (Osawa et al., 2005). Skin characterized by quiescent hair follicles in telogen demonstrated Dct but no apparent Tyrp1 at the hair germ (Figures S1A–B).
Figure 1. Melanocyte stem cells (MCSCs) can act as melanoma cells of origin and adult MCSC activation is required for melanoma initiation.
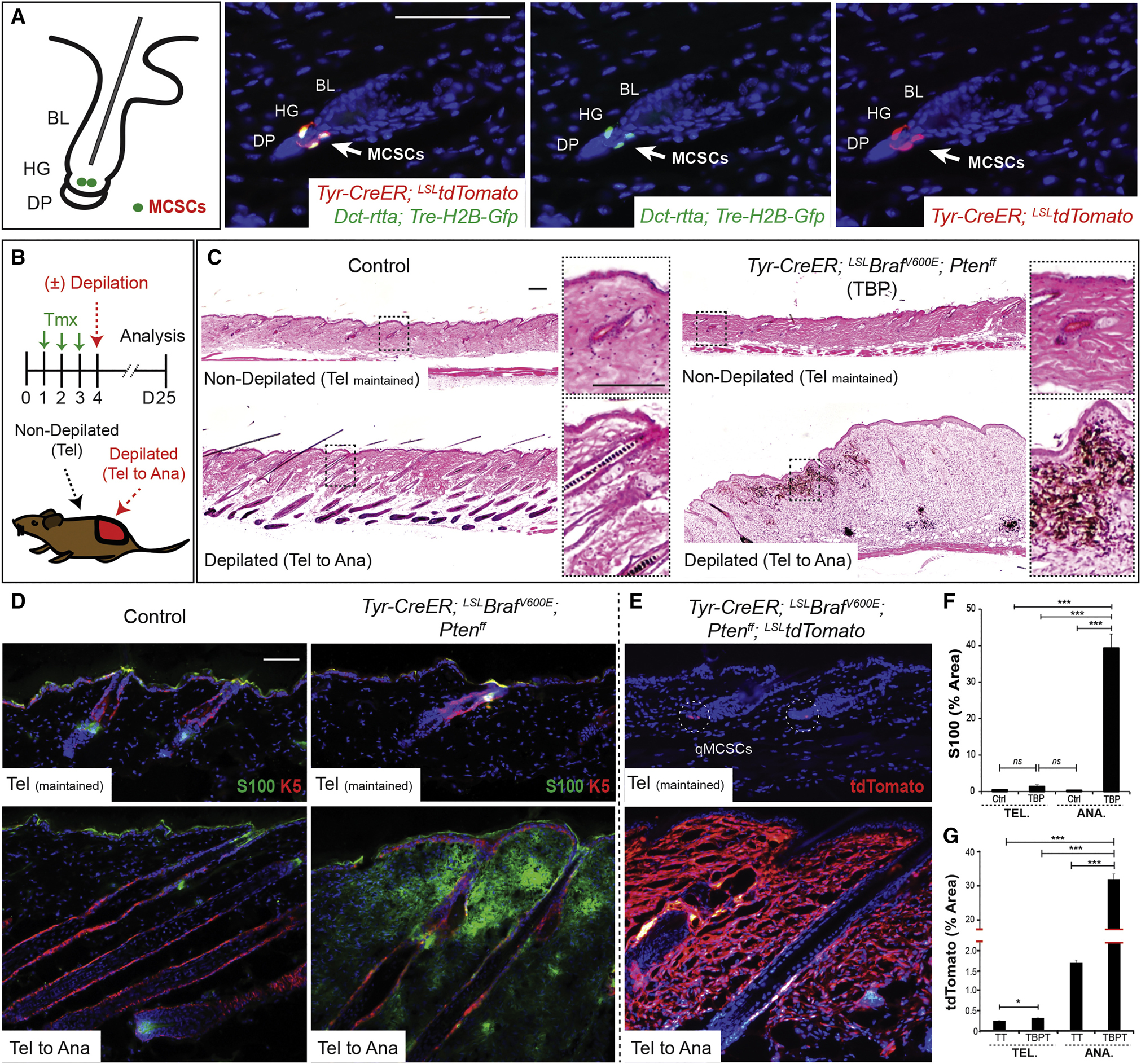
(A) Location of adult MCSCs, shown by co-expression of H2B-GFP and tdTomato in MCSCs at the hair germ (HG). BL, bulge; DP, dermal papillae. (B) Experimental scheme. Tmx, tamoxifen; Tel, telogen; Ana, anagen. (C-G) Histological phenotypes, and immunostaining for melanoma marker S100 and tdTomato lineage tracing in control, Tyr-CreER; LSL-tdTomato (TT) and TBP or TBP; LSL-tdTomato (TBPT) mice. Sequential photomicrographs were reconstructed for the panoramic images of histology (C). S100, n ≥ 60 fields, 8 mice; tdTomato, n ≥ 160 fields, 6 mice per group. IF counterstaining, DAPI. Scale bars, 100 μm. Data are represented as mean ± SEM. Also see Figures S1 and S2.
Tyrosinase (Tyr) expression is not commonly associated with MCSCs (Nishimura et al., 2002). However, the Tyr-CreER transgene has been shown to label MCSCs located in the hair follicle stem cell compartment (the bulge) during anagen (Bosenberg et al., 2006). To assess the utility of Tyr-CreER in targeting quiescent MCSCs, we compared the relative labeling overlap between Tyr-CreER; LSL-tdTomato (inducible by tamoxifen) and Dct-rtta; Tre-H2B-Gfp (inducible by doxycycline) (Zaidi et al., 2011) in MCSCs during telogen and anagen. Based on co-labeling of tdTomato with GFP, we confirmed that the Tyr-CreER effectively targets quiescent MCSCs during telogen (93 ± 4 % of GFP cells, Figure 1A) and anagen (Figure S1B) utilizing the quadruple combination of Tyr-CreER; LSL-tdTomato and Dct-rtta; Tre-H2B-Gfp in a single mouse. Furthermore, consistent with a previous report (Bosenberg et al., 2006), tdTomato cells maintain residence in the melanocyte stem cell niche during subsequent hair cycles and continue to produce differentiated melanocytes (Figure S1C). These results suggested that the Tyr-CreER allele can efficiently induce the expression of oncogenic mutant alleles together with lineage tracing markers in quiescent MCSCs through inducible Cre-recombination.
Melanocytic tumor initiation requires MCSC activation upon oncogenic expression
Genetic alterations in Braf (BrafV600E and other mutations, ~60%) and Pten (deletion, ~25%) are frequently observed in cutaneous melanomas (Hodis et al., 2012; Vultur and Herlyn, 2013), and the Tyr-CreER; LSL-BrafV600E; Ptenflox/flox (TBP) mouse model has been used in pre-clinical studies (Damsky et al., 2011; Dankort et al., 2009). To determine whether MCSCs can act as melanoma cells of origin and if malignant melanocytic tumor initiation relies on adult stem cell activation state, we utilized the Tyr-CreER allele to express BrafV600E combined with Pten loss of function in young adult mice (Dankort et al., 2007; Groszer et al., 2001).
To determine the necessity of MCSC activation for tumor initiation, we designed an experimental scheme to assess this dependency in individual mice (Figure 1B). By depilating only one half of the dorsal skin in TBP mice at 7 weeks of age, when adult MCSCs are quiescent, we were able to activate a specific subset of the MCSCs, leaving the untreated skin in a quiescent state. Skin regions with activated MCSCs demonstrated macroscopically evident melanocytic tumors within 30 days of tamoxifen treatment and depilation, and microscopic tumors were found within 15–20 days (Figure 1C). Similar to previous studies (Damsky et al., 2011; Dankort et al., 2009), these melanocytic tumors, if left untreated, can rapidly develop large and pigmented asymmetric tumors with irregular borders which also ulcerate requiring euthanasia (Figure S1D). Histologically, early tumors (Figure 1C) display melanocytic hyperplasia characterized by collections of heavily pigmented and unpigmented spindled and epithelioid melanocytes splaying through dermal collagen bundles and wrapping around adnexal structures in the dermis, loosely resembling cellular blue nevi. However, as previously reported (Damsky et al., 2011; Dankort et al., 2009), later lesions display extension of pleomorphic, highly atypical melanocytes into all layers of the skin (Figure S1D). The atypical features include effacement of the dermal-epidermal junction by atypical lentiginous melanocytes at the junction with overlying pagetoid spread of melanocytes, numerous erosions and ulcerations of the epidermis, and sheets of atypical epithelioid and spindled melanocytes infiltrating the dermis and subcutis. The constituent dermal component displays numerous apoptotic cells, mitoses, and atypical mitoses, and tumors also display a nuclear proliferation rate of 15–20%. These clinical and histological features are compatible with the histologic diagnosis of melanoma, and resemble animal-type melanoma as well as human blue nevus-like melanoma.
In remarkable contrast, MCSCs maintained in a quiescent state did not demonstrate significant evidence of tumor initiation, despite Cre mediated recombination of the Braf and Pten alleles (Figures 1C and S1E–F). This finding was further confirmed by visualization and quantification of the total area labeled by melanoma markers and the lineage tracing allele LSL-tdTomato in depilated versus non-depilated skin areas in TBP mice (Figures 1D–G and S1G–H).
In addition, we similarly assessed whether MCSC activation is required for benign melanocytic tumor initiation. Tyr-CreER; LSL-BrafV600E animals generate benign hyperpigmentation lesions (Damsky et al., 2015), and Tyr-CreER; LSL-BrafV600E; Ptenwt/flox animals form a reduced number of malignant melanocytic tumors (Xie et al., 2015). Both demonstrated a dependence on MCSC activation. During the same period, quiescent MCSCs were not responsive to oncogenic BrafV600E-driven benign nevogenesis and remained at the hair germs without apparent ectopic differentiation (Figures S2A–B). However, activated MCSCs produced prematurely differentiated and pre-growth arrested melanocytes at ectopic locations directly adjacent to the hair follicular bulb (Figures S2A–B) similar to previous observations (Damsky et al., 2015). In contrast, wild type MCSCs show no pigmentation and mature differentiated melanocytes generate pigmentation for the hair shaft within hair bulb. As shown previously (Damsky et al., 2015; Dankort et al., 2009), these hair follicle associated benign melanocytes ultimately remained in the dermis during the next catagen and telogen period.
Kras mutations are found in around 2% of human melanoma cases (Milagre et al., 2010). To confirm the melanoma initiation dependency on MCSC activation, we used an alternative oncogene by substituting the BrafV600E with the KrasG12D driver mutation previously reported for melanoma models (Milagre et al., 2010). Although melanomas were evident upon MCSC activation in Tyr-CreER; LSL-KrasG12D; Ptenflox/flox (TKP) mice, KrasG12D-driven melanoma development was significantly suppressed by MCSC quiescence (Figures S2C–D), similar to BrafV600E-driven melanocytic tumor initiation.
Taken together, these results demonstrate that while MCSCs can act as melanocytic tumor cells of origin, adult stem cell quiescence enables melanocytic tumor suppression in MCSCs expressing a mutational load known to be sufficient for benign or malignant melanocytic tumor formation. Tumor-competent but quiescent MCSCs still require additional activation steps for tumor initiation, which could include extrinsic stimuli that induces stem cell activation.
Ultraviolet-B radiation is an extrinsic stimulus regulating melanoma initiation from quiescent MCSCs
Ultraviolet-B (UVB) exposure is one of the major risk factors for cutaneous melanomas (Lo and Fisher, 2014). UVB can accelerate epidermal hyperplasia and cutaneous inflammation, and is also known to induce the migration of follicular MCSCs to the interfollicular epidermis (IFE) in both mice and humans (Chou et al., 2013). We hypothesized that UVB-mediated recruitment of follicular MCSCs to the IFE may act as an activation stimulus sufficient to initiate melanoma formation from quiescent MCSCs harboring a pre-existing intrinsic oncogenic load.
To determine whether UVB could elicit melanomagenesis from tumor-prone quiescent MCSCs in telogen, we first optimized the acute UVB dose which efficiently induces MCSC migration but does not initiate anagen induction (Figures 2A–B). While this acute UVB exposure (180 mJ cm−2) temporarily induced detectable epidermal and dermal hyper-proliferation, UVB-exposed skin returned to homeostasis within a few days without activation of the hair cycle (Figure 2A). This UVB dose, however, was sufficient to induce follicular MCSC migration into the IFE, and MCSC translocation was confirmed by inducible lineage tracing markers throughout the UVB-exposed skin using both Dct-rtta and Tyr-CreER (Figures 2A–B).
Figure 2. Ultraviolet radiation stimulates melanoma initiation by inducing the activation and translocation of quiescent melanoma-competent MCSCs.
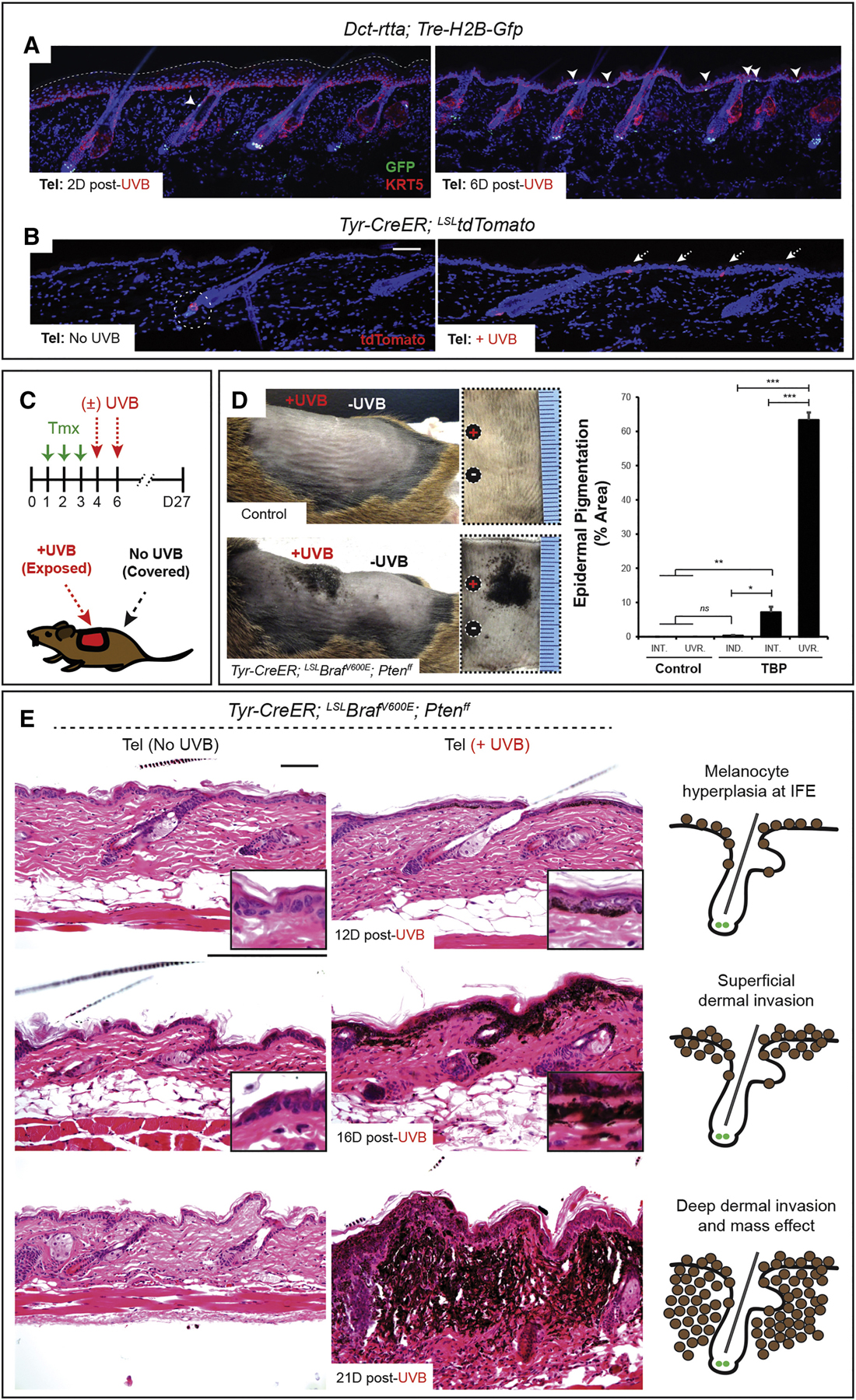
(A-B) The lineage tracing alleles, H2B-GFP and tdTomato, show MCSC translocation during telogen upon ultraviolet-B (UVB) exposure. (C) Experimental scheme. Tmx, tamoxifen. (D) Macroscopic epidermal pigmentation in control and TBP mice with/without UVB exposure. Mean ± SEM. n = 10 UVB-irradiated control, n = 7 non-UVB TBP and n = 10 UVB-irradiated TBP mice. INT., non-UVB internal control; UVR., UVB-irradiated; IND., independent control skin.
(E) Histological progression of melanoma in TBP mice with/without UVB irradiation. IF counterstaining, DAPI. Scale bars, 100 μm. Also see Figures S3 and S4.
We then tested whether regional UVB exposure could initiate melanomagenesis by MCSC activation and translocation evenly throughout the skin, by exposing 40–50% of the shaved dorsal skin to UVB and covering the all other skin regions (Figure 2C). Strikingly, UVB-exposed skin in TBP mice exhibited macroscopic melanocytic tumor formation within 12–16 days of UVB exposure, whereas covered skin containing quiescent MCSCs remained relatively clear of melanocytic tumors (Figures 2D–E). UVB-induced melanomas histologically looked similar to induced tumors during anagen, but the involvement of the epidermis by melanocytes typically happened earlier. This tumorigenic event was further quantified using total area of epidermal pigmentation (Figure 2D), and confirmed by labeling by the lineage tracing tdTomato marker and immunostaining for melanoma markers (Figures S3A–C).
To better define the initial transformation process in this UVB-induced melanoma model, we identified when MCSCs transition to a proliferative state. In wild type MCSCs, proliferation occurs within the hair germ two days post-UVB exposure (Figures S3D–E). Translocation to the IFE follows proliferation resulting in Trp2+ cells in the epidermis, but these relocated cells do not proliferate further, instead differentiating into Tyrp1+ cells (Figure S3F). In contrast, migrating/migrated tumor-prone MCSCs continue to expand, ultimately resulting in melanoma cells throughout the IFE (Figure S3G) without anagen induction (Figures S4A–B). Similarly, UVB-mediated melanomagenesis from MCSCs was also demonstrated in TKP mice (Figures S4C–E). These data indicate that MCSCs expressing a mutational load sufficient for melanoma development may remain in quiescence until exposure to the extrinsic stimulus UVB, through which MCSC activation and translocation to the epidermis can initiate malignant melanocytic tumorigenesis.
It is possible that UVB induced melanomas could be due to DNA damage which in turn generates tumor promoting mutations. However, unlike in the hyper-proliferative epidermal cells, a sustained DNA damage response was not observed in MCSCs in the UVB exposed skin (Figure S4F). Furthermore, the presence of widespread, generalized MCSC translocation and melanoma tumor induction rather than focal to multifocal aggregates of transmigrated MCSCs or localized tumors were found throughout UVB exposed skin. These data strongly suggest that melanoma tumor initiation from MCSCs is due to stem cell activation and translocation instead of random mutation events.
Remarkably, UVB-mediated early melanomas originating from follicular MCSCs in TBP and TKP mice (Figures 2E and S4E) histologically resembled human cutaneous melanomas arising from the epidermis (Vultur and Herlyn, 2013). This phenotype contrasts with many previously described cutaneous melanoma mouse models in which melanomas initiate from the dermis (Walker et al., 2011). Upon ultraviolet radiation, tumor-prone MCSCs first migrate to the epidermis, then exhibit expansion and hyperpigmentation within the epidermis (Figures 3A, S3G and S4E), and finally invade the underlying dermis (Figures 3B–C), as shown in human cutaneous melanoma progression (Vultur and Herlyn, 2013). In contrast, depilation-induced melanoma initiation in our experimental model demonstrated malignant melanocytic tumor cells arising directly from the follicular stem cell compartment and invading the dermis during MCSC activation (Figures 3D–E). While normal melanocytes were strictly located in the hair bulb (Figure 3D), premature differentiation of active state MCSCs produced malignant melanocytes showing aggressive invasiveness and growth in the dermis (Figure 3E). Thus, these observations indicate these UVB induced melanomas originating from MCSCs may serve as a new mouse model recapitulating many of the important stages of human melanoma development.
Figure 3. Ultraviolet radiation efficiently induces MCSC-originating melanomas which arise in the epidermis and progress to the dermis.
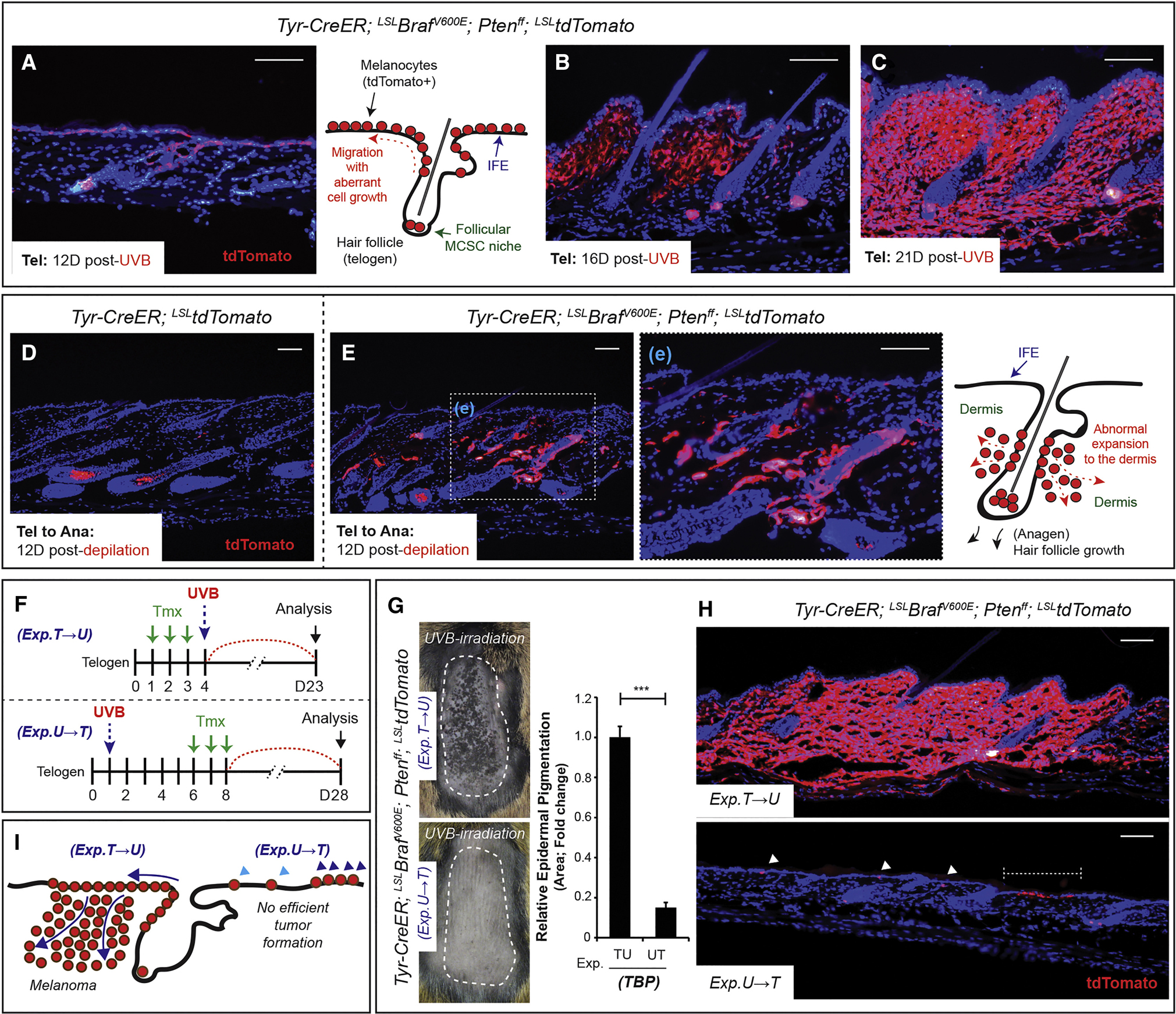
(A-C) Lineage tracing allele tdTomato shows the progression of UVB-initiated melanoma from tumor-prone MCSCs during telogen. (D-E) Lineage tracing allele tdTomato shows normal melanocytogenesis and malignant melanocytic tumorigenesis in control (TT) and TBPT mice by MCSC activation through anagen induction. (F) Experimental scheme. T, Tmx, tamoxifen; U, UVB during telogen. (G) Macroscopic phenotypes with quantification of epidermal pigmentation from TBPT mice. Mean ± SEM. n = 6 / group. (H-I) Lineage tracing of tdTomato and the summary of microscopic phenotypes. Scale bars, 100 μm. IF counterstaining, DAPI.
Given that melanoma-competent MCSCs are unable to induce melanoma while arrested in a quiescent state (Figures 1C and S2D), non-proliferating Tyrp1+ translocated melanocytes may similarly resist oncogenic stimuli. To test this, tamoxifen was administered to TBPT mice following UVB mediated translocation, rather than prior to UVB, resulting in BrafV600E expression and Pten loss in migrated melanocytes instead of actively migrating MCSCs (Figure 3F). Consistent with this hypothesis, migrated melanocytes were relatively unable to induce melanocytic tumors (Figures 3G–I).
MCSC quiescence is a melanoma suppressor during anagen and catagen
Like hair follicle stem cells, MCSCs are only briefly active in early anagen, then become quiescent and remain so during the remainder of the hair cycle (Figure 4A) (Rabbani et al., 2011). To test whether quiescent MCSCs in anagen also resist melanoma initiation, depilated skin was treated with tamoxifen to induce an oncogenic load in quiescent MCSCs during anagen (Figure 4A). Intriguingly, quiescent MCSCs were refractory to melanomagenesis during anagen and subsequently catagen (the hair follicle regression stage spanning the time period between anagen and telogen), demonstrating no evidence of cutaneous melanocytic tumor formation (Figure 4B) in the similar experimental period of melanoma formation from active MCSCs (Figure 1B). These results support the conclusion that MCSCs initiate melanocytic tumors only upon proliferative activation, whereas quiescent MCSCs and post-mitotic, terminally differentiated melanocytes present within the hair bulb during anagen resist melanocytic tumor development.
Figure 4. Ultraviolet-B exposure initiates melanomagenesis through activation of quiescent melanoma-prone MCSCs.

(A) Experimental scheme and hypothesis showing a lack of efficient melanomagenesis during MCSC quiescence (Q) periods. Tmx, tamoxifen; Tel, telogen; Ana, anagen; Cat, catagen; A, active status of MCSCs. (B) Histological phenotypes with tdTomato lineage tracing. (C) Experimental scheme and hypothesis for the role of UVB exposure in melanomagenesis through MCSC activation during the quiescent period of telogen (circled A, UVB-mediated activation). (D) Melanomagenesis via UVB irradiation during late anagen, and demonstration of melanoma initiation by tdTomato lineage tracing. (E) Early UVB-induced melanocytic hyperplasia throughout the interfollicular epidermis (IFE) demonstrated by lineage tracing. (F) Histology and tdTomato lineage tracing demonstrating UVB-induced melanomagenesis from quiescent tumor-prone MCSCs during late anagen. (G) Summary of UVB effects in melanomagenesis originating from quiescent melanoma-competent MCSCs. Activation of MCSCs is required for melanomagenesis (1, 2 and 4), UVB can significantly activate quiescent melanoma-competent MCSCs in telogen (3) and anagen (5) to induce melanomagenesis in a manner similar to human tumor initiation and progression. IF counterstaining, DAPI. Scale bars, 100 μm.
UVB can initiate melanomagenesis from quiescent MCSCs regardless of hair cycle stage
Although MCSCs are quiescent after the initial transition to anagen (Figure 4A), UVB is sufficient to induce MCSC activation and migration at any stage of the hair cycle. Given this independence, we hypothesized that UVB could induce melanomagenesis in this mouse model during anagen (Figure 4C). When TBP mice were exposed to UVB during late anagen, tumor-prone quiescent MCSCs were significantly able to produce melanocytic hyperplasia (Figures 4D–F). Similar to the melanoma progression upon UVB exposure during telogen detailed above, tumor-competent MCSCs were able to translocate and initiate melanomagenesis throughout the IFE (Figure 4E). Like human melanomas, UVB-initiated malignant melanocytic tumors induced during anagen were histologically associated with the epidermis (Figures 4F).
In summary, MCSCs cycle coincident with hair follicle cycling, and MCSCs only in an active state at the telogen to anagen transition can produce malignant melanocytes upon oncogenic expression (Figure 4G). While adult stem cell quiescence can work as a tumor suppressor in tumor-competent MCSCs (late anagen, catagen and telogen), UVB radiation is sufficient to initiate melanomagenesis from tumor-prone quiescent MCSCs regardless of hair cycle stage via MCSC activation and translocation (Figure 4G).
MCSC-originating melanoma initiation can be suppressed by attenuation of acute inflammation
To define the gene expression changes in depilation- and UVB-induced melanomas arising from MCSCs, we isolated unperturbed tumor-prone quiescent MCSCs, depilation-mediated and UVB-mediated early melanocytic tumor cells based on tdTomato expression for RNAseq analysis (Figure 5A). Though many cellular mechanisms were suggested by the differential expression profile during early melanomagenesis, including changes in cell cycle genes, the enriched classification suggested a significant potential role of inflammation-mediated processes in UVB-mediated melanomagenesis (Figures 5B–E). Altered gene expression was found in numerous genes related to leukocyte migration, such as chemokines and matrix metalloproteinases (Figures 5E). This suggests that acute inflammation may perturb the cellular activation status of MCSCs thus leading to melanoma tumor initiation. In support of this, it has been reported that inflammatory conditions can enhance cancer initiation, such as pancreatitis in pancreatic cancers (Westphalen et al., 2016) and prostatitis in prostate adenocarcinomas (Kwon et al., 2014). Therefore, we hypothesized that acute localized inflammation may induce melanoma initiation from tumor-competent quiescent MCSCs.
Figure 5. Transcriptomic analysis demonstrates differentially expressed genes during early melanomagenesis.
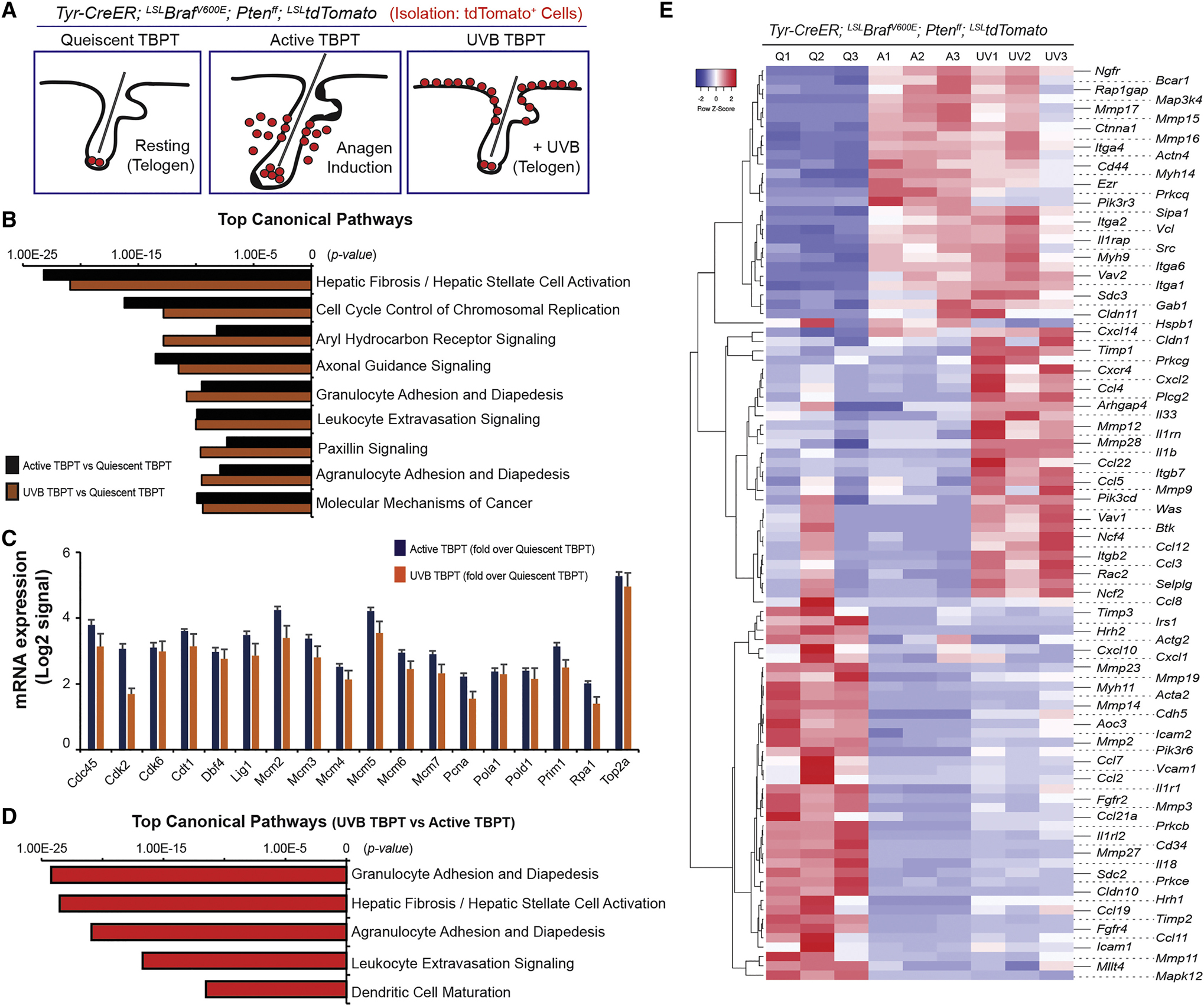
(A) Cells were isolated for quiescent tumor-prone MCSCs (Quiescent TBPT), depilation-induced active melanocytes (Active TBPT), and UVB-induced active melanocytes (UVB TBPT) from TBPT mice. (B) Top canonical pathways were enriched for quiescent TBPT versus Active TBPT, and quiescent TBPT versus UVB-TBPT. (C) mRNA expression associated with cell cycle control of chromosomal replication measured by RNAseq. (D) Top canonical pathways were enriched for UVB-mediated versus depilation-induced active melanocytes from TBPT mice. (E) Heatmap of 98 genes differentially expressed related to leukocyte migration (adhesion and extravasation) between tumor-prone quiescent MCSCs (Q1-Q3), depilation-induced active (A1-A3) and UVB-mediated active early malignant melanocytes (UV1-UV3) from TBPT mice.
Topical treatment of 12-O-tetradecanoylphorbol-13-acetate (TPA) is known to cause cutaneous inflammation and epidermal hyperplasia without mutagenesis, and has been widely used in combination with the carcinogen 7,12-Dimethylbenz[a]anthracene (DMBA) to induce SCCs (Nassar et al., 2015). To test whether acute regional inflammation via TPA could induce melanomagenesis from tumor-prone quiescent MCSCs, we first examined if TPA could alter MCSC status. TPA treatment could induce epidermal hyperplasia and immune cell recruitment (Figure S5A). Furthermore, we observed MCSC translocation during the MCSC quiescence period by topical TPA treatment (Figure 6A). As hypothesized, topical TPA significantly triggered melanoma initiation in TBP and TKP mice, whereas un-treated skin regions did not exhibit any melanocytic tumorigenesis (Figures 6B–D and S5B–C). Similar to UVB exposure, TPA-induced melanoma development was closely related to the epidermis as it induces MCSC migration into the IFE (Figures 6D and S5C), while depilation-initiated melanomas arose directly from the follicular stem cell compartment without a notable association with the epidermis (Figures 3E and S5C).
Figure 6. Melanoma formation from quiescent melanoma-competent MCSCs can be attenuated by suppression of inflammation and sunscreen application.

(A) MCSC translocation by topical TPA treatment shown by tdTomato lineage tracing. (B) Experimental scheme. (C) Macroscopic phenotypes in TBP mice with/without topical TPA treatment, and quantification of epidermal pigmentation. n = 10 TPA-treated control, n = 7 non-TPA TBP and n = 7 TPA-treated TBP mice. (D) Histological phenotypes of TPA-induced melanoma during quiescent MCSC, telogen period, and co-immunostaining for Sox10, a melanocyte stem cell/melanoma marker, with Ki67, a marker of proliferation. (E) Experimental scheme. Dex., dexamethasone. (F) Relative number of MCSC migration was quantified via tdTomato lineage tracing in TT mice, compared to the total number of hair follicles (≥ 500 hair follicles / each). Mean ± SEM, n = 10 / group. (G) Experimental scheme. (H) Macroscopic phenotypes with the quantification of epidermal pigmentation. n = 6 low-dose (80 mJ cm−2) UVB, n = 10 per each group (UVB only or UVB + Dex.). Data are represented as Mean ± SEM. Also see Figures S5 and S6.
To test the converse hypothesis, that suppression of UVB-mediated inflammation can decrease melanoma initiation from tumor-prone MCSCs, the anti-inflammatory steroid dexamethasone was administered during the course of UVB exposure and low-dose UVB exposure was tested (Figures 6E–H). Attenuation of acute inflammation by dexamethosone treatment notably suppressed MCSC migration to the IFE in wild type animals (Figures 6F and S6A), and importantly, decreased the incidence of MCSC-originating cutaneous melanoma formation upon UVB exposure (Figures 6H and S6B). Taken together, these results identify UVB-induced inflammation as one of the major factors which can mediate MCSC activation and translocation, and also suggest a potential method through which melanomas could develop from tumor-prone quiescent MCSCs in skin regions that are not regularly exposed to sunlight.
Sunscreen is believed to prevent skin cancer by suppressing many of the harmful effects mediated by UV radiation, such as DNA damage and inflammation. In addition to attenuated melanoma formation by anti-inflammation, we also found that melanoma initiation is UVB-dose dependent (Figures 6H and S6B). Therefore, we hypothesized that sunscreen may also prevent melanoma initiation from quiescent melanoma-competent MCSCs by inhibiting activation and/or migration of MCSCs to the IFE. To test this hypothesis, 3 commercially available sunscreens (SPF 50+) were screened for melanoma initiation prevention using our UVB inducible model system. To allow sunscreen to penetrate or coat the skin layers, sunscreen was applied 15 minutes prior to UVB irradiation. Interestingly, sunscreen application significantly inhibited UVB-mediated MCSC translocation (Figure S6C), potentially through the indirect effect of suppressing UVB-mediated cutaneous hyperplasia and/or immune cell recruitment. To explore the benefit of commercially available sunscreen in melanoma prevention, we applied sunscreen to 60–70% of the dorsal skin before UVB irradiation in mice harboring tumor-prone MCSCs. Notably, the inhibition of UVB-mediated MCSC activation and translocation by sunscreen application (Figure S6C) could also prevent macroscopic malignant melanocytic tumor initiation from melanoma-competent quiescent MCSCs (Figure S6D).
Hmga2 expression in the microenvironment significantly promotes efficient MCSC translocation and melanoma initiation
The presented data demonstrate that upon expression of a melanoma sufficient oncogenic combination, MCSC activation is required for melanoma initiation. The activation step can be synchronized with hair follicle cycle, but UVB exposure or topical TPA treatment can activate MCSCs without the requirement of a perturbed hair cycle. We hypothesized that although UVB might have a direct effect in MCSC activation, UVB could also indirectly regulate MCSC activation and/or translocation through the perturbation of cutaneous microenvironment.
It has been reported that ultraviolet radiation alters the expression profiles of various transcriptional regulators, such as transcription factors involved in the nuclear factor-kappaB and transforming growth factor-β signaling (Kaufman and Fuchs, 2000; Sesto et al., 2002). Among many transcriptional regulators, the expression of Hmga2 has been shown to be altered in human keratinocytes by UVB irradiation (Sesto et al., 2002), and we found significant overexpression of Hmga2 in UVB-exposed skin tissues (Figure 7A). It is known that Hmga2 is expressed in embryonic tissues, and undifferentiated and proliferating mesenchymal cells (D’Armiento et al., 2016). However, while Hmga2 is highly overexpressed in cutaneous epithelial tumors, its expression level is low in normal keratinocytes (White et al., 2016). Interestingly, we found a significant overexpression of Hmga2 in both temporarily hyper-proliferative epidermis and dermis following acute UVB exposure (Figure 7A). However, unlike cutaneous SCCs (Boumahdi et al., 2014; White et al., 2016), depilation- or UVB-induced hyper-proliferating melanocytes showed no significant increase in expression (Figure 7B). Therefore, we hypothesized that Hmga2 overexpression in the cutaneous microenvironment plays a significant role in contributing to UVB-mediated MCSC activation and migration into the IFE.
Figure 7. Loss of Hmga2 function in the microenvironment significantly suppresses MCSC activation/translocation and melanomagenesis in quiescent melanoma-competent MCSCs.
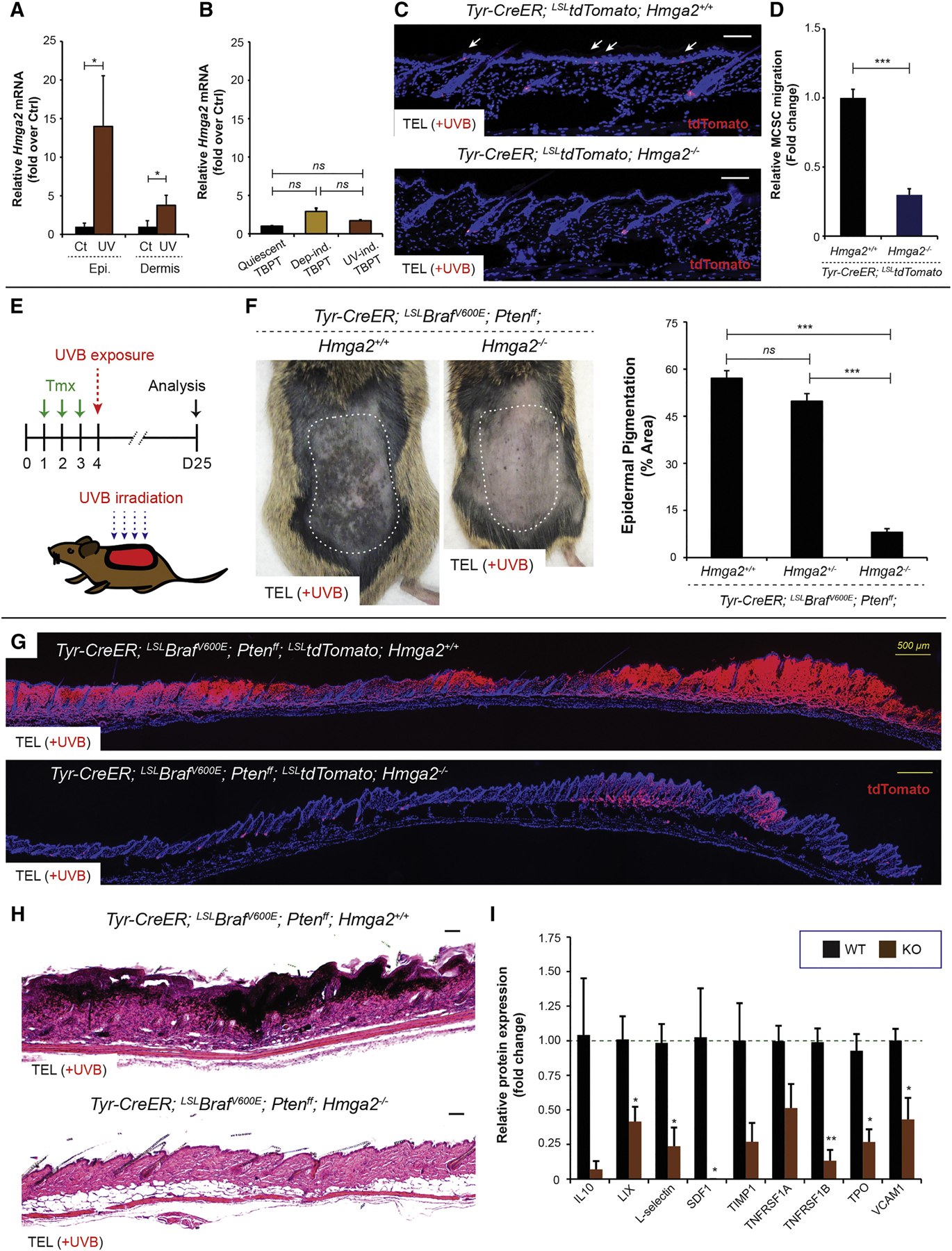
(A) Relative Hmga2 mRNA expression between control and UVB-exposed epidermis or dermis was measured using ΔCt method. n = 5. (B) Relative mRNA expression of Hmga2 was measured by RNAseq using sorted quiescent tumor-prone MCSCs (Quiescent TBPT), and depilation-induced (Dep-ind. TBPT) or UVB-induced early melanoma cells (UV-ind. TBPT). n = 3. ns = not significant. (C) tdTomato lineage tracing demonstrates no significant MCSC migration (white arrows) by UVB irradiation in Hmga2−/− mice. (D) Relative MCSC migration between TT Hmga2+/+ and TT Hmga2−/−. n = 6. (E) Experimental scheme. Tmx, tamoxifen. (F) Suppressed UVB-induced melanomagenesis in Hmga2−/− mice. Macroscopic phenotypes and quantification of epidermal pigmentation show significantly suppressed UVB-induced melanomagenesis in Hmga2−/− mice. n = 10 TBP Hmga2+/+, n = 11 TBP Hmga2+/− and n = 11 TBP Hmga2−/−. (G-H) tdTomato lineage tracing and histological phenotypes in TBP Hmga2+/+ and TBP Hmga2−/−. Sequential photomicrographs were reconstructed for the panoramic images for tdTomato lineage tracing. Counter staining, DAPI. Scale bars, 100 μm and 500 μm (yellow bars). (I) Relative protein expression level for differentially expressed chemokines/cytokines in the skin post-UVB irradiation between Hmga2+/+ and Hmga2−/−. n = 4. Data are represented as Mean ± SEM. Also see Figure S7.
First, to determine whether Hmga2 is necessary for MCSC migration, Hmga2 global knockout animals (Zhou et al., 1995) were subjected to UVB irradiation and MCSC translocation efficiency was determined through the lineage tracing tdTomato marker (Figure 7C). UVB-mediated MCSC translocation was significantly suppressed by Hmga2 loss of function in Tyr-CreER; LSL-tdTomato; Hmga2−/− mice, whereas Tyr-CreER; LSL-tdTomato; Hmga2+/+ mice showed efficient MCSC translocation after UVB exposure (Figures 7C–D). Therefore, to test whether Hmga2 loss of function in the skin can also inhibit UVB-mediated melanoma initiation, we bred Hmga2−/− animals to TBP mice and exposed these animals to UVB (Figure 7E). Strikingly, UVB irradiated TBPT; Hmga2−/− skin demonstrated a decreased incidence of melanomas (Figures 7F–H). While control TBPT; Hmga2+/+ mice showed macroscopic and aggressive melanoma formation throughout UVB-exposed skin, TBPT; Hmga2−/− skin showed a significantly lower incidence of macroscopic melanoma-related pigmentation (Figures 7F). TBPT; Hmga2+/+ skin showed malignant melanocytes evident throughout the entire UVB-exposed skin (Figures 7G–H), with almost every hair follicle associated with the presence of abnormal melanocytes during early melanomagenesis (Figures S3G and S4E). On the other hand, although some areas showed melanoma development in TBPT; Hmga2−/− skin, many MCSCs still remained at the hair germs without ectopic pigmentation or induction of melanoma markers following UVB exposure (Figures 7G and S7A).
To determine whether this loss of melanoma initiation was due to Hmga2 deletion in MCSCs or due to extrinsic environmental mechanisms as hypothesized, tamoxifen induced TBPT; Hmga2+/+ and TBPT; Hmga2−/− MCSCs were sorted by FACS and transplanted into recipient mice (Figure S7B). Consistent with a cell extrinsic mechanism for Hmga2 in regulating melanoma initiation, both TBPT; Hmga2+/+ and TBPT; Hmga2−/− MCSCs were sufficient to generate melanomas with equivalent incidence (Figure S7B–C).
Though these data support a cell extrinsic role for Hmga2 in melanoma initiation from UVB exposed tumor-prone MCSCs, Hmga2 could be important for melanoma growth after initiation. To test this, a melanoma cell line (C.MM) from UVB exposed TBPT mice was established through transplantation (Figure S7D), similar to a previous report (Jenkins et al., 2014). CRISPR/Cas9-mediated gene editing was utilized in C.MM cells to generate targeted loss of Hmga2, and CRISPR/Cas9 efficiency was confirmed at the protein level by immunoblotting (Figure S7E). Then, control and Hmga2-edited cells were transplanted, and tumor growth was monitored (Figure S7F). Both groups showed similar tumor sizes and proliferation rate, as assessed by Ki67 labeled cells, indicating no significant effect of Hmga2 in in vivo melanoma growth (Figures S7F–G).
Finally, to better understand the potential mechanism regulating decreased MCSC translocation and melanoma initiation in Hmga2−/− mice, a cytokine/chemokine screen was performed using an antibody array. Among various candidates, six factors were identified as differentially expressed at the protein level with statistical significance (Figure 7I). These included SDF1, TNFRSF1B, LIX, L-Selectin, TPO, and VCAM1 (Figure 7I). Although not significant, IL-10, TIMP1 and TNFRSF1A also showed a decreased trend. Interestingly, SDF1, also known as CXCL12, has been previously implicated in melanoblast migration (Belmadani et al., 2009), and its receptor Cxcr4 was also found significantly upregulated in UVB activated melanocytes through transcriptomics (Figure 5E and S7H). SDF1, LIX (also known as CXCL5) and TNF-α signaling pathways can promote cell migration (Xiang et al., 2017), and their potential in melanoma development and/or progression has also been implicated (Katerinaki et al., 2003; Kim et al., 2006; Singh et al., 2009). Therefore, these factors represent a potential link between Hmga2 and inflammatory pathways in melanoma initiation, and suggest a direction for future studies in MCSC-originating melanomagenesis.
Taken together, these results indicate that loss of Hmga2 function in the cutaneous microenvironment inhibits melanoma initiation by indirectly suppressing UVB-mediated tumor-competent MCSC activation and translocation, and this may be mediated fully or in part by a change in the UVB-induced inflammatory profile (Figure S7I).
DISCUSSION
It is known that the oncogenic BrafV600E driver mutation induces cellular senescence-like state, but additional mechanisms such as PI3K/AKT/mTOR pathways can help benign nevus cells overcome this growth-arrest (Damsky et al., 2015). Around 20–25% cutaneous melanomas are known to arise from pre-existing benign nevi, and many studies have focused on defining the molecular mechanisms underlying the transition from a benign cell to a malignant cell. However, the majority (≥ 75%) of melanomas arise from normal looking clear skin without this precursor lesion (Bevona et al., 2003; Marks et al., 1990). Furthermore, although BrafV600E is the most common driver mutation in melanocytic tumors, the mutation frequency is much lower in melanocytic neoplasms (40–45%) compared to benign nevi (80%) (Kato et al., 2016). These inconsistent statistical observations suggest the possibility that melanomas can arise not only from precursor benign melanocytes, but also from melanocytes or melanocyte stem cells with oncogenic combinations in normal looking, clear skin. Furthermore, we observed that BrafV600E expression is not always sufficient to induce benign melanocytic tumors, especially when adult MCSCs are in the quiescent state. This indicates that for melanoma development, driver and passenger mutations could be accumulated in quiescent adult stem cells over time without the requirement or presence of preceding benign tumors.
Recent studies have demonstrated adult stem cells as cancer cells of origin, but extrinsic factors regulating tumor initiation steps from the cellular origin remain unclear (White and Lowry, 2015). Here we demonstrate that ultraviolet radiation serves as an extrinsic stimulus which can accelerate tumor formation from tumor-competent MCSCs. While numerous molecular mechanisms may be involved in UVB-mediated melanomagenesis, we show the potential role of UVB-induced regional acute inflammation. In addition, we found that internal (untreated) control skin in both UVB and TPA treatment mice showed higher uncontrolled melanoma incidence compared to independent control melanoma mouse skin (Figures 2D and 6C). These results suggest that secreted factors from UVB-exposed skin and/or induced melanocytic tumor cells can potentially affect other skin regions, which in turn activate quiescent tumor-competent MCSCs for melanoma formation. Secreted factors, such as the chemokines/cytokines identified here, may affect MCSCs directly or indirectly in the surrounding microenvironment. Further studies will define these factors for effective MCSC-originating melanoma prevention.
Hmga2 is a chromatin remodeling transcriptional regulator that has long been suggested as an oncogene in many cancers including melanomas (Raskin et al., 2013) and widely implicated in cancer progression including tumor metastasis (Morishita et al., 2013). While its role in melanoma metastasis has been examined (Zhang et al., 2015), the functional necessity of Hmga2 in MCSC activation or early melanoma formation has never been examined. Importantly, we found the increased expression of Hmga2 in the skin, potentially due of its interplay with DNA damage response genes (Singh et al., 2015). A recent study reported that Hmga2 is dispensable during cutaneous SCC formation (White et al., 2016), although its role is generally known to be oncogenic and associated with the stemness of cutaneous SCCs (Boumahdi et al., 2014). However, our study demonstrated that Hmga2 loss of function in the skin significantly suppressed melanoma formation through inhibiting UVB-mediated activation and translocation of tumor-competent MCSCs. Importantly, although our acute UVB-mediated MCSC activation was prevented by sunscreen application, it is also known that potentially chronic long-term UVB exposure can eventually induce additional mutations in melanocytes which may cause cutaneous melanoma development (Viros et al., 2014). Therefore, Hmga2 and related molecular pathways need to be considered for combinational preventative therapy together with sunscreen application for skin cancer prevention, especially for melanomas.
In summary, this report defines the role of MCSCs as melanoma cells of origin and identifies UVB as an extrinsic stimulus capable of initiating melanomagenesis via an acute inflammation-dependent process. Moreover, Hmga2 loss of function in the cutaneous microenvironment can greatly reduce MCSC activation and translocation, and UVB-initiated melanomas, thus providing new strategies for MCSC-originating melanoma prevention.
STAR★ METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents should be directed to, and will be fulfilled by, the Lead Contact, Andrew White (acw93@cornell.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All animal experiments and related procedures were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee at Cornell University. In conducting research using animals, the investigator adhered to the laws of the United States and regulations of the Department of Agriculture. Mice acquired from Jackson Labs, the National Cancer Institute Mouse Models of Human Cancers Consortium repository and the Chada lab (Zhou et al., 1995) were maintained by Cornell Center for Animal Resources and Education. All genotyping was done as previously published (Anand and Chada, 2000; Lesche et al., 2002) or recommended by Jackson Labs and NCI (Table S1). For co-expression of tdTomato and H2B-GFP in Tyr-CreER; LSL-tdTomato; Dct-rtta; Tre-H2B-Gfp mice, drinking water containing doxycycline (200 μg ml−1 dissolved in sterilized water) was continuously provided from 5 days before intraperitoneal (IP) injection of 2 mg of tamoxifen per day (200 μl of 10 mg ml−1 dissolved in filtered sunflower seed oil 3 days). In pilot studies, we confirmed that MCSCs expressing H2B-GFP in the hair germ during telogen and in the bulge during anagen are Trp2 positive, and dMCs at hair bulbs during anagen are Trp2 and Tyrp1 positive. For melanocytic tumorigenesis, mice were treated by IP injection of 2 mg of tamoxifen per day (200 μl of 10 mg ml−1 dissolved in filtered sunflower seed oil) at the designated time periods for each experiment. Since Hmga2 knockout induces a pygmy mouse phenotype (Zhou et al., 1995), 100 μg of tamoxifen per 1 g body weight per day (10 μl of 10 mg ml−1 dissolved in filtered sunflower seed oil per 1 g body weight) was administered intraperitoneally prior to each experiment for the study of Hmga2 loss of function in MCSC translocation and melanomagenesis. Seven weeks postnatal young adult mice were included for each experiment, but any mice showing pre-existing macroscopic pigmentation at the shaved dorsal skin of interest, due to leaky Cre activation, were excluded from the studies. All animals shown were maintained on a mixed C57BL6/FVB background. All experimental phenotypes described in the study are representative of a minimum of n = 6 animals (mixed genders of at least 3 males and 3 females) as denoted in each experimental legend.
Primary murine melanoma cells
Isolated murine primary melanoma cells from TBPT mice (mixed genders) were cultured in DMEM/F12 containing 10% FBS and antibiotics (Pen/Strep). C.MM cell line obtained through transplantation of murine melanoma cells in NOD-scid IL2Rgnull mice was cultured in DMEM containing 10% FBS and Pen/Strep. Lentivirial transduced C.MM cell lines were cultured in DMEM containing 10% FBS, Pen/Strep and puromycin. All primary cells were cultured in 5% CO2 cell culture incubator at 37°C.
METHOD DETAILS
Chemical depilation
After shaving mouse dorsal skin using a small animal clipper, chemical depilation was done using hair remover lotion. During depilation, mice were shortly anesthetized using isoflurane for careful application of Nair cream onto the dorsal skin region of interest. The level of oxygen and isoflurane was maintained by Matrx VIP 3000® vaporizer. To prevent any cutaneous irritation by Nair, the skin was gently wiped with wet tissues immediately after hair removal, and then dried.
UVB, dexamethasone and sunscreen application
UVB irradiation was performed using UVB bench lamps, and the amount of UVB delivered was measured using a calibrated UV light meter. During the telogen period, tamoxifen was intraperitoneally administered for 3 days prior to UVB irradiation in mice containing the Tyr-CreER allele. In Dct-rtta; Tre-H2B-Gfp mice, drinking water including doxycycline was provided at least two days before UVB exposure. To perform UVB irradiation on the skin region of interest, mice were anesthetized using isoflurane, and then 180 mJ cm−2 UVB was irradiated onto the shaved dorsal skin. Internal control skin was also shaved at the time of irradiation, but was protected by a covering of UV resistant material. For UVB irradiation during late anagen, chemical depilation was performed 14 days before intraperitoneal tamoxifen injection. Then, 180 mJ cm−2 UVB irradiation was performed under the anesthesia. For dose-dependent experiments, low-dose irradiation exposures included 40 mJ cm−2 and 80 mJ cm−2 UVB. To attenuate UVB-induced acute inflammation, 10 μg g−1 dexamethasone was subcutaneously injected 1 day prior to and post UVB-irradiation. In this study, 3 different commercially available broad spectrum SPF50 (lotion) sunscreens recommended by the Skin Cancer Foundation were used. The details of each sunscreen are as follows: type A, Banana Boat® SPF50 (active ingredients containing titanium dioxide and zinc oxide); type B, Aveeno® (active ingredients including avobenzone, homosalate, octisalate, octocrylene, oxybenzone); type C, Aveeno® (active ingredients containing titanium dioxide and zinc oxide).
TPA treatment
After tamoxifen IP injection, mice were treated with 4 μg of TPA dissolved in 200 μl ethanol topically on the shaved skin region of interest at the designated time periods for each experiment.
Tissue immunostaining
Formalin-fixed paraffin-embedded (FFPE) tissue sections were cut at 5 μm and frozen tissues were cut at 6–8 μm for haematoxylin and eosin staining, and immunostaining. For tdTomato imaging, tissues were fixed in 10% neutral buffered formalin (Thermo Fisher Scientific) at 4°C overnight before embedding in OCT (Tissue-Tek, Sakura). Immunofluorescence staining on fresh frozen sections and immunohistochemistry (IHC) on FFPE tissue sections were performed as previously described (White et al., 2011). IHC was performed with antigen retrieval using antigen unmasking solution (citric acid based, Vector) for 30 min at 90°C. Antibodies were used as anti-S100 (1:200), anti-y-H2A.X (1:500), anti-Tenascin C (1:400), anti-SOX10 (1:200), anti-GFP (1:600), anti-Tyrp1 (1:200), anti-Trp2 (1:200), anti-F4/80 (1:400), anti-K5 (1:600), anti-CD45 (1:400), anti-Gr-1 (1:400), anti-Ki67 (1:200, #900-C01-B38 and 1:400, #14–5698), together with suitable Alexa-fluor or biotinylated secondary antibodies for IF and IHC. The IHC signal was amplified and detected using VECTASTAIN Elite ABC HRP Kit and AEC Peroxidase Substrate Kit followed by mounting using ImmunoHistoMount™ (Sigma). Prolong Gold antifade mountant with DAPI were used for IF staining.
FACS, RNAseq and bioinformatics
To compare gene expression profiles from tumor-prone quiescent MCSCs with chemical depilation and UVB-induced early active malignant melanocytes, total RNA was isolated from sorted cells. Through FACS, tdTomato positive cells were sorted from Tyr-CreER; LSLBrafV600E; Ptenff; LSLtdTomato mice, around 15 days after each treatment such as chemical depilation or UVB irradiation. FACS was performed as previously explained (White et al., 2014). Briefly, dorsal skin from Tyr-CreER; LSLBrafV600E; Ptenff; LSLtdTomato mice was digested with collagenase type I and IV (20mg ml−1 dissolved in serum free DMEM/F12) for 2 hr at 37°C. Digested skin tissues were mechanically dissociated using a serological pipette and filtered with a 100 μm cell strainer. Cells were washed and collected in cold PBS after centrifugation at 220 g for 7 min at 4°C, and then filtered twice through a 40 μm cell strainer for FACS. tdTomato positive cells sorted through BD FACS Aria (BD Biosciences) were directly collected in Trizol LS reagent and homogenized by vortexing. Total RNA isolation was subsequently performed following the manufacturer’s instruction including steps with chloroform, isopropanol and ethanol wash. During the isolation, linear acrylamide (Thermo Fisher Scientific) was used as a co-precipitant. Isolated RNA was stored at −80°C for future experiments, and the quality of RNA was measured on a fragment analyzer at Cornell Biotechnology Resource Center. Then, RNAseq was conducted through Cornell RNA Sequencing Core facility. To prepare Illumina library preparation, TruSeq-barcoded RNAseq libraries were generated using the NEBNext Ultra Directional RNA Library Prep Kit with 500 ng total RNA input. Prior to pooling, each library was quantified using a Qubit 2.0 dsDNA HS kit, and the size distribution was also determined on a Fragment Analyzer. Libraries were sequenced on a NextSeq500 instrument (Illumina). At least 25M single-end 75bp reads were generated per library. For the RNAseq data, reads were trimmed for low quality and adaptor sequences with cutadapt v1.8 (Martin, 2011) and mapped to the reference transcriptome/genome (UCSC mm10) using tophat v2.1 (Kim et al., 2013). For gene expression analysis, cufflinks v2.2 was used to generate FPKM values and analyse differential gene expression (Trapnell et al., 2013). Heatmaps for gene expression were generated using Heatmapper (Babicki et al., 2016). Canonical pathways were enriched through the use of QIAGEN’s Ingenuity® Pathway Analysis (IPA®, QIAGEN Redwood City, www.qiagen.com/ingenuity).
PCR and real time quantitative RT-PCR
Mouse tail snips were collected according to IACUC approved procedures and digested in 0.05N NaOH (Sigma) at 95°C for 1.5 hours, then briefly vortexed and neutralized with 1M Tris-HCl, pH 8 (Sigma). Genotyping was performed by PCR using G-Bioscience Taq Polymerase and supplied 10x PCR buffer in a solution with dNTPs, RediLoad™ loading buffer (Invitrogen) with appropriate forward and reverse primers as described by the Jackson Labs or NCI. PCR product was run on 2% Agarose gel and imaged using BioRad ChemiDoc™ XRS+ for subsequent analysis. To confirm genomic recombination in the cells of interest, regional melanoma to use as a positive control was induced by the topical application of 4-hydroxytamoxifen (5mM dissolved in 100% ethanol) followed by chemical depilation. Skin and induced melanoma tissues were harvested and DNA collected using Invitrogen Pure Link® Genomic DNA mini kit. tdTomato positive or negative cells were directly sorted into Trizol-LS and DNA isolated by the manufacturer’s instructions. PCR was then performed as previously described (Dankort et al., 2007; Groszer et al., 2001; Lesche et al., 2002). PCR primers to determine genetic recombination include Braf-F, Braf-R, Pten-1, Pten-2 and Pten-3 (Table S1). Epidermal keratinocytes and dermis tissues were separated using Trypsin-EDTA (Thermo Fisher Scientific) as previously published (Soteriou et al., 2016).
qPCR was carried out using SYBR green after Trizol-based RNA extraction followed by cDNA synthesis using SuperScript III First-Strand Synthesis System. Hmga2 expression was assessed using Hmga2-F and -R primers as previously published (Boumahdi et al., 2014), and its relative expression was normalized to β-actin using primers Actb-F and -R (Table S1).
In vitro genome engineering
To perform clustered regularly interspaced short palindrome repeats (CRISPR)-associated nuclease Cas9 (CRISPR/Cas9)-mediated in vitro genome editing in murine melanoma cells, we first isolated murine melanoma cells from TBPT mice. For more efficient in vitro cell culture of murine melanoma cells, we further generated a Cornell Murine Melanoma (C.MM) cell line via transplantation in NOD-scid IL2Rgnull mice, similar to a previous report (Jenkins et al., 2014). Then, in vitro genome editing via lentiviral transduction was performed as previously reported (Sanjana et al., 2014). Briefly, sgRNAs for genome-wide, targeted loss of Hmga2 function were cloned into lentiCRISPRv2 vectors. The lentiCRISPRv2 plasmid was linearized using Esp3I and guide sequences ligated in using the Rapid DNA Ligation Kit. The modified plasmid was then transformed into One Shot™ Stbl3™ Chemically Competent E. coli, and grown in LB media with 100μg/mL Ampicillin. The plasmid was harvested using the E.Z.N.A® Plasmid Midi Kit. In the current study, sgRNAs targeting three different sequences for murine Hmga2 were used: CACCTTCTGGGCTGCTTTAG, CCCCTCTAAAGCAGCCCAGA and ATCCTCGCAAGAGTCCGCAG. Lentiviral packaging was done in HEK293T cells (kindly provided by Dr. Scott Coonrod) by co-transfection with packaging plasmids psPAX2 and pMD2.G using Lipofectamine 3000 and OptiMEM (Invitrogen) in DMEM, GlutaMAX™. Lentiviral supernatant was filtered (0.45 μm membrane). Before the transduction, Polybrene (Millipore) was also added to enhance the infection rate. Since embryonic tissues highly express Hmga2, the high efficiency of gene editing was initially tested in MEF cells (generously provided by Dr. Sergiy Libert). Then, C.MM cells were cultured in DMEM with 10% fetal bovine serum (FBS, Hyclone) and Pen/Strep. At 48 hours post-lentiviral transduction in C.MM cells, puromycin was added into the growth media. After 12 days selection period, control (control lentiviral vector) and experimental (targeted loss of Hmga2 function) cell pellets were harvested by centrifugation at 300 g, 4°C to further verify the protein expression level of Hmga2 through immunoblotting. Both control and experimental transduced cell lines were maintained in the selection media, DMEM including 10% FBS, Pen/Strep and puromycin, until in vivo transplantation experiments. In the conduct of research utilizing recombinant DNA, the investigator adhered to NIH Guidelines for research involving recombinant DNA molecules.
Western blotting
Melanoma cell pellets were prepared by centrifugation at 300 g, and then total cells were lysed in 1x cell lysis buffer (Cell Signaling technology) on ice. 1 mM PMSF was added into the lysis buffer immediately before use. After removing insoluble material by centrifugation at 10,000 g at 4°C for 5 min, total protein concentration was determined using BCA assay as manufacturer’s instruction with a microplate reader. Cell lysate (20 μg) was diluted in SDS-PAGE gel electrophoresis sample buffer (Bio-Rad) and boiled at 95°C for 5 min. Denatured proteins were resolved on SDS-PAGE gels (TGX™ FastCast™ Acrylamide Solutions, Bio-Rad) and transferred onto PVDF membranes (Bio-Rad). Blocking was done with 5% milk, and then membranes were incubated with primary antibodies; anti-Cas9 (1:1,000), anti-HMGA2 (1:1,000) and anti-tubulin (1:5,000) overnight at 4°C. After washing, membranes were incubated with secondary antibodies (peroxidase-conjugated, suitable for each primary antibody) for 1 hr at room temperature. The signal was detected using Bio-Rad ChemiDoc™ XRS+ System after adding SuperSignal West Pico chemiluminescence.
MCSC and melanoma allo-transplantation
Every allo-transplantation experiment was done in NOD-scid IL2Rgnull mice. To determine the relative melanomagenic capabilities of melanoma-prone MCSCs from either TBPT; Hmga2+/+ or TBPT; Hmga2−/− mouse dorsal skin, cell sorting via FACS was performed during telogen 3 days after tamoxifen intraperitoneal injection. tdTomato positive or negative cells (5 × 104 cells) mixed with Matrigel (1:1 mixture, total volume 100 μL) were subcutaneously injected into the recipient mice. In vivo melanoma formation was determined 45 days post-transplantation. Similarly, in vitro cultured control or Hmga2 gene edited experimental C.MM cell lines were harvested and mixed with Matrigel (5 × 105 cells, 50% Matrigel in total 100 μL) before in vivo transplantation. In vivo melanoma growth was monitored for 21 days. C.MM tumor volume was determined using a modified ellipsoid formula, 0.5 x (length x width2).
Cytokine/chemokine screening
Mouse dorsal skin was irradiated by UVB and total protein lysates were then collected from the skin using cell lysis buffer with tissue homogenizer 24 hours post-UVB irradiation. All steps were performed on ice. Then, using BCA assay, protein concentration was determined for skin lysates. The same amount of protein lysate (500 ng) was used to quantitatively detect 62 mouse chemokine/cytokine protein candidates between groups using RayBio® C-Series Mouse Cytokine Antibody Array following the manufacturer’s instructions.
QUANTIFICATION AND STATISTICAL ANALYSIS
Microscopy images were taken using a Leica DM2500 upright microscope with a DFC7000T camera for fluorescence and bright field microscopy operated by Leica Application Suite X. Image analysis was carried out using Image J. % area of melanoma markers or lineage tracing (green or red channel) was quantified compared to the total area of DAPI positive regions. The relative number of Ki67 positive cells were determined compared to the total number of DAPI positive cells. Pair-wise comparisons between two groups were performed by two-tailed statistical analysis using Student’s t-test. The statistical significance in the contingency table regarding MCSC-originating melanoma development between two groups was determined by two-tailed, Fisher’s exact test. Statistical significances were considered if p < 0.01 (*); p < 0.001 (**); p < 0.0001 (***). Experimental data are demonstrated as the mean ± standard error of the mean (SEM). Statistical analysis was done using GraphPad Prism. Sample size and statistical details can be found in the figure legends.
DATA AND SOFTWARE AVAILABILITY
The RNAseq data in this paper have been deposited at the Gene Expression Omnibus (NCBI) with GEO series accession number GSE102597.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Antibodies | ||
| Rabbit monoclonal anti-phospho-Histone H2A.X (Ser139) | Cell Signaling Technology | Cat#9718S; RRID: AB_2118009 |
| Rat monoclonal anti-Tenascin C | R & D systems | Cat#MAB2138; RRDI: AB_2203818 |
| Rabbit monoclonal anti-SOX10 | Abcam | Cat#ab155279; RRID: AB_2650603 |
| Rabbit polyclonal anti-S100 A6 Ab-9 | Thermo Fisher Scientific | Cat#RB-1805-A; RRID: AB_149337 |
| Chicken polyclonal anti-GFP | Abcam | Cat#ab13970; RRID:AB_300798 |
| Rabbit polyclonal anti-TRP1 | Santa Cruz Biotechnology | Cat#sc-25543; RRID:AB_2211150 |
| Goat polyclonal anti-TRP2 | Santa Cruz Biotechnology | Cat#sc-10451; RRID:AB_793582 |
| Rat monoclonal anti-F4/80 | Novus | Cat#NB600–404; RRID:AB_10003219 |
| Chicken polyclonal anti-Keratin5 | BioLegend | Cat#SIG-3475; RRID:AB_10720202 |
| Rat monoclonal anti-CD45 | BioLegend | Cat#103101; RRID:AB_312966 |
| Rat monoclonal anti-Ly-6G/Ly-6C (Gr-1) | BioLegend | Cat#108403; RRID:AB_313368 |
| Rabbit monoclonal anti-Ki-67 | Rockland | Cat#900-C01-B38; RRID:AB_2142230 |
| Rat monoclonal anti-Ki-67 | Thermo Fisher Scientific | Cat#14–5698-82; RRID:AB_10854564 |
| Mouse monoclonal anti-Cas9 | Thermo Fisher Scientific | Cat#MA1–201; RRID:AB_2610640 |
| Rabbit polyclonal anti-HMGI-C | Santa Cruz Biotechnology | Cat#sc-30223; RRID:AB_2248305 |
| Rabbit monoclonal anti-HMGA2 | Cell Signaling Technology | Cat#8179S; RRID:AB_11178942 |
| Rat monoclonal anti-Tubulin | Novus | Cat#NB 600–506; RRID:AB_343284 |
| Chicken anti-goat IgG (H+L) cross-adsorbed secondary antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat#A-21467; RRID:AB_2535870 |
| Chicken anti-goat IgG (H+L) cross-adsorbed secondary antibody, Alexa Fluor 594 | Thermo Fisher Scientific | Cat# A-21468; RRID:AB_2535871 |
| Donkey anti-goat IgG (H+L) cross-adsorbed secondary antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat#A-11055; RRID:AB_2534102 |
| Donkey anti-goat IgG (H+L) cross-adsorbed secondary antibody, Alexa Fluor 594 | Thermo Fisher Scientific | Cat#A-11058; RRID:AB_2534105 |
| Donkey anti-rabbit IgG (H+L) highly cross-adsorbed secondary antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat#A-21206; RRID:AB_2535792 |
| Donkey anti-rabbit IgG (H+L) highly cross-adsorbed secondary antibody, Alexa Fluor 594 | Thermo Fisher Scientific | Cat#A21207; RRID:AB_141637 |
| Goat anti-rabbit IgG (H+L) cross-adsorbed secondary antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat#A-11008; RRID:AB_143165 |
| Goat anti-rabbit IgG (H+L) cross-adsorbed secondary antibody, Alexa Fluor 594 | Thermo Fisher Scientific | Cat#A-11012; RRID:AB_2534079 |
| Donkey anti-rat IgG (H+L) highly cross-adsorbed secondary antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat#A-21208; RRID:AB_2535794 |
| Donkey anti-rat IgG (H+L) highly cross-adsorbed secondary antibody, Alexa Fluor 594 | Thermo Fisher Scientific | Cat#A-21209; RRID:AB_2535795 |
| Goat anti-rat IgG (H+L) cross-adsorbed secondary antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat#A-11006; RRID:AB_2534074 |
| Goat anti-rat IgG (H+L) cross-adsorbed secondary antibody, Alexa Fluor 594 | Thermo Fisher Scientific | Cat#A-11007; RRID:AB_2534075 |
| Goat anti-chicken IgY (H+L) secondary antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat#A-11039; RRID:AB_2534096 |
| Goat anti-chicken IgY (H+L) secondary antibody, Alexa Fluor 594 | Thermo Fisher Scientific | Cat#A-11042; RRID:AB_2534099 |
| Biotinylated goat anti-rabbit IgG antibody | Vector Laboratories | BA-1000; RRID:AB_2313606 |
| Donkey anti-rabbit IgG (H&L) antibody peroxidase conjugated | Rockland | Cat#611–703-127; RRID:AB_218614 |
| Donkey anti-rat IgG (H&L) antibody peroxidase conjugated | Rockland | Cat#612–703-002; RRID:AB_219797 |
| Donkey anti-mouse IgG (H&L) antibody peroxidase conjugated | Rockland | Cat#710–703-124; RRID:AB_217688 |
| Bacterial and Virus Strains | ||
| One Shot™ Stbl3™ Chemically Competent E. Coli | Invitrogen | Cat#C737303 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Doxycycline hyclate | Alfa Aesar | Cat#J60579 |
| Tamoxifen | Sigma-Aldrich | Cat#T5648 |
| 4-Hydroxytamoxifen | Sigma-Aldrich | Cat#T176 |
| Phorbol 12-myristate 13-acetate | AdipoGen | Cat#A00083/B |
| Dexamethasone | TCI America | Cat#D1961 |
| Esp3I | ThermoFisher Scientific | Cat#ER0451 |
| DTT | Amresco | Cat#0281–25G |
| Deoxynucleotide Mix | G-Biosciences | Cat#786–442 |
| Taq DNA Polymerase | G-Biosciences | Cat#786–447 |
| Ethidium Bromide | Amresco | Cat#X328–10mL |
| Matrigel® Matrix Basement Membrane | Corning | Cat#354234 |
| Puromycin dihydrocholoride, 99% | Alfa Aesar | Cat#J61278 |
| Penicillin-Streptomycin | Thermo Fisher Scientific | Cat#15140122 |
| Collagenase type I | Gibco | Cat#17100–017 |
| Collagenase type IV | Gibco | Cat#17104–019 |
| TR zol™ LS | Invitrogen | Cat#10296028 |
| Lipofectamine™ 3000 | Invitrogen | Cat#L3000–001 |
| Isoflurane | Piramal Healthcare | Cat#NDC66794–013 |
| Nair™ | Church & Dwight | |
| Critical Commercial Assays | ||
| Rapid DNA Ligation Kit | Thermo Fisher Scientific | Cat#K1422 |
| EZNA Plasmid Midi Kit | Omega Bio-Tek | Cat#D6904 |
| PureLink™ Genomic ONA Mini Kit | Invitrogen | Cat#K1820–01 |
| SuperScript® II First-Strand Synthesis SuperMix | Invitrogen | Cat#18080–400 |
| PerfeCTa® SYBR® Green SuperMix | Quanta BioSciences | Cat#95054–100 |
| VECTASTAIN Elite ABC HRP Kit | Vector Laboratories | Cat#PK-6100 |
| ImmPACT AEC Peroxidase (HRP) Substrate | Vector Laboratories | Cat#SK-4205 |
| BCA assay | Thermo Fisher Scientific | Cat#23227 |
| SuperSignal West Pico chemiluminescence | Thermo Fisher Scientific | Cat#34087 |
| NEBNext® Ultra™ Directional RNA Library Prep Kit | New England BioLabs | Cat#E7420 |
| Qubit™ dsONA HS Assay Kit | Thermo Fisher Scientific | Cat#Q32851 |
| Mouse Cytokine Antibody Array C3 | RayBiotech | Cat#AAM-CYT-3–8 |
| Deposited Data | ||
| RNAseq data | This paper | GEO: GSE102597 |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | ATCC® CRL-3216™ |
| Murine melanoma cell | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| NOD-scid IL2Rgnull | Jackson Laboratories | Stock#: 005557 |
| B6.Cg-Tg(Tyr-cre/ERT2)13Bos/J | Jackson Laboratories | Stock#: 012328 |
| B6.129P2(Cg)-Braftm1Mmcm/J | Jackson Laboratories | Stock#: 017837 |
| C;129S4-Ptentm1Hwu/J | Jackson Laboratories | Stock#: 004597 |
| B6;129S6-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J | Jackson Laboratories | Stock#: 007908 |
| LSL K-ras G12D | NCI | Stock#: 01XJ6 |
| iDCT-GFP | NCI | Stock#: 01XT4 |
| Hmga2−/+ | Zhou et al., 1995 | |
| Oligonucleotides | ||
| See Table S1 for oligonucleotide sequences | ||
| Recombinant DNA | ||
| lentiCRISPRv.2 | Addgene | Cat#52961 |
| psPAX2 | Addgene | Cat#12260 |
| pMD2.G | Addgene | Cat#12259 |
| Software and Algorithms | ||
| Heatmapper | Babicki et al., 2016 | http://www.heatmapper.ca |
| Ingenuity® Pathway Analysis | QIAGEN | www.qiagen.com/ingenuity |
| Cutadapt | Martin, 2011 | https://cutadapt.readthedocs.io/en/stable/ |
| TopHat | Kim et al., 2013 | http://ccb.jhu.edu/software/tophat/index.shtml |
| Cufflinks | Trapnell et al., 2013 | http://cole-trapnell-lab.github.io/cufflinks/ |
| CRISPR Design | Zhang Lab, MIT 2015 | http://crispr.mit.edu |
| Leica Application Suite X | Leica Biosystems | |
| GraphPad Prism | GraphPad Software | http://www.graphpad.com/scientific-software/prism/ |
| Photoshop | Adobe | http://www.adobe.com/# |
| Illustrator | Adobe | http://www.adobe.com/# |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Other | ||
| Prolong™ Gold Antifade Mountant with OAP | Invitrogen | Cat#P36935 |
| Sunflower Seed Oil | Sigma-Aldrich | Cat#S5–007 |
| UV Light Meter | SPER Scientific | Cat#850009 |
| UVB bench lamps | UVP | |
| DME/F12 | Hyclone | Cat#SH30023.02 |
| Opti-MEM | Gibco | Cat#51985–034 |
| DMEM | Corning | Cat#10–013-CV |
| DMEM, GlutaMAX™ | Gibco | Cat#10566–016 |
| Small animal clipper | Wahl | Chro Mini |
| Matrx VIP 3000® vaporizer | Midmark | |
ACKNOWLEDGEMENTS
We thank Drs. Tudorita Tumbar, Robert Weiss, Alex Nikitin and Mark Roberson for providing scientific comments, Dr. Kiran Chada for Hmga2 knockout animals, Dr. Scott Coonrod for HEK293T cells, Nicole Grbic, Silas Ferraro, Jiaming Huang and Carly Eubanks for technical assistance. This work was supported by the Office of the Assistant Secretary of Defense for Health Affairs, through the Peer Reviewed Cancer Research Program under Award No. W81XWH-16-1-0272. Opinions, interpretations, conclusions and recommendations are those of the authors and are not necessarily endorsed by the Department of Defense. This study was also supported by a Cornell Center for Vertebrate Genomics scholar award to H.M.; and a NIH-NIAMS award (5R01AR057409) to W.E.L.
REFERENCES
- Anand A, and Chada K (2000). In vivo modulation of Hmgic reduces obesity. Nat Genet 24, 377–380. [DOI] [PubMed] [Google Scholar]
- Belmadani A, Jung H, Ren D, and Miller RJ (2009). The chemokine SDF-1/CXCL12 regulates the migration of melanocyte progenitors in mouse hair follicles. Differentiation 77, 395–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevona C, Goggins W, Quinn T, Fullerton J, and Tsao H (2003). Cutaneous melanomas associated with nevi. Arch Dermatol 139, 1620–4; discussion 1624. [DOI] [PubMed] [Google Scholar]
- Bosenberg M, Muthusamy V, Curley DP, Wang Z, Hobbs C, Nelson B, Nogueira C, Horner JW, Depinho R, and Chin L (2006). Characterization of melanocyte-specific inducible Cre recombinase transgenic mice. Genesis 44, 262–267. [DOI] [PubMed] [Google Scholar]
- Boumahdi S, Driessens G, Lapouge G, Rorive S, Nassar D, Le Mercier M, Delatte B, Caauwe A, Lenglez S, Nkusi E, et al. (2014). SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature 511, 246–250. [DOI] [PubMed] [Google Scholar]
- Chou WC, Takeo M, Rabbani P, Hu H, Lee W, Chung YR, Carucci J, Overbeek P, and Ito M (2013). Direct migration of follicular melanocyte stem cells to the epidermis after wounding or UVB irradiation is dependent on Mc1r signaling. Nat Med 19, 924–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Armiento J, Shiomi T, Marks S, Geraghty P, Sankarasharma D, and Chada K (2016). Mesenchymal Tumorigenesis Driven by TSC2 Haploinsufficiency Requires HMGA2 and Is Independent of mTOR Pathway Activation. Cancer Res 76, 844–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damsky W, Micevic G, Meeth K, Muthusamy V, Curley DP, Santhanakrishnan M, Erdelyi I, Platt JT, Huang L, Theodosakis N, et al. (2015). mTORC1 activation blocks BrafV600E-induced growth arrest but is insufficient for melanoma formation. Cancer Cell 27, 41–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damsky WE, Curley DP, Santhanakrishnan M, Rosenbaum LE, Platt JT, Gould Rothberg BE, Taketo MM, Dankort D, Rimm DL, McMahon M, et al. (2011). β-catenin signaling controls metastasis in Braf-activated Pten-deficient melanomas. Cancer Cell 20, 741–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dankort D, Filenova E, Collado M, Serrano M, Jones K, and McMahon M (2007). A new mouse model to explore the initiation, progression, and therapy of BRAFV600E-induced lung tumors. Genes Dev 21, 379–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE, You MJ, DePinho RA, McMahon M, and Bosenberg M (2009). Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet 41, 544–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groszer M, Erickson R, Scripture-Adams DD, Lesche R, Trumpp A, Zack JA, Kornblum HI, Liu X, and Wu H (2001). Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science 294, 2186–2189. [DOI] [PubMed] [Google Scholar]
- Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat J-P, Nickerson E, Auclair D, Li L, Place C, et al. (2012). A landscape of driver mutations in melanoma. Cell 150, 251–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins MH, Steinberg SM, Alexander MP, Fisher JL, Ernstoff MS, Turk MJ, Mullins DW, and Brinckerhoff CE (2014). Multiple murine BRaf(V600E) melanoma cell lines with sensitivity to PLX4032. Pigment Cell Melanoma Res 27, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katerinaki E, Evans GS, Lorigan PC, and MacNeil S (2003). TNF-alpha increases human melanoma cell invasion and migration in vitro: the role of proteolytic enzymes. Br J Cancer 89, 1123–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato S, Lippman SM, Flaherty KT, and Kurzrock R (2016). The conundrum of genetic “drivers” in benign conditions. J Natl Cancer Inst 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman CK, and Fuchs E (2000). It’s got you covered. NF-kappaB in the epidermis. J Cell Biol 149, 999–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Mori T, Chen SL, Amersi FF, Martinez SR, Kuo C, Turner RR, Ye X, Bilchik AJ, Morton DL, et al. (2006). Chemokine receptor CXCR4 expression in patients with melanoma and colorectal cancer liver metastases and the association with disease outcome. Ann Surg 244, 113–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon O-J, Zhang L, Ittmann MM, and Xin L (2014). Prostatic inflammation enhances basal-to-luminal differentiation and accelerates initiation of prostate cancer with a basal cell origin. Proc Natl Acad Sci U S A 111, E592–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang D, Mascarenhas JB, and Shea CR (2013). Melanocytes, melanocyte stem cells, and melanoma stem cells. Clin Dermatol 31, 166–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapouge G, Youssef KK, Vokaer B, Achouri Y, Michaux C, Sotiropoulou PA, and Blanpain C (2011). Identifying the cellular origin of squamous skin tumors. Proc Natl Acad Sci U S A 108, 7431–7436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesche R, Groszer M, Gao J, Wang Y, Messing A, Sun H, Liu X, and Wu H (2002). Cre/loxP-mediated inactivation of the murine Pten tumor suppressor gene. Genesis 32, 148–149. [DOI] [PubMed] [Google Scholar]
- Lo JA, and Fisher DE (2014). The melanoma revolution: from UV carcinogenesis to a new era in therapeutics. Science 346, 945–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks R, Dorevitch AP, and Mason G (1990). Do all melanomas come from “moles”? A study of the histological association between melanocytic naevi and melanoma. Australas J Dermatol 31, 77–80. [DOI] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10. [Google Scholar]
- Milagre C, Dhomen N, Geyer FC, Hayward R, Lambros M, Reis-Filho JS, and Marais R (2010). A mouse model of melanoma driven by oncogenic KRAS. Cancer Res 70, 5549–5557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AJ, and Mihm MC (2006). Melanoma. N Engl J Med 355, 51–65. [DOI] [PubMed] [Google Scholar]
- Morishita A, Zaidi MR, Mitoro A, Sankarasharma D, Szabolcs M, Okada Y,D’Armiento J, and Chada K (2013). HMGA2 is a driver of tumor metastasis. Cancer Res 73, 4289–4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassar D, Latil M, Boeckx B, Lambrechts D, and Blanpain C (2015). Genomic landscape of carcinogen-induced and genetically induced mouse skin squamous cell carcinoma. Nat Med 21, 946–954. [DOI] [PubMed] [Google Scholar]
- Nishimura EK, Jordan SA, Oshima H, Yoshida H, Osawa M, Moriyama M, Jackson IJ, Barrandon Y, Miyachi Y, and Nishikawa S-I (2002). Dominant role of the niche in melanocyte stem-cell fate determination. Nature 416, 854–860. [DOI] [PubMed] [Google Scholar]
- Osawa M, Egawa G, Mak S-S, Moriyama M, Freter R, Yonetani S, Beermann F, and Nishikawa S-I (2005). Molecular characterization of melanocyte stem cells in their niche. Development 132, 5589–5599. [DOI] [PubMed] [Google Scholar]
- Rabbani P, Takeo M, Chou W, Myung P, Bosenberg M, Chin L, Taketo MM, and Ito M (2011). Coordinated activation of Wnt in epithelial and melanocyte stem cells initiates pigmented hair regeneration. Cell 145, 941–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raskin L, Fullen DR, Giordano TJ, Thomas DG, Frohm ML, Cha KB, Ahn J, Mukherjee B, Johnson TM, and Gruber SB (2013). Transcriptome profiling identifies HMGA2 as a biomarker of melanoma progression and prognosis. J Invest Dermatol 133, 2585–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesto A, Navarro M, Burslem F, and Jorcano JL (2002). Analysis of the ultraviolet B response in primary human keratinocytes using oligonucleotide microarrays. Proc Natl Acad Sci U S A 99, 2965–2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, and Jemal A (2015). Cancer statistics, 2015. CA Cancer J Clin 65, 5–29. [DOI] [PubMed] [Google Scholar]
- Singh I, Ozturk N, Cordero J, Mehta A, Hasan D, Cosentino C, Sebastian C, Krüger M, Looso M, Carraro G, et al. (2015). High mobility group protein-mediated transcription requires DNA damage marker γ-H2AX. Cell Res 25, 837–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S, Nannuru KC, Sadanandam A, Varney ML, and Singh RK (2009). CXCR1 and CXCR2 enhances human melanoma tumourigenesis, growth and invasion. Br J Cancer 100, 1638–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soteriou D, Kostic L, Sedov E, Yosefzon Y, Steller H, and Fuchs Y (2016). Isolating Hair Follicle Stem Cells and Epidermal Keratinocytes from Dorsal Mouse Skin. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasetti C, and Vogelstein B (2015). Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 347, 78–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, and Pachter L (2013). Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol 31, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viros A, Sanchez-Laorden B, Pedersen M, Furney SJ, Rae J, Hogan K, Ejiama S, Girotti MR, Cook M, Dhomen N, et al. (2014). Ultraviolet radiation accelerates BRAF-driven melanomagenesis by targeting TP53. Nature 511, 478–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vultur A, and Herlyn M (2013). SnapShot: melanoma. Cancer Cell 23, 706–706.e1. [DOI] [PubMed] [Google Scholar]
- Walker GJ, Soyer HP, Terzian T, and Box NF (2011). Modelling melanoma in mice. Pigment Cell Melanoma Res 24, 1158–1176. [DOI] [PubMed] [Google Scholar]
- Westphalen CB, Takemoto Y, Tanaka T, Macchini M, Jiang Z, Renz BW, Chen X, Ormanns S, Nagar K, Tailor Y, et al. (2016). Dclk1 Defines Quiescent Pancreatic Progenitors that Promote Injury-Induced Regeneration and Tumorigenesis. Cell Stem Cell 18, 441–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White AC, and Lowry WE (2015). Refining the role for adult stem cells as cancer cells of origin. Trends Cell Biol 25, 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White A, Flores A, Ong J, and Lowry WE (2016). Hmga2 is dispensable for cutaneous squamous cell carcinoma. Exp Dermatol 25, 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White AC, Tran K, Khuu J, Dang C, Cui Y, Binder SW, and Lowry WE (2011). Defining the origins of Ras/p53-mediated squamous cell carcinoma. Proc Natl Acad Sci U S A 108, 7425–7430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White AC, Khuu JK, Dang CY, Hu J, Tran KV, Liu A, Gomez S, Zhang Z, Yi R, Scumpia P, et al. (2014). Stem cell quiescence acts as a tumour suppressor in squamous tumours. Nat Cell Biol 16, 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Powers S, Zhu W, and Hannun YA (2016). Substantial contribution of extrinsic risk factors to cancer development. Nature 529, 43–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Z, Zhou Z-J, Xia G-K, Zhang X-H, Wei Z-W, Zhu J-T, Yu J, Chen W, He Y, Schwarz RE, et al. (2017). A positive crosstalk between CXCR4 and CXCR2 promotes gastric cancer metastasis. Oncogene. [DOI] [PubMed] [Google Scholar]
- Xie X, Koh JY, Price S, White E, and Mehnert JM (2015). Atg7 Overcomes Senescence and Promotes Growth of BrafV600E-Driven Melanoma. Cancer Discov 5, 410–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi MR, Hornyak TJ, and Merlino G (2011). A genetically engineered mouse model with inducible GFP expression in melanocytes. Pigment Cell Melanoma Res 24, 393–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Huang C, Fu C, Tian Y, Hu Y, Wang B, Strasner A, Song Y, and Song E (2015). Cordycepin (3’-deoxyadenosine) suppressed HMGA2, Twist1 and ZEB1-dependent melanoma invasion and metastasis by targeting miR-33b. Oncotarget 6, 9834–9853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Benson KF, Ashar HR, and Chada K (1995). Mutation responsible for the mouse pygmy phenotype in the developmentally regulated factor HMGI-C. Nature 376, 771–774. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNAseq data in this paper have been deposited at the Gene Expression Omnibus (NCBI) with GEO series accession number GSE102597.