

SUMMARY
Although features of ribosome assembly are shared between species, our understanding of the diversity, complexity, dynamics, and regulation of ribosome production in multicellular organisms remains incomplete. To gain insights into ribosome biogenesis in human cells, we perform a genome-wide loss-of-function screen combined with differential labeling of pre-existing and newly assembled ribosomes. These efforts identify two functionally uncharacterized genes, C1orf109 and SPATA5. We provide evidence that these factors, together with CINP and SPATA5L1, control a late step of human pre-60S maturation in the cytoplasm. Loss of either C1orf109 or SPATA5 impairs global protein synthesis. These results link ribosome assembly with neurodevelopmental disorders associated with recessive SPATA5 mutations. Based on these findings, we propose that the expanded repertoire of ribosome biogenesis factors likely enables multicellular organisms to coordinate multiple steps of ribosome production in response to different developmental and environmental stimuli.
Graphical Abstract
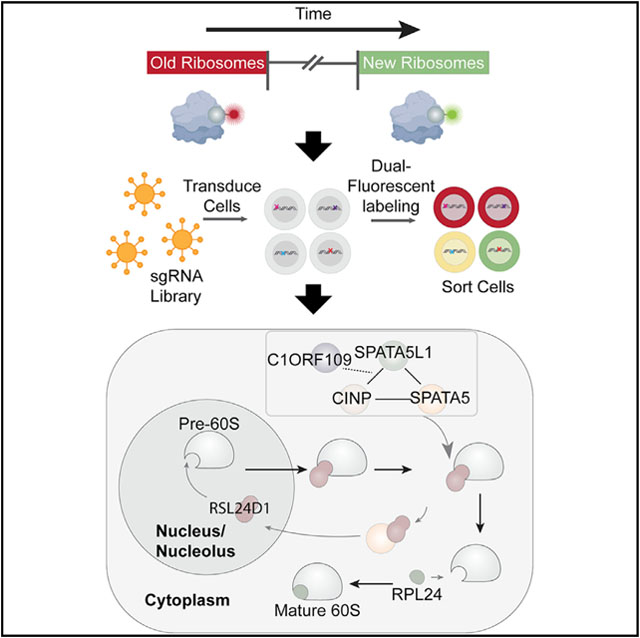
In brief
Ni et al. describe a live-cell labeling technique to track the production and movement of old and new ribosomes. Through a CRISPR screen, they identify C1ORF109 and SPATA5 as two ribosome biogenesis factors. They further reveal that SPATA5 allelic variants associated with neurodevelopmental defects impair ribosome maturation.
INTRODUCTION
The homeostatic balance between protein synthesis and turnover is tightly associated with the growth, health, and longevity of almost all organisms (Hipp et al., 2019; Klaips et al., 2018; Labbadia and Morimoto, 2015; Steffen and Dillin, 2016). In multicellular organisms, protein synthesis can vary greatly between different cell types, even in cells from the same lineage. Protein production depends on ribosomes, and changes in normal ribosome levels have important consequences: enhanced ribosome biogenesis often drives tumor growth, while defects in ribosome assembly and function cause a group of diseases collectively known as ribosomopathies, which share several common phenotypes including anemia and a broad range of craniofacial defects (Farley-Barnes et al., 2019; Mills and Green, 2017). In addition, ribosome malfunction contributes to age-related phenotypes and various neurodegenerative disorders (Farley-Barnes et al., 2019).
Ribosome biogenesis represents a complex process involving multiple steps that must be tightly coordinated with different cellular functions and activities (Klinge and Woolford, 2019). Ribosome assembly depends on rDNA transcription, rRNA processing, synthesis of 80 ribosomal proteins, assembly of these proteins with 4 different rRNAs, and molecular trafficking between the nucleolus, nucleoplasm, and cytoplasm. Most of what we know about eukaryotic ribosome biogenesis comes from genetic and biochemical studies performed using S. cerevisiae (Klinge and Woolford, 2019). Although the basic features of ribosome assembly are shared between yeast and humans, ribosome biogenesis has become more complex through evolution (Bohnsack and Bohnsack, 2019). The significance of this enhanced complexity remains poorly understood. Human ribosomes are larger than their yeast counterparts, and many additional factors associate with pre-ribosomes and mature ribosomes in human cells. The study of these factors has revealed interesting differences between the ribosome assembly pathways in yeast and human cells (Wild et al., 2010). For example, 286 human ribosome biogenesis factors were identified in a previous study and 74 of these do not have a clear yeast homolog (Tafforeau et al., 2013). In addition, several signaling pathways and oncogenes, often acting in a cell-specific manner, feed into the regulation of ribosome biogenesis across different cell types (Bohnsack and Bohnsack, 2019). The catalog of factors that directly participate in ribosome biogenesis or regulate this process in human cells likely remains incomplete. In particular, the factors that control late steps of pre-60S ribosomal subunit maturation in the cytoplasm have not been well characterized (Bohnsack and Bohnsack, 2019).
In this study, we developed a “Ribo-SNAP” technique that allows for the differential labeling of pre-existing and newly produced ribosomes in living cells. This technique allows us to track and quantify the level of old and new ribosomes, and the maturation rate of pre-ribosomes across different human cells. This work reveals unexpected variability in ribosome assembly rates across different cell types. Using Ribo-SNAP labeling in a genome-scale CRISPR-Cas9 knockout screen, we identified C1ORF109, SPATA5, SPATA5L1, and CINP, which together promote the recycling of RSL24D1 from cytoplasmic pre-60S ribosomal subunits back to the nucleolus, a step that is carried out by a single protein in yeast. Allelic variants of SPATA5, SPATA5L1, and CINP have been linked with microcephaly, intellectual disability, hearing loss, and seizures in human patients. We find two of these allelic variants in SPATA5 compromise ribosome biogenesis, implicating defects in ribosome production and mRNA translation in these disorders. Results presented here provide further evidence for the growing complexity of ribosome biogenesis in multicellular organisms.
RESULTS
Heterochronic labeling of ribosomes in different cell types
To characterize ribosome assembly dynamics under different conditions, we incorporated a SNAP tag into the endogenous ribosomal protein L28 (RPL28) gene in HEK293T, HCT116, and H9 human embryonic stem cells (hESCs) using CRISPR-based strategies (Figures 1A and S1). The SNAP tag is a self-labeling enzymatic tag, engineered from the human O-6-methylguanine-DNA methyltransferase (Hoelzel and Zhang, 2020) that can form a covalent link with various fluorophores in living cells. Using sucrose gradient fractionation, we found SNAP-tagged RPL28 incorporated into the 60S subunit and readily contributed to monosomes and polysomes (Figure S1). Moreover, proliferation rates and global translation after incorporation of the tag match those of parental controls (Figure S1).
Figure 1. Differential labeling of ribosomes reveals variation of ribosome biogenesis and turnover across different cell types and conditions.
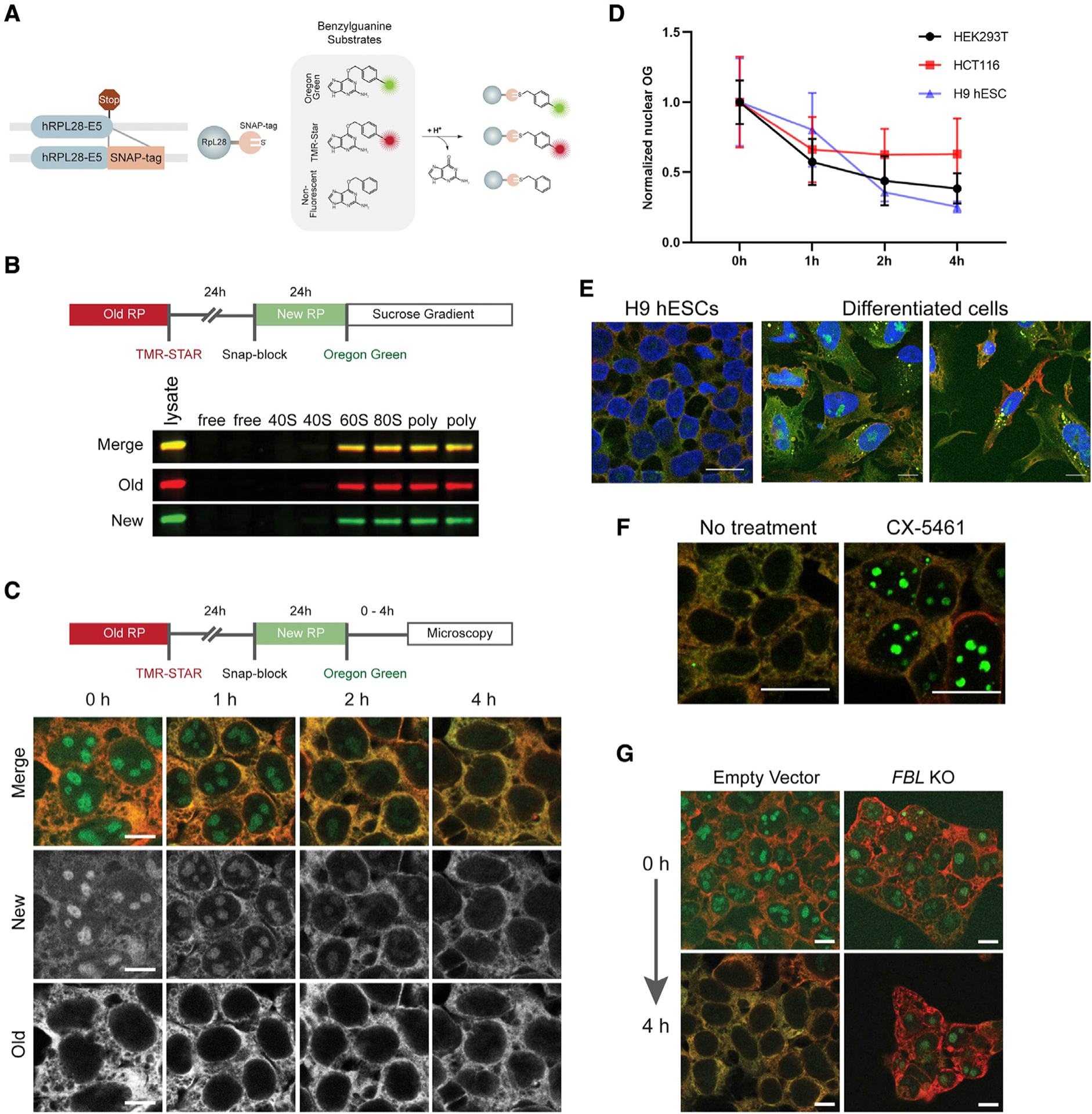
(A) Schematic detailing the tagging of the RPL28 open reading frame with a SNAP tag at the 3′ end of exon 5 (E5). The SNAP-tagged proteins can be indelibly labeled with a number of different benzylguanine substrates. They can also be “blocked” through the use of unconjugated benzylguanine.
(B) An outline of a standard pulse-labeling experiment in which ribosomes older than 48 h are labeled with TMR-Star (red) and ribosomes 0–24 h old are labeled with Oregon green. Cell lysates were subjected to sucrose gradient fractionation. Old and newly labeled RPL28 incorporated into the 60S, monosome, and polysome fractions.
(C) Imaging of pulse-labeled Ribo-SNAP cells. After the final labeling step, the cells are incubated for variable lengths of time. At the start of the experiment, newly labeled RPL28 is clearly observed in the nucleoli of cells, whereas old ribosomes are cytoplasmic, as expected. The nucleolar levels of newly labeled RPL28 continue to drop over the course of 4 h.
(D) The rate at which the levels of nuclear RPL28 decreases varies between HEK293T, HCT116, and hESCs. Values were normalized to the mean of the 0-h group of each cell type. Mean ± SD is shown.
(E) H9 hESCs carrying the RPL28 SNAP tag reporter or their randomly differentiated progeny were pulse labeled for old and new ribosomes and examined after a 4-h chase. Levels of labeled ribosomes appear relatively homogeneous within hESCs but their differentiated progeny displayed a range of old and new ribosome levels. Moreover, differentiated cells exhibited variable amounts of newly labeled nucleolar RPL28 after a 4-h chase period.
(F) Treatment of cells with the Pol I and TOP2B inhibitor CX-5461 results in retention of newly labeled RPL28 within nucleoli after a 4-h chase period.
(G) Cells carrying SNAP-tagged RPL28 were transduced with sgRNAs targeting FBL and subjected to pulse labeling. Disruption of FBL resulted in retention of newly labeled RPL28 within nucleoli after a 4-h chase period. Scale bars, 10 μm in (C, E, and G) and 20 μm in (F).
We sequentially applied three different SNAP tag substrates (TMR-Star [red fluorescent], non-fluorescent, and Oregon green [green fluorescent]) to separately label pre-existing and newly produced ribosomes (Figures 1B and 1C). The SNAP tag performs a suicide reaction and cannot react with other substrates after its first labeling, which ensures that red and green labeling mark chronologically distinct ribosome populations. Generally, we used a strategy whereby red-labeled ribosomes were at least 48 h old and green-labeled ribosomes were 0–24 h old. Assaying fluorescently labeled old and new ribosomes using sucrose gradient fractionation confirmed the rapid incorporation of newly labeled RPL28 into functional 60S subunits, 80S monosomes, and polysomes under normal conditions (Figure 1B). Moreover, the relative levels of old and new ribosomes appeared similar across the 80S and polysome fractions. These results suggest that ribosomes that are at least 48 h old are functionally equivalent to newly synthesized ribosomes.
As expected, red-labeled old RPL28 resided in the cytoplasm while green-labeled new RPL28 was observed in nucleoli immediately after the labeling procedure. Subsequent chase periods showed that the vast majority of nucleolar RPL28 was rapidly exported out into the cytoplasm within 1 h of labeling (Figure 1C). These levels continued to drop over the subsequent 3 h. The rate at which newly labeled RPL28 trafficked into and out of the nucleolus differed between HEK293T, HCT116, and hESCs (Figures 1D, S2A, and S2B). Moreover, pulse labeling revealed different levels of newly labeled RPL28 in the nucleoli of hESCs and their differentiated progeny (Figures 1E and S2B). Levels of old ribosomes also differed between these cells, likely reflecting differences in cell division rates or ribosome turnover. RNA sequence data from the genotype-tissue expression (GTEx) database shows that expression levels of different ribosome biogenesis factors vary greatly between different tissues, especially when compared with translation factor and ribosomal protein mRNAs (Figure S3). These results suggest that ribosome assembly rates are regulated at multiple levels to control the total number of functional ribosomes within a cell.
Heterochronic labeling reveals perturbations in ribosome assembly and function
We next tested how perturbations in ribosome production and activity influenced the behavior of old and newly labeled RPL28. Ribosome biogenesis depends on ongoing rRNA transcription. Treating cells with the Pol I and TOP2B inhibitor CX-5461 (Drygin et al., 2011; Pan et al., 2021) resulted in retention of newly labeled RPL28 within nucleoli (Figure 1F), further suggesting that trafficking of RPL28 from the nucleolus to the cytoplasm depends on ribosome assembly. Treatment of cells with sodium arsenite (NaAsO2) results in stress granule assembly and a rapid loss of protein synthesis. To see if NaAsO2 exposure alters 60S subunit use, we labeled pre-existing ribosomes with TMR-Star and exposed these cells to NaAsO2 for 24 h (Figures S2C and S2D). The cells were then washed, and ribosomes formed after the treatment were labeled with Oregon green. While NaAsO2-exposed ribosomes were found in polysome fractions, they also accumulated in the 60S fractions and were reduced in the 80S fraction. By contrast, ribosomes assembled after NaAsO2 treatment were largely incorporated into polysomes. These results suggest that newly made 60S subunits rapidly substitute for their damaged counterparts to support the ongoing demands for translation elongation competent ribosomes.
We next examined the dynamic behavior of new and old ribosomes in different mutant backgrounds. Disruption of ribosome assembly factors or rRNA-processing machinery would be predicted to slow down or halt specific aspects of ribosome biogenesis, potentially leading to the retention of new RPL28 within nucleoli. To test this, we targeted two ribosome biogenesis factors: fibrillarin (FBL) and TSR2 ribosome maturation factor (TSR2). FBL is an essential rRNA 2′-O-methyltransferase that acts at an early step in ribosome biogenesis (Rodriguez-Corona et al., 2015), whereas TSR2 promotes the release of the small subunit protein RPS26 from Importin upon entry into the nucleus and participates in 20S pre-rRNA processing (Schutz et al., 2014). TSR2 has been linked with different ribosomopathies, including Diamond-Blackfan anemia (Aubert et al., 2018). CRISPR-mediated mutation of FBL resulted in prolonged retention of RPL28 within nucleoli, even after a 4-h chase period, reflecting defects in ribosome assembly and subsequent defects in pre-60S export out of the nucleus (Figures 1G and S4). By contrast, knockout of TSR2 resulted in a reduction of new RPL28 levels, as reflected by the loss of Oregon green signal, but did not cause retention of nucleolar RPL28 (Figure S2E). Thus, as expected, loss of TSR2 does not appear to interfere with ongoing assembly and trafficking of pre-60S subunits. Of note, FBL or TSR2 mutant cells retain higher levels of mature ribosomes in the absence of new ribosome production.
CRISPR screen reveals regulators of ribosome biogenesis
To gain insights into the regulatory landscape that controls ribosome levels in rapidly dividing cells, we conducted a loss-of-function CRISPR-based screen, using our SNAP-tagged RPL28 HEK293T cell line (Figure 2A). Through the course of our studies, we found knockout of core ribosome assembly factors resulted in cell lethality 4–6 days after transduction of sgRNA expression. To identify new genes that control ribosome biogenesis levels, we elected to limit our selection to 8 days after delivery of the CRISPR library. Using the Human CRISPR Knockout Pooled Library (Brunello), RPL28-SNAP cells were transduced and selected for expression of sgRNAs. Starting on day 6 after transduction, old (≥48 h) and new (≤24 h) ribosomes were pulse labeled with red and green fluorophores, respectively. Cells within the top 0.5% of the “brighter red and dimmer green” population were sorted. We specifically focused on hits that exhibited less green fluorescence and more red fluorescence, reasoning that this shift in old and new ribosomes would reveal new factors that regulate ribosome biogenesis. We used next-generation sequencing (NGS) to verify high overall representation of the entire library in the initial population of infected cells. sgRNAs present within the sorted cell populations were identified using NGS and these data were analyzed by MAGeCK (Li et al., 2014). This analysis revealed 35 high-confidence hits (Figures 2B and 2C), including multiple aminoacyl-tRNA synthetases (CARS, KARS, AARS, LARS, VARS, NARS, HARS, and SARS), tRNA/5S rRNA transcription and processing factors (GTF3C5, GTF3C1, POLR3K, and RPP21), multiple components of the RNA exosome (EXOSC2 and EXOSC8), and genes involved in translation elongation (eEF1A, eEF1G, and eEF2). We expected to identify genes involved in mRNA translation given the nature of our pulse-labeling assay. Cell imaging and flow cytometry confirmed that loss of representative genes results in both loss of new ribosome labeling and a retention of old ribosomes, relative to control cells (Figures 2D and S4).
Figure 2. Loss-of-function screen identifies genes that contribute to ribosome biogenesis and turnover.
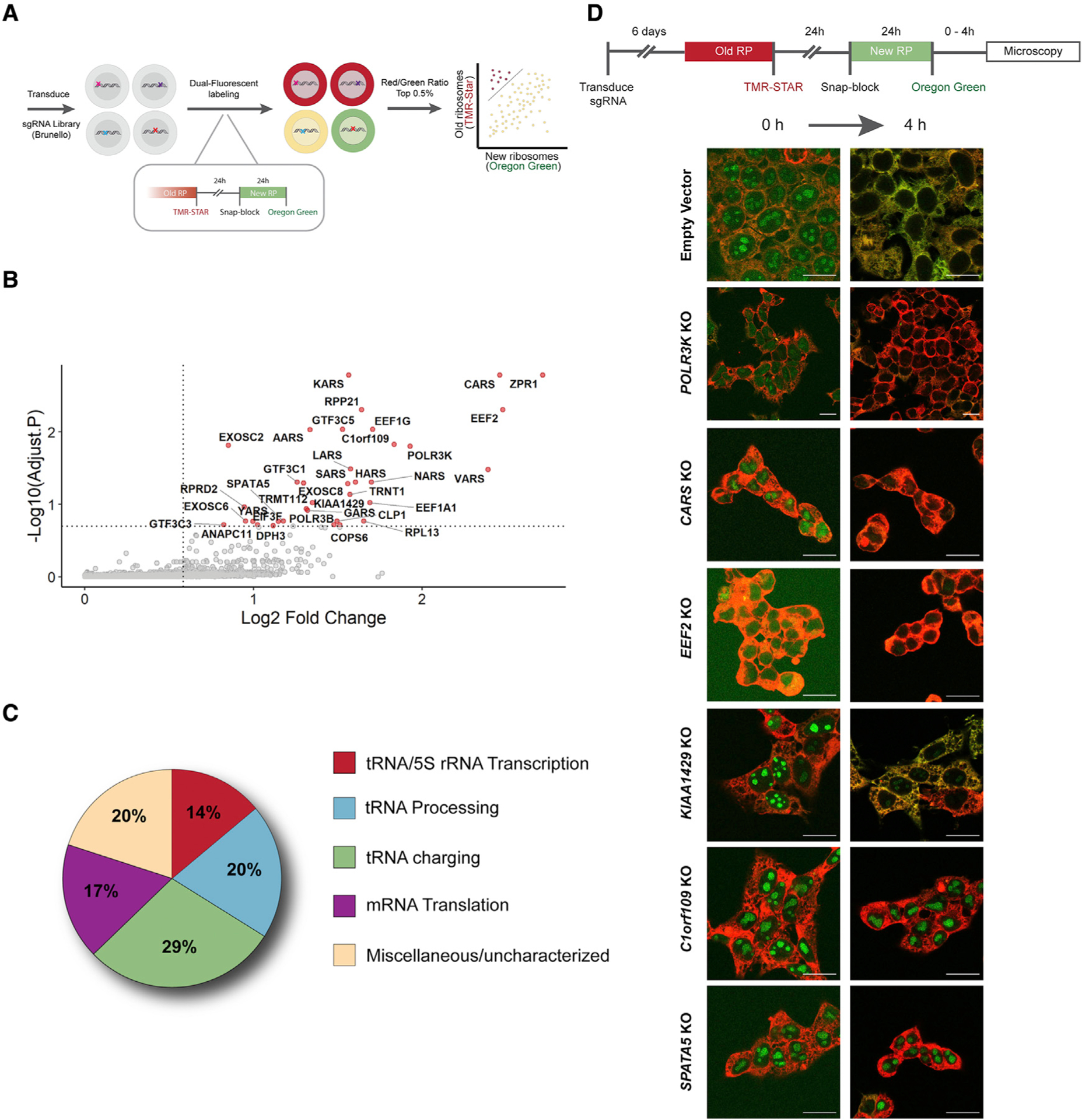
(A) Overview of CRISPR-Cas9 screen design.
(B) MAGeCK analysis identified positive hits that result in lower levels of newly labeled RPL28 and increased levels of old ribosomes.
(C) Functional categorization of genes identified in the screen.
(D) Validation of representative hits from each of the functional categories using HEK293T cells. Strong retention of newly labeled nucleolar RPL28 after a 4-h chase period distinguishes C1orf109 and SPATA5 from other positive hits. Scale bars, 20 μm.
C1ORF109 and SPATA5 control a late step in ribosome maturation
During our screen validation efforts, fluorescence microscopy revealed that newly labeled RPL28 persisted in the nucleoli of C1orf109 and SPATA5 mutant cells for over 4 h (Figure 2D). C1ORF109 has been linked with cell proliferation, but little is known about its molecular function (Liu et al., 2012). While SPATA5 has not been biochemically characterized, it belongs to the AAA ATPase family of proteins and is distantly related to the yeast protein Drg1 (also known as AFG2) (Kressler et al., 2012). AAA ATPases play important roles in the assembly of ribosomes by using ATP hydrolysis to drive changes in protein-protein interactions and protein conformation (Prattes et al., 2019). For example, during ribosome assembly in yeast, RLP24 serves as a placeholder within the pre-60S ribosomal subunit. Once the pre-60S subunit has been exported to the cytoplasm, Drg1 helps to remove RLP24, allowing for recruitment of RPL24 to the pre-60S subunit (Awad et al., 2019; Kappel et al., 2012; Kressler et al., 2012; Pertschy et al., 2004, 2007; Zakalskiy et al., 2002). Drg1 performs this function as a single protein. A recent study characterized the cryo-EM structure of Drg1 and suggested potential homology between this protein and SPATA5 (Prattes et al., 2021).
Given their shared RPL28 nucleolar retention phenotypes, we reasoned that C1ORF109 and SPATA5 may work together during ribosome biogenesis. To investigate the function of these genes further, we assayed rRNA processing and transcription in C1orf109 and SPATA5 mutant cells. We used northern blot analysis of total RNA using a combination of ITS1 and ITS2 probes that distinguish different intermediate processed forms of rRNA (Figures 3A and S5). These experiments revealed that loss of C1orf109 and SPATA5 results in an increase of unprocessed 47S/45S pre-rRNA and a reduction of 12S pre-rRNA. We also observed a decrease in the ratio of mature 28S to 18S (Figures 3A and 3B), which suggests a potential defect in 60S maturation. A previous study had also noted a defect in rRNA processing upon RNAi knockdown of SPATA5 (Tafforeau et al., 2013). Subsequent 5-ethynyl uridine (EU) pulse-labeling experiments indicated that disruption of C1orf109 and SPATA5 both resulted in a reduction of labeled nascent nucleolar/nuclear RNA (Figures 3C and S5D), indicating that the increased levels of 47S/45S rRNA in C1orf109 and SPATA5 mutant cells observed by northern blot reflect a processing defect and not increased rRNA transcription.
Figure 3. C1orf109 and SPATA5 mutant cells display defects in ribosome assembly and protein translation.
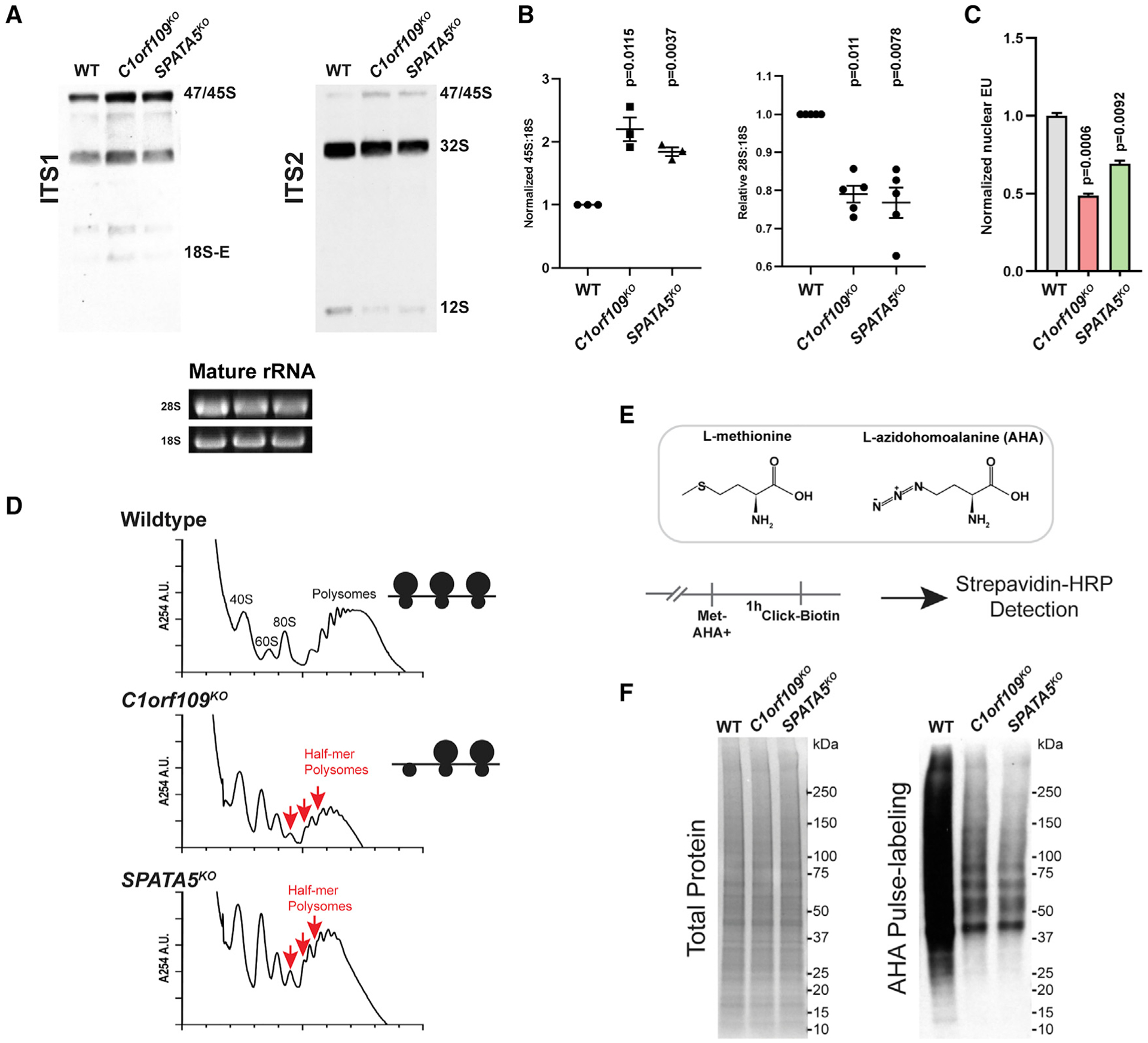
(A) Northern blot analysis for pre-rRNA species reveals that disruption of C1orf109 and SPATA5 results in an increase of pre-47S/45S rRNA and a reduction in 12S rRNA in HEK293T cells.
(B) Quantification of northern blot results. n = 5 biological replicates (left) and n = 3 biological replicates (right). Mean ± SEM is shown. p values were calculated using a ratio-paired t test.
(C) Quantification of EU labeling in wild-type, C1orf109KO, and SPATA5KO (n = 3 biological replicates), Values were normalized to the mean of wild type. Mean ± SEM is shown. p values were calculated by nested one-way ANOVA with Dunnett’s multiple comparisons test.
(D) Polysome profiles from wild-type, C1orf109KO, and SPATA5KO cells. Red arrows indicate half-mer polysomes, which reflect defective 60S biogenesis.
(E) Schematic describing AHA pulse labeling of ongoing protein synthesis.
(F) Total protein as assayed by Ponceau (left) and biotin labeling of AHA-labeled proteins (right) in wild-type (WT) control, C1orf109KO, and SPATA5KO cells.
To further assess the impact of C1orf109 and SPATA5 loss on ribosome biogenesis and function, we examined polysome profiles from wild-type and mutant cells. These experiments revealed that C1orf109 or SPATA5 mutant cells exhibit an imbalance between the 40S and the 60S subunits and formation of half-mers, which represent polysomes with stalled 48S initiation complexes (Figure 3D). Half-mers are commonly observed in mutants that exhibit reduced levels of functional 60S subunits (Moore et al., 2010; Rotenberg et al., 1988). The appearance of half-mers in C1orf109 and SPATA5 mutant extracts suggest that these two proteins promote pre-60S maturation.
Since both reduction of ribosome maturation and formation of half-mers could be indicators of impaired mRNA translation, we performed a series of pulse-labeling experiments using azidohomoalanine (AHA) to test if loss of C1orf109 or SPATA5 leads to impaired global protein synthesis. Activation of AHA by tRNAMet-synthetase allows azides to become incorporated into proteins, which can then be labeled through click chemistry (Figure 3E). Pulse-labeling control and mutant samples revealed that loss of C1orf109 and SPATA5 resulted in lower levels of translation, as expected for proteins needed for ribosome biogenesis (Figure 3F).
Given the distant sequence homology between Drg1 and SPATA5, we tested whether loss of either C1orf109 or SPATA5 results in retention of RSL24D1 (human ortholog of RLP24) on pre-60S subunits. Strikingly, the subcellular localization of RSL24D1 shifted from the nucleolus to the cytoplasm in C1orf109 and SPATA5 mutant HEK293T cells (Figure 4A), suggesting that the recycling of RSL24D1 is disrupted in the absence of these two factors. We next examined whether cytoplasmic RSL24D1 continued to associate with pre-60S ribosomal subunits. We employed a SNAP pull-down, whereby we incubated cytoplasmic lysates from control and mutant cells expressing SNAP-tagged RPL28 with benzylguanine beads (Figure 4B). Consistent with the immunofluorescent analysis, the levels of RSL24D1 in cytoplasmic lysates increased in C1orf109 and SPATA5 mutant cells (Figure 4C). Moreover, high levels of RSL24D1 precipitated with SNAP-tagged ribosomes, indicating that this protein remains inappropriately associated with cytoplasmic pre-60S subunits in the absence of C1orf109 and SPATA5 (Figure 4C).
Figure 4. C1orf109 and SPATA5 mutant cells exhibit defects in the recycling of RSL24D1.
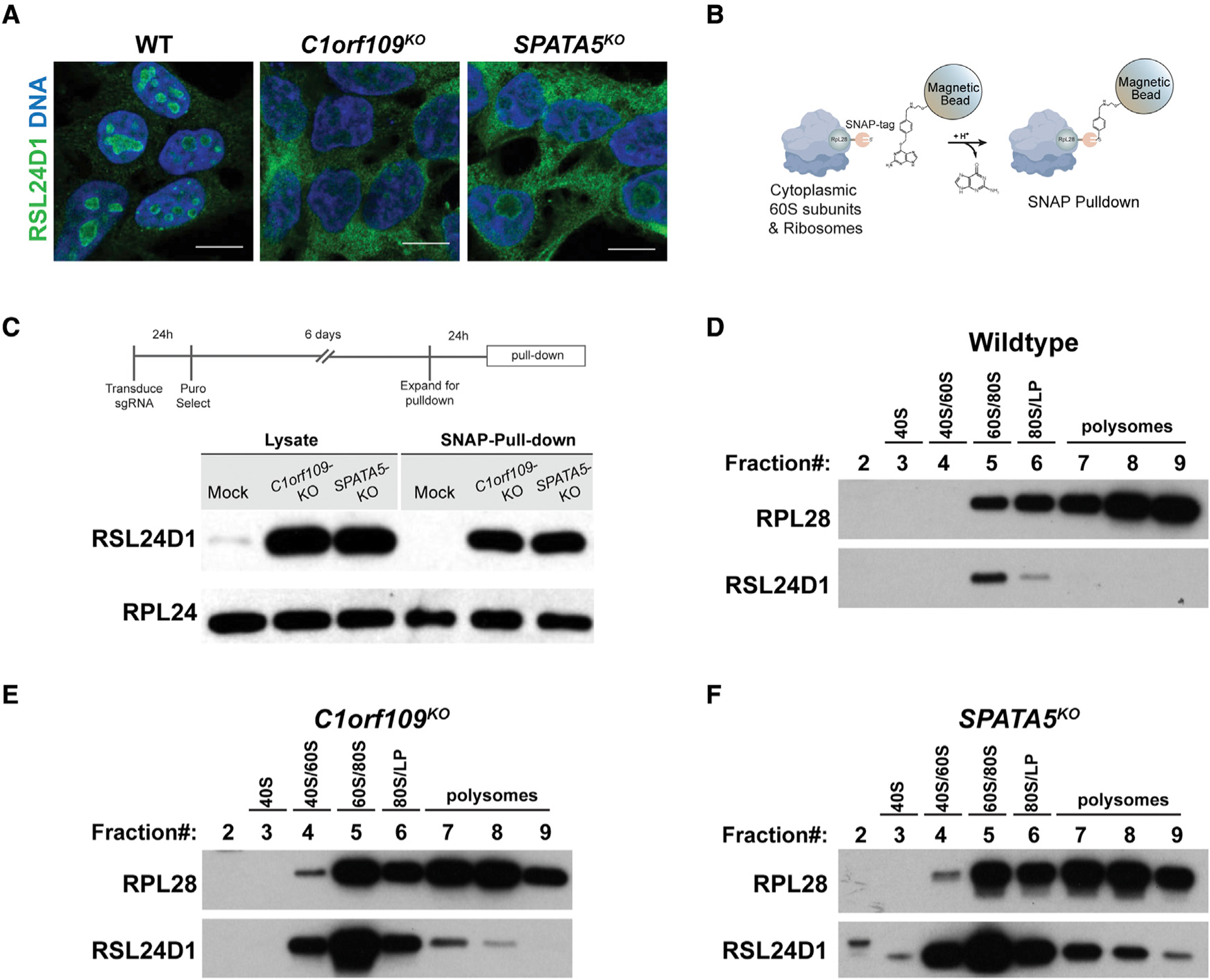
(A) Wild-type (WT) controls and HEK293T cells transduced with sgRNAs targeting C1orf109 and SPATA5 stained for RSL24D1 (green) and DNA (blue). While most RSL24D1 localizes to the nucleoli of control cells, disruption of C1orf109 and SPATA5 results in ectopic localization of RSL24D1 to the cytoplasm. Scale bars, 20 μm.
(B) Overview of SNAP-tagged ribosome pull-down.
(C) Western blot analysis of cytoplasmic extracts and Ribo-SNAP pull-down samples probed with either RSL24D1 or RPL24 antibodies. Loss of C1orf109 and SPATA5 results in a dramatic increase in cytoplasmic localization and ribosome associated RSL24D1.
(D) Western blot of sucrose gradient fractions from WT cells probed with antibodies against RPL28 and RSL24D1. Most RSL24D1 was observed in the 60S/80S fraction, with a sharp drop-off in the 80S/light polysome (LP) fraction.
(E) Western blot of sucrose gradient fractions from C1orf109KO cells probed with antibodies against RPL28 and RSL24D1.
(F) Western blot of sucrose gradient fractions from SPATA5KO cells probed with antibodies against RPL28 and RSL24D1.
To further characterize the behavior of RSL24D1 and its association with the pre-60S subunit, we performed sucrose gradient fractionation in wild-type and mutant cells. In wild-type cells, RSL24D1 fractioned with pre-60S subunits and was not observed to fractionate with monosomes or polysomes (Figure 4D). By contrast, RSL24D1 exhibited a dramatic increase in the fractions occupied by 60S subunits, monosomes, and polysomes in SPATA5 and C1orf109 mutant cells (Figures 4E and 4F). However, RSL24D1 was absent from the heaviest polysome fractions in mutant cells, indicating that RSL24D1 containing pre-60S subunits likely do not participate in productive translation.
Disease-linked allelic variants of SPATA5 compromise ongoing ribosome biogenesis
Allelic variants of SPATA5 in humans have been linked with several neurodevelopmental disorders including microcephaly, hearing loss, epilepsy, and intellectual disability (Buchert et al., 2016; Forstbauer et al., 2012; Kurata et al., 2016; Puusepp et al., 2018; Szczaluba et al., 2017; Tanaka et al., 2015; Zanus et al., 2020). A previous study suggested SPATA5 phenotypes may be caused by changes in mitochondrial morphology and activity (Puusepp et al., 2018). However, data presented here indicate that SPATA5 plays an essential role in ribosome assembly, suggesting that pathogenic mutations may compromise ribosome biogenesis and subsequent protein synthesis. To test this hypothesis, we compared the ability of wild-type and two variant transgenes (Figure 5A) to rescue the SPATA5 knockout phenotypes. We assayed RPL28 levels and localization, as well as the subcellular distribution of RSL24D1 (Figures 5B–5F). The expression of all three transgenes was higher than physiological SPATA5 levels (Figure S5F), but the two pathogenic variants were expressed at slightly lower levels, suggesting that these proteins may be less stable or subject to quality control. Labeling old and new ribosomes showed that, while a wild-type SPATA5 transgene largely rescued the retention of newly labeled RPL28 within nucleoli of SPATA5 knockout cells, cells expressing the Thr330del and D628G human variants displayed low levels of nucleolar RPL28 even after a 4-h chase period (Figures 5C and 5E). Moreover, we observed a modest increase of cytoplasmic and a reduction in nucleolar RSL24D1 upon rescue with the allelic variant transgenes compared with the wild-type control (Figures 5D and 5F). Expression of both allelic variants also resulted in a modest increase in the amount of RSL24D1 that remained associated with cytoplasmic pre-60S subunits relative to the wild-type transgene (Figure 5G). These data suggest that the Thr330del and D628G variants represent weak loss-of-function alleles of SPATA5 in the context of pre-60S subunit maturation.
Figure 5. Allelic variants of SPATA5 exhibit ribosome assembly defects.
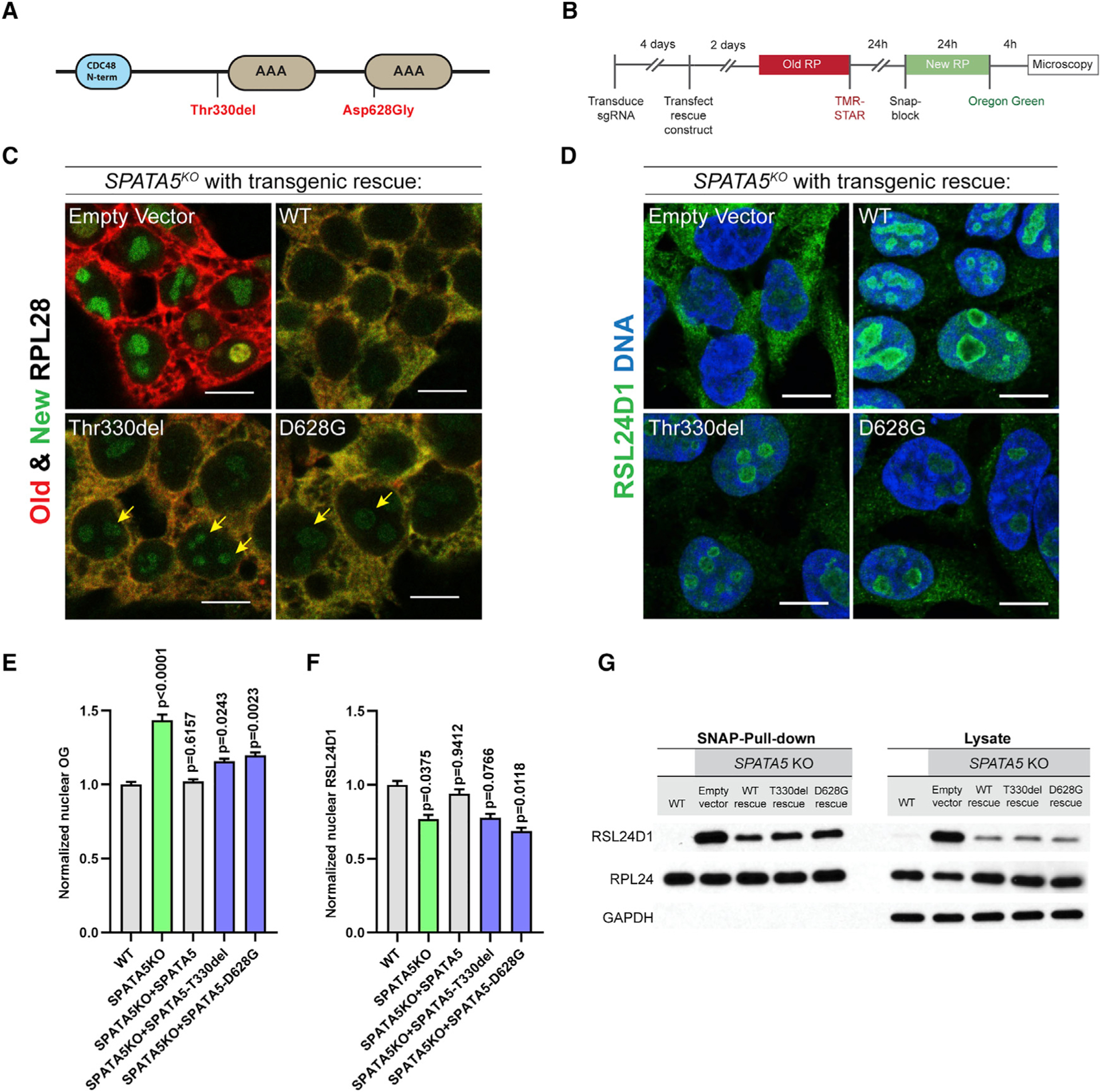
(A) Schematic of SPATA5 domain structure. The two allelic variants indicated in red have been linked with microcephaly, hearing loss, and intellectual disability in humans.
(B) Schematic describing how the labeling of SNAP-tagged RPL28 was conducted.
(C) HEK293T cells were transduced with an sgRNA targeting SPATA5, transfected with plasmids carrying transgenes corresponding to wild-type SPATA5 or the disease-linked (Thr330del and D628G) allelic variants, and old ribosomes were pulse labeled with TMR-Star (red) and new RPL28 proteins were labeled with Oregon green (green), as indicated. The wild-type transgene rescued the RPL28 nucleolar retention phenotype caused by loss of SPATA5, whereas cells expressing the two allelic variants continued to display low levels of newly labeled nucleolar RPL28 after a 4-h chase period (yellow arrows).
(D) Staining cells for RSL24D1 (green) and DNA (blue) showed that, in contrast to wild-type transgenic controls, expression of the Thr330del and D628G variants did not fully rescue the localization defects of RSL24D1 in SPATA5 mutant cells.
(E) Quantification of Oregon green (OG)-labeled new SNAP-tagged RPL28 within nuclei in the indicated genetic backgrounds. n = 3 biological replicates.
(F) Quantification of nuclear RSL24D1 staining in the indicated genetic backgrounds. n = 3 biological replicates. Statistical comparisons in (E and F) are between wild-type cells and cells with each of the indicated rescuing transgenes. Mean ± SEM is shown. p values were calculated by nested one-way ANOVA with Dunnett’s multiple comparisons test.
(G) Western blot analysis of cytoplasmic extracts and Ribo-SNAP pull-down samples probed with RSL24D1 and RPL24 antibodies. Wild-type transgenes could partially rescue the increase of cytoplasmic RSL24D1 and its aberrant prolonged association with the 60S subunit in SPATA5 mutant cells. By contrast, the amount of cytoplasmic and ribosome bound RSL24D1 were modestly elevated upon expression of the Thr330del and D628G allelic variants relative to the wild-type control. Scale bars, 10 μm.
C1orf109 and SPATA5 work together with SPATA5L1 and CINP
Previous large-scale proteomic analyses (Huttlin et al., 2017; Szklarczyk et al., 2019) linked C1ORF109 and SPATA5 with two other proteins: SPATA5-like 1 (SPATA5L1) and cyclin-dependent kinase 2 interacting protein (CINP) (Figure S6A). SPATA5L1 is a second AAA ATPase of unknown function (Kressler et al., 2012), but recently published results show that allelic variants in this gene are linked with phenotypes strikingly similar to those exhibited by SPATA5 patients (Richard et al., 2021). CINP is a component of the DNA replication complex and functions to regulate checkpoint signaling in response to DNA damage (Grishina and Lattes, 2005; Lovejoy et al., 2009). We performed immunoprecipitation experiments in the presence of DSP crosslinking and found that C1ORF109 interacts with all three other proteins (Figure 6A). To determine whether these four proteins interact with ribosomes, we employed the SNAP-tag-based pull-down strategy using benzylguanine beads. Ribosomes were precipitated from cells carrying SNAP-tagged RPL28 and FLAG-tagged C1ORF109. While SPATA5, SPATA5L1, and CINP physically interacted with ribosomes (Figure 6B), C1ORF109 did not under the conditions tested, consistent with results from the C1ORF109 IP. However, loss of C1orf109 resulted in a substantial decrease in SPATA5, SPATA5L1, and CINP levels (Figure S6B). Together, these data suggest that physical interactions between all four proteins help to stabilize them within cells. Similar to loss of C1orf109 and SPATA5, knockout of both SPATA5L1 and CINP resulted in retention of nucleolar RPL28 after 4 h of labeling and a redistribution of RSL24D1 (Figures 6C and 6D), indicative of a ribosome biogenesis defect. However, this retention was much more sporadic across the population of cells, likely reflecting inefficiency of the specific guide RNAs used for Cas9 targeting and providing a reasonable explanation for why these genes were not identified in our initial screen.
Figure 6. SPATA5 and C1ORF109 interact with CINP and SPATA5L1.
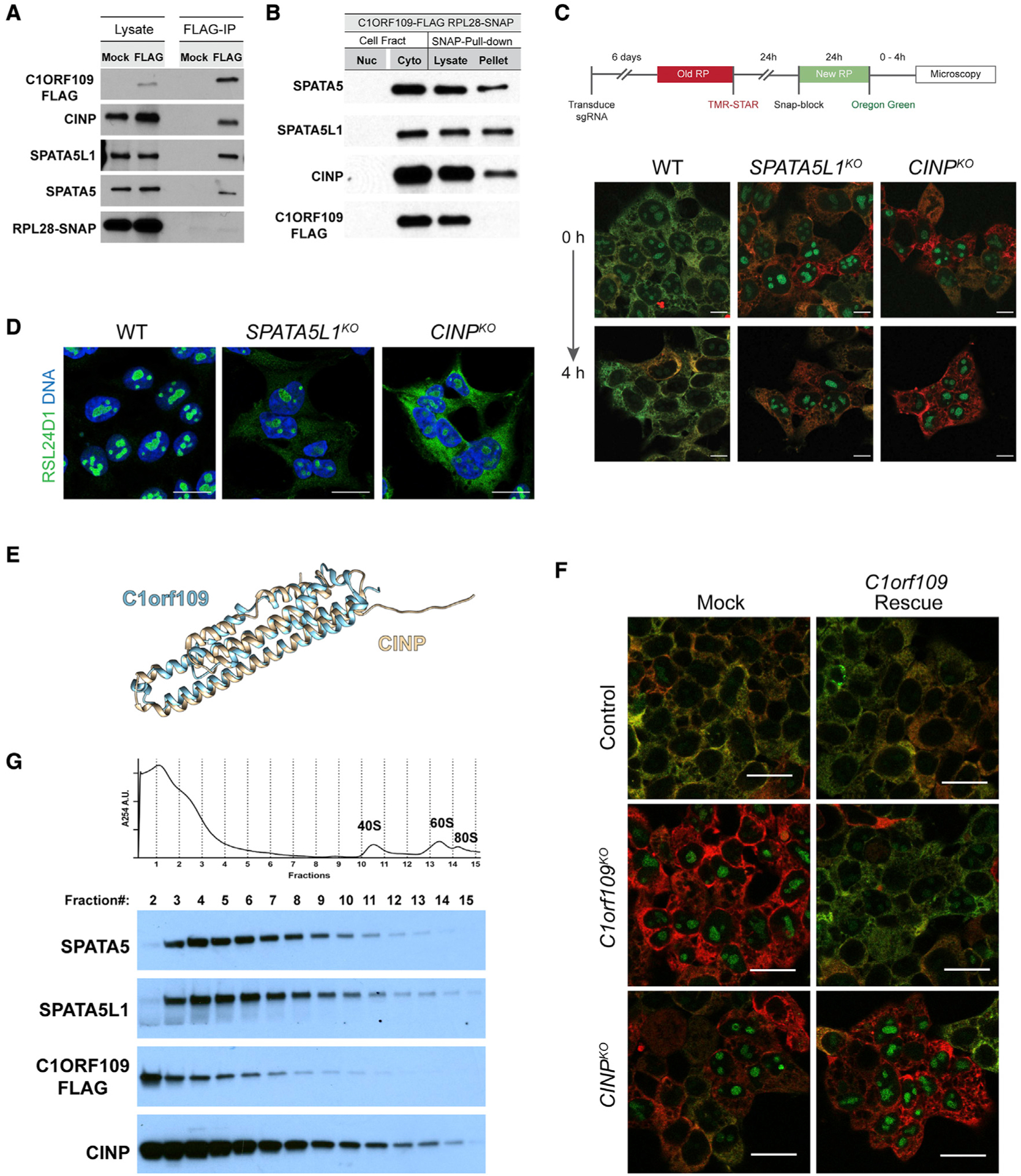
(A) Immunoprecipitation of FLAG-tagged C1ORF109 from HEK293T cell extracts crosslinked with DSP reveals that C1ORF109 physically interacts with SPATA5, SPATA5L1, and CINP, but not with RPL28.
(B) Pull-down of SNAP-tagged RPL28 shows that SPATA5, SPATA5L1, and CINP interact with ribosomes, but C1ORF109 does not.
(C) Knockout of SPATA5L1 or CINP leads to defects in the trafficking of newly labeled RPL28 from the nucleolus to the cytoplasm.
(D) Control, SPATA5L1KO, and C1orf109KO cells stained for RSL24D1 (green) and DNA (blue).
(E) Matchmaker alignment of CINP and C1ORF109 AlphaFold predicted structures.
(F) Control, CINPKO, and C1orf109KO cells transfected with a control or C1orf109 rescue construct stained for old and new ribosomes.
(G) Cytoplasmic lysates from C1ORF109–3xFLAG expressing cells subjected to sucrose gradient fractionation and probed for SPATA5, SPATA5L1, C1ORF109, and CINP.
To gain further insights into how SPATA5, SPATA5L1, CINP, and C1ORF109 cooperate to promote pre-60S maturation, we compared their evolutionary history and predicted structures. As mentioned above, SPATA5 and SPATA5L1 both belong to the AAA ATPase family of enzymes and share a common domain structure, including a CDC48 N-terminal domain followed by two AAA domains (Figure 5A). Although related to one another (Figure S6C), AlphaFold predictions (Senior et al., 2020) highlight differences between the N termini of SPATA5 and SPATA5L1 (Figure S6D), further suggesting that these proteins carry out non-overlapping functions in removing RSL24D1 from pre-60S subunits. Phylogenetic analysis shows that CINP and C1ORF109 are likely related to one another. Orthologs of these proteins are not present in yeast, but CINP-like proteins are found in Drosophila and C. elegans, among other species. Matchmaker alignments of AlphaFold predictions (Meng et al., 2006; Senior et al., 2020) indicate that CINP and C1ORF109 share remarkably similar structures (Figure 6E), with the one notable difference being that CINP has a 14 AA N-terminal extension not found in C1ORF109. Given the similarities in their predicted structures, we wondered whether C1ORF109 and CINP could compensate for one another. While C1ORF109 overexpression rescued the ribosome biogenesis defects in C1orf109 knockout cells, it did not rescue these same defects in CINP mutant cells (Figure 6F). Similarly, overexpression of SPATA5L1, CINP, or C1orf109 did not rescue phenotypes caused by loss of the SPATA5 (Figure S7), suggesting that these proteins carry out discrete functions during pre-60S maturation.
Next, we performed sucrose gradient fractionation over a much steeper gradient followed by western blot analysis to further characterize potential higher-order interactions between SPATA5, SPATA5L1, C1ORF109, and CINP (Figure 6G). SPATA5 and SPATA5L1 displayed a remarkably similar distribution across the gradient leading to heavier fractions that contain 40S subunits, 60S subunits, and 80S monosomes. Both appeared largely absent from the first and lightest fractions. By contrast, C1ORF109 and CINP were both present in the lightest fractions. Like SPATA5 and SPATA5L1, CINP was distributed over a broad range of fractions, consistent with the observation that CINP physically associates with pre-60S subunits, while C1ORF109 was not. While these data suggest that SPATA5, SPATA5L1, CINP, and C1ORF109 interactions may be varied and potentially dynamic, further biochemical and structural studies will be needed to more fully understand complex assembly and the specific roles that each protein plays in promoting the dissociation of RSL24D1 from pre-60S subunits.
DISCUSSION
Ribosome biogenesis is generally considered a constitutive process, and current models have most regulation of ribosome production occurring at the level of rRNA transcription or the expression of ribosomal protein genes. Here, we provide evidence for the dynamic regulation of multiple steps of ribosome assembly in a cell-specific manner. For example, the rates of both new ribosomal protein import into the nucleolus and pre-60S subunit export out of the nucleus differs between HEK293T cells, HCT116 cells, hESCs, and their differentiating progeny. Previous studies revealed that multicellular organisms have evolved additional ribosome biogenesis factors not present in yeast and other unicellular organisms (Tafforeau et al., 2013). In addition, the expression levels of human ribosome assembly factors vary broadly across different tissues (Figure S3). These observations, combined with other results from this study, suggest that higher eukaryotes may have evolved many ways to fine-tune particular steps of ribosome biogenesis to fit specific cellular requirements during development and stress response. The enhanced complexity of the ribosome assembly process potentially endows cells from multicellular organisms with the ability to modulate their capacity for protein synthesis in ways distinct from their unicellular ancestors.
The catalog of factors that regulate and participate in ribosome biogenesis in humans and other multicellular organisms likely remains incomplete. Previous screening efforts (Tafforeau et al., 2013; Wild et al., 2010) targeted specific genes based on biochemical identification of nucleolar proteins or visual criteria. These efforts revealed hundreds of human ribosome biogenesis factors. The list of high significance hits from our screen shows little overlap with those identified in the Wild et al. (2010), although both studies identified genes involved in common processes such as translation initiation. We also found that loss of translation elongation factors (EEF1A1 and EEF1G), the RNA exosome (EXOSC2/6/8), tRNA-modifying enzymes (TRMT112), and SPATA5 disrupted ribosome production, similar to findings reported in Tafforeau et al. (2013). While our significant hit list is relatively small compared with these two previous studies, we did identify genes with no known link to ribosome biogenesis, including C1ORF109. We anticipate that varying the timing of the pulse-labeling assay after sgRNA transduction and screening additional cell types would lead to the discovery of additional ribosome biogenesis factors, particularly those that do not display enriched localization to nucleoli.
Work presented here provides insights into the cytoplasmic maturation of pre-60S subunits in human cells. More specifically, we find that SPATA5, C1ORF109, SPATA5L1, and CINP function together to promote the recycling of RSL24D1 from cytoplasmic pre-60S ribosomal subunits back to the nucleolus (Figure 7). In yeast, this step is carried out by the single protein Drg1. While our data indicate that SPATA5 and SPATA5L1 physically interact with ribosomes and are both needed for the recycling of RSL24D1, a detailed understanding of how these two AAA ATPases work together, whether they form hexameric rings similar to yeast Drg1, whether such rings are homomeric, heteromeric, or double ring structures, and how C1ORF109 and CINP foster the formation and activity of these potential complexes will require additional study.
Figure 7. Model for the function of SPATA5, C1ORF109, SPATA5L1, and CINP.
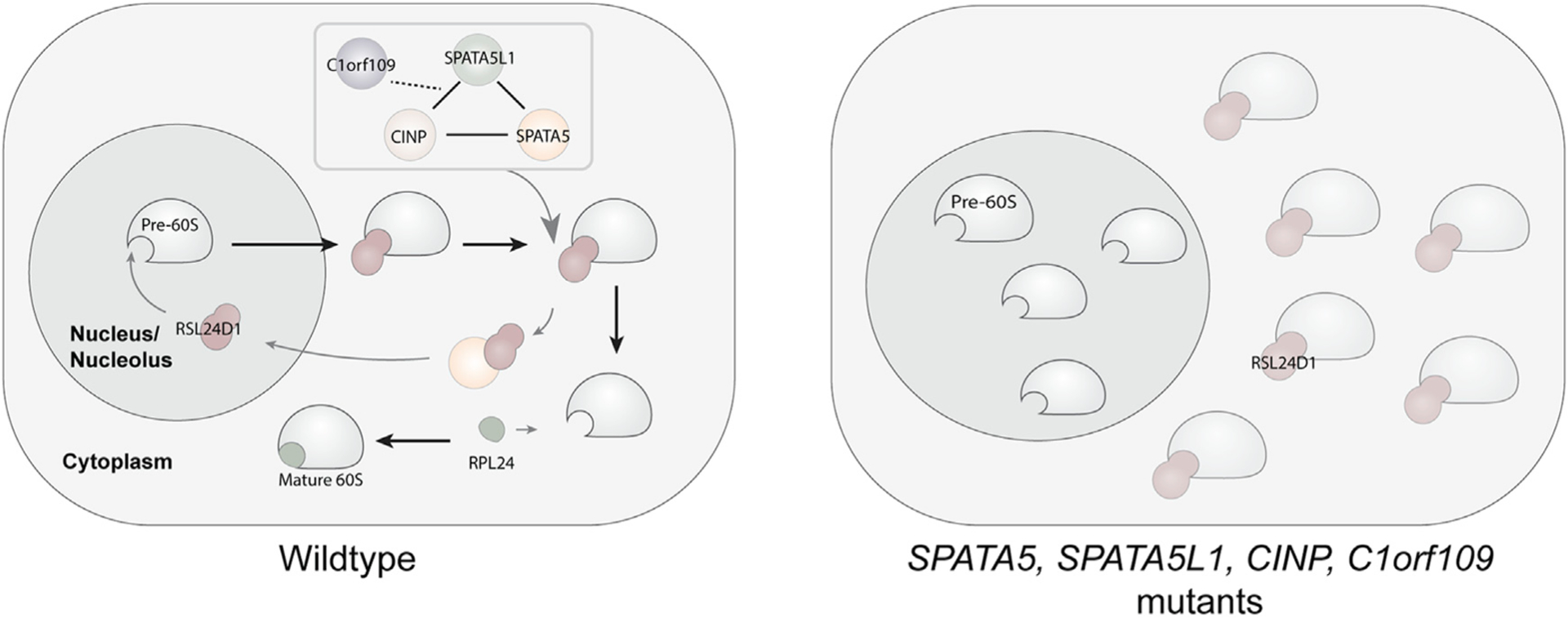
Our data suggest a model whereby SPATA5, C1ORF109, SPATA5L1, and CINP cooperate to promote the recycling of RSL24D1 off of cytoplasmic pre-60S subunits back to the nucleolus, thereby promoting 60S maturation. In the absence of these factors, this recycling is disrupted, resulting in the aberrant cytoplasmic retention of RSL24D1 and a decrease in fully functional 60S ribosomal subunits.
Our data suggest that defects in ribosome biogenesis and protein synthesis may underlie the phenotypes associated with SPATA5 variants in human patients. Our rescue experiments show that two allelic variants of SPATA5 associated with disease in humans display minor defects in ribosome biogenesis. SPATA5, SPATA5L1, CINP, and C1orf109 mRNA expression levels appear low in many regions of the brain based on RNA sequence data from genotype-tissue expression (GTEx) databases, perhaps making these cells acutely sensitive to minor perturbations in late steps in cytoplasmic ribosome maturation. Allelic variants of human CINP and SPATA5L1 have also been linked with microcephalic and neurodevelopmental phenotypes, strikingly similar to those associated with SPATA5 (Karaca et al., 2015; Landrum et al., 2018; Richard et al., 2021). A separate study also provides evidence that SPATA5L1 variants are linked with autism (Matsunami et al., 2014). Mutations in ribosomal proteins and specific ribosome assembly factors result in a group of diseases, collectively known as ribosomopathies (Farley-Barnes et al., 2019). While these disorders result in a range of phenotypes, they are most often associated with anemia and craniofacial defects. Work in several model systems suggests that these defects arise, at least in part, because of a nucleolar stress response and p53-induced cell death (Jones et al., 2008; Noack Watt et al., 2016). Given that SPATA5, SPATA5L1, and CINP regulate a late step in pre-60S maturation, the reasons why allelic variants of these genes cause a spectrum of disorders distinct from other ribosome mutations remains an open question. Defining the specific developmental stage and cell types that are particularly sensitive to loss of SPATA5 function remains important work for the future. Recent results show that increasing global mRNA translation can partially suppress specific ribosomopathy phenotypes in mice (Tiu et al., 2021). Similar experiments that test whether short-term activation of protein synthesis at specific times during fetal development can improve nervous system development in models of SPATA5 disease should be conducted in the future.
Pulse-labeling ribosomal proteins allows one to trace the half-lives of ribosomes. Previous work has shown that ribosome turnover varies between different cell types (Retz and Steele, 1980; Stoykova et al., 1983; Toyama et al., 2013). In rapidly dividing cells, such as those examined in this study and others (An et al., 2020), old ribosomes quickly become diluted through cell division. The differentiated progeny of hESCs exhibit some differences in the levels of old ribosomes (Figure 1) and it will be of interest to examine ribosome half-lives more systematically in different post-mitotic cells. Our initial analysis presented here suggests that ribosomes at least 48 h old show the same ability to enter polysomes as newly generated ribosomes. In the future, pulse-labeling strategies can be used to look for potential differences between heterochronic ribosomes over longer timescales and at higher resolution. Furthermore, we have focused on the biology of the pre-60S and mature 60S subunits. Similar strategies can be used to study 40S subunit assembly and function. The further functional characterization of ribosome assembly dynamics and ribosome turnover promises to provide new insights into how translation machinery is regulated by diverse developmental processes across species and how perturbations in ribosome homeostasis contribute to a broad range of human diseases.
Limitations of the study
Using the Ribo-SNAP system, we were able to label heterochronic ribosomes in living cells. However, this work only used RPL28-SNAP as a reporter. Further work tagging multiple different ribosomal proteins in both the 40S and 60S subunits would enhance the rigor of the conclusions and reveal additional insights into ribosome dynamics. The CRISPR screen was performed after 8 days of puromycin selection. Although selection ensured maximum knockout efficiency and helped us to identify previously uncharacterized ribosome biogenesis factors, the length of the selection resulted in the drop-out of many essential genes that contribute to ribosome biogenesis. A CRISPR screen with shorter puromycin selection would likely identify more ribosome biogenesis factors. Finally, additional work is needed to carefully assess potential connections between the neurodevelopmental phenotypes associated with SPATA5 variants and ribosome biogenesis.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Michael Buszczak (michael.buszczak@utsouthwestern.edu).
Materials availability
All reagents generated in this study are available upon request from the lead contact with no restrictions.
Data and code availability
The raw and processed CRISPR screen data have been deposited at GEO and are publicly available (GSE197277).
This paper does not report original code.
Any additional data and information required to analyze the data reported in this paper are available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
HEK293T, HEK293T-RPL28-SNAP (endogenous RPL28 fused to SNAP) and LentiX-HEK293T cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) (ATCC) supplemented with 10% fetal bovine serum (ATCC) and 1% Penicillin-Streptomycin (Gibco) and maintained in a 5% CO2 incubator at 37°C. HCT116 and HCT116-RPL28-SNAP (endogenous RPL28 fused to SNAP) cells were grown in McCoy’s 5A (Modified) Medium (Gibco) supplemented with 10% fetal bovine serum (ATCC) and 1% Penicillin-Streptomycin (Gibco) and maintained in a 5% CO2 incubator at 37°C. Human ES cell line H9 (WA09) was obtained from WiCell and authenticated by short tandem repeat (STR) profiling. H9 and H9-RPL28-SNAP (endogenous RPL28 fused to SNAP) were cultured in mTeSR Plus media (STEMCELL) on plates coated with Matrigel (Corning) at 37°C with 5% CO2. The cell lines were routinely tested to be free of mycoplasma contamination.
METHOD DETAILS
Plasmid constructs generated in this study
For the generation of RPL28-snap knock-in cell lines, guide RNA (sgRNA) targeting the C-terminal region of human RPL28 gene is from previous published work (An and Harper, 2018). The guide sequence for RPL28 (5′-CTTTCTCACCTGCCTCGAC-3′) was synthesized as paired-oligos, annealed and assembled into pSpCas9(BB)-2A-GFP (PX458) into the BbsI site. Donor vectors were constructed by assembling a snap-tag coding sequence with upstream and downstream homology arms (synthesized as a single double strand DNA gBlock by IDT) into the pUC19 vector by Gibson assembly. For the knock-out experiments by lentiviral transduction, the sgRNA was synthesized, annealed and assembled into LentiCRISPRv2 into the Esp3I site. For transgenic rescue, the cDNAs of rescue constructs were synthesized as double strand DNA gBlocks by IDT and then cloned into the pCMV2 vector by Gibson assembly.
Generation of RPL28-snap knock-in cell lines
HEK293T and HCT116 cells were transfected with sgRNA and donor vectors (1:3 by molarity) by Lipofectamine 3000(Thermofisher). 24 h after transfection, cells were sorted and GFP positive cells were collected. After 7 days’ culture, cells were treated with 3 μM SNAP-Cell TMR-Star (NEB S9105S) for 30 min, followed by wash. Cells were sorted and the TMR positive cells were collected into 96-well plates with 1 cell/well. 14 days after the single cell sorting, the colonies were screened for knock-in by genotyping and western blot with antibodies for RPL28(Abcam 138125). For H9 hESCs, linearized RPL28-SnapTag double stranded oligonucleotide (dsODN) donor was produced by PCR amplification from donor vector. H9 hESCs were electroporated with a NEPA21 Type II Super Electroporator (Bulldog-Bio) using the manufacturer’s recommended parameters. After electroporation, cells were collected in 2 mL of mTeSR Plus media supplemented with ROCK inhibitor (Y-27632). After 48 h, EGFP + cells were sorted and plated at a clonal density (1 × 103 cells/mL). Resulting colonies were manually isolated and expanded for genotyping and western blot analysis.
Cell proliferation assay
Wild type HEK293T cells and HEK293T-RPL28-SNAP cells were dislodged, counted and seeded into 6 well plates at 2.0 × 105 cells/well. Cells were counted 24, 48, 72 h after seeding by Countess Automated Cell Counter (Thermo Scientific) to generate the growth curve.
Random monolayer differentiation of RPL28-SnapTag H9 hESCs
RPL28-SnapTag H9 hESCs were dissociated into single cells using Versene (Gibco) and plated at a density of 1.0 × 105 cells/mL in mTeSR Plus media supplemented with ROCK inhibitor (Y-27632) on dishes coated with Matrigel. After 48 h, the media was replaced with DMEM (SIGMA) supplemented with 10% fetal bovine serum (FBS) and refreshed every other day. After an additional 12 days of culture, the cells were dissociated using TrypLE Express (Gibco) and plated onto Matrigel-coated glass coverslips for a 48 h growth before Ribo-SNAP labeling.
Embryoid body formation
Embryoid bodies (EBs) were generated using AggreWell-800 plates (Stem Cell Technologies) per manufacturer’s instructions in mTeSR Plus media with ROCK inhibitor (Y-27632). After 48 h, media was removed and formed EBs were transferred to an ultra-low attachment 24-well plate (Corning) in 2 mL of DMEM supplemented with 10% FBS. After 1 week, EBs were transferred to Matrigel-coated glass coverslips and cultured for an additional week in DMEM media supplemented with 10% FBS, before Ribo-SNAP labeling.
Ribo-SNAP labeling
Cells were incubated with 3μM snap-cell TMR-STAR (NEB) in complete medium for 30 min at 37°C to label pre-existing ribosomes. After labeling, cells were washed 3 times with complete medium and incubated with complete medium for 30 min to remove unreacted snap substrate. Cells were then grown in complete medium for 24 h. At the time of blocking, cells were incubated with 20 μM snap-cell Block (NEB) in complete medium for 30 min at 37°C to block unlabeled snap-tag. After blocking, cells were washed 2 times with complete medium and were grown in complete medium until Oregon green labeling. At the time of new ribosome labeling, cells were incubated with 5 μM snap-cell Oregon Green (NEB) in complete medium for 30 min at 37°C to label newly synthesized ribosomes. After labeling, cells were washed 3 times with complete medium and incubated with complete medium for 30 min to remove unreacted snap substrate. The chase starts at the time of the final medium change.
Polysome profiling and fractionation
Cells were grown to ~80% confluency at the time of assay. Cells were washed once with DPBS and then lysed on cold chamber in lysis buffer (20 mM Tris-Cl pH7.5, 5 mM MgCl2, 150 mM NaCl, 1 mM DTT, 100 μg/mL cycloheximide (CHX, sigma), 1xcOmplete Protease Inhibitor Cocktail (Roche), 0.1% NP-40). For 100 mm dish, 500μL lysis buffer was used. The lysate was collected into 1.5 mL tube and stay on ice for 15 min. Lysate was then centrifuged at 21130 g at 4°C for 10 min to remove nuclei, mitochondria, and debris. RNA concentration of lysates was quantified by nanodrop and 200 μg of RNA were loading onto 10%–50% sucrose gradient (20 mM Tris-Cl pH7.5, 5 mM MgCl2, 150 mM NaCl, 1 mM DTT, 100 μg/mL CHX) and centrifuged in a TH-641 rotor (Thermo Scientific) at 34000 rpm for 2 h at 4°C. Gradients were fractionated and collected using a BioLogic LP Chromatography System (BIO-RAD). Protein in fractions was methanol-precipitated and resuspended in 2xLaemmi buffer (BIO-RAD) with 2-mercaptoethanol (BIO-RAD) and denatured at 70°C for 10 min. For in-gel fluorescence assay, 15μL denatured samples were loaded to 4%–15% Mini-PROTEAN TGX Precast Protein Gels, 12-well and run for 30 min with 200 V. The gel was rinsed twice in deionized waster and imaged by iBright FL1500 Imaging System (Invitrogen).
Confocal microscopy of ribo-snap labeled cells
Cells were plated on poly-D-Lys coated coverslip 24 h before labeling. After ribo-snap labeling, cells were fixed with 4% formaldehyde (v/v) in DPBS for 15 min, followed by 3× wash with DPBS. Coverslips were mounted on slides with ProLong Gold Antifade Mountant with DAPI (Invitrogen) and imaged by ZEISS LSM 800.
Virus production
293T cells were plated at a density of 1.0 × 106 cells/well in a 6-well plate. Cells were grown for 20 h after plating. The medium was changed 1 h before transfection. Transfection was performed using Lipofectamine 3000. For one well in a 6-well plate, the transfection mixture contains 0.5 ug transfer vector (Brunello library or single LentiCRISPRv2-sgRNA plasmid), 0.3 ug pSPAX2 and 0.2 μg pMG2.G, with 2 μL P3000, 3 μL Lipo3000 and 250 μL Opti-MEM (Gibco 31985070). The mixture was incubated at room temperature for 15 min and was then added dropwise to the cells. Virus was harvested 48 h after transfection. The virus-containing medium was collected and centrifuged at 300× g for 3 min and the supernatant was then passed through 0.45 μM SFCA filter. The filtered virus-medium was then aliquoted and stored at −80°C.
Knock out single gene by using LentiCRISPRv2
Lentivirus was produced as mentioned in virus production. sgRNA sequences used in this research are listed in Table S1 designed according to (Doench et al., 2016). The virus was transduced into HEK293T or HCT116 cells by mixing 106 cells with 1 mL viral medium with 8 μg/mL polybrene in a total of 2 mL. The mix was plated onto 1 well in a 12 well plate. 24 h after transduction, cells were dislodged, counted, and plated in complete medium with 2 μg/mL puromycin (HEK293T) or 1 μg/mL puromycin (HCT116) at 5.5 × 105 cells/well in 6 well plate for a 6-day selection.
Pooled sgRNA screen
Lentivirus was produced as mentioned in virus production. The library was transduced by spinfection into 1.6 × 108 HEK293T.24 h after the end of spinfection, cells were dislodged, pooled and plated in complete medium with 2 μg/mL puromycin for a 7-day selection. On day 8, cells were dislodged, pooled, counted and then labeled sequentially by snap cell TMR-STAR (3 μM), snap cell Block (20 μM), snap cell Oregon Green (5 μM) as indicated in Figure 2B. 4 h after snap cell Oregon Green labeling, cells were dislodged and resuspended in DPBS + 2%FBS for sorting. Approximately 8 × 107 cells were sorted at University of Texas Southwestern Moody Foundation Flow Cytometry Facility using a FACS Aria SORP (BD Biosciences). Cells with top 0.5% red/green ratio were collected. Approximately 3 × 105 sorted cells and 4 × 107 unsorted cells were pelleted and frozen at −80°C until genomic DNA preparation. Sorted cells were lysed in STE buffer (10 mM Tris pH8.0, 100 mM NaCl, 1 mM EDTA, 0.4%SDS, 0.2 mg/mL Proteinase K). 5 μL RNa-seA (2 mg/mL) was added after Proteinase K reaction. Lysate were then separated by phenol-chloroform-isoamyl alcohol (PCI) and the aqueous layer was collected for ethanol precipitation. Precipitated DNA was resuspended in 30uL H2O. Unsorted cells were lysed in Tissue & Cell Lysis Solution (Lucigen) and the protein was removed by centrifuge after adding MPC Protein Precipitation Solution. The supernatant was collected, precipitated by isopropanol, and resuspended by 1.2 mL H2O/3 × 107 cells. Guides were amplified from genomic DNA by ExTaq following the protocol from previously published work (Sanson et al., 2018). Briefly, a 100 μL reaction mixture (10 μL 10× reaction buffer, 8 μL dNTP, 0.5 μL P5 primer mix, 1.5 μL ExTaq polymerase, 10 μL P7 primer, 10 μg of genomic DNA) was used per reaction. The thermal cycler parameters are: 1. 95°C, 1 min, 2. 95°C 30 s (denaturation), 3. 53°C 30 s (annealing) 4. 72°C 30 s (extension), back to step 2, total of 28 cycles, 5. 72°C 10 min, 6. 4°C forever. The PCR products were pooled and mixed with AMPure XP (Beckman) and then washed twice with 70% Ethanol, then eluted by water. Purified libraries were sequenced by NextSeq® 500(Illumina). NGS reads were analyzed by following MAGeCKFlute pipeline (Li et al., 2014; Wang et al., 2019) (See Data S1).
Flow cytometry of ribo-snap labeled cells
Cells were grown in 24 well plate to maintain 20%–80% confluency during ribo-snap labeling. After labeling and chasing, cells were dislodged and resuspended in flow buffer (2%FBS in DPBS). The suspension passed through 70 micron cell strainer (FALCON). Cells were analyzed by flow cytometry using a LSRFortessa (BD Biosciences). Data were analyzed using Cytobank (Kotecha et al., 2010).
AHA incorporation and click chemistry for protein synthesis assay
Cells were grown to ~80% confluency at the time of assay. Complete medium was replaced by AHA medium formulated with DMEM, high glucose, no glutamine, no methionine, no cystine (Gibco), 10% dialyzed FBS (Gibco), 1% GlutaMAX (Gibco), 1% Penicillin-Streptomycin (Gibco), 62.6 mg/L L-Cysteine (Sigma), 250uM L-Azidohomoalanine (AHA, Click Chemistry Tools). For non-AHA control, the AHA was replaced by 30 mg/L L-Methionine (Sigma). For Anisomycin (ANS) treatment, AHA medium was further added with 10 μg/mL Anisomycin (Cell Signaling). After 3 h of incubation, cells were scraped in cold DPBS, washed twice with cold DPBS and lysed in RIPA buffer. Protein concentration of the lysate was quantified by BCA and input protein concentrations were normalized to 2.1 mg/mL for downstream click chemistry. 50μL of the normalized cell lysate was used for click reaction with biotin-Alkyne (Click Chemistry Tools) by using Click-&-Go Click Chemistry Reaction Buffer Kit (Click Chemistry Tools). The reaction mixture was then methanol precipitated and was resuspended in 2xLaemmi buffer (BIO-RAD) with 2-mercaptoethanol (BIO-RAD) and denatured at 95°C for 10 min. Final protein concentration is 1 mg/mL. The sample was then resolved by SDS–PAGE, and total protein signal was detected by One-Step Blue Protein Gel Stain (Biotium). The biotin signal was detected by HRP-Conjugated Streptavidin (Thermo Scientific).
Crosslinking immunoprecipitation
HEK293T cells were transfected with or without pCW57.1-C1ORF109-FLAG vector by using Lipo3000. Mock transfected and C1orf109-FLAG transfected cells were grown to 80% confluency in 100 mm dish. Cells were washed once in DPBS and were incubated with DPBS with 2 mM DSP (Thermo Scientific) for 30 min at room temperature. Crosslinking reaction was quenched by adding stop solution (1 M Tris-Cl, pH 7.5) to a final concentration of 20 mM. Cells were lysed in FLAG-IP buffer (50 mM Tris-Cl pH 7.5, 150 mM NaCl, 1% Triton X-100, 1xcOmplete Protease Inhibitor Cocktail). After lysis, lysate was centrifuged at 15000 g for 15 min at 4°C to remove insoluble components. Protein concentration of the lysate was quantified by BCA and input protein concentrations were normalized to 0.5 mg/mL with FLAG-IP buffer. An aliquot from each sample was saved as input sample and ~500 μg total protein was incubated with 50 μL FLAG affinity gel (Sigma) for 2 h at room temperature. After incubation, samples were washed 2 times with 1xTBS, 2 times with FLAG-IP buffer. Samples were then eluted using FLAG-IP buffer with 500ug/mL FLAG peptide (Sigma) at room temperature for 30 min. Samples were denatured with 4xLaemmi buffer (BIO-RAD) with 2-mercaptoethanol.
Nuclear and cytoplasmic partitioning
Cells were grown to 80% confluency in 100 mm dish. Cells were washed twice in DPBS and were then lysed on ice by adding 600μL/dish lysis buffer (20 mM Tris-Cl pH7.5, 5 mM MgCl2, 150 mM NaCl, 1xcOmplete Protease Inhibitor Cocktail, 0.1% NP-40). Lysate was collected into 1.5 mL tube with 15× pipetting. A small aliquot of lysate was checked on microscope to ensure complete lysis of cells without disrupting nucleus. Lysate was then centrifuged at 4700 g for 30 s to pellet nuclei. The supernatant was transferred into a new tube and was the centrifuged at 21130 g at 4°C for 10 min and the supernatant was collected as “cytoplasmic fraction”. The pelleted nuclei were washed once with DPBS and then lysed in 200uL RIPA buffer with 3× sonication.
Ribosome pulldown
Cells were grown to 80% confluency in 100 mm dish. Cells were washed once in DPBS and were then lysed on ice by adding 500 μL/dish lysis buffer (20 mM Tris-Cl pH7.5, 5 mM MgCl2, 150 mM NaCl, 1 mM DTT, 1xcOmplete Protease Inhibitor Cocktail, 0.1% NP-40). Lysate was collected into 1.5 mL tube and stay on ice for 15 min. Lysate was then centrifuged at 21130 g at 4°C for 10 min to remove nuclei, mitochondria, and debris. RNA concentration of lysates was quantified by nanodrop and diluted to 150 μg/mL. 50μL of lysate from each sample was saved as cytoplasmic ribosome input. 600U RNaseT1(Thermo Scientific) was added to 150 μg RNA and then mixed with 100μL SNAP-Capture Magnetic Beads (NEB). SNAP-Capture Magnetic Beads was pre-washed by lysis buffer twice. The mixture was incubated at room temperature for 1 h and then 100U SUPERaseIn RNase Inhibitor (Invitrogen) was added to quench the reaction. The mixture was further incubated for 1 h at room temperature, for a total of 2 h incubation. After incubation, beads were washed 3 times with lysis buffer and then eluted with 50 μL 2xLaemmi buffer with 2-mercaptoethanol at 95°C for 10 min.
Northern blot analysis of pre-rRNA
Total RNAs were purified with Trizol reagent and the RNA pellet was dissolved in RNase free water. RNA concentration was quantified by nanodrop and then diluted to 1600 ng/μL. 10 μg of RNA for each sample was used for pre-rRNA analysis. The northern blot was performed by using NorthernMax kit (Invitrogen). Probes that were used in this study are listed in Table S2.
5-EU incorporation assay for RNA synthesis
Cells were plated on poly-D-Lys coated coverslip 24 h before labeling. At the time of the assay, cells were incubated with complete medium with 0.5 mM EU for 1 h. Cells were then washed twice with DPBS, fixed with 4% formaldehyde (v/v) in DPBS for 15 min and permeabilized with 0.5% Triton X-100 in DPBS for 15 min. After permeabilization, cells were washed once with 3%BSA in DPBS, and then incubated with click-reaction cocktail made with Click-&-Go Cell Reaction Buffer Kit and 2.5μM AFDye 488 Azide (Click Chemistry Tools) for 30 min. After reaction, cells were washed once with 3%BSA in DPBS and once with DPBS. Coverslips were mounted on slides with ProLong Gold Antifade Mountant with DAPI and imaged by ZEISS LSM 800. Random regions were imaged, and the quantification is as described in “Quantitative image analysis”.
Immunofluorescence
Cells were grown, fixed and washed the same as described in “Confocal microscopy of ribo-snap labeled cells”. After DPBS wash, cells were permeabilized with 0.1% Triton X100 in DPBS for 10 min then then incubated with blocking buffer (5% BSA, 0.1% Tween-20 in DPBS) for 60 min. Primary antibodies were diluted in antibody dilution buffer (1% BSA, 0.3% Triton X-100 in DPBS) and samples were incubated with antibodies at 4°C overnight. After incubation, samples were washed 3 times with DPBS and incubated with secondary antibodies with antibody dilution buffer for 60 min at dark. Samples were then washed 3 times with DPBS and the coverslips were mounted on slides with ProLong Gold Antifade Mountant with DAPI.
Transgenic rescue
Cells were transduced by lentivirus and selected by puromycin as described in “Knock out single gene by using LentiCRISPRv2”. On day 4 of the selection, cells were transfected with rescue vectors by using Lipo3000. 24 h after transfection, cells were dislodged and plated in complete medium with puromycin for downstream assays.
Phylogenetic analysis
Homologs of CINP, C1ORF109, SPATA5 and SPATA5L were collected using BLAST and aligned using Muscle (Edgar, 2004). All positions with more than 25% gaps were eliminated after alignment. Evolutionary analyses were conducted in MEGA X (Kumar et al., 2018). The evolutionary history was inferred by using the Maximum Likelihood method with the LG model (Le and Gascuel, 2008). The tree with the highest log likelihood is shown, and branch confidence values (500 bootstrap replicates) are shown. Branch lengths are measured in the number of substitutions per site.
QUANTIFICATION AND STATISTICAL ANALYSIS
General statistical analysis
No statistical method was used to determine whether the data met assumptions of the statistical approach. All statistical data were calculated using GraphPad Prism 8. Statistical details on number of biological replicates (n) and the statistical test applied can be found in figure legends and the following sections.
Quantitative image analysis
Images were taken with 40× objective lens and were directedly processed in Fiji as follows: (1) subtract background, (2) gaussian blur, (3) split channels. For DAPI channel, the images were then processed: (1) threshold-auto, (2) fill holes, (3) erode, (4) watershed, (5) erode, (6) analyze particles, to generate region of interests (ROIs). The green channel images were then measured with ROIs for average intensity. The individual intensity data points were then normalized to the mean of control groups. The n for quantitative image analysis is number of different batches of biological samples. Mean ± S.E.M. is show in corresponding plots. p values were calculated by nested one-way ANOVA with Dunnett’s multiple comparisons test.
rRNA and northern blot quantification
The intensity of the RNA band was quantified using Fiji. The n for quantitative image analysis is number of different batches of biological samples. Mean ± S.E.M. is show in corresponding plots. p values were calculated using a ratio-paired t-test.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| RPL28 Polyclonal Antibody | Invitrogen | PA5-62192; RRID: AB_2646739 |
| Anti-RPL24 antibody | Abcam | ab126172; RRID: AB_11127305 |
| RSL24D1 Polyclonal antibody | Proteintech | 25190-1-AP; RRID: AB_2879950 |
| RSL24D1 Polyclonal antibody | Invitrogen | PA5-64058; RRID: AB_2646835 |
| Recombinant Anti-AFG2 antibody | Abcam | ab189519 |
| Recombinant Anti-CINP antibody | Abcam | ab183519 |
| SPATA5L1 Monoclonal Antibody (16A2) | Invitrogen | MA5-34952; RRID: AB_2848858 |
| Monoclonal ANTI-FLAG® M2 antibody | Sigma | F1804; RRID: AB_262044 |
| ANTI-FLAG® M2 Affinity Gel | Sigma | A2220; RRID: AB_10063035 |
| Anti-SNAP-tag® Antibody (Polyclonal) | NEB | P9310S; RRID: AB_10631145 |
| Anti-Glyceraldehyde-3-Phosphate Dehydrogenase Antibody, clone 6C5 | Sigma | MAB374; RRID: AB_2107445 |
| Chemicals, peptides, and recombinant proteins | ||
| Anisomycin | Sigma | A9789 |
| Puromycin | Sigma | P8833 |
| Cycloheximide | Sigma | C4859 |
| Sodium Arsenite | Sigma | 5092650001 |
| CX5461 | Sigma | S7400 |
| SUPERase•In™ RNase Inhibitor | Invitrogen | AM2694 |
| RNase T1 | Thermo Scientific | EN0541 |
| DSP | Thermo Scientific | 22586 |
| SNAP-Cell TMR-Star | NEB | S9105S |
| SNAP-Cell Block | NEB | S9106S |
| SNAP-Cell Oregon Green | NEB | S9104S |
| SNAP-Capture Magnetic Beads | NEB | S9145S |
| Lipofectamine 3000 | Thermo Scientific | L3000008 |
| Click-&-Go™ Click Chemistry Reaction Buffer Kit | clickchemistrytools | 1001 |
| Click-&-Go™ Cell Reaction Buffer Kit | clickchemistrytools | 1263 |
| L-Azidohomoalanine (AHA) | clickchemistrytools | 1066 |
| AZDye 488 Azide | clickchemistrytools | 1275 |
| Biotin Alkyne | clickchemistrytools | 1266 |
| NorthernMax™ Kit | Invitrogen | AM1940 |
| DMEM, high glucose, no glutamine, no methionine, no cystine | Gibco | 21013024 |
| GlutaMAX™ Supplement | Gibco | 35050061 |
| Fetal Bovine Serum, dialyzed | Gibco | A3382001 |
| L-Cysteine | Sigma | C7352 |
| HRP-Conjugated Streptavidin | Thermo Scientific | N100 |
| Deposited data | ||
| Raw CRISPR screen data | This study | GSE197277 |
| Analyzed CRISPR screen data_gene list | This study | GSE197277 |
| Analyzed CRISPR screen data_guide list | This study | GSE197277 |
| Experimental models: Cell lines | ||
| Lenti-X 293T | Takarabio | 632180 |
| 293T-RPL28-SNAP | This study | N/A |
| 293T-RPL28-SNAP-pCW57.1-C1orf109-3xFLAG-Tet-off | This study | N/A |
| HCT116-RPL28-SNAP | This study | N/A |
| H9-RPL28-SNAP | This study | N/A |
| Oligonucleotides | ||
| sgRNA for RPL28 knock-in: CTTTCTCACCTGCCTCGAC | (An and Harper, 2018) | N/A |
| See Table S1 for sgRNA for CRISPRKO | (Doench et al., 2016) | N/A |
| See Table S2 for Northern probes | (Tafforeau et al., 2013) | N/A |
| Recombinant DNA | ||
| pCW57.1-C1orf109-3xFLAG-Tet-off | This study | N/A |
| pCMV2-C1orf109 | This study | N/A |
| pCMV2-SPATA5 | This study | N/A |
| pCMV2-SPATA5-T330del | This study | N/A |
| pCMV2-SPATA5-D628G | This study | N/A |
| Software and algorithms | ||
| GraphPad Prism 8 | GraphPad | N/A |
| FlowJo v10 | FlowJo | N/A |
| MAGeCKFlute | (Wang et al., 2019) | N/A |
Highlights.
Ribo-SNAP enables live tracking of old and new ribosomes at single-cell resolution
C1ORF109, CINP, SPATA5, and SPATA5L1 are essential for ribosome maturation
Microcephaly associated SPATA5 mutations affect ribosome biogenesis
ACKNOWLEDGMENTS
We thank E. Olson for sharing the HEK293T cell line, J. Mendell for sharing the HCT116 cell line and technical support for the sgRNA screen, and the Brown and Goldstein laboratory for sharing the Brunello library. We acknowledge the UTSW Flow Cytometry Facility and Moody Foundation Flow Cytometry Facility at UT Southwestern for performing cell sorting/flow cytometry analysis for this project. We also acknowledge the Next-Generation Sequencing Core of McDermott Center for performing next-generation sequencing. We thank J. Mendell, H. Zhu, P. Douglas, M. Conacci Sorrell, and members of the Buszczak laboratory for discussion, suggestions, and critically reviewing the manuscript. We thank O. Cheng for help with illustrations and J. Schoggins for sharing data prior to publication. This work was supported by grants from NIGMS (GM125812 and GM144043) and funding from the UT Southwestern Simmons Comprehensive Cancer Center to M.B. J.W. is a New York Stem Cell Foundation-Robertson Investigator and Virginia Murchison Linthicum Scholar in Medical Research and funded by CPRIT (RR170076), NIH (GM138565-01A1 and OD028763), and Welch (854671). D.A.S. is partially supported by a trainee fellowship from the Hamon Center for Regenerative Science and Medicine.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110597.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- An H, and Harper JW (2018). Systematic analysis of ribophagy in human cells reveals bystander flux during selective autophagy. Nat. Cell Biol 20, 135–143. 10.1038/s41556-017-0007-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An H, Ordureau A, Korner M, Paulo JA, and Harper JW (2020). Systematic quantitative analysis of ribosome inventory during nutrient stress. Nature 583, 303–309. 10.1038/s41586-020-2446-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubert M, O’Donohue MF, Lebaron S, and Gleizes PE (2018). Pre-ribosomal RNA processing in human cells: from mechanisms to congenital diseases. Biomolecules 8, 123. 10.3390/biom8040123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awad D, Prattes M, Kofler L, Rossler I, Loibl M, Pertl M, Zisser G, Wolinski H, Pertschy B, and Bergler H (2019). Inhibiting eukaryotic ribosome biogenesis. BMC Biol. 17, 46. 10.1186/s12915-019-0664-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnsack KE, and Bohnsack MT (2019). Uncovering the assembly pathway of human ribosomes and its emerging links to disease. EMBO J. 38, e100278. 10.15252/embj.2018100278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchert R, Nesbitt AI, Tawamie H, Krantz ID, Medne L, Helbig I, Matalon DR, Reis A, Santani A, Sticht H, and Abou Jamra R (2016). SPATA5 mutations cause a distinct autosomal recessive phenotype of intellectual disability, hypotonia and hearing loss. Orphanet. J. Rare Dis 11, 130. 10.1186/s13023-016-0509-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol 34, 184–191. 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drygin D, Lin A, Bliesath J, Ho CB, O’Brien SE, Proffitt C, Omori M, Haddach M, Schwaebe MK, Siddiqui-Jain A, et al. (2011). Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 71, 1418–1430. 10.1158/0008-5472.CAN-10-1728. [DOI] [PubMed] [Google Scholar]
- Edgar RC (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5, 113. 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farley-Barnes KI, Ogawa LM, and Baserga SJ (2019). Ribosomopathies: old concepts, new controversies. Trends Genet. 35, 754–767. 10.1016/j.tig.2019.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forstbauer LM, Brockschmidt FF, Moskvina V, Herold C, Redler S, Herzog A, Hillmer AM, Meesters C, Heilmann S, Albert F, et al. (2012). Genome-wide pooling approach identifies SPATA5 as a new susceptibility locus for alopecia areata. Eur. J. Hum. Genet 20, 326–332. 10.1038/ejhg.2011.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishina I, and Lattes B (2005). A novel Cdk2 interactor is phosphorylated by Cdc7 and associates with components of the replication complexes. Cell Cycle 4, 1120–1126. [PubMed] [Google Scholar]
- Hipp MS, Kasturi P, and Hartl FU (2019). The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol 20, 421–435. 10.1038/s41580-019-0101-y. [DOI] [PubMed] [Google Scholar]
- Hoelzel CA, and Zhang X (2020). Visualizing and manipulating biological processes by using HaloTag and SNAP-tag Technologies. Chembiochem, 1935–1946. 10.1002/cbic.202000037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Bruckner RJ, Paulo JA, Cannon JR, Ting L, Baltier K, Colby G, Gebreab F, Gygi MP, Parzen H, et al. (2017). Architecture of the human interactome defines protein communities and disease networks. Nature 545, 505–509. 10.1038/nature22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones NC, Lynn ML, Gaudenz K, Sakai D, Aoto K, Rey JP, Glynn EF, Ellington L, Du C, Dixon J, et al. (2008). Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function. Nat. Med 14, 125–133. 10.1038/nm1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappel L, Loibl M, Zisser G, Klein I, Fruhmann G, Gruber C, Unterweger S, Rechberger G, Pertschy B, and Bergler H (2012). Rlp24 activates the AAA-ATPase Drg1 to initiate cytoplasmic pre-60S maturation. J. Cell Biol 199, 771–782. 10.1083/jcb.201205021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaca E, Harel T, Pehlivan D, Jhangiani SN, Gambin T, Coban Akdemir Z, Gonzaga-Jauregui C, Erdin S, Bayram Y, Campbell IM, et al. (2015). Genes that affect brain structure and function identified by rare variant analyses of Mendelian neurologic disease. Neuron 88, 499–513. 10.1016/j.neuron.2015.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaips CL, Jayaraj GG, and Hartl FU (2018). Pathways of cellular proteostasis in aging and disease. J. Cell Biol 217, 51–63. 10.1083/jcb.201709072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinge S, and Woolford JL Jr. (2019). Ribosome assembly coming into focus. Nat. Rev. Mol. Cell Biol 20, 116–131. 10.1038/s41580-018-0078-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotecha N, Krutzik PO, and Irish JM (2010). Web-based analysis and publication of flow cytometry experiments. Curr. Protoc. Cytom 10, Unit10. 17. 10.1002/0471142956.cy1017s53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kressler D, Hurt E, Bergler H, and Bassler J (2012). The power of AAA-AT-Pases on the road of pre-60S ribosome maturation–molecular machines that strip pre-ribosomal particles. Biochim. Biophys. Acta 1823, 92–100. 10.1016/j.bbamcr.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Li M, Knyaz C, and Tamura K (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol 35, 1547–1549. 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurata H, Terashima H, Nakashima M, Okazaki T, Matsumura W, Ohno K, Saito Y, Maegaki Y, Kubota M, Nanba E, et al. (2016). Characterization of SPATA5-related encephalopathy in early childhood. Clin. Genet 90, 437–444. 10.1111/cge.12813. [DOI] [PubMed] [Google Scholar]
- Labbadia J, and Morimoto RI (2015). The biology of proteostasis in aging and disease. Annu. Rev. Biochem 84, 435–464. 10.1146/annurev-biochem-060614-033955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W, et al. (2018). ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–D1067. 10.1093/nar/gkx1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le SQ, and Gascuel O (2008). An improved general amino acid replacement matrix. Mol. Biol. Evol 25, 1307–1320. 10.1093/molbev/msn067. [DOI] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, and Liu XS (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554. 10.1186/s13059-014-0554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SS, Zheng HX, Jiang HD, He J, Yu Y, Qu YP, Yue L, Zhang Y, and Li Y (2012). Identification and characterization of a novel gene, c1orf109, encoding a CK2 substrate that is involved in cancer cell proliferation. J. Biomed. Sci 19, 49. 10.1186/1423-0127-19-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovejoy CA, Xu X, Bansbach CE, Glick GG, Zhao R, Ye F, Sirbu BM, Titus LC, Shyr Y, and Cortez D (2009). Functional genomic screens identify CINP as a genome maintenance protein. Proc. Natl. Acad. Sci. U S A 106, 19304–19309. 10.1073/pnas.0909345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunami N, Hensel CH, Baird L, Stevens J, Otterud B, Leppert T, Varvil T, Hadley D, Glessner JT, Pellegrino R, et al. (2014). Identification of rare DNA sequence variants in high-risk autism families and their prevalence in a large case/control population. Mol. Autism 5, 5. 10.1186/2040-2392-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng EC, Pettersen EF, Couch GS, Huang CC, and Ferrin TE (2006). Tools for integrated sequence-structure analysis with UCSF Chimera. BMC Bioinformatics 7, 339. 10.1186/1471-2105-7-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills EW, and Green R (2017). Ribosomopathies: there’s strength in numbers. Science 358. 10.1126/science.aan2755. [DOI] [PubMed] [Google Scholar]
- Moore J.B.t., Farrar JE, Arceci RJ, Liu JM, and Ellis SR (2010). Distinct ribosome maturation defects in yeast models of Diamond-Blackfan anemia and Shwachman-Diamond syndrome. Haematologica 95, 57–64. 10.3324/haematol.2009.012450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noack Watt KE, Achilleos A, Neben CL, Merrill AE, and Trainor PA (2016). The roles of RNA polymerase I and III subunits Polr1c and Polr1d in craniofacial development and in zebrafish models of Treacher collins syndrome. PLoS Genet. 12, e1006187. 10.1371/journal.pgen.1006187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan M, Wright WC, Chapple RH, Zubair A, Sandhu M, Batchelder JE, Huddle BC, Low J, Blankenship KB, Wang Y, et al. (2021). The chemotherapeutic CX-5461 primarily targets TOP2B and exhibits selective activity in high-risk neuroblastoma. Nat. Commun 12, 6468. 10.1038/s41467-021-26640-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertschy B, Saveanu C, Zisser G, Lebreton A, Tengg M, Jacquier A, Liebminger E, Nobis B, Kappel L, van der Klei I, et al. (2007). Cytoplasmic recycling of 60S preribosomal factors depends on the AAA protein Drg1. Mol. Cell Biol 27, 6581–6592. 10.1128/MCB.00668-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertschy B, Zisser G, Schein H, Koffel R, Rauch G, Grillitsch K, Morgenstern C, Durchschlag M, Hogenauer G, and Bergler H (2004). Diazaborine treatment of yeast cells inhibits maturation of the 60S ribosomal subunit. Mol. Cell Biol 24, 6476–6487. 10.1128/MCB.24.14.6476-6487.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prattes M, Grishkovskaya I, Hodirnau V-V, Rössler I, Klein I, Hetzmannseder C, Zisser G, Gruber CC, Gruber K, Haselbach D, and Bergler H (2021). Structural basis for inhibition of the AAA-ATPase Drg1 by diazaborine. Nat. Commun 12, 3483. 10.1038/s41467-021-23854-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prattes M, Lo YH, Bergler H, and Stanley RE (2019). Shaping the nascent ribosome: AAA-ATPases in eukaryotic ribosome biogenesis. Biomolecules 9, 715. 10.3390/biom9110715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puusepp S, Kovacs-Nagy R, Alhaddad B, Braunisch M, Hoffmann GF, Kotzaeridou U, Lichvarova L, Liiv M, Makowski C, Mandel M, et al. (2018). Compound heterozygous SPATA5 variants in four families and functional studies of SPATA5 deficiency. Eur. J. Hum. Genet 26, 407–419. 10.1038/s41431-017-0001-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retz KC, and Steele WJ (1980). Ribosome turnover in rat brain and liver. Life Sci. 27, 2601–2604. 10.1016/0024-3205(80)90546-9. [DOI] [PubMed] [Google Scholar]
- Richard EM, Bakhtiari S, Marsh APL, Kaiyrzhanov R, Wagner M, Shetty S, Pagnozzi A, Nordlie SM, Guida BS, Cornejo P, et al. (2021). Bi-allelic variants in SPATA5L1 lead to intellectual disability, spastic-dystonic cerebral palsy, epilepsy, and hearing loss. Am. J. Hum. Genet 108, 2006–2016. 10.1016/j.ajhg.2021.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Corona U, Sobol M, Rodriguez-Zapata LC, Hozak P, and Castano E (2015). Fibrillarin from archaea to human. Biol. Cell 107, 159–174. 10.1111/boc.201400077. [DOI] [PubMed] [Google Scholar]
- Rotenberg MO, Moritz M, and Woolford JL Jr. (1988). Depletion of Saccharomyces cerevisiae ribosomal protein L16 causes a decrease in 60S ribosomal subunits and formation of half-mer polyribosomes. Genes Dev. 2, 160–172. 10.1101/gad.2.2.160. [DOI] [PubMed] [Google Scholar]
- Sanson KR, Hanna RE, Hegde M, Donovan KF, Strand C, Sullender ME, Vaimberg EW, Goodale A, Root DE, Piccioni F, and Doench JG (2018). Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat. Commun 9, 5416. 10.1038/s41467-018-07901-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutz S, Fischer U, Altvater M, Nerurkar P, Pena C, Gerber M, Chang Y, Caesar S, Schubert OT, Schlenstedt G, and Panse VG (2014). A RanGTP-independent mechanism allows ribosomal protein nuclear import for ribosome assembly. Elife 3, e03473. 10.7554/eLife.03473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senior AW, Evans R, Jumper J, Kirkpatrick J, Sifre L, Green T, Qin C, Zidek A, Nelson AWR, Bridgland A, et al. (2020). Improved protein structure prediction using potentials from deep learning. Nature 577, 706–710. 10.1038/s41586-019-1923-7. [DOI] [PubMed] [Google Scholar]
- Steffen KK, and Dillin A (2016). A ribosomal perspective on proteostasis and aging. Cell Metab. 23, 1004–1012. 10.1016/j.cmet.2016.05.013. [DOI] [PubMed] [Google Scholar]
- Stoykova AS, Dudov KP, Dabeva MD, and Hadjiolov AA (1983). Different rates of synthesis and turnover of ribosomal RNA in rat brain and liver. J. Neurochem 41, 942–949. 10.1111/j.1471-4159.1983.tb09038.x. [DOI] [PubMed] [Google Scholar]
- Szczaluba K, Szymanska K, Kosinska J, Pollak A, Murcia V, Kedra A, Stawinski P, Rydzanicz M, Demkow U, and Ploski R (2017). Isolated hearing impairment caused by SPATA5 mutations in a family with variable phenotypic expression. Adv. Exp. Med. Biol 980, 59–66. 10.1007/5584_2016_206. [DOI] [PubMed] [Google Scholar]
- Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, et al. (2019). STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613. 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tafforeau L, Zorbas C, Langhendries JL, Mullineux ST, Stamatopoulou V, Mullier R, Wacheul L, and Lafontaine DL (2013). The complexity of human ribosome biogenesis revealed by systematic nucleolar screening of Pre-rRNA processing factors. Mol. Cell 51, 539–551. 10.1016/j.molcel.2013.08.011. [DOI] [PubMed] [Google Scholar]
- Tanaka AJ, Cho MT, Millan F, Juusola J, Retterer K, Joshi C, Niyazov D, Garnica A, Gratz E, Deardorff M, et al. (2015). Mutations in SPATA5 are associated with microcephaly, intellectual disability, seizures, and hearing loss. Am. J. Hum. Genet 97, 457–464. 10.1016/j.ajhg.2015.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiu GC, Kerr CH, Forester CM, Krishnarao PS, Rosenblatt HD, Raj N, Lantz TC, Zhulyn O, Bowen ME, Shokat L, et al. (2021). A p53-dependent translational program directs tissue-selective phenotypes in a model of ribosomopathies. Dev. Cell 56, 2089–2102.e2011. 10.1016/j.devcel.2021.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama BH, Savas JN, Park SK, Harris MS, Ingolia NT, Yates JR 3rd, and Hetzer MW (2013). Identification of long-lived proteins reveals exceptional stability of essential cellular structures. Cell 154, 971–982. 10.1016/j.cell.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Wang M, Zhang W, Xiao T, Chen C-H, Wu A, Wu F, Traugh N, Wang X, Li Z, et al. (2019). Integrative analysis of pooled CRISPR genetic screens using MAGeCKFlute. Nat. Protoc 14, 756–780. 10.1038/s41596-018-0113-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild T, Horvath P, Wyler E, Widmann B, Badertscher L, Zemp I, Kozak K, Csucs G, Lund E, and Kutay U (2010). A protein inventory of human ribosome biogenesis reveals an essential function of exportin 5 in 60S subunit export. PLoS Biol. 8, e1000522. 10.1371/journal.pbio.1000522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakalskiy A, Hogenauer G, Ishikawa T, Wehrschutz-Sigl E, Wendler F, Teis D, Zisser G, Steven AC, and Bergler H (2002). Structural and enzymatic properties of the AAA protein Drg1p from Saccharomyces cerevisiae. Decoupling of intracellular function from ATPase activity and hexamerization. J. Biol. Chem 277, 26788–26795. 10.1074/jbc.M201515200. [DOI] [PubMed] [Google Scholar]
- Zanus C, Costa P, Faletra F, Musante L, Russo A, Grazian L, and Carrozzi M (2020). Description of a peculiar alternating ictal electroclinical pattern in a young boy with a novel SPATA5 mutation. Epileptic Disord. 22, 659–663. 10.1684/epd.2020.1204. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw and processed CRISPR screen data have been deposited at GEO and are publicly available (GSE197277).
This paper does not report original code.
Any additional data and information required to analyze the data reported in this paper are available from the lead contact upon request.