

Abstract
The mouse closed head injury (CHI) model of traumatic brain injury (TBI) produces widespread demyelination. Myelin content is restored by minocycline (MINO) plus n-acetylcysteine (NAC) or MINO alone when first dosed at 12 hours after CHI. In a rat controlled cortical impact model of TBl, a first dose of MINO plus NAC one hour after injury protects resident oligodendrocytes that induce remyelination. In contrast, MINO less effectively protects oligodendrocytes and remyelination is mediated by oligodendrocyte precursor cell proliferation and differentiation. MINO plus NAC or MINO alone is hypothesized to work similarly in the CHI model as in the controlled cortical impact model even when first dosed at 12-hours post-CHI. We tested this hypothesis by examining the time course of the changes in the oligodendrocyte antigenic markers CC1, 2′,3′-Cyclic-nucleotide 3′-phosphodiesterase and phospholipid protein between 2 and 14 days post-CHI in mice treated with saline, NAC, MINO or MINO plus NAC. CHI produced a long-lasting loss of these markers that was not altered by NAC treatment. In contrast, oligodendrocyte marker expression was maintained by MINO plus NAC between 2 and 14 days post-injury. MINO alone did not prevent the early loss of oligodendrocyte markers, but marker expression significantly increased by 14-days post-injury. These data suggest that MINO plus NAC or MINO alone when first dosed 12 hours after CHI increase myelin content using similar mechanisms seen when first dosed 1 hour after closed head injury. These data also suggest that drugs protect oligodendrocytes with a clinically useful therapeutic time window.
Keywords: traumatic brain injury, corpus callosum, glia, therapeutic time window
TBI damages both axons and oligodendrocytes that retract their myelin sheaths early after injury [12, 21]. The corpus callosum is a white matter tract commonly damaged after TBI due to shear forces produced by the independent movements of the two cerebral hemispheres [20]. The extent of demyelination and remyelination after injury is determined by the response of oligodendrocytes and oligodendrocyte precursor cells [21]. If oligodendrocytes do not die, they remyelinate nearby axons [1]. If oligodendrocyte are lost, remyelination can occur following proliferation and differentiation of oligodendrocyte precursor cells into newly myelinating oligodendrocytes [1, 21].
The effect of MINO plus NAC on demyelination and remyelination has been examined in two TBI animal models. The rat CCI model produces a focal contusion and gray and white matter injury both proximal and distal to the impact site [25]. In the mouse CHI model, the contusion is smaller than in the CCI model and the gray and white matter injury is more diffuse [9]. MINO plus NAC or MINO alone limits myelin loss in both the CHI and CCI models [5, 9]. In the CCI model, MINO plus NAC prevented oligodendrocytes loss and produced remyelination mediated by surviving oligodendrocytes. In contrast, MINO alone induced remyelination subsequent to oligodendrocyte loss by oligodendrocyte precursor cell proliferation and differentiation. MINO plus NAC or MINO alone maintained myelin content in mice 14 days post-injury even when first dosed at 12 hours post-CHI [19]. We tested if MINO plus NAC or MINO alone had similar actions on oligodendrocytes in the CHI model as in the CCI model when first first dosed 12 hours after CHI.
Materials and methods
Experimental TBI and drug treatments
CHI was performed on male C57/BL6 mice (15 to 17 old, 26-28gr, Jackson Laboratories). Mouse were encoded so experimenters were unaware of its injury or treatment status. All animal experiments were carried out in accordance with the National Institutes of Health guide for the care and use of Laboratory animals (NIH Publications No. 8023, revised 1978) and approved by the Institutional Animal Care and Use Committee of the SUNY-Downstate Medical Center (protocol #14-10406). CHI was produced as described by Grin’kina, et al. [9]. Five groups (sham-CHI saline, CHI saline, CHI NAC, CHI MINO, and CHI MINO plus NAC) tested the hypothesis that the actions on oligodendrocytes seen with MINO plus NAC or MINO dosed first 1 hour after CCI in rats would be similar to dosing 12 hours after CHI in mice. After drug treatments, mice were sacrificed for histological analysis at 2, 4, 7, and 14 days after sham-CHI or CHI (Fig. 1). Sangobowale, et al. 2018 determined the optimal concentrations of MINO (22.5 mM) and NAC (75 mM) that enhanced behavioral outcomes in the mouse CHI model [19]. These are the drug concentrations used in this study. Sham-injured mice received identical treatment without the impact. The ability of the mouse to spontaneously right itself was assessed after injury and the average righting reflex of injured mice was 8.64 ± 0.47 minutes [19].
Figure 1.
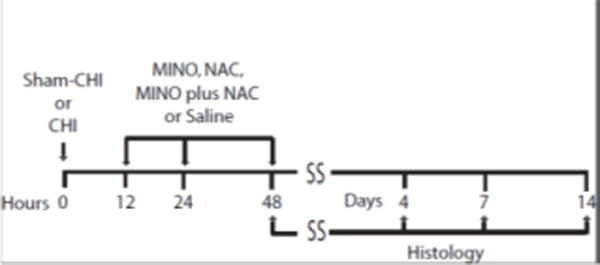
Experimental Design A schematic of the experimental design is shown. The time line has hours for units on the left and days for units on the right. Mice received either Sham-CHI or CHI followed by 3 doses of saline, MINO, NAC or MINO plus NAC at 12, 24 and 48 hours. Mice were sacrificed for histology at 2, 4, 7, or 14 days after Sham-CHI or CHI.
Histology
Saline, MINO, NAC or MINO plus NAC (Sigma, St. Louis, MO) were first first dosed intraperitoneally 12 hours after CHI. A 12-hour time point was selected since this treatment maintained myelin content [19]. A second and third dose was given intraperitoneally 24 and 48 hours after sham-CHI or CHI (Fig. 1). Two, 4, 7, and 14 days after sham-CHI or CHI, brains were fixed by transcardial perfusion paraformaldehyde (4% w/v)), embedded in paraffin and parasagittal sections prepared (Histowiz, Brooklyn, NY). The corpus callosum was analyzed in parasagittal sections ipsilateral to the impact site (from bregma; DV -2.4; ML, 1.5-1.9 mm; DV, −0.9) [16]. Oligodendrocyte soma were stained with antibodies that bind the N-terminus of the APC protein that also recognizes CC1 protein (Millipore, 1:100) [2, 3, 14]. Antibodies against proteolipid protein (PLP) stained myelinating processes of oligodendrocytes (Abcam, 1:100). The soma and processes of myelinating oligodendrocytes were stained using 2′,3′-Cyclic-nucleotide 3′-phosphodiesterase (CNPase, Developmental Studies Hybridoma Bank, 1:500) [18]. Apoptotic cells were stained with an antibody against cleaved caspase 3 (Cell Signalling, 1:300). CNPase and PLP immunoreactivity was assayed as described by Haber, et al [10]. Adjacent regions were stained with only primary or secondary antibodies to ensure antibody specificity. CC1+ cells were counted in a 100 mm2 area of corpus callosum. NIH image J software measured the area of CNP and PLP antibody immunoreactivity [11].
Statistics
Two-way ANOVA assessed group differences on the factors of treatment and day and 1-way ANOVA analyzed within treatment groups differences on the factor of day. Tukey’s post-hoc test analyzed pairwise differences. IBM SPSS™ (V.23) performed all analyses with significance set at 0.05. All values are mean ± SEM.
Results
CC1+ cells were assessed in the corpus callosum of sham-CHI saline, CHI saline, CHI NAC, CHI MINO, and CHI MINO plus NAC ipsilateral to the impact site (Fig. 2A). There was a significant effect of treatment but not of day with no interaction between treatment and day (Fig. 2B) (treatment F4,73 = 36.4, p < 0.0001; day, F3,73 = 5.80, p > 0.5; interaction, F9,142 = 0.37, p > 0.5). Post-hoc analysis indicated that the number of CC1+ cells in the sham-CHI saline group was significantly more than in the CHI saline or CHI NAC groups. These data suggest that CHI induced loss of CC1+ cells that was unaffected by NAC. The Sham-CHI saline and CHI MINO plus NAC groups had similar numbers of CC1+ cells. The CHI MINO group had more CC1+ cells than the CHI saline group, but fewer CC1+ cells than the CHI MINO plus NAC group. These data suggest protection of resident oligodendrocytes by MINO plus NAC or MINO alone, yet MINO plus NAC in protecting oligodendrocytes was more effective than MINO alone. This conclusion was further supported by analyzing the time course of the change in CC1+ cells within each group. There was no significant effect of day in the CHI MINO plus NAC group (F3,15 = 0.17, p > 0.5). In contrast, the effect of day was significant within the CHI MINO group (F3,15 = 3.53, p < 0.05). Post-hoc analysis indicated CC1+ cell number in the CHI MINO group on days 2 and 4 post-CHI was significantly lower than at days 7 and 14 (p < 0.05). These data further suggest that MINO plus NAC provided greater protection of oligodendrocytes than MINO alone.
Figure 2.
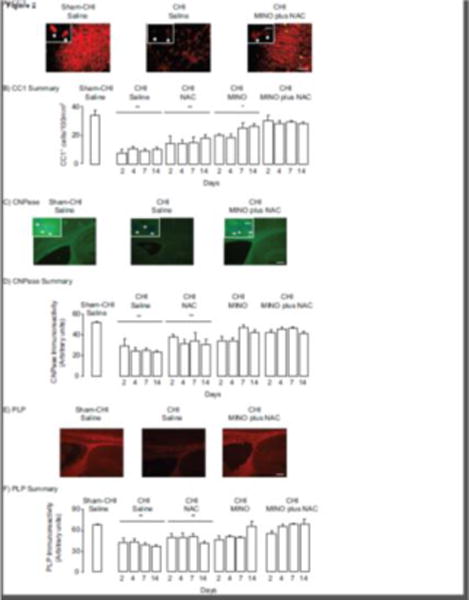
Changes in oligodendrocyte marker expression between 2 and 14 days after CHI. All images are of the corpus callosum ipsilateral to the impact site. Panel A, Representative images of CCI immunoreactivity. Panels A and C contain inserts of higher power images, respectively, of CC1 and CNPase positive cells. Panel B, Summary of changes in CCI+ cells. The Sham CHI saline or CHI MINO plus NAC groups had significantly more cells than the CHI saline, CHI NAC, and CHI MINO groups (**p < 0.01, *p < 0.05). Panel C, Representative images of CNPase immunoreactivity. Panel D, Summary of changes in CNPase immunoreactivity. The Sham CHI saline or CHI MINO plus NAC groups had significantly more CNPase immunoreactivity than the CHI saline, and CHI NAC groups (**p < 0.01). Panel E. Representative images of PLP immunoreactivity, Panel F, Summary of changes in PLP immunoreactivity, The Sham CHI saline or CHI MINO plus NAC groups had significantly more PLP immunoreactivity than the CHI saline, and CHI NAC groups (**p < 0.01). Scale Bars for panel A, 20 μm, for panels C and E; 250 μm. Scale Bar for inserts, 10 μm.
CNPase immunoreactivity was also assessed in corpus callosum (Fig. 2C). CNPase immunoreactivity had a significant effect of treatment but no effect of day and no significant interaction between treatment and day (Fig. 2D) (treatment, F4,73 = 17.89, p < 0.0001; day, F3,73 = 1.84, p > 0.1; interaction. F9,142 = 40.63, p > 0.5). Post-hoc analysis indicated that CNPase immunoreactivity in the Sham-CHI saline group was significantly higher than in the CHI saline or CHI NAC groups. These data suggest that CHI diminished CNPase immunoreactivity that was unaffected by NAC. CNPase immunoreactivity in the CHI MINO plus NAC groups was similar to the Sham-CHI saline group. CNPase immunoreactivity in the MINO alone group differed significantly from both the CHI MINO plus NAC and the Sham-CHI saline groups. These data suggest that MINO plus NAC more effectively prevented the loss of CNPase immunoreactivity than MINO alone. These results were supported by analysis of the time course of changes in CNPase activity within each group. The CHI MINO plus NAC group had no significant effect of day (F3,15 = 0.68, p > 0.5). In contrast the CHI MINO group had a significant effect of day (F3,15 = 4.92, p < 0.02). Post-hoc analysis indicated significantly lower CNPase immunoreactivity in the CHI MINO group on day 2 post-CHI than on day 14. These data suggest that MINO plus NAC maintained CNPase immunoreactivity between 2 and 14 days post-CHI at sham CHI levels while CNPase immunoreactivity rapidly diminished in injured mice treated with MINO alone. By day 14, the CHI MINO group significantly increased CNPase immunoreactivity.
PLP immunoreactivity was also assessed in the corpus callosum (Fig. 2E). PLP immunoreactivity had significant effects of treatment and day with no interactions between treatment and day (Fig 2F) (treatment, F4,73 = 4.01, p < 0.01; day, F3,73 = 7.60, p < 0.005; interaction, F9,142 = 1.26, p > 0.1). PLP immunoreactivity in the Sham-CHI saline group significantly differed from the CHI saline and CHI NAC groups. These data suggest that CHI reduces PLP expression; NAC had no effect on this loss of PLP immunoreactivity. Within the CHI MINO plus NAC group, there was no significant effect of day (F3,15 = 0.17, p > 0.5). The effect of day, however, was significant within the CHI MINO group (F3,15 = 3.53, p < 0.05) with significantly less PLP immunoreactivity on day 4 than day 14 post- CHI. These data suggest that MINO plus NAC prevent loss of PLP immunoreactivity while MINO alone did not. MINO treatment did significantly increase PLP immunoreactivity between 4 and 14 days post-CHI.
Cleaved caspase 3 is a marker of apoptotic cells [10]. A first dose of MINO plus NAC one hour after CCI lowered the number of CC3+ cells 2 days after injury [10]. CC3+ cells in the corpus callosum were examined 2 and 4 days after sham-CHI or CHI (Figure 3). There was a significant effect of treatment (ANOVA, F4,15 = 21.46, p<0.0001) with MINO plus NAC significantly lowering the number of CC3+ cells on both days 2 and 4.
Figure 3.
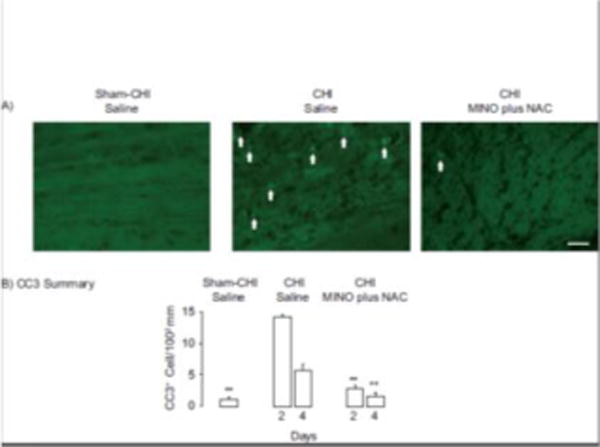
MINO plus NAC lowers the number of apoptotic cells in the corpus callosum. Panel A, Representative images of CC3 immunoreactivity. Arrows indicate CC3+ cells. Panel B, Summary of changes of CC3 immunoreactivity. The sham CHI saline and CHI MINO plus NAC groups had significantly fewer CC3 positive cells than the CHI saline group (**p<0.01). Scale Bar, 100 μm.
Discussion
The major findings of this study are: 1) CHI produced a persistent loss of oligodendrocytes in the corpus callosum; 2) A first dose of MINO plus NAC at 12 hours post CHI prevented apoptosis and protected oligodendrocytes between days 2 to 14 days after CHI (Figs 2 and 3), 3) A first dose of MINO at 12 hours did not prevent the early loss of oligodendrocytes after CHI; oligodendrocytes did recover by 14 days post-CHI. These conclusions were supported by the time course of the expression of three separate oligodendrocyte antigenic markers (Fig. 2). A first dose of MINO plus NAC or MINO alone increased also luxol fast blue staining 14 days post-CHI [19]. These data, with the oligodendrocytes studies presented, here suggest that MINO plus NAC or MINO promote remyelination albeit by different mechanisms (Fig. 2).
Demyelination occurs after TBI and in animal models of TBI [1, 13, 20, 21]. Remyelination fails due to loss of oligodendrocytes and the inability of oligodendrocyte precursor cells to proliferate and differentiate into myelinating oligodendrocytes [1, 5]. Oligodendrocytes were protected by a first dose of MINO plus NAC beginning 1 hour after CHI in rats [10]. MINO alone did not protect oligodendrocytes, but supported oligodendrocyte precursor cell proliferation and differentiation [10]. The data in this study suggests that these drug actions were retained if the first dose of MINO or MINO plus NAC was delayed 12 hours after CHI (Fig. 2). MINO plus NAC or MINO alone, however, no longer maintained myelin content when first dosed at 24 after CHI [19]. These data suggest that the ability to remyelinate is not lost after TBI and can be reactivated with drugs within 12 hours after injury. It is unknown why MINO plus NAC no longer maintains myelin content when first dosed at 24 hours after CHI, but is likely that the drug combination, at this time point, no longer protects oligodendrocytes.
MINO alone restored oligodendrocytes and improved myelin content when first dosed 12 hours after CHI (Fig. 2) [19]. MINO alone has had differing effects in other models of white matter damage and repair. MINO alone improved remyelination in the experimental autoimmune encephalitis model of multiple sclerosis [6, 17]. In contrast, conflicting effects of MINO on remyelination were reported in the cuprizone model of demyelination-remyelination [15, 22, 23].
An unanswered question is why MINO plus NAC and MINO alone use different mechanisms to promote remyelination. Inflammation induces oligodendrocyte death following CHI [1]. Inflammation also modulates remyelination [7, 24] Both MINO and NAC have anti-inflammatory actions [4, 8]. Studies using the rat CCI model suggest that MINO plus NAC increases microglial activation followed by a rapid inhibition of activation. [10]. These actions were not seen with the individual drugs [10]. Similar results were obtained with MINO plus NAC in the mouse CHI model (Ho, J., Prela, O. and Bergold, P.J., unpublished results). Oligodendrocytes were protected by MINO plus NAC but not MINO or NAC alone (Fig. 2). We are testing the hypothesis that modulation of inflammation after CHI underlies the protection of oligodendrocytes by MINO plus NAC.
Highlights.
Closed head injury induces a rapid and persistent loss of oligodendrocytes in the corpus callosum.
Minocycline plus N-acetylcysteine produces a persistent protection of oligodendrocytes after closed head injury.
Minocycline alone produces a partial protection of oligodendrocytes; oligodendrocyte number recovers by 14 days post-injury.
N-acetylcysteine has no effect on the loss of oligodendrocytes.
These effects were seen when the drugs were first dosed 12 hours after closed head injury suggesting that act with a clinically useful time window.
Acknowledgments
This work was supported by a grant (RO1070512) to P.J.B.
Abbreviations
- CCI
controlled cortical impact
- CC3
Cleaved caspase 3
- CHI
closed head injury
- CNPase
2′,3′-Cyclic-nucleotide 3′-phosphodiesterase
- MINO
minocycline
- NAC
n-acetylcysteine
- PLP
proteolipid protein
- TBI
traumatic brain injury
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Armstrong RC, Mierzwa AJ, Marion CM, Sullivan GM. White matter involvement after TBI: Clues to axon and myelin repair capacity. Exp Neurol. 2016;275:328–333. doi: 10.1016/j.expneurol.2015.02.011. [DOI] [PubMed] [Google Scholar]
- 2.Bhat RV, Axt KJ, Fosnaugh JS, Smith KJ, Johnson KA, Hill DE, Kinzler KW, Baraban JM. Expression of the APC tumor suppressor protein in oligodendroglia. Glia. 1996;17:169–174. doi: 10.1002/(SICI)1098-1136(199606)17:2<169::AID-GLIA8>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 3.Brakeman JSF, Gu SH, Wang XB, Dolin G, Baraban JM. Neuronal localization of the adenomatous polyposis coli tumor suppressor protein. Neurosci. 1999;91:661–672. doi: 10.1016/s0306-4522(98)00605-8. [DOI] [PubMed] [Google Scholar]
- 4.Chen G, Shi J, Hu Z, Hang C. Inhibitory effect on cerebral inflammatory response following traumatic brain injury in rats: a potential neuroprotective mechanism of N-acetylcysteine. Mediators Inflam. 2008;2008:716458. doi: 10.1155/2008/716458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen S, Pickard JD, Harris NG. Time course of cellular pathology after controlled cortical impact injury. Exp Neurol. 2003;182:87–102. doi: 10.1016/s0014-4886(03)00002-5. [DOI] [PubMed] [Google Scholar]
- 6.Chen XX, Ma L, Jiang Y, Chen S, Zhu C, Liu M, Ma X, Zhu D, Liu Y, Peng F, Wang Q, Pi R. Minocycline up-regulates the expression of brain- derived neurotrophic factor and nerve growth factor in experimental autoimmune encephalomyelitis. Eur J Pharmacol. 2012;686:124–129. doi: 10.1016/j.ejphar.2012.04.043. [DOI] [PubMed] [Google Scholar]
- 7.Foote AF, Blakemore WF. Inflammation stimulates remyelination in areas of chronic demyelination. Brain. 2005;128:528–539. doi: 10.1093/brain/awh417. [DOI] [PubMed] [Google Scholar]
- 8.Garrido-Mesa N, Zarzuelo A, Galvez J. Minocycline: far beyond an antibiotic. Br J Pharmacol. 2013;169:337–352. doi: 10.1111/bph.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grin’kina NM, Li Y, Haber M, Sangobowale M, Nikulina E, Le’Pre C, El Sehamy AM, Dugue R, Ho JS, Bergold PJ. Righting Reflex Predicts Long-Term Histological and Behavioral Outcomes in a Closed Head Model of Traumatic Brain Injury. PLOS ONE. 2016;11:e0161053. doi: 10.1371/journal.pone.0161053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haber M, James J, Kim J, Sangobowale M, Irizarry R, Ho JS, Nikulina E, Grin’kina NM, Ramadani A, Hartman I, Bergold PJ. Minocycline plus N-acteylcysteine induces remyelination, synergistically protects oligodendrocytes, and modifies neuroinflammation in a rat model of mild traumatic brain injury. J Cereb Blood Flow Metab. 2018 Jan; doi: 10.1177/0271678X17718106. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Inman CF, Rees LEN, Barker E, Haverson K, Stokes CR, Bailey M. Validation of computer-assisted, pixel-based analysis of multiple-colour immunofluorescence histology. J Immuno Meth. 2005;302:156–167. doi: 10.1016/j.jim.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 12.Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, Stewart W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain. 2013;136:28–42. doi: 10.1093/brain/aws322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kinnunen KM, Greenwood R, Powell JH, Leech R, Hawkins PC, Bonnelle V, Patel MC, Counsell SJ, Sharp DJ. White matter damage and cognitive impairment after traumatic brain injury. Brain. 2011;134:449–463. doi: 10.1093/brain/awq347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lang J, Maeda Y, Bannerman P, Xu J, Horiuchi M, Pleasure D, Guo F. Adenomatous Polyposis Coli Regulates Oligodendroglial Development. J Neurosci. 2013;33:3113–3130. doi: 10.1523/JNEUROSCI.3467-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li WW, Setzu A, Zhao C, Franklin RJ. Minocycline-mediated inhibition of microglia activation impairs oligodendrocyte progenitor cell responses and remyelination in a non-immune model of demyelination. J Neuroimmunol. 2005;158:58–66. doi: 10.1016/j.jneuroim.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 16.Paxinos G, Franklin KBJ. The Mouse Brain in Stereotaxic Coordinates. Academic Press; New York: 2013. [Google Scholar]
- 17.Popovic N, Schubart A, Goetz BD, Zhang SC, Linington C, Duncan ID. Inhibition of autoimmune encephalomyelitis by a tetracycline. Ann Neurol. 2002;51:215–223. doi: 10.1002/ana.10092. [DOI] [PubMed] [Google Scholar]
- 18.Raasakka A, Kursula P. The myelin membrane-associated enzyme 2′,3′-cyclic nucleotide 3′-phosphodiesterase: on a highway to structure and function. Neurosci Bull. 2014;30:956–966. doi: 10.1007/s12264-013-1437-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sangobowale MA, Grin’kina NM, Whitney K, Nikulina E, St Laurent-Ariot K, Ho JS, Bayzan N, Bergold PJ. Minocycline plus N-acetylcysteine reduce behavioral deficits and improve histology with a clinically useful time window. J Neurotrauma. 2018 Feb; doi: 10.1089/neu.2017.5348. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 20.Shenton ME, Hamoda HM, Schneiderman JS, Bouix S, Pasternak O, Rathi Y, Vu MA, Purohit MP, Helmer K, Koerte I, Lin AP, Westin CF, Kikinis R, Kubicki M, Stern RA, Zafonte R. A Review of Magnetic Resonance Imaging and Diffusion Tensor Imaging Findings in Mild Traumatic Brain Injury. Brain Imaging Behav. 2012;6:137–192. doi: 10.1007/s11682-012-9156-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shi H, Hu X, Leak RK, Shi Y, An C, Suenaga J, Chen J, Gao Y. Demyelination as a Rational Therapeutic Target for Ischemic or Traumatic Brain Injury. Exp Neurol. 2015;272:17–25. doi: 10.1016/j.expneurol.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Skripuletz T, Miller E, Moharregh-Khiabani D, Blank A, Pul R, Gudi V, Trebst C, Stangel M. Beneficial effects of minocycline on cuprizone-induced cortical demyelination. Neurochem Res. 2010;35:1422–1433. doi: 10.1007/s11064-010-0202-7. [DOI] [PubMed] [Google Scholar]
- 23.Tanaka T, Murakami K, Bando Y, Yoshida S. Minocycline reduces remyelination by suppressing ciliary neurotrophic factor expression after cuprizone-induced demyelination. J Neurochem. 2013;127:259–270. doi: 10.1111/jnc.12289. [DOI] [PubMed] [Google Scholar]
- 24.Watzlawik J, Warrington AE, Rodriguez M. Importance of oligodendrocyte protection, BBB breakdown and inflammation for remyelination. Expert Rev Neurother. 2010;10:441–457. doi: 10.1586/ern.10.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiong Y, Mahmood A, Chopp M. Animal models of traumatic brain injury. Nat Rev Neurosci. 2013;14:128–142. doi: 10.1038/nrn3407. [DOI] [PMC free article] [PubMed] [Google Scholar]