

ABSTRACT
Monitoring the prevalence of SARS-CoV-2 variants is necessary to make informed public health decisions during the COVID-19 pandemic. PCR assays have received global attention, facilitating a rapid understanding of variant dynamics because they are more accessible and scalable than genome sequencing. However, as PCR assays target only a few mutations, their accuracy could be reduced when these mutations are not exclusive to the target variants. Here we introduce PRIMES, an algorithm that evaluates the sensitivity and specificity of SARS-CoV-2 variant-specific PCR assays across different geographical regions by incorporating sequences deposited in the GISAID database. Using PRIMES, we determined that the accuracy of several PCR assays decreased when applied beyond the geographic scope of the study in which the assays were developed. Subsequently, we used this tool to design Alpha and Delta variant-specific PCR assays for samples from Illinois, USA. In silico analysis using PRIMES determined the sensitivity/specificity to be 0.99/0.99 for the Alpha variant-specific PCR assay and 0.98/1.00 for the Delta variant-specific PCR assay in Illinois, respectively. We applied these two variant-specific PCR assays to six local sewage samples and determined the dominant SARS-CoV-2 variant of either the wild type, the Alpha variant, or the Delta variant. Using next-generation sequencing (NGS) of the spike (S) gene amplicons of the Delta variant-dominant samples, we found six mutations exclusive to the Delta variant (S:T19R, S:Δ156/157, S:L452R, S:T478K, S:P681R, and S:D950N). The consistency between the variant-specific PCR assays and the NGS results supports the applicability of PRIMES.
IMPORTANCE Monitoring the introduction and prevalence of variants of concern (VOCs) and variants of interest (VOIs) in a community can help the local authorities make informed public health decisions. PCR assays can be designed to keep track of SARS-CoV-2 variants by measuring unique mutation markers that are exclusive to the target variants. However, the mutation markers may not be exclusive to the target variants because of regional and temporal differences in variant dynamics. We introduce PRIMES, an algorithm that enables the design of reliable PCR assays for variant detection. Because PCR is more accessible, scalable, and robust for sewage samples than sequencing technology, our findings will contribute to improving global SARS-CoV-2 variant surveillance.
KEYWORDS: PCR assays, SARS-CoV-2 variants, in silico analysis, PRIMES, wastewater-based epidemiology
INTRODUCTION
SARS-CoV-2 has had an unprecedented impact on public health globally. However, despite the availability of vaccines, emerging new variants, which may have better infectivity, transmissibility, and immune evasion, threaten global public health again (1, 2). Monitoring the introduction and prevalence of variants of concern (VOCs) and variants of interest (VOIs) in a community can help the local authorities make informed decisions regarding public health (3–5). In particular, wastewater-based epidemiology (WBE) has been applied across the globe to monitor SARS-CoV-2 circulating in a community (6–9). WBE could complement clinical diagnosis, because WBE allows health authorities to monitor transmission levels in communities, including asymptomatic patients, without requiring excessive resources (10).
Although sequencing is considered the gold standard to identify SARS-CoV-2 lineages and mutations, PCR assays have attracted global attention for variant detection due to several advantages (11). First, PCR is a more accessible tool because the instruments and reagents are more affordable. Second, PCR is more scalable because it can analyze dozens or hundreds of samples in only a couple of hours, while sequencing takes a much longer time (>12 h) (12). Third, PCR is more robust for sewage samples that have low concentrations of SARS-CoV-2 genomes and contain different types of impurities (13). These advantages are beneficial for ramping up capacity for SARS-CoV-2 surveillance and facilitating deployment in regions lacking access to sequencing facilities to initiate their variant monitoring systems.
PCR assays are composed of about 20- to 30-bp-long primers or probes designed to detect single or multiple loci that characterize a target variant. Importantly, PCR assays can only examine sequences that are less than 100 bp long, while sequencing produces reads that span longer genome regions (>1,000 bp). Meanwhile, each variant or sublineage of SARS-CoV-2 is defined by a group of different mutations located throughout the genome. Therefore, distinct variants may have the same mutations, reducing specificity when used in PCR assays (https://covariants.org).
As new variants of SARS-CoV-2 emerge and fade away throughout the world, a number of different lineages (1,340 lineages as of August 2021) have been reported (14). Due to the evolutionary relationship of these lineages, they often share characteristic mutations. As such, PCR assays targeting only a few mutations (typically 1 to 3 mutations) have difficulty detecting samples from a specific lineage of interest with high specificity and sensitivity. In addition, while most lineages are limited to where they emerged, outbreaks of some lineages occasionally spread across the borders and become global concerns (such as VOCs and VOIs). Thus, regional and temporal differences in variant dynamics have to be considered for PCR assays.
In this study, we introduce PRIMES (PRIMer Efficacy Sleuth), a computational tool that can be used to analyze sequences available in open-source databases such as GISAID (14, 15) to predict the sensitivity and specificity of a PCR assay to detect specific pathogen lineages of interest (Fig. 1a). Moreover, for a given set of mutations characterizing the target variant, PRIMES can also identify a subset of variant-specific mutations for designing PCR assays with high specificity and sensitivity. Using PRIMES, we show multiple examples of previous PCR assays (13, 15, 16) that were successfully applied to certain study areas that might not work for other regions (Fig. 1b). We also demonstrate that the PCR assays designed using PRIMES successfully identify the dominant lineages in sewage samples from Champaign County, IL, USA. We conclude that PCR assays should be designed or modified considering regional and temporal variations and that in silico analyses using open-source databases can improve the sensitivity and specificity of PCR assays. These findings will allow PCR assays to be applied more reliably for SARS-CoV-2 variant surveillance.
FIG 1.

(a) Schematic describing the workflow for designing PCR assays while considering regional and temporal variations in GISAID samples. After selecting a target variant, we identified variant-specific mutations, which we ranked in terms of sensitivity and specificity by using the introduced tool PRIMES. Finally, we designed primers for mutations with high sensitivity and specificity for the geographical region of interest. (b) Illustration showing the effect of regional lineages on the accuracy of variant-specific PCR assays.
RESULTS
Analysis of previously developed PCR assays.
Here, we use PRIMES to analyze the efficacy of previously developed PCR assays targeting specific spike (S) protein mutations in the SARS-CoV-2 genome. First, we analyzed PCR assays targeting S:Δ69/70 to detect the Alpha variant (13, 15, 17). S:Δ69/70 means the Δ69/70 mutation in the spike gene. These PCR assays were verified with synthetic RNA controls and local sewage samples from Israel. We then simulated the application of these assays to sequences deposited in GISAID for Israel (n = 13,932 from January 2021 to October 2021). Figure 2a shows that the Alpha variant was dominant (most prevalent variant) from January 2021 until May 2021, after which most samples were from other lineages. PRIMES predicts that the PCR assays targeting S:Δ69/70 (15) correctly assigned GISAID samples to the Alpha variants with a sensitivity of 0.95 and a specificity of 0.93. This finding can be attributed to the observation that the target mutation of these PCR assays, S:Δ69/70, is mostly exclusive to the Alpha variant in Israel, where this PCR assay was developed. However, although S:Δ69/70 was once a key mutation for the Alpha variant (B.1.1.7 first reported in February 2020), B.1.258.17 (first reported in August 2020) and B.1.620 (first reported in February 2021) and other lineages are also known to have the same mutation. Although these SARS-CoV-2 lineages are not significant in Israel (29/13,932), they have a significant prevalence in certain regions at some points in time due to local outbreaks. For example, the B.1.258.17 lineage accounted for 21.8% of all the sequences in GISAID from Slovenia for the period between January 2021 and October 2021.
FIG 2.

In silico analysis of PCR assay targeting the S:Δ69/70 mutation (15) to detect the Alpha variant for GISAID samples from Israel (n = 13,932) (a), Slovenia (n = 25,528) (b), Central African Republic (n = 49) (c), and Congo (n = 183) (d). Dotted lines in the graphs on the left indicate the number of sequences used for the in silico analyses.
While this PCR assay targeting S:Δ69/70 was applied only to wastewater samples from Israel in the previous study (15), we can use PRIMES to predict the sensitivity and specificity of this PCR assay for GISAID samples from any other region. Figure 2b shows our analysis of samples from Slovenia (n = 25,528, from January 2021 to October 2021), where the prevalence of lineage B.1.258.17 was significant until May 2021 and dominating in January 2021 and February 2021. On the other hand, the Alpha variant had less than 10% prevalence in January 2021 and February 2021. However, our analysis of the PCR assays shows that the Alpha variant would have been dominant from January 2021 until June 2021. This error came from the fact that these assays would have incorrectly assigned genomes belonging to the B.1.258 and B.1.258.17 lineages to the Alpha variant. The false positives for the Alpha variant would continue until June 2021, when the B.1.258.17 lineage faded out. Thus, while the estimated sensitivity of the assay for the Alpha variant in Slovenia is 0.89, the specificity is estimated to be only 0.68. We also found that PCR assays targeting S:Δ69/70 could lead to significant numbers of false positives when applied to samples from the Central African Republic (Fig. 2c shows that the estimated specificity is only 0.46 due to samples from B.1.620) and Republic of Congo (Fig. 2d shows that the estimated specificity is only 0.64 due to B.1.620 and B.1.631).
We conducted a similar analysis for another PCR assay targeting the mutation S:Δ144/145 to detect the Alpha variant (13). This assay was applied to samples from wastewater treatment plants and selected residential buildings across the United States to track the occurrence of the Alpha variant over time in 19 communities. Figure S1a in the supplemental material shows that this assay works well for GISAID sequences from the United States, with an estimated sensitivity and specificity of 0.90 and 0.98, respectively. However, several lineages, including C.1.2, B.1.620, B.1.1.318, B.1.525 (or the Eta variant), B.1.637, B.1.625, and AZ.2 also have the same mutation, S:Δ144/145, targeted by this assay. This PCR assay can produce false results if any of the aforementioned lineages have a significant prevalence in the studied area. For example, we analyzed GISAID sequences collected from Gabon (n = 254, from January 2021 to May 2021). Figure S1b suggests that Gabon had significant sequences from the Eta variant and the B.1.1.318 lineage from February 2021 to May 2021, both of which have the target mutation S:Δ144/145. As a result, even though the number of sequences from the Alpha variant increased from February 2021 to April 2021 and then decreased in May 2021, the PCR assay would have predicted a continuous increase in the prevalence of the Alpha variant from February 2021 to May 2021 (Fig. S1b). The estimated specificity of this assay for detecting the Alpha variant in GISAID sequences from Gabon is only 0.74. We see a similar result by analyzing GISAID sequences from Togo (n = 157, from January 2021 to April 2021) in Fig. S1c. Many countries in Africa, including Nigeria and Ghana, were also expected to have lower specificity for the PCR assay targeting S:Δ144/145 because of B.1.1.338 and B.1.525 lineages (Table S1).
This propensity for false-positive results is not limited to PCR assays developed to detect samples from the Alpha variant. We demonstrate this by considering a recent PCR assay targeting mutation S:T478K of the Delta and Delta plus lineages (16). However, this mutation is also present in the B.1.1.519 lineage. This lineage accounted for only around 1.2% of sequences from the United States (n = 1,187,412, from January 2021 to October 2021), so the PCR assay targeting S:T478K was expected to work well for Illinois, USA, showing an estimated sensitivity of 0.94 and an estimated specificity of 0.97 (Fig. 3a). However, the B.1.1.519 lineage was dominant in Mexico (n = 28,956) and explained 30% of the total GISAID sequences from January 2021 to October 2021 (Fig. 3b). Therefore, our analysis shows that the PCR assay targeting S:T478K would estimate that the Delta variant was dominant from January 2021 to October 2021, when in reality, Delta variant sequences were collected and later deposited in GISAID starting in May 2021 (Fig. 3b).
FIG 3.

In silico analysis of PCR assay targeting the S:T478K mutation (16) to detect the Delta variant in the United States (n = 1,187,412) (a) and Mexico (n = 28,956) (b). Dotted lines in the graphs on the left indicate the number of sequences used for the in silico analyses.
The examples of regional and temporal characteristics affecting the accuracy of PCR assays for the detection of SARS-CoV-2 samples of specific lineages of interest are not limited to the cases mentioned above. Globally, only a few VOCs and VOIs accounted for more than 1% of the total sequences in GISAID, while most of other SARS-CoV-2 lineages explain less than 1%. However, as we examine narrower regions, we may find outbreaks of certain lineages that could be overlooked when we focus on the prevalence on a global scale. For example, as of October 2021, B.1.526 lineage accounted for 1% of reported sequences in the world. However, the prevalence of the lineage increases as we narrow down the study area to the local level: 4% in the United States, 17% in New York State, and 30% in Bronx County. In the case of the B.1.429 lineage, the prevalences are 1%, 4%, 11%, and 38% in the world, the United States, California State, and Riverside County, respectively. The number of B.1.258 lineages was less than 0.5% of total sequences worldwide, but this lineage accounted for 54% of cases in Cyprus.
We document the SARS-CoV-2 lineages with the same mutation that our target variant possesses in Table S1. When we use PCR assays targeting certain mutations, this table will help identify the lineages that would interfere with our PCR assay. In Table S2, we also tabulated countries where each of the SARS-CoV-2 lineages summarized in Table S1 accounted for more than 1% of the total sequences. This table explains whether the lineages that would interfere with your PCR assays are dominant in the study areas. By interpreting Table S1 and Table S2 together, we can find various examples where certain PCR assays would not work reliably.
In conclusion, we used PRIMES to estimate the performance of previously developed PCR assays on sequences from various countries. Strikingly, our analysis shows that several previously developed variant-specific PCR assays would not be as accurate for samples collected from locations and periods beyond those included in the original study due to the presence of other lineages sharing mutations that characterized the lineage of interest. These findings motivated us to establish PCR assays for variant detection based on the characteristics of sequences reported from our target study area (Illinois, USA).
Design of variant-specific PCR assays considering regional and temporal characteristics.
We describe the proposed workflow for designing variant-specific PCR assays considering regional and temporal variant dynamics using PRIMES (Fig. 1a). Our goal is to design variant-specific PCR assays to track variants with significant prevalence in the United States, with a particular focus on the state of Illinois.
First, we investigated the prevalence of SARS-CoV-2 lineages in our regions of interest to select lineages that we need to track. To this end, we downloaded 1,187,412 SARS-CoV-2 sequences from GISAID collected between January 2021 and October 2021 in the United States, including 20,165 sequences collected in Illinois. These sequences were assigned to the most likely lineage using Pangolin (Fig. 4a and b). Focusing on the variant dynamics in the state of Illinois (Fig. 4a), we observed that the B.1.2 lineage was dominant from January (61%) to February (44%), eventually giving way to the Alpha variant. The Alpha variant became dominant in April (44%), May (61%), and June (62%). Then, the Delta variant samples replaced the Alpha variant samples and became the dominant lineage in the state (95% in July and >99% in August, September, and October). Other VOIs and VOCs, including Epsilon, Iota, and Beta variants, accounted for only 2.2%, 1.5%, and 0.4% of total sequences, respectively. Similar trends were observed in sequences collected throughout the United States (Fig. 4b). Based on the variant dynamics of our regions of interest, we decided to design PCR assays to enable monitoring of the two major variants, the Alpha and Delta variants.
FIG 4.

(a and b) Variant dynamics determined by Pangolin using GISAID samples from the state of Illinois in the United States (n = 20,165) (a) and from the United States (n = 1,187,412) (b). (c and d) Focusing on the spike protein mutations in the Delta variant, we showed the sensitivity and specificity of assigning the Delta variant based on the presence of each mutation in GISAID samples from Illinois (c) and the United States (d). (e and f) Estimated assignment of GISAID samples from Illinois (e) and the United States (f) to variants using the primer designed to target mutation S:P681R.
Second, we designed PCR assays to find unique mutations exclusive to our lineage of interest. We utilized the website https://covariants.org to list nonsynonymous mutations that define target variants (Table S1). We focused on mutations of the spike gene, which has a higher frequency of mutation than other SARS-CoV-2 genes (18). Previous studies have shown that primers targeting mutations in the spike gene enable accurate detection of SARS-CoV-2 lineages in sewage samples with a low virus concentration (17). As a result, for the Alpha variant, we identified nine mutations in the spike gene: S:Δ69/70, S:Δ144, S:N510Y, S:A570D, S:D614G, S:P681H, S:T716I, S:S982A, and S:D1118H. For the Delta variant, we identified seven mutations: S:T19R, S:Δ156/157, S:L452R, S:T484K, S:D614G, S:P681R, and S:D950N.
Third, we used PRIMES to compute the sensitivity and specificity of lineage assignments performed using each of the selected mutations. We assumed that if the specificity and sensitivity of the mutations were higher than 0.99, the mutations were exclusive to the target variant. This criterion allowed us to identify the ideal target mutation for the design of the PCR assay that would yield high specificity and sensitivity in our regions of interest. For the Alpha variant, we found three acceptable mutations, S:A570D, S:T716I, and S:S982A (Fig. S2b), and we chose the S:A570D mutation because the PCR assay targeting S:A570D has already been verified to work for sewage samples; we adopted this mutation in our analysis (13). For the Delta variant, we found three mutations, S:L452R, S:P681R, and S:Δ156/157, in GISAID samples from the state of Illinois. However, if we look at all GISAID samples from the United States, the sensitivity for the S:L452R and S:Δ156/157 mutations to characterize the Delta variant drops below 0.97. Thus, regional variation can lead to a drastic change in the performance of variant-specific PCR assays. Since our goal was to develop PCR assays that are also effective in other states in the United States, we instead chose S:P681R, which has high sensitivity and specificity in both Illinois (sensitivity is 0.99, and specificity is 0.99) and the United States (sensitivity is 0.99, and specificity is 0.99). Importantly, this mutation has higher sensitivity and specificity in both regions of interest than the mutation S:T478K (sensitivity is 0.99 and specificity is 0.97 in Illinois, while sensitivity is only 0.96 and specificity is only 0.98 in the United State [Fig. 4c and d]), which was previously targeted to monitor the Delta and Delta plus variants (16).
The fourth step was to design the allele-specific primers for the selected mutations. Since both of our selected target mutations are single nucleotide polymorphisms (SNPs), we designed allele-specific quantitative PCR (qPCR) assays in which either a forward or a reverse primer targets the SNP at the 3′ end with a mismatch near the SNP location to improve the specificity of the assays (13). All reverse transcription (RT)-qPCR assays were designed using PrimerQuest (Integrated DNA Technologies [IDT], USA) to have a melting temperature (Tm) of 59 to 63°C for primers and GC contents of 30% to 60%.
Finally, we can estimate the efficacy of the candidate RT-qPCR assays using PRIMES. Specifically, we determined the sensitivity and specificity of our assays on the GISAID samples collected from the regions of interest by searching for sequences of a forward primer and a reverse primer in each query sequence. Note that the sequences of reverse primers were converted to reverse sequences to have all sequences, including primers and viruses, on the same strand. If the viral sequence includes the forward and reverse sequences, we assumed that the PCR assay would detect the viral sequence (an illustrative example is shown in Fig. S3). Some lineages should be expected to lower the sensitivity or specificity of our assays based on Table S1 (e.g., B.1.1.189, C38, and B.1.636 for Alpha variant detection and AU.3, AU.2, P.1.8, B.1.617.3, A.23.1, B.1.617.1, B.1.551, B.1.466.2, B.1.1.528, Q.4, B.1.623, B.1.1.25, C.36, and AY.28 for Delta variant detection), but importantly, those lineages were not detected or had very low prevalences in our regions of interest. The estimated sensitivity and specificity for PCR assays designed to detect viruses from the Alpha and Delta variants were all high for our study scope and in Illinois in particular (sensitivity is 0.99 and specificity is 0.99 for detection of Alpha variant, and sensitivity is 0.98 and specificity is 1.00 for detection of the Delta variant in Illinois) (see Fig. S4 for sensitivity and specificity of detecting the two variants in GISAID samples from all of the United States). These values are higher than the sensitivity and specificity estimated for the previously developed PCR assays in their regions of interest (Fig. 2 and 3). In the following section, we demonstrate this performance of our PCR assays with synthetic RNA controls and actual sewage samples collected in our community.
Verification of PRIMES-designed PCR assays by synthetic RNA controls.
Operational failures of PCR assays due to inappropriate primer design or PCR inhibitors are not considered by PRIMES. Therefore, PCR assays designed by PRIMES must be verified by in vitro experiments. We applied the RT-qPCR assays designed with PRIMES to synthetic RNA controls for the wild type (WT), the Alpha and Delta variants to experimentally confirm the sensitivity (i.e., the limit of quantification [LOQ] and limit of detection [LOD]) and specificity (i.e., cross-reactivity). Regarding sensitivity, we found that the LOQs for total SARS-CoV-2, Alpha variant, and Delta variant were all 10 gene copies (gc)/μL or 50, 30, and 30 gc/reaction mixture, respectively (Fig. 5a). Also, the LODs of RT-qPCR assays for total SARS-CoV-2, Alpha variant, and Delta variant were 1.0, 1.3, and 1.3 gc/μL or 5.0, 3.9, and 3.8 gc/reaction mixture, respectively (Fig. 5b). We used LODs as thresholds to report RT-qPCR results, so the data below the LODs were considered negative for target genes. Because LODs for our assays were close to the theoretical LODs of RT-qPCR (3.0 gc/reaction mixture) (19), we concluded that our RT-qPCR assays are sensitive to detect RNA of target variants.
FIG 5.

Determination of sensitivities (i.e., LOQs and LODs) of RT-qPCR assays for total SARS-CoV-2, Alpha variant, and Delta variant. (a) Dashed lines indicate the coefficient of variation at 0.35. (b) The trend lines for positive rate (solid lines) were calculated using equation 6. The LODs were the RNA concentration at which the positive rate was 0.95 (dashed lines).
As for cross-reactivity, we found that when the concentrations of the synthetic RNA control were high (i.e., 104 and 105 gc/μL), we detected quantitation cycle (Cq) values from WT RNA controls. This finding suggests that the presence of the WT caused false positives for Alpha variant detection (Fig. 6a). However, the Cq value differences between the Alpha variant and the WT were greater than 11, which is about a 1,000-fold difference in RNA concentrations. This difference in Cq values is equivalent to less than 0.1% error when quantifying the Alpha variant, and thus we considered this error acceptable for our study. When the synthetic RNA control concentrations were low (i.e., less than 103 gc/μL), the Cq values from the WT were lower than the LOD, and these values will be disregarded in this study. Thus, false positives were not detected. We found similar results from the specificity experiments for the Delta variant assay (Fig. 6b). When the concentrations of the synthetic RNA controls were high (i.e., 104 and 105 gc/μL), the Cq value differences for the Delta variant and WT were greater than 13. At the lower concentration (i.e., less than 103 gc/μL), the Cq values from the WT (i.e., false positives) were less than the LOD. Because the measured cross-reactivities with the WT were negligible, we concluded that our RT-qPCR assays are specific for measuring target variants.
FIG 6.

Cross-reactivity of PCR assays for Alpha and Delta variants. RT-qPCR assays for the Alpha (a) and Delta (b) variants were applied to the corresponding variant and the WT to determine the specificity (i.e., cross-reactivity with the WT).
We further confirmed the applicability of the PRIMES-designed PCR assay to determine predominant variants in mixtures of synthetic RNA controls. The results from the experiments with mixtures of synthetic RNA controls are presented in Fig. 7a and b. The y axis shows the prevalences, calculated as the ratio of each variant’s concentration to the total SARS-CoV-2 concentration. The variant showing the highest prevalence became the dominant variant. If none of the two targets (i.e., Alpha and Delta variants) has a prevalence higher than 0.5, the “others,” which comprises all SARS-CoV-2 lineages other than our target variants (i.e., the Alpha and Delta variant), becomes the dominant variant. With the highest total virus concentrations (104 gc/μL), our RT-qPCR assays successfully assigned the correct dominant variant to all experimental cases (P < 0.001) (Fig. 7a). For example, in the case of the mixtures between the WT and the Alpha variant, we assigned the Alpha variant to the RNA mixtures whose actual prevalences of Alpha variant were 0.7 and 0.9. In contrast, we assigned “others” when the prevalences of the Alpha variant were 0.1 and 0.3. Similarly, we also assigned the dominant variant correctly to the mixtures of the Alpha and Delta variants. Specifically, we assigned the Alpha variant as dominant when the actual prevalences of the Alpha variant were 0.7 and 0.9. At the same time, the Delta variant was assigned as dominant to the other two mixtures whose prevalences of the Alpha variant were 0.1 and 0.3. In addition, we found that the PCR assays assigned the dominant variant correctly when the total virus concentrations were 101 gc/μL for all mixing ratios (Fig. 7b). However, the statistical analysis showed that the comparisons of prevalences determined by the RT-qPCR were significant only when the mixing ratios were 0.9:0.1 or vice versa for mixtures of the WT and the Alpha variant or the Alpha variant and the Delta variant. Note that when the total virus concentrations were 101 gc/μL, concentrations of each synthetic RNA control ranged from 1 × 10° to 9 × 10° gc/μL depending on the mixing ratios, which were less than their LOQs (101 gc/μL). Based on these findings, we concluded that our RT-qPCR assays could find the dominant variant when the total SARS-CoV-2 concentrations were higher than the LOQs (101 gc/μL) and the prevalence of target variants was higher than 0.9 (Fig. 7b). When the concentrations of the SARS-CoV-2 N gene, Alpha and Delta variants become higher than the respective LOQ values (101 gc/μL), our RT-qPCR assays can assign the dominant variant when its prevalence is higher than 0.7 (Fig. 7a).
FIG 7.

Dominant variants of the mixtures of synthetic RNA controls determined by RT-qPCR assays. Total SARS-CoV-2 concentrations were determined by the N gene concentrations at 104 gc/μL (a) and 101 gc/μL (b). Prevalences on the y axis indicate the ratio of the concentration of each variant to the total virus concentration. The x axis shows the mixing ratios of different synthetic RNA controls (W, A, and D represent WT, Alpha variant, and Delta variant, respectively). The label at the top of each graph (others, Alpha, or Delta) indicates the dominant variant determined by the RT-qPCR assays. A one-sample t test or a two-sample t test was conducted to compare the prevalences between the Alpha variant and 0.5 or the prevalences between the Alpha and Delta variants (nonsignificant [ns], P > 0.05; *, 0.001 < P < 0.05; **, P < 0.001), respectively.
Application of PCR assays to sewage samples and confirmation by NGS.
We applied our PCR assays to six different local sewage samples. We first obtained RNA extracts from those sewage samples. The total SARS-CoV-2 concentrations (i.e., N gene) of these RNA extracts ranged from 1.4 × 101 to 1.8 × 102 gc/μL (Table 1). After accounting for recovery efficiencies and concentration factors, the SARS-CoV-2 concentrations (i.e., N gene) of these sewage samples ranged from 1.3 × 103 to 6.0 × 104 gc/L (see equation 3 below). These concentrations agree with the SARS-CoV-2 concentrations of sewage samples analyzed previously (20). We then determined the prevalence of variants based on the ratios of the Alpha variant concentration (determined by PRIMES-designed PCR) to the total SARS-CoV-2 (N gene). We found that sample 1 has a prevalence of the Alpha variant of 0.85. Based on the results with the synthetic RNA mixtures, we assigned the Alpha variant as the dominant variant to sample 1. Similarly, we assigned the Delta variant to samples 5 and 6 because of their prevalences of 0.92 and 0.73, respectively. On the other hand, none of the Alpha and Delta variants presented a prevalence higher than 0.5 for samples 2, 3, and 4, so we assigned “others” to these three samples.
TABLE 1.
Applications of RT-qPCR assays to local sewage samples
| Sample | Total SARS-CoV-2 concentration of RNA extracts (gc/μL) | Concentration (% prevalence) |
Recovery efficiency | Concentration factor | Total SARS-CoV-2 concentration of sewage sample (gc/L) | Variant decision |
|
|---|---|---|---|---|---|---|---|
| Alpha variant | Delta variant | ||||||
| 1 | 2.7 × 101 | 2.3 × 101 (85) | Below LOD | 0.58 × 10-2 | 0.6 × 10-4 | 2.6 × 103 | Alpha |
| 2 | 1.4 × 101 | Below LOD | Below LOD | 0.58 × 10-2 | 0.5 × 10-4 | 1.3 × 103 | Others |
| 3 | 9.9 × 101 | Below LOD | Below LOD | 0.74 × 10-2 | 1.7 × 10-4 | 2.3 × 104 | Others |
| 4 | 5.6 × 101 | Below LOD | 3.4 × 100 (6)a | 2.37 × 10-2 | 4.1 × 10-4 | 9.6 × 103 | Others |
| 5 | 1.7 × 102 | Below LOD | 1.5 × 102 (92) | 0.67 × 10-2 | 0.9 × 10-4 | 4.3 × 104 | Delta |
| 6 | 1.8 × 102 | Below LOD | 1.3 × 102 (73) | 0.71 × 10-2 | 1.2 × 10-4 | 6.0 × 104 | Delta |
Below the LOQ.
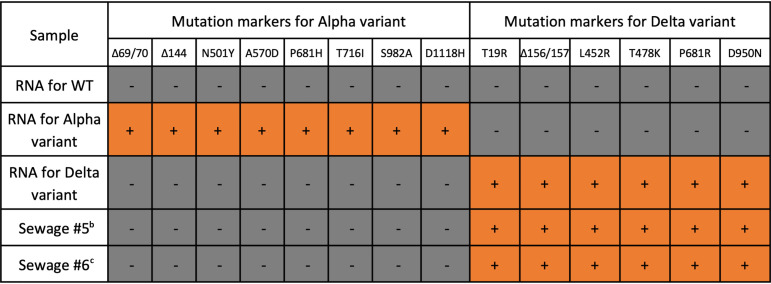
To further confirm whether the RT-qPCR results were correct, we conducted NGS analysis to examine eight mutation markers for the Alpha variant (S:Δ69/70, S:Δ144, S:N501Y, S:A570D, S:P681H, S:T716I, S:S982A, and S:D1118H) and six mutation markers for the Delta variant (S:T19R, S:Δ156/157, S:L452R, S:T478K, S:P681R, and S:D950N) on the spike gene of two sewage samples (samples 5 and 6) and three synthetic RNA controls (WT, Alpha variant, and Delta variant). Even though we amplified the entire spike gene with the three pairs of primers, samples 1, 2, 3, and 4 were not appropriate for sequencing due to the low SARS-CoV-2 concentrations (<102 gc/μL) (21). For samples 5 and 6 classified to the Delta variant by the RT-qPCR assays, we detected all six mutations for the Delta variant. In comparison, none of the eight mutations for the Alpha variant were detected in these samples. We believe that these NGS analyses were reliable because we detected all mutation markers with the corresponding synthetic RNA controls. For example, we detected the eight Alpha variant mutations from the Alpha variant RNA samples and found the six Delta variant mutations from Delta variant RNA controls (Table 2). Therefore, the agreement between the NGS analysis and RT-qPCR assays supports that our RT-qPCR can assign the most likely variant for the local sewage samples.
TABLE 2.
Comparisons between RT-qPCR assays and NGS analysisa
|
+ symbols in orange cells represents mutations that were detected, while − symbols in gray cells indicate mutations that were not detected.
bGISAID accession ID is EPI_ISL_10113885.
cGISAID accession ID is EPI_ISL_10113884.
DISCUSSION
PCR assays have advantages for SARS-CoV-2 variant detection in sewage samples over sequencing technologies because of low cost, fast turnaround, and robustness with environmental samples. However, PCR assays can examine only a few mutations due to size constraints of primer and probe sequences, compromising their accuracy since distinct SARS-CoV-2 lineages may share target mutations. We used the PRIMES algorithm to show that the current variant-specific PCR assays have diminished accuracy when applied outside the region where they were developed. These findings suggest that consideration of regional and temporal dynamics of variants is important to secure the sensitivity and specificity of PCR assays that target only a limited number of mutations. Subsequently, we used PRIMES and open-source databases (e.g., GISAID, Pangolin, or outbreak.info) to design PCR primers to determine the dominant SARS-CoV-2 variants (i.e., Alpha and Delta variants) in local sewage samples. Note that viral load in feces significantly varies depending on an individual’s characteristics, such as type of variants, vaccination, and so forth (22). Therefore, the dominant variant in sewage may not necessarily indicate that the variant is also dominant in a community.
The regional and temporal variations are especially critical for SARS-CoV-2 detection because various SARS-CoV-2 lineages with different genotypes have been reported worldwide. Commercial PCR kits for variant detection are currently available. However, these kits also target a few mutation markers originating from SARS-CoV-2 lineages of interest (23–25). As we showed above, targeting a single mutation might make the assay less accurate in certain regions due to the presence of other lineages that have the same mutation. In addition, our findings are not limited to PCR assays but are also relevant for other types of molecular assays such as loop-mediated isothermal amplification (LAMP), PfAgo-based assays, and CRISPR (clustered, regularly interspaced short palindromic repeats)-based assays that are designed to detect specific RNA sequences for virus detection (26–28). For example, a LAMP assay that targets N genes for SARS-CoV-2 testing might have low accuracy when applied outside of Germany or the United States, where the assays were developed and verified with clinical samples (26, 29). This low-accuracy issue might happen because their primers include sequences for N:A119S, a mutation marker for the Zeta variant (P.2 lineage). The Zeta variant was dominant in some South American countries (Suriname, Paraguay, Uruguay, and Brazil). The Food and Drug Administration (FDA) also recommended that mutations present in the sequences which molecular diagnostic tests target for virus detection should be monitored by in silico analysis (30). Our PRIMES tool allows users and developers of molecular diagnostic assays to follow this recommendation.
Perfect loci for targeting viral mutations are not realistic because viruses evolve randomly, so one that looks perfect could be affected by emerging variants. For example, the S:Δ69/70 mutation used to be a unique mutation for the Alpha variant, but the Eta variant, which appeared later, also has this mutation. Thus, if S:Δ69/70 is considered an exclusive mutation for the Alpha variant, the Eta variant will be false positive for the Alpha variant. In addition, sublineages in the target variant may not have one of the mutation markers for the target variant. For example, less than 0.5% of Q.4 (one of the sublineages for the Alpha variant and reported in December 2020) is known to have an S:P681H mutation. The S:P681H mutation is one of the mutation markers for the Alpha variant. Thus, if the S:P681H mutation is targeted for the Alpha variant, Q.4 will cause false negatives. These examples demonstrate that PCR assays could have different sensitivities and specificities depending on various lineages of SARS-CoV-2 that coexist with the target lineage.
Global genomic databases for emergent variants have greatly improved since the onset of COVID-19 pandemics (31). Before COVID-19, influenza virus sequences are archived in GISAID. Quickly mutating pathogens such as influenza virus and coronavirus should be monitored because they have pandemic potential. As we showed in this study, assays targeting these pathogens need to keep up with their evolution, and the developed methodology facilitates genomic surveillance of any quickly mutating pathogen.
In this study, we developed a PRIMES algorithm that calculates the sensitivity and specificity of SARS-CoV-2 variant-specific PCR assays in silico for prespecified geographical regions. Using PRIMES, we designed two PCR assays for detecting the Alpha and Delta variants. We verified those variant-specific PCR assays with in vitro experiments using synthetic RNA controls. We also showed that these assays could detect the dominant variants in actual sewage samples, and these PCR results were confirmed by NGS analysis of the spike gene amplicons. The PRIMES-designed PCR assays can also be applied to assign dominant variants in human specimens. Because RNA levels in human specimens are higher than those in sewage in general, the false positives from the variant-specific qPCR assays (i.e., allele-specific qPCR assays) may result in measurements above the LOD, thereby affecting RNA quantification. However, we confirmed that these errors account for less than 0.1% of RNA quantification (Fig. 6), so the errors are not expected to impact the assignment of the dominant variant. In summary, the PRIMES-designed PCR assays will contribute to improving the capacity for SARS-CoV-2 variant surveillance. This tool will be especially helpful for underserved regions, because PCR assays are more accessible and scalable tools than sequencing-based SARS-CoV-2 variant surveillance.
MATERIALS AND METHODS
Analysis and design of PCR assays using PRIMES.
The most widely used computational tool for assigning lineages to SARS-CoV-2 genomes is the Phylogenetic Assignment of Named Global Outbreak Lineages (Pangolin; https://pangolin.cog-uk.io/). Pangolin is a lineage designation pipeline that takes a FASTA file as input, containing one or more query sequences. Each query sequence is first aligned to the SARS-CoV-2 reference genome (Wuhan-Hu-1; GenBank accession no. NC_045512.2) using minimap2 v2.17 (32). After trimming of the noncoding regions at the 5′ and 3′ ends of the aligned sequences, the sequences are assigned to the most likely lineage out of all currently designated lineages by use of an underlying machine learning model referred to as PangoLEARN. The current version of PangoLEARN is a decision tree trained on data from GISAID that were manually curated with lineages.
By considering the lineage designation of Pangolin as ground truth, we performed an in silico analysis of the efficacy of PCR assays using PRIMES (available at https://github.com/elkebir-group/primes). Specifically, we searched for an exact match of the target sequence (containing a mutation targeted by the PCR assay) in each GISAID sequence and then estimated the overall specificity and sensitivity of the PCR assay defined as follows:
| (1) |
| (2) |
where true positives is the number of virus sequences that include the target sequence and also belong to the lineage of interest according to Pangolin. False negatives is the number of virus sequences that do not include the target sequence but belong to the lineage of interest. True negatives is the number of virus sequences that do not include the target sequence and do not belong to the lineage of interest as well. Finally, false positives is the number of virus sequences that include the target sequence but do not belong to the lineage of interest. Note that our analysis assumes that the PCR assays do not tolerate mismatches in the target sequence.
While the in silico estimates of the sensitivity and specificity are valuable in their own right, they can also be used to design effective variant-specific PCR assays. Specifically, for a lineage of interest and a set of characteristic mutations, we use PRIMES to identify the set of mutations that should be targeted by PCR assays with high specificity and sensitivity. We employed this approach to design PCR assays to detect the presence SARS-CoV-2 of both Alpha (e.g., B.1.1.7) and Delta (e.g., B.1.617.2) variants in sewage samples.
Sewage sample processing.
We followed the guidelines for minimum information for publication of quantitative real-time PCR experiments (MIQE) to ensure the credibility and reproducibility of our data (33). Detailed information on the MIQE is summarized in Table S3 in the supplemental material. Also, detailed information from sample collection to data analysis is described in Table S4. Briefly, we used ISCO automatic samplers (catalog no. 6712; Teledyne ISCO, USA) to collect 3-day composite sewage samples (about 2 L) from the sewer distribution system across Champaign County, IL, USA. MgCl2 was added to the sewage samples at a final concentration of 50 mM to facilitate the coagulation of viruses and sewage sludge. We kept sewage samples on ice while moving them to our laboratory in 2 h. We gently removed the supernatant upon arrival and added 200 μL of bovine coronavirus (BCoV) to the remaining solution (about 50 mL) to determine virus recovery efficiency. The recovery efficiency of BCoV ranged from 0.58% to 2.37%, which is similar to those previously reported (34). After 5 min of incubation at room temperature, we centrifuged the mixture at 10,000 rpm (13,900 × g) for 30 min (Sorvall Legend RT Plus; Thermo Fisher Scientific, USA). The supernatant was discarded again, and the sludge (about 1 g) was taken to harvest viruses. Then, we extracted viral RNA from the sludge using a viral RNA extraction minikit (Qiagen, Germany) by following the manufacturer’s procedure. The RNA extracts were purified using an RNA purification kit (RNeasy MinElute cleanup kit; Qiagen, German) to reduce the PCR inhibition. It took less than 9 h from sample collection to RNA extraction. The RNA samples were stored at −80°C until downstream analyses were ready. The same sample preparation processes were applied to drainages discharged from a food processing industry whenever we processed sewage samples. There are no sources of human feces that merged with these drainages, which were therefore used for negative controls. Indeed, we did not detect any SARS-CoV-2 from these negative controls. Therefore, we are confident that there were no false positives for SARS-CoV-2 in our sewage samples. With the concentrations of RNA extracts, we used equations 3 to 5 to determine the virus concentrations (C) in sewage samples:
| (3) |
| (4) |
| (5) |
Determination of LODs and LOQs.
We first determined the limit of detection (LOD) and the limit of quantification (LOQ) for Alpha and Delta variants with serial dilutions of the synthetic RNA controls. We prepared 10-fold serial dilutions of synthetic RNA controls and determined a positive sample fraction at each concentration. The number of replicates for concentrations near the LOD was 20, while the number of samples for the higher concentration was 4. We used a sigmoidal function (equation 6) to determine the trend lines for fraction-positive samples with different concentrations and calculated the LODs (35):
| (6) |
where X is gene copy (gc/μL), Y is positive rate, and both a and b are constants. The LOQ was defined as the lowest concentration with a coefficient of variation (CV) of less than 35% (35). We calculated the CV using equation 7:
| (7) |
where E is qPCR efficiency and SD is standard deviation of Cq values.
PCR assays for SARS-CoV-2 variant detection in synthetic RNA control.
We applied the RT-qPCR assays to 10-fold serial dilutions of synthetic RNA controls to determine LOQs and LODs. For example, we applied the RT-qPCR assay for the Alpha variant to 10-fold serial dilutions of Alpha variant RNA controls. LOQ was defined as the lowest concentration with a coefficient of variation (CV) of less than 35% (35). LOD was defined as the concentration at which RNA samples test positive (i.e., Cq < 40) with 95% probability.
We applied each RT-qPCR assay for the Alpha or Delta variant to the synthetic controls of its target variant and the WT to determine the cross-reactivity. This process is important, because our assays for the Alpha and Delta variants were designed to detect only a SNP of target variants among other lineages that do not have the same SNP. In this experiment, we mixed synthetic RNA controls of the WT with the Alpha variant or the Alpha and Delta variants because these two mixtures represented the transitions where one dominant variant was replaced by the other one in our community. For instance, the “others” (mainly B.1.2) were dominant until February 2021, and the Alpha variant raced to be the dominant variant around March 2021. Also, the Alpha variant was dominant in April and May in 2021, but the Delta variant competed with the Alpha variant around June 2021 (Fig. 4b). The total SARS-CoV-2 concentrations (i.e., N gene concentrations) of the mixtures were 104 and 101 gc/μL, which is a reasonable concentration range of SARS-CoV-2 in local sewage samples (20). Also, we mixed the two different RNA controls at four different ratios (i.e., 9:1, 7:3, 3:7, and 1:9) to mimic different scenarios of variant dynamics.
PCR assays for SARS-CoV-2 variant detection in sewage samples.
We conducted six different RT-qPCR assays to analyze sewage samples. Three assays targeted different loci of the SARS-CoV-2 genome for virus quantification and dominant variant detection. The other three assays were applied to measure bovine coronavirus (BCoV), pepper mild mottle virus (PMMoV), and Tulane virus (TV), which were used for calculation of virus recovery efficiency, normalization of SARS-CoV-2 to human feces, and inhibition tests, respectively. BCoV was added to the sludge as described previously. PMMoV was used as an internal control to represent the presence of human feces. We detected more than 100-fold-higher concentrations of PMMoV than SARS-CoV-2 N gene in the sewage samples (>108 PMMoV gc/g sludge); therefore we concluded that our sewage samples contained human feces from local residents living in the sewersheds. The RNA extracts were diluted 2-fold in molecular biology-grade water (Millipore Sigma, USA) before the quantification. We spiked 10 μL of RNA extract or 10 μL of molecular biology-grade water with 1 μL TV RNA, followed by analysis of the TV RNA in those two types of samples. We found differences in Cq values between the RNA extract and the negative controls (i.e., molecular biology-grade water) that were less than ±1, which indicated a negligible impact of PCR inhibitors on our samples (36). We used TaqMan-based RT-qPCR for the N1 gene detection, as suggested by the CDC, and SYBR-based RT-qPCR for the other five assays (Table 3). The SYBR-based RT-qPCR started with mixing 3 μL of viral genome with 5 μL of 2 × iTaq universal SYBR green reaction mix, 0.125 μL of iScript reverse transcriptase from the iTaq universal SYBR green reaction mix (Bio-Rad Laboratories, USA), 0.3 μL of 10 μM forward primer for each virus, 0.3 μL of 10 μM reverse primer for each virus, and 1.275 μL of molecular biology-grade water (Corning, NY, USA). The PCR cocktail for the one-step RT-qPCR was placed in 96-well plates (catalog no. 4306737; Applied Biosystems, USA) and analyzed by a qPCR system (QuantStudio 3; Thermo Fisher Scientific, USA). The thermocycle began with 10 min at 50°C and 1 min at 90°C, followed by 40 cycles of 30 s at 60°C and 1 min at 90°C. The annealing temperature was determined based on the optimal temperature of antibody-mediated hot-start iTaq DNA polymerase (iTaq universal SYBR green one-step kit; Bio-Rad Laboratories, USA). We analyzed melting curves and found no primer-dimers from our RT-qPCR analyses. The TaqMan-based RT-qPCR was initiated by mixing 5 μL of viral genome with 5 μL of TaqMan fast virus 1-step master mix (catalog no. 4444432; Applied Biosystems, USA), 1.5 μL of primer/probe mixture for the N1 gene (2019-nCoV RUO kit; Integrated DNA Technologies, USA), and 8.5 μL of water. The 20 μL of mixture was analyzed by the same qPCR system used for the SYBR-based RT-qPCR, except for a different thermal cycle (5 min at 50°C and 20 s at 95°C, followed by 45 cycles of 3 s at 95°C and 30 s at 55°C). We used synthetic RNA controls to get standard curves for the WT, Alpha variant, and Delta variant (TWIST Bioscience, USA; part numbers 102024, 103907, and 104533, respectively). The PCR standard curves were obtained for every RT-qPCR analysis with 10-fold serial dilutions of synthetic RNA controls, and PCR efficiencies for RT-qPCR were higher than 85% (R2 > 0.99). The SYBR signal was normalized to the ROX reference dye. The cycle of quantification (Cq) values were determined automatically by QuantStudio Design & Analysis Software (v1.5.1). Based on the melting curves, the primers were specifically bound to the target genome. The numbers of technical replicates were 4 for synthetic RNA controls and 3 for sewage samples except for LOD and LOQ determination, for which 20 replicates were analyzed.
TABLE 3.
Information for RT-qPCR assays applied to sewage samples
| Target species | Target gene or mutation | Primer name | Sequence (5′–3′) | GC content (%) | Tm (°C) | Amplicon size (bp) (location) | Purpose |
|---|---|---|---|---|---|---|---|
| SARS-CoV-2 | Na | CDC_N1_Forward | GACCCCAAAATCAGCGAAAT | 45.0 | 61.1 | 73 (28287–28358) | Total SARS-CoV-2 |
| CDC_N1_Reverse | TCTGGTTACTGCCAGTTGAATCTG | 45.8 | 64.5 | ||||
| CDC_N1_Probe | ACCCCGCATTACGTTTGGTGGACC | 58.3 | 70.3 | ||||
| S:A570Db | Alpha_Forward | ACAATTTGGCAGAGACATCGA | 42.9 | 62.3 | 85 (23251–23335) | Alpha variant | |
| Alpha_Reverse | AGAACATGGTGTAATGTCAAGAATC | 36.0 | 61.7 | ||||
| S:P681Rf | Delta_Forward | ATCAGACTCAGACTAATTCACG | 40.9 | 59.6 | 87 (23583–23669) | Delta variant | |
| Delta_Reverse | TTTCTGCACCAAGTGACATA | 40.0 | 59.7 | ||||
| PMMOVc | PMMOV_Forward | GAGTGGTTTGACCTTAACGTTTGA | 41.7 | 63.4 | 68 (1878–1945) | Normalization to feces | |
| PMMOV_Reverse | TTGTCGGTTGCAATGCAAGT | 45.0 | 63.6 | ||||
| BCoVd | BCoV_Forward | CTAGTAACCAGGCTGATGTCAATACC | 46.2 | 64.2 | 88 (29799–29886) | Recovery efficiency | |
| BCoV_Reverse | GGCGGAAACCTAGTCGGAATA | 52.4 | 63.5 | ||||
| TVe | TV_Forward | GTGCGCATCCTTGAGACAAT | 50.0 | 63.0 | 133 (879–1011) | Inhibition test | |
| TV_Reverse | TTGGAGCCGGGTAGAAACAT | 50.0 | 63.5 | ||||
Taqman-based RT-qPCR was used.
Lee et al., 2021 (13).
Haramoto et al., 2013 (37); coding sequences for replicase protein were targeted (GenBank accession no. MN496154.1).
Cho et al., 2013 (38); N gene is targeted (GenBank accession no. LC494177.1).
Fuzawa et al., 2020 (39); FLA45_gp1 gene is targeted (GenBank accession no. NC_043512.1).
The specificity of the primer pair targeting this mutation was checked by the primer-blast tool (National Center of Biotechnology Information). We confirmed that our primers do not target any sequences of their host cells (Homo sapiens; taxonomy ID 9606).
Next-generation sequencing to assign SARS-CoV-2 lineages.
The PCR results were confirmed by sequencing the spike gene of three controls (wild type, Alpha variant, Delta variant) and two sewage samples (samples 5 and 6) on the Illumina MiSeq platform. A set of three pairs of in-house-designed primers were used to amplify the spike of RNA samples using the SuperScript III one-step RT-PCR system with Platinum Taq high-fidelity DNA polymerase (ThermoFisher). Amplicons were purified using a QIAquick PCR purification kit (Qiagen), quantified using a Qubit fluorometer, and subject to library preparation using a Nextera XT kit and sequencing on MiSeq.
Data availability.
All the sequence data analyzed in this study are publicly available at GISAID (https://www.gisaid.org/). The analyzed and processed real data results are available at https://github.com/elkebir-group/primes-data.
Code availability.
The code has been deposited on Github at https://github.com/elkebir-group/primes.
ACKNOWLEDGMENTS
We acknowledge funding from the Grainger College of Engineering and the Jump ARCHES program of OSF Healthcare in conjunction with the University of Illinois. Sequencing was funded in part by the Food and Drug Administration Veterinary Laboratory Investigation and Response Network (FOA PAR-17-141) under grant no. 1U18FD006673-01. M.E.-K. acknowledges the National Science Foundation (grant no. CCF-2027669 and CCF-2046488).
We also acknowledge Bill Brown for sampling site selection, Hayden Wennerdahl, Kip Stevenson, Laura Keefer, and Art Schmidt for sampling deployment, and Yuqing Mao, Matthew Robert Loula, Aashna Patra, Kristin Joy Anderson, Mikayla Diedrick, Hubert Lyu, Hamza Elmahi Mohamed, Jad R. Karajeh, Runsen Ning, Rui Fu, Kate O’Brien, and Kyukyoung Kim for sewage sampling and processing.
Footnotes
Supplemental material is available online only.
Contributor Information
Mohammed El-Kebir, Email: melkebir@illinois.edu.
Thanh H. Nguyen, Email: thn@illinois.edu.
Christopher A. Elkins, Centers for Disease Control and Prevention
REFERENCES
- 1.Harvey WT, Carabelli AM, Jackson B, Gupta RK, Thomson EC, Harrison EM, Ludden C, Reeve R, Rambaut A, Peacock SJ, Robertson DL, COVID-19 Genomics UK (COG-UK) Consortium. 2021. SARS-CoV-2 variants, spike mutations and immune escape. Nat Rev Microbiol 19:409–424. 10.1038/s41579-021-00573-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lazarevic I, Pravica V, Miljanovic D, Cupic M. 2021. Immune evasion of SARS-CoV-2 emerging variants: what have we learnt so far? Viruses 13:1192. 10.3390/v13071192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grubaugh ND, Hodcroft EB, Fauver JR, Phelan AL, Cevik M. 2021. Public health actions to control new SARS-CoV-2 variants. Cell 184:1127–1132. 10.1016/j.cell.2021.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peccia J, Zulli A, Brackney DE, Grubaugh ND, Kaplan EH, Casanovas-Massana A, Ko AI, Malik AA, Wang D, Wang M, Warren JL, Weinberger DM, Arnold W, Omer SB. 2020. Measurement of SARS-CoV-2 RNA in wastewater tracks community infection dynamics. Nat Biotechnol 38:1164–1167. 10.1038/s41587-020-0684-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Larsen DA, Wigginton KR. 2020. Tracking COVID-19 with wastewater. Nat Biotechnol 38:1151–1153. 10.1038/s41587-020-0690-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahmed W, Angel N, Edson J, Bibby K, Bivins A, O'Brien JW, Choi PM, Kitajima M, Simpson SL, Li J, Tscharke B, Verhagen R, Smith WJM, Zaugg J, Dierens L, Hugenholtz P, Thomas KV, Mueller JF. 2020. First confirmed detection of SARS-CoV-2 in untreated wastewater in Australia: a proof of concept for the wastewater surveillance of COVID-19 in the community. Sci Total Environ 728:138764. 10.1016/j.scitotenv.2020.138764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prado T, Fumian TM, Mannarino CF, Resende PC, Motta FC, Eppinghaus ALF, Chagas do Vale VH, Braz RMS, de Andrade J da SR, Maranhão AG, Miagostovich MP. 2021. Wastewater-based epidemiology as a useful tool to track SARS-CoV-2 and support public health policies at municipal level in Brazil. Water Res 191:116810. 10.1016/j.watres.2021.116810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gonzalez R, Curtis K, Bivins A, Bibby K, Weir MH, Yetka K, Thompson H, Keeling D, Mitchell J, Gonzalez D. 2020. COVID-19 surveillance in Southeastern Virginia using wastewater-based epidemiology. Water Res 186:116296. 10.1016/j.watres.2020.116296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saththasivam J, El-Malah SS, Gomez TA, Jabbar KA, Remanan R, Krishnankutty AK, Ogunbiyi O, Rasool K, Ashhab S, Rashkeev S, Bensaad M, Ahmed AA, Mohamoud YA, Malek JA, Abu Raddad LJ, Jeremijenko A, Abu Halaweh HA, Lawler J, Mahmoud KA. 2021. COVID-19 (SARS-CoV-2) outbreak monitoring using wastewater-based epidemiology in Qatar. Sci Total Environ 774:145608. 10.1016/j.scitotenv.2021.145608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hart OE, Halden RU. 2020. Computational analysis of SARS-CoV-2/COVID-19 surveillance by wastewater-based epidemiology locally and globally: feasibility, economy, opportunities and challenges. Sci Total Environ 730:138875. 10.1016/j.scitotenv.2020.138875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang H, Miller JA, Verghese M, Sibai M, Solis D, Mfuh KO, Jiang B, Iwai N, Mar M, Huang C, Yamamoto F, Sahoo MK, Zehnder J, Pinsky BA. 2021. Multiplex SARS-CoV-2 genotyping reverse transcriptase PCR for population-level variant screening and epidemiologic surveillance. J Clin Microbiol 59:e00859-21. 10.1128/JCM.00859-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guglielmi G. 2020. The explosion of new coronavirus tests that could help to end the pandemic. Nature 583:506–509. 10.1038/d41586-020-02140-8. [DOI] [PubMed] [Google Scholar]
- 13.Lin Lee W, Imakaev M, Armas F, McElroy KA, Gu X, Duvallet C, Chandra F, Chen H, Leifels M, Mendola S, Floyd OR, Powell MM, Wilson STJ, Berge KL, J Lim CY, Wu F, Xiao A, Moniz K, Ghaeli N, Matus M, Thompson J, Alm EJ. 2021. Quantitative SARS-CoV-2 alpha variant B.1.1.7 tracking in wastewater by allele-specific RT-qPCR. Environ Sci Technol Lett 8:675–682. 10.1021/acs.estlett.1c00375. [DOI] [Google Scholar]
- 14.O'Toole Á, Hill V, Pybus OG, Watts A, Bogoch II, Khan K, Messina JP, COVID-19 Genomics UK (COG-UK) Consortium, Network for Genomic Surveillance in South Africa (NGS-SA), Brazil-UK CADDE Genomic Network, Tegally H, Lessells RR, Giandhari J, Pillay S, Tumedi KA, Nyepetsi G, Kebabonye M, Matsheka M, Mine M, Tokajian S, Hassan H, Salloum T, Merhi G, Koweyes J, Geoghegan JL, de Ligt J, Ren X, Storey M, Freed NE, Pattabiraman C, Prasad P, Desai AS, Vasanthapuram R, Schulz TF, Steinbrück L, Stadler T, Swiss Viollier Sequencing Consortium, Parisi A, Bianco A, García de Viedma D, Buenestado-Serrano S, Borges V, Isidro J, Duarte S, Gomes JP, Zuckerman NS, Mandelboim M, Mor O, Seemann T, Arnott A, et al. 2021. Tracking the international spread of SARS-CoV-2 lineages B.1.1.7 and B.1.351/501Y-V2. Wellcome Open Res 6:121. 10.12688/wellcomeopenres.16661.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yaniv K, Ozer E, Shagan M, Lakkakula S, Plotkin N, Bhandarkar NS, Kushmaro A. 2021. Direct RT-qPCR assay for SARS-CoV-2 variants of concern (alpha, B.1.1.7 and beta, B.1.351) detection and quantification in wastewater. Environ Res 201:111653. 10.1016/j.envres.2021.111653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin Lee W, Gu X, Armas F, Chandra F, Chen H, Wu F, Leifels M, Xiao A, Jun Desmond Chua F, Kwok GW, Jolly S, Lim CY, Thompson J, Alm EJ. 6 August 2021. Quantitative SARS-CoV-2 tracking of variants Delta, Delta plus, Kappa and Beta in wastewater by allele-specific RT-qPCR. medRxiv 10.1101/2021.08.03.21261298. [DOI] [Google Scholar]
- 17.Carcereny A, Martínez-Velázquez A, Bosch A, Allende A, Truchado P, Cascales J, Jesús J, Romalde L, Lois M, Polo D, Sánchez G, Pérez-Cataluñ A, Díaz-Reolid A, Antón A, Gregori J, Garcia-Cehic D, Quer J, Palau M, Ruano CG, Pintó RM, Guix S, 2021. Monitoring emergence of the SARS-CoV-2 B.1.1.7 variant through the Spanish National SARS-CoV-2 Wastewater Surveillance System (VATar COVID-19). Environ Sci Technol 55:11756–11766. 10.1021/acs.est.1c03589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Q, Wu J, Nie J, Zhang L, Hao H, Liu S, Zhao C, Zhang Q, Liu H, Nie L, Qin H, Wang M, Lu Q, Li X, Sun Q, Liu J, Zhang L, Li X, Huang W, Wang Y. 2020. The impact of mutations in SARS-CoV-2 spike on viral infectivity and antigenicity. Cell 182:1284–1294.e9. 10.1016/j.cell.2020.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ståhlberg A, Kubista M. 2014. The workflow of single-cell expression profiling using quantitative real-time PCR. Expert Rev Mol Diagn 14:323–331. 10.1586/14737159.2014.901154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pecson BM, Darby E, Haas CN, Amha YM, Bartolo M, Danielson R, Dearborn Y, Di Giovanni G, Ferguson C, Fevig S, Gaddis E, Gray D, Lukasik G, Mull B, Olivas L, Olivieri A, Qu Y, SARS-CoV-2 Interlaboratory Consortium. 2021. Reproducibility and sensitivity of 36 methods to quantify the SARS-CoV-2 genetic signal in raw wastewater: findings from an interlaboratory methods evaluation in the U.S. Environ Sci (Camb) 7:504–520. 10.1039/d0ew00946f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Izquierdo-Lara R, Elsinga G, Heijnen L, Oude Munnink BB, Schapendonk CME, Nieuwenhuijse D, Kon M, Lu L, Aarestrup FM, Lycett S, Medema G, Koopmans MPG, De Graaf M. 2021. Monitoring SARS-CoV-2 circulation and diversity through community wastewater sequencing, the Netherlands and Belgium. Emerg Infect Dis 27:1405–1415. 10.3201/eid2705.204410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kissler SM, Fauver JR, Mack C, Tai CG, Breban MI, Watkins AE, Samant RM, Anderson DJ, Metti J, Khullar G, Baits R, MacKay M, Salgado D, Baker T, Dudley JT, Mason CE, Ho DD, Grubaugh ND, Grad YH. 2021. Viral dynamics of SARS-CoV-2 variants in vaccinated and unvaccinated persons. N Engl J Med 385:2489–2491. 10.1056/NEJMc2102507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Caza M, Hogan CA, Jassem A, Prystajecky N, Hadzic A, Wilmer A. 2021. Evaluation of the clinical and analytical performance of the Seegene allplex™ SARS-CoV-2 variants I assay for the detection of variants of concern (VOC) and variants of interests (VOI). J Clin Virol 144:104996. 10.1016/j.jcv.2021.104996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chan CT-M, Leung JS-L, Lee L-K, Lo HW-H, Wong EY-K, Wong DS-H, Ng TT-L, Lao H-Y, Lu KK, Jim SH-C, Yau MC-Y, Lam JY-W, Ho AY-M, Luk KS, Yip K-T, Que T-L, To KK-W, Siu GK-H. 2022. A low-cost TaqMan minor groove binder probe-based one-step RT-qPCR assay for rapid identification of N501Y variants of SARS-CoV-2. J Virol Methods 299:114333. 10.1016/j.jviromet.2021.114333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirotsu Y, Omata M. 2021. Detection of R.1 lineage severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) with spike protein W152L/E484K/G769V mutations in Japan. PLoS Pathog 17:e1009619. 10.1371/journal.ppat.1009619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ganguli A, Mostafa A, Berger J, Aydin M, Sun F, Valera E, Cunningham BT, King WP, Bashir R. 2020. Rapid isothermal amplification and portable detection system for SARS-CoV-2. Proc Natl Acad Sci USA 117:22727–22735. 10.1073/pnas.2014739117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Broughton JP, Deng X, Yu G, Fasching CL, Servellita V, Singh J, Miao X, Streithorst JA, Granados A, Sotomayor-Gonzalez A, Zorn K, Gopez A, Hsu E, Gu W, Miller S, Pan C-Y, Guevara H, Wadford DA, Chen JS, Chiu CY. 2020. CRISPR-Cas12-based detection of SARS-CoV-2. Nat Biotechnol 38:870–874. 10.1038/s41587-020-0513-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xun G, Lane ST, Petrov VA, Pepa BE, Zhao H. 2021. A rapid, accurate, scalable, and portable testing system for COVID-19 diagnosis. Nat Commun 12:2905. 10.1038/s41467-021-23185-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thi VLD, Herbst K, Boerner K, Meurer M, Kremer LP, Kirrmaier D, Freistaedter A, Papagiannidis D, Galmozzi C, Stanifer ML, Boulant S, Klein S, Chlanda P, Khalid D, Miranda IB, Schnitzler P, Kräusslich H-G, Knop M, Anders S. 2020. A colorimetric RT-LAMP assay and LAMP-sequencing for detecting SARS-CoV-2 RNA in clinical samples. Sci Transl Med 12:eabc7075. 10.1126/scitranslmed.abc7075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.U.S. FDA. 2021. Genetic variants of SARS-CoV-2 may lead to false negative results with molecular tests for detection of SARS-CoV-2—letter to clinical laboratory staff and health care providers. U.S. FDA, Rockville, MD. [Google Scholar]
- 31.Maier W, Bray S, van den Beek M, Bouvier D, Coraor N, Miladi M, Singh B, De Argila JR, Baker D, Roach N, Gladman S, Coppens F, Martin DP, Lonie A, Grüning B, Kosakovsky Pond SL, Nekrutenko A. 2021. Ready-to-use public infrastructure for global SARS-CoV-2 monitoring. Nat Biotechnol 39:1178–1179. 10.1038/s41587-021-01069-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34:3094–3100. 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT. 2009. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55:611–622. 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- 34.Feng S, Roguet A, McClary-Gutierrez JS, Newton RJ, Kloczko N, Meiman JG, McLellan SL. 2021. Evaluation of sampling, analysis, and normalization methods for SARS-CoV-2 concentrations in wastewater to assess COVID-19 burdens in Wisconsin communities. ACS Est Water 1:1955–1965. 10.1021/acsestwater.1c00160. [DOI] [Google Scholar]
- 35.Forootan A, Sjöback R, Björkman J, Sjögreen B, Linz L, Kubista M. 2017. Methods to determine limit of detection and limit of quantification in quantitative real-time PCR (qPCR). Biomol Detect Quantif 12:1–6. 10.1016/j.bdq.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oh C, Kim K, Araud E, Wang L, Shisler JL, Nguyen TH. 2022. A novel approach to concentrate human and animal viruses from wastewater using receptors-conjugated magnetic beads. Water Res 212:118112. 10.1016/j.watres.2022.118112. [DOI] [PubMed] [Google Scholar]
- 37.Haramoto E, Kitajima M, Kishida N, Konno Y, Katayama H, Asami M, Akiba M. 2013. Occurrence of pepper mild mottle virus in drinking water sources in Japan. Appl Environ Microbiol 79:7413–7418. 10.1128/AEM.02354-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Il Cho Y, Han JI, Wang C, Cooper V, Schwartz K, Engelken T, Yoon KJ. 2013. Case-control study of microbiological etiology associated with calf diarrhea. Vet Microbiol 166:375–385. 10.1016/j.vetmic.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fuzawa M, Bai H, Shisler JL, Nguyen TH. 2020. The basis of peracetic acid (PAA) inactivation mechanisms for rotavirus and Tulane virus under conditions relevant for vegetable sanitation. Appl Environ Microbiol 86:1–47. 10.1128/AEM.01095-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 to S4 and Tables S1 to S4. Download aem.02289-21-s0001.pdf, PDF file, 1.5 MB (1.5MB, pdf)
Data Availability Statement
All the sequence data analyzed in this study are publicly available at GISAID (https://www.gisaid.org/). The analyzed and processed real data results are available at https://github.com/elkebir-group/primes-data.