

ABSTRACT
The phylum “Candidatus Omnitrophica” (candidate division OP3) is ubiquitous in anaerobic habitats but is currently characterized only by draft genomes from metagenomes and single cells. We had visualized cells of the phylotype OP3 LiM in methanogenic cultures on limonene as small epibiotic cells. In this study, we enriched OP3 cells by double density gradient centrifugation and obtained the first closed genome of an apparently clonal OP3 cell population by applying metagenomics and PCR for gap closure. Filaments of acetoclastic Methanosaeta, the largest morphotype in the culture community, contained empty cells, cells devoid of rRNA or of both rRNA and DNA, and dead cells according to transmission electron microscopy (TEM), thin-section TEM, scanning electron microscopy (SEM), catalyzed reporter deposition-fluorescence in situ hybridization (CARD-FISH), and LIVE/DEAD imaging. OP3 LiM cells were ultramicrobacteria (200 to 300 nm in diameter) and showed two physiological stages in CARD-FISH fluorescence signals: strong signals of OP3 LiM cells attached to Bacteria and to Archaea indicated many rRNA molecules and an active metabolism, whereas free-living OP3 cells had weak signals. Metaproteomics revealed that OP3 LiM lives with highly expressed secreted proteins involved in depolymerization and uptake of macromolecules and an active glycolysis and energy conservation by the utilization of pyruvate via a pyruvate:ferredoxin oxidoreductase and an Rnf complex (ferredoxin:NAD oxidoreductase). Besides sugar fermentation, a nucleotidyl transferase may contribute to energy conservation by phosphorolysis, the phosphate-dependent depolymerization of nucleic acids. Thin-section TEM showed distinctive structures of predation. Our study demonstrated a predatory metabolism for OP3 LiM cells, and therefore, we propose the name “Candidatus Velamenicoccus archaeovorus” gen. nov., sp. nov., for OP3 LiM.
IMPORTANCE Epibiotic bacteria are known to live on and off bacterial cells. Here, we describe the ultramicrobacterial anaerobic epibiont OP3 LiM living on Archaea and Bacteria. We detected sick and dead cells of the filamentous archaeon Methanosaeta in slowly growing methanogenic cultures. OP3 LiM lives as a sugar fermenter, likely on polysaccharides from outer membranes, and has the genomic potential to live as a syntroph. The predatory lifestyle of OP3 LiM was supported by its genome, the first closed genome for the phylum “Candidatus Omnitrophica,” and by images of cell-to-cell contact with prey cells. We propose naming OP3 LiM “Candidatus Velamenicoccus archaeovorus.” Its metabolic versatility explains the ubiquitous presence of “Candidatus Omnitrophica” 3 in anoxic habitats and gives ultramicrobacterial epibionts an important role in the recycling and remineralization of microbial biomass. The removal of polysaccharides from outer membranes by ultramicrobacteria may also influence biological interactions between pro- and eukaryotes.
KEYWORDS: “Candidatus Omnitrophica”, predatory bacteria, LPS, methanogenic enrichment culture, candidate division OP3, limonene, lipopolysaccharide, methanogens
INTRODUCTION
Life may have begun in a primordial soup (1) with the evolution of an RNA world (2). Once the dissolved organic carbon in this soup was depleted, chemical energy was available only in the form of reduced inorganic compounds or as particulate organic matter in the form of cells. Geochemically produced hydrogen has been identified as a potential energy source for ancient acetogenic and methanogenic organisms (3). The origin and evolution of predation have been underestimated as important processes in early times of the earth (4) but are now established as a widespread mode of interaction among living organisms in many ecosystems (5, 6). Predators have been identified in groundwater, rivers, estuaries, the open ocean, sewage, soils, plant roots, and animal feces (7, 8). Predators have been classified as obligate (unable to grow in the absence of prey) or facultative (able to grow as a pure culture without the presence of prey) (8).
Bacterial predators attack their prey in groups (Myxobacteria) or individually. Epibiotic species attach to the prey, and some species penetrate the periplasm or the cytoplasm (8, 9). Members of the genus Bdellovibrio and related organisms, summarized as “Bdellovibrio and like organisms” (BALOs), are the most-studied group of predatory bacteria (10). They prey exclusively on Gram-negative cells and have a dimorphic life cycle. Motile cells with a single polar flagellum find prey cells and attach to the outer membrane (7). After an irreversible attachment (11), invading BALOs such as Bdellovibrio bacteriovorus enter the prey’s periplasmic space and proliferate at the expense of the prey’s cytoplasmic content. Motile progeny cells release themselves from the remnants of the prey cell to start a new cycle (11).
Epibiotic predators remain attached to the outer membrane while deriving nourishment from the prey (11). This lifestyle has been described for Bdellovibrio exovorus, formerly Bdellovibrio sp. strain JSS and a novel predator of Caulobacter crescentus, and Micavibrio aeruginosavorus (12, 13). The eukaryotic microalga Chlorella has an epibiotic predator, Vampirovibrio chlorellavorus (14). Recently, the ultramicrobacterium “Nanosynbacter lyticus” of the phylum “Candidatus Saccharibacteria” (formerly candidate division TM7) was isolated in coculture with its host/prey, an Actinomyces strain (15).
Absence of molecular oxygen characterized early evolution. The anaerobe Vampirococcus, which was defined by microscopic cell counts and electron micrographs of cells on anoxygenic phototrophs in environmental samples (16), has stimulated discussion on the early evolution of predators. Recently, a phototrophic enrichment from an athalassic salt lake was found to contain anoxygenic phototrophs related to Halochromatium and epibiotic predators which were described as “Candidatus Vampirococcus lugosii” (17). Its genome revealed that it is a member of the phylum “Candidatus Absconditabacteria.”
Here, we describe a novel anaerobic predatory bacterium with a coccal morphology. We had observed cells of candidate division OP3, also named phylum “Candidatus Omnitrophica” (18), in high abundances in a methanogenic enrichment culture on limonene. The phylotype OP3 LiM originated from a 16S rRNA gene clone library. Catalyzed reporter deposition-fluorescence in situ hybridization (CARD-FISH) with the specific OP3 LiM probe OP3-565 revealed that 18% of all cells in the enrichment culture were OP3 LiM cells (19). The micrographs showed small round cells, either free living or attached to larger cells. Physical cell separations and a range of visualizations as well as metagenomes and metaproteomes provided insight into the biology of OP3 LiM cells. Based on our observations, the name “Candidatus Velamenicoccus archaeovorus” gen. nov. sp. nov. is proposed for OP3 LiM.
RESULTS
Maintenance of a methanogenic enrichment culture on limonene.
Methanogenic enrichment cultures (20) were used as inocula in 1999 in liquid dilution series. An inoculum of 1 μL grew, and this culture was maintained in 12 parallel lineages by an annual transfer of 10% (vol/vol), which corresponds to three to four generations each year. These cultures showed active methanogenesis for more than 2 years. Template dilution OP3 LiM-specific PCR and CARD-FISH experiments with probe OP3-565 guided the selection of a lineage containing a high cell number of OP3 LiM for experiments.
Visualization of Methanosaeta in methanogenic limonene enrichment cultures.
Methanosaeta was identified in 16S rRNA gene amplicon libraries as one of the abundant methanogenic Archaea in the limonene enrichment culture. FISH studies using the Methanosaetaceae-specific probe MX-825 (21) confirmed the filamentous cell morphology, which was also evident in electron micrographs. Methanosaeta cells are located within a filamentous sheath in chambers separated by spacer plugs (22). Sheath and cells were visible in filaments of enrichment cultures in transmission electron micrographs (TEM) (Fig. 1; also, see Fig. S1 in the supplemental material). The enrichment was morphologically very diverse, also containing smaller filaments (Fig. S2 to S4). TEM images also showed large sheaths without cells (Fig. 2). To investigate the absence of cells or of cellular components in Methanosaeta filaments, we combined DNA staining by DAPI (4′,6-diamidino-2-phenylindole) with rRNA visualization by FISH, lipid staining by Nile red, and a relief view of the filament morphology by differential interference contrast microscopy applying confocal laser scanning microscopy. Methanosaeta filaments contained healthy cells with a full biovolume and the presence of lipids, rRNA, and DNA. However, the filaments also had cells with a reduced biovolume, and either rRNA or rRNA and DNA were not present according to the staining intensity. In the absence of a cell biovolume in the differential interference contrast (DIC) graph, a faint Nile red stain identified the shape of cells in the filament. We annotated these shapes as empty cells (Fig. 3; Fig. S5 to S7). For control experiments, Methanosaeta concilii GP6T was obtained as Methanothrix soehngenii DSM3671. Cells in stationary phase showed loss of rRNA in only a few Nile red-stained cells. Nile red also stained spacer plugs, albeit with less sensitivity than lipids. In some cases, overstaining revealed the presence of pairs of spacer plugs between normal-sized cells, suggesting the presence of small cells as precursors of filament breakage (Fig. S8 and S9). Methanosaeta performs, besides its equal cell division into two viable cells, an unequal cell division into a normal and a short cell, followed by lysis of the short daughter cell, a biological strategy to control the filament length (22).
FIG 1.
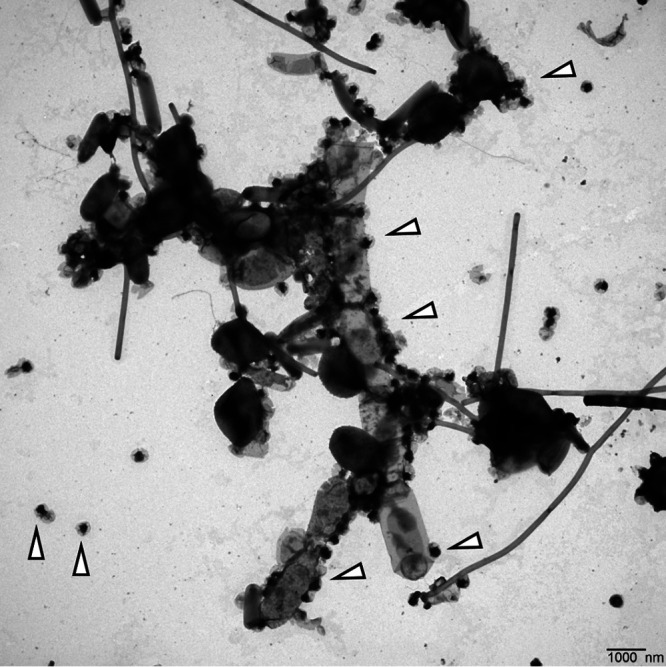
Transmission electron micrograph of a resuspended cell pellet of the methanogenic limonene enrichment culture. We assigned the large filament to Methanosaeta, based on the spacer disc at the lower end of the filament and the presence and structure of a sheath (see Fig. S1). The cells may have been shrunk by the TEM preparation. The small black cells with a gray cape were assigned to the ultramicrobacterium OP3 LiM (triangles indicate OP3 LiM cells). Bar, 1 μm.
FIG 2.
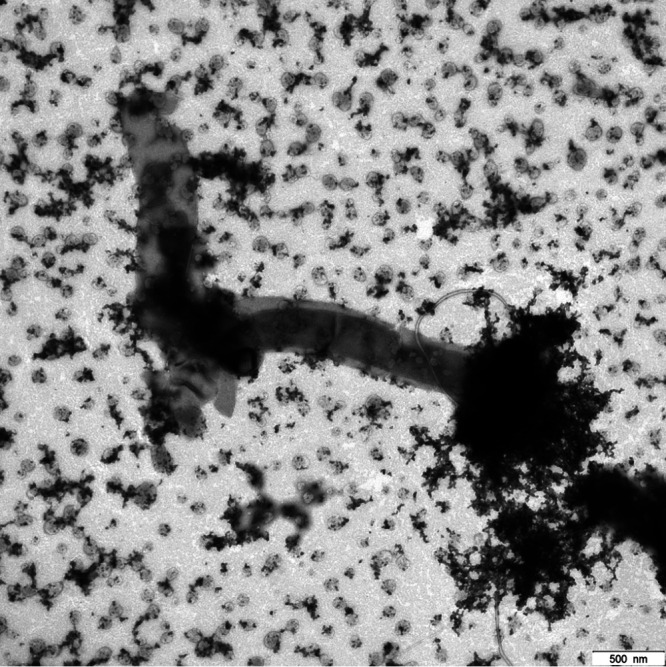
Transmission electron micrograph of a dried culture droplet of the methanogenic limonene enrichment culture showing sheaths with cells and without cells. We assigned the large filament to Methanosaeta. The small cells were assigned to OP3 LiM and other ultramicrobacteria in the culture. Bar, 500 nm.
FIG 3.

Differential interference contrast micrograph (A) and superresolution structured illumination microscopy (SR-SIM) overlay image (B) of a filament assigned to Methanosaeta. Staining was achieved using Nile red (red) for lipids, probe Arch915 labeled with four 6-carboxyfluorescein molecules (green) for rRNA, and DAPI (blue) for DNA. Bar, 5 μm. For individual and dual overlay SR-SIM images, see Fig. S5.
A decrease in cellular content of Methanosaeta cells was also observed in thin-section TEM. For visibility in TEM images, cells require the presence of electron-diffracting heavy metal ions that are bound to anionic groups of proteins, lipids, and nucleic acids. Thus, the darkness of the cellular content in the image is a proxy for cellular integrity. Methanosaeta filaments were identified by their unique morphology and contained cells that appeared as full, intermediate, or empty with respect to the cellular content (Fig. 4; Fig. S10). Some cells contained faintly stained intracellular local structures of unknown biological identity, likely neutral storage compounds or unknown infectious viruses. Very short cells within filaments were annotated as short daughter cells enabling filament segregation. These cells did not contain cytoplasm, and we expected them to be dead based on their biological function. In addition, we detected large cells in filaments without cellular contents. To confirm their physiological status, we investigated the integrity of Methanosaeta membranes with a LIVE/DEAD stain. In confocal laser scanning micrographs, Methanosaeta cells showed a condensed DNA using optimized laser intensities. The green stain colored the whole cell at higher laser intensities, which is equivalent to overexposed images (Fig. 5). Besides live cells, Methanosaeta filaments also contained dead cells of ordinary size, accounting for 11% of all Methanosaeta cells in a 3-year-old, still-methanogenic, active enrichment culture. We annotated shorter cells as recently divided cells. The LIVE/DEAD stain also visualized very short dead cells with a very small thickness between normal-sized cells (Fig. 5). The presence of these short cells was also visualized in thin-section TEM images, where they appeared to be empty (Fig. 4; Fig. S10), and gained additional support from pairs of spacer plugs that were stained by Nile red (Fig. S8 and S9). We suggest that these short dead cells result from unequal cell division preceding filament segregation.
FIG 4.
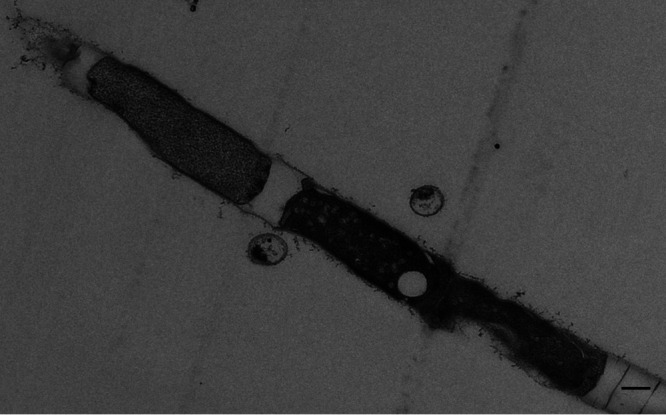
TEM image of a thin section preparation showing a Methanosaeta filament with microcells and damaged regular cells. Bar, 300 nm.
FIG 5.

LIVE/DEAD staining of a filament assigned to Methanosaeta in images with optimal exposure (A) and overexposure (B) to visualize regions of weak staining. In the DIC micrograph, dead (red) cells had less biovolume than live (green) cells. Overexposure showed a weak green staining of cells and short red cells separating green cells similar to spacer plugs. We assigned the short dead cells to unequally produced daughter cells that precede filament separation of Methanosaeta based on the observation of two spacer plugs between cells visible by Nile red staining (Fig. S8 and S9) and Methanosaeta microcells in thin-section TEM images (Fig. S11, S15, and S16). Bar, 5 μm.
Methanosaeta cells have a sheath made of protein and carbohydrates (22). To exclude a problem with accessibility of FISH probes to cellular rRNA molecules because of the rigid sheath, we tested harsh cell lysis treatments with proteinase K to improve the detection of archaeal rRNA in all cells of filaments. Small cells were lysed, but the detection efficiency of rRNA-containing cells in filaments did not increase, similar to the results of Kubota et al. (23). We conclude that the lack of CARD-FISH signals of cells in Methanosaeta filaments was caused by the absence of rRNA molecules in the cells, but not by a sheath that is impermeable to probes. In nature, dead cells within filaments may be widespread, as Methanosaeta filaments in environmental samples had low in situ hybridization detection rates of cells independent of the applied detection technique (FISH, CARD-FISH, or hybridization chain reaction [HCR]-FISH) (24). In summary, our visualization experiments confirmed previous observations of the Methanosaeta cell biology and detected the presence of sickly and dead cells of ordinary cell size in Methanosaeta filaments. Here, we present initial evidence that a biological cause is responsible for the sickliness and death of Methanosaeta.
OP3 LiM cell enrichment in density gradients yielded an ultramicrobacterium.
The small size of OP3 LiM cells in CARD-FISH images (19) suggested a separation of cell populations by density gradients. The density centrifugation of concentrated cells from the limonene-degrading methanogenic enrichment culture yielded two visible bands close to the top and to the bottom in a Percoll gradient (Fig. S11). PCR and CARD-FISH analyses specific for OP3 LiM detected the highest relative abundance of OP3 cells in the gradient in a macroscopically clear layer above the visible bottom band of cells. This macroscopically clear fraction was collected from 10 gradients and further concentrated in a second Percoll gradient, yielding a fraction with over 80% OP3 LiM cells according to CARD-FISH. The twice-enriched cells were used for transmission electron microscopy and an OP3 LiM-enriched metagenome.
TEM images showed a dominant morphotype of small cells surrounded by a surface structure weakly stained with uranyl acetate (Fig. 6A). The cell size was 200 to 300 nm in diameter, which is in the range of ultramicrobacteria. The abundance of this morphotype correlated with the abundance of OP3 LiM cells detected by CARD-FISH experiments in the cell population. This observation related the small cells with a cape in TEM images to the phylotype OP3 LiM.
FIG 6.

TEM images of OP3 LiM cells obtained from the second density gradient (A) (the small particles are colloidal silica particles 15 to 30 nm in diameter originating from Percoll) and in the limonene enrichment culture as free-living cells (B) or attached to other cells (C and D). Bars, 100 nm (A), 200 nm (B) and 500 nm (C and D).
TEM images of enrichment cultures were prepared together with those of the OP3 LiM-enriched gradient samples, and on the basis of the characteristic morphotype, they revealed that OP3 LiM cells were mainly attached to larger cells (Fig. 6B to D). The attached lifestyle detected earlier by FISH (19) was confirmed in these first TEM pictures recorded in 2012. TEM images in 2017 showed the same ultramicrobacterial morphotype, characterized by a veil around a strongly stained cell (Fig. 1; Fig. S1 to S4).
As ultramicrobacteria are too small to be visualized by phase-contrast microcopy, we initially used scanning electron microscopy to confirm the presence of ultramicrobacteria on Methanosaeta filaments (Fig. S12 and S13). Differential interference contrast microscopy can resolve 100-nm objects and has become the preferred technology to quickly demonstrate the presence of ultramicrobacteria in samples of the limonene enrichment cultures (Fig. 3 and 5; Fig. S6 and S7).
Thin-section TEM images visualized cell-to-cell interactions.
TEM of 70-nm thin sections provided insights into the cell-to-cell interactions in the methanogenic enrichment culture (Fig. 7; Fig. S14 to S16). Methanosaeta was identified by its morphotype. A second morphotype of small, irregular, pentagonal cells with strong cellular staining and small dark dots on the outer surface (Fig. 7B) was annotated as an methanogenic archaeon with an S-layer on the surface, identified as Methanoculleus-related cells based on an archaeal 16S rRNA gene library of the enrichment (19) and published descriptions of Methanoculleus strains (25). The dominant cell type in thin-section TEM images was an ultramicrobacterium with uneven staining of the cytoplasm and with strong staining of an intracellular complex. These cells were annotated as OP3 LiM. The cells interacted with other cells in an attack mode, with the intracellular complex close to the attack site (Fig. 7). Similar images of predation were reported in the first “Candidatus Vampirococcus” publication, which described it as “sucking the innards of its prey in a fashion reminiscent of vampires” (16). Prey cells include bacterial cells (Fig. 7A) as well as Methanoculleus (Fig. 7B). Stationary-phase cultures contained many remains of cell membranes, which suggested intensive predation in the enrichment culture. The cell morphotype assigned to OP3 LiM (Fig. S15 and S16) was still present in these stationary-phase cultures.
FIG 7.
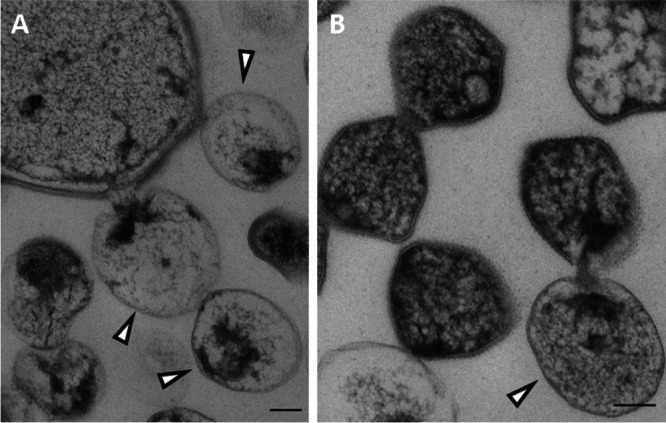
TEM images of thin sections of the methanogenic limonene enrichment culture showing the attack by OP3 LiM ultramicrobacteria of a large bacterial cell (A) and of a small archaeal cell (B). Ultramicrobial cells with a strongly stained part of the cytosol were assigned to OP3 LiM (triangles) based on the frequency of occurrence of this cell type in the thin-section TEM images. The small irregular cells with intense staining and a characteristic surface structure, likely S-layer protein, were assigned to archaeal cells, likely Methanoculleus-related species. Bars, 100 nm.
Visualization of the phylotype OP3 LiM.
CARD-FISH detection of OP3 LiM cells with the probe OP3-565 was improved by introducing four helper oligonucleotides (26). In cultures, small coccal OP3 LiM cells presented up to 30% of DAPI-stained cells in two different signal intensities (Fig. S16). Stronger detection signals were observed for attached OP3 LiM cells, suggesting a larger ribosome content and a higher metabolic activity of the attached cells than the free-living cells. The latter had weaker signals, suggesting a state of low metabolic activity and evidence of starvation. OP3 LiM cells attached to archaeal and bacterial cells, according to double-hybridization CARD-FISH experiments with probes for OP3 LiM cells and for Archaea or for Bacteria, respectively (Fig. 8; Fig. S17). This is possible, as the bacterial probe mix EUB338I-III does not detect OP3 LiM due to mismatches in the targeted 16S rRNA sequence (19). A range of morphologies was identified as Bacteria, including vibrios of different sizes, large coccoid cells, and short thin rod-shaped cells (Fig. S17).
FIG 8.
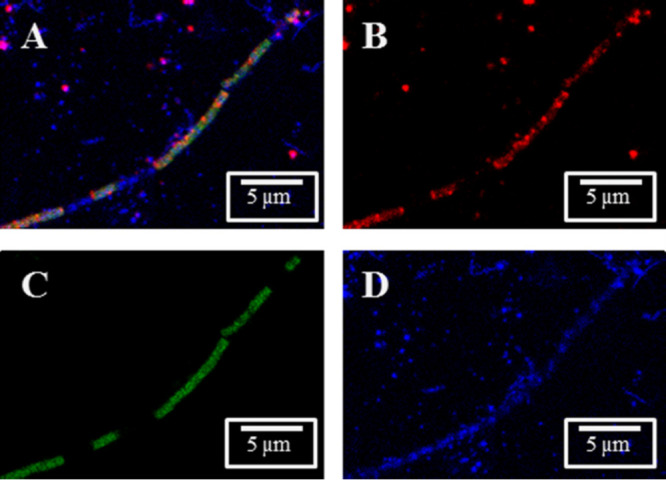
Detection of rRNA from OP3 LiM by fluorescence in situ hybridization. Archaea and OP3 LiM were detected in a methanogenic enrichment culture with probes ARCH-915 and OP3-565, respectively, in confocal laser scanning microscopy images: overlay (A) and individual signals of OP3 LiM rRNA (B, red), rRNA of Archaea (C, green), and DNA (D, blue, obtained by DAPI staining). Bars, 5 μm.
OP3 LiM cells were frequently attached to Methanosaeta filaments, either as single cells (Fig. 8) or as a group of cells (Fig. S17 and S18). Besides staining small cells of OP3 LiM, the CARD-FISH signal of probe OP3-565 faintly stained Methanosaeta cells (Fig. 8). To exclude an archaeal target sequence for the probe, BLASTn was performed with the probe sequence against sequences affiliated with Methanosaeta and the Methanosaeta concilii GP-6 genome. The best match was a 13-mer oligonucleotide with a G+C content of 54%. The predicted melting temperature was below the hybridization temperature of the CARD-FISH experiments, which excluded a false-positive signal and suggested that the weak signal of the probe OP3-565 originated from the hybridization to rRNA of OP3 LiM present on or in the archaeal cells.
Secondary electron micrographs from the scanning electron microscope showed healthy and sickly Methanosaeta filaments as well as ultramicrobacteria on the filaments (Fig. S12 and S13). Scanning electron microscopy (SEM) in combination with CARD-FISH detected some intact Methanosaeta cells in filaments together with OP3 LiM cells attached to a degraded part of the filament (Fig. S19).
Closed genome of OP3 LiM.
In density gradients, OP3 LiM cells enriched in density gradients were the basis for 454 and Illumina metagenomes. A first closed draft genome had uncertainties in repetitive regions, indicated by 454 read mappings. Combinatorial PCRs integrated additional contigs of repetitive elements and confirmed unusual regions, including the presence of three repeats of about 2,000 bp with small sequence variations mixed with two completely identical sequences of 2,506 bp within the open reading frame (ORF) of the very large multienzyme surface protein.
The assembled OP3 LiM genome is a single chromosome of 1,974,201 bp with a GC content of 52.9%. We placed the genome start point at the origin of replication (ori) at the N terminus of dnaA. The asymmetry of the nucleotide composition between leading and lagging strands indicated the first base of the start codon of dnaA coding for the chromosomal replication initiator protein DnaA (27) as base 1. The terminus is about 950,000 bp away from ori, based on the shift points of the GC skew graphs (28). Gene orientation is highly ordered. Eighty-five percent of the genes are oriented in the direction of DNA replication, and only 15% are oriented opposite to the direction of DNA replication. We circulated the genome in a GC-homopolymeric region close to the terminus; however, reads covering both regions adjacent to the GC-homopolymer were not obtained in both 454 and Illumina MiSeq read sets. The genome has 1,851 protein-coding open reading frames, 45 tRNA genes, and one rRNA operon, with a group I intron in the 23S rRNA gene. The intron encodes a homing endonuclease in the peripheral stem-loop regions of the group I ribozyme (29, 30). The identified insertion position 1,917 (referring to the Escherichia coli sequence) has been reported as a preferred insertion site (31), e.g., in Coxiella burnetii (32), Thermotoga subterranea (32), Thiomargarita sp. clone NAM092 (33), Synechococcus sp. strain C9 (29), and groundwater-associated bacteria (31). The 16S rRNA gene of the genome has a sequence identity of 99% to 100% to 16S rRNA gene clone sequences obtained in the initial characterization of the limonene enrichment culture (19). Only one other 16S rRNA gene sequence is known with an identity above 97% with the genomic gene sequence, a clone sequence from a methanogenic benzene degradation (GenBank number KT028835). The identity to 16S rRNA genes of validly described bacteria was below 80%, confirming a genetic distance on the level of phyla. Besides 45 tRNA genes, the genome codes for two noncoding RNA molecules, assigned to SsrS and Rnp, likely with regulating (inhibitory) functions. The genome encodes a type I restriction-modification cluster (hsdMSR), a type II restriction-modification system, and a subunit of a type III restriction-modification system. Three candidate loci for clustered regularly interspaced short palindromic repeats (CRISPR) providing acquired immunity against foreign genetic elements (34) were identified at positions 875578 to 875727, 1062302 to 1062425, and 1625552 to 1625657. Each CRISPR locus was separated by only one spacer.
Proteomic insights into the physiology of OP3 LiM.
A second method to enrich OP3 LiM cells was differential centrifugation. A 10,000S pellet collected all large and aggregated cells (10kS cells). Small cells in the supernatant were collected in a 100S pellet that after resuspension was separated by a second 10,000S centrifugation into aggregated cells (second 10,000S pellet [100S aggregates]) and free-living cells (supernatant of second 10,000S centrifugation [100S cells]). SEM images of the different fractions confirmed the size fractionation. Small single cells accounted for more than 99% of all cells in the 100S aggregate and 100S cell fractions. All fractions (10kS cells, 100S aggregates, and 100S cells) were sources of metaproteomes. A total of 1,279 proteins encoded in the OP3 LiM genome were identified in the proteomes of two biological replicates and gave insight into the metabolism (Table S1). Most frequently detected was a 18-stranded beta-sheet outer membrane pore protein that facilitates the uptake and export of macromolecules. Three other large proteins with transmembrane helices are among the most frequently detected proteins, a glycosylphosphatidylinositol (GPI) membrane-anchored actin/protein binding protein with Kelch repeats (499 kDa), a protein with unknown function (326 kDa), and a very large multienzyme surface protein (4,384 kDa). Its gene codes for 39,678 amino acids, containing hundreds of protein domains. Most were predicted to be outside the cell, anchored by 42 predicted transmembrane helices. Among 44 conserved enzyme domains were 7 glycosyl transferases, 3 glycosyl hydrolases, and a sugar epimerase, likely catalyzing the degradation of external polysaccharides. Three peptidases and a DnaJ chaperone may act on proteins. A phosphatase, a dehydrogenase, a methyltransferase, and an acetyltransferase completed the identified range of degradative enzyme domains. A signal receiver domain and a cellular signaling response via cyclic guanine nucleotides by a diguanylate cyclase/phosphodiesterase (GGDEF and EAL domains) may serve as an environmental signal sensor. The nucleotide metabolism may be influenced by three ATP-binding domains, a protein kinase and two nucleotide kinases, two ppGpp synthases/hydrolases, and two single-strand-DNA (ssDNA)-binding and two double-strand-DNA (dsDNA)-binding domains. The gene for the secreted very large multienzyme surface protein is located on the leading strand directly after the genes for proteins involved in DNA replication and in a secretion pathway. This is the expected position for highly expressed proteins minimizing collisions of replicating and transcribing polymerases (35, 36). Together with a pilus secretin, this large protein was induced 3-fold in attached OP3 LiM cells (10kS cells) in comparison to free-living cells (100S cells), suggesting a function as an “attack” protein. We propose that the very large surface protein stains as a coat that is visible in TEM images.
Among the detected transporter proteins were sugar ABC transporters. Monosaccharide degradation likely involves a class II fructose-bisphosphate aldolase. A pyruvate:ferredoxin oxidoreductase may provide reducing equivalents for energy conservation via a membrane-integrated Rnf complex (ferredoxin:NAD oxidoreductase). The proteome supported a polysaccharide fermentation to acetate, carbon dioxide, and eventually hydrogen or formate. OP3 LiM has the potential to conserve energy from the phosphoester bonds in nucleic acids. An abundant polyribonucleotide nucleotidyl transferase is expected to move a nucleoside monophosphate from RNA onto phosphate, thus synthesizing a nucleoside diphosphate. Other potentially energy-conserving enzymes were pyruvate phosphate dikinase and a proton-translocating pyrophosphatase.
Based on N-terminally encoded signal peptides in genes, the periplasm also contains a peptidyl prolyl cis-trans isomerase, a Clp protease and an ATPase that has, in addition to the CpaF/VirB11-like ATPase domain, an N-terminal CobQ/CobB/MinD/ParA nucleotide-binding domain, suggesting participation in nucleic acid transfer. Although OP3 has no flagellum, an abundant protein was MotD, a stator ring protein for a flagellum forming a pore in the inner membrane that is known to be filled by a type III secretion system and accessory proteins. The second stator protein, MotB, was also expressed. Other abundant proteins were an outer membrane protein assembly factor, the Sec translocase SecD/SecF, and the bacterial and archaeal forms of the chromosome segregation protein SMC, which binds nucleic acids. The bacterial actin MreB was also highly expressed.
We searched for other proteins potentially involved in the uptake of nucleic acids or other macromolecules. Besides the aforementioned extracellular nucleotide-binding CpaF-related ATPase, three PilT ATPases were present to export/import macromolecules. Of the pil genes, CpaB, PilM, PilO, PilQ, and two PilZ proteins were expressed. A PilA-related pseudopilin, PulG, was the most abundant pilin. OP3 LiM has an active Sec secretion system and a type II secretion/type IV pilus assembly system. Several loci contained pil-related genes, including one locus in front of the very large multienzyme surface protein with several export proteins of a type II secretion system.
A FIC protein was expressed. FIC proteins modify proteins by posttranslational AMPylation, which results in the activation of toxins of pathogenic bacteria and inhibition of host/prey small GTPases, thereby reducing metabolic activities (37). The FIC protein was shown to induce hibernation (38). It may be responsible for the low 16S rRNA content of OP3 LiM cells in the free-living state.
The OP3 LiM proteomes highlighted the presence of large outer membrane pores and of polymer-binding and -depolymerizing domains located extracellularly and in the periplasm. The metabolism of OP3 LiM was dominated by sugar fermentation. However, the genome also encodes a syntrophic life on hydrogen or formate, with carbon dioxide fixation on the Wood–Ljungdahl pathway using formate dehydrogenase, the reduction of a formyl group on tetrahydrofolate, and methyl transfer to an acetyl coenzyme A (acetyl-CoA) synthase, but these enzymes were not highly expressed. Among the four hydrogenases, an uptake hydrogenase (hydrogen:NAD oxidoreductase) was the most highly expressed. Interestingly, high expression of serine hydroxymethyltransferase suggested serine and glycine as precursors of C1 compounds. Building blocks (amino acids, ribose, nucleotides, and fatty acids) were synthesized de novo according to expressed enzymes. OP3 LiM may eventually salvage these monomers from the environment, but it did not seem to depend on salvage pathways. d-Alanine and d-glutamate racemases as well as peptidoglycan biosynthetic enzymes were expressed. Together with the presence of peptidoglycan-binding domains in several secreted proteins, this suggested the presence of a murein sacculus in OP3 LiM. Genes for the synthesis of the LPS core glycolipid suggested the well-known lipid asymmetry in the outer membrane.
DISCUSSION
Methanosaeta is likely one of the most abundant microbes on Earth (39). Individual cells of this anaerobic acetoclastic archaeon are strongly separated from the environment and from the neighboring cells in a filament. They are enclosed in an amorphous matrix and shielded by a protein- and sugar-containing sheath and by spacer plugs that grow out from the sheath during cell division (22). Together with the demand for only one transporter for substrate (acetate), this morphology offers few entrance points for predators, which can be interpreted as a necessity to balance the slow growth of the archaeon in the environment. In this study, using a variety of microscopic investigations and specific stains for RNA, DNA and lipids as well as the integrity of the cellular lipid layer, we found that Methanosaeta cells were individually attacked. We detected cells with a decreased biovolume, a decreased concentration of cellular components in general, and decreased concentrations of RNA and DNA. Dead cells were present in filaments. This predation pressure on individual Methanosaeta cells plausibly explains the evolution of spacer plugs and the isolation of cells. The filament can continue to live although individual cells are dead.
These observations ask for the identification of the biological predator. Phages have been observed in methanogenic reactors (40) but not convincingly shown in Methanosaeta cells. Our thin-section electron micrographs show nonstaining structures within Methanosaeta cells. The absence of stain indicates a neutral or cationic surface that does not bind the heavy metal cations used for staining. Whether these structures are polysaccharide storage compounds or viral coats may be answered in future studies. In this study, we focused on the characterization of the phylotype candidate division OP3 LiM. It is a chemoheterotroph attaching to and living off Bacteria and Archaea. As an ultramicrobacterium (0.2 to 0.3 μm in diameter), it is barely or not visible by phase-contrast microscopy (optical resolution > 0.3 μm). In addition, it is not detectable by the standard FISH probes EUBI to -III for Bacteria (19). To detect it, we used a range of electron-microscopic techniques in combination with in situ hybridizations.
The presence of the candidate division OP3 in many anaerobic habitats and cultures has been established in hundreds of studies using gene-based, cultivation-independent detection by 16S rRNA amplicon sequences and metagenome-assembled genomes (MAGs). For example, over 100 MAGs for the phylum “Candidatus Omnitrophica” (18) have been established, but a closed genome was lacking due to the absence of a pure strain. We developed a physical enrichment of OP3 LiM cells and obtained after PCR-based integration of repetitive regions in the genome the first closed genome for the phylum. Metaproteomes enriched in attached OP3 LiM cells and in free-living OP3 LiM cells gave insights into the biology of OP3 LiM. It may grow preferentially as a sugar fermenter.
Most bacteria are Gram-negative; they have two membranes, and the outer layer of the outer membrane consists of lipopolysaccharides (LPS) with a lipid anchor, a polysaccharide core, and a long polysaccharide chain directed to the environment. The biosynthesis of LPS is well understood, but it is a one-way synthesis: lipopolysaccharides are not recycled by their producers. They provide the substrate for ultramicrobial and small epibionts, for example, “Nanosynbacter lyticus” from the phylum “Candidatus Saccharibacteria,” the TM7 phylum (41, 42). The expressed proteins identified in this study suggested that OP3 LiM also grazes the polysaccharide side chains of LPS, using glycosyl transferase and hydrolase domains present in the very large multienzyme surface protein which is 3-fold induced in the metaproteome enriched in attached OP3 cells. The utilization of surface polysaccharides is not limited to epibiotic (ultramicro)bacteria. Cellulosomes of clostridia have been described as extracellular, surface-associated “highly efficient nanomachines” that evolved to perfect depolymerization multienzyme complexes for plant cell wall complex carbohydrates (43). The degradation of LPS polysaccharides by epibionts like “Nanosynbacter lyticus” and OP3 LiM has many understudied ecological consequences. For example, in humans, the highly antigenic lipopolysaccharide core of pathogenic bacteria is camouflaged by polysaccharide side chains, and the degradation of these sugars by epibionts exposes the core lipid to the immune system. In addition, lipopolysaccharides have not been used as the substrate to isolate anaerobic microorganisms. Future studies will reveal whether lipopolysaccharide-fermenting ultramicrobacteria can be isolated on membrane particles.
Beyond carbohydrate-active enzymes, OP3 LiM has a range of degradative enzyme domains in its very large multienzyme surface protein. This attack protein may open an entry to the cytoplasm of the prey, as is visible on thin-section electron micrographs. Unusual proteins encoded in the genome and highly expressed proteins suggested a draft of the predation process. To paralyze the preýs metabolism, noncoding RNA molecules and excreted ATPases may block essential metabolic steps and deplete the host´s energy charge. The abundant 18-strand beta-barrel pore in the outer membrane has the size to allow import and export macromolecules. Several PilT ATPases may act in the energy-driven uptake of macromolecules, probably through a stator in the inner membrane. The MreB fragment, a bacterial actin usually absent in coccal cells but present in OP3 LiM, may serve as the foothold and cytoplasmatic endpoint of a transport mechanism. An energy-economical depolymerization of nucleic acids is ensured by a nucleotidyltransferase.
This study provides a closed genome of high quality of the OP3 LiM population in a limonene-degrading methanogenic enrichment culture. The smallest inoculum was 1 μL in a dilution series, potentially presenting a population bottleneck. The 16S rRNA gene sequence indicated a large phylogenetic distance from the next related species. The phylogenetic probe OP3-565 showed the morphology of the identified cells and guided the physical enrichment of OP3 LiM cells and correlation of a conspicuous morphotype in electron micrographs to OP3 LiM. Metaproteomes provided insights to infer the putative metabolism. Based on these observations, we propose naming the OP3 LiM cells “Candidatus Velamenicoccus archaeovorus” gen. nov., sp. nov.
Taxonomic note.
We deposited the genome of OP3 LiM as “Candidatus Vampirococcus archaeovorus” strain LiM on 22 January 2019 at NCBI (CP019384.1). Moreira et al. (17) reported in 2021 an anaerobic predatory organism of anoxygenic phototrophic bacteria that fits the original description of the genus Vampirococcus better than OP3 LiM. The 16S rRNA gene identity of these two organisms is 67.8%. In consequence, we have updated the NCBI database file of CP019384 and changed the name to “Candidatus Velamenicoccus archaeovorus” strain LiM.
Description of “Candidatus Velamenicoccus” gen. nov.
Candidatus Velamenicoccus (O.L. n. velamen, covering; N.L. masc. n. coccus [from Gr. masc. n. kokkos, grain, seed], berry, coccus; N.L. masc. n. Velamenicoccus, a coccus with a covering).
Members of the genus “Ca. Velamenicoccus” are obligately anaerobic, chemoheterotrophic bacteria. The only representative so far is the species “Ca. Velamenicoccus archaeovorus,” a predatory bacterium. The genus “Ca. Velamenicoccus” belongs to a lineage within the candidate division OP3 or phylum “Candidatus Omnitrophica.”
Description of “Candidatus Velamenicoccus archaeovorus” sp. nov.
Candidatus Velamenicoccus archaeovorus (Gr. masc. adj. archaios [Latin transliteration archaeos], ancient; L. v. voro, to eat, to devour; N.L. masc. adj. archaeovorus, archaea [ancient microorganisms] devouring).
The species is represented by the phylotype and strain LiM and its genome (GenBank number CP019384). The genome of 1.97 Mb has a GC content of 52.9%. OP3 LiM was highly enriched in a limonene-degrading methanogenic enrichment culture. Cells are coccoid. They are ultramicrobacteria 0.2 μm in diameter and occur free living and attached to other microorganisms. The bacterium is maintained in slowly growing Methanosaeta-rich methanogenic enrichment cultures in freshwater medium with low concentrations of limonene as the carbon source at 28°C. It can be visualized by the FISH probe OP3-565.
MATERIALS AND METHODS
Cultivation of methanogenic enrichment cultures.
The methanogenic enrichment culture originated from a wastewater sample taken in 1997 (20). From a dilution-to-extinction series prepared in 1999, a culture that had been inoculated with 1 μL enrichment culture became the origin of all cultures investigated in this study. Twelve lineages were established in 2005 and maintained with an annual transfer of 10% (vol/vol) inoculum. The cultures contained 300 mL freshwater methanogenic medium including 2 mM acetate and 1 mM cysteine, 30 mL 2,2,4,6,8,8-heptamethylnonane (HMN), and 1.5 mL of R-(+)-limonene in 500-mL borosilicate bottles (19, 20). OP3 LiM-specific PCR and CARD-FISH were performed to select lineages for the experiments.
Cell separation by Percoll density gradient centrifugation.
Cell biomass from 100 mL of enrichment culture was pelleted at 15,500 × g for 10 min in a Beckman 70.1 Ti ultracentrifuge rotor (Beckman, Palo Alto, CA). The pellet was resuspended in a mixture of 45 mL Percoll (GE Healthcare, Freiburg, Germany) and 5 mL 1.5 M NaCl. Portions of a 10-mL suspension were centrifuged at 36,680 × g for 60 min in the aforementioned rotor. Gradient fractions of 1 mL were assayed for the presence of OP3 LiM cells by applying OP3 LiM-specific PCR with a template dilution series and CARD-FISH with the probe OP3-565. Fractions enriched in OP3 LiM cells had densities of 1.05 ± 0.05 g/cm3 and were combined from several separations. The enriched fractions were further concentrated with a second gradient centrifugation using the aforementioned conditions. OP3 LiM cells were macroscopically visible as a band at a density of 1.05 g/cm3 and were collected for an OP3 LiM-enriched metagenome and electron microscopy.
Cell separation by differential centrifugation.
For the separation of large cells and aggregates from small cells in enrichment cultures, a Beckman SW28 ultracentrifuge rotor was used at 7,600 rpm (7,643 × g) for 20 min, corresponding to a sedimentation coefficient of 10,000S. The pellet (10kS cells) was resuspended in 1 mL 10 mM Tris–1 mM EDTA, pH 8.0 (TE). The supernatant was centrifuged at 27,000 rpm (96,467 × g) for 160 min, corresponding to a sedimentation coefficient of 100S. The pellet of each tube was resuspended in 0.5 mL TE, and a 10,000S pellet of aggregated cells and a few large cells (100S aggregates) was obtained at 12,400 rpm (16,331 × g, 10,000 S) for 3 min. Cell suspensions (10kS cells and 100S aggregates) and the supernatant of the last centrifugation (100S cells) were stored at −80°C. Alternatively, 100S cells were pelleted using a Beckmann Ti 50.2 ultracentrifuge rotor at 184,048 × g for 55 min (100S cell pellet).
Transmission electron microscopy.
For negative-staining transmission electron microscopy, bacterial cultures were adsorbed onto carbon film, washed in 20 mM Tris HCl–1 mM EDTA, pH 6.9, and stained for 1 min with 4% (wt/vol) aqueous uranyl acetate (44). After transfer onto copper grids and air drying, samples were examined in a Zeiss EM902A transmission electron microscope (TEM) (Zeiss, Oberkochen, Germany) operated at 80 kV at calibrated magnifications. Cells were analyzed using the program MeasureIT (Olympus Soft Imaging System GmbH, Münster, Germany).
Thin-section TEM.
Bacterial cultures were centrifuged in 1.5-mL portions at 16,000 × g for 5 min or, in later experiments, at 2,500 × g for 5 min (at 28°C) and frozen in their medium together with hexadecane or resuspended with a few microliters of 20% Ficoll in medium before high-pressure freezing on an HPM010 (Abra Fluid AG, Switzerland). The planchettes were freeze substituted in an AFS2 machine (Leica Microsystems, Wetzlar, Germany). Ficoll-containing samples were freeze substituted with 0.1% uranyl acetate as described by Ronchi et al. (45). Hexadecane-containing samples were freeze substituted in 1% osmium tetroxide and 0.1% uranyl acetate as according to Cohen et al. (46). In separate experiments, EPON infiltration was performed in steps, as follows. Samples were rinsed four times at 0°C for 15 min and then infiltrated with 30% (vol/vol) Epon in acetone for 3 h. The temperature was raised to 10°C, and cells were infiltrated with 50% (vol/vol) Epon in acetone mix for 3 h. The temperature was raised to room temperature and cells were infiltrated with a 70% (vol/vol) Epon-acetone mix overnight. Cells were then infiltrated with 100% Epon, and the solution was exchanged after 4 h and left overnight. After a final exchange in the morning, the cells were placed in the oven at 60°C for 48 h. Thin sections were cut at 70 nm on a diamond knife (Diatome, Switzerland), floated onto Formvar-coated slot grids (G2500PD; Plano GmbH, Wetzlar, Germany), poststained with uranyl acetate and Reynolds lead citrate, and imaged on either a JEOL 2100 Plus (200-kV) or a CM120 Philips Biotwin (120-kV) electron microscope.
Scanning electron microscopy.
Cells fixed with formaldehyde (1.3% [wt/vol] in 1× phosphate-buffered saline, pH 7.4 [PBS]) were spotted on silicon wafers (5 by 7 mm; Plano, Wetzlar, Germany) and incubated for 60 min at room temperature. Then cells were dehydrated in an ethanol series (30%, 50%, 70%, 80%, and 96% [vol/vol], each for 10 min) and critical point dried (Leica EM CPD 300; Leica, Vienna, Austria). Secondary electron micrographs were obtained with a Quanta FEG 250 (FEI, Hillsboro, OR, USA).
Extraction of nucleic acids.
OP3 LiM-enriched fractions were extracted for genomic DNA according to Martín-Platero et al. (47). After in-house quality control by spectroscopy and agarose gel electrophoresis, sample quality control by capillary electrophoresis and sequencing was performed by the Max Planck Genome Center, Cologne, Germany (http://mpgc.mpipz.mpg.de/home/). For PCR analyses, DNA was extracted from biomass of 1-mL samples of enrichment culture using the FastDNA spin kit for soil (MP Biomedicals, Santa Ana, CA, USA) according to the manufacturer’s instructions.
OP3 LiM-specific PCR.
OP3 LiM was detected using as the template 1.0 ng extracted genomic DNA or 1.0 μL of culture with an optical density at 600 nm (OD600) of 0.2 to 0.5 with freeze-thaw-fractured cells (48) together with a 1.7 μM concentration each of primers OP3-565F and OP3-1481R (Table 1), 15.0 μL 2× GoTaq master mix (Promega, Madison, WI, USA), and 13 μL water. The PCR protocol was 4 min at 94°C, 30 cycles of 94°C for 1 min, 62°C for 1 min, and 72°C for 3 min, and finally 72°C for 10 min. After amplicon analysis by separation in a 1% (wt/vol) agarose gel and ethidium bromide staining, sequences were obtained from dideoxynucleotide-terminated oligonucleotides. The sequencing reaction was performed for 60 cycles with an initial denaturing step of 20 s at 96°C, a denaturation of 10 s at 96°C, 5 s at 62°C, and 4 min at 62°C. The products were purified by molecular sieve chromatography using Sephadex G50 Superfine (GE Healthcare Life Sciences, Freiburg, Germany) and were separated on an ABI Prism 3130 XL genetic analyzer (Applied Biosystems, Foster City, CA, USA).
TABLE 1.
PCR primers used in this study
| Primer | Sequence (5′–3′) | Position | Description | Reference |
|---|---|---|---|---|
| OP3-565F | GGGTGTAAAGGGCAGGTA | 608–626a | OP3-specific 16S rRNA gene forward primer | This study |
| OP3-1481R | TACGACTTAGCGCCAGTC | 1525–1543a | OP3-specific 16S rRNA gene reverse primer | This study |
| 8-27F | AGAGTTTGATCCTGGCTCAG | 8–27b | Bacterial universal 16S rRNA gene forward primer | 71 |
| 907R | CCGTCAATTCMTTTGAGTTT | 907–926b | Bacterial universal 16S rRNA gene reverse primer | 72 |
OP3 LiM 16 rRNA location.
E. coli 16S rRNA location.
Fluorescence in situ hybridization of 16S rRNA.
For cell identity visualization, we used the catalyzed reporter deposition-fluorescence in situ hybridization (CARD-FISH) technique (49). For brighter signals of OP3 LiM cells, clone sequences (GenBank accession numbers FN646451.1, FN646447.1, FN646441.1, FN646440.1, and FN646435.1) were used to manually design helper oligonucleotides, two for each adjacent side of probe OP3-565 (Table 2). They were used at the probe concentration. One mL of enrichment cultures was fixed with formaldehyde (1.3% [wt/vol] in 1× PBS) for 60 min at room temperature. Fixed cultures were filtered on 0.2-μm isopore membrane filters (Millipore, Darmstadt, Germany) and washed three times with 15 mL 1× PBS (pH 7.4). After air drying, permeabilization was performed for 60 min at 37°C by lysozyme treatment (10 mg/mL). Probes (Table 3) were hybridized for 160 min at 46°C. After staining with DAPI (1 μg/mL), cells were visualized using an epifluorescence microscope (Nikon Eclipse 50i; Nikon, Tokyo, Japan) or a confocal laser scanning microscope (CLSM; Zeiss LSM 780, Zeiss, Oberkochen, Germany). The visualization of two populations required two separate CARD-FISH experiments with two probes and differently labeled tyramides for the first and second signal amplification, respectively. Horseradish peroxidase present at the first probe was inactivated after the signal amplification by incubation of the filter with 3% H2O2 for 10 min at room temperature. After rinsing with 1 L water and air drying, filters were stored overnight at −20°C, and the second CARD-FISH experiment was performed the next day. Cells were counterstained with 1 μg/mL DAPI and optionally 5 μg/mL Nile red in water. To visualize spacer plugs, the sample was washed a few seconds in water and then quickly in pure ethanol. Cells were embedded in Citifluor-Vectashield (4:1 [vol/vol]).
TABLE 2.
Helpersa (nonlabeled) used and designed in this study
| Helper | Sequence (5′–3′) |
|---|---|
| H548-A | AATAAATCCGAGTAACGC |
| H548-C | AATCAATCCGAGTAACGC |
| H583-TC | CTCCCCACTTGTCAGGCCGCC |
| H583-CT | CCTCCCACTTGTCAGGCCGCC |
All helpers were used in a mix.
TABLE 3.
HRP labeled oligonucleotide probes used in this studya
| Probe name | Probe sequence (5′–3′) | Position | Target group | FA concn(%)b | Reference |
|---|---|---|---|---|---|
| EUB338 Ic | GCTGCCTCCCGTAGGAGT | 338–355 | Most Bacteria | 35 | 73 |
| EUB338 IIc | GCAGCCACCCGTAGGTGT | 338–355 | Planctomycetales | 35 | 74 |
| EUB338 IIIc | GCTGCCACCCGTAGGTGT | 338–355 | Verrucomicrobiales | 35 | 74 |
| ARCH-915 | GTGCTCCCCCGCCAATTCCT | 915–934 | Archaea | 35 | 75 |
| OP3-565 | TACCTGCCCTTTACACCC | 608–626d | Candidate OP3 LiM | 30 | 19 |
Alternatively, ARCH-915 was labeled with carboxyfluorescein at the bases shown in italics: GUG CTC CCC CGC CAA TTC CT.
Formamide (FA) concentration in the hybridization buffer.
Used in a mix.
OP3 LiM 16S rRNA location.
LIVE/DEAD staining of cells.
Enrichment samples of 1 mL were centrifuged for 15 min at 4,000 × g. The cell pellet was resuspended in 1 mL of anoxic 0.85% (wt/vol) NaCl and mixed with 3 μL of the LIVE/DEAD dye mix (LIVE/DEAD BacLight viability kit [Invitrogen]) yielding 0.03 μmol/mL propidium iodide and 0.005 μmol/mL SYTO 9. After 15 min of incubation in the dark, 5 μL was pipetted on a glass slide (ground edges, 90°; Scientific Menzel-Gläser, Thermo Fisher, Schwerte, Germany) and covered with a coverslip (Menzel-Gläser Cover slips number 1.5). The edges of the coverslip were sealed with clear nail polish. The slides were directly imaged under the CLSM (Zeiss LSM 780) using differential interference contrast to increase contrast in the bright-field image. Propidium iodide was excited with the 561-nm laser line and detected in the window from 570 to 668 nm. For SYTO 9, the 488-nm laser line and the detection window from 499 to 552 nm were used. Laser intensities were adjusted for each sample to avoid overexposure.
SR-SIM.
Substrate incubation samples were visualized on a Zeiss ELYRA PS.1 (Carl Zeiss, Jena, Germany) using 561-, 488-, and 405-nm lasers and BP 573-613, BP 502-538, and BP 420-480+LP 750 optical filters. Z-stack images were taken with a Plan-Apochromat 63×/1.4 numerical aperture oil objective and processed with the software ZEN (Carl Zeiss, Jena, Germany). Superresolution structured illumination microscopy (SR-SIM) images are taken by exciting the sample using nonuniform wide-field illumination. The laser light passes through an optical grating, generating a striped sinusoidal interference pattern. This pattern then combines with the sample information originating from structures below the diffraction limit to generate moiré fringes. The image was detected by an electron-multiplying charge-coupled device (EMCCD) camera and contains high-spatial-frequency sample information shifted to a lower-spatial-frequency band that is transmitted through the objective. Mathematical reconstructions from raw image slices then allow reconstruction of a high-resolution image with doubled resolution in the x-y plane (50).
Genome assembly from an OP3 LiM-enriched metagenome.
DNA of an OP3 LiM-enriched sample from the second Percoll gradient was sequenced by 454 Titanium pyrosequencing technology (450-bp reads; 454 GS FLX; Roche, Basel, Switzerland) as well as by MiSeq technology (2 × 250-bp reads; Illumina, San Diego, CA, USA) by the Max Planck Genome Center, Cologne, Germany (http://mpgc.mpipz.mpg.de/home/). 454 pyrosequencing yielded 491,907 reads. An assembly of 454 reads with Newbler v. 2.3 (51) resulted in a metagenome of 5,779 contigs with 16,026,544 bp. Analysis of tetranucleotide frequencies using JSpecies (52) indicated that five of the six largest contigs covering 1,650,908 bp were part of the OP3 LiM genome. For further analyses, the raw reads of the 454 sequencing were processed in mothur 1.29.1 (53), resulting in 426,697 quality-controlled reads. A MiSeq read set of the same biological sample was used to finish the genome. A total of 9,888,618 paired-end reads were quality-controlled using FastQC (www.bioinformatics.babraham.ac.uk/projects/fastqc/). Raw reads were processed with dynamic trimming with SolexaQA v.2.2. (54) and normalization with Khmer 1.0 (55). The assembly by SPAdes 3.1.0 (56) contained 28,618 contigs with 61,364,565 bp. The assembly was inspected using QUAST v2.3 (57).
Contigs of the Newbler assembly were binned using Metawatt-2.1 (58). Twenty-two contigs of two Planctomycetales bins with 1,949,258 bp were selected as targets of a mapping of processed 454 and MiSeq reads within Geneious R8 (Biomatters, Auckland, New Zealand). In addition, MiSeq raw reads were processed with tools of the BBMap package (version 32.27; http://sourceforge.net/projects/bbmap/). Contigs of the two bins were extended by read mapping using 454 and MiSeq quality-controlled reads and then de novo assembled by Geneious R8 (Biomatters, Auckland, New Zealand). The assembly was improved by 14 rounds of read mapping with BBMap (version 32.27), with assembly of the mapping reads in SPAdes 3.5 taking the actual assembly as trusted and binning of contigs with Metawatt 2.1. When the improvement in the assembly became zero, the MiSeq read data set was trimmed with a different stringency and then used for the next mapping. Almost all bin information was assembled in one contig. Finally, assemblies were based on the Newbler or the first Geneious assembly as the trusted assembly to correct errors introduced in the assembly process. The comparison of the final assemblies revealed a linear presentation of a circular genome with different start points.
Manual visual inspection of mapping results of 454 and MiSeq reads obtained with Geneious R8 and with the Burrows-Wheeler Aligner (BWA) algorithm (Sequencher 5.3; Gene Codes, Ann Arbor, MI, USA) revealed questionable regions in the genome. These regions were repetitive elements which were identified using REPuter (59) and dot plot visualization in Geneious R8 (Biomatters, Auckland, New Zealand). Flanking sequences of repetitive elements were manually identified in the visualization of sequence diversity of mapped reads. This indicated a false-positive assembly of two or more repetitive elements into one in the contig. Sequence comparison using dot plot visualization identified repetitive elements of the draft genome also in two small contigs of the 454 assembly assigned by Metawatt 2.1 to the OP3 LiM bin. Both contigs were de novo assembled from 454 reads that mapped to the Newbler contigs using Sequencher 5.3 and Geneious R8 in several mapping rounds to verify and extent the Newbler assembly for these two contigs. In silico read walking from one flanking sequence to the other site across the repetitive elements failed due to read length shortage (repetitive elements were longer than 454 reads) and additional repetitive elements within the genetic content between the repetitive elements.
To clarify the physical sequence order around the triple repetitive element with a second repetitive element within the region, combinatory PCRs were performed with primers developed with Primer3 v.4.0.0 and located on the flanking sites of repetitive elements. PCR conditions were 4 min at 94°C, 41 cycles of 94°C for 1 min, 55°C for 1 min and 72°C for 4 min, and finally 72°C for 10 min. Amplicons were purified using a PCR purification kit or from agarose gels using a gel extraction kit (both from Qiagen, Hilden, Germany) and sequenced as described above. Sequences of amplicons were obtained using 60 cycles at 96°C for 10 s and 58°C for 5 s with ramping of 1°C per s, and 60°C for 4 min using BigDye Terminator chemistry (Applied Biosystems, Foster City, CA, USA) and analyzed on a 3130xl genetic analyzer (Applied Biosystems). Based on in silico read mappings and in vitro amplicons and their sequences, the genetic content was assembled between the repetitive elements and integrated into the large contig, thus providing a closed genome of OP3 LiM. The OP3 LiM genome was verified by mapping processed reads onto the OP3 LiM genome in Geneious R9 and visual inspection.
Genome annotation.
The OP3 LiM genome was annotated using several pipelines and manual annotations. The NCBI Prokaryotic Genome Annotation (60) was refined using JCoast (61) and Geneious with results of an in-house annotation based on GenDB (62), Rapid Annotations using Subsystems Technology (RAST) (63), and online resources of NCBI. CRISPR structures were searched using CRISPRFinder (64). Artemis release 16.0.0 (65) was applied for the nucleotide composition. RNAmmer 1.2 was used to predict 5S, 16S, and 23S rRNA genes in the genome sequence (66). The number of tRNA genes was identified using ARAGON v1.2.38 (67).
Linearization at ori.
The OP3 LiM genome was circularized and linearized at the origin of replication (ori), with the start codon of the chromosomal replication initiator protein DnaA gene as base 1. The GC skew (68, 69) and the pattern of ORF orientation supported this decision, as analyzed with GenSkew (http://genskew.csb.univie.ac.at) and ORF prediction programs. However, the genome contains a homopolymer of 21 guanines as a potential telomere.
Metaproteomic analysis.
From two cultures representing biological replicates, cell pellets were obtained by differential centrifugation (10kS cells, 100S aggregates, and 100S cells). They were resuspended in TE buffer (10 mM Tris-HCl, 10 mM EDTA [pH 7.5], containing cOmplete protease inhibitor [Roche]) and extracted by sonication (3 times for 30 s each). The protein content was determined by photometric measurement of the absorption at 595 nm using Nanoquant (Carl Roth, Karlsruhe, Germany). To increase the protein concentration, necessary for direct loading on a gel, samples of 10kS cells and 100S aggregates were concentrated in a vacuum centrifuge. Proteins of the samples were separated by size by using SDS-PAGE. Each gel lane was sliced into 10 equal pieces, and the proteins were digested with trypsin. Peptides were eluted in an ultrasonic bath for 15 min, concentrated, and finally purified with ZipTips with C18 resin (Millipore, Billerica, MA, USA). The peptide mix was separated on a nano-high-performance liquid chromatograph (nano-HPLC) (Easy-nLCII HPLC system; Thermo Fisher Scientific, Dreieich, Germany) and analyzed by tandem mass spectrometry (MS/MS) in an LTQ Orbitrap Velos mass spectrometer (Thermo Fisher Scientific) (70). For protein identification, tandem mass spectra were extracted using the Sorcerer-SEQUEST platform, version 3.5 (Sage-N Research, Milpitas, CA), searching the MS/MS data against a metaproteome database generated from the metagenomes and common laboratory contaminants. Search parameters were a parent ion tolerance of 10 ppm, fragment ion mass tolerance of 1.00 Da, and oxidation of methionine (15.99 Da) as variable modification (maximum of three modifications per peptide). MS/MS-based peptide and protein identifications were validated with Scaffold V4.4.8 (Proteome Software, Portland, OR). Peptide false discovery rates (FDRs) were set to 1%, and protein FDRs were set to 5% throughout all experiments. Quantification of each protein was performed as total spectral counts (TSC) for each protein, because the molecular weight of ORFs varied largely.
Data availability.
The genome of OP3 LiM was deposited under number CP019384 in GenBank. The mass spectrometry proteomics data were deposited with the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifiers PXD025008 and 10.6019/PXD025008.
ACKNOWLEDGMENTS
We thank the Max Planck-Genome-center Cologne (http://mpgc.mpipz.mpg.de/home/) for performing NGS sequencing.
This study was funded by the Max Planck Society. Jana Kizina and Almud Lonsing are members of the International Max Planck Research School of Marine Microbiology (MarMic).
Concept: J.H., investigation: J.K., S.F.A.J., G.A.M., A.L., C.P., A.K., R.S.-M., K.S., J.H., data curation: J.K., S.M., J.H., resources and supervision: R.S.-M., E.R., S.L., M.R., T.S., J.H., visualization and writing original draft: J.K., J.H., writing - review and editing: all authors.
Footnotes
Supplemental material is available online only.
Contributor Information
Jens Harder, Email: jharder@mpi-bremen.de.
Nicole R. Buan, University of Nebraska-Lincoln
REFERENCES
- 1.Bada JL. 2013. New insights into prebiotic chemistry from Stanley Miller's spark discharge experiments. Chem Soc Rev 42:2186–2196. 10.1039/c3cs35433d. [DOI] [PubMed] [Google Scholar]
- 2.Robertson MP, Joyce GF. 2012. The origins of the RNA world. Cold Spring Harb Perspect Biol 4:a003608. 10.1101/cshperspect.a003608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martin WF, Sousa FL, Lane N. 2014. Evolution. Energy at life's origin. Science 344:1092–1093. 10.1126/science.1251653. [DOI] [PubMed] [Google Scholar]
- 4.Bengtson S. 2002. Origins and early evolution of predation, p 289–317. In Kowalewski M, Kelley PH (ed), The fossil record of predation. The Paleontological Society papers 8. The Paleontological Society, McLean, VA. 10.1017/S1089332600001133. [DOI] [Google Scholar]
- 5.Jurkevitch E. 2007. A brief history of short bacteria: a chronicle of Bdellovibrio (and like organisms) research, p 1–9. In Jurkevitch E (ed), Predatory prokaryotes. Springer, Berlin, Germany. [Google Scholar]
- 6.Pérez J, Moraleda-Muñoz A, Marcos-Torres FJ, Muñoz-Dorado J. 2016. Bacterial predation: 75 years and counting! Environ Microbiol 18:766–779. 10.1111/1462-2920.13171. [DOI] [PubMed] [Google Scholar]
- 7.Jurkevitch E. 2007b. Predatory behaviors in bacteria-diversity and transitions. Microbe 2:67–73. 10.1128/microbe.2.67.1. [DOI] [Google Scholar]
- 8.Soo RM, Woodcroft BJ, Parks DH, Tyson GW, Hugenholtz P. 2015. Back from the dead; the curious tale of the predatory cyanobacterium Vampirovibrio chlorellavorus. PeerJ 3:e968. 10.7717/peerj.968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tudor JJ, McCann MP. 2007. Genomic analysis and molecular biology of predatory prokaryotes, p 153–189. In Jurkevitch E (ed), Predatory prokaryotes. Springer, Berlin, Germany. [Google Scholar]
- 10.Jurkevitch E, Davidov Y. 2006. Phylogenetic Diversity and Evolution of Predatory Prokaryotes. P 11–56. In Jurkevitch E (ed), Predatory prokaryotes. Springer, Berlin, Germany. [Google Scholar]
- 11.Mahmoud KK, Koval SF. 2010. Characterization of type IV pili in the life cycle of the predator bacterium Bdellovibrio. Microbiology (Reading) 156:1040–1051. 10.1099/mic.0.036137-0. [DOI] [PubMed] [Google Scholar]
- 12.Wang Z, Kadouri DE, Wu M. 2011. Genomic insights into an obligate epibiotic bacterial predator: Micavibrio aeruginosavorus ARL-13. BMC Genomics 12:453. 10.1186/1471-2164-12-453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koval SF, Hynes SH, Flannagan RS, Pasternak Z, Davidov Y, Jurkevitch E. 2013. Bdellovibrio exovorus sp. nov., a novel predator of Caulobacter crescentus. Int J Syst Evol Microbiol 63:146–151. 10.1099/ijs.0.039701-0. [DOI] [PubMed] [Google Scholar]
- 14.Ganuza E, Sellers CE, Bennett BW, Lyons EM, Carney LT. 2016. A novel treatment protects Chlorella at commercial scale from the predatory bacterium Vampirovibrio chlorellavorus. Front Microbiol 7:848. 10.3389/fmicb.2016.00848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bedree JK, Bor B, Cen L, Edlund A, Lux R, McLean JS, Shi W, He X. 2018. Quorum sensing modulates the epibiotic-parasitic relationship between Actinomyces odontolyticus and its Saccharibacteria epibiont, a Nanosynbacter lyticus strain, TM7x. Front Microbiol 9:2049. 10.3389/fmicb.2018.02049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guerrero R, Pedros-Alio C, Esteve I, Mas J, Chase D, Margulis L. 1986. Predatory prokaryotes: predation and primary consumption evolved in bacteria. Proc Natl Acad Sci USA 83:2138–2142. 10.1073/pnas.83.7.2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moreira D, Zivanovic Y, López-Archilla AI, Iniesto M, López-Garcia P. 2021. Reductive evolution and unique predatory mode in the CPR bacterium Vampirococcus lugosii. Nature Comm 12:2454. 10.1038/s41467-021-22762-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, Cheng JF, Darling A, Malfatti S, Swan BK, Gies EA, Dodsworth JA, Hedlund BP, Tsiamis G, Sievert SM, Liu WT, Eisen JA, Hallam SJ, Kyrpides NC, Stepanauskas R, Rubin EM, Hugenholtz P, Woyke T. 2013. Insights into the phylogeny and coding potential of microbial dark matter. Nature 499:431–436. 10.1038/nature12352. [DOI] [PubMed] [Google Scholar]
- 19.Rotaru AE, Schauer R, Probian C, Mussmann M, Harder J. 2012. Visualization of candidate division OP3 cocci in limonene-degrading methanogenic cultures. J Microbiol Biotechnol 22:457–461. 10.4014/jmb.1110.10055. [DOI] [PubMed] [Google Scholar]
- 20.Harder J, Foss S. 1999. Anaerobic formation of the aromatic hydrocarbon p-cymene from monoterpenes by methanogenic enrichment cultures. Geomicrobiol J 16:295–305. 10.1080/014904599270550. [DOI] [Google Scholar]
- 21.Raskin L, Stromley JM, Rittmann BE, Stahl DA. 1994. Group-specific 16S rRNA hybridization probes to describe natural communities of methanogens. Appl Environ Microbiol 60:1232–1240. 10.1128/aem.60.4.1232-1240.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patel GB, Sprott GD, Humphrey RW, Beveridge TJ. 1986. Comparative analyses of the sheath structures of Methanothrix concilii GP6 and Methanospirillum hungatei strains GP1 and JF1. Can J Microbiol 32:623–631. 10.1139/m86-117. [DOI] [Google Scholar]
- 23.Kubota K, Imachi H, Kawakami S, Nakamura K, Harada H, Ohashi A. 2008. Evaluation of enzymatic cell treatments for application of CARD-FISH to methanogens. J Microbiol Methods 72:54–59. 10.1016/j.mimet.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 24.Yamaguchi T, Kawakami S, Hatamoto M, Imachi H, Takahashi M, Araki N, Yamaguchi T, Kubota K. 2015. In situ DNA-hybridization chain reaction (HCR): a facilitated in situ HCR system for the detection of environmental microorganisms. Environ Microbiol 17:2532–2541. 10.1111/1462-2920.12745. [DOI] [PubMed] [Google Scholar]
- 25.Mikucki JA, Liu Y, Delwiche M, Colwell FS, Boone DR. 2003. Isolation of a methanogen from deep marine sediments that contain methane hydrates, and description of Methanoculleus submarinus sp. nov. Appl Environ Microbiol 69:3311–3316. 10.1128/AEM.69.6.3311-3316.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fuchs BM, Glöckner FO, Wulf J, Amann R. 2000. Unlabeled helper oligonucleotides increase the in situ accessibility to 16S rRNA of fluorescently labeled oligonucleotide probes. Appl Environ Microbiol 66:3603–3607. 10.1128/AEM.66.8.3603-3607.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wolański M, Donczew R, Zawilak-Pawlik A, Zakrzewska-Czerwińska J. 2014. oriC-encoded instructions for the initiation of bacterial chromosome replication. Front Microbiol 5:735. 10.3389/fmicb.2014.00735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frank AC, Lobry JR. 1999. Asymmetric substitution patterns: a review of possible underlying mutational or selective mechanisms. Gene 238:65–77. 10.1016/s0378-1119(99)00297-8. [DOI] [PubMed] [Google Scholar]
- 29.Haugen P, Bhattacharya D, Palmer JD, Turner S, Lewis LA, Pryer KM. 2007. Cyanobacterial ribosomal RNA genes with multiple, endonuclease-encoding group I introns. BMC Evol Biol 7:159. 10.1186/1471-2148-7-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nesbø CL, Doolittle WF. 2003. Active self-splicing group I introns in 23S rRNA genes of hyperthermophilic bacteria, derived from introns in eukaryotic organelles. Proc Natl Acad Sci USA 100:10806–10811. 10.1073/pnas.1434268100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown CT, Hug LA, Thomas BC, Sharon I, Castelle CJ, Singh A, Wilkins MJ, Wrighton KC, Williams KH, Banfield JF. 2015. Unusual biology across a group comprising more than 15% of domain Bacteria. Nature 523:208–211. 10.1038/nature14486. [DOI] [PubMed] [Google Scholar]
- 32.Raghavan R, Miller SR, Hicks LD, Minnick MF. 2007. The unusual 23S rRNA gene of Coxiella burnetii: two self-splicing group I introns flank a 34-base-pair exon, and one element lacks the canonical omegaG. J Bacteriol 189:6572–6579. 10.1128/JB.00812-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Flood BE, Fliss P, Jones DS, Dick GJ, Jain S, Kaster AK, Winkel M, Mußmann M, Bailey J. 2016. Single-cell (meta-)genomics of a dimorphic Candidatus Thiomargarita nelsonii reveals genomic plasticity. Front Microbiol 7:603. 10.3389/fmicb.2016.00603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Horvath P, Barrangou R. 2010. CRISPR/Cas, the immune system of bacteria and archaea. Science 327:167–170. 10.1126/science.1179555. [DOI] [PubMed] [Google Scholar]
- 35.Mao X, Zhang H, Yin Y, Xu Y. 2012. The percentage of bacterial genes on leading versus lagging strands is influenced by multiple balancing forces. Nucleic Acids Res 40:8210–8218. 10.1093/nar/gks605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen X, Zhang J. 2013. Why are genes encoded on the lagging strand of the bacterial genome? Genome Biol Evol 5:2436–2439. 10.1093/gbe/evt193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Veyron S, Peyroche G, Cherfils J. 2018. FIC proteins: from bacteria to humans and back again. Pathog Dis 76:fty012. 10.1093/femspd/fty012. [DOI] [PubMed] [Google Scholar]
- 38.Harms A, Stanger FV, Scheu PD, de Jong IG, Goepfert A, Glatter T, Gerdes K, Schirmer T, Dehio C. 2015. Adenylylation of gyrase and Topo IV by FicT toxins disrupts bacterial DNA topology. Cell Rep 12:1497–1507. 10.1016/j.celrep.2015.07.056. [DOI] [PubMed] [Google Scholar]
- 39.Smith KS, Ingram-Smith C. 2007. Methanosaeta, the forgotten methanogen? Trends Microbiol 15:150–155. 10.1016/j.tim.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 40.Chien IC, Meschke JS, Gough HL, Ferguson JF. 2013. Characterization of persistent virus-like particles in two acetate-fed methanogenic reactors. PLoS One 8:e81040. 10.1371/journal.pone.0081040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bor B, McLean JS, Foster KR, Cen L, To TT, Serrato-Guillen A, Dewhirst FE, Shi W, He X. 2018. Rapid evolution of decreased host susceptibility drives a stable relationship between ultrasmall parasite TM7x and its bacterial host. Proc Natl Acad Sci USA 115:12277–12282. 10.1073/pnas.1810625115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bor B, Collins AJ, Murugkar PP, Balasubramanian S, To TT, Hendrickson EL, Bedree JK, Bidlack FB, Johnston CD, Shi W, McLean JS, He X, Dewhirst FE. 2020. Insights obtained by culturing Saccharibacteria with their bacterial hosts. J Dent Res 99:685–694. 10.1177/0022034520905792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fontes CM, Gilbert HJ. 2010. Cellulosomes: highly efficient nanomachines designed to deconstruct plant cell wall complex carbohydrates. Annu Rev Biochem 79:655–681. 10.1146/annurev-biochem-091208-085603. [DOI] [PubMed] [Google Scholar]
- 44.Valentine RC, Shapiro BM, Stadtman ER. 1968. Regulation of glutamine synthetase. XII. Electron microscopy of the enzyme from Escherichia coli. Biochemistry 7:2143–2152. 10.1021/bi00846a017. [DOI] [PubMed] [Google Scholar]
- 45.Ronchi P, Mizzon G, Machado P, D’Imprima E, Best BT, Cassella L, Schnorrenberg S, Motero MG, Jechlinger M, Ephrussi A, Leptin M, Mahamid J, Schwab Y. 2021. High-precision targeting workflow for volume electron microscopy. J Cell Biol 220:e202104069. 10.1083/jcb.202104069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cohen M, Santarella R, Wiesel N, Mattaj I, Gruenbaum Y. 2008. Electron microscopy of lamin and the nuclear lamina in Caenorhabditis elegans. Methods Cell Biol 88:411–429. 10.1016/S0091-679X(08)00421-4. [DOI] [PubMed] [Google Scholar]
- 47.Martín-Platero AM, Valdivia E, Maqueda M, Martínez-Bueno M. 2007. Fast, convenient, and economical method for isolating genomic DNA from lactic acid bacteria using a modification of the protein “salting-out” procedure. Anal Biochem 366:102–104. 10.1016/j.ab.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 48.Hahnke RL, Harder J. 2013. Phylogenetic diversity of Flavobacteria isolated from the North Sea on solid media. Syst Appl Microbiol 36:497–504. 10.1016/j.syapm.2013.06.006. [DOI] [PubMed] [Google Scholar]
- 49.Pernthaler A, Pernthaler J, Amann R. 2004. Sensitive multi-color fluorescence in situ hybridization for the identification of environmental microorganisms, p 711–726. In Kowalchuk G, de Bruijn FJ, Head IM, Akkermans DL, van Elsas JD (ed), Molecular microbial Ecology Manual, 2nd ed, vol 1. Kluwer Academic Publishers, Dordrecht, Boston, London. [Google Scholar]
- 50.Schermelleh L, Heintzmann R, Leonhardt H. 2010. A guide to super-resolution fluorescence microscopy. J Cell Biol 190:165–175. 10.1083/jcb.201002018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer ML, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu P, Begley RF, Rothberg JM. 2005. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376–380. 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Richter M, Rosselló-Móra R. 2009. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci USA 106:19126–19131. 10.1073/pnas.0906412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cox MP, Peterson DA, Biggs PJ. 2010. SolexaQA: at-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics 11:485. 10.1186/1471-2105-11-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Crusoe MR, Alameldin HF, Awad S, Boucher E, Caldwell A, Cartwright R, Charbonneau A, Constantinides B, Edvenson G, Fay S, Fenton J, Fenzl T, Fish J, Garcia-Gutierrez L, Garland P, Gluck J, González I, Guermond S, Guo J, Gupta A, Herr JR, Howe A, Hyer A, Härpfer A, Irber L, Kidd R, Lin D, Lippi J, Mansour T, McA'Nulty P, McDonald E, Mizzi J, Murray KD, Nahum JR, Nanlohy K, Nederbragt AJ, Ortiz-Zuazaga H, Ory J, Pell J, Pepe-Ranney C, Russ ZN, Schwarz E, Scott C, Seaman J, Sievert S, Simpson J, Skennerton CT, Spencer J, Srinivasan R, Standage D, et al. 2015. The khmer software package: enabling efficient sequence analysis. F1000Res 4:900. 10.12688/f1000research.6924.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gurevich A, Saveliev V, Vyahhi N, Tesler G. 2013. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29:1072–1075. 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Strous M, Kraft B, Bisdorf R, Tegetmeyer HE. 2012. The binning of metagenomic contigs for microbial physiology of mixed cultures. Front Microbiol 3:410. 10.3389/fmicb.2012.00410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, Giegerich R. 2001. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res 29:4633–4642. 10.1093/nar/29.22.4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Angiuoli SV, Gussman A, Klimke W, Cochrane G, Field D, Garrity G, Kodira CD, Kyrpides N, Madupu R, Markowitz V, Tatusova T, Thomson N, White O. 2008. Toward an online repository of Standard Operating Procedures (SOPs) for (meta)genomic annotation. Omics 12:137–141. 10.1089/omi.2008.0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Richter M, Lombardot T, Kostadinov I, Kottmann R, Duhaime MB, Peplies J, Glöckner FO. 2008. JCoast—a biologist-centric software tool for data mining and comparison of prokaryotic (meta)genomes. BMC Bioinformatics 9:177. 10.1186/1471-2105-9-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Meyer F, Goesmann A, McHardy AC, Bartels D, Bekel T, Clausen J, Kalinowski J, Linke B, Rupp O, Giegerich R, Pühler A. 2003. GenDB—an open source genome annotation system for prokaryote genomes. Nucleic Acids Res 31:2187–2195. 10.1093/nar/gkg312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O. 2008. The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Grissa I, Vergnaud G, Pourcel C. 2007. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res 35:W52–57. 10.1093/nar/gkm360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B. 2000. Artemis: sequence visualization and annotation. Bioinformatics 16:944–945. 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- 66.Lagesen K, Hallin P, Rødland EA, Staerfeldt HH, Rognes T, Ussery DW. 2007. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35:3100–3108. 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Laslett D, Canback B. 2004. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res 32:11–16. 10.1093/nar/gkh152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bao Q, Tian Y, Li W, Xu Z, Xuan Z, Hu S, Dong W, Yang J, Chen Y, Xue Y, Xu Y, Lai X, Huang L, Dong X, Ma Y, Ling L, Tan H, Chen R, Wang J, Yu J, Yang H. 2002. A complete sequence of the T. tengcongensis genome. Genome Res 12:689–700. 10.1101/gr.219302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Necşulea A, Lobry JR. 2007. A new method for assessing the effect of replication on DNA base composition asymmetry. Mol Biol Evol 24:2169–2179. 10.1093/molbev/msm148. [DOI] [PubMed] [Google Scholar]
- 70.Heinz E, Williams TA, Nakjang S, Noël CJ, Swan DC, Goldberg AV, Harris SR, Weinmaier T, Markert S, Becher D, Bernhardt J, Dagan T, Hacker C, Lucocq JM, Schweder T, Rattei T, Hall N, Hirt RP, Embley TM. 2012. The genome of the obligate intracellular parasite Trachipleistophora hominis: new insights into microsporidian genome dynamics and reductive evolution. PLoS Pathog 8:e1002979. 10.1371/journal.ppat.1002979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lane DJ, Pace B, Olsen GJ, Stahl DA, Sogin ML, Pace NR. 1985. Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc Natl Acad Sci USA 82:6955–6959. 10.1073/pnas.82.20.6955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lane DJ. 1991. 16S/23S rRNA sequencing, p 115–175. In Stackebrandt E, Goodfellow M (ed), Nucleic acid techniques in bacterial systematics. John Wiley and Sons, New York, NY. [Google Scholar]
- 73.Amann RI, Krumholz L, Stahl DA. 1990. Fluorescent-oligonucleotide probing of whole cells for determinative, phylogenetic, and environmental studies in microbiology. J Bacteriol 172:762–770. 10.1128/jb.172.2.762-770.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Daims H, Brühl A, Amann R, Schleifer KH, Wagner M. 1999. The domain-specific probe EUB338 is insufficient for the detection of all Bacteria: development and evaluation of a more comprehensive probe set. Syst Appl Microbiol 22:434–444. 10.1016/S0723-2020(99)80053-8. [DOI] [PubMed] [Google Scholar]
- 75.Stahl DA, Amann R. 1991. Development and application of nucleic acid probes in bacterial systematics, p 205–248. In Stackebrandt E, Goodfellow M (ed), Nucleic acid techniques in bacterial systematics. John Wiley and Sons Ltd., New York, NY. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 to S19 and Table S1. Download aem.02407-21-s0001.pdf, PDF file, 8.2 MB (8.2MB, pdf)
Data Availability Statement
The genome of OP3 LiM was deposited under number CP019384 in GenBank. The mass spectrometry proteomics data were deposited with the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifiers PXD025008 and 10.6019/PXD025008.