

Abstract
Background:
There is strong rationale for interference with T cell co-stimulation in IgG4-related disease (IgG4-RD), but the literature to evaluate this is limited to a single case report.
Methods:
We conducted a ten-subject proof-of-concept trial of abatacept in active IgG4-RD. All subjects met the ACR/EULAR Classification Criteria for IgG4-RD. Subjects received subcutaneous abatacept 125 mg weekly for 24 weeks. Concurrent glucocorticoid treatment was permitted but if used had to be discontinued by week four. The primary endpoint, complete remission at 24 weeks, was defined as an IgG4-RD Responder Index score of 0. Peripheral blood mononuclear cells were collected at baseline, four weeks, and 12 weeks. B and T cell subsets were quantified using a 25-parameter flow cytometry panel.
Findings:
The subjects’ median age was 68 years; seven subjects were male and nine were Caucasian. Baseline organ involvement was diverse with a median of 5 organs affected at the time of enrollment. The median serum IgG4 concentration was 597 mg/dL (IQR 304–913 mg/dL). Three subjects received concomitant prednisone at baseline. Six subjects (60%) had a disease response by week 12, five of whom maintained this response at week 24. Abatacept was stopped in the remaining five subjects (50%) due to flare (N = 1) or lack of response by week 12 (N = 4). Three subjects (30%) achieved the primary endpoint.
Baseline proportions of unswitched memory B cells predicted responsiveness to abatacept. Reductions in serum IgE, circulating plasmablasts, and activated type 2 T follicular helper (TFH2) cells correlated with response to treatment. One adverse event (grade two thrombocytopenia) was attributed to abatacept.
Interpretation:
Abatacept was associated with variable treatment responses in IgG4-RD. Half of the subjects achieved sustained treatment responses to abatacept alone, without glucocorticoids. Correlates of clinical response included reductions in serum IgE, circulating plasmablasts, and activated TFH2 cells. Response to abatacept was predicted by higher proportions of unswitched memory B cells at baseline.
INTRODUCTION
IgG4-related disease (IgG4-RD) is a chronic, immune-mediated, fibrotic disease characterized by tumor-like masses composed of infiltrating immune cells and deposited extra-cellular matrix.1 The condition was first recognized in the context of autoimmune pancreatitis but has since been shown to affect a variety of organ systems. IgG4-RD most commonly involves the major salivary glands, lacrimal glands, orbits, bile ducts, retroperitoneal tissues, aorta, kidneys, and lungs.2 In contrast to many other fibrotic diseases, IgG4-RD demonstrates marked clinical responsiveness to high-dose glucocorticoids and B cell depleting therapy.3,4 However, without effective treatment, insidious progression of this disease often causes irreversible end-organ damage and failure.5
Some of the immunologic mechanisms underlying IgG4-RD have been elucidated.1 Reports have implicated activated B cells and IgG4-expressing plasmablasts as sources of autoantibody production and potential drivers of this disease.1,6–8 The relevance of B cells to IgG4-RD is supported by the clinical improvement observed following B cell depleting therapy.4 T follicular helper (TFH) cells, which interact directly with B cells and induce affinity maturation and isotype switching, are consistently expanded in the blood of subjects with IgG4-RD. TFH cells also accumulate in the tissue sites of disease and track with various markers of disease activity, such as extent of organ involvement and serum IgG4 concentration.9–11 Thus, there exists a strong mechanistic rationale for targeting costimulatory signals needed for T cell activation and T-B collaboration as a therapeutic approach in IgG4-RD.
Glucocorticoids are regarded as the standard of care for IgG4-RD.3 Glucocorticoid responsiveness is essentially universal in IgG4-RD.12 Most subjects achieve disease remissions on these drugs, but the relapse rate upon tapering or discontinuation of glucocorticoids is high.3 Moreover, the frequent presence of comorbidities, especially diabetes mellitus related to underlying pancreatic damage, makes long-term maintenance with glucocorticoids an untenable treatment option for many subjects.3 Currently, there are no approved therapies for IgG4-RD.
A single case report from Japan provided intriguing evidence for abatacept in a subject with multi-organ IgG4-RD whose disease was refractory to B cell depletion.14 Abatacept is a fusion protein of human IgG1 and the extracellular domain of CTLA4. The drug binds to CD80 and CD86 on antigen presenting cells, thereby blocking co-stimulatory signal delivery to naïve T cells. In this proof-of-concept trial, we investigated abatacept in a group of subjects with active IgG4-RD.
METHODS
The trial was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines. The study protocol was approved by the institutional review board of Mass General Brigham. All subjects provided written informed consent prior to enrollment. The investigators designed the study and gathered and analyzed the data. Bristol-Myers Squibb provided abatacept and financial support for the trial.
Study Design and Enrollment Criteria
We conducted a prospective, open-label, proof-of-concept study to evaluate the potential efficacy of abatacept in subjects with active IgG4-RD (ClinicalTrials.gov NCT03669861). The CONSORT diagram is shown in Figure 1. Subjects were required to be at least 18 years of age, to fulfill the 2019 ACR/EULAR classification criteria for IgG4-RD, and to have active disease as defined by an IgG4-RD Responder Index (RI) ≥ 2 at the time of screening (Table 1).12 The IgG4-RD RI was designed to quantify longitudinal changes in disease activity.13 Subjects may or may not have received previous IgG4-RD treatment. Disease duration at baseline was defined by the date of symptom onset or radiologic abnormality attributed to IgG4-RD. Any concomitant synthetic disease-modifying anti-rheumatic drugs (DMARDs), e.g., methotrexate or mycophenolate mofetil, were discontinued at screening. Subjects could have been treated with B cell targeted therapies (i.e., rituximab, ocrelizumab, or obexelimab) as long as the last administration was ≥ six months prior to study enrollment. Subjects with IgG4-related renal disease and a serum creatinine >2.0 mg/dL at screening were excluded.
Figure 1: CONSORT diagram.
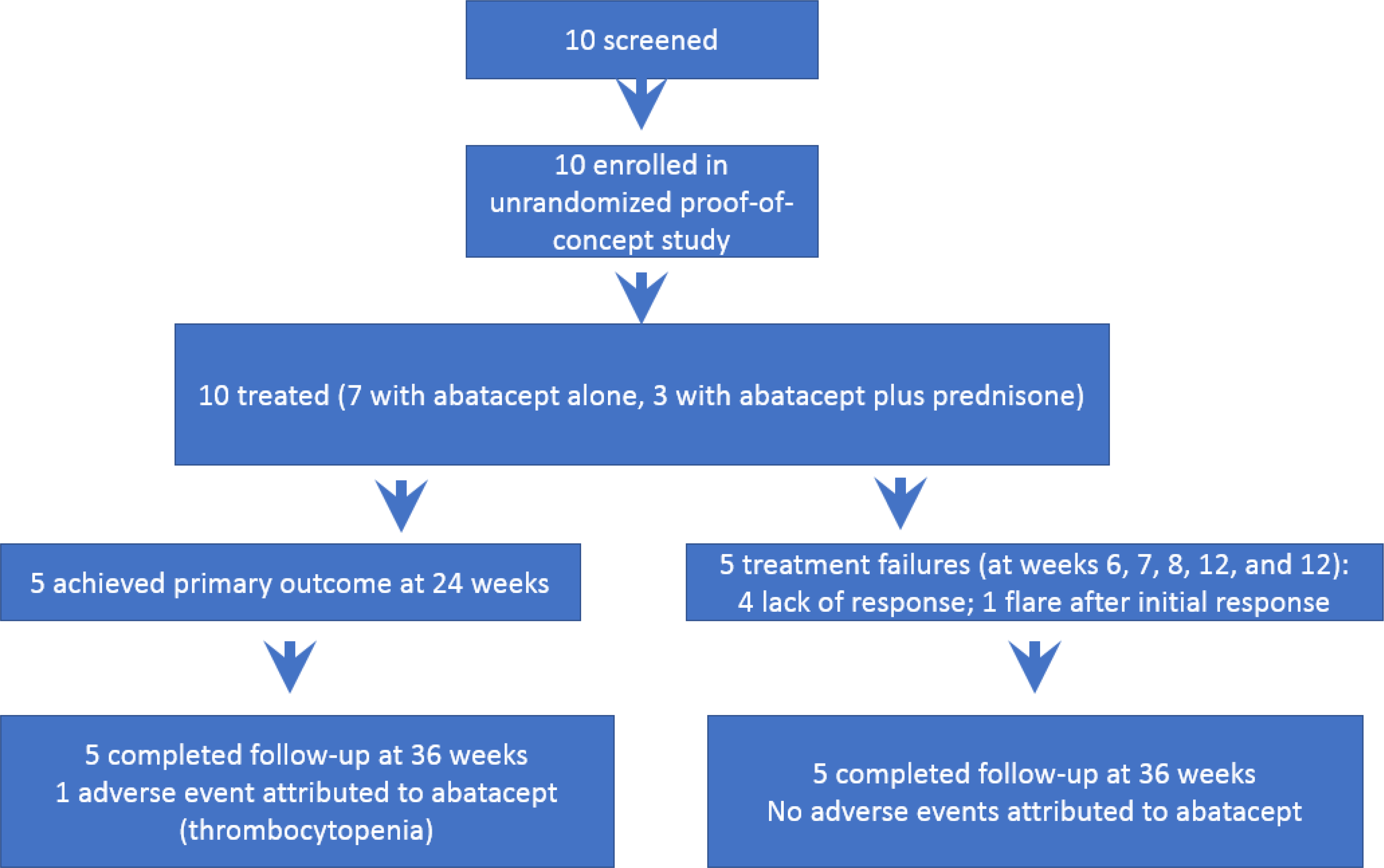
Table 1:
Baseline Characteristics
| IgG4-RD Subjects (n = 10) | |
|---|---|
| Age, years (median) | 68 |
| Male gender, n (%) | 7 (70%) |
| Female gender, n (%) | 3 (30%) |
| Caucasian, n (%) | 9 (90%) |
| Hispanic, n (%) | 1 (10%) |
| Duration of IgG4-RD, months, median (IQR) | 34.5 (3–68) |
| Number of active organs affected, median (IQR) | 5 (3–5) |
| IgG4-RD Responder Index score, median (IQR) | 6 (4–8) |
| Serum IgG4, mg/dL, median (IQR) | 597 (304 to 913) |
| Serum IgE, IU/mL, median (IQR) | 481 (195–802) |
Treatment Regimen
The treatment protocol consisted of subcutaneous abatacept 125 mg administered at baseline and then weekly for a total of 24 doses. Glucocorticoids could be used at baseline at the discretion of the investigators, but all subjects treated with glucocorticoids had to discontinue these drugs entirely by week four.
Disease Assessments and Study Endpoints
After screening and baseline, subjects were evaluated at weeks 1, 2, 4, 8, 12, 16, 20, and 24 (treatment phase). Additional safety follow-up visits occurred at week 28 and 36. The primary study endpoint, complete remission of IgG4-RD at 24 weeks, was defined as: 1) IgG4-RD RI score of 0; 2) prednisone dose of 0 mg/day beyond week four; and, 3) no recurrence of disease activity since the baseline visit. Imaging studies, guided by organ involvement at baseline, were repeated at 24 weeks in order to assess disease activity at the time of the primary endpoint. Imaging studies could also be performed at other timepoints at the discretion of the investigating physician. Recurrent disease activity was defined by an increase in the IgG4-RD RI that required therapy outside the trial protocol. Subjects unable to discontinue glucocorticoids by week four or who required reinstitution of glucocorticoid therapy at any time during the study were counted as treatment failures but could remain in the study at the discretion of the investigator. If the IgG4-RD RI had not improved by week eight or if new or progressive organ dysfunction developed after week four, subjects were deemed treatment failures and could begin glucocorticoid or alternative immunosuppressive therapy at the investigator’s discretion.
Disease response at 12 and 24 weeks was assessed as a secondary outcome measure. Disease response was defined as: 1) improvement of ≥ 1 point in the IgG4-RD RI score over baseline; 2) no glucocorticoid use following the week four visit; and, 3) no disease flares as assessed by the IgG4-RD RI. Disease response at week 4 was not assessed because patients could be treated with prednisone until that time. Disease response at week 36 was not assessed because the study medication was discontinued by week 24 and disease response patterns had already been defined at 24 weeks. The other secondary outcome measures assessed were time to remission, number of disease flares, IgG4 and IgE concentrations, peripheral eosinophilia, serum complement (C3 and C4) values, and the physician global assessment of disease activity. Data on the cumulative glucocorticoid doses and the Glucocorticoid Toxicity Index were collected but not reported because of the short glucocorticoid taper employed in the trial. Data on the Symptom Severity Index, an exploratory assessment of a disease-specific patient-reported outcome measure, will be reported separately.
Mechanistic Studies
To understand how a clinical response to abatacept may predict changes in immunologic parameters, we used flow cytometry to quantify the relative proportions of immune cells in the blood of subjects both before and during treatment. Given the known mechanism of interference with T cell co-stimulation by abatacept, we focused our studies on relevant adaptive immune cells. Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll density gradient centrifugation within 6 hours of phlebotomy and cryopreserved in vapor-phase liquid nitrogen. Blood was collected from each subject at weeks 0, 4, and 12, corresponding with clinical blood draws. Samples were studied using multi-color flow cytometry to quantify the proportions of B and T cell subsets both before and after treatment with abatacept.
Because some subjects were treated concurrently with glucocorticoids at enrollment, the week 12 time point was used as a consistent post-treatment timepoint to compare to the baseline assessment across subjects. If the patient withdrew from the trial before week 12, we used the last timepoint prior to withdrawal. All flow cytometric analyses of the cell subsets are displayed as the percent change from baseline. Subjects were categorized as responders if they demonstrated either a partial or complete clinical response based on the IgG4-RD-RI, as defined above. Full details of the mechanistic study procedures, including a detailed gating strategy (Appendix p 3–5), are included in the supplement. The surface markers used to define each cell type are provided (Appendix p 4).
Statistical analysis
An intention-to-treat analysis was performed. All clinical data were analyzed using descriptive methods. Medians and inter-quartile ranges (IQR) were reported for continuous variables. Counts and percentages were reported for categorical variables. For flow cytometric analyses, subjects who experienced a disease response to abatacept (n = 6) were combined and compared to subjects with no clinical response (n = 4). Mann-Whitney test and linear regression were used for statistical analyses of flow cytometry data where appropriate.
RESULTS
Subjects.
Ten subjects were enrolled in the trial from December 5, 2018 to August 22, 2019. The last subject visit was conducted on April 9, 2020. Baseline characteristics are shown in Table 1. The median age of the cohort was 68 years. Seven subjects were male and nine were Caucasian. The median disease duration at the time of enrollment was 34.5 months (IQR 3–68 months). Three subjects had new-onset disease diagnosed in the preceding three months. The seven other subjects had been diagnosed with IgG4-RD at least two years before the baseline visit. Individual subject data are displayed in Table 2.
Table 2:
Individual patient data
| Subject | Sex | Age (years) | Disease Duration (months) | Organs Affected | Serum IgG4 (mg/dL) |
Serum IgE (IU/mL) |
Concurrent Prednisone Treatment | Complete Remission (Week 24) | Disease Response (Week 12) | Disease Response (Week 24) | ABA doses received (#) | Previous Treatment History | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (Baseline) | (Week 24) | (Baseline) | (Week 24) | |||||||||||
| 1 | F | 63 | 84 | Lacrimal, salivary, pancreas | 170 | NA | 127 | NA | No | No | No | No | 6 | Prednisone & RTX |
| 2 | M | 65 | 240 | Lungs, PVM, RPF, orbit, salivary | 787.4 | NA | 544 | NA | Yes | No | No | No | 7 | Prednisone, RTX, & ocrelizumab |
| 3 | M | 66 | 68 | Orbit, lungs, salivary, ENT, LN | 591.2 | 811.6 | 807 | 593 | No | Yes | Yes | Yes | 24 | Obexelimab, prednisone & RTX |
| 4 | M | 76 | 27 | Lungs, aorta, RP, pancreas | 298.4 | 163.3 | 802 | 470 | No | Yes | Yes | Yes | 24 | Prednisone & obexelimab |
| 5 | F | 73 | 25 | Orbit, lungs, salivary | 1359.5 | 1441.3 | 324 | 187 | No | No | Yes | Yes | 24 | RTX |
| 6 | M | 48 | 42 | Orbit, skin, salivary, LN, lungs | 602.7 | NA | 527 | NA | No | No | No | No | 12 | Prednisone & RTX |
| 7 | M | 69 | 52 | Lacrimal, salivary, pancreas, bile duct, kidney | 346.3 | 428.7 | 9,154 | 4,241 | No | Yes | Yes | Yes | 24 | Prednisone & RTX |
| 8 | F | 40 | 1 | Orbit, lungs, Salivary, ENT, LN | 304 | NA | 174 | NA | No | No | Yes | No | 12 | None |
| 9 | M | 69 | 2 | Lacrimal, salivary, kidney | 912.7 | NA | 434 | NA | Yes | No | No | No | 8 | None |
| 10 | M | 79 | 3 | Orbit, lung, lacrimal, salivary, pancreas | 1199.4 | 495.2 | 195 | 88 | Yes | No | Yes | Yes | 24 | None |
Key: ABA= abatacpet; RTX = rituximab; PVM = paravertebral mass; RPF = retroperitoneal fibrosis; LN = lymph node; ENT = ears, nose, throat
Baseline organ involvement and laboratory parameters
The median number of active organs affected was 5 (IQR 3–5). The median IgG4-RD RI score was 6 (IQR 4–8). As detailed in Table 2, nine subjects had salivary gland involvement, seven had lung involvement, six had orbital involvement, four had lacrimal gland involvement and four had pancreatic involvement. The median serum IgG4 concentration at baseline was 597 mg/dL (IQR 304–913 mg/dL; reference range 4–86 mg/dL). The median serum IgE concentration was 481 IU/mL (IQR 195–802 IU/mL; reference range < 100 IU/mL).
Previous treatment for IgG4-RD
Seven subjects had been previously treated with B cell-targeted therapy (six with rituximab, one with ocrelizumab, two with obexelimab). One subject (subject #2) had been responsive to rituximab, administered every six months, for 8 years, but had grown refractory to B cell depletion. That subject had also failed ocrelizumab one year prior to entry.
Efficacy assessment
Three subjects (subjects 2, 9, and 10) received prednisone combined with abatacept at baseline. This prednisone was tapered off by week 4 as required by the trial protocol. Six subjects achieved at least a partial disease response at week 12, and five of those maintained their disease responses at week 24. Only one subject who achieved a disease response at week 24 and complete remission was treated with prednisone. Among the 5 subjects who were treatment failures, the times to treatment failure were 6, 7, 12, 12, and 8 weeks. The primary outcome, complete remission at week 24, was achieved by three subjects (Table 2). Abatacept was stopped early in five subjects because of either a disease flare while on treatment (n = 1) or suboptimal response (n = 4). Physician Global Assessments corresponded well to changes in disease activity, rising with disease flares and treatment failures (data not shown). Data on the Symptom Severity Index are not shown.
Laboratory values
All ten participants had elevated serum IgG4 levels at baseline, the magnitude of which varied from 170 to 1,359 mg/dL (Table 2). Three of the six (50%) subjects with a disease response demonstrated a decline in serum IgG4 concentration (Table 2). One of the four (25%) subjects who did not achieve a disease response demonstrated a decline in serum IgG4, but that subject was treated with prednisone at baseline. The other three non-responders had increases in their serum IgG4 levels. All ten participants had elevated serum IgE at baseline. Serum IgE concentrations declined in all six subjects (100%) who demonstrated a disease response with abatacept (Tables 2 and 3). The baseline serum IgE declined from a median of 802 IU/mL to 470 IU/mL among the six responders. Among the four non-responders, there was no consistent pattern to serum IgE concentrations in follow-up. This analysis was confounded partly by prednisone use before measurement of IgE in follow-up.
Table 3:
Efficacy Results
| IgG4-RD subjects (n = 10) | |
|---|---|
| Complete remission at week 24, n (%) | 3 (30%) |
| Disease response at week 12, n (%) | 6 (60%) |
| Disease response at week 24, n (%) | 5 (50%) |
| Decline in serum IgG4 among responders, n (%) | 3/6 (50%) |
| Decline in serum IgE of responders, n (%) | 6/6 (100%) |
Five patients overall and three of the six responders had a peripheral eosinophilia at baseline. All three treatment responders demonstrated improvement in their peripheral eosinophilia during follow-up. Only two patients (both abatacept non-responders) had hypocomplementemia at baseline. Their hypocomplementemia did not respond clearly to abatacept treatment but eventually resolved once glucocorticoids were administered following treatment failure.
Effects of Abatacept on B and T Cell Compartments
We used a comprehensive flow cytometry panel to understand what changes among adaptive immune cells tracked with clinical responsiveness to abatacept. Naïve T cells are highly dependent on co-stimulatory signals for initial activation. By blocking co-stimulation and preventing the differentiation of naïve CD4+ T cells into other effector subsets, we observed a relative accumulation of naïve CD4+ T cells in the blood of most subjects following treatment with abatacept. Although this demonstrated the expected immunologic effect of therapy targeting co-stimulation, we observed no significant difference in changes in naïve CD4+ T cell proportions between the subjects who responded clinically to those who did not (Figure 2A). Among subjects who showed at least a partial clinical response to abatacept treatment, we observed a relative reduction in overall numbers of activated CD4+ T cells, using HLA-DR as a surface marker of recent activation (Figure 2B). This observation, however, did not achieve statistical significance (p = 0.11).
Figure 2: Immunologic effects of abatacept on B and T cells.
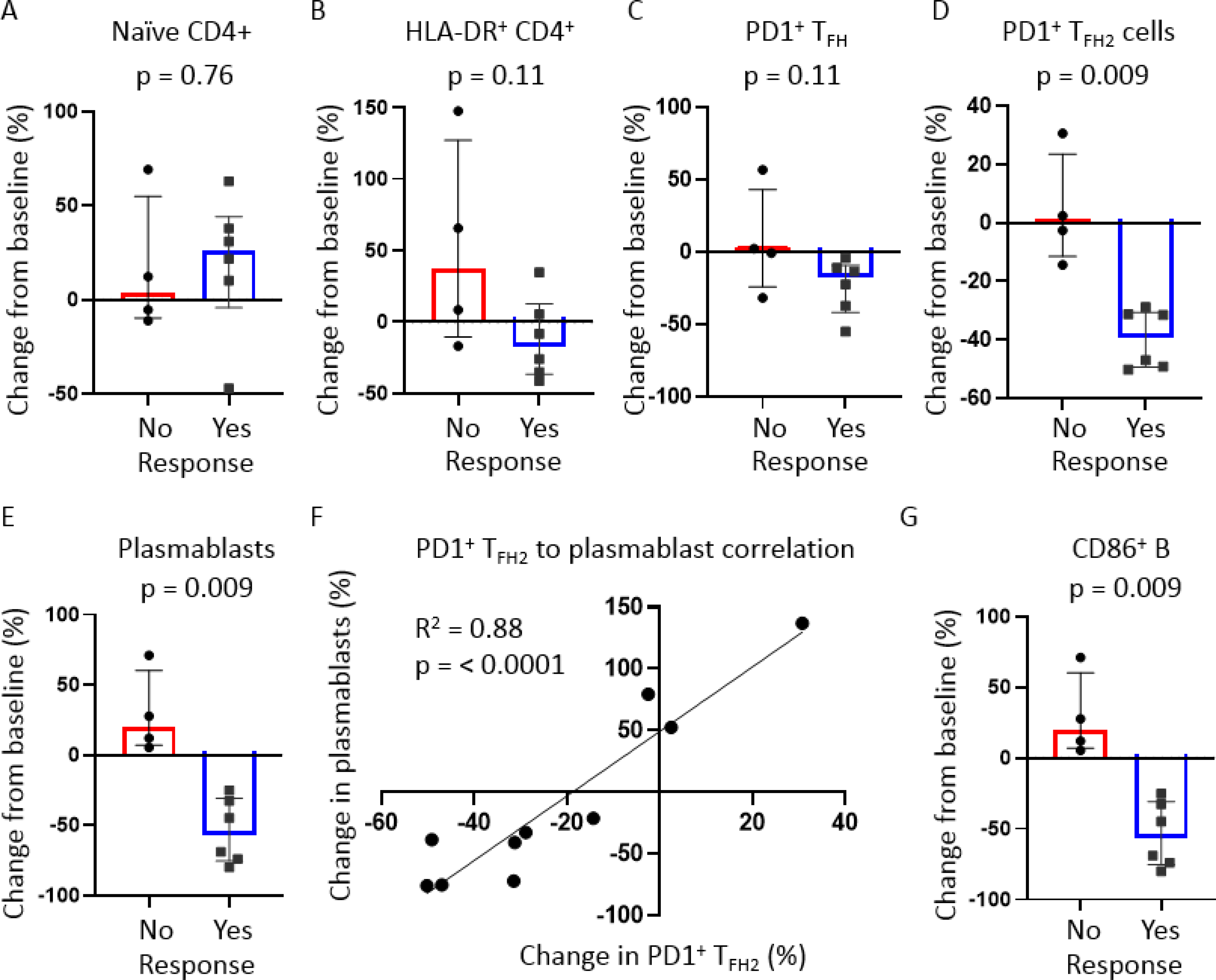
Dot plots of flow cytometry data displaying changes in relative proportions of respective T and B cell subsets from pre-to post-treatment time points. Blood samples from week 12 (or the latest time point prior to withdrawal if withdrawn prior to week 12) were used as post-treatment samples. Subjects were stratified based on clinical response to abatacept with ‘Yes’ indicating either a partial or complete clinical response. Dot plots displayed include A) naïve CD4+ T cells; B) Activated (HLA-DR+) CD4+ T cells; C) Effector (PD1+) circulating follicular helper T (TFH) cells; D) effector (PD1+) type 2 TFH; E) Plasmablasts (CD19+IgD−CD27+CD20LoCD38Hi); and G) CD86+ B cells. For all dot plot analyses, p-values were calculated by Mann-Whitney test. Bars represent medians and inter-quartile ranges. F) xy plot showing strong correlation between the pre- and post-treatment changes in plasmablasts and PD1+ TFH2 cells. p-value was calculated by linear regression. p-values <0.05 were considered significant.
Literature supports the efficacy of B cell depleting therapy in the treatment of IgG4-RD4 and an important role for TFH cells in orchestrating the B cell response seen in this disease9–11. We were therefore interested in understanding if interference of naïve CD4+ to TFH cell differentiation following treatment with abatacept tracked with clinical response and alterations in B cell differentiation. We observed a greater reduction in total activated circulating TFH cells among subjects with a disease response (Figure 2C), but this comparison also did not achieve statistical significance. However, when we examined changes in type 2 TFH cell proportions (type 2 TFH cells, or TFH2), which have more specifically been implicated in the pathogenesis of IgG4-RD, we found a consistent and marked decline in relative TFH2 cell numbers in the blood of clinical responders to abatacept compared to non-responders (Figure 2D). We did not observe any significant differences in proportions of other CD4+ T cell subsets including CD4+ cytotoxic T lymphocytes, regulatory T cells, and other TFH cell subsets (Appendix p 6). There was a trend to significance in the reduction of regulatory T cells among abatacept responders (Appendix p 6).
We observed a striking and essentially categorical reduction in relative numbers of circulating plasmablasts among the responders (Figure 2E). Changes in plasmablasts strongly correlated with changes in TFH2 cells within each subject following treatment with abatacept (R2 = 0.88, p = < 0.0001) (Figure 2F). We observed a marked reduction in circulating CD86+ B cells among the responders. In contrast, consistent expansion of these cells was observed among the subjects who did not achieve disease responses (Figure 2G). We did not observe any significant differences between groups in proportional changes in any other CD19+ B cell subset following treatment with abatacept (Appendix p 6).
Immunologic Predictors of Clinical Response to Abatacept
We observed significantly higher numbers of unswitched memory B cells in the baseline blood samples of responders (Figure 3A). To explore possible cellular predictors of abatacept responsiveness, we compared baseline proportions of relevant adaptive immune cells in the blood of responders and non-responders. Higher frequencies of activated TFH cells at baseline predict poor clinical responsiveness to treatment with abatacept in the context of type 1 diabetes mellitus.15 In contrast to the experience reported in type 1 diabetes, we did not observe an increase in activated circulating TFH cells in the blood of non-responders (Figure 3B–C). We also did not detect differences in baseline CD4+ naïve T cells, HLA-DR+ CD4+ T cells, CD19+ naïve B cells, or plasmablasts between the two subject groups (data not shown).
Figure 3: Immunologic predictors of clinical response to abatacept.
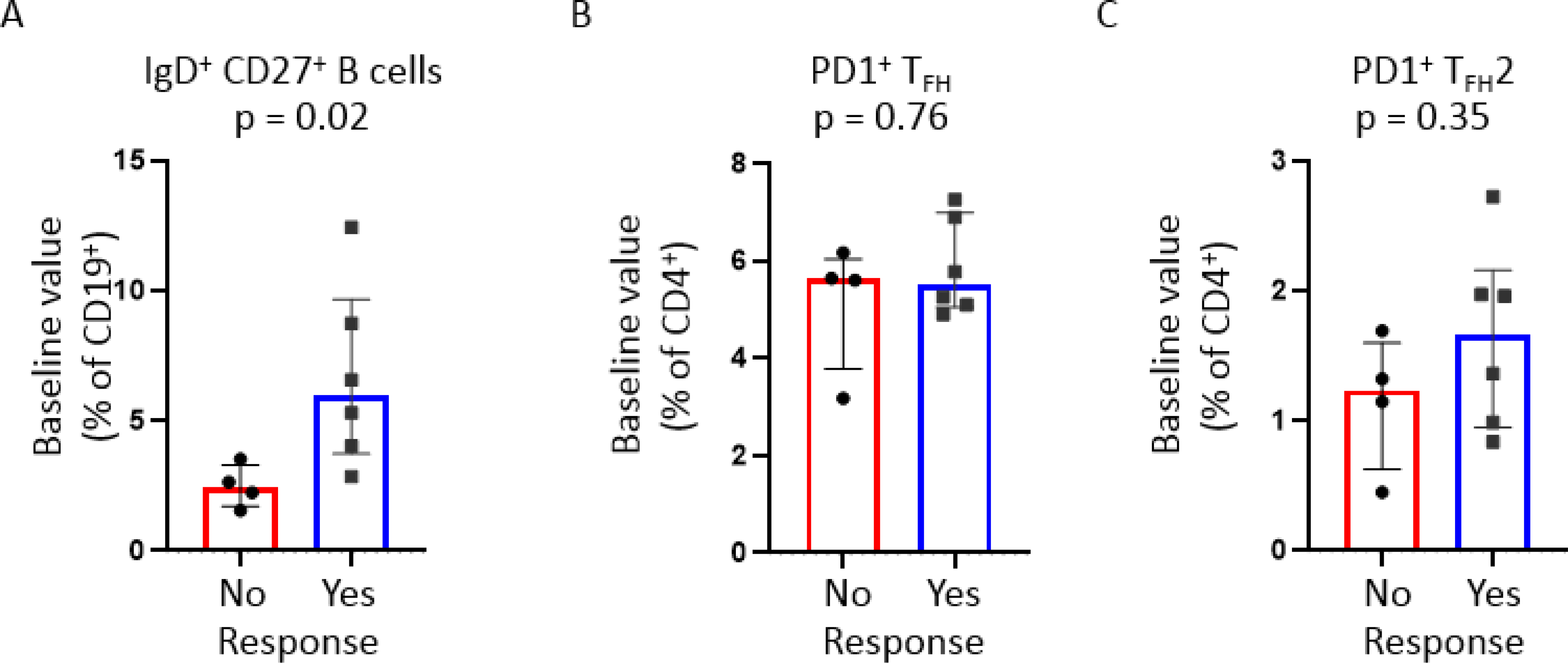
Dot plots of flow cytometry data displaying baseline relative proportions of respective B and T cell subsets. Subjects were stratified based on clinical response to abatacept with ‘Yes’ indicating either a partial or complete clinical response. Dot plots displayed include A) switched memory (CD19+IgD+CD27+ B cells); B) Effector (PD1+) circulating follicular helper T (TFH) cells; and C) effector (PD1+) type 2 TFH. p-values were calculated by Mann-Whitney test. Bars represent medians and inter-quartile ranges. p-values <0.05 were considered significant.
Adverse Events
Four adverse events were reported. One adverse event, thrombocytopenia, began to develop slowly after the initiation of abatacept in a patient who had not previously had thrombocytopenia. The platelet count declined at every trial visit until it became a grade 2 thrombocytopenia at 24 weeks. The platelet count rebounded promptly after abatacept was discontinued and the thrombocytopenia was attributed to abatacept. The other adverse events reported – myocardial infarction, episcleritis, and non-specific abdominal pain – were judged to be unrelated to abatacept.
DISCUSSION
In this prospective, single-arm, single-center, open-label proof-of-concept trial evaluating abatacept for the treatment of IgG4-RD, abatacept was associated with a variable treatment response. Some subjects had excellent clinical responses that correlated well with measures of immunologic activity, yet others showed no response to abatacept clinically or immunologically. Three subjects met the primary outcomes of complete remission at 24 weeks. Six subjects demonstrated disease responses at week 12, and five of those sustained these responses at week 24. Although abatacept was not effective in all subjects in this trial and some subjects clearly failed this treatment approach, it is important to note that all subjects except one who met the primary outcome and all subjects except one who achieved disease responses were treated only with abatacept and no glucocorticoids. Subjects who failed to achieve a disease response may have been on the higher end of the disease severity spectrum as suggested by lower proportions of unswitched memory B cells at baseline among non-responders. These data suggest that abatacept may have utility for remission induction without glucocorticoids in subjects with relatively mild disease, and also raises the possibility that it might have a role in the maintenance of disease remissions in some patients.
There are no approved therapies for the treatment of IgG4-RD and glucocorticoids are considered the first-line therapy across the world.3 Case series have suggested the efficacy of rituximab, but this therapy is not approved in IgG4-RD4. The published experience with B cell depletion suggests that this treatment approach is efficacious in a high percentage of subjects, but responses are not sustained and subjects invariably require additional courses of treatment.4 In the era of COVID-19, maintaining continuous B cell depletion can be a decidedly unfavorable outcome of the treatment of immune-mediated disease.15 Many of the subjects in our trial had been previously treated with rituximab with waning clinical response and one subject had become refractory altogether to B cell depletion. Thus, the modest clinical results in this trial are important, as they provide some evidence that another DMARD may be efficacious in this disease. Careful subject selection – perhaps on the basis of the finding of normal proportions of unswitched memory B cells at baseline – may be essential to future trial planning.
We identified important differences between the responders and non-responders in our mechanistic studies. Among the responders, higher pre-treatment proportions of unswitched memory B cells predicted clinical responsiveness to abatacept. In contrast, baseline values of activated TFH2 cells and plasmablasts were not helpful in predicting response. Among disease responders, there was a striking reduction in the relative numbers of both plasmablasts and TFH2 cells (but not other TFH subsets). From these studies, it is not clear why some subjects showed a compelling immunologic response to abatacept with the accumulation of naïve CD4+ T cells and reductions in the concentrations of activated CD4+ T cells, circulating TFH cells, and plasmablasts, whereas other subjects lacked such responses. The differences between responders and non-responders were most evident in the levels of plasmablasts and circulating TFH2. The fact that baseline plasmablast and TFH2 proportions did not predict response suggests that it is not simply the expansion of cell populations susceptible to interference with co-stimulation that determines response to abatacept. Greater understanding of these observations add important insights into the nature of IgG4-RD. Although pre-treatment expansion of TFH cells has been reported to predict poor clinical responsiveness to abatacept in the context of type 1 diabetes mellitus15, we did not observe the same phenomenon in this study in the setting of IgG4-RD.
This trial calls into question the value of serum IgG4 concentrations as a surrogate marker for disease activity. Although the serum IgG4 concentration was elevated in all 10 subjects at baseline, there was no consistent decline in serum IgG4 among subjects treated with abatacept. These results are consistent with our previous demonstration of a delayed and gradual decline in serum IgG4 following B cell depletion therapy, suggesting that in fact most IgG4 in the peripheral blood of patients with IgG4-RD is produced by long-lived plasma cells rather than more recently generated plasmablasts. These observations have the effect of limiting the utility of serum IgG4 concentrations as a disease biomarker in certain clinical situations – including costimulatory blockade - because long-lived plasma cells are unlikely to be affected by this intervention.6 Our data suggest that serum IgE concentrations may be a useful correlate of clinical response in patients who have elevations of IgE at baseline, but this question requires further study.
This trial is the first time a therapeutic approach targeting T cell activation has been evaluated in the treatment of IgG4-RD. Given abatacept’s focused mechanism of action and the mechanistic studies performed, this study helps elucidate the immunologic basis for the disease and whether B cells or T cells are the pathogenic driver of disease. The observations from this trial, consistent with the literature regarding the pathogenesis of IgG4-RD, support the notion that both B and T cells are of pathogenic relevance in IgG4-RD. The clinical manifestations of IgG4-RD in this trial were varied, with a median of five organs affected among trial participants. Most subjects exhibited classic head and neck manifestations; i.e., “Mikulicz disease” (enlargement in some combination of the lacrimal, parotid, and submandibular glands). More than half of the subjects in the trial also exhibited lung abnormalities, however, and there were also subjects with pancreatic, orbital, renal, bile duct, retroperitoneal, and aortic involvement.
The trial also has some limitations. Its open-label, proof-of-concept design and relatively small size could have led to investigator bias in the assessment of outcomes and a failure to appreciate the full potential efficacy of abatacept in IgG4-RD, particularly in disease subsets. Although the trial subjects manifested a wide array of organ system involvement and detailed mechanistic studies were performed, larger studies will be required to understand fully the role of co-stimulation blockade in IgG4-RD and to define responses is disease subsets.
In conclusion, the treatment response of IgG4-RD to abatacept in this proof-of-concept trial was variable. Half of the subjects achieved good treatment responses upon treatment with abatacept alone, without requiring concomitant glucocorticoids. Correlates of clinical response included reductions in serum IgE, circulating plasmablasts, and activated TFH2 cells. Response to abatacept was predicted by baseline proportions of unswitched memory B cells. Findings from this trial may inform futures studies of other immunological therapies for the treatment of IgG4-RD.
Supplementary Material
RESEARCH IN CONTEXT.
Evidence before this study:
There are no approved therapies for IgG4-related disease (IgG4-RD). Glucocorticoids are regarded as the standard of care for this disease. B cell depletion appears to be effective in a high percentage of subjects, but treatment responses are not sustained and subjects generally require additional courses of therapy. T follicular helper (TFH) cells are consistently expanded in the blood and accumulate in the tissues of subjects with IgG4-RD. Thus, there exists a strong mechanistic rationale for targeting costimulatory signals needed for T cell activation as a therapeutic approach in IgG4-RD. Abatacept is a fusion protein that blocks co-stimulation of T cells. In this proof-of-concept study, we evaluated abatacept in subjects with active IgG4-RD. We coupled our clinical outcomes assessments with flow cytometry-based mechanistic studies.
Added value of this study:
This is the first study to target T cells as a therapeutic approach in IgG4-RD. We observed variable treatment responses to abatacept but were able to identify both predictors and correlates of good treatment responses. Approximately half achieved good treatment responses upon treatment with abatacept alone. Correlates of clinical response included reductions in serum IgE, circulating plasmablasts, and activated TFH2 cells. Response to abatacept was predicted by higher proportions of unswitched memory B cells at baseline.
Implications of all the available evidence:
Findings from this trial may inform futures studies of other immunological therapies for the treatment of IgG4-RD.
Acknowledgments
This is an investigator-initiated trial funded by Bristol-Myers Squibb (BMS). This work was supported by the NIH Autoimmune Centers of Excellence UM1 AI1144295 to JHS and U19 AI 110495 to SP. CAP was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (K08 AR079615) and a Rheumatology Research Foundation Scientist Development Award.
Funding:
This is an investigator-initiated trial funded by Bristol-Myers Squibb (BMS) with additional support from NIH.
Role of the funding source:
BMS and NIH had no role in the writing of the manuscript or in the decision to submit for publication. BMS and NIH had no role in data collection, analysis, or interpretation; trial design; subject recruitment; or any aspect pertinent to the study.
Footnotes
Declaration of Interests
JHS and CAP have received grants and consulting fees (< $10,000) from BMS. ZSW has received grant funding from BMS to study abatacept in COVID-19, and consulting fees from Principia/Sanofi, VielaBio and Medpace, all unrelated to this study. MAM, LH, ADF, HL, HAC, SP declare no competing interests.
Data sharing:
De-identified subject-level data can be shared upon request.
Clinical trial registration number: NCT03669861
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1).Perugino CA, Stone JH. IgG4-related disease: an update on pathophysiology and implications for clinical care. Nat Rev Rheumatol. 2020. Dec;16(12):702–714. doi: 10.1038/s41584-020-0500-7. Epub 2020 Sep 16. [DOI] [PubMed] [Google Scholar]
- 2).Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012. Feb 9;366(6):539–51. doi: 10.1056/NEJMra1104650. [DOI] [PubMed] [Google Scholar]
- 3).Khosroshahi A, Wallace ZS, Crowe JL et al. Second International Symposium on IgG4-Related Disease. International Consensus Guidance Statement on the Management and Treatment of IgG4-Related Disease. Arthritis Rheumatol. 2015. Jul;67(7):1688–99. doi: 10.1002/art.39132. [DOI] [PubMed] [Google Scholar]
- 4).Carruthers MN, Topazian MD, Khosroshahi A et al. Rituximab for IgG4-related disease: a prospective, open-label trial. Ann Rheum Dis. 2015. Jun;74(6):1171–7. doi: 10.1136/annrheumdis-2014-206605. Epub 2015 Feb 9. [DOI] [PubMed] [Google Scholar]
- 5).Zhang W, Stone JH. Management of IgG4-related disease. Lancet Rheumatol 2019; 1: e55–65 [DOI] [PubMed] [Google Scholar]
- 6).Mattoo H, Mahajan VS, Della-Torre E et al. De novo oligoclonal expansions of circulating plasmablasts in active and relapsing IgG4-related disease. J Allergy Clin Immunol. 2014. Sep;134(3):679–87. doi: 10.1016/j.jaci.2014.03.034. Epub 2014 May 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7).Mattoo H, Mahajan VS, Maehara T et al. Clonal expansion of CD4(+) cytotoxic T lymphocytes in subjects with IgG4-related disease. J Allergy Clin Immunol. 2016. Sep;138(3):825–838. doi: 10.1016/j.jaci.2015.12.1330. Epub 2016 Mar 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8).Heeringa JJ, Karim AF, van Laar JAM et al. Expansion of blood IgG4+ B, TH2, and regulatory T cells in subjects with IgG4-related disease. J Allergy Clin Immunol. 2018. May;141(5):1831–1843.e10. doi: 10.1016/j.jaci.2017.07.024. Epub 2017 Aug 19. [DOI] [PubMed] [Google Scholar]
- 9).Akiyama M, Suzuki K, Yamaoka K et al. Number of Circulating Follicular Helper 2 T Cells Correlates With IgG4 and Interleukin-4 Levels and Plasmablast Numbers in IgG4-Related Disease. Arthritis Rheumatol. 2015. Sep;67(9):2476–81. doi: 10.1002/art.39209. [DOI] [PubMed] [Google Scholar]
- 10).Akiyama M, Yasuoka H, Yamaoka K et al. Enhanced IgG4 production by follicular helper 2 T cells and the involvement of follicular helper 1 T cells in the pathogenesis of IgG4-related disease. Arthritis Res Ther. 2016. Jul 13;18:167. doi: 10.1186/s13075-016-1064-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11).Maehara T, Mattoo H, Mahajan VS et al. The expansion in lymphoid organs of IL-4+ BATF+ T follicular helper cells is linked to IgG4 class switching in vivo. Life Sci Alliance. 2018. Jan;1(1):e201800050. doi: 10.26508/lsa.201800050. Epub 2018 Apr 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12).Wallace ZS, Naden RP, Chari S et al. The 2019 American College of Rheumatology/European League Against Rheumatism classification criteria for IgG4-related disease. Ann Rheum Dis. 2020. Jan;79(1):77–87. doi: 10.1136/annrheumdis-2019-216561. Epub 2019 Dec 3. [DOI] [PubMed] [Google Scholar]
- 13).Wallace ZS, Khosroshahi A, Carruthers MD, et al. An International, Multi-Specialty Validation Study of the IgG4-Related Disease Responder Index. Arthritis Care Res. 2018. Nov;70(11):1671–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14).Yamamoto M, Takahashi H, Takano K et al. Efficacy of abatacept for IgG4-related disease over 8 months. Ann Rheum Dis. 2016. Aug;75(8):1576–8. doi: 10.1136/annrheumdis-2016-209368. Epub 2016 May 4. [DOI] [PubMed] [Google Scholar]
- 15).Edner NM, Heuts F, Thomas N, Wang CJ, Petersone L, Kenefeck R, Kogimtzis A, Ovcinnikovs V, Ross EM, Ntavli E, Elfaki Y, Eichmann M, Baptista R, Ambery P, Jermutus L, Peakman M, Rosenthal M, Walker LSK. Follicular helper T cell profiles predict response to costimulation blockade in type 1 diabetes. Nat Immunol. 2020. Oct;21(10):1244–1255. doi: 10.1038/s41590-020-0744-z. Epub 2020 Aug 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16).Sparks JA, Wallace ZS, Seet AM et al. Associations of baseline use of biologic or targeted synthetic DMARDs with COVID-19 severity in rheumatoid arthritis: Results from the COVID-19 Global Rheumatology Alliance physician registry. Ann Rheum Dis. 2021. May 28:annrheumdis-2021–220418. doi: 10.1136/annrheumdis-2021-220418. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17).Wallace ZS, Mattoo H, Mahajan VS et al. Predictors of disease relapse in IgG4-related disease following rituximab. Rheumatology (Oxford). 2016. Jun;55(6):1000–8. doi: 10.1093/rheumatology/kev438. Epub 2016 Feb 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.