

Summary
Background
Patients with melanoma that progresses on ipilimumab and, if BRAFV600 mutant-positive, a BRAF or MEK inhibitor or both, have few treatment options. We assessed the efficacy and safety of two pembrolizumab doses versus investigator-choice chemotherapy in patients with ipilimumab-refractory melanoma.
Methods
We carried out a randomised phase 2 trial of patients aged 18 years or older from 73 hospitals, clinics, and academic medical centres in 12 countries who had confirmed progressive disease within 24 weeks after two or more ipilimumab doses and, if BRAFV600 mutant-positive, previous treatment with a BRAF or MEK inhibitor or both. Patients had to have resolution of all ipilimumab-related adverse events to grade 0–1 and prednisone 10 mg/day or less for at least 2 weeks, an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, and at least one measurable lesion to be eligible. Using a centralised interactive voice response system, we randomly assigned (1:1:1) patients in a block size of six to receive intravenous pembrolizumab 2 mg/kg or 10 mg/kg every 3 weeks or investigator-choice chemotherapy (paclitaxel plus carboplatin, paclitaxel, carboplatin, dacarbazine, or oral temozolomide). Randomisation was stratified by ECOG performance status, lactate dehydrogenase concentration, and BRAFV600 mutation status. Individual treatment assignment between pembrolizumab and chemotherapy was open label, but investigators and patients were masked to assignment of the dose of pembrolizumab. We present the primary endpoint at the prespecified second interim analysis of progression-free survival in the intention-to-treat population. This study is registered with ClinicalTrials.gov, number NCT01704287. The study is closed to enrolment but continues to follow up and treat patients.
Findings
Between Nov 30, 2012, and Nov 13, 2013, we enrolled 540 patients: 180 patients were randomly assigned to receive pembrolizumab 2 mg/kg, 181 to receive pembrolizumab 10 mg/kg, and 179 to receive chemotherapy. Based on 410 progression-free survival events, progression-free survival was improved in patients assigned to pembrolizumab 2 mg/kg (HR 0·57, 95% CI 0·45–0·73; p<0·0001) and those assigned to pembrolizumab 10 mg/kg (0·50, 0·39–0·64; p<0·0001) compared with those assigned to chemotherapy. 6-month progression-free survival was 34% (95% CI 27–41) in the pembrolizumab 2 mg/kg group, 38% (31–45) in the 10 mg/kg group, and 16% (10–22) in the chemotherapy group. Treatment-related grade 3–4 adverse events occurred in 20 (11%) patients in the pembrolizumab 2 mg/kg group, 25 (14%) in the pembrolizumab 10 mg/kg group, and 45 (26%) in the chemotherapy group. The most common treatment-related grade 3–4 adverse event in the pembrolizumab groups was fatigue (two [1%] of 178 patients in the 2 mg/kg group and one [<1%] of 179 patients in the 10 mg/kg group, compared with eight [5%] of 171 in the chemotherapy group). Other treatment-related grade 3–4 adverse events include generalised oedema and myalgia (each in two [1%] patients) in those given pembrolizumab 2 mg/kg; hypopituitarism, colitis, diarrhoea, decreased appetite, hyponatremia, and pneumonitis (each in two [1%]) in those given pembrolizumab 10 mg/kg; and anaemia (nine [5%]), fatigue (eight [5%]), neutropenia (six [4%]), and leucopenia (six [4%]) in those assigned to chemotherapy.
Interpretation
These findings establish pembrolizumab as a new standard of care for the treatment of ipilimumab-refractory melanoma.
Introduction
In the past 4 years, melanoma treatment has remarkably improved because of the successful clinical development of therapies targeting the MAPK pathway and the development of immune checkpoint inhibitors that reactivate the anticancer immune response.1 The CTLA-4 inhibitor ipilimumab improved overall survival in two randomised trials leading to its regulatory approval for the treatment of advanced melanoma in many countries.2,3 However, when disease progresses on or after ipilimumab, and BRAF inhibitor-based therapy if disease is BRAFV600 mutated (observed in approximately 40–50% of melanomas4), standard systemic treatment options are limited to cytotoxic chemotherapy or interleukin 2.
When T cells infiltrate tumours, they produce interferon-mediated signals that lead cells in the tumour microenvironment to express PD-L1, which inhibits T cells through engagement of the PD-1 receptor.5 Preclinical studies provided the rationale for the use of antibodies that block PD-1 or PD-L1 to treat cancer.6,7 Studies have provided clinical evidence of anti-tumour activity in melanoma, renal cell carcinoma, and lung and bladder cancer, among others.8–13
In a phase 1b trial, the anti-PD-1 antibody pembrolizumab (formerly MK-3475 and lambrolizumab) showed anti-tumour activity and a manageable safety profile in patients with advanced melanoma, including those with disease refractory to ipilimumab and, when indicated, BRAF inhibitors.10,11 Here, we present results of KEYNOTE-002, the definitive assessment of pembrolizumab in this population.
Methods
Study design and participants
KEYNOTE-002 is an international, randomised, controlled, phase 2 clinical trial comparing two pembrolizumab doses with investigator-choice chemotherapy. Eligible patients were aged 18 years or older and had histologically or cytologically confirmed unresectable stage III or stage IV melanoma not amenable to local therapy; confirmed disease progression14 within 24 weeks of the last ipilimumab dose (minimum two doses, 3 mg/kg once every 3 weeks); previous BRAF or MEK inhibitor therapy or both (if BRAFV600 mutant-positive); resolution or improvement of ipilimumab-related adverse events to grade 0–1 and prednisone dose 10 mg/day or less for at least 2 weeks before the first dose of study drug; Eastern Cooperative Oncology Group (ECOG) performance status 0 or 1; measurable disease per Response Evaluation Criteria in Solid Tumors, version 1.1 (RECIST v1.1); and values within the prespecified range for absolute neutrophil count (≥1500 cells per mL), platelets (≥100 000 cells per mL), haemoglobin (≥90 g/L), serum creatinine (≤1·5 upper limit of normal [ULN]), serum total bilirubin (≤1·5 ULN or direct bilirubin ≤ULN for patients with total bilirubin concentrations >1·5 ULN), aspartate and alanine aminotransferases (≤2·5 ULN or ≤5 ULN for patients with liver metastases), international normalised ratio or prothrombin time (≤1·5 ULN if not using anticoagulants), and activated partial thromboplastin time (≤1·5 ULN if not using anticoagulants). Patients had a washout period of at least 4 weeks between the last dose of the most recent therapy and the first dose of pembrolizumab. We excluded patients with known active brain metastases or carcinomatous meningitis, active autoimmune disease, active infection requiring systemic therapy, known history of HIV infection, active hepatitis B virus or hepatitis C virus infection, a history of grade 4 ipilimumab-related adverse events or grade 3 ipilimumab-related adverse events lasting longer than 12 weeks, or previous treatment with any other anti-PD-1 or anti-PD-L1 therapy.
The study was conducted in accordance with the protocol, good clinical practice standards, and the Declaration of Helsinki. The protocol and subsequent amendments were approved by the appropriate institutional review board or ethics committee at each participating institution. All patients provided voluntary written informed consent. An external data monitoring committee reviewed interim trial results to ensure patient safety and to recommend whether the trial should continue in accordance with the protocol.
Randomisation and masking
Patients were randomly assigned (1:1:1) to pembrolizumab 2 mg/kg or pembrolizumab 10 mg/kg given intravenously every 3 weeks or investigator-choice chemotherapy (paclitaxel plus carboplatin, paclitaxel, carboplatin [eliminated with protocol amendment one], dacarbazine, or oral temozolomide). Randomisation was stratified by ECOG performance status (0 vs 1), lactate dehydrogenase concentration (normal vs raised [ie, ≥110% upper limit of normal]), and BRAF status (wild type vs V600 mutant-positive). Block randomisation with a block size of six in each stratum was used. After all screening procedures were complete, a centralised interactive voice-response system with or without web functionality was used to allocate patients to treatment. Before entering information into the system, the investigator was to identify which chemotherapy regimen would be given in the event the patient was allocated to the chemotherapy group. Individual treatment assignment between pembrolizumab and chemotherapy was open label; investigators and patients were masked to assignment to pembrolizumab dose. A designated pharmacist at each site who was unmasked prepared the pembrolizumab dose so that it could be administered to the patient in a masked fashion. The sponsor was masked to all treatment assignments in the statistical analyses, as well as treatment-level analysis results. A non-Merck unmasked statistician generated interim analysis reports for external data monitoring committee review for the first and second interim analyses. After the second interim analysis, the external data monitoring committee indicated that the progression-free survival superiority objective had been met and recommended unmasking the study for reporting purposes but continuing the study for overall survival at the final analysis. At the second interim analysis and after the data monitoring committee’s recommendation, the treatment group was unmasked; investigators and patients remained masked to the pembrolizumab dose.
Procedures
Patients were given their first dose of study treatment within 3 days of treatment assignment. Chemotherapy was given per the approved product information and standard practice protocols at each institution and continued until evidence of disease progression. Patients in the chemotherapy group with documented and verified disease progression at or after week 12 who met the relevant eligibility criteria could cross over to receive pembrolizumab after a washout period of at least 28 days from the last dose of chemotherapy; patients who crossed over were randomly assigned to one of the two pembrolizumab doses in a double-blind manner. Pembrolizumab was continued until disease progression, unacceptable toxicity, consent withdrawal, physician decision, or other reason. If the week 12 scan showed disease progression, patients were allowed to continue treatment until progression was confirmed at a subsequent scan to account for atypical response patterns.14 For purposes of statistical analysis, progressive disease per RECIST v1.1 was defined as the first documented disease progression, irrespective of confirmation at a second assessment. The criteria for removal of a patient from the study and details of permitted interruptions of study treatment are available in the study protocol (appendix).
Tumour assessments were done before starting study treatment (baseline), at week 12, every 6 weeks through to week 48, and every 12 weeks thereafter. After study treatment discontinuation, patients were contacted every 12 weeks to assess survival. Adverse events, laboratory values, and vital signs were assessed regularly throughout the study and graded per the Common Terminology Criteria for Adverse Events, version 4.0. We assessed patient-reported health-related quality of life and disease symptoms using the European Organisation for the Research and Treatment of Cancer (EORTC) Quality of Life Questionnaire C30 (QLQ-C30).
Outcomes
The primary endpoint at the second interim analysis was progression-free survival, the time from randomisation to first documented disease progression per RECIST v1.1 by independent central review or death from any cause, whichever occurred first. All scans were evaluated by independent central review. The independent radiologists were masked to treatment assignments, identifying patient characteristics, and investigator-assessed findings. Overall survival, the time from randomisation to death from any cause, will be the primary endpoint at final analysis. Progression-free survival was also assessed by investigator review. Both progression-free survival assessed per RECIST v1.1 by investigator review and progression-free survival assessed per modified RECIST v1.1 (where confirmation of disease progression on a scan ≥4 weeks after initial evidence of disease progression was required14) by investigator review were done as sensitivity analyses. Secondary endpoints included the proportion of patients who had an objective response, the proportion of patients who had a complete or partial response as assessed per RECIST v1.1 by central review (patients without post-baseline disease assessment were considered to be non-responders); response duration, the time from best overall response of complete or partial response until disease progression; and safety. Prespecified exploratory endpoints included change from baseline to week 12 in the global health status and quality-of-life score of the EORTC QLQ-C30 questionnaire, with other functional and symptom subscales as supportive evidence. Per the prespecified analysis plan, the EORTC QLQ-C30 data collected at weeks 24 and 36 would be analysed only if the completion rate in the chemotherapy treatment group was higher than 50% at week 24 or 36.
Statistical analysis
The sample size of the study was determined on the basis of the overall survival endpoint at the final analysis. We estimated that with 370 events (deaths) among a total of 510 patients in the three treatment groups (170 patients per group), and assuming that the median overall survival in the chemotherapy group is 6 months and the hazard ratio between pembrolizumab and chemotherapy is 0·65, the study would have at least 90% power to detect the overall survival difference in at least one pembrolizumab group using the Hochberg testing procedure15 at a 2% α level (one sided).
Before recommendations from the external data monitoring committee to unmask the study treatment allocation, both the first and second interim analyses were done by an unmasked statistician. The first planned interim analysis on Nov 13, 2013, focused on detecting whether one pembrolizumab dose was inferior to the other, with the possible decision of discontinuing one pembrolizumab dose, and safety. The external data monitoring committee did not identify any concerns when they reviewed the first interim analysis results, and the study proceeded as planned. The second interim analysis (reported here) was planned to occur after at least 270 progression-free survival events (about 180 events between one pembrolizumab group and the chemotherapy group) occurred, at which time 210 deaths were anticipated. At the second interim analysis, the study had 92% power to detect a hazard ratio (HR) of 0·55 for progression-free survival at a one-sided α of 0·25% between one pembrolizumab group and the chemotherapy group. The study would be considered positive at this interim analysis if the one-sided p value for progression-free or overall survival was lower than 0·0025 in either pembrolizumab group compared with the chemotherapy group; based on 270 progression-free survival events, the observed HR that would meet the criterion for a positive trial was 0·66. Because the study completed enrolment 3 months earlier than expected and the death rate was lower than was expected, the timing of the second interim analysis was driven by the targeted number of deaths. Therefore, the number of progression-free survival events at the time of the second interim analysis was higher than that prespecified in the statistical analysis plan. As of the May 12, 2014, data cutoff date, there was 215 deaths and 410 progression-free survival events. Although overall survival was not the primary endpoint of the second interim analysis, comparisons for each pembrolizumab group compared with the chemotherapy group were tested at an α of 0·25%. Because the prespecified threshold for declaring superiority of pembrolizumab over chemotherapy was met for progression-free but not overall survival, the data monitoring committee recommended that the study could be unmasked but that follow-up should continue as planned. Final overall survival analysis will be done when 370 deaths have occurred.
All statistical analyses were done with SAS (version 9.3). We did all efficacy analyses in the intention-to-treat population. Safety was assessed in all patients who received at least one dose of study treatment. Kaplan-Meier methods were used to estimate progression-free and overall survival, duration of response, and time to the first treatment-related grade 3–4 adverse event. We analysed between-group comparisons of progression-free and overall survival with the stratified log-rank test. We calculated HRs and associated 95% CIs with a stratified Cox proportional hazard model with Efron’s method of tie handling.16 The stratified Miettinen and Nurminen method17 was used to compare the the proportion of patients achieving an objective response between treatment groups. The same stratification factors used for randomisation were used in all stratified efficacy analyses. A constrained longitudinal data analysis model was used to analyse differences in EORTC QLQ-C30 score changes between treatment groups. This study is registered with ClinicalTrials.gov, number NCT01704287.
Role of the funding source
Merck Sharp & Dohme, a subsidiary of Merck & Co, sponsored this study. Along with academic advisers, representatives of the sponsor participated in designing the study. Data collected by the investigators and their site personnel were analysed and interpreted by senior academic authors and representatives of the sponsor. All authors had full access to the data used to prepare this manuscript. The corresponding author wrote the first draft of the manuscript, with subsequent involvement of all authors, including those employed by the study sponsor. The corresponding author had the final responsibility to submit for publication.
Results
Between Nov 30, 2012, and Nov 13, 2013, we enrolled 540 patients from 73 sites in 12 countries (full site list available in the appendix). We randomly allocated patients to pembrolizumab 2 mg/kg (n=180), pembrolizumab to pembrolizumab 10 mg/kg (n=181), or chemotherapy (n=179 [42 patients to paclitaxel plus carboplatin, 28 to paclitaxel, 13 to carboplatin, 45 to dacarbazine, and 43 to temozolomide; eight did not receive treatment]; figure 1). Patient characteristics were well balanced across study groups (table 1). Visceral metastases (stage M1c) and elevated lactate dehydrogenase concentrations, poor prognostic factors for advanced melanoma, were observed in 446 (83%) and 218 (40%) of the 540 patients, respectively, at baseline. Most patients received two or more previous lines of therapy for advanced disease, including chemotherapy in just under half of the patients; a quarter of patients had received BRAF or MEK inhibitor (table 1). As of the May 12, 2014, data cutoff date, median follow-up duration was 10 months (IQR 8–12). Of the 179 patients allocated to the chemotherapy group, 86 (48%) crossed over to pembrolizumab treatment, with 46 randomly assigned to receive 2 mg/kg and 40 to receive 10 mg/kg.
Figure 1:
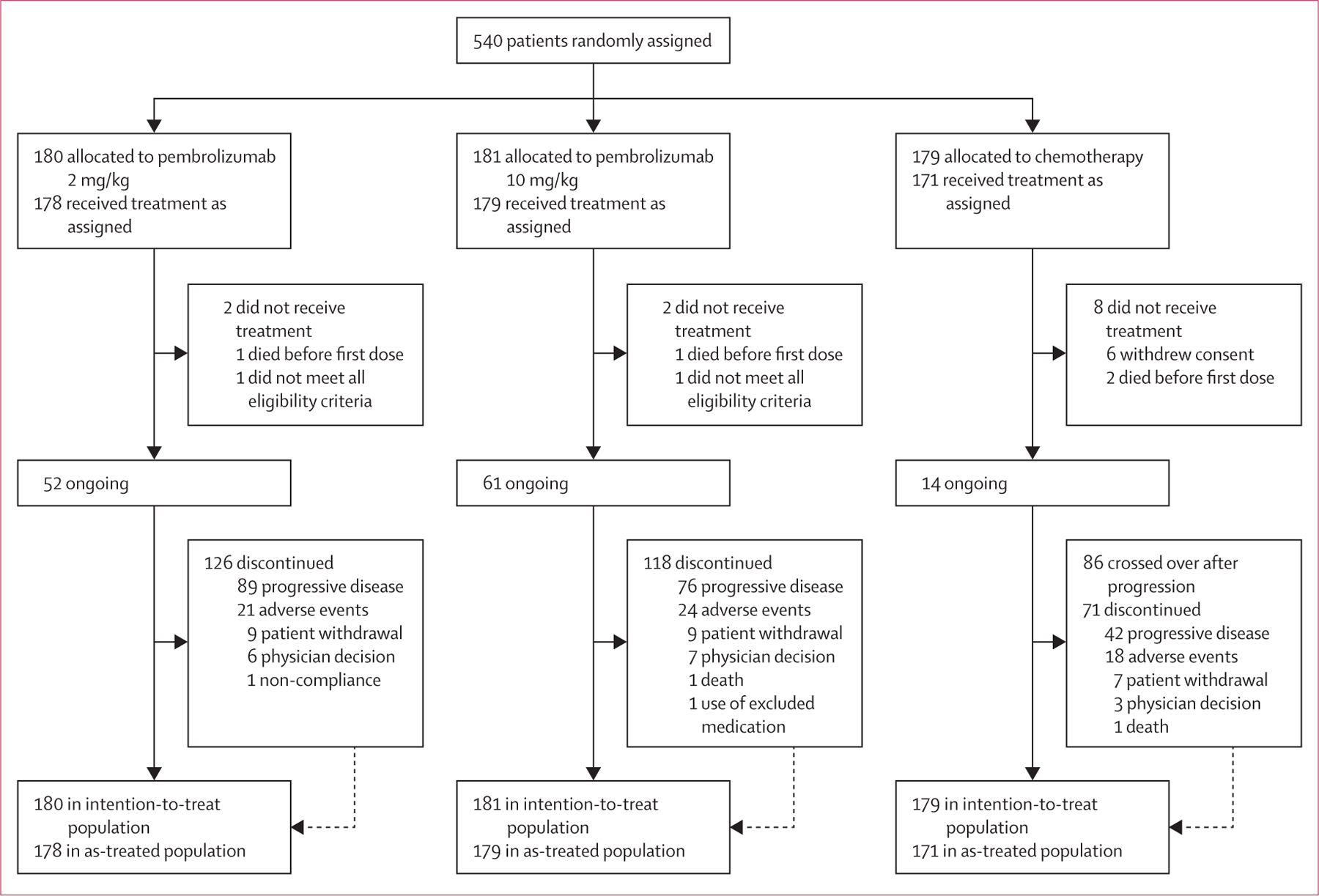
Study profile
Table 1:
Baseline characteristics in the intention-to-treat population
| Pembrolizumab 2 mg/kg (n=180) | Pembrolizumab 10 mg/kg (n=181) | Chemotherapy control (n=179) | |
|---|---|---|---|
| Median age (years) | 62 (15–87) | 60 (27–89) | 63 (27–87) |
| Men | 104 (58%) | 109 (60%) | 114 (64%) |
| Women | 76 (42%) | 72 (40%) | 65 (36%) |
| Ethnic origin | |||
| White | 176 (98%) | 179 (99%) | 172 (96%) |
| Other | 4 (2%) | 2 (1%) | 6 (3%) |
| Missing | 0 | 0 | 1 (<1%) |
| ECOG performance status | |||
| 0 | 98 (54%) | 98 (54%) | 99 (55%) |
| 1 | 80 (44%) | 83 (46%) | 80 (45%) |
| Missing | 2 (1%) | 0 | 0 |
| BRAFV600 status | |||
| Mutant | 44 (24%) | 40 (22%) | 41 (23%) |
| Wild type | 136 (76%) | 141 (78%) | 138 (77%) |
| Lactate dehydrogenase concentration | |||
| Normal | 99 (55%) | 105 (58%) | 107 (60%) |
| Raised | 77 (43%) | 73 (40%) | 68 (38%) |
| Unknown | 4 (2%) | 3 (2%) | 4 (2%) |
| Median size of target lesions | 95 (10–428) | 101 (12–560) | 102 (11–568) |
| M staging of extent of metastasis | |||
| M0 | 1 (<1%) | 1 (<1%) | 2 (1%) |
| M1a | 9 (5%) | 13 (7%) | 15 (8%) |
| M1b | 22 (12%) | 17 (9%) | 15 (8%) |
| M1c | 148 (82%) | 150 (83%) | 147 (82%) |
| Number of lines of previous systemic therapies | |||
| 0* | 1 (<1%) | 0 | 0 |
| 1 | 40 (22%) | 56 (31%) | 47 (26%) |
| 2 | 79 (44%) | 66 (36%) | 78 (44%) |
| ≥3 | 60 (33%) | 59 (33%) | 54 (30%) |
| Previous therapy | |||
| Ipilimumab | 180 (100%) | 181 (100%) | 179 (100%) |
| Interleukin 2 | 21 (12%) | 16 (9%) | 12 (7%) |
| Immunotherapy, excluding ipilimumab and interleukin 2 | 25 (14%) | 18 (10%) | 23 (13%) |
| Chemotherapy | 90 (50%) | 84 (46%) | 86 (48%) |
| BRAF or MEK inhibitor | 46 (26%) | 45 (25%) | 43 (24%) |
Data are median (range) or n (%). ECOG=Eastern Cooperative Oncology Group.
Patients with no previous systemic therapies received neoadjuvant or adjuvant therapy only.
Based on 410 total progression-free survival events (RECIST v1.1, central review), the study met the pre-specified criteria to show significant improvement in progression-free survival, with hazard ratios of 0·57 (95% CI 0·45–0·73) for pembrolizumab 2 mg/kg and 0·50 (95% CI 0·39–0·64) for 10 mg/kg compared with chemotherapy (p<0·0001 for both; table 2). We tested the assumption of proportional hazards with Schoenfeld residuals18 and the test was not rejected. Median progression-free survival was around the time of first scheduled post-baseline tumour assessment in all study groups because more than half of patients had progressed by week 12 (figure 2A, table 2). By 6 months, about a third of patients assigned to pembrolizumab were progression free (34% assigned to 2 mg/kg and 38% assigned to 10 mg/kg), whereas only 16% assigned to chemotherapy had not progressed (table 2). At 9 months, about a quarter of patients assigned to pembrolizumab were progression free (24% assigned to 2 mg/kg and 29% assigned to 10 mg/kg) compared with 8% assigned to chemotherapy (table 2).
Table 2:
Summary of efficacy in the intention-to-treat population
| Pembrolizumab 2 mg/kg (n=180) | Pembrolizumab 10 mg/kg (n=181) | Chemotherapy control (n=179) | |
|---|---|---|---|
| Progression-free survival assessed per RECIST v1.1, by independent central review | |||
| Number of events* | 129 (72%) | 126 (70%) | 155 (87%) |
| Median duration (months) | 2·9 (2·8–3·8) | 2·9 (2·8–4·7) | 2·7 (2·5–2·8) |
| Proportion progression free at 6 months | 34% (27–41) | 38% (31–45) | 16% (10–22) |
| Proportion progression free at 9 months | 24% (17–31) | 29% (23–37) | 8% (4–14) |
| Restricted mean duration based on 12 months of follow-up (months; post-hoc analysis) | 5·4 (4·7–6·0) | 5·8 (5·1–6·4) | 3·6 (3·2–4·1) |
| HR for death or disease progression,† pembrolizumab vs chemotherapy | 0·57 (0·45–0·73); p<0·0001‡ | 0·50 (0·39–0·64); p<0·0001‡ | Ref |
| HR for death or disease progression,† pembrolizumab 10 mg/kg vs 2 mg/kg | Ref | 0·91 (0·71–1·16)§ | ·· |
| Progression-free survival assessed per RECIST v1.1, by investigator review | |||
| Number of events* | 122 (68%) | 112 (62%) | 157 (88%) |
| Median duration (months) | 3·7 (2·9–5·4) | 5·4 (3·8–6·8) | 2·6 (2·4–2·8) |
| Proportion progression free at 6 months | 39% (32–46) | 45% (37–52) | 15% (10–21) |
| Proportion progression free at 9 months | 32% (25–40) | 36% (29–44) | 10% (6–15) |
| Restricted mean duration based on 12 months of follow-up (months; post-hoc analysis) | 5·8 (5·2–6·4) | 6·5 (5·8–7·1) | 3·7 (3·2–4·1) |
| HR for death or disease progression,† pembrolizumab vs chemotherapy | 0·49 (0·38–0·62); p<0·0001‡ | 0·41 (0·32–0·52); p<0·0001‡ | Ref |
| HR for death or disease progression,† pembrolizumab 10 mg/kg vs 2 mg/kg | Ref | 0·81 (0·63–1·05); p=0·12¶ | ·· |
| Progression-free survival assessed per modified RECIST v1.1, by investigator review | |||
| Number of events | 117 (65%) | 108 (60%) | 154 (86%) |
| Median duration (months) | 4·2 (3·1–6·2) | 5·6 (4·2–7·7) | 2·6 (2·5–2·8) |
| Proportion progression free at 6 months | 43% (35–50) | 48% (40–55) | 17% (12–23) |
| Proportion progression free at 9 months | 35% (27–43) | 38% (30–46) | 10% (6–16) |
| HR for death or disease progression,† pembrolizumab vs chemotherapy | 0·45 (0·35–0·.57); p<0·0001‡ | 0·39 (0·30–0·51); p<0·0001‡ | Ref |
| HR for death or disease progression,† pembrolizumab 10 mg/kg vs 2 mg/kg | Ref | 0·82 (0·63–1·07); p=0·15¶ | ·· |
| Best overall response assessed per RECIST v1.1, by independent central review | |||
| Complete response | 4 (2%) | 5 (3%) | 0 |
| Partial response | 34 (19%) | 41 (23%) | 8 (4%) |
| Stable disease | 32 (18%) | 31 (17%) | 33 (18%) |
| Progressive disease | 84 (47%) | 86 (48%) | 111 (62%) |
| Not evaluable|| | 26**(14%) | 18 (10%) | 27 (15%) |
| Overall response assessed per RECIST v1.1, by independent central review | |||
| Number of patients who responded (% [95% CI]) | 38 (21% [15–28]) | 46 (25% [19–32]) | 8 (4%; [2–9]) |
| Difference in overall response,†† pembrolizumab vs control | 13% (7–21); p<0・0001 | 18% (11–27); p<0・0001 | Ref |
| Diff erence in overall response,†† pembrolizumab 10 mg/kg vs 2 mg/kg | Ref | 6% (–3 to 14); p=0・21¶ | ・・ |
| Duration of response | |||
| Median duration (weeks) | Not reached (25 to not reached) | Not reached (36 to not reached) | 37 (12–41) |
Data in parentheses are % or 95% CIs, unless otherwise indicated. ECOG=Eastern HR=hazard ratio. RECIST v1.1=Response Evaluation Criteria in Solid Tumors, version 1.1.
A breakdown of progression-free survival events is provided in the appendix.
HRs and associated 95% CIs were based on Cox regression models with treatment as a covariate stratified by ECOG performance status (0 vs 1), lactate dehydrogenase concentration (normal vs raised), and BRAFV600 status (mutant vs wild type).
One-sided p value on the log-rank test.
No p value available for the comparison of pembrolizumab doses because the study was not powered for such a comparison.
Nominal p value, not powered for hypothesis testing.
Accounts for patients who withdrew consent, were withdrawn by the investigator, died, or started new anticancer therapy before the first tumour assessment and therefore did not have response evaluated, or patients with tumour assessments, but overall response based on the scans was not evaluable.
Includes one patient with no disease.
Difference calculated based on Miettinen and Nurminen method covariate stratified by ECOG performance status (0 vs 1), lactate dehydrogenase concentration (normal vs raised), and BRAFV600 status (mutant vs wild type).
Figure 2: Kaplan-Meier estimates of progression-free survival assessed in the intention-to-treat population.
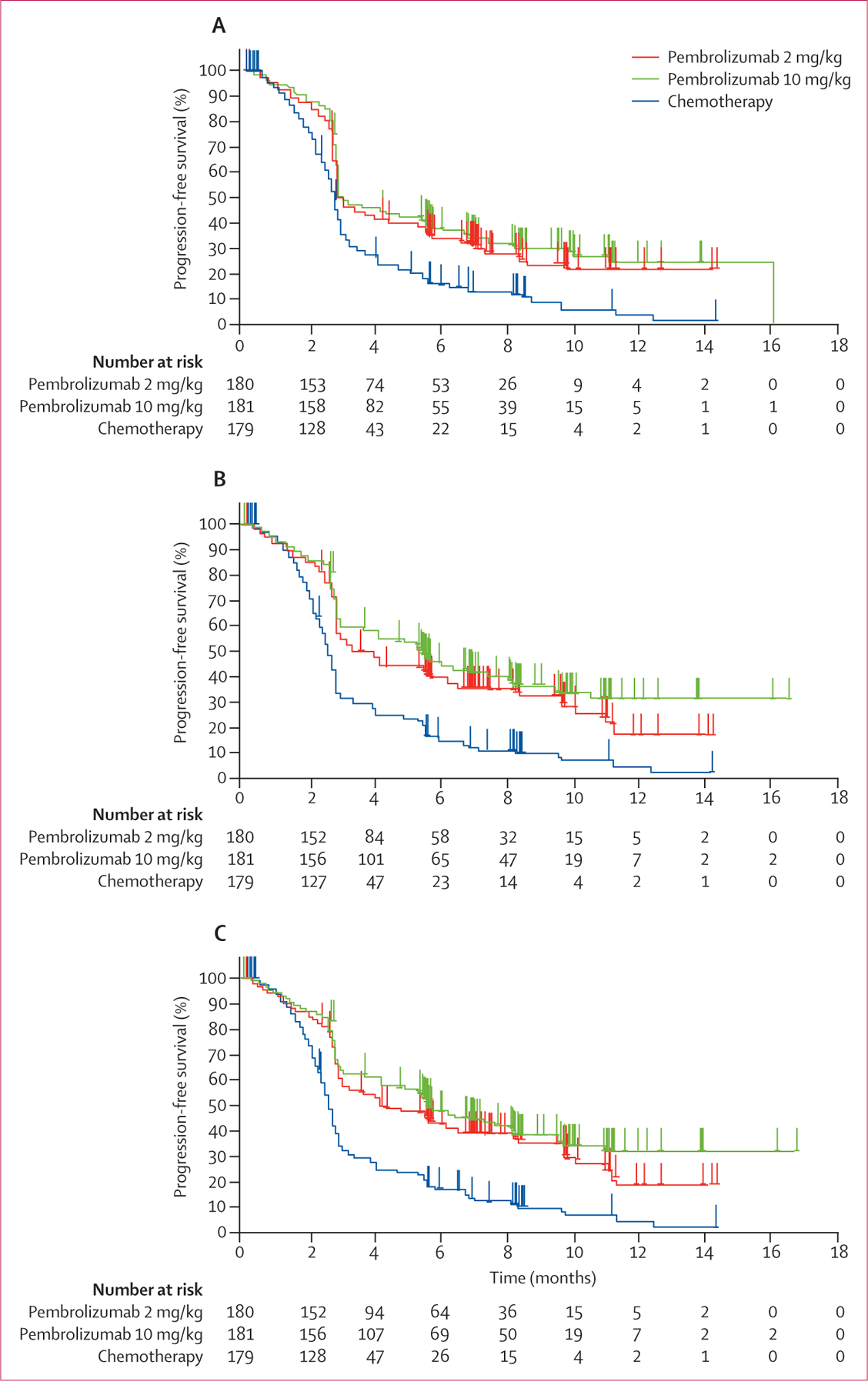
Kaplan-Meier curves for RECIST v1.1-assessed progression by independent central review (A), RECIST v1.1, by investigator review (B), and modified RECIST v1.1, by investigator review (C). RECIST v1.1=Response Evaluation Criteria in Solid Tumors, version 1.1.
In a post-hoc analysis, restricted mean progression-free survival time based on the area under the Kaplan-Meier curve up to 12 months19 was 5·4 months (95% CI 4·7–6·0) for the pembrolizumab 2 mg/kg group, 5·8 months (5·1–6·4) for the pembrolizumab 10 mg/kg group, and 3·6 months (3·2–4·1) for the chemotherapy group. Pembrolizumab also significantly improved progression-free survival when assessed by investigator review, with a larger treatment effect for pembrolizumab (table 2) and a greater separation of the curves (figure 2B), although possible investigator bias in a partly open-label trial such as this might explain the greater effect size. A protocol-specified supportive analysis of progression-free survival per investigator review that assessed progression only if it was observed in two consecutive scans also showed a significant treatment effect favouring pembrolizumab (table 2, figure 2C). The superiority of both pembrolizumab doses was evident in all prespecified patient subgroups (figure 3).
Figure 3: Prespecified subgroup analysis of progression-free survival.
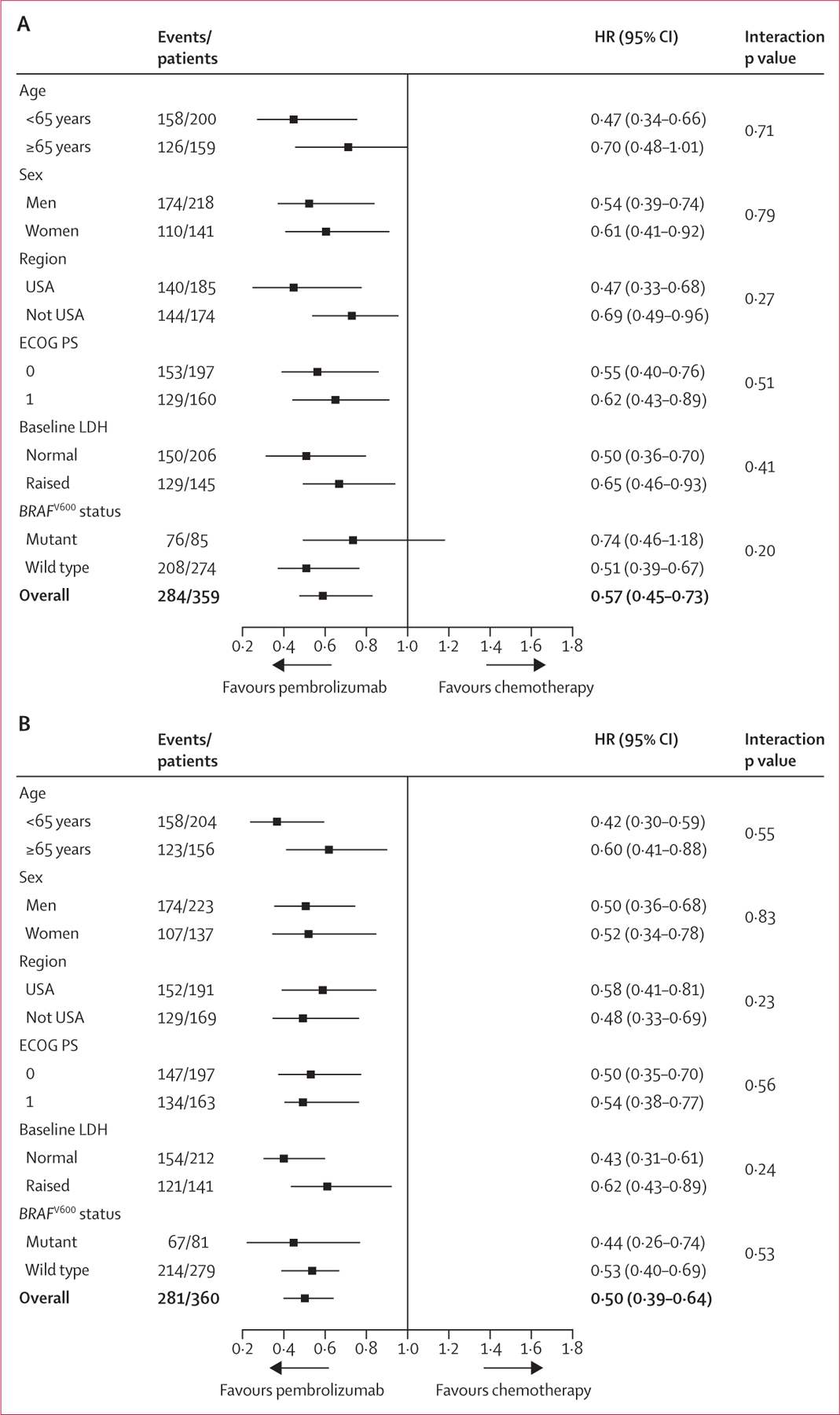
Progression-free survival assessed by RECIST v1.1, by independent central review in the intention-to-treat population; pembrolizumab 2 mg/kg versus chemotherapy (A) and pembrolizumab 10 mg/kg versus chemotherapy (B). HR=hazard ratio. ECOG PS=Eastern Cooperative Oncology Group performance status. LDH=lactate dehydrogenase.
Immature overall survival data, which were evaluated at a small α level at this interim analysis, did not meet the prespecified 0·25% superiority threshold for each pembrolizumab dose compared with chemotherapy (data not shown). Final overall survival will be assessed after 370 deaths.
On independent central review, 38 (21%) patients in the pembrolizumab 2 mg/kg group and 46 (25%) in the 10 mg/kg group responded to treatment as per RECIST v1.1 criteria, compared with eight (4%) in the chemotherapy group (p<0·0001 for each pembrolizumab dose vs chemotherapy). Median time to response was 13 weeks (IQR 12–18) in the pembrolizumab 2 mg/kg group, 15 weeks (12–18) in the pembrolizumab 10 mg/kg group, and 13 weeks (12–18) in the chemotherapy group. At the time of analysis 35 (92%) of 38 responders in the pembrolizumab 2 mg/kg group, 40 (87%) of 46 responders in the pembrolizumab 10 mg/kg group, and five (63%) of eight responders in the chemotherapy group remained progression free. Median duration of response was not reached in either pembrolizumab group and was 37 weeks (IQR 12–41) in the chemotherapy group (table 2). Based on the prespecified futility rule, the proportion of responses did not differ between the two pembrolizumab groups. As such, neither pembrolizumab dose was discontinued at the first interim analysis. 35 (19%) of 180 patients in the pembrolizumab 2 mg/kg group and 37 (20%) of 181 patients in the pembrolizumab 10 mg/kg group were treated beyond initial evidence of disease progression.
Of the 540 patients enrolled, 528 received at least one dose of study treatment and were assessed for safety (figure 1). Median time on treatment was 113 days (range 1–499) in the pembrolizumab 2 mg/kg group, 145 days (1–505) in the pembrolizumab 10 mg/kg group, and 61 days (1–335) in the chemotherapy group (appendix). There were no treatment-related deaths. One death in a man aged 75 years treated with pembrolizumab 2 mg/kg was reported by the investigator to be possibly related to treatment. The day after the first pembrolizumab dose, the patient presented with a bleeding pelvic mass causing severe pain. He was also found to have melaena, possibly from existing small bowel metastases. The patient died 3 days later. Given his age and overall poor clinical condition, comprehensive investigations were not done, and the cause of death was listed as unknown. After the analysis cutoff date, the causality of this death was changed by the investigator to be unrelated to treatment.
Incidence of grade 3–4 treatment-related adverse events was higher in those given chemotherapy (45 [26%] of 171 patients) than in those given pembrolizumab (20 [11%] of 178 patients] in the pembrolizumab 2 mg/kg group and 25 [14%] of 179 patients in the 10 mg/kg group). Grade 3–4 treatment-related adverse events occurred earlier in the chemotherapy group (appendix). The most common grade 3–4 treatment-related adverse events in the pembrolizumab 2 mg/kg treatment group were fatigue, generalised oedema, and myalgia (two [1%] each). The most common grade 3–4 treatment-related adverse events observed in those given pembrolizumab 10 mg/kg were hypopituitarism, colitis, diarrhoea, decreased appetite, hyponatraemia, and pneumonitis (two [1%] each). The most most common grade 3–4 treatment-related adverse events observed in patients given chemotherapy were anaemia (nine [5%]), fatigue (eight [5%]), neutropenia (six [4%]), and leucopenia (six [4%]).
Treatment interruption as a result of treatment-related adverse events was needed in 15 (8%) of 178 patients treated with pembrolizumab 2 mg/kg, 15 (8%) of 179 patients treated with pembrolizumab 10 mg/kg, and 30 (18%) of 171 patients treated with chemotherapy. Treatment-related adverse events led to permanent treatment discontinuation in five (3%) patients given pembrolizumab 2 mg/kg, 12 (7%) given pembrolizumab 10 mg/kg, and 10 (6%) patients given chemotherapy. Treatment-related serious adverse events occurred at a similar frequency across treatment groups (table 3). The most common serious treatment-related adverse events observed in the combined pembrolizumab treatment groups were diarrhoea and pneumonitis (three [1%] of 357 patients each). Treatment-related adverse events more common with chemotherapy were alopecia, anaemia, decreased appetite, fatigue, nausea, and vomiting; those more frequent with pembrolizumab were rash and pruritus (table 3). Consistent with previous experience, the adverse event profiles of the 2 mg/kg and 10 mg/kg doses were generally similar.10,11 Adverse events of a potentially immune-mediated nature were infrequent, and none were of grade 4 or 5 severity (appendix). These events were generally manageable with supportive care, withholding treatment, or corticosteroid therapy.
Table 3:
Treatment-related adverse events
| Pembrolizumab 2 mg/kg (n=178) |
Pembrolizumab 10 mg/kg (n=179) |
Chemotherapy control (n=171) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Grade 1–2 | Grade 3 | Grade 4 | Grade 1–2 | Grade 3 | Grade 4 | Grade 1–2 | Grade 3 | Grade 4 | |
| Summary | |||||||||
| Any | 101 (57%) | 18 (10%) | 1 (<1%) | 107 (60%) | 23 (13%) | 2 (1%) | 93 (54%) | 34 (20%) | 11 (6%) |
| Serious* | 3 (2%) | 9 (5%) | 1 (<1%) | 3 (2%) | 14 (8%) | 2 (1%) | 3 (2%) | 8 (5%) | 6 (4%) |
| Led to discontinuation† | 0 | 3 (2%) | 1 (<1%) | 2 (1%) | 9 (5%) | 1 (<1%) | 6 (4%) | 2 (1%) | 2 (1%) |
| Observed in 5% or more of patients in any treatment group | |||||||||
| Fatigue | 38 (21%) | 2 (1%) | 0 | 51 (28%) | 1 (<1%) | 0 | 54 (32%) | 8 (5%) | 0 |
| Pruritus | 37 (21%) | 0 | 0 | 42 (23%) | 0 | 0 | 6 (4%) | 0 | 0 |
| Nausea | 8 (4%) | 0 | 0 | 15 (8%) | 1 (<1%) | 0 | 52 (30%) | 3 (2%) | 1 (<1%) |
| Decreased appetite | 8 (4%) | 0 | 0 | 15 (8%) | 2 (1%) | 0 | 26 (15%) | 0 | 0 |
| Anaemia | 4 (2%) | 1 (<1%) | 0 | 7 (4%) | 0 | 0 | 26 (15%) | 9 (5%) | 0 |
| Diarrhoea | 15 (8%) | 0 | 0 | 17 (9%) | 2 (1%) | 0 | 11 (6%) | 3 (2%) | 0 |
| Rash | 21 (12%) | 0 | 0 | 18 (10%) | 0 | 0 | 8 (5%) | 0 | 0 |
| Alopecia | 5 (3%) | 0 | 0 | 1 (<1%) | 0 | 0 | 24 (20%) | 1 (<1%) | 0 |
| Vomiting | 1 (<1%) | 1 (<1%) | 0 | 9 (5%) | 1 (<1%) | 0 | 22 (13%) | 3 (2%) | 1 (<1%) |
| Arthralgia‡ | 12 (7%) | 1 (<1%) | 0 | 10 (6%) | 1 (<1%) | 0 | 8 (5%) | 1 (<1%) | 0 |
| Constipation | 5 (3%) | 0 | 0 | 9 (5%) | 0 | 0 | 14 (8%) | 0 | 0 |
| Myalgia | 7 (4%) | 2 (1%) | 0 | 7 (4%) | 0 | 0 | 9 (5%) | 1 (<1%) | 0 |
| Asthenia | 5 (3%) | 1 (<1%) | 0 | 7 (4%) | 1 (<1%) | 0 | 9 (5%) | 1 (<1%) | 0 |
| Hypothyroidism | 9 (5%) | 0 | 0 | 13 (7%) | 0 | 0 | 0 | 0 | 0 |
| Vitiligo | 10 (6%) | 0 | 0 | 9 (5%) | 0 | 0 | 2 (1%) | 0 | 0 |
| Dry skin | 9 (5%) | 0 | 0 | 9 (5%) | 0 | 0 | 2 (1%) | 0 | 0 |
| Thrombocytopenia | 2 (1%) | 0 | 0 | 0 | 1 (<1%) | 0 | 12 (7%) | 2 (1%) | 2 (1%) |
| Neutropenia | 1 (<1%) | 0 | 0 | 1 (<1%) | 0 | 0 | 8 (5%) | 4 (2%) | 2 (1%) |
| Peripheral neuropathy | 2 (1%) | 0 | 0 | 0 | 0 | 0 | 12 (7%) | 2 (1%) | 0 |
| Maculopapular rash | 4 (2%) | 1 (<1%) | 0 | 9 (5%) | 1 (<1%) | 0 | 0 | 0 | 0 |
| Leucopenia | 0 | 0 | 0 | 0 | 0 | 0 | 8 (5%) | 4 (2%) | 2 (1%) |
| Paraesthesia | 1 (<1%) | 0 | 0 | 2 (1%) | 0 | 0 | 11 (6%) | 0 | 0 |
Adverse events are listed in order of descending frequency in the total study population (patients who had at least one study drug dose).
One additional patient treated with pembrolizumab 10 mg/kg had two serious treatment-related adverse events of unspecifi ed severity.
One additional patient in the pembrolizumab 2 mg/kg treatment group died because of an adverse events considered to be possibly related to treatment at the time of data cutoff (full details provided in text).
One additional patient treated with pembrolizumab 10 mg/kg had arthralgia of unspecifi ed severity.
Baseline EORTC QLQ-C30 global health status and quality-of-life scores were similar across treatment groups. The least squares mean change from baseline to week 12 in the score was –2·60 (95% CI –6·15 to 0·96) in the pembrolizumab 2 mg/kg group, –2·55 (–5·99 to 0·89) in the 10 mg/kg group, and –9·13 (–12·86 to –5·39) in the chemotherapy group (appendix). The least squares mean change significantly differed between the 2 mg/kg pembrolizumab and chemotherapy groups (6·53, 95% CI 1·53–11·53; p=0·011) and the 10 mg/kg pembrolizumab and chemotherapy groups (6·57, 1·65–11·50; p=0·009). Patients treated with pembrolizumab had consistently smaller decrements in the individual function and symptoms scales (appendix). The global health status quality-of-life score deteriorated by 10 points or more in approximately 7% to 12% fewer patients in the pembrolizumab treatment groups than in the chemotherapy group at week 12 (64 [38%] of 167 patients in the chemotherapy group, 56 [32%] of 176 patients in the pembrolizumab 2 mg/kg group, and 47 [27%] of 177 patients in the pembrolizumab 10 mg/kg group; appendix).
Discussion
In this population of patients with advanced melanoma that progressed on ipilimumab and, in many cases, on chemotherapy or MAPK pathway inhibitors, pembrolizumab reduced the risk of disease progression or death compared with investigator-choice standard-of-care chemotherapy. Although median progression-free survival values were similar, around the time of the first scheduled tumour assessment at week 12 in all treatment groups, the Kaplan-Meier curves separated dramatically thereafter, indicating the treatment effect.
The ability of ipilimumab-refractory disease to respond to pembrolizumab is probably a reflection of the different mechanisms by which anti-CTLA-4 and anti-PD-1 therapies stimulate an anti-tumour T-cell response. CTLA-4 blockade broadens the immune response, evidenced by an increased T-cell receptor repertoire leading to increased tumour infiltration,20–23 whereas PD-1 blockade induces intratumoural T-cell proliferation without detectable changes in the peripheral immune repertoire.12,24–27
Five times more patients had an objective response with pembrolizumab than did those assigned to chemotherapy. This marked improvement is particularly meaningful because of the superior response durability observed with pembrolizumab, although longer follow-up is warranted to fully appreciate response duration. Most patients in this trial had disease that progressed on several lines of therapy in the metastatic setting, including chemotherapy in a half of patients. In addition, all patients with BRAFV600-mutant-positive melanoma had received treatment with a MAPK pathway inhibitor. Overall, less than 5% of this population responded when allocated to investigator’s choice of chemotherapy. These results support the Food and Drug Administration’s accelerated approval of pembrolizumab in this poor-prognosis population.
Although our study was not powered to assess the equivalence of the two pembrolizumab doses, the results of the two interim analyses do not suggest a significant, clinically meaningful, difference between the doses. The higher number of responses in the pembrolizumab 10 mg/kg treatment group than in the 2 mg/kg group might be due to random variation. The absence of a significant difference between the pembrolizumab doses is consistent with findings in a similar population treated in the KEYNOTE-001 study.10,11 Taken together, there is no evidence that one pembrolizumab dosing regimen is superior to another. Therefore, the minimally effective pembrolizumab dose of 2 mg/kg given every 3 weeks is recommended for further use.
Pembrolizumab was well tolerated compared with chemotherapy, with fewer treatment-related and grade 3–4 adverse events in the pembrolizumab groups despite an approximately two-fold longer duration of exposure. Potentially immune-mediated adverse events such as hypothyroidism, hypophysitis, colitis, pneumonitis, hepatitis, and nephritis were observed with pembrolizumab, although they were infrequent and mostly of grade 1 or 2 severity. Of note, there were no cases of new-onset type 1 diabetes mellitus. Potentially immune-mediated events were generally manageable with immunosuppressive therapy and treatment interruption or discontinuation as appropriate. The benefit of pembrolizumab is further strengthened by the favourable health-related quality-of-life scores compared with chemotherapy.
Two recent studies of nivolumab (an anti-PD-1 antibody) have reported an improvement in objective responses compared with chemotherapy in patients who had previously received ipilimumab28 and improvement in overall survival compared with chemotherapy in patients with BRAFV600 wild-type melanoma.29 Furthermore, pembrolizumab given to patients who were ipilimumab treatment-naive showed improvement in responses, progression-free survival, and overall survival compared with ipilimumab.30 Combined with our study findings, these data support the remarkable activity of PD-1 blockade in patients with advanced melanoma. Evaluation of the effect of PD-1 blockade on overall survival relative to standard-of-care treatment is continuing in KEYNOTE-002 and other studies. The assessment of biomarkers and studies of combination regimens are also ongoing to improve further the clinical outcomes of patients with advanced melanoma. Analyses of biomarkers and the relation between PD-L1 expression and outcomes in this study are continuing and will be reported with the final overall survival analysis. Overall, targeting of the PD-1 axis is a major advancement in melanoma and establishes pembrolizumab as a new standard treatment after progression on ipilimumab and other therapies.
Supplementary Material
Research in context.
Evidence before this study
Throughout the writing process and most recently on April 30, 2015, we did an extensive search of PubMed for studies of PD-L1 and PD-1 in advanced cancers, including melanoma. This search was not limited by date. Search terms were “PD-1 OR PD-L1 OR MK-3475 OR lambrolizumab OR nivolumab OR BMS-936558 OR MPDL3280A OR BMS-936559”. Two studies (Weber et al, 2015, and Robert et al, 2015) report fi ndings for nivolumab that corroborate our data. We also searched PubMed to identify treatment options for ipilimumab-refractory melanoma, without any language restrictions. Apart from cytotoxic chemotherapy, we identifi ed no standard treatment options for ipilimumab-refractory melanoma. Although some studies have shown efficacy for other anti-PD-1 and PD-L1 inhibitors after ipilimumab, these were not controlled and the sample sizes were small.
Added value of this study
Our study is, to the best of our knowledge, the largest reported randomised, controlled trial of an anti-PD-1 or anti-PD-L1 drug for ipilimumab-refractory melanoma. Results of this study confirm the efficacy and safety of pembrolizumab and the absence of a significant difference in outcomes between pembrolizumab doses of 2 mg/kg and 10 mg/kg given once every 3 weeks, as observed in an earlier phase 1b study of patients with ipilimumab-refractory melanoma. More important, the study shows the superiority of pembrolizumab over cytotoxic chemotherapy, the current standard-of-care therapy, to improve progression-free survival in ipilimumab-refractory melanoma. Our data are arguably of greater clinical relevance than are the data reported for nivolumab in this population because our study included patients with BRAFV600-mutant melanoma and we report progression-free survival and patient-reported outcomes data.
Implications of all the available evidence
Overall, these findings establish pembrolizumab as a new standard of care for melanoma and support the accelerated approval granted by the US Food and Drug Administration for the use of pembrolizumab 2 mg/kg once every 3 weeks by patients with unresectable or metastatic melanoma whose disease progressed after ipilimumab and, if BRAFV600 mutant-positive, a BRAF inhibitor.
Acknowledgments
This study and assistance with manuscript editing were funded by Merck Sharp & Dohme. We thank the patients and their families and caregivers; all primary investigators and their site personnel (see appendix); Tricia Brown and Melanie Leiby (The APO Group, Yardley, PA, USA), for assistance with manuscript editing; James R Anderson (University of Nebraska Medical Center, Omaha, NE, USA), Ronald Blum (Albert Einstein College of Medicine and Beth Israel Medical Center, New York, NY, USA), Janice Dutcher (Cancer Research Foundation, New York, NY, USA), and Lawrence Flaherty (Barbara Ann Karmanos Cancer Institute, Detroit, MI, USA), for serving on the external data monitoring committee; Jeff Maca (Quintiles, Morrisville, SC, USA), for serving as the unmasked study statistician; Eric Rubin and Alise Reicin (Merck Sharp & Dohme), for critical review of the manuscript and supervision of the study group; Cong Chen (Merck) for critical review of the manuscript and statistical expertise; and Clara Benner, Linda Gammage, Claudia Geis, Klaas Koekenbier, Amanda McDonald, Kathleen Oxberry, Lei Pang, Theodora Paraskevopoulos, Andrea Perrone, Kathryn Schneck, Sigalit Segal, Sophie Vauclair, and Jen Villetard (all of Merck) for critical study support.
Funding
Merck Sharp & Dohme.
Footnotes
Declaration of interests
AR reports fees to his institution from Merck for serving as an advisory board member. RD reports grant support from Bristol-Myers Squibb (BMS) and advisory board honoraria from BMS, GlaxoSmithKline (GSK), Merck Sharp & Dohme (MSD), Novartis, and Roche. DS reports grant support and study fees to his institution from Merck; personal fees for serving as an advisory board, steering committee, and speaker’s bureau member and for travel and hotel support from BMS, Merck, Novartis, and Roche-Genentech; and personal fees for advisory board and speaker’s bureau membership from Amgen and Boehringer Ingelheim. OH reports grant support from and personal fees for serving as a speaker for BMS and Merck. CR reports serving as an advisory board member for Amgen, BMS, GSK, Merck, Novartis, and Roche. FSH reports personal fees, non-financial support, and clinical trial support to his institution from Merck, grant and non-financial support and clinical trial support to his institution from BMS, and clinical trial support to his institution from Genentech. KDL reports grant support from Merck. LDC reports receiving funding to his institution from Merck for conduct of clinical trials; grant support to his institution from BMS and Genentech; and serving as a speaker programme member for BMS and Genentech. CUB reports advisory roles for BMS, GSK, MSD, Novartis, and Roche, and receipt of a research grant from Novartis. SJO’D reports receiving clinical research support from and serving as a consultant and advisory board member for Merck. PAA reports grants and personal fees from BMS, Roche-Genentech, and Ventana; personal fees and non-financial support from MSD; and personal fees from Amgen, Novartis, and GSK, all outside of the submitted work. AKSS reports grant support from BMS and personal fees for serving as a consultant for BMS and Roche-Genentech. CL reports personal fees for serving as an advisory board member from Amgen, Biontech, BMS, Celgene, MSD, and Roche; personal fees for lectures from BMS, MSD, and Roche; and personal fees for consulting from Biontech, Celgene, and Roche. TKE reports personal fees from BMS, GSK, Leo Pharma GmbH, MSD, Philogen, and Roche. TCG reports personal fees from Merck. MSC reports personal fees outside the submitted work from BMS and Merck for serving as an adviser and from BMS, GSK, Novartis, and Roche for honoraria. SJM reports support from MSD for clinical trial support and serving as an advisory board member for MSD. SMG reports personal fees and non-financial support from BMS for travel support and providing expert testimony; from MSD for travel support and advisory board membership; and research funding from the University Hospital Zurich. RG reports grant support from BMS, GSK, Merck, Novartis, and Roche, and personal fees from BMS and Roche. JMK reports personal fees from BMS, Celgene, GSK, Merck, Vical, and Ziopharm. JDW reports grant and non-financial support and personal fees from BMS and MSD. AE reports personal fees for serving on scientific advisory boards for Amgen, BMS, GSK, MedImmune, and MSD. XNL, WZ, AMZ, JL, SE, and SPK are employees of MSD. XNL, WZ, SE, and SPK hold stock in MSD. AD reports serving on advisory boards for and receiving research funding from Amgen, Genentech, GSK, OncoSec, and Roche. IP, JS, AP, KAM SSA, JAS, and RS-F declare no competing interests.
Contributor Information
Antoni Ribas, University of California, Los Angeles, Los Angeles, CA, USA.
Igor Puzanov, Vanderbilt-Ingram Cancer Center, Nashville, TN, USA.
Reinhard Dummer, University of Zürich, Zürich, Switzerland.
Dirk Schadendorf, University Hospital Essen, Essen, Germany.
Omid Hamid, The Angeles Clinic and Research Institute, Los Angeles, CA, USA.
Caroline Robert, Gustave Roussy and Paris-Sud University, Villejuif, France.
F Stephen Hodi, Dana-Farber Cancer Institute, Boston, MA, USA.
Jacob Schachter, Sheba Medical Center, Tel Hashomer, Israel.
Anna C Pavlick, New York University Cancer Institute, New York, NY, USA.
Karl D Lewis, University of Colorado Denver, Aurora, CO, USA.
Lee D Cranmer, University of Arizona Cancer Center, Tucson, AZ, USA.
Christian U Blank, Netherlands Cancer Institute, Amsterdam, Netherlands.
Steven J O’Day, Beverly Hills Cancer Center, Beverly Hills, CA, USA.
Paolo A Ascierto, Istituto Nazionale Tumori Fondazione G. Pascale, Napoli, Italy.
April K S Salama, Duke Cancer Institute, Durham, NC, USA.
Kim A Margolin, Seattle Cancer Care Alliance/University of Washington, Seattle, WA, USA.
Carmen Loquai, University Medical Center, Mainz, Germany.
Thomas K Eigentler, Universitätsklinikum Tübingen, Tübingen, Germany.
Tara C Gangadhar, Abramson Cancer Center of the University of Pennsylvania, Philadelphia, PA, USA.
Matteo S Carlino, Crown Princess Mary Cancer Centre, Westmead and Blacktown Hospitals, and Melanoma Institute Australia, Westmead, NSW, Australia.
Sanjiv S Agarwala, St Luke’s Cancer Center, Bethlehem, PA, USA, Temple University, Philadelphia, PA, USA.
Stergios J Moschos, University of North Carolina, Chapel Hill, NC, USA.
Jeffrey A Sosman, Vanderbilt-Ingram Cancer Center, Nashville, TN, USA.
Simone M Goldinger, University of Zürich, Zürich, Switzerland.
Ronnie Shapira-Frommer, Sheba Medical Center, Tel Hashomer, Israel.
Rene Gonzalez, University of Colorado Denver, Aurora, CO, USA.
John M Kirkwood, University of Pittsburgh, Pittsburgh, PA, USA.
Jedd D Wolchok, Memorial Sloan Kettering Cancer Center, New York, NY, USA.
Alexander Eggermont, Gustave Roussy and Paris-Sud University, Villejuif, France.
Xiaoyun Nicole Li, Merck & Co, Kenilworth, NJ, USA.
Wei Zhou, Merck & Co, Kenilworth, NJ, USA.
Adriane M Zernhelt, Merck & Co, Kenilworth, NJ, USA.
Joy Lis, Merck & Co, Kenilworth, NJ, USA.
Scot Ebbinghaus, Merck & Co, Kenilworth, NJ, USA.
S Peter Kang, Merck & Co, Kenilworth, NJ, USA.
Adil Daud, University of California, San Francisco, San Francisco, CA, USA (Prof A Daud MBBS).
References
- 1.McArthur GA, Ribas A. Targeting oncogenic drivers and the immune system in melanoma. J Clin Oncol 2013; 31: 499–506. [DOI] [PubMed] [Google Scholar]
- 2.Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363: 711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011; 364: 2517–26. [DOI] [PubMed] [Google Scholar]
- 4.Ascierto PA, Kirkwood JM, Grob JJ, et al. The role of BRAF V600 mutation in melanoma. J Transl Med 2012; 10: 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012; 12: 252–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blank C, Brown I, Peterson AC, et al. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res 2004; 64: 1140–45. [DOI] [PubMed] [Google Scholar]
- 7.Okazaki T, Honjo T. PD-1 and PD-1 ligands: from discovery to clinical application. Int Immunol 2007; 19: 813–24. [DOI] [PubMed] [Google Scholar]
- 8.Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012; 366: 2455–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012; 366: 2443–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 2013; 369: 134–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robert C, Ribas A, Wolchock JD, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet 2014; 384: 1109–17. [DOI] [PubMed] [Google Scholar]
- 12.Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014; 515: 563–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Powles T, Eder JP, Fine GD, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 2014; 515: 558–62. [DOI] [PubMed] [Google Scholar]
- 14.Wolchok JD, Hoos A, O’Day S, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res 2009; 15: 7412–20. [DOI] [PubMed] [Google Scholar]
- 15.Hochberg Y A sharper Bonferroni procedure for multiple tests of significance. Biometrika 1988; 75: 800–02. [Google Scholar]
- 16.Efron B The efficacy of Cox’s likelihood function for censored data. J Am Stat Assoc 1977; 72: 557–65. [Google Scholar]
- 17.Miettinen O, Nurminen M. Comparative analysis of two rates. Stat Med 1985; 4: 213–26. [DOI] [PubMed] [Google Scholar]
- 18.Schoenfeld D Partial residuals for the proportional hazards regression model. Biometrika 1982; 69: 239–41. [Google Scholar]
- 19.Uno H, Claggett B, Tian L, et al. Moving beyond the hazard ratio in quantifying the between-group difference in survival analysis. J Clin Oncol 2014; 32: 2380–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robert L, Tsoi J, Wang X, et al. CTLA4 blockade broadens the peripheral T-cell receptor repertoire. Clin Cancer Res 2014; 20: 2424–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cha E, Klinger M, Hou Y, et al. Improved survival with T cell clonotype stability after anti-CTLA-4 treatment in cancer patients. Sci Transl Med 2014; 6: 238ra70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kvistborg P, Philips D, Kelderman S, et al. Anti-CTLA-4 therapy broadens the melanoma-reactive CD8+ T cell response. Sci Transl Med 2014; 6: 254ra128. [DOI] [PubMed] [Google Scholar]
- 23.Huang RR, Jalil J, Economou JS, et al. CTLA4 blockade induces frequent tumor infiltration by activated lymphocytes regardless of clinical responses in humans. Clin Cancer Res 2011; 17: 4101–09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robert L, Harview C, Emerson R, et al. Distinct immunological mechanisms of CTLA-4 and PD-1 blockade revealed by analyzing TCR usage in blood lymphocytes. Oncoimmunology 2014; 3: e29244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014; 515: 568–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spranger S, Spaapen RM, Zha Y, et al. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med 2013; 5: 200ra116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bald T, Landsberg J, Lopez-Ramos D, et al. Immune cell-poor melanomas benefit from PD-1 blockade after targeted type I IFN activation. Cancer Discov 2014; 4: 674–87. [DOI] [PubMed] [Google Scholar]
- 28.Weber JS, D’Angelo SP, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol 2015; 16: 375–84. [DOI] [PubMed] [Google Scholar]
- 29.Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015; 372: 320–30. [DOI] [PubMed] [Google Scholar]
- 30.Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med 2015; published online April 18. DOI: 10.1056/NEJMoa1503093. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.