

Abstract
MYC family oncoproteins are regulators of metabolic reprogramming that sustains cancer cell anabolism. Normal cells adapt to nutrient-limiting conditions by activating autophagy, which is required for amino acid (AA) homeostasis. Here we report that the autophagy pathway is suppressed by Myc in normal B cells, in premalignant and neoplastic B cells of Eμ-Myc transgenic mice, and in human MYC-driven Burkitt lymphoma. Myc suppresses autophagy by antagonizing the expression and function of transcription factor EB (TFEB), a master regulator of autophagy. Mechanisms that sustained AA pools in MYC-expressing B cells include coordinated induction of the proteasome and increases in AA transport. Reactivation of the autophagy-lysosomal pathway by TFEB disabled the malignant state by disrupting mitochondrial functions, proteasome activity, amino acid transport, and amino acid and nucleotide metabolism, leading to metabolic anergy, growth arrest and apoptosis. This phenotype provides therapeutic opportunities to disable MYC-driven malignancies, including AA restriction and treatment with proteasome inhibitors.
Introduction
Phenotypes acquired during malignant transformation include sustained proliferation, increases in cell mass, the resistance to cell death, evasion of the immune system, and invasion and metastasis, amongst others (1). All these phenotypes require metabolic reprogramming, which includes the switch to aerobic glycolysis, glutaminolysis and increased transport of glucose, amino acids and other nutrients needed to generate ATP and sustain high levels of protein, nucleic acid, and fatty acid synthesis for the rapidly dividing cancer cell. Accordingly, a major focus of cancer research is defining metabolic strategies and targets that can be exploited for therapeutics.
Key regulators of cancer cell metabolic reprogramming are MYC family oncoproteins (e.g., MYC and MYCN) (2), which are activated in a large cast of malignancies (3) and function as basic helix-loop-helix-leucine zipper (bHLH-Zip) transcription factors that control a large cast of target genes harboring E-boxes (CACGTG) (4). These targets include those involved in glycolysis (5,6), glutamine metabolism (7) and mitochondrial biogenesis (8), which allow the cancer cell to sustain its anabolic state.
Proper control of amino acid (AA) homoeostasis is critical for cell growth and survival. For example, AA are needed for the translation of proteins yet are also catabolized for synthesis of lipids, nucleotides, glutathione, one-carbon units, and energy via the TCA cycle, and this is especially important during nutrient deprivation (9,10). The highly anabolic state of cancer cells requires they maintain sufficient pools of free amino acids. Accordingly, several mechanisms to sustain AA pools are upregulated in cancer cells, including the autophagy-lysosome circuit (11), macropinocytosis (12), the proteasome (13), amino acid transport (7) and de novo synthesis (14). For example, oncogenic KRAS-driven cancers up-regulate and require the autophagy pathway and macropinocytosis to sustain growth (11,15–17). Furthermore, MYC-driven tumors rely on increased uptake of glutamine, and on glutamine catabolism by glutaminase to glutamate that feeds into the TCA cycle (7,18).
Transcription factor EB (TFEB), a MYC-related micropthalmia-transcription (MiT) bHLH-Zip transcription factor (19), is a master regulator of the autophagy-lysosome pathway that sustains AA pools via breakdown of proteins (20,21). TFEB induces genes involved in this pathway by binding to CLEAR sites (TCACGTGA) (22) which also contain the CACGTG E-boxes recognized by MYC. Indeed, like MYC (23), TFEB is required for transformation by oncogenic RAS (24) and TFEB is also activated in human cancer, by recurrent t(6;11)(p21;q12) translocations in renal carcinoma (19). However, recent studies by us and others have shown MYC suppresses the expression of TFEB and other MIT/TFE family members and autophagy in tumor cell lines and myeloid progenitors, as well as in AML cells and a model of MYC-driven medulloblastoma (25,26).
Given the global effects of MYC on metabolic programming we reasoned that the autophagy-lysosomal pathway would be necessary for the development and maintenance of MYC-driven tumors. Here we report that MYC represses the autophagy pathway via antagonism of TFEB, and that this repressive circuit is necessary for maintenance of the malignant state, as restoring autophagy leads to metabolic anergy. Notably, this evolutionary trajectory of MYC-induced tumors creates a high reliance on sufficient AA pools, and to induction of the proteasome and amino acid transport that affords actionable means for treating malignancies with MYC involvement.
Materials and Methods
Select methods are presented in this section. A more detailed methods section is provided as Supplementary Methods within the Supplementary Material of this article.
Mouse studies
Eμ-rtTA2 transgenic mice were generated by cloning the coding region for rtTA2 into the pEμSR plasmid (27) and transgenic founders were identified by PCR. For the described experiments, 6-week old WT C57BL/6 and double transgenic Eμ-Myc;Eμ-rtTA2 or Eμ-Myc;Rosa26-rtTA2 littermates of either sex were randomly allocated to treatment cohorts. Tfebfl/fl and Atg7fl/fl conditional knockout mice have been reported (28,29). All animal studies were approved by the IACUC of Scripps Florida and by the IACUC of Moffitt Cancer Center/University of South Florida.
Cell culture
P493–6 human B lymphoma cells were cultured in RPMI in the presence of Tetracycline (Tet; 0.1 μg/ml). Eμ-Myc;Eμ-rtTA2 or Eμ-Myc;Rosa26-rtTA2 lymphomas were harvested, homogenized and cultured as a single cell suspension in 45% IMDM (with 25mM HEPES), 45% DMEM, 4 mM L-glutamine, 25 μM β-mercaptoethanol (Millipore-Sigma), 1 mM sodium pyruvate and 5 ng/ml mouse IL-7 (R&D Systems). Namalwa Burkitt lymphoma (BL) cells were purchased from ATCC and maintained in RPMI and authenticated after verification of 14 locus matching reference at Applied Biosystems. Atg7−/− mouse embryo fibroblasts (MEFs) (29) were grown in 1x DMEM containing 4 mM L-Glutamine, and 1% NEAA. 293T cells, which were used to generate stocks of MSCV-based retroviruses, were cultured in DMEM.
Lymphoma transplant studies
Eμ-Myc transgenic mice were bred to CD19-Cre mice to produce Eμ-Myc;CD19-Cre offspring, which were then bred to Tfebfl/fl. or Atg7fl/fl mice to produce the desired Eμ-Myc;CD19-Cre;Tfebfl/fl or Eμ-Myc;CD19-Cre;Atg7fl/fl cohorts, as well as the corresponding Tfeb or Atg7 heterozygous and wild-type counterparts. Mice were monitored daily for illness and tumor development. Sick animals were sacrificed, and tumors were collected.
For lymphoma transplant studies, intravenously injected 6-week-old C57BL/6 or NOD.CB17-Prkdcscid/J mice were monitored daily for signs of morbidity. For doxycycline (Dox) studies, mice were switched to a Dox-containing chow (Envigo, TD.05298) three days following transplants. For Bortezomib (Millipore Sigma, 504314) studies, the mice were intravenously injected with 0.25 mg/kg weekly. Control mice developed hind limb paralysis by 21–26 days, a hallmark of advanced disease in this lymphoma transplant model.
Expression profile analysis
GSE37792 and GSE32239: The CEL files were downloaded from GEO and normalized using IRON (30). GSE40782, GSE37222, and Immgen/GSE15907: The normalized Series Matrix File was downloaded from GEO. GSE4475: CEL files were downloaded from GEO, normalized using IRON, and were then debatched using ComBat (31). EGAS00001002606: The FPKM gene were summarized, preprocessed to remove lowly expressed and variable genes (30,31), and then quantile normalized data from Reddy et al. (32) were used. GSE51008: The RPKM normalized data were downloaded from GEO. Multiple Myeloma Research Foundation (MMRF): Data was downloaded from GDC (gdc.cancer.gov) both as FPKM and FPKM-UQ and were analyzed. All downloaded data was log2 transformed before analysis.
Principal Component Analysis (PCA) was used to summarize the expression of all TFEB target genes using the first principal component (PC1) as described in Berglund et al. (33), and PC1 was then compared to MYC expression. The list of 408 MYC dependent genes in B cells was derived using the RNA-Seq and ChIP-Seq data from (34); TFEB target genes were derived from data in (20); genes for amino acid transporters and proteasome components were derived from the Kyoto Encyclopedia of Genes and Genomes (KEGG). All gene lists are included in Supplementary Data Table S1.
To represent the effects of activation of TFEB on the expression of all MYC target genes, a PCA model was calculated using 413 MYC target genes for the six RNA-seq samples. The first principal component, PC1, explains 75.6% of the variation and was used to represent the overall change of MYC target genes.
Gene set enrichment analysis
GSEA was performed using GSEA (35) as implemented at www.gsea-msigdb.org/ and the different gene sets reported therein was used. Functional Annotation Clustering was performed using Pathway Interaction Database (PID) and KEGG using default settings.
RNA-seq analyses
RNA was extracted from Eμ-Myc;Eμ-rtTA2 lymphoma cells transduced with control (Vector) or TFEBSA-ERT2-expressing retrovirus that were treated with 4-OHT for 4 days as described above. Quality of RNA was confirmed using an Agilent TapeStation RNA ScreenTape (Agilent Technologies) and fluorometrically quantified using the Qubit RNA BR Assay Kit (ThermoFisher Scientific). The samples were then processed for RNA-sequencing (RNA-seq) using the NuGen Ovation Mouse RNA-Seq System (NuGen, Inc.). 100 ng of RNA was used to generate double-stranded cDNA and a ribosomal RNA-depleted strand-specific library following the manufacturer’s protocol (NuGEN, Inc.). Quality control steps including TapeStation library assessment and quantitative RT-PCR for library quantification. The libraries were then sequenced on the Illumina NextSeq 500 v2.5 sequencer with a 2 X 75-base paired-end high output run to generate an average of 39 million read pairs per sample. Sequencing reads were subjected to pre- and post-alignment QC measures before mapping against mouse reference genome mm10 using STAR-2.5.3a. Gene-level quantification was determined using HTSeq 0.6.1 by summation of raw counts of reads aligned to the region associated with each gene according to refSeq gene model. Read counts reported are normalized to library size estimates using the R package DESeq2 v1.6.3. Differential gene expression for treatment effects were assessed using DESeq2. Genes with Benjamini-Hochberg corrected p-value of less than ≤0.05 were considered significantly differentially expressed. The Gene Expression Omnibus (GEO) accession number for the RNA-seq data reported in this paper is GSE153570.
XF metabolic analysis
Pretreated Eμ-Myc;Eμ-rtTA2 lymphoma or Namalwa BL cells were plated in XFe96 microplates in unbuffered DMEM or RPMI containing 10 mM glucose, 1 mM sodium pyruvate, and 2 mM L-Glutamine (ThermoFisher) for mitochondrial stress test, glycolytic rate, or real-time ATP production assays at a n = 4–8 as per the manufacturer’s recommendations. For the XF PMP assays, cells were plated in 1x MAS-BSA solution containing 1 nM XF PMP Reagent (Agilent). All data analysis was performed in the Wave Software using the Mitochondrial Stress Test, Glycolytic Rate Assay, and ATP Production Rate Reports.
Statistical analysis
Data are reported as mean values ± standard deviation. Unpaired Student’s t-tests were performed utilizing the GraphPad Prism 8 Software. Bonferroni correction was applied when a set of comparisons were carried out. For comparison of survival curves, a Log-rank (Mantel-Cox) test Log-rank test was used. For RNA-Seq data, samples were normalized to universal mRNA content and Student’s t-tests were carried out under heteroscedastic parameters. For all tests defined above, statistical significance was defined by a two-tailed P≤0.05. For metabolomics analysis, samples were normalized by universal metabolite abundance and Student’s t-tests were carried out under heteroscedastic parameters, and statistical significance was defined by a two-tailed p≤0.1. MATLAB (R2020a) was used for PCA analysis, to generate Dot-Plot figures and Box-plots for gene expression and RNA-seq data.
Reagents
A detailed list of primers, antibodies, and primers used for this study are included in Supplementary Data Table S2.
Data availability
RNA-seq data have been deposited at NCBI as GEO (accession number GSE153570). The data underlying all findings of this study are available from the author upon request and are provided as separate source data files.
Results
Repression of TFEB and its targets is a hallmark of MYC-driven lymphoma
To initially assess if MYC controlled the autophagy-lysosomal pathway, three independent expression profiling datasets from the Eμ-Myc transgenic mouse (34,36,37), a validated model of human B cell lymphoma with MYC involvement (38), were queried for expression of target genes of TFEB, which coordinately controls expression of components of this recycling center (20,21). Wild type (WT) B220+ splenic B cells express high levels of Tfeb and many Tfeb target genes (Fig. 1A). Notably, the expression of Tfeb and nearly all Tfeb target genes are significantly suppressed in Eμ-Myc lymphoma (Fig. 1A), and in premalignant (age 4–8 weeks) B220+ splenic Eμ-Myc B-cells (Supplementary Fig. S1A and S1B). Further, Tfeb target genes expressed in WT B220+ bone marrow (BM) B cells are generally downregulated in malignant Eμ-Myc BM B cells (Supplementary Fig. S1C; Supplementary Data Table S1). Reduced expression of Tfeb and Tfeb targets genes in Eμ-Myc BM B cells was verified via RT-qPCR analysis of BM B cells from WT and Eμ-Myc littermates (Fig. 1B). To assess if this phenotype is manifest in human B cells and is MYC dependent, the expression of TFEB and its targets was assessed in P493–6 B lymphoma cells that harbor a TET repressible MYC transgene (39). Again, TFEB and TFEB target genes were inversely regulated in a low MYC (+ TET) versus high MYC (− TET) state (Fig. 1C), suggesting MYC antagonizes TFEB expression and function.
Figure 1.
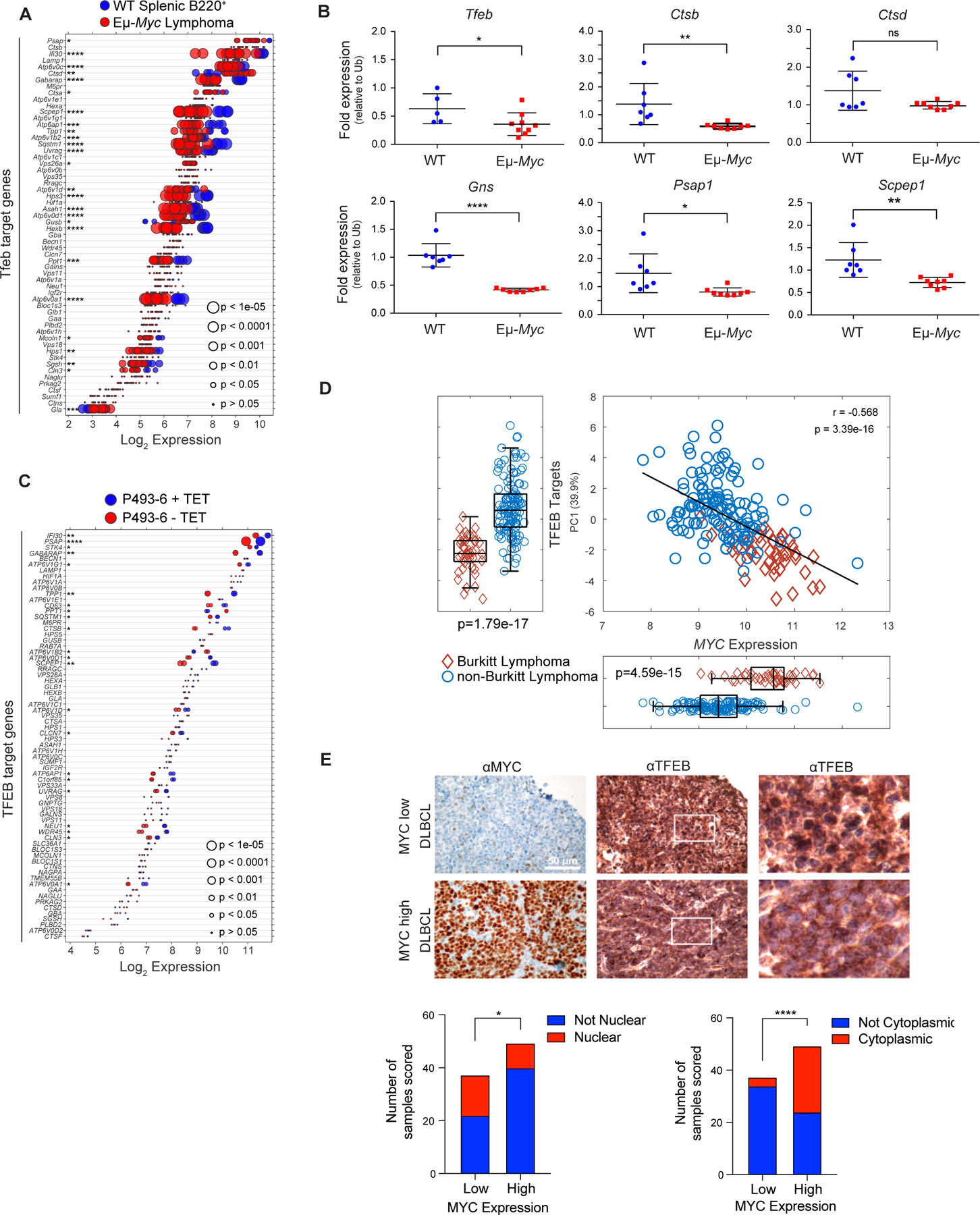
MYC suppresses the TFEB transcriptional program in B cell lymphoma. A, Gene expression profile (GSE32239) comparing splenic B220+ B cells from wild-type (WT; n = 4) and Eμ-Myc lymphomas (n = 13). Log2 gene expression of TFEB target genes are shown and are presented as a dot plot that is ordered based on expression. Each dot represents one sample, and the size corresponds to its statistical significance as shown. B, Quantitative real-time PCR (qRT-PCR) analyses of BM B220+ B cells from WT (blue symbols, n = 6) and premalignant Eμ-Myc mice (red symbols, n = 9) of Tfeb and select Tfeb target genes. C, Gene expression profile of TFEB target genes in human P493–6 B lymphoma cells (GSE40782) under either a MYC off state (n = 2) or MYC on state (n = 2). Log2 gene expression was plotted in a dot plot ordered based on expression; each dot represents one sample, and its size corresponds to its statistical significance as shown. D, Gene expression profiling comparing (D) 44 human Burkitt lymphoma and 129 human non-Burkitt B cell lymphoma samples (GSE4475) for the Log2 expression of MYC and TFEB target genes. The overall correlation of MYC expression to TFEB target genes is also shown. E, Immunohistochemistry staining of DLBCL samples classified as having low (n = 38) or high (n = 50) MYC expression with MYC and TFEB antibodies. TFEB staining was then classified and scored as either being localized to the nuclear (bottom left) or the cytoplasm (bottom right). Left and middle panels, scale bar is 50 μM. Right panels, magnified area of white outlined region of middle panels. Statistical analysis: B, Student’s t-test was performed; F, Fisher’s exact test was performed. *, p ≤ 0.05, ****, p ≤ 0.0001.
To determine if MYC alters the expression of TFEB and its targets in human B cell lymphoma, expression analyses were performed on Burkitt lymphoma (BL) that harbor MYC/Immunoglobulin gene chromosomal translocations and that express high MYC levels versus non-BL B cell lymphomas (40). Again, there are significant reductions in most TFEB targets (and there is a moderate correlation considering all TFEB target genes) in BL versus non-BL B cell lymphoma (Fig. 1D). Finally, this inverse relationship is manifest in multiple myeloma where TFEB target genes are generally downregulated in multiple myeloma samples expressing high levels of MYC in comparison to patient samples that express low levels of MYC (Supplementary Fig. S1D and S1E). Thus, there is an inverse relationship between MYC and TFEB and TFEB target genes in human hematological tumors with MYC involvement.
TFEB functions are also regulated by mTORC1, which phosphorylates TFEB on serine-211, sequestering TFEB in the cytosol by binding to 14–3-3 proteins (41). Under nutrient deprived conditions mTORC1 kinase activity is shut off and this leads to TFEB nuclear localization. While queries of DLBCL databases did not reveal statistically significant correlations between MYC and TFEB expression, we assessed if the localization of TFEB is affected in primary DLBCL and if this is linked to MYC expression. Indeed, lymphomas that express high levels of MYC protein generally express reduced levels of nuclear (i.e., active) TFEB (Fig. 1E, bottom left) and increased levels of cytoplasmic (i.e., inactive) TFEB (Fig. 1E, bottom right). In contrast, DLBCL that express low MYC levels generally have high levels of nuclear TFEB (Fig. 1E, bottom left).
A Myc-TFEB circuit is manifest in lymphopoiesis and is regulated by mitogens
c-Myc and N-Myc play essential roles in B cell progenitor development, and in the proliferative response of B cells to IL-7 (42,43). Analyses of mouse B cell development (44) revealed both c-Myc and N-Myc are expressed at high levels in early proliferating B cell progenitors, and that c-Myc expression drops as B cells differentiate (Fig. 2A). The expression of Tfeb and its targets is the inverse that of c-Myc or N-Myc, where low levels of TFEB and its targets are manifest in progenitors and higher levels are expressed in differentiated B cells (Fig. 2A; Supplementary Data Table S1). A similar pattern is observed in human B cells (45), where peripheral blood CD27−IgD+ B cells have high MYC and low TFEB levels whereas their expression pattern is the reverse in more differentiated CD27+IgD− B cells and in CD27−IgD− memory B cells (Supplementary Fig. S2A).
Figure 2.
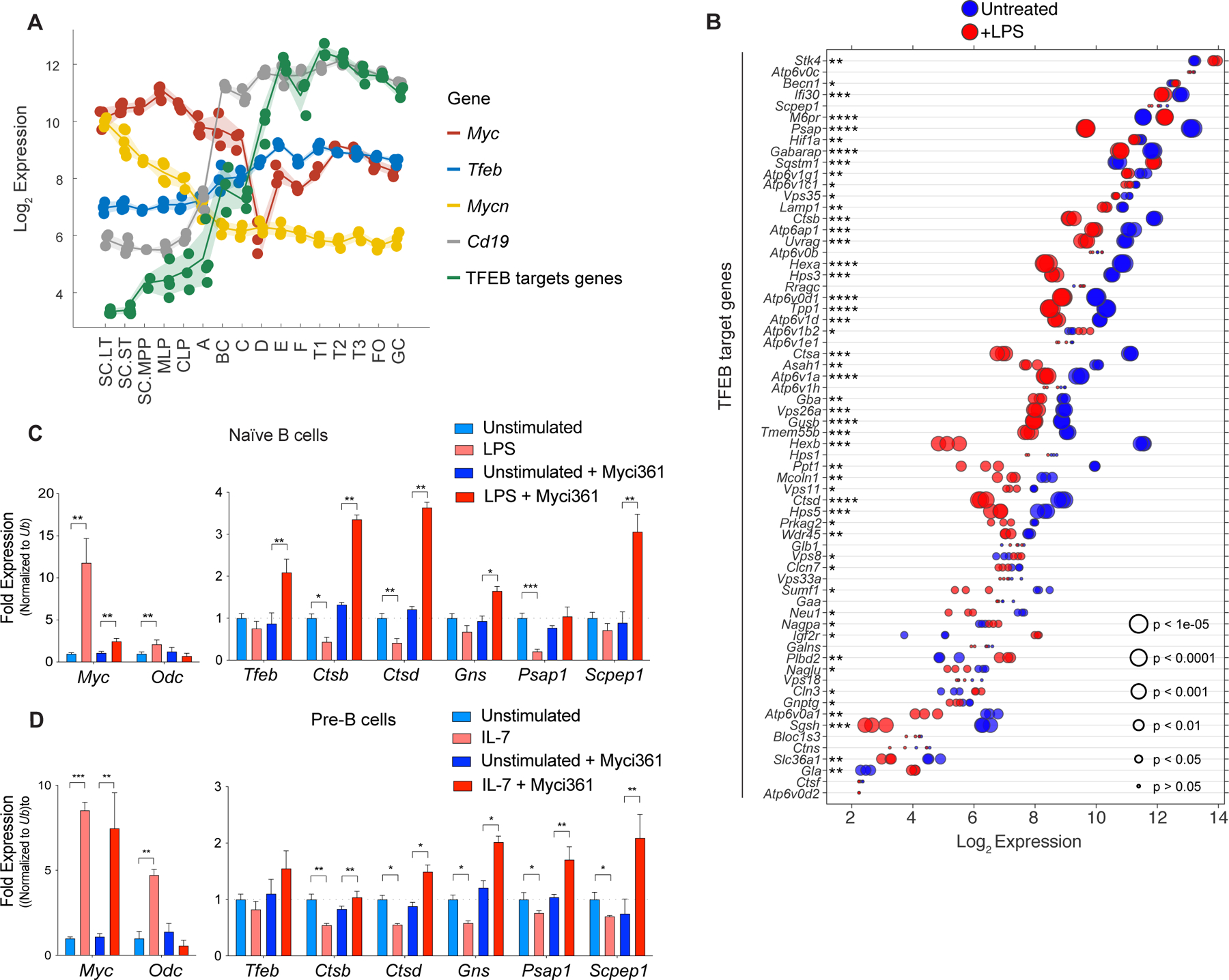
A MYC-TFEB circuit is manifest during B cell development and mitogenic signaling. A, Log2 expression levels of Tfeb, Tfeb target genes, Myc, Mycn and CD19 transcripts at different stages of mouse B cell development (Immgen Dataset). B, Gene expression profile of LPS-stimulated naïve mouse splenic B cells (GSE37222). Log2 gene expression of Tfeb target genes was plotted in a dot plot ordered based on expression. Each dot represents one sample, and dot size corresponds to its statistical significance as shown. C, qRT-PCR analyses of expression of Myc, Odc, Tfeb and the indicated TFEB target genes in naïve mouse splenic B220+ B cells (Unstimulated) or following stimulation of these cells with LPS for 4 hr (+ LPS), or naïve mouse splenic B220+ B cells pretreated with the Myc inhibitor Myci361 for 2 hr followed by stimulation of these cells with LPS for 4 hr (+ LPS) versus those not treated with LPS (Unstimulated). D, qRT-PCR analyses of Myc, Odc, Tfeb and TFEB target genes in pre-B cells that were deprived of IL-7 for 18 hr (Unstimulated) and then treated with IL-7 (+ IL7) for 6 hr in the absence or presence of the Myc inhibitor Myci361. Statistical analysis: C-D, Student’s t-tests were performed. Data are represented as mean ± SD (n = 3). *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001.
Expression of MYC normally relies on mitogenic signaling, and interleukin-7 (IL-7) or lipopolysaccharide (LPS) treatment of primary B cells induces Myc expression (43). To assess control of Tfeb and its targets by B cell mitogens, we first analyzed expression datasets from naïve mouse splenic B cells activated with LPS (46). Interestingly, levels of most TFEB target genes were repressed by LPS treatment (Fig. 2B). PCR expression analyses of LPS-treated splenic mouse B220+ B cells confirmed these findings (Fig. 2C).
Myc binding to and regulation of its target genes requires its dimerization with the bHLH-Zip protein Max (47). As expected, treatment of pre-B cells with the MYC/MAX dimerization inhibitor Myci361 (48) blocked LPS-mediated induction of the Myc target gene ornithine decarboxylase (Odc) without affecting the induction of c-Myc by LPS (Fig. 2D). Notably, Myci361 treatment induced Tfeb expression and blocked LPS-directed repression of Tfeb targets (Fig. 2C). Similarly, qRT-PCR analyses of primary bone marrow derived pre-B cells showed that IL-7 treatment led to robust induction of c-Myc but to repression of Tfeb target genes (Fig. 2D), and again IL-7 directed repression of Tfeb target genes (and the induction of Odc) was blocked by pretreatment with Myc inhibitors (Fig. 2D; Supplementary Fig. S2B). Similarly, treatment of Eμ-Myc lymphoma cells with Myc inhibitors led to up-regulation of Tfeb and its target genes while suppressing the expression of Odc (Supplementary Fig. S2C and S2D). Thus, MYC suppresses Tfeb and antagonizes the control of Tfeb target genes in primary mouse B cells and B cell lymphoma.
Lysosomal biogenesis and autophagic flux are repressed by MYC
Repression of Tfeb and its target genes by Myc and mitogenic signaling suggested suppression of the autophagy-lysosomal circuit. To test this, lysosomal mass and function were first assessed using LysoTracker staining in models where Myc expression can be manipulated, including naïve mouse splenic B cells +/− LPS, primary BM derived pre-B cells +/− IL7, and P493–6 human B lymphoma cells +/− tetracycline (TET). LysoTracker staining revealed reduced lysosomal mass in a MYC-on state in all three models (Fig. 3A). In contrast, treatment of LPS-stimulated splenic B cells or IL-7-stimulated pre-B cells with Myc inhibitors led to increases in lysosomal mass (Fig. 3B).
Figure 3.
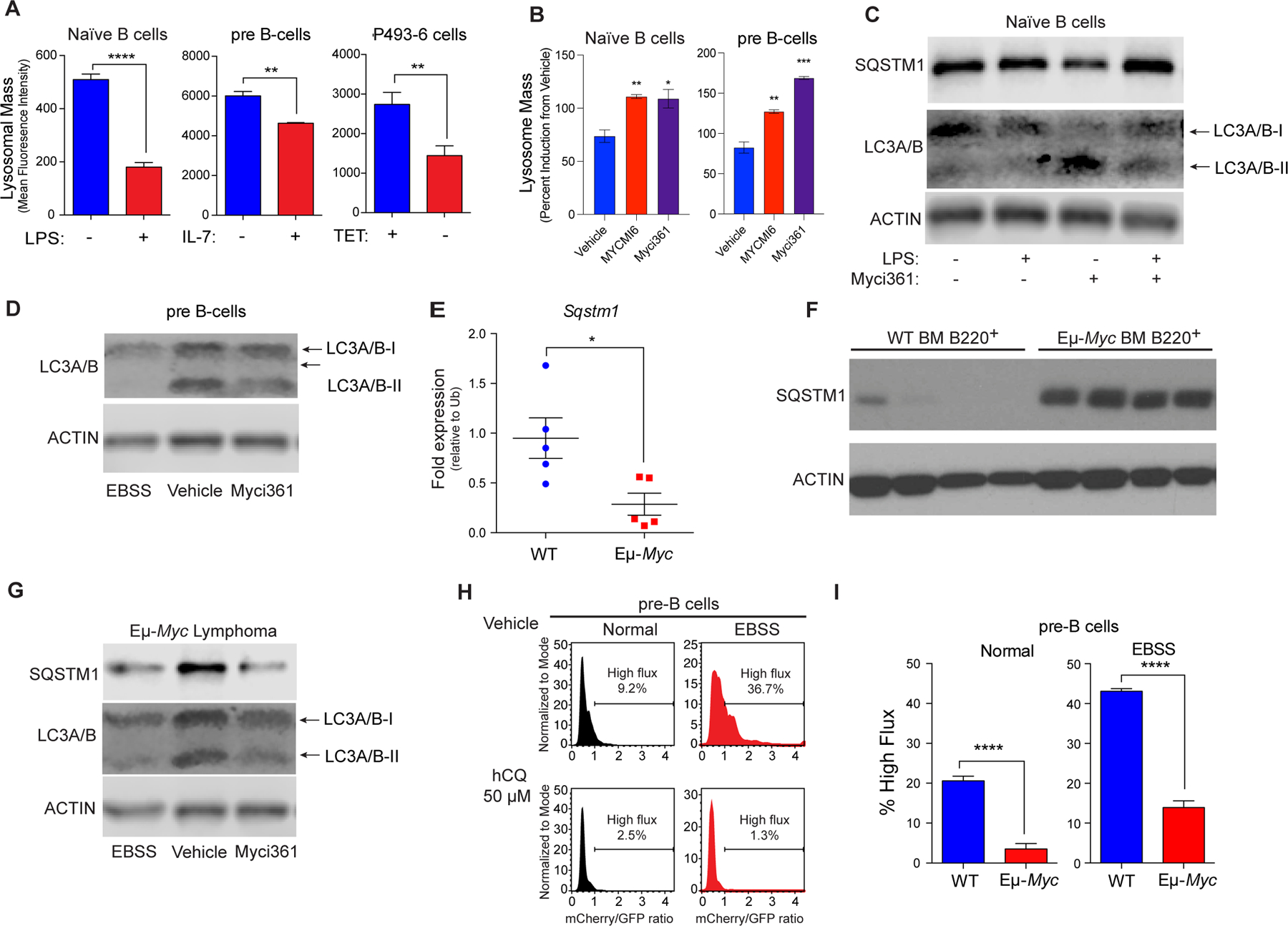
MYC blocks autophagic flux. A, Mean fluorescent Intensity (MFI) of Lysotracker staining in (left to right): untreated vs. LPS-treated (6 hr) naïve splenic mouse B cells; IL-7 deprived (18 hr) pre-B cells that were then re-stimulated with IL-7 for 6 hr; and P493–6 B lymphoma cells treated with TET for 48 hr and released from TET for 4 hr (n = 3 for all). B, MFI of LysoTracker staining of naïve B or pre-B cells treated with vehicle or with the Myc inhibitors Myci361 or MYCMI6 for 2 hr prior to stimulation with either LPS or IL-7, respectively. C, Western blot analysis of SQSTM1, and LC3A/B isoforms in naïve splenic B220+ B cells that were left untreated or pretreated with Myci361 for 2hr followed by treatment with or without LPS for 4 hr. D, Western blot analysis of LC3A/B isoforms in pre-B cells (cultured in IL-7 medium) that were treated with vehicle or with Myci361 for 2 hr, or that were shifted to EBSS medium for 6 hr. E and F, qRT-PCR (E) and Western blot (F) analysis of SQSTM1 levels in WT versus premalignant Eμ-Myc BM B220+ cells (n = 5 for E). G, Levels of SQSTM1 and LC3A/B isoforms levels in Eμ-Myc Lymphoma after treatment with the Myc inhibitor Myci361 for 2 hr or following culture in EBSS media for 6 hr. Actin was used as a loading control for all immunoblots. H and I, Autophagic flux analyses in (H) pre-B cells treated with 50 μM hydroxychloroquine (hCQ), as measured in cells cultured in normal vs. EBSS media for 6 hr, or in (I) BM-derived WT vs. Eμ-Myc pre-B cells. The ratio of GFP to mCherry from the retrovirally expressed mCherry-GFP-LC3 fusion protein was calculated and the percent of cells having high flux is shown (n = 3). Statistical analysis: A, B, E, and I, Student’s t-tests were performed. *, p ≤ 0.05; ***, p ≤ 0.001; ****, p ≤ 0.0001.
To assess if modulating Myc expression or function was associated with functional changes in levels of proteins degraded by the autophagosome-lysosome circuit, we evaluated levels of SQSTM1/p62, a receptor for ubiquitylated cargo on autophagosomes (49), and the levels of LC3A/B-I and LC3A/B-II, the phosphatidyl-ethanolamine-modified form of LC3 that is degraded following fusion autophagosomes with lysosomes. As predicted, treatment of naïve splenic B cells with LPS reduced the protein levels of SQSTM1/p62 and of LC3A/B-I and LC3A/B-II, consistent with increased autophagy flux, and co-treatment with the Myci361 inhibitor impaired LPS-mediated reductions in SQSTM1/p62 and in LC3A/B-I and LC3A/B-II (Fig. 3C). Further, treatment of pre-B cells cultured in IL-7 with the Myci361 inhibitor reduced levels of LC3A/B-II, consistent with the induction of the autophagy pathway as seen in these cells cultured in Earle’s balanced salt solution (EBSS) media that lacks amino acids and thus activates autophagic flux (29) (Fig. 3D). Notably, although there were reductions in Sqstm1 transcripts in Eµ-Myc versus WT BM B220+ B cells (Fig. 3E) there were marked increases in SQSTM1/p62 protein levels in Eµ-Myc B cells (Fig. 3F), and levels of SQSTM1/p62 and LC3A/B-II that are manifest in Eµ-Myc B lymphoma cells were reduced following treatment with Myci361 or by culture in EBSS media (Fig. 3G). Finally, there were also reductions in numbers of LC3 punctae following MYC induction in p493–6 B lymphoma cells (Supplementary Fig. S3A).
To directly assess if autophagic flux is repressed in Myc-expressing B cells, primary WT and Eµ-Myc pre-B cells were transduced with a retroviral vector expressing the fusion reporter GFP-mCherry-LC3 (50) (Supplementary Fig. S3B). GFP+ cells were then cultured in replete medium or in EBSS media to activate autophagic flux. The increases in autophagic flux induced by brief culture (2 hr) in EBSS, as documented by increases in the ratio of mCherry to eGFP, were dependent on autophagy, as they were blocked by treatment of WT pre-B cells with hydroxychloroquine (hCQ) (Fig. 3H) and were not observed in mouse embryo fibroblasts (MEFs) lacking Atg7 (Supplementary Fig. S3C). Notably, basal rates of autophagic flux were reduced 5-fold in Eµ-Myc versus WT pre-B cells, and reductions in autophagic flux were sustained when these cells were shifted to EBSS (Fig. 3I; Supplementary Fig. S3D). Thus, autophagic flux is suppressed in Myc-driven B cell lymphoma.
TFEB functions as a tumor suppressor that disables Myc-driven lymphoma
To assess the significance of Myc-directed suppression of Tfeb, Eμ-Myc lymphoma cells were transduced with a control retrovirus (vector) or a retrovirus that expresses a mutant form of TFEB, TFEBS211A (here designated TFEBSA) that cannot be phosphorylated and inactivated by mTORC1 (51), and which we fused in frame to the estrogen-binding domain of the estrogen receptor (ER) modified such that it is activated by treatment of cells with the ER agonist 4-hydroxytamoxifen (TFEBSA-ERT2). As predicted, induction of TFEBSA-ERT2 activity by 4-OHT treatment led to increases in lysosomal mass in Eμ-Myc lymphoma cells (Fig. 4A); thus, TFEBSA can induce lysosome biogenesis. Increased lysosomal mass was also observed in Eμ-Myc lymphoma cells transduced with a retrovirus constitutively expressing TFEBSA and iGFP and that were then immediately sorted for GFP expression (Fig. 4B). Activation of TFEBSA in Eμ-Myc lymphoma cells led to significant effects on the transcriptome as 956 genes were found to be statistically differentially regulated via RNA-seq analysis (Supplementary Fig. S4A and Supplementary Data Table S3). Further, qRT-PCR and RNA-seq analyses of vector versus TFEBSA-ERT2-expressing lymphoma cells revealed that 4-OHT activation of TFEBSA consistently induced TFEB target genes in Eμ-Myc lymphoma cells (Fig. 4C and D, and Supplementary Data Table S3), while repressing the activity of most Myc target genes (Supplementary Figs. S4B and S4C). Gene set enrichment analysis of the Pathway Interaction Database (PID) and Kyoto Encyclopedia of Gene and Genomes (KEGG) Database of the RNA-seq data revealed that 4-OHT activation of TFEBSA particularly upregulated Myc-repressed pathways, Caspase activation, and lysosome biogenesis, while downregulating Myc-activation pathways and growth promoting pathways such as MAPK, JAK/STAT, and E2F signaling (Figs. 4E and F). Notably, PCA revealed that the activation of TFEBSA-ERT2 with 4-OHT in Eμ-Myc lymphoma profoundly suppressed Myc target genes (Fig. 4G) but did not affect levels of Myc protein (Fig. 4H); thus, the effects of TFEBSA in Eμ-Myc lymphoma cells are not due to suppressing the Myc transgene per se but are rather due to functional antagonism of Myc signaling.
Figure 4.
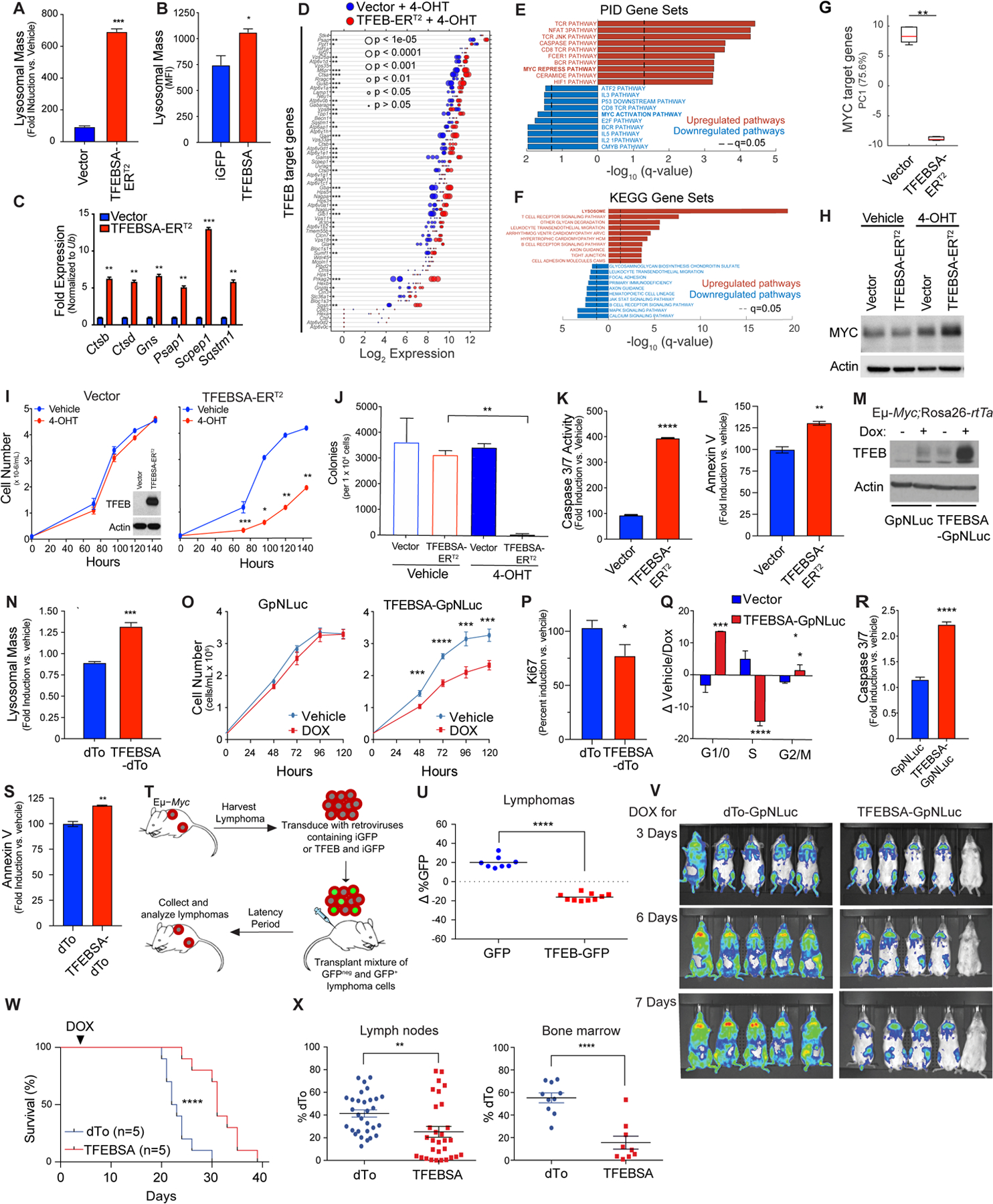
TFEB functions as a tumor suppressor in Myc-driven lymphoma. A, Eμ-Myc lymphoma cells expressing vector or TFEBSA-ERT2 were treated with vehicle or 25 nM 4-OHT for 4 days and analyzed for lysosomal mass by flow cytometry after staining cells with Lysotracker DND99. B, Eμ-Myc lymphoma cells were transduced for 48 hr with retroviruses expressing GFP or TFEBSA plus GFP, and GFP+ cells were analyzed for lysosomal mass. C and D, Eμ-Myc lymphoma cells expressing vector or TFEBSA-ERT2 were treated as in (A), and then analyzed for the indicated TFEB target genes by qRT-PCR (C) or RNA-seq (D) (n = 3 for both). For (D) the Log2 gene expression profile of TFEB target genes is shown as a dot plot that is ordered based on expression; each dot represents one sample, and its size corresponds to its statistical significance as shown. E and F, Gene set enrichment analysis of significant, differentially expressed genes from RNA-seq analysis were performed using the (E) Pathway Interaction Database (PID) and (F) Kyoto Encyclopedia of Genes and Genomes (KEGG) databases. G, Box plot comparing the change of canonical MYC target genes (PC1) in Vector + 4-OHT vs. TFEBSA-ERT2 + 4-OHT RNA-seq data. H, Immunoblot analysis of Myc protein levels in Eμ-Myc lymphoma cells expressing vector or TFEBSA-ERT2 and treated as in (A). I-M, Eμ-Myc lymphoma cells expressing vector or TFEBSA-ERT2 were assessed for: (I) cell proliferation over 6 days (inset shows TFEBSA-ERT2 protein expression); (J) colony-forming potential in methylcellulose (day 14); and (K and L) apoptosis after 4 days of 4-OHT treatment, by measuring Caspase 3/7 and Annexin V staining, respectively (I-L, n = 3). M-S, Western blot analysis of TFEBSA and Actin protein levels (M) in Eμ-Myc;Eμ-rtTA2 lymphoma cells expressing vector (GpNLuc or dTo) or TFEBSA (TFEBSA-GpNLuc or TFEBSA-dTo) two days after treatment with vehicle or Dox; (N) Lysosomal mass of these cells treated +/− Dox (48 hr); (O) cell numbers over 120 hr; (P) cell proliferation index, as determined by Ki67 staining (48 hr); (Q) cell cycle analysis with propidium iodide (48 hr); and (R, S) apoptosis (Caspase 3/7 activity and Annexin V staining after 48 hr). T and U, Eμ-Myc lymphoma cells were transduced with retrovirus expressing GFP or GFP plus TFEBSA. (T) A 50:50 mix of control or TFEB-expressing GFP+:GFPNeg Eμ-Myc lymphoma cells from each transduction was transplanted i.v. into congenic recipient mice (n = 8). (U) Lymphomas arising in transplanted animals in (S) were assessed for percent of GFP+ lymphoma cells. V, In vivo imaging of Nod/Scid mice transplanted with Eμ-Myc;Eμ-rtTA2 lymphoma cells expressing vector (GpNLuc) or TFEBSA-GpNLuc at day 3, 6 and 7 after doxycycline (Dox) chow was provided ad libitum. W, Survival of syngeneic mice transplanted with Eμ-Myc;Eμ-rtTA2 lymphoma cells expressing vector (dTo) or TFEBSA-dTo; Dox chow was provided at day 3 post-transplant (arrow). X, Percent dTo+ B220+ cells isolated from lymph nodes or bone marrow of diseased recipient mice in (V). Statistical analysis: A-C, I-L, N-S, U and X: Students t-tests were performed. W, Chi-square test was performed. *, p ≤0.05; ** p ≤0.01; ***, p ≤ 0.001, ****, p ≤ 0.0001.
Surprisingly, activation of TFEBSA-ERT2 with 4-OHT significantly impaired the growth of Eμ-Myc lymphoma (Fig. 4I). Further, 4-OHT activation of TFEBSA-ERT2 abolished long term 3D growth of Eμ-Myc lymphoma cells in methylcellulose (Fig. 4J) and triggered apoptosis, as measured by marked increases in caspase-3/7 activity and Annexin V staining (Fig. 4K and L).
Collectively, these studies suggested TFEB acts as a tumor suppressor in the context of MYC-driven lymphoma. To test this hypothesis, Eμ-Myc transgenic mice were crossed to Rosa26-rtTA2 transgenic mice that ubiquitously express the reverse tetracycline transactivator (rtTA2) (52), and lymphomas arising in Eμ-Myc;Rosa26-rtTA2 double transgenics were transduced with retroviruses that express a doxycycline (Dox)-inducible TFEBSA transgene along with the imaging reporters dTomato (dTo) or GpNLuc (53). As expected, Dox-mediated induction of TFEBSA (Fig. 4M) led to increased lysosomal mass, impaired cell growth rates, decreased levels of the proliferation marker Ki67, an accumulation of cells in the G1 phase of the cell cycle (at the expense of cells in S phase), and an increase in the apoptotic index (Fig. 4N–S).
Two syngeneic transplant models were used to test the tumor suppressor activity of TFEB in vivo. First, Eμ-Myc lymphomas were transduced with control GFP or TFEB-iGFP retrovirus, and each recipient mouse received a 50:50 mix of GFP+ and GFPNeg lymphoma cells (Fig. 4T). When disease was manifest the percentage of GFP+ B220+ cells in tumors in the two cohorts was determined. Notably, there was a selection against TFEB-iGFP-expressing lymphoma cells versus GFP-only expressing lymphoma cells (Fig. 4U). Second, Eμ-Myc;Eμ-rtTA2 lymphomas were transduced with retroviruses that constitutively express GpNLuc and that inducibly express just dTo or dTo plus TFEBSA. GFP+ lymphoma cells were isolated by flow cytometry and transplanted into syngeneic mice. After three days recipients were shifted to Dox chow to induce TFEBSA transgene expression and disease was monitored by IVIS imaging. Induction of TFEBSA expression impaired tumor progression (Fig. 4V) and recipient mice bearing TFEBSA-expressing lymphoma cells survived significantly longer than those transplanted with the control dTo virus-transduced lymphomas that retained dTo expression (Fig. 4W). Finally, in accord with the notion that TFEB functions as a tumor suppressor, many of the tumors arising in lymph nodes and BM of recipient mice transplanted with TFEBSA-expressing lymphoma lost expression of dTo and thus also TFEB, consistent with silencing or loss of the virus (Fig. 4X).
To determine if the effects observed in Eμ-Myc lymphoma were applicable to human MYC-driven B cell lymphoma, we transduced Namalwa cells, a BL cell line that overexpresses MYC, with a lentivirus expressing a control short-hairpin targeting Renilla luciferase or a short-hairpin targeting MYC (26). Reductions in the level of MYC mRNA led to increases in the expression of TFEB and TFEB target genes and to elevated lysosomal mass (Supplementary Fig. S4D and S4E). To assess if the effects of TFEBSA were applicable to Namalwa cells, these BL cells were also engineered to express TFEBSA-ERT2 or TFEBSA. Notably, activation of TFEBSA-ERT2 with 4-OHT, or constitutive expression of TFEBSA, elevated lysosomal mass (Supplementary Fig. S4F and S4G), without affecting MYC protein levels (Supplementary Fig. S4H). Further, TFEBSA-ERT2 activation impaired the growth of Namalwa cells in cell culture and especially in methylcellulose (Supplementary Fig. S4I and S4J). In this model the inhibitory effects of TFEBSA were however cytostatic, as there were no changes in viability following activation of TFEBSA (Supplementary Fig. S4K). Nonetheless, in Namalwa BL cells that were engineered to express the Dox-inducible TFEBSA system, the induction of TFEBSA levels (Supplementary Fig. S4L) induced lysosomal mass, repressed cell growth, and led to G1 arrest (Supplementary Fig. S4M–S4O). Finally, in nude mice subcutaneously transplanted with Namalwa BL cells, activation of TFEBSA-ERT2 following oral gavage with tamoxifen improved overall survival versus recipient mice bearing TFEBSA-ERT2-expressing tumors treated with vehicle (Supplementary Fig. S4P). Thus, TFEB also acts as a tumor suppressor in Burkitt lymphoma.
To test if super-activation of Myc in Eμ-Myc lymphomas could bypass effects of TFEBSA activity, TFEBSA-ERT2- or vector-only expressing Eμ-Myc lymphomas were transduced with retroviruses expressing MYC-ER and GFP or only GFP, and cells were sorted for GFP expression (Supplementary Fig. S4Q). Cell number, viability and lysosomal mass were then examined in GFP+ cells treated with 4-OHT. While MYC-ER activation modestly suppressed lysosomal mass (Supplementary Fig. S4R), this did not revert inhibitory effects of TFEBSA on cell proliferation or survival (Supplementary Fig. S4S and S4T); thus, phenotypes driven by TFEBSA are dominant over those controlled by MYC in Myc-driven lymphomas.
Finally, the suppression of the autophagy-lysosome pathway by Myc suggested that loss-of-function mutations in the autophagy pathway would have little impact on lymphoma development. To test this, conditional Atg7 (Atg7fl/fl) and Tfeb (28) (Tfebfl/fl) knockout mice were crossed to Eµ-Myc;CD19-Cre mice to selectively delete these genes in B cells. Notably, lymphoma onset and overall survival were similar in the wild type, heterozygous and null Atg7 or Tfeb cohorts for each model (Supplementary Fig. S4U and S4V). Thus, the autophagy-lysosome pathway is dispensable for the development of Myc-driven lymphoma.
Induction of the proteasome is a hallmark of Myc-driven lymphoma
The autophagy pathway is thought to be essential for maintenance of amino acids (AA) pools (54). Given Myc-dependent suppression of autophagy, we assessed if there were changes in intracellular AA pools in WT versus premalignant Eµ-Myc pre-B cells. Levels of AA were equal to, or were significantly increased (Ile, Val, Leu, Gln, Glu, Gly and Arg), in primary Eµ-Myc pre-B cells versus WT pre-B cells (Supplementary Fig. S5A). Thus, compensatory mechanisms must maintain AA pools in Myc-expressing B cells.
Macropinocytosis is a major source of AA pools for K-Ras-driven cancers (12,15). However, macropinocytosis was not elevated in Eμ-Myc B cells versus wild type B cells (Supplementary Fig. S5B). Thus, we assessed if alterations in the proteasome were manifest in Myc-expressing B cells. Notably, there were significant increases in the expression of many regulatory and catalytic proteasome genes in premalignant and neoplastic Eμ-Myc B cells versus WT B cells (Fig. 5A; Supplementary Fig. S5C; Supplementary Data Table S1), and in LPS-stimulated naïve splenic B cells (Fig. 5B). This pattern was also manifest in human P493–6 lymphoma cells in the MYC-on state (Supplementary Fig. S5D), and a similar trend was evident in multiple myeloma with MYC involvement (Supplementary Fig. S5E and S5F). Importantly, elevated expression of proteasome components was associated with increases in proteasome activity in Eμ-Myc versus WT B220+ BM B cells, and in Eμ-Myc versus WT pre-B cells (Fig. 5C and D). Conversely, the proteasome activity of pre-B cells grown in IL-7 was reduced following treatment with Myc-Max dimerization inhibitors (Fig. 5E). Finally, the activity of the proteasome was also induced by mitogenic stimulation of primary naïve B cells (Fig. 5F). Thus, the proteasome and the autophagy-lysosome pathway are coordinately and inversely regulated by Myc and mitogenic signaling in B cells.
Figure 5.
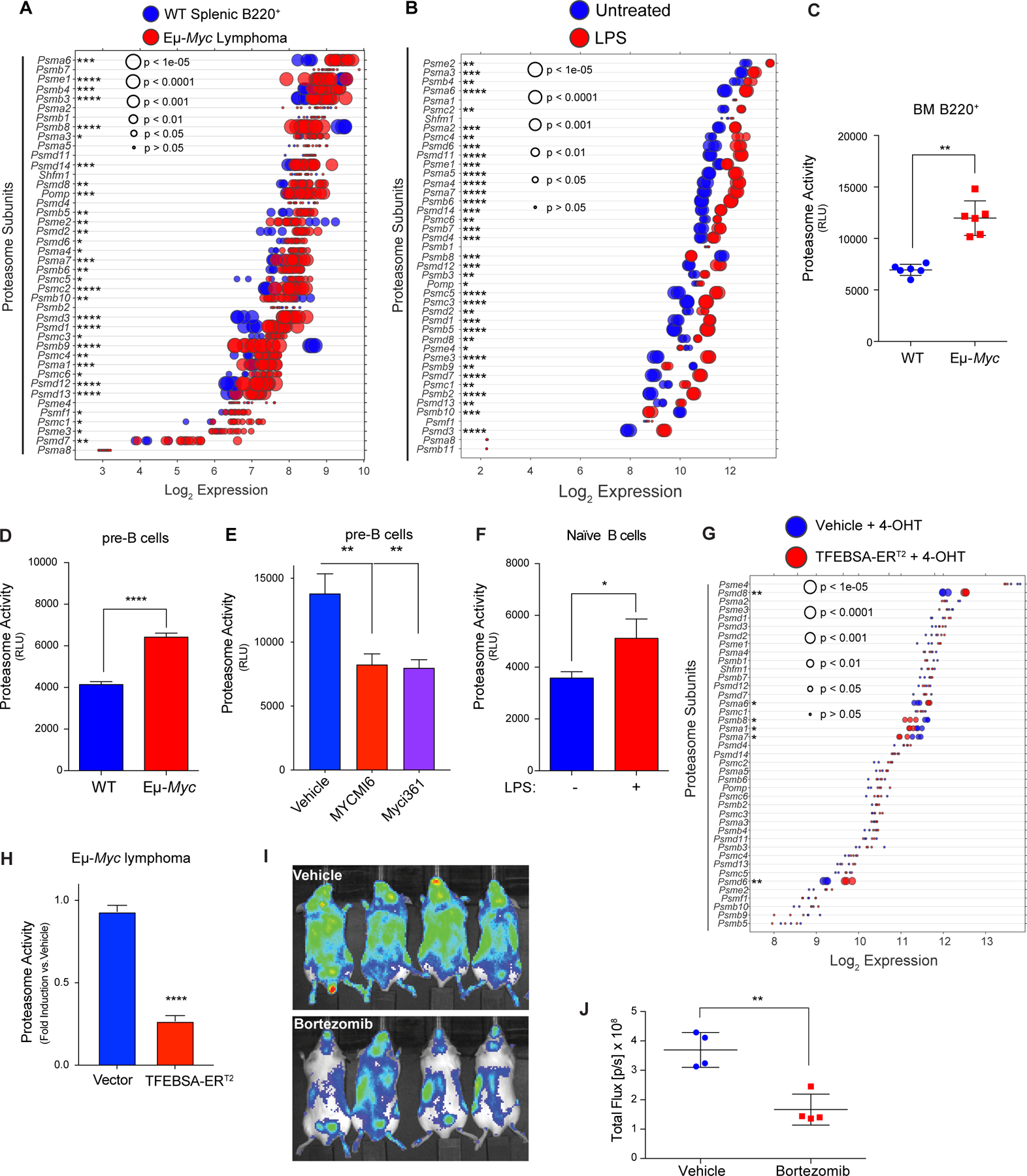
MYC induces expression and activity of the proteasome in B cell lymphoma. A and B, Gene expression profile (A) of proteasome subunits in splenic B220+ B cells from wild-type (WT) and from Eμ-Myc lymphomas (GSE32239), and (B) of proteasome subunits in untreated vs. LPS (4 hr) stimulated naïve B cells (GSE37222). Log2 gene expression profile of TFEB target genes is shown as a dot plot ordered based on expression; each dot represents one sample, and its size corresponds to its statistical significance as shown. C-F, Proteasome activity was measured using Proteasome-Glo® in: (C) WT vs. Eμ-Myc B220+ BM cells (n = 6); (D) WT vs. Eμ-Myc pre-B cells cultured in IL7; (E) WT pre-B cells treated with vehicle or with the Myc inhibitors Myci361 or MYCMI6 for 2 hr; and (F) naïve mouse splenic B cells that were untreated (MYC-Off) or LPS-stimulated (4 hr, MYC-On) (D-F, n = 3). G, Log2 gene expression of proteasome-associated genes in Eμ-Myc lymphoma cells expressing vector or TFEBSA-ERT2 4 days after vehicle or 4-OHT treatment is shown as a dot plot ordered based on expression; each dot represents one sample, and its size corresponds to its statistical significance as shown (n = 3 for each cohort). H, Proteasome activity, measured using Proteasome-Glo®, in Eμ-Myc lymphoma cells expressing vector or TFEBSA-ERT2 after treatment with vehicle or 4-OHT for 4 days (n = 3). I and J, In vivo imaging of NOD-SCiD mice intravenously transplanted with Eμ-Myc;Eμ-rtTA2 lymphoma cells expressing either vector (GpNLuc) or TFEBSA-GpNLuc that were treated with vehicle or 0.25 mg/kg bortezomib (i.p. weekly) for 10 days (I). Average bioluminescence for treated with vehicle or Bortezomib for 10 days (J). Statistical analysis: C-F and J, Students t-tests were performed. *, p ≤0.05; ** p ≤0.01; ****, p ≤ 0.0001.
Notably, induction of TFEBSA-ERT2 activity following treatment with 4-OHT suppressed the expression of some of the proteasome components that are elevated in Eμ-Myc lymphoma cells (Fig. 5G) and significantly reduced proteasome activity in both Eμ-Myc lymphoma and Namalwa BL cells (Fig. 5H; Supplementary Fig. S5G). Finally, proteasome activity in Namalwa BL cells was also suppressed following knockdown of MYC expression via Dox-inducible expression of MYC-targeting shRNA (Supplementary Fig. S5H).
Proteasome inhibition leads to lethal shortages in AA pools (13). Accordingly, up-regulation of the proteasome in Myc-expressing B cells and in MYC-driven lymphoma suggested these tumors would be hypersensitive to proteasome inhibitors. Indeed, Eμ-Myc lymphomas are highly sensitive to low doses (1–5 nM) of bortezomib, which abolished their proliferation (Supplementary Fig. S5I). Further, bortezomib treatment and imaging of syngeneic recipient mice transplanted with Eμ-Myc lymphomas expressing the imaging reporter GpNLuc revealed these tumors were highly sensitive to proteasome inhibition (Fig. 5I and J). Thus, up-regulation of the proteasome in MYC-driven lymphoma evokes actionable vulnerabilities to proteasome inhibitors.
Up-regulation of amino acid transport in Myc-driven lymphoma
Myc induces transcription of Asct2 (SLC1A5) and LAT1 (SLC7A5) solute transporters that direct uptake of glutamine (Asct2) or of neutral branched chain (Leu, Ile, Val) and aromatic (Tyr and Trp) amino acids (LAT1) (7). We therefore reasoned Myc-expressing B cells might also up-regulate the expression of AA transporters. Expression analyses revealed that select amino acid transporters were significantly up-regulated in premalignant and neoplastic Eμ-Myc B cells, in p493–6 B-lymphoma cells in the MYC-on state, and in multiple myeloma having MYC involvement (Fig. 6A; Supplementary Fig. S6A–D; Supplementary Data Table S1). These include Slc1a4 (Asct1; Ser, Ala, Cys, Thr), Slc1a5 (Asct2; Gln), Slc3a2 (CD98, the heavy chain for LAT1), Slc7a1 (Arg, Lys), Slc7a5 (LAT1; Leu, Ile, Val, Trp, Tyr), Slc36a4 (Pro, Trp), and Slc38a2 (Gln). qRT-PCR analyses confirmed up-regulation of Slc1a4, Slc7a1 and Slc7a5 in Eμ-Myc versus WT B220+ BM B cells (Supplementary Fig. S6E). Amino acid transporters were also up-regulated by mitogenic stimulation of primary naïve B cells (Fig. 6B). Notably, these increases in expression had functional consequences, where there are increases in uptake of 14C-labeled AA (all twenty AA) in Eμ-Myc B220+ BM and pre-B cells versus WT B cells (Fig. 6C and D). Finally, AA uptake is also mitogen dependent in naïve splenic B cells (Fig. 6E).
Figure 6.
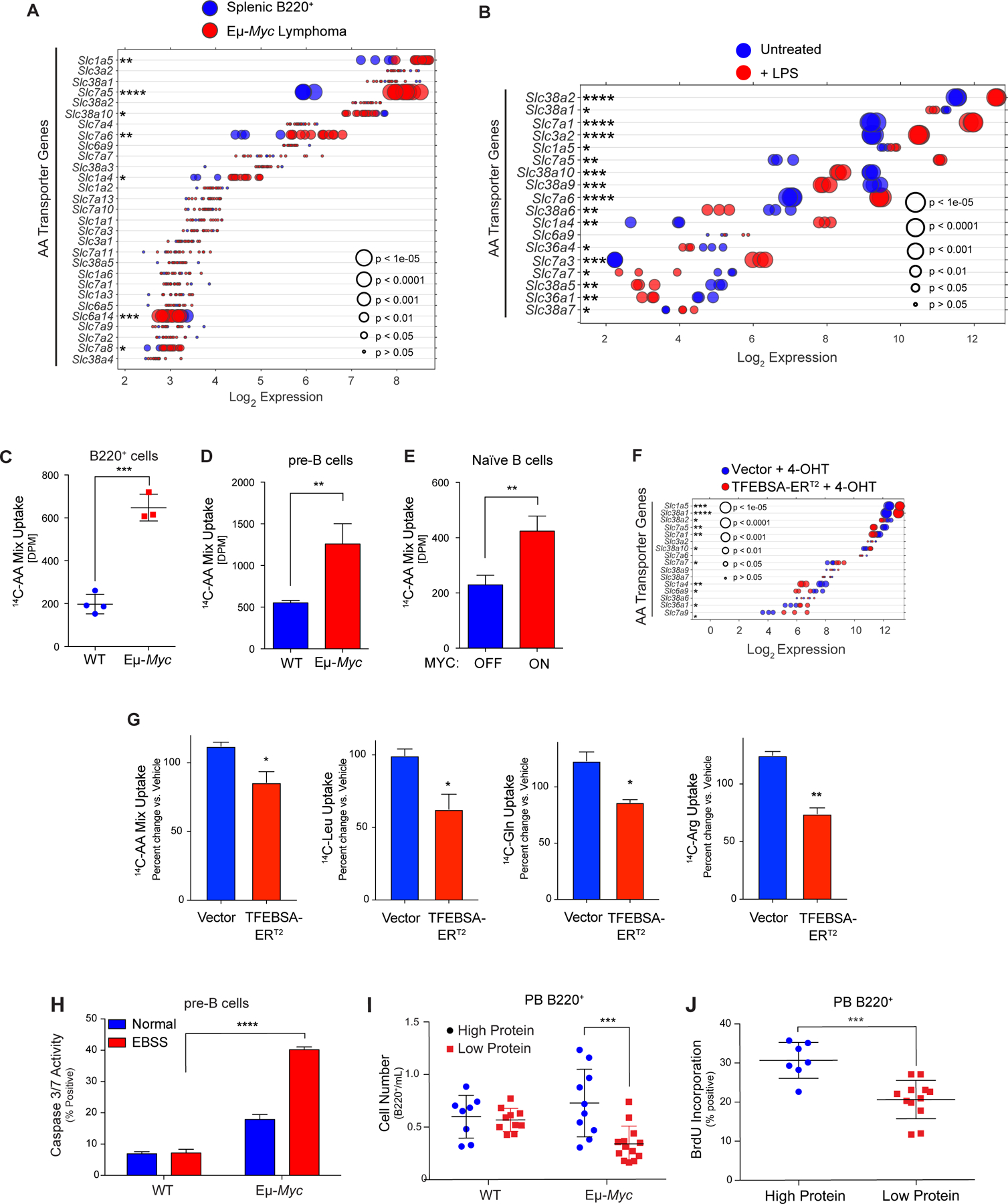
MYC induces expression and activity of select amino acid transporters in B lymphoma. A and B, Gene expression of amino acid transporters in (A) of splenic WT and neoplastic Eμ-Myc B cells (GSE32239), and (B) untreated and LPS-stimulated mouse splenic B cells (GSE37222). Log2 mRNA levels are shown in a dot plot that is ordered based on expression; each dot represents one sample, and the size corresponds to its statistical significance as shown. C-E, Uptake of 14C-labeled amino acids in (C) WT vs. Eμ-Myc B220+ BM cells; (D) WT vs. Eμ-Myc pre-B cells cultured in IL-7; and (E) untreated (MYC-Off) or LPS-stimulated (4 hr, MYC-ON) primary splenic B cells. F, Log2 gene expression profile of genes encoding select amino acid transporters are shown in a dot plot ordered based on their expression in Eμ-Myc lymphoma cells expressing vector or TFEBSA-ERT2 4 days after treatment with vehicle or 4-OHT; each dot represents one sample, and its size corresponds to its statistical significance as shown. G, Uptake of indicated 14C-labeled amino acids in Eμ-Myc lymphoma cells expressing vector or TFEBSA-ERT2 after treatment with vehicle or 4-OHT (4 days). H, Pre-B cells isolated from WT vs. Eμ-Myc mice were plated in normal or EBSS media for 3 hr and apoptosis measured via Caspase3/7 activity. I, WT and Eμ-Myc littermate mice were switched to a chow containing either high (20%) or low (6%) protein three weeks after birth. The number of peripheral blood (PB) B220+ cells were determined after 1 week. J, 6-week-old WT and Eμ-Myc littermate mice were switched to a chow containing either high (20%) or low (6%) protein for one week and were then injected i.v. with BrdU and incorporation was measured in PB B220+ cells. Statistical analysis: C-E and G-J, Students t-tests were performed.*, p ≤0.05; ** p ≤0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001.
Again, activation of TFEBSA-ERT2 inversely regulated the expression of AA transporters manifest in Eμ-Myc lymphoma cells (Fig. 6F) and significantly suppressed transport of total AA in Eμ-Myc lymphoma and Namalwa BL cells, as well as that of 14C-labeled Leu, 14C-Gln, and 14C-Arg in Eμ-Myc lymphoma B cells (Fig. 6G; Supplementary Fig. S6F).
Elevated AA transport in Myc-expressing B cells suggested these cells might be vulnerable to amino acid deprivation. To test this, WT and Eμ-Myc pre-B cells were deprived of AA by culture in EBSS media supplemented with IL-7. Notably, culture in EBSS media triggered rapid apoptotic death of Eμ-Myc pre-B cells (Fig. 6H). Further, shifting WT and premalignant Eμ-Myc transgenic littermates to a low protein (5% protein) chow for 1 week revealed this selectively reduced numbers and proliferation of peripheral blood B220+ Eμ-Myc B cells versus WT peripheral blood B cells, and selectively augmented the in vivo apoptotic index of Eμ-Myc B cells (Fig. 6I and J; Supplementary Fig. S6G). These findings support the notion that MYC-driven malignancies are highly reliant on sufficient amino acid pools.
TFEB-directed tumor suppression is associated with metabolic anergy
Myc is a master regulator of cancer cell metabolism and MYC-induced lymphomas are sensitive to agents or strategies that disrupt glycolysis (6), amino acid homeostasis (Figs. 5 and 6, and (55)) and glutamine catabolism (56). To evaluate if the tumor suppressor functions of TFEB were linked to these metabolic processes, we assessed effects of TFEBSA-ERT2 activation on basal and maximal capacity for aerobic glycolysis and oxidative phosphorylation (OXPHOS) using the glycolytic rate assay (GRA) and the mitochondrial stress test (MST). Interestingly, TFEBSA-ERT2 activation in Eμ-Myc lymphomas significantly reduced basal levels of both glycolysis and OXPHOS, and the ability to maximize the capacity for both pathways (Fig. 7A and B). In addition, TFEBSA activation impaired glucose uptake in Eμ-Myc lymphomas as determined by uptake of 2-deoxy-glucose (2-DG) (Fig. 7C). In Namalwa BL cells TFEBSA-ERT2 activation only reduced basal levels of aerobic glycolysis (Fig. 7D and E), which may explain the cytostatic effects of TFEB in this model. Regardless, TFEBSA-ERT2 activation significantly reduced rates of total ATP production in Eμ-Myc and Namalwa lymphoma cells (Fig. 7F–H).
Figure 7.
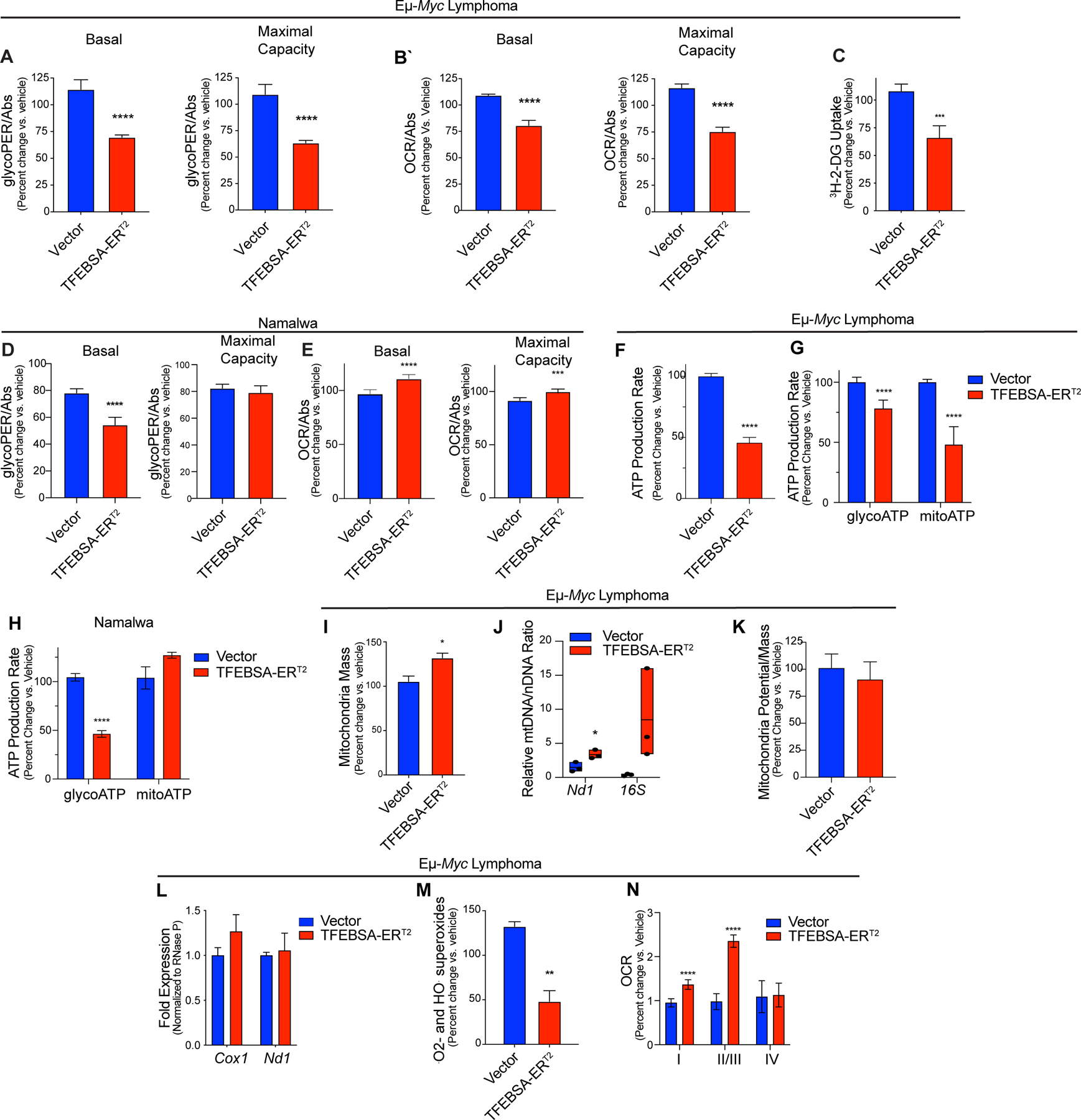
Induction of TFEB compromises the metabolism in MYC-driven B cell lymphoma. A-C, The basal and maximal capacity for aerobic glycolysis (A, glycolytic rate assays, GRA), basal and maximal capacity for oxidative phosphorylation (B, mitochondrial stress tests), and uptake of 3H-2-Deoxy-D-Glucose (C) were measured in Eμ-Myc lymphoma cells expressing vector or TFEBSA-ERT2 after treatment with vehicle or 4-OHT for 4 days (n = 6–8). D and E, The basal and maximal capacity for aerobic glycolysis (D), and basal and maximal capacity for oxidative phosphorylation (E) were measured using the XFe96 Analyzer in Namalwa BL cells expressing vector or TFEBSA-ERT2 after treatment with vehicle or 4-OHT for 6 days (n = 3). F-H, Total (F), glycolytic (glycoATP) and mitochondrial (mitoATP) (G and H) real-time ATP production were determined in Eμ-Myc lymphoma (F and G) or Namalwa BL (H) cells expressing vector or TFEBSA-ERT2 after treatment with vehicle or 4-OHT for 4 days (F and G, n = 6–8) or 6 days (H, n = 3) using the XFe96 Analyzer. I-M, Eμ-Myc lymphoma expressing vector or TFEBSA-ERT2 were treated with vehicle or 4-OHT for 4 days and assessed for: (I) mitochondrial mass by staining with MitoTracker Green; (J) the relative ratio of mitochondrial (Nd1, 16S) to nuclear (HK2) gene DNA content by qPCR; (K) mitochondrial membrane potential; (L) the expression of mitochondrial Nd1 and Cox1 genes by qRT-PCR; and (M) mitochondrial superoxide levels (CellRox) (n = 3). N, Analysis of ETC Complex I, II, III, and IV activity in mitochondria isolated from Eμ-Myc lymphoma expressing vector or TFEBSA-ERT2 after treatment with vehicle or 4-OHT for 4 days (n = 6–8). Basal OCR readings were used to determine for Complex I activity. Injections of 2 μM Rotenone and 10 mM Succinate were performed to analyze Complex II/III activity. Complex IV activity was analyzed by injecting 2 μM Antimycin A, 10 mM Ascorbate (A4034), and 100 μM TMPD (T7394). Statistical analysis: A-N, Students t-tests were performed.*, p ≤0.05; ** p ≤0.01; ***; p ≤ 0.001, ****, p ≤ 0.0001.
Given TFEBSA activation induces autophagy (Fig. 4) and suppresses OXPHOS (Fig. 7B) in Eμ-Myc lymphoma, we tested if the latter was due to induction of mitophagy. However, independent measures of mitochondrial parameters revealed TFEBSA activation slightly induced mitochondrial mass and had no effect on mitochondrial potential (Fig. 7I–K). In addition, activation of TFEBSA did not affect mRNA levels of the mitochondrial genes Cox1 and Nd1 (Fig. 7L). Nonetheless and consistent with impaired OXPHOS, TFEBSA activation reduced levels of mitochondrial superoxides in Eμ-Myc lymphoma cells (Fig. 7M). Interestingly, measurements of electron transport chain (ETC) complex activity in mitochondria isolated from Eμ-Myc lymphoma cells revealed TFEBSA augments Complex I, II, and III activity when all essential nutrients are provided (Fig. 7N), implying that TFEB activation rather limits nutrient availability and/or catabolism.
Integrated and parallel analyses of RNA-seq and global metabolomics using LC-MS/MS and MetaboAnalyst and GeneGo Metacore provided mechanistic insights into how TFEB activation disables Eμ-Myc lymphoma metabolism. Analyses of significantly upregulated or downregulated metabolites (p <0.1; Fig. 8A–G; Supplementary Data Tables S4, S5, and S6) revealed TFEBSA activation significantly and highly impacted several amino acid pathways (Fig. 8D–I). Further, the inhibitory effects of TFEBSA activation on glycolysis and the TCA cycle were reflected in major changes in these metabolites (Fig. 8C, D and G). Finally, other metabolic pathways significantly affected by TFEB activation include aminoacyl tRNA and ribose biosynthesis, and pyrimidine and purine metabolism (Fig. 8A, B, H and I). Thus, TFEB activation affects central metabolic processes in Eμ-Myc lymphomas that extend beyond regulation of AA homeostasis, glycolysis and the TCA cycle.
Figure 8.
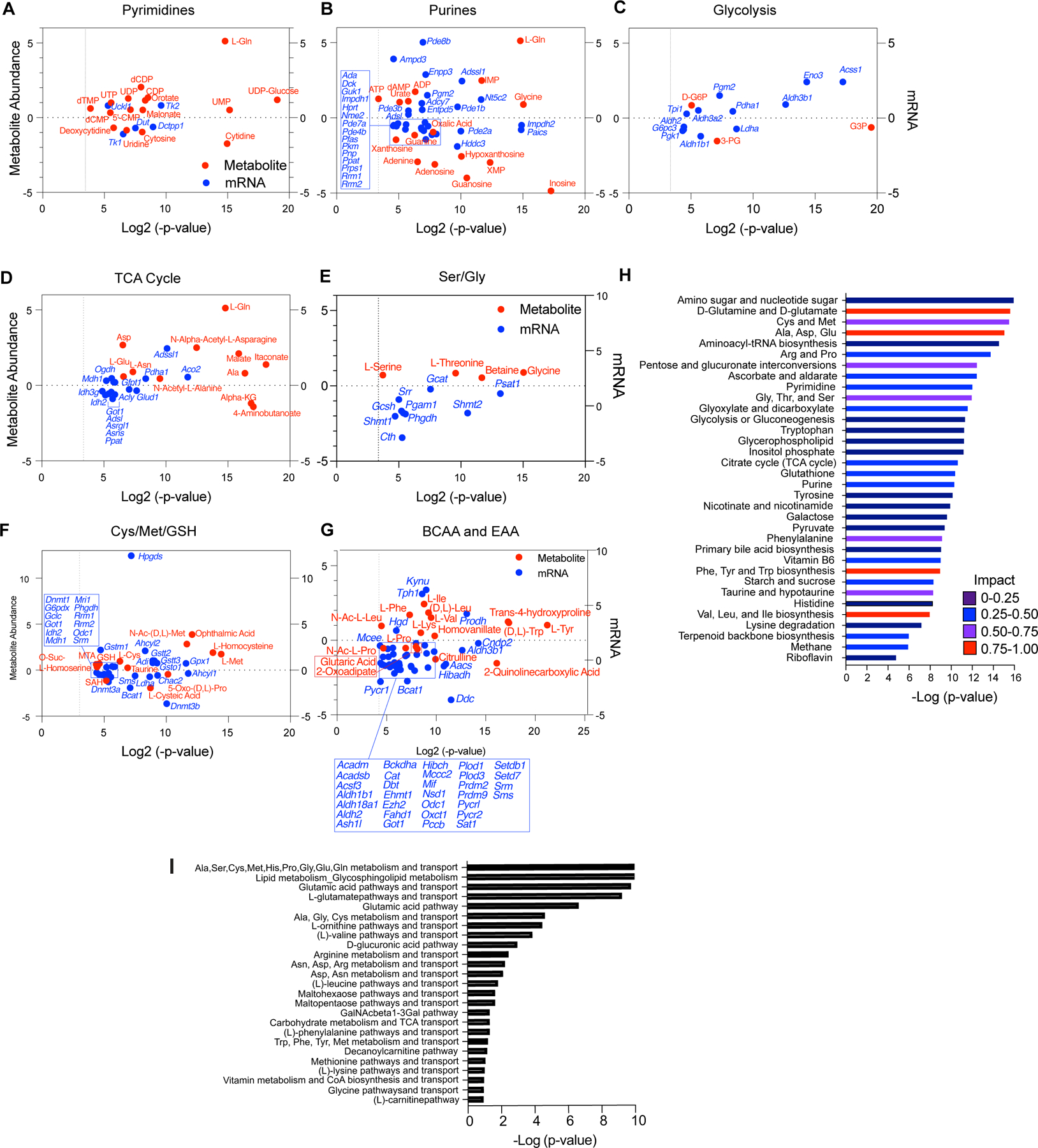
TFEB provokes metabolic anergy in MYC-induced B cell lymphoma. A-G, Differential and statistically significant metabolites and genes (p ≤ 0.1 and p ≤ 0.05, respectively), were grouped based on the indicated KEGG metabolic pathways, and are illustrated as two-axis dot plots to show the metabolite abundance (left-axis of plots) and changes in mRNA expression (right-axis of plots) that are up-regulated or down-regulated following the induction of TFEB activity in Eμ-Myc lymphoma cells. H, Untargeted metabolomic profiling via LC-MS/MS was performed in Eμ-Myc lymphoma expressing vector or TFEBSA-ERT2 after treatment with vehicle or 4-OHT for 4 days (n = 4). LC-MS spectra was analyzed using MZMine2. These data were then uploaded into MetaboAnalyst, and samples were normalized by the sum of all metabolites and log2 transformed, followed by a functional enrichment analysis to assess metabolic pathway enrichment using MetaboAnalyst. I, Normalized log2 transformed metabolomic data from Eμ-Myc lymphoma expressing TFEBSA-ERT2 treated with vehicle or 4-OHT were uploaded to GeneGo MetaCore to assess metabolic pathways affected by TFEB activation.
Grouping genes significantly up- or down-regulated (p <0.05) with significantly altered metabolic pathways defined by KEGG (Supplementary Data Table S7) revealed that, in agreement with the GRA data (Fig. 7A and B), TFEB activation led to suppression of glycolytic genes (Ldha, Pgk) and the metabolite 2-phosphoglycerate (2-PG). Conversely, there was an accumulation of glyceraldehyde 3-phosphate (G3P) and glucose-6-phosphate (G6P) (Fig. 8C), consistent with reduced glycolytic flux. Further, reduced OXPHOS provoked by TFEB activation correlated with a marked elevation of most amino acids, including Gln, which could reflect the induction of autophagy and/or reduced amino acid catabolism. Consistent with the latter, activation of TFEBSA led to: (i) an accumulation of most (17/20) amino acids (Ala, Asp, Asn, Glu, Gln, Ile, Lys, Phe, Trp, Tyr, Val, Leu, Ser, Gly, Thr, Cys, Met, Fig. 8D–G); (ii) reductions in their essential catabolic products, specifically α-ketoglutarate, S-adenosyl-L-homocysteine (SAH) and citrulline (Fig. 8D–G); (iii) the suppression of genes directing amino acid catabolism (Idh2, Asns, Phgdh, Shmt1, Shmt2, Bcat1, Got1, Odc) (Fig. 8D–G); and (iv) increased glutathione levels (Fig. 8F) that is associated with the buildup of Ser, Gly, and Cys (Fig. 8E and F).
The basic leucine zipper transcription factor ATF4 regulates genes that maintain amino acid pools during nutrient deprivation (57). However, expression of ATF4 and ATF4 target genes was unaffected by TFEBSA activation in Eμ-Myc lymphoma (Supplementary Data Table S8); thus, suppressive effects of TFEBSA on amino acid metabolism appear independent of an ATF4 response.
Myc induces transcription of genes directing purine and pyrimidine biosynthesis (58). Genes downregulated by TFEBSA include Prps1 and Dck that direct production of purine nucleotides and the nucleoside salvage pathway, and this correlates with decreased levels of adenine, adenosine, guanosine, and inosine (Fig. 8A and B). Further, elevated expression of Pgm2 (Fig. 8B) and Enpp3 (Fig. 8B), which harness purine production, might reflect a compensatory response to the decreased levels of purines (Fig. 8B and H). Similar findings were evident in pyrimidine synthesis, where TFEB activation suppressed levels of cytidine, uridine, and cytosine, and the expression of Tk1 that controls dTTP production and that is upregulated in cancers (59) (Fig. 8A). Finally, downregulation of Dnmt1, Dnmt3a, Dnmt3b genes along with accumulation of Met (Fig. 8F) suggest TFEB activation may also have global effects on epigenetic control in Eμ-Myc lymphoma, as observed in AML cells (26). Thus, TFEB tumor suppressor functions are linked to marked disruptions in glucose, amino acid and nucleotide metabolism that led to metabolic anergy.
Discussion
Cancer cells constantly adapt to environmental changes in the tumor, including alterations in blood flow, oxygen and essential nutrients necessary to meet the high energetic demands of the growing tumor. Glucose and glutamine appear principal sources for energy of cancer cells, yet there is also a clear requirement to maintain sufficient pools of intracellular AA and nucleotides to support high rates of transcription and translation and increased cell mass that are associated with the anabolic state of tumor cells (60,61). Notably, the studies herein establish surprising alterations in amino acid and nucleotide metabolism manifest in Myc-driven malignancies that are linked to the suppression of the TFEB-autophagy circuit. Importantly, restoring this circuit disables maintenance of the tumorigenic state by provoking metabolic anergy, including the collapse of oxidative phosphorylation and glycolysis, amino acid catabolism, and nucleotide synthesis.
Proper control of intracellular amino acid pools is required for development, homeostasis, cell growth, metabolism and survival (62). Amino acid pools in cancer cells are controlled by at least six inputs – biosynthesis, glutamine anapleurosis, AA uptake via dedicated transporters, macropinocytosis, the proteasome, and the autophagy-lysosome system. Notably, these circuits are up-regulated in human tumors, where: (i) amino acid biosynthesis is elevated in several tumor types (60); (ii) Myc up-regulates expression of glutamine (Asct2/Slc1a5) and branched chain/large neutral amino acid transporters (LAT1/Slc7a5) (7,63); and (iii) Ras-driven malignancies display up-regulation of, and a reliance on, the autophagy pathway (11,12,15,17).
Here we show that in normal and malignant B cells Myc sustains AA pools by up-regulating the expression of: (i) select amino acid transporters and AA transport; and (ii) components on the proteasome responsible for increasing its catalytic activity. Myc is also revealed to suppress the autophagy-lysosomal pathway that is essential to sustain intracellular AA pools in other tumor types, and this phenotype is a hallmark of B cell lymphomas with MYC involvement. Notably, the suppression of TFEB, and the skewed reliance of Myc-driven tumors on the proteasome and AA transport for sustaining intracellular amino acid pools, evokes easily actionable therapeutic opportunities for treating malignancies with MYC involvement, including mTORC1 inhibitors such as Everolimus that will reactivate the autophagy pathway, proteasome inhibitors and/or restricted protein diets (55,64).
Mechanistically, Myc suppresses the expression of TFEB and TFEB target genes, in accord with our findings in AML and of others in tumor cell lines and medulloblastoma (25,26). Indeed, MYC generally inversely regulates TFEB transcription targets in all hematological model systems tested, repressing genes activated by TFEB and inducing those repressed by TFEB. Further, we show TFEB antagonizes MYC in the same fashion, where TFEB inversely regulates MYC transcription programs. Heretofore, antagonism of MYC transcription functions has been relegated to related MXD (MXD1–4) and MNT bHLH-Zip transcription factors (65). As we show here, at least in the context of B cells, TFEB also has such antagonistic roles, and we predict this is manifest in other tumors with MYC involvement.
TFEB controls autophagy, which is thought to be required for proper control of amino acid and nucleotide pools (54), and loss of the pathway compromises tumorigenicity in the context of RAS-driven malignancies (11). In striking contrast, we show here that MYC represses autophagy and that this pathway is dispensable for the development and maintenance of MYC-driven lymphoma. Thus, inhibition of the autophagy pathway in tumor types having MYC involvement is likely to have no therapeutic benefit. However, as restoring TFEB function provokes a synthetic lethal response in MYC-driven lymphoma, agonists of TFEB may hold promise for treating MYC-driven malignancies.
Supplementary Material
Significance:
MYC suppresses TFEB and autophagy and controls amino acid homeostasis by upregulating amino acid transport and the proteasome, and reactivation of TFEB disables the metabolism of MYC-driven tumors.
Acknowledgments
We thank Dr. Jerry M. Adams for providing the pEµSRα transgenic plasmid; Dr. Gina DeNicola for critical review of the study; Dr. Sandeep Dave for providing access to RNA-Seq dataset of DLBCL patient samples; Kelly Psilos, Rea Güertler, Dr. Antony Packiam-Dayalan, and Emma Fallahi for technical assistance and advice; Rosemary Lyda and Tiffany Razabdousky of the Scripps-Florida and Moffitt Animal Resource Centers, respectively, for their technical assistance; Sean Yoder and the Moffitt Genomics Core for their help with RNA-Seq analyses; Jodi Kroeger of the Moffitt Flow Cytometry Core, and the Southeast Center for Integrated Metabolomics (SECIM) supported by NIH grant U24DK097209.
This work was supported by grants CA154739, CA167093 and CA241713 (to J.L. Cleveland), by the Cortner-Couch Endowed Chair for Cancer Research from the University of South Florida School of Medicine (to J.L. Cleveland), a Swiss National Science Foundation Postdoctoral Fellowship (to F.X. Schaub), by NRSA F32 CA203217 (to M.R. Fernandez), and K08 CA237627 (to S. Yun). These studies were also supported in part by the Flow Cytometry, Molecular Genomics, the Proteomics & Metabolomics Core, and the Biostatistics & Bioinformatics Shared Resource at the H. Lee Moffitt Cancer Center & Research Institute, by NCI Comprehensive Cancer Center Grant P30 CA076292, and by monies from the State of Florida to the H. Lee Moffitt Cancer Center & Research Institute.
Footnotes
Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors’ Disclosures
Andrea Ballabio is a Co-Founder of CASMA Therapeutics. There are no other disclosures.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74 [DOI] [PubMed] [Google Scholar]
- 2.Morrish F, Neretti N, Sedivy JM, Hockenbery DM. The oncogene c-Myc coordinates regulation of metabolic networks to enable rapid cell cycle entry. Cell Cycle 2008;7:1054–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer 2008;8:976–90 [DOI] [PubMed] [Google Scholar]
- 4.Carroll PA, Freie BW, Mathsyaraja H, Eisenman RN. The MYC transcription factor network: balancing metabolism, proliferation and oncogenesis. Front Med 2018;12:412–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim JW, Zeller KI, Wang Y, Jegga AG, Aronow BJ, O’Donnell KA, et al. Evaluation of myc E-box phylogenetic footprints in glycolytic genes by chromatin immunoprecipitation assays. Mol Cell Biol 2004;24:5923–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doherty JR, Yang C, Scott KE, Cameron MD, Fallahi M, Li W, et al. Blocking lactate export by inhibiting the Myc target MCT1 Disables glycolysis and glutathione synthesis. Cancer Res 2014;74:908–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A 2008;105:18782–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morrish F, Hockenbery D. MYC and mitochondrial biogenesis. Cold Spring Harb Perspect Med 2014;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011;476:346–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsun ZY, Possemato R. Amino acid management in cancer. Semin Cell Dev Biol 2015;43:22–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev 2011;25:460–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013;497:633–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suraweera A, Munch C, Hanssum A, Bertolotti A. Failure of amino acid homeostasis causes cell death following proteasome inhibition. Mol Cell 2012;48:242–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hsieh TJ, Lin T, Hsieh PC, Liao MC, Shin SJ. Suppression of Glutamine:fructose-6-phosphate amidotransferase-1 inhibits adipogenesis in 3T3-L1 adipocytes. J Cell Physiol 2012;227:108–15 [DOI] [PubMed] [Google Scholar]
- 15.Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res 2015;75:544–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y, Wang XD, Lapi E, Sullivan A, Jia W, He YW, et al. Autophagic activity dictates the cellular response to oncogenic RAS. Proc Natl Acad Sci U S A 2012;109:13325–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev 2011;25:717–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009;458:762–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davis IJ, Hsi BL, Arroyo JD, Vargas SO, Yeh YA, Motyckova G, et al. Cloning of an Alpha-TFEB fusion in renal tumors harboring the t(6;11)(p21;q13) chromosome translocation. Proc Natl Acad Sci U S A 2003;100:6051–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, et al. A gene network regulating lysosomal biogenesis and function. Science 2009;325:473–7 [DOI] [PubMed] [Google Scholar]
- 21.Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, et al. TFEB links autophagy to lysosomal biogenesis. Science 2011;332:1429–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palmieri M, Impey S, Kang H, di Ronza A, Pelz C, Sardiello M, et al. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet 2011;20:3852–66 [DOI] [PubMed] [Google Scholar]
- 23.Soucek L, Whitfield JR, Sodir NM, Masso-Valles D, Serrano E, Karnezis AN, et al. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev 2013;27:504–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Urbanelli L, Magini A, Ercolani L, Sagini K, Polchi A, Tancini B, et al. Oncogenic H-Ras up-regulates acid beta-hexosaminidase by a mechanism dependent on the autophagy regulator TFEB. PLoS One 2014;9:e89485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Annunziata I, van de Vlekkert D, Wolf E, Finkelstein D, Neale G, Machado E, et al. MYC competes with MiT/TFE in regulating lysosomal biogenesis and autophagy through an epigenetic rheostat. Nat Commun 2019;10:3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yun S, Vincelette ND, Yu X, Watson GW, Fernandez MR, Yang C, et al. TFEB links MYC signaling to epigenetic control of myeloid differentiation and acute myeloid leukemia. Blood Cancer Discov 2021;2:162–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bodrug SE, Warner BJ, Bath ML, Lindeman GJ, Harris AW, Adams JM. Cyclin D1 transgene impedes lymphocyte maturation and collaborates in lymphomagenesis with the myc gene. EMBO J 1994;13:2124–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 2012;31:1095–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Joo JH, Dorsey FC, Joshi A, Hennessy-Walters KM, Rose KL, McCastlain K, et al. Hsp90-Cdc37 chaperone complex regulates Ulk1- and Atg13-mediated mitophagy. Mol Cell 2011;43:572–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Welsh EA, Eschrich SA, Berglund AE, Fenstermacher DA. Iterative rank-order normalization of gene expression microarray data. BMC Bioinformatics 2013;14:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007;8:118–27 [DOI] [PubMed] [Google Scholar]
- 32.Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 2017;171:481–94 e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berglund AE, Welsh EA, Eschrich SA. Characteristics and Validation Techniques for PCA-Based Gene-Expression Signatures. Int J Genomics 2017;2017:2354564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sabo A, Kress TR, Pelizzola M, de Pretis S, Gorski MM, Tesi A, et al. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature 2014;511:488–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nilsson JA, Keller UB, Baudino TA, Yang C, Norton S, Old JA, et al. Targeting ornithine decarboxylase in Myc-induced lymphomagenesis prevents tumor formation. Cancer Cell 2005;7:433–44 [DOI] [PubMed] [Google Scholar]
- 37.Rounbehler RJ, Fallahi M, Yang C, Steeves MA, Li W, Doherty JR, et al. Tristetraprolin impairs myc-induced lymphoma and abolishes the malignant state. Cell 2012;150:563–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, et al. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 1985;318:533–8 [DOI] [PubMed] [Google Scholar]
- 39.Zeller KI, Zhao X, Lee CW, Chiu KP, Yao F, Yustein JT, et al. Global mapping of c-Myc binding sites and target gene networks in human B cells. Proc Natl Acad Sci U S A 2006;103:17834–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hummel M, Bentink S, Berger H, Klapper W, Wessendorf S, Barth TF, et al. A biologic definition of Burkitt’s lymphoma from transcriptional and genomic profiling. N Engl J Med 2006;354:2419–30 [DOI] [PubMed] [Google Scholar]
- 41.Napolitano G, Esposito A, Choi H, Matarese M, Benedetti V, Di Malta C, et al. mTOR-dependent phosphorylation controls TFEB nuclear export. Nat Commun 2018;9:3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Alboran IM, O’Hagan RC, Gartner F, Malynn B, Davidson L, Rickert R, et al. Analysis of C-MYC function in normal cells via conditional gene-targeted mutation. Immunity 2001;14:45–55 [DOI] [PubMed] [Google Scholar]
- 43.Morrow MA, Lee G, Gillis S, Yancopoulos GD, Alt FW. Interleukin-7 induces N-myc and c-myc expression in normal precursor B lymphocytes. Genes Dev 1992;6:61–70 [DOI] [PubMed] [Google Scholar]
- 44.Heng TS, Painter MW, Immunological Genome Project C. The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol 2008;9:1091–4 [DOI] [PubMed] [Google Scholar]
- 45.Monaco G, Lee B, Xu W, Mustafah S, Hwang YY, Carre C, et al. RNA-Seq Signatures Normalized by mRNA Abundance Allow Absolute Deconvolution of Human Immune Cell Types. Cell Rep 2019;26:1627–40 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 2012;151:68–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blackwood EM, Eisenman RN. Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 1991;251:1211–7 [DOI] [PubMed] [Google Scholar]
- 48.Han H, Jain AD, Truica MI, Izquierdo-Ferrer J, Anker JF, Lysy B, et al. Small-Molecule MYC Inhibitors Suppress Tumor Growth and Enhance Immunotherapy. Cancer Cell 2019;36:483–97 e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lamark T, Kirkin V, Dikic I, Johansen T. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 2009;8:1986–90 [DOI] [PubMed] [Google Scholar]
- 50.Gump JM, Staskiewicz L, Morgan MJ, Bamberg A, Riches DW, Thorburn A. Autophagy variation within a cell population determines cell fate through selective degradation of Fap-1. Nat Cell Biol 2014;16:47–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal 2012;5:ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hochedlinger K, Yamada Y, Beard C, Jaenisch R. Ectopic expression of Oct-4 blocks progenitor-cell differentiation and causes dysplasia in epithelial tissues. Cell 2005;121:465–77 [DOI] [PubMed] [Google Scholar]
- 53.Schaub FX, Reza MS, Flaveny CA, Li W, Musicant AM, Hoxha S, et al. Fluorophore-NanoLuc BRET Reporters Enable Sensitive In Vivo Optical Imaging and Flow Cytometry for Monitoring Tumorigenesis. Cancer Res 2015;75:5023–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Carroll B, Korolchuk VI, Sarkar S. Amino acids and autophagy: cross-talk and co-operation to control cellular homeostasis. Amino Acids 2015;47:2065–88 [DOI] [PubMed] [Google Scholar]
- 55.Maddocks ODK, Athineos D, Cheung EC, Lee P, Zhang T, van den Broek NJF, et al. Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 2017;544:372–6 [DOI] [PubMed] [Google Scholar]
- 56.Xiang Y, Stine ZE, Xia J, Lu Y, O’Connor RS, Altman BJ, et al. Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. J Clin Invest 2015;125:2293–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ye J, Kumanova M, Hart LS, Sloane K, Zhang H, De Panis DN, et al. The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J 2010;29:2082–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu YC, Li F, Handler J, Huang CR, Xiang Y, Neretti N, et al. Global regulation of nucleotide biosynthetic genes by c-Myc. PLoS One 2008;3:e2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jagarlamudi KK, Shaw M. Thymidine kinase 1 as a tumor biomarker: technical advances offer new potential to an old biomarker. Biomark Med 2018;12:1035–48 [DOI] [PubMed] [Google Scholar]
- 60.Hosios AM, Hecht VC, Danai LV, Johnson MO, Rathmell JC, Steinhauser ML, et al. Amino Acids Rather than Glucose Account for the Majority of Cell Mass in Proliferating Mammalian Cells. Dev Cell 2016;36:540–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Truitt ML, Ruggero D. New frontiers in translational control of the cancer genome. Nat Rev Cancer 2016;16:288–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yuan HX, Xiong Y, Guan KL. Nutrient sensing, metabolism, and cell growth control. Mol Cell 2013;49:379–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hayashi K, Jutabha P, Endou H, Anzai N. c-Myc is crucial for the expression of LAT1 in MIA Paca-2 human pancreatic cancer cells. Oncol Rep 2012;28:862–6 [DOI] [PubMed] [Google Scholar]
- 64.Wall M, Poortinga G, Stanley KL, Lindemann RK, Bots M, Chan CJ, et al. The mTORC1 inhibitor everolimus prevents and treats Emu-Myc lymphoma by restoring oncogene-induced senescence. Cancer Discov 2013;3:82–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Diolaiti D, McFerrin L, Carroll PA, Eisenman RN. Functional interactions among members of the MAX and MLX transcriptional network during oncogenesis. Biochim Biophys Acta 2015;1849:484–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data have been deposited at NCBI as GEO (accession number GSE153570). The data underlying all findings of this study are available from the author upon request and are provided as separate source data files.