

Abstract
Solid-state Li batteries require solid electrolytes which have high Li+ conductivity and good chemical/mechanical compatibility with Li metal anodes and high energy cathodes. Structure/function correlations which relate local bonding to macroscopic properties are needed to guide development of new solid electrolyte materials. This study combines diffraction measurements with solid-state nuclear magnetic resonance spectroscopy (ssNMR) and neutron pair distribution function (nPDF) analysis to probe the short-range vs. long-range structure of glass-ceramic Li3PS4-based solid electrolytes. This work demonstrates how different synthesis conditions (e.g., solvent selection and thermal processing) affect the resulting polyanionic network. More specifically, structures with high P coordination numbers (e.g., PS43– and P2S74–) correlate with higher Li+ mobility compared to other polyanions (e.g., (PS3)nn– chains and P2S64–). Overall, this work demonstrates how ssNMR and nPDF can be used to draw key structure/function correlations for solid-state superionic conductors.
Keywords: lithium thiophosphate, NMR, pair distribution function, solid electrolytes, solvent-mediated synthesis
Graphical Abstract
1. Introduction
Li-based solid-state batteries (SSBs) have gained significant interest due to their potential for improved energy density and safety compared to Li-ion systems containing liquid electrolytes. A major challenge for SSBs is development of solid electrolytes (SEs) which meet several key requirements including: (i) high Li+ conductivity (ca. 1–10 mS/cm at room temperature), (ii) good compatibility with Li metal anodes and high energy cathodes, and (iii) ability to be scalably processed into thin separators (<30 µm thick) for practical devices.1,2 A wide range SE classes including oxides, sulfides, and polymers have been developed, but no single material has been able to satisfy all the requirements for Li metal batteries.3,4
A promising class of SEs include lithium thiophosphates which have room temperature Li+ conductivities exceeding 1 × 10–4 S/cm and can be prepared using scalable solvent-mediated routes. The structure and properties of these materials are highly dependent on the synthesis conditions and thermal treatment,5,6 and oftentimes the materials are glass-ceramics containing both crystalline and amorphous domains. While the structures of crystalline phases (e.g., ß-Li3PS47, Li7P3S118, and Li6PS5Cl9) have been solved using diffraction techniques, relatively little is known about the local structure of their amorphous counterparts. Furthermore, the connection between local structure and SE performance (e.g., Li+ conductivity and resistance to Li dendrite growth) is still poorly understood. As such, identification of structure/function correlations, especially for amorphous and composite materials, is needed to aid development of lithium thiophosphate SEs.
Two methods which are well-suited for studying local bonding in amorphous materials include pair distribution function (PDF) analysis and solid-state nuclear magnetic resonance (ssNMR) spectroscopy. While X-ray scattering methods are relatively insensitive to light elements such as H and Li, neutron PDF is particularly advantageous for studying Li-based SEs due to lithium’s large neutron scattering cross section. Similarly, ssNMR is well-suited to correlate Li+ dynamics and local structure10 with experimentally measured Li+ conductivities. These techniques have been applied to various amorphous and glass-ceramic materials including sulfide/thiophosphate SEs11–14 and Lipon15,16. The present work extends application of ssNMR and nPDF techniques to study ß-Li3PS4 and Li3PS4-based composite SEs.
Our team recently reported a solvent-mediated synthesis route to produce amorphous composite SEs containing Li3PS4 and a poly(ethylene oxide) (PEO) binder.5 Incorporating polymer binders while maintaining high Li+ conductivity is one possible route to enable thin separators (<30 µm) for practical SSBs. Several polymer binders for sulfide-based composite SEs have been reported including nitrile butadiene rubber17,18, poly(tert-butyl acrylate)19, poly(vinylidene fluoride)-co-hexafluoropropylene20, PEO20, and polystyrene-block-polyethylene-ran-butylene-block-polystyrene20. Overall, these studies have shown that the composite’s structure and ionic conductivity are highly sensitive to the solvent/binder selection. Most prior work on composite SEs incorporates the polymer binder after thermally annealing the ceramic phase. On the other hand, the present study utilizes a unique one-pot synthesis approach in which the Li3PS4 precursors (i.e., Li2S and P2S5) are blended with the polymer binder prior to annealing, and this approach may enable improved mixing of the two phases. While Li3PS4/PEO composites are explored in the present study, this one-pot synthesis approach can easily be extended to other binders to optimize the composite’s structure and performance.
In our prior work on Li3PS4/PEO composite SEs, we utilized X-ray photoelectron spectroscopy and Raman spectroscopy to understand how the polyanionic network evolves during thermal treatment. However, assignment of spectroscopic bands is somewhat ambiguous, and there is some discrepancy in the literature on how to assign key polyanionic groups including PS43–, P2S74–, P2S62–, (PS3)nn– chains, and P2S64–. To supplement our original study, herein we apply ssNMR and nPDF to evaluate local bonding in ß-Li3PS4 and amorphous Li3PS4 + 1% PEO prepared through solvent-mediated routes.
2. Materials and Methods
2.1. Solid Electrolyte Synthesis
ß-Li3PS4 and amorphous Li3PS4/PEO composite SEs were prepared using a solvent-mediated route as described previously.5 Tetrahydrofuran (THF) and acetonitrile (AN) were used as the solvents during synthesis of ß-Li3PS4 and amorphous Li3PS4/PEO composites, respectively. The THF reagent (99.8%, Acros Organics) did not contain a reductive stabilizer (e.g., butylated hydroxytoluene), and the solvent was stored and dispensed in an Ar glovebox to mitigate peroxide formation. SE powders were dried for at least 12 h under vacuum at 45–140 °C. All materials were handled in an Ar-filled glovebox and characterized in containers sealed under Ar. Ionic conductivity was measured using AC impedance spectroscopy in a blocking cell configuration as described previously.5
2.2. XRD
XRD measurements were performed on a Scintag XDS 2000 powder diffractometer with Cu Kα radiation (λ = 1.5406 Å) in the 2θ range of 10−80°. The operating voltage and current of the X-ray generator were 38 kV and 32−35 mA, respectively. Powders were mounted on glass slides and covered with Kapton tape to mitigate air exposure during XRD measurements.
2.3. NMR
Solid state MAS NMR (ssNMR) experiments were conducted using a 7.05 T Varian-S direct drive wide bore spectrometer with an operating frequency of 301.4 MHz for protons, using a 3.2 mm MAS Chemagnetics broadband probe. A single pulse experiment (SPE) was applied to 1H, 31P and 7Li to evaluate the materials’ structure. All samples were handled under Ar atmosphere and packed into 3.2 mm thick-walled zirconia rotors, and the MAS rate was 15 kHz. For 1H spectra, pulse widths were 7 µs using 128 scans with a 20 second recycle delay. For 31P spectra, pulse widths were 2.8 µs using 64 scans with 60 – 300 second recycle delays. For 7Li spectra, pulse widths were 2.5 µs, with 64 scans with 20 second recycle delay. Tetramethylsilane (TMS), 1M H3PO4, and Li trifluoromethanesulfonate were used as external references for 1H, 31P, and 7Li spectra, respectively.
2.4. Neutron Scattering
Time-of-flight (TOF) neutron scattering experiments were performed using the NOMAD instrument (BL-1B) at the Spallation Neutron Source (SNS, Oak Ridge National Laboratory). Samples were sealed in vanadium cans, and data were collected at 300 K. The raw data were normalized against a vanadium rod after removing the background signal (empty V cans) from the multiple banks (#2 to #5) diffraction data. The neutron total scattering structure, S(Q), was obtained by sine Fourier transformation of the reduced pair distribution function, G(r), at a Qmax of 28 Å–1. The Rietveld refinements of ß-Li3PS4 and Li3PS4-based composite SEs were performed using TOPAS v621. The d-spacing of the TOF diffraction data was converted by TOF = Zero + Difc*d + Difa*d2. Zero and Difc were fixed as constants after refining against the neutron diffraction data of the NIST standard Si 640e. Difa was refined against the multiple banks (#2 to #5) diffraction data to accommodate the sample displacements and peak shifts induced by absorption. Absorption correction was performed by employing an empirical Lobanov formula.22 For low-resolution banks (#2 and #3), the peak profiles were described by a convolution of a back-to-back exponential function and a symmetrical Gaussian function. For high-resolution banks (#4 and #5), the moderator-induced peak profiles were described by a modified Ikeda-Carpenter-David function.
3. Results and Discussion
Solvent-mediated synthesis routes can produce a wide range of glass-ceramic lithium thiophosphate SEs. The present study focuses on two Li3PS4-based SEs including ß-Li3PS4 and amorphous composites containing 1 wt% PEO binder. Lab-source powder XRD patterns (see Figure S1) demonstrate that the Bragg peaks of the ß-Li3PS4 sample indexed with the expected phase, and the composites were largely amorphous except for a small amount of unreacted Li2S. The amorphous nature of the Li3PS4+1% PEO composites is due to using acetonitrile (AN) as the solvent which inhibits formation of crystalline ß-Li3PS4.5 XRD patterns in Figure S1 show that Li3PS4 with/without PEO prepared from AN have very similar structures.
Table 1 lists the Li+ conductivity at 25°C and activation energies of these SEs as determined using AC impedance spectroscopy. ß-Li3PS4 exhibited the highest Li+ conductivity and lowest activation energy with values that are consistent with previous reports.23,24 Annealing the composites at a moderate temperature (140°C) increased the Li+ conductivity several orders of magnitude due to loss of coordinated solvent and rearrangement of the polyanionic network.5
Table 1.
Room temperature (RT) ionic conductivity (σLi+) and activation energy (EA) for ß-Li3PS4 and composite Li3PS4+1% PEO samples. Corresponding Nyquist and activation plots were reported in our previous study.5 Full-width half maxima (FWHM) of the 7Li peaks (determined via ssNMR) correlate well with the observed σLi+ values.
| Sample | Tanneal (°C) | σLi+ at RT (S/cm) | EA (eV) | 7Li FWHM (Hz) |
|---|---|---|---|---|
| Li3PS4 + 1% PEO | 45 | 4.5 × 10−9 | 1.35 | 618 |
| Li3PS4 + 1% PEO | 140 | 8.7 × 10−6 | 0.42 | 239 |
| ß-Li3PS4 | 140 | 1.2 × 10−4 | 0.33 | 165 |
The long-range structure of ß-Li3PS4 and Li3PS4 + 1% PEO samples was further characterized using neutron diffraction (see Figure 1). Interestingly, all patterns exhibited the following features: (i) sloping backgrounds from hydrogen absorption, where proton sources include coordinated solvent (THF for ß-Li3PS4 and AN for the composites) and PEO binder, (ii) Bragg reflections due to crystalline phases, and (iii) broad features due to amorphous domains. R-values derived from Rietveld refinement of the neutron diffraction data are summarized in Table S1. Small R-values were obtained despite the low signal-to-noise ratio, and we have high confidence in these refinements because: (i) Bank 5 data (2θ = 154°, Figure 1) clearly shows the presence of Li2S and (ii) the presence of β-Li3PS4 and γ-Li3PS4 is supported by the Bank 2 data (2θ = 31°, Figure S2) and 31P ssNMR results (Figure 2b, discussed later in the text). Future work will explore sample preparation using deuterated solvents to mitigate incoherent scattering by 1H and improve the quality of the neutron diffraction data.
Figure 1.
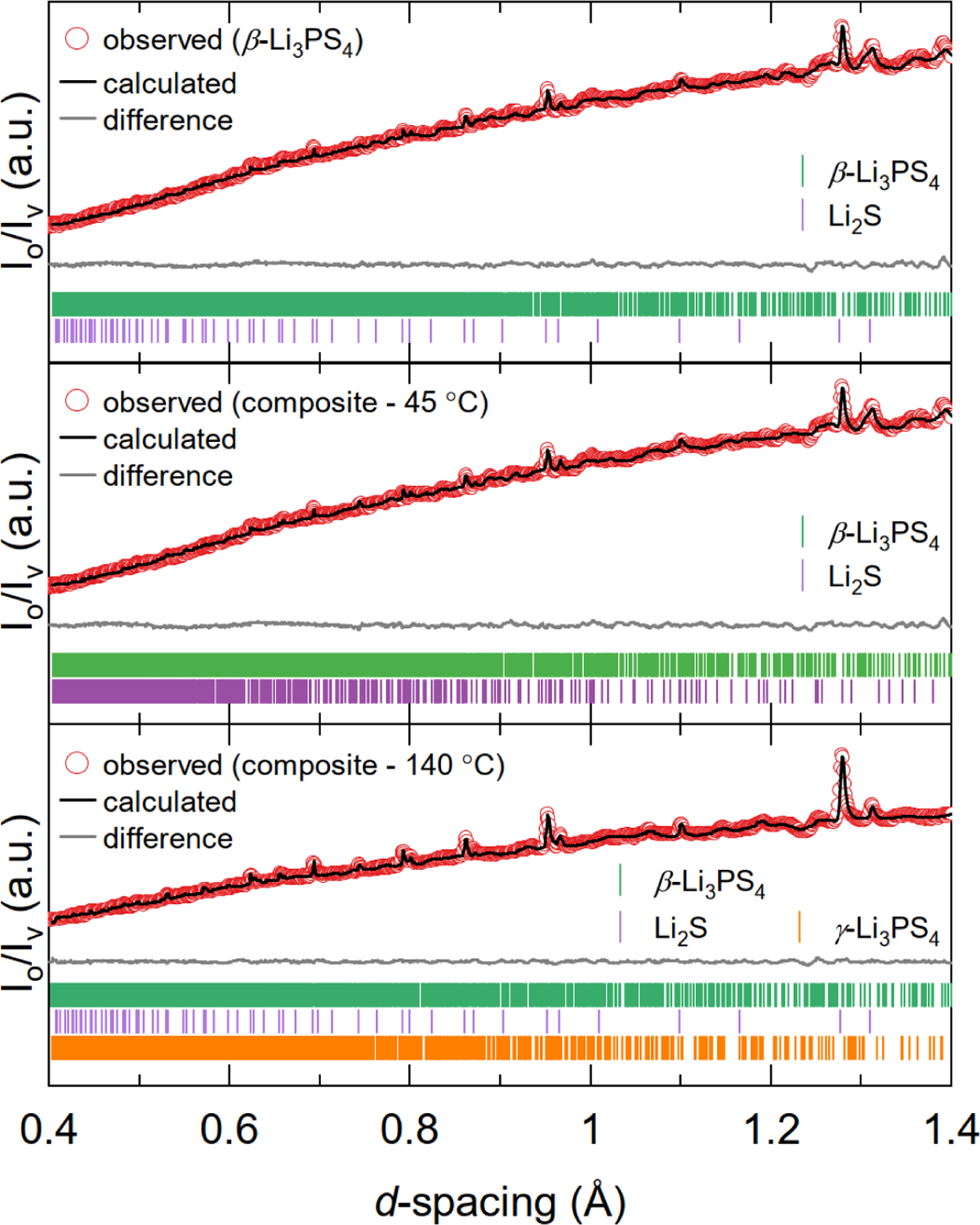
Rietveld refinement of neutron diffraction data (Bank 5; 2θ = 154°) for ß-Li3PS4 and Li3PS4 + 1% PEO (composites) samples.
Figure 2.
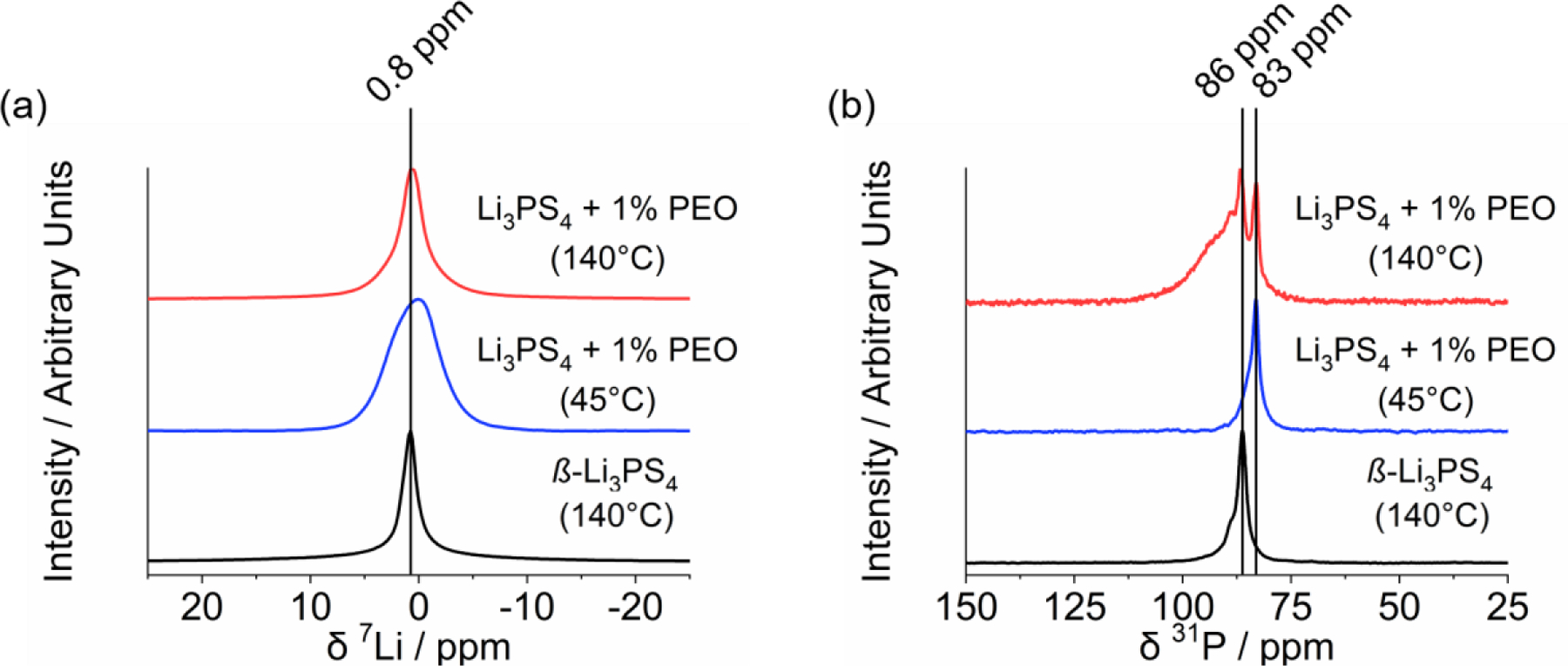
ssNMR of ß-Li3PS4 and amorphous Li3PS4 + 1% PEO SE powders showing (a) 7Li and (b) 31P spectra.
The ß-Li3PS4 sample contained predominantly the expected phase along with 10% unreacted Li2S (see Table 2) which likely lowered the sample’s Li+ conductivity.25 As such, future work should explore decreasing the Li2S/P2S5 molar ratio to eliminate Li2S impurities in the final product. For the composites with 1% PEO, similar results were observed except that γ-Li3PS4 was obtained after annealing at 140 °C. Notably, this phase was not detected in the lab-source XRD measurements due to experimental limitations (e.g., use of Kapton film in a reflection geometry which compromised data quality). These refinements only reflect the relative amounts of crystalline phases in each sample, so ssNMR and nPDF analyses were also performed to probe the local structure of amorphous components.
Table 2.
Relative amounts of crystalline phases detected in ß-Li3PS4 and amorphous Li3PS4 + 1% PEO samples as determined by Rietveld refinement of the neutron diffraction data shown in Figure 1.
| Sample | ß-Li3PS4 (%) | γ-Li3PS4 (%) | Li2S (%) |
|---|---|---|---|
| Li3PS4 + 1% PEO, 45°C | 87 | 0 | 13 |
| Li3PS4 + 1% PEO, 140°C | 43 | 46 | 11 |
| ß-Li3PS4 | 90 | 0 | 10 |
ssNMR data for the Li3PS4-based SEs is shown in Figure 2. Single pulse 7Li spectra of all materials exhibited a peak centered around 0.8 ppm. Compared to ß-Li3PS4, the increased linewidth and sideband intensities of the composites (see Figure S3) indicates the presence of less mobile Li+ ions in lower symmetry environments, and the FWHM of these peaks correlate well with the measured conductivity values (see Table 1).
In addition to Li+ dynamics from the 7Li spectra, the 31P NMR spectra (Figure 2b) provide key information on the materials’ polyanionic structures. Thiophosphate glass-ceramics generally contain one or more of the following tetrahedrally-coordinated P units: PS43–, P2S62–, P2S64–, P2S74– and/or (PS3)nn– chains which can be distinguished via NMR by their unique chemical shifts. The ß-Li3PS4 contained a prominent peak at 86 ppm which is consistent with the presence of isolated PS43– polyanions as expected.26,27 The weak shoulder at 89 ppm for the ß-Li3PS4 sample signifies the presence of another P environment such as P2S74–27 or γ-Li3PS412. Since no crystalline γ-Li3PS4 was detected in this material (see neutron diffraction data in Figure 1), the material is more accurately described as a glass-ceramic containing crystalline (ß-Li3PS4) and amorphous (Li4P2S7-like) domains. The 31P spectra of the samples with 1% PEO showed dramatic changes upon thermal annealing at 45 vs. 140°C. When dried at 45°C, the spectrum showed a single peak at 83 ppm which can be attributed to: (i) oxygen substitution in the thiophosphate polyanions (e.g., PS3O3–)12 due to reactions with the PEO binder and/or (ii) metathiophosphate (PS3)nn– chains.27,28 Significant amounts of PS3O3– in these composites are unlikely since they only contained 1 wt% PEO. Moreover, assignment (ii) is consistent with the nPDF analysis discussed later. After annealing at 140°C, a band at 87 ppm (due to PS43– tetrahedra) appeared along with a broad shoulder that extended from ca. 88 to 110 ppm. This broad band is attributed to several P coordination environments including γ-Li3PS4, P2S74–, and/or P2S64–.12,26,27 Interestingly, no peaks ~55 ppm due to P2S62– polyanions29 were observed, which is contradictory to the Raman band assignment in our prior work.5
1H NMR spectra were collected on the composite samples (see Figure S4) to probe interactions between the lithium thiophosphate phase(s), coordinated acetonitrile, and PEO binder. Pure PEO is a semi-crystalline polymer, and its 1H spectrum showed the following features: (i) a sharp peak at 4.7 ppm corresponding to amorphous domains with high chain mobility and (ii) a broad component ~10 ppm due to crystalline domains with limited chain mobility.30 On the other hand, the 1H spectra of composite Li3PS4 + 1% PEO samples contained broad features which indicates complexation between the polymer’s ether groups and Li+ ions. Interestingly, the composite sample heated at 140°C contained sharp peaks at 0.3 and −0.2 ppm which are attributed to coordinated acetonitrile not removed during thermal processing. The absence of these peaks in the 45°C sample is unclear at this time, but one possible explanation is the presence of “protected” isolated regions of AN for this sample.
Following the findings obtained through 1H, 7Li and 31P ssNMR, neutron pair distribution function (nPDF) analysis was performed to further reveal the local structure of these materials. Specifically, PDF is powerful tool that allows one to precisely track the P–P, S–S, and P–S correlations for disordered glass-ceramics.11,29,31,32 Figure 3 shows each sample’s reduced nPDF, G(r), which represents the probability of finding a neighboring atom at a distance r from a given atom. Approximate distances between two atoms (P–P, S–S, and P–S) associated with each polyanionic structure observed in the ssNMR measurements (i.e., PS43–, P2S74–, (PS3)nn–, and P2S64–) are highlighted in Figure 3a. ß-Li3PS4 exhibited a strong peak at ~2.0 Å (red line) which is consistent with the P–S bond length in PS43– tetrahedra29,33, the dominant polyanion in this structure. This finding is further supported by the peak near 3.3 Å (black line) which corresponds to the distance between adjacent S atoms in PS43–.29,33.
Figure 3.
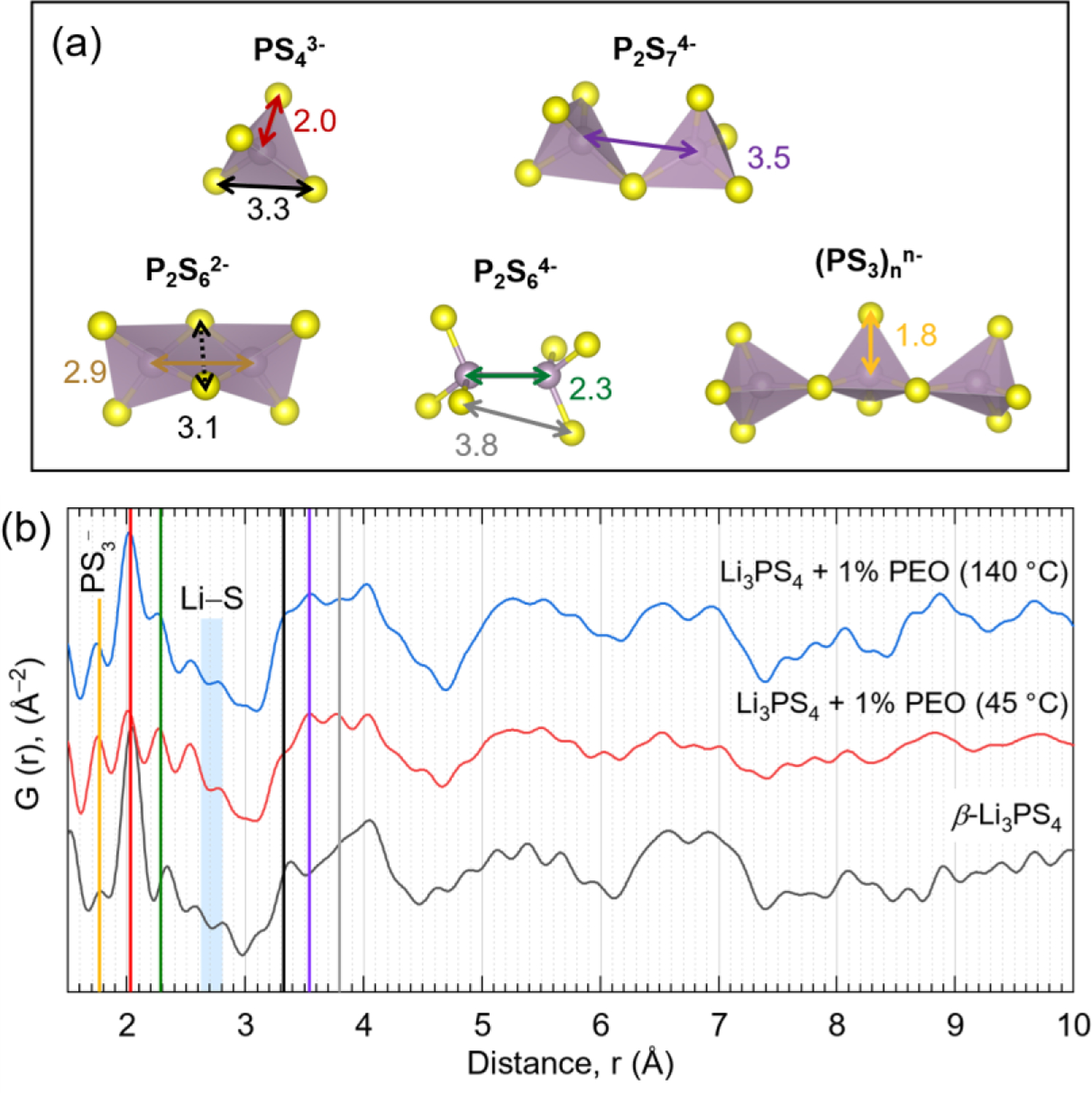
(a) Various P–S polyanionic structures with approximate bond lengths. (b) Neutron pair distribution function (nPDF) for ß-Li3PS4 and Li3PS4 + 1% PEO samples measured at 300K. Vertical lines correspond to expected bond lengths described in (a).
Compared to ß-Li3PS4, the composites contained more diverse P–P bonding correlations (e.g., P2S64– and P2S74– indicated by the green and purple lines at ~2.3 Å and ~3.5 Å, respectively)33 which evolved during thermal treatment. Notably, annealing at higher temperature (from 45°C to 140°C) coincided with decreased intensity of the 1.8 Å peak and increased intensity of the 2 Å peak. Here, the peak centered around 1.8 Å (yellow line) is attributed to terminal P–S bonds (i.e., involving non-bridging S atoms) in (PS3)nn– chains. This assignment is supported by the trend in analogous phosphates wherein terminal P–O bonds in metaphosphate chains (PO3–) are typically ~0.1 Å shorter than in orthophosphates (PO43–) 34,35. These findings suggest that the (PS3)nn– chains are broken into isolated PS43– units when annealing at moderate temperature (140 °C). A quantitative analysis on the relative distribution of these amorphous and crystalline phases would require detailed modeling efforts which is outside the scope of this study.
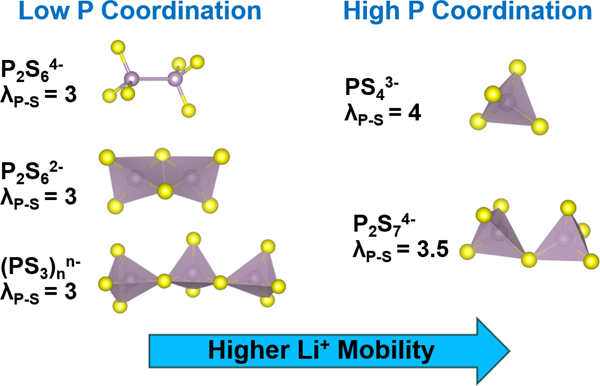
Overall, the ssNMR and neutron scattering results demonstrate the structural complexity of lithium thiophosphate SEs prepared through solvent-mediated routes. In addition to the expected PS43– tetrahedra, the ß-Li3PS4 sample contained other P bonding environments (e.g., P2S74–) which indicates the presence of amorphous domains not detected by diffraction methods. Furthermore, the composite samples with 1 wt% PEO binder contained a broad range of P environments (e.g., PS43–, P2S74–,(PS3)nn– chains, and P2S64–) whose distribution depends on thermal treatment. These differences, along with complexation between the PEO binder and Li+ ions, are consistent with the lower Li+ conductivity of the composite samples. In general, these results suggest that polyanions with higher P coordination numbers (λP-S) increase Li+ mobility as illustrated schematically in Figure 4.
Figure 4.
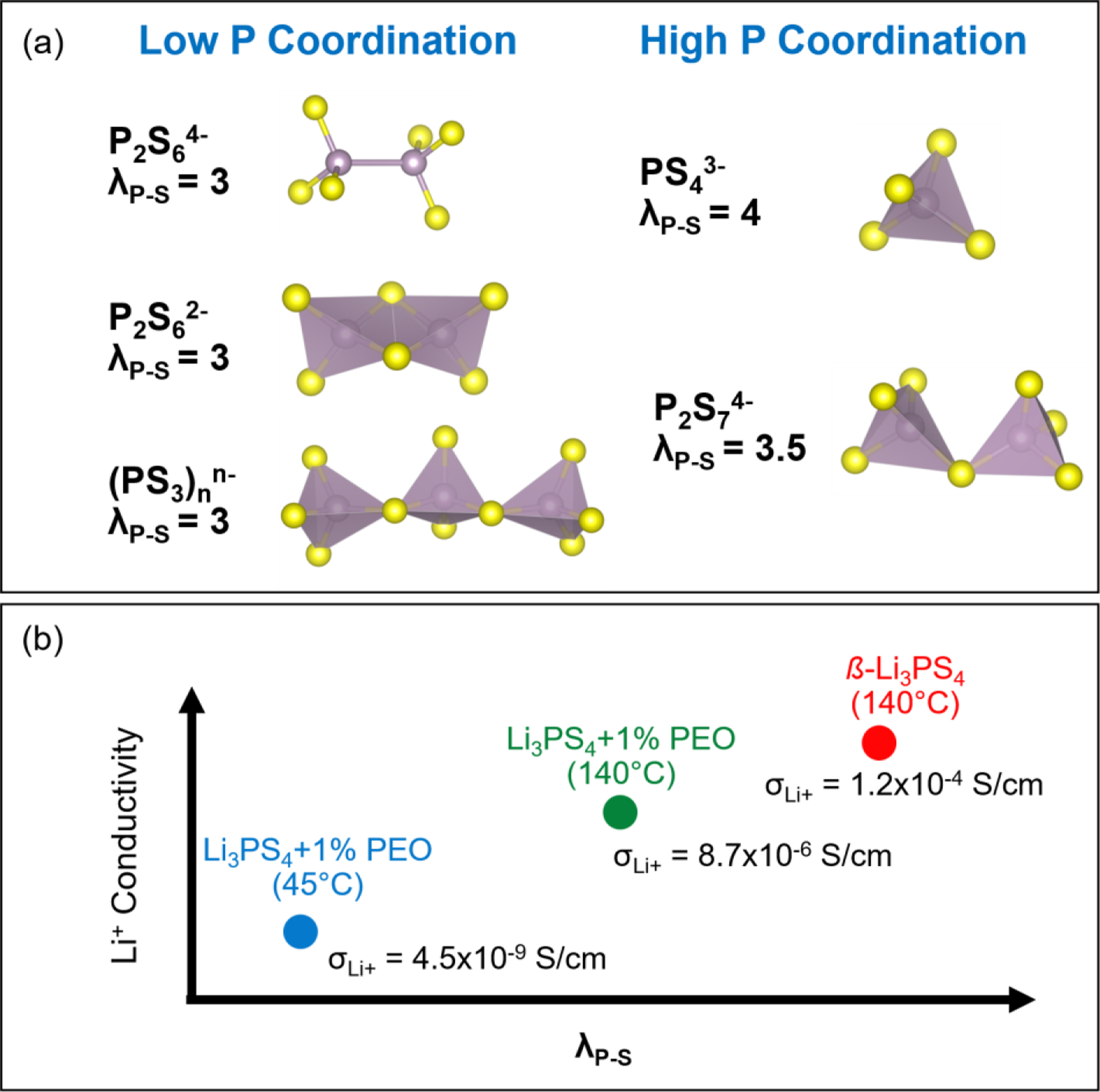
(a) Structure of various thiophosphate polyanions and corresponding λP–S values which denote the number of S atoms bonded with P. (b) Qualitative illustration of how polyanionic structure impacts Li+ conductivity for the SEs investigated in this study. σLi+ values listed in (b) were measured at room temperature (see Table 1).
4. Conclusions
Development of superionic solid conductors using scalable synthesis routes is critical to enable all-solid-state batteries with improved energy density and safety compared to conventional Li-ion systems. Despite growing activity in the field, integration of SEs into devices with Li metal anodes and high voltage cathodes remains a challenge.
The present work utilizes ssNMR and neutron scattering to understand the local structure of ß-Li3PS4 and amorphous composites containing Li3PS4 + 1% PEO. These findings highlight the complexity of these glass-ceramics which contain both amorphous and crystalline domains. While the predominant polyanion in ß-Li3PS4 was PS43– tetrahedra, the material also contained some unexpected P coordination environments such as P2S74–. For the composite samples, 1H NMR spectra suggested there was complexation between the binder’s ether groups and Li+ cations in the thiophosphate phase. The polyanionic structure of these composites was highly sensitive to thermal treatment. Drying at 45°C yielded (PS3)nn– metathiophosphate chains, whereas annealing at 140°C coincided with formation of amorphous PS43– and P2S74– structures. These polyanions with higher P coordination numbers correlated with significantly higher Li+ conductivity (e.g., 4.5 × 10–9 vs. 8.7 × 10–6 S/cm at room temperature for Li3PS4 + 1% PEO annealed at 45 and 140 °C, respectively). Interestingly, neutron scattering measurements showed significant 1H absorption for all samples, indicating that thermally processing at 140°C did not fully remove coordinated solvent (THF for ß-Li3PS4 and AN for amorphous Li3PS4 + 1% PEO composites) which likely decreased the materials’ Li+ conductivity. One way to better understand how trace solvent affects SE structure is to use deuterated solvents which will improve neutron diffraction data quality by mitigating incoherent scattering.
Overall, the results in this work highlight structure/function correlations for glass-ceramic SEs produced through solvent-mediated synthesis routes. The polyanionic network in these materials has a dramatic impact on their Li+ conductivity, and the final structure depends on the relative stability of solvated complexes and equilibrium among these intermediates. As such, interactions among the precursors, solvent, and polymer binder (in the case of composite SEs) requires careful consideration for development of high-performance SEs through solution-based routes. Several future research directions to increase the Li+ conductivity of the Li3PS4-based composites include5: (i) optimizing the Li2S/P2S5 molar ratio to eliminate Li2S impurities which likely reduce the overall ionic conductivity, (ii) incorporating Li-based salts in the PEO matrix to enable Li+ conduction through the binder, and (iii) exploring new solvent/binder combinations which do not inhibit crystallization of ß-Li3PS4.
Supplementary Material
NMR and nPDF are used to probe the local structure of Li3PS4-based solid electrolytes.
The effects of different compositions and thermal treatments on structure and Li+ conductivity are reported.
Polyanions with greater P-S coordination (e.g., PS43– and P2S74–) correlate with higher Li+ mobility.
Acknowledgments
Research conducted at Oak Ridge National Laboratory, managed by UT Battelle, LLC, for the U.S. Department of Energy (DOE) was sponsored by the Office of Energy Efficiency and Renewable Energy (EERE) in the Vehicle Technologies Office (VTO) through the Advanced Battery Materials Research (BMR) Program. Neutron scattering experiments were performed on the NOMAD beamline at the Spallation Neutron Source, a DOE Office of Science User Facility operated by Oak Ridge National Laboratory. The NMR work at Hunter College was also supported by the U.S. Office of Naval Research (grant #N00014–20-1–2186). D.M. acknowledges financial support from the National Institutes of Health RISE program at Hunter College (grant GM060665).
This manuscript has been authored in part by UTBattelle, LLC, under contract DE-AC05–00OR22725 with the US Department of Energy (DOE). The publisher, by accepting the article for publication, acknowledges that the US government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this manuscript, or allow others to do so, for US government purposes. DOE will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://energy.gov/downloads/doepublic-access-plan).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data Availability
The raw/processed data required to reproduce these findings cannot be shared at this time due to technical or time limitations.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Liu J, Bao Z, Cui Y, Dufek EJ, Goodenough JB, Khalifah P, Li Q, Liaw BY, Liu P, Manthiram A, Meng YS, Subramanian VR, Toney MF, Viswanathan VV, Whittingham MS, Xiao J, Xu W, Yang J, Yang X-Q, Zhang J-G, Nature Energy (2019) 4 (3), 180 [Google Scholar]
- 2.Hao F, Han F, Liang Y, Wang C, Yao Y, MRS Bull (2018) 43 (10), 775 [Google Scholar]
- 3.Manthiram A, Yu X, Wang S, Nat. Rev. Mater (2017) 2 (4), 16103 [Google Scholar]
- 4.Samuthira Pandian A, Medina PA, Yahyazadeh S, Nguyen KV, Miller R, Aetukuri NP, La Y-H, ACS Applied Energy Materials (2020) 3 (12), 12709 [Google Scholar]
- 5.Self EC, Hood ZD, Brahmbhatt T, Delnick FM, Meyer HM, Yang G, Rupp JLM, Nanda J, Chem. Mater (2020) 32 (20), 8789 [Google Scholar]
- 6.Delnick FM, Yang G, Self EC, Meyer HM, Nanda J, J. Phys. Chem. C (2020) 124 (50), 27396 [Google Scholar]
- 7.Homma K, Yonemura M, Kobayashi T, Nagao M, Hirayama M, Kanno R, Solid State Ionics (2011) 182 (1), 53 [Google Scholar]
- 8.Yamane H, Shibata M, Shimane Y, Junke T, Seino Y, Adams S, Minami K, Hayashi A, Tatsumisago M, Solid State Ionics (2007) 178 (15–18), 1163 [Google Scholar]
- 9.Kraft MA, Culver SP, Calderon M, Bocher F, Krauskopf T, Senyshyn A, Dietrich C, Zevalkink A, Janek J, Zeier WG, J. Am. Chem. Soc (2017) 139 (31), 10909. [DOI] [PubMed] [Google Scholar]
- 10.Vijayakumar M, Emery J, Bohnke O, Vold R, Hoatson G, Solid State Ionics (2006) 177 (19–25), 1673 [Google Scholar]
- 11.Shiotani S, Ohara K, Tsukasaki H, Mori S, Kanno R, Sci Rep (2017) 7 (1), 6972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gobet M, Greenbaum S, Sahu G, Liang C, Chem. Mater (2014) 26 (11), 3558 [Google Scholar]
- 13.Bischoff C, Schuller K, Dunlap N, Martin SW, J. Phys. Chem. B (2014) 118 (7), 1943. [DOI] [PubMed] [Google Scholar]
- 14.Stöffler H, Zinkevich T, Yavuz M, Senyshyn A, Kulisch J, Hartmann P, Adermann T, Randau S, Richter FH, Janek J, Indris S, Ehrenberg H, J. Phys. Chem. C (2018) 122 (28), 15954 [Google Scholar]
- 15.Stallworth PE, Vereda F, Greenbaum SG, Haas TE, Zerigian P, Goldner RB, J. Electrochem. Soc (2005) 152 (3) [Google Scholar]
- 16.Lacivita V, Westover AS, Kercher A, Phillip ND, Yang G, Veith G, Ceder G, Dudney NJ, J. Am. Chem. Soc (2018) 140 (35), 11029. [DOI] [PubMed] [Google Scholar]
- 17.Emley B, Liang Y, Chen R, Wu C, Pan M, Fan Z, Yao Y, Materials Today Physics (2021) 18 [Google Scholar]
- 18.Lee K, Kim S, Park J, Park SH, Coskun A, Jung DS, Cho W, Choi JW, J. Electrochem. Soc (2017) 164 (9), A2075 [Google Scholar]
- 19.Lee J, Lee K, Lee T, Kim H, Kim K, Cho W, Coskun A, Char K, Choi JW, Adv. Mater (2020) 32 (37), e2001702. [DOI] [PubMed] [Google Scholar]
- 20.Tan DHS, Banerjee A, Deng Z, Wu EA, Nguyen H, Doux J-M, Wang X, Cheng J. h., Ong SP, Meng YS, Chen Z, ACS Applied Energy Materials (2019) 2 (9), 6542 [Google Scholar]
- 21.Coelho A, J. Appl. Crystallogr (2018) 51 (1), 210 [Google Scholar]
- 22.Lobanov NN, and Veiga L. a. d., 6th European Powder Diffraction Conference, Abstract P12–16 (Aug. 22–25, 1998) [Google Scholar]
- 23.Liu Z, Fu W, Payzant EA, Yu X, Wu Z, Dudney NJ, Kiggans J, Hong K, Rondinone AJ, Liang C, J. Am. Chem. Soc (2013) 135 (3), 975. [DOI] [PubMed] [Google Scholar]
- 24.Wang H, Hood ZD, Xia Y, Liang C, J. Mater. Chem. A (2016) 4 (21), 8091 [Google Scholar]
- 25.Choi S, Lee S, Park J, Nichols WT, Shin D, Appl. Surf. Sci (2018) 444, 10 [Google Scholar]
- 26.Seino Y, Nakagawa M, Senga M, Higuchi H, Takada K, Sasaki T, J. Mater. Chem. A (2015) 3 (6), 2756 [Google Scholar]
- 27.Eckert H, Zhang Z, Kennedy JH, Chem. Mater (2002) 2 (3), 273 [Google Scholar]
- 28.Dietrich C, Koerver R, Gaultois MW, Kieslich G, Cibin G, Janek J, Zeier WG, Phys. Chem. Chem. Phys (2018) 20 (30), 20088. [DOI] [PubMed] [Google Scholar]
- 29.Dietrich C, Weber DA, Sedlmaier SJ, Indris S, Culver SP, Walter D, Janek J, Zeier WG, J. Mater. Chem. A (2017) 5 (34), 18111. [DOI] [PubMed] [Google Scholar]
- 30.Johansson A, Wendsjö Å, Tegenfeldt Jörgen, Electrochim. Acta (1992) 37 (9), 1487 [Google Scholar]
- 31.Dietrich C, Sadowski M, Sicolo S, Weber DA, Sedlmaier SJ, Weldert KS, Indris S, Albe K, Janek J, Zeier WG, Chem. Mater (2016) 28 (23), 8764 [Google Scholar]
- 32.Dietrich C, Weber DA, Culver S, Senyshyn A, Sedlmaier SJ, Indris S, Janek J, Zeier WG, Inorg. Chem (2017) 56 (11), 6681. [DOI] [PubMed] [Google Scholar]
- 33.Ohara K, Mitsui A, Mori M, Onodera Y, Shiotani S, Koyama Y, Orikasa Y, Murakami M, Shimoda K, Mori K, Fukunaga T, Arai H, Uchimoto Y, Ogumi Z, Sci Rep (2016) 6, 21302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cruickshank DWJ, J. Chem. Soc (1961), 5486 [Google Scholar]
- 35.Corbridge DEC, Bulletin de la Société française de Minéralogie et de Cristallographie (1971) 94 (3), 271 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.