

Abstract
Motivation
The interactions between proteins and other molecules are essential to many biological and cellular processes. Experimental identification of interface residues is a time-consuming, costly and challenging task, while protein sequence data are ubiquitous. Consequently, many computational and machine learning approaches have been developed over the years to predict such interface residues from sequence. However, the effectiveness of different Deep Learning (DL) architectures and learning strategies for protein–protein, protein–nucleotide and protein–small molecule interface prediction has not yet been investigated in great detail. Therefore, we here explore the prediction of protein interface residues using six DL architectures and various learning strategies with sequence-derived input features.
Results
We constructed a large dataset dubbed BioDL, comprising protein–protein interactions from the PDB, and DNA/RNA and small molecule interactions from the BioLip database. We also constructed six DL architectures, and evaluated them on the BioDL benchmarks. This shows that no single architecture performs best on all instances. An ensemble architecture, which combines all six architectures, does consistently achieve peak prediction accuracy. We confirmed these results on the published benchmark set by Zhang and Kurgan (ZK448), and on our own existing curated homo- and heteromeric protein interaction dataset. Our PIPENN sequence-based ensemble predictor outperforms current state-of-the-art sequence-based protein interface predictors on ZK448 on all interaction types, achieving an AUC-ROC of 0.718 for protein–protein, 0.823 for protein–nucleotide and 0.842 for protein–small molecule.
Availability and implementation
Source code and datasets are available at https://github.com/ibivu/pipenn/.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Protein interactions are crucial in many biological and cellular processes (Jones and Thornton, 1996), such as transcription, signal transduction or enzymatic activity. Proteins, through their binding interfaces, interact with each other and a variety of other molecules, giving rise to all manner of cell functions. Knowledge about these interfaces provides essential clues about the mechanisms underlying associated activities. This molecular level knowledge can be obtained by experimental and computational methods, and is applied in many scientific and therapeutic areas (Sperandio, 2012).
Protein interaction prediction refers to a set of computational methods that aim to predict different protein interaction types: protein–protein (PPI), protein–small molecule, protein–nucleotide (DNA/RNA), as summarized in Supplementary Figure S1 (Cui et al., 2019; Wang et al., 2019; Zhang and Kurgan, 2018). Such methods may utilize various classic Machine Learning (ML) (Cheng et al., 2008; Hou et al., 2017, 2021) and Deep Learning (DL) architectures (Hanson et al., 2018; Shi et al., 2021). Input features may be based on information from protein structure and/or sequence. Notwithstanding the enormous progress that has been made in the area of structure prediction, no reliable structural information is available (e.g. Su et al., 2021; Tunyasuvunakool et al., 2021) for many organisms, types of proteins and protein regions. Moreover, the usefulness of predicted structures for interface prediction may be limited (e.g. Xie and Xu, 2021). Therefore, we aim to predict protein bindings of the mentioned interaction types at residue level, using only information related to protein sequence. We explore the following question: Which DL architecture and composition of architectural building blocks are able to improve the performance of sequence-based interface predictors?
DL architectures are composed of multiple building blocks, such as initialization, regularization, loss, activation, each containing various parameters. The simplest neural architecture considered here is the fully connected Artificial Neural Network (ANN): every neuron in a layer is connected to all neurons in the next layer. This configuration is general purpose and structure agnostic. The input to this network is a single protein residue at a time, which means their sequence context is not considered.
Convolutional Neural Network (CNN) architectures are already extensively used in various flavors in computational biology (e.g. Shi et al., 2021), including protein interaction site prediction (e.g. Cui et al., 2019). A CNN consists of three types of hidden layers: convolutional layers, pooling layers and fully connected layers. For predictions in a protein sequence, neurons are organized sequentially (1D spatial form) and every neuron is connected to the neurons of a local region (receptive field) in the previous layer. Pooling layers perform down-sampling to speed up computation, and fully connected layers perform the actual classification task based on the abstract representations of the original protein sequence input.
Dilated Convolutional Networks (DCN) achieve large receptive fields by gradually increasing the dilation rate in subsequent layers (Yu and Koltun, 2016). This allows DCNs to remain relatively shallow, require few parameters and converge quickly. DCNs typically maintain high output resolutions without up-sampling, and have been successfully applied in many areas (Ho and Lin, 2018), including computational genomics (Gupta and Rush, 2017; Kelley et al., 2018).
The U-Net is the most commonly used CNN architecture, especially when the amount of training data is limited (Ronneberger et al., 2015). In a U-Net, high-dimensional information is first reduced (contracted) to a smaller latent space, and subsequently increased (expanded) to the original dimensionality. Visually, this gives rise to a U-shape, from which this architecture derives its name. Though more typically used in image recognition tasks, we find U-Nets—like CNNs in general—equally applicable for 1D (sequence-based) predictions.
The ResNet residual learning framework is a variation on CNN, which aims to solve the vanishing or exploding gradient problem, while retaining the ability to learn complex features (He et al., 2016a).
Recurrent Neural Networks (RNN) are particularly suitable for learning nonlinear dependencies in sequential data, such as protein sequences. No limit is imposed on the size of the input series of amino acid symbols, but each symbol is represented by additional layers. To mitigate the vanishing/exploding gradient in the resulting many-layer models (Hochreiter and Schmidhuber, 1997) and retain a computationally efficient training, Gated Recurrent Unit (GRU) was proposed by Cho et al. (2014). Later, Chung et al. (2014) showed that the performance of GRU is on par with the more complex LSTM (Hochreiter and Schmidhuber, 1997) on sequence modeling tasks, while taking much less time to train.
Recently, hybrid architectures of CNNs and RNNs are increasingly employed in computational biology. The hybrid models aim to get the best of both worlds: the spatial aspects of CNNs combined with the temporal aspects of RNNs. For instance, Hanson et al. (2018) combined residual convolutional network with LSTM to predict a protein’s residue–residue contacts (contact map prediction task). They use ResNet to capture spatial relationships between local residues, and LSTM to capture long-range relations between non-local residues. In the field of genomics, Quang and Xie (2016) integrated CNNs with bidirectional LSTMs for modeling of the properties and functions of non-coding DNA sequences. To learn a regulatory grammar in the DNA motifs, they use convolution layers to capture local patterns in the motif sequences, and recurrent layers to capture long-term dependencies between the motifs.
Here, we explore the effectiveness of each of the above neural net architectures: ann, dnet, unet, rnet, rnn and cnet. We expect they will each capture overlapping but distinct patterns in protein sequence data, yielding different predictions for which residues are part of an interface. We therefore also include an ensemble architecture, which combines the outputs of these six neural nets into one. All models, collectively referred to as PIPENN, are trained on five different training sets. Their performance is benchmarked with 11 test sets, including the standardized benchmark dataset introduced by Zhang and Kurgan (2018), referred to as ZK448. The latter is also used to assess and compare the performance of the PIPENN ensemble method with competing models. To our knowledge, we are the first to apply DL architectures to different types of protein interface prediction at this scale, and obtain the best prediction results to date.
2 Materials and methods
2.1 Datasets
To perform experiments on different types of protein interaction data, i.e. PPI, small molecule and nucleotide (DNA/RNA), we used five datasets for training (see Supplementary Table S1) and 11 independent datasets for testing (see Supplementary Table S3). The HHC dataset, containing homo- and heteromeric proteins annotated with PPI interface residues, was already available in our group from Hou et al. (2015, 2017). Another part was obtained from Zhang and Kurgan (2018): the benchmark test set ZK448, which contains proteins annotated with residues for all types of protein interaction. The final part was newly constructed: the BioDL dataset that contains proteins annotated with residues for all types of protein interaction, as described below and illustrated in Figure 1.
Fig. 1.
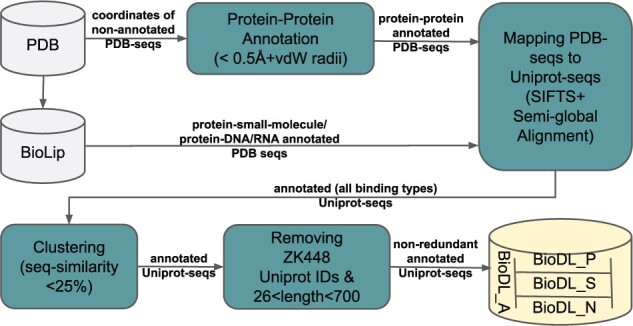
Generation of the BioDL dataset from the PDB and BioLip databases
For small molecule and nucleotide interactions, we retrieved the whole BioLip (Yang et al., 2013) database and extracted the interaction annotation data. For PPIs, we downloaded the coordinates of 138 729 protein structures of 2.5 Å resolution or lower, excluding fragments, from the PDB (Berman et al., 2000) on 3 April 2019. Following the annotation criterion in BioLip, we annotated a residue as interacting if the distance between one of its atoms and any atom of the ligand is less than the sum of their Van der Waals radii plus 0.5 Å.
The newly derived annotations and the existing annotations from BioLip are associated with PDB sequences. In order to test our models on the benchmark test set ZK448, where annotations are associated with Uniprot sequences, we mapped PDB sequences to Uniprot as follows. We used SIFTS (Velankar et al., 2013) to retrieve all Uniprot sequences corresponding to the PDB entries. Entries with missing or conflicting Uniprot IDs were discarded. We mapped residues between PDB and Uniprot sequences by alignment with harsh penalties (mismatches—5, gap opening—20 and extension—50). Alignments where <80% of interaction site residues were mapped, or with more than two inserts, five deletions, three mismatches in the interaction sites, or five mismatches were discarded.
Subsequently, BLASTClust was used to cluster the obtained Uniprot sequences at 25% sequence similarity. All clusters containing a Uniprot ID used in ZK448 were removed and thereafter randomly one sequence from each cluster was chosen. From this non-redundant dataset, based on the criterion used by Wang et al. (2017), proteins having sequences longer than 700 and shorter than 26 amino acids were removed. The final dataset was split into a training set BioDL_A_TR (95%) and a testing set BioDL_A_TE (5%), containing annotations for all three types of interactions. We further split these into proteins annotated with PPI (BioDL_P_TR & _TE), small molecule (BioDL_S_TR & _TE) and nucleotide (BioDL_N_TR & _TE) interactions. See Supplementary Tables S1 and S3 for dataset statistics of the training (TR) and test (TE) sets, respectively.
2.2 Data features
For each residue in our BioDL dataset, we record the amino acid type (AA); its conservation, represented by a Position Specific Scoring Matrix (PSSM) score; if it is part of a domain; and the length of the sequence it belongs to. Furthermore, we predict its accessible surface area (SA) and secondary structure (SS) from sequence. We include four training/testing labels for each residue, tracking whether the residue is part of a known interface with (i) other proteins, (ii) DNA or RNA, (iii) small molecule ligands or (iv) any of the above.
PSSM profiles were generated for each sequence using PSI-BLAST (Altschul et al., 1997), retrieving max. 500 sequence homologs from the NR70 database, using three iterations and an E-value threshold of 0.001. As a result, we obtained 20 PSSM values (one per amino acid type) for each residue. The PSSM scores were normalized using the sigmoid function. We used NetSurfP to predict from sequence the Absolute/Relative Surface Accessibility (ASA/RSA) and the propensities for α-helix (PA), β-sheet (PB) and coil (PC) (Petersen et al., 2009). To indicate whether or not a residue belongs to a conserved protein domain, we employed the protein domain information from the Pfam (Mistry et al., 2021) database for the Uniprot sequences.
Finally, we included four windowed aggregates for each of the features. Each aggregated feature is the unweighted average of the feature value over a specific number of the neighboring residues: 3, 5, 7 or 9. They are abbreviated as <window size>_wm_<feature name>, e.g. 9_wm_PB refers to the window mean of the predicted β sheet probabilities (PB) across a window of nine adjacent residues, annotating the one in the center. This leads to a total of 128 features, as detailed in Supplementary Table S13.
2.3 Learning architectures
The input layer of our ANN-based architecture ann consists of a number of neurons, each representing a feature of one amino acid (see Supplementary Fig. S4). The network has eight hidden layers and its output is a binary classification predicting whether or not an amino acid is part of an interface. In order to capture as much as possible information about a residue, the number of neurons in the first hidden layer is the highest, decreasing gradually in the subsequent layers to achieve more general representations. However, it has a large number of parameters (weights) and relatively long convergence time.
We designed three different CNN architectures dnet, unet and rnet. The input layers for these consist of one neuron per feature per amino acid in the protein sequence. dnet is based on dilated CNN, has five convolutional layers, zero pooling layer and one fully connected layer (Supplementary Fig. S5). The dilation rate increases from 1 to 16. unet is based on the U-Net architecture and the contraction block starts with 1024 (length of padded protein sequence) and is gradually down-sampled by max-pooling to 32 at the bottleneck (Supplementary Fig. S6). For up-sampling transposed convolutions are used (Dumoulin and Visin, 2016). rnet is a residual CNN that allows us to experiment with deeper CNNs (Supplementary Fig. S7). We use a slightly modified version of the full pre-activation configuration (He et al., 2016b). In rnet, a residual block consists of two sequential full pre-activation configurations, each containing a Dropout, BatchNormalization and Parametric Rectified Linear Unit (PReLU) followed by a 1D convolution. We use eight such residual blocks.
Our recurrent NN architecture rnn consists of two GRU layers, each containing 1024 cells with each cell having an output dimension of 128 that goes to the next cell (Supplementary Fig. S8). Finally, our cnet (Supplementary Fig. S8) is a hybrid architecture that simply combines our rnet residual and rnn recurrent architectures.
2.4 Architecture building blocks and parameters
DL architectures are composed of multiple building blocks, each containing various parameters. Different compositions enable learning architectures to provide great flexibility and a broad application area. The choice for building blocks and parameter values strongly influences the performance that may be achieved; however, this depends both on the architecture and on the dataset, making this one of the main challenges in DL. Below, we briefly motivate each of our choices.
Initialization method (IM)
We experimented with uniform and normal variants of the Glorot (Glorot and Bengio, 2010) and He (He et al., 2015) IMs. He-Uniform initialization proved most suitable for our architectures.
Regularization method (RM)
To overcome overfitting, we explored combinations of RMs: Lasso (L1), Ridge (L2), BatchNormalization, Dropout and Early-Stopping. A combination of BatchNormalization, Dropout (20%) and Early-Stopping (based on the AUC metric) yielded best results.
Loss function (LF)
We explored a number of LFs: Mean Squared Error (MSE), Jaccard, Tversky and CrossEntropy. After running several experiments and due to having significant class imbalance in our data, we decided to use the CrossEntropy loss with an additional term that compensates for class imbalance.
Activation function (AF)
Our choice of AF is based on a neuron’s position in the network, the computational speed of calculating its gradient, and its differentiability. We used Sigmoid at the last layer of all our architectures. For the hidden layers, using Tanh for the rnn architecture and PReLU for all other architectures provided best performance.
Encoding scheme (ES)
We performed experiments with One-Hot and ProtVec to encode amino acid sequences. One-Hot represents each amino acid simply as a 20D sparse vector. ProtVec constructs 3-gram words of amino acids for each protein sequence from Swiss-Prot, and trains a skip-gram Neural Network on these data (Asgari and Mofrad, 2015). The output of the network is an embedding space of 100D dense vectors, which we used for as input features for our models. We achieved better performance with the One-Hot representation.
2.5 Training and testing procedures
We split each of the training datasets into two parts: train-set and val-set (Supplementary Table S1). The train-set part (80%) is used exclusively for training, and the val-set part (20%) is used for three purposes: (i) for gaining insight into the performance of an architecture, (ii) for terminating the training when the performance does not show any improvement and (iii) for training of the ensnet architecture. The training procedure is as follows. After all six architectures are trained on a train-set, their trained models are applied on the corresponding val-set. The obtained predictions of each model become the training data for the ensnet architecture, see overview in Figure 2.
Fig. 2.
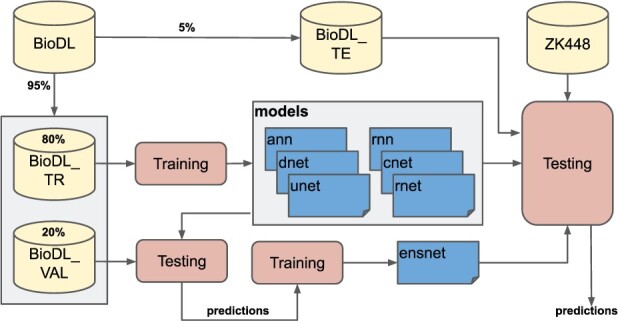
Training and testing procedure of our predictors (Section 2.5 for the explanation of the procedure)
For all our architectures and datasets, we used the same hyperparameters, software platform and infrastructure. The hyperparameters that influence the performance and convergence time of the architectures are: batch size (8), optimization algorithm (Adam), learning rate (1e–4), maximum number of epochs (300), padding constants, bias vector usage and the float size (64). We used the Keras API of Tensorflow-2.1.0 to build our architectures and trained them on a Linux machine having 32 CPUs, 2 GPUs and 256 GB memory (see Supplementary Table S2 for the run-time statistics).
We evaluated the performance of our trained models on the completely independent test sets shown in Supplementary Table S3, using Accuracy (ACC), Specificity (SPEC), Precision (PREC), Sensitivity/Recall (SENS), Balanced F-score (F1), Matthews Correlation Coefficient (MCC), average precision on the PR-curve (AP) and area under the ROC curve (AUC). We calculate P-values for differences in AUC-ROC using the approach by Hanley and McNeil (1982). Except for AP and AUCs, all metrics use the confusion matrix. MCC and AP are insensitive to class imbalance (on average about 11% of residues are interface, see Supplementary Table S1). All results shown are based on the Equal method (Zhang and Kurgan, 2018), by which a cutoff point is selected where FP and FN are equal. Hence, the values of PREC, SENS and F1 are always the same. Output (predictions) of the models is unpadded before applying the metrics. Performance plots were created using the Plotly package.
2.6 Feature importance
For scoring and ranking the importance of features, we used the KernelExplainer from the SHAP package (Lundberg and Lee, 2017). For each sample in the test set the contribution of each feature to the predicted outcome of a model is estimated, which is called an SHAP value.
3 Results
3.1 BioDL is sufficiently large for DL
DL architectures learn best when trained on a large dataset. We constructed the BioDL dataset based on BioLip and PDB, for training and testing of our various architectures. In total, over 6800 proteins are included, containing over 2 million residues of which 10.7% are interface residues; see Supplementary Table S1 for details. Runtimes for prediction are dominated by the feature generation, notably running PSI-Blast to generate the PSSMs, which takes up to 5 min per input protein.
Within BioDL we have collected annotations of three specific types of protein interaction: PPI in BioDL_P, small molecule ligands in BioDL_S and nucleic acid interaction in BioDL_N. A fourth set annotates any of these three interactions: BioDL_A. For better comparison with our previous work using Random Forest models, we also include the HHC dataset from Hou et al. (2017, 2019). Each of these datasets are further split into training (_TR) and test (_TE) sets as shown in Figure 2; see Supplementary Table S3 for an overview and statistics of training sets. For HHC_TE, we also report separately for homomeric (Homo_TE) and heteromeric (Hetero_TE) PPI. Training duration can be found in Supplementary Table S2. As a further independent test set, we also used ZK448_TE from Zhang and Kurgan (2018), which also allows comparison with their benchmark results. The ZK448_TE, like BioDL, contains protein (ZK448_P_TE), small molecule (ZK448_S_TE) and nucleotide interaction annotations (ZK448_N_TE), as well as all combined (ZK448_A_TE). See Supplementary Table S3 for an overview and statistics of test sets.
The overlap between the BioDL training sets is shown in a Venn diagram in Supplementary Figure S2; out of 6832 proteins in the training set there are only 85 with annotations for all three interaction types. This pattern is similar in the BioDL and ZK448 test sets (Supplementary Fig. S3). We make distinctions between the models that were trained on the BioDL_A_TR dataset containing all interaction types, i.e. generic, and those trained on a type-specific interaction data, i.e. BioDL_P_TR for PPI, BioDL_S_TR for small molecules and BioDL_N_TR for nucleic acids. We suffix those models correspondingly: _a, _p, _s and _n. Models trained on the PPI-specific HHC dataset are suffixed with _hhc.
3.2 Building block composition matters
We tested our predictors with different compositions of the architectural building blocks introduced in Section 2.4. These were trained on the smaller HHC PPI dataset for efficiency. Table 1 shows the metrics for different choices of building blocks for the dnet_hhc predictor. Other architectures and datasets show very similar trends (data not shown). dnet_hhc (the first row) corresponds to the composition with the highest performance in AUC: HeUniform kernel initialization, CrossEntropy loss function, 1D spatial form, PReLU activation function, Padding for unifying the input shape, One-Hot amino acid encoding and Dropout and BatchNormalization for regularization. Subsequent rows show the effect of substituting (→), adding (+) or removing (–) blocks. GlorotNormal kernel initialization yields highest AP; MeanSquaredError loss function highest accuracy (ACC), F1 and MCC. It is worth mentioning that for the best composition in dnet_hcc we did not use the MaxPooling. All other variations yield lower performances, and the impact of omitting Dropout, Padding and BatchNormalization is the strongest.
Table 1.
Impact of different architectural building blocks on the performance of the dnet_hhc PPI predictor trained on HHC_TR and tested on HHC_TE
| Model | ACC | SPEC | F1 | MCC | AP | AUC | |
|---|---|---|---|---|---|---|---|
| dnet_hhc | 0.784 | 0.868 | 0.403 | 0.272 | 0.381 | 0.733 | 0 |
| hu → gn | 0.783 | 0.868 | 0.401 | 0.269 | 0.398 | 0.730 | –0.003 |
| ce → mse | 0.785 | 0.868 | 0.404 | 0.273 | 0.391 | 0.728 | –0.005 |
| 1d → 2d | 0.781 | 0.866 | 0.394 | 0.261 | 0.379 | 0.723 | –0.010* |
| pre → rel | 0.780 | 0.866 | 0.392 | 0.258 | 0.390 | 0.720 | –0.013* |
| + mp | 0.784 | 0.868 | 0.403 | 0.272 | 0.387 | 0.718 | –0.015* |
| oh → pv | 0.774 | 0.862 | 0.373 | 0.235 | 0.358 | 0.714 | –0.019** |
| − bn | 0.770 | 0.860 | 0.364 | 0.224 | 0.327 | 0.696 | –0.037** |
| − pa | 0.750 | 0.848 | 0.309 | 0.157 | 0.267 | 0.661 | –0.072** |
| − do | 0.752 | 0.849 | 0.314 | 0.163 | 0.291 | 0.646 | –0.087** |
Note: ‘hu → gn’: kernel initialization GlorotNormal instead of HeUniform used; ‘ce → mse’: loss function MeanSquaredError instead of CrossEntropy used; ‘1d → 2d’: spatial form 2D instead of 1D used; ‘pre → rel’: activation function RELU instead of PRELU used; ‘+ mp’: MaxPooling layer used; ‘ov → pv’: ProtVec encoding instead of One-Hot used; ‘− bn’: no BatchNormalization layer used; ‘− pa’: no padding used; ‘− do’: no Dropout layer used. Highest score per metric indicated in bold.
P < 0.05.
P < 0.0005.
3.3 Ensembling improves performance
We compared the performance of our ensemble predictor with those of our other DL predictors. Table 2 shows that ensnet_p improves the AUC by 0.016 (P < 0.0006) wrt the best scoring other DL predictor (dnet_p) on the PPI data, and it achieves the highest sensitivity (TPR) for any error (FPR) in the ROC plot (Fig. 3A). Moreover, the P/R plot in Figure 3B shows that rnet_p obtains the highest precision at low recall values but ensnet_p achieves the highest precision at high recall values. To further investigate the reason behind this improvement, we explored the relation between accuracy of the ensnet_p and the other predictors in scatter plots of MCC scores of ensnet_p versus the average and standard deviation of MCC scores of our six other predictors. Our analyses suggest that ensnet especially improves predictions for individual proteins where the average performance of the other models was already relatively high (Supplementary Fig. S12).
Table 2.
Performance of the ensemble PPI predictor ensnet_p compared with all other predictors trained on BioDL_P_TR and tested on BioDL_P_TE
| Model | ACC | SPEC | F1 | MCC | AP | AUC |
|---|---|---|---|---|---|---|
| ensnet_p | 0.840 | 0.909 | 0.339 | 0.249 | 0.302 | 0.755 |
| dnet_p | 0.834 | 0.905 | 0.312 | 0.218 | 0.276 | 0.739 |
| rnn_p | 0.833 | 0.905 | 0.310 | 0.215 | 0.276 | 0.736 |
| rnet_p | 0.833 | 0.905 | 0.309 | 0.215 | 0.279 | 0.735 |
| cnet_p | 0.832 | 0.904 | 0.303 | 0.208 | 0.273 | 0.733 |
| ann_p | 0.833 | 0.905 | 0.309 | 0.214 | 0.270 | 0.729 |
| unet_p | 0.829 | 0.903 | 0.292 | 0.196 | 0.253 | 0.717 |
Note: Highest score per metric indicated in bold; AUC differences >0.10 are P < 0.05.
Fig. 3.
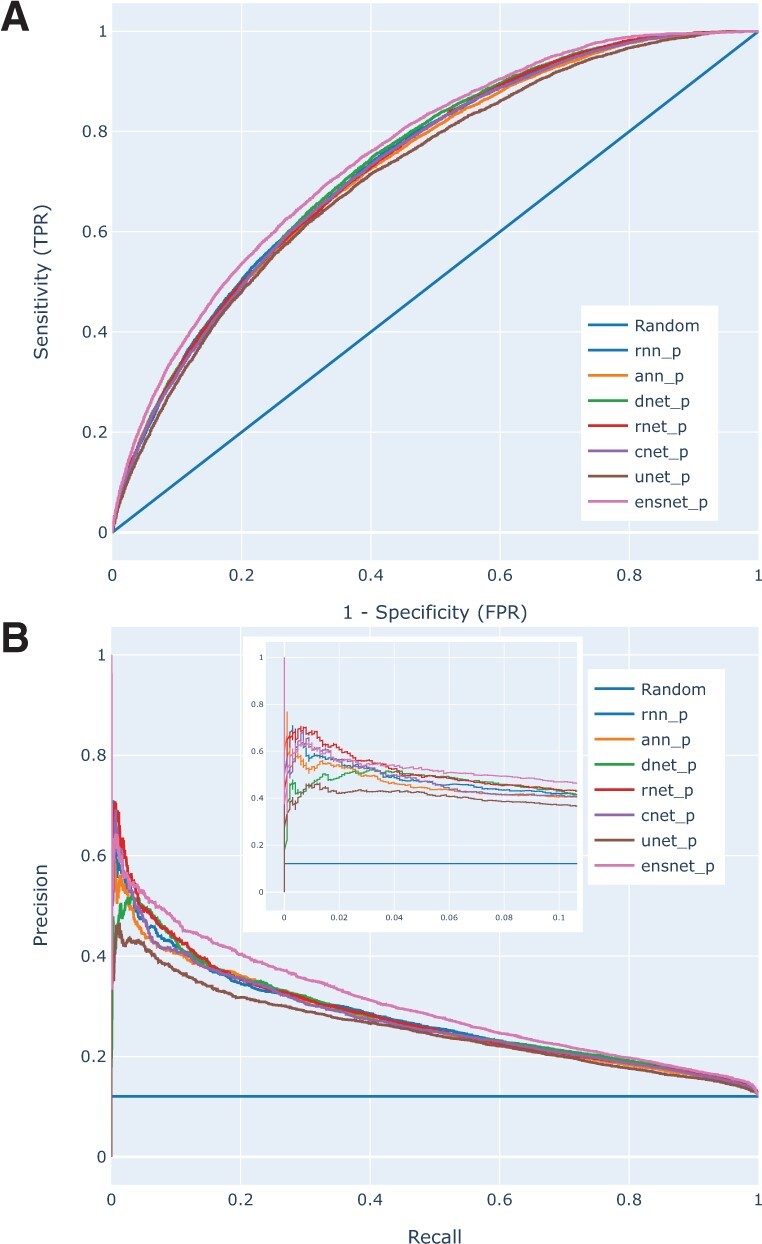
(A) ROC and (B) P/R plots of all six architecture models and the ensemble models, trained on BioDL_P_TR and tested on BioDL_P_TE PPI data. The ensnet_p clearly outperforms the six architecture models in the ROC plot, and in the P/R plot only rnet_p and rnn_p yield somewhat higher precision (∼0.6) at very low recall (0.01–0.02)
3.4 Type-specific predictions are more accurate
In Table 3, we compare the generic ensnet_a with type-specific ensnet_p, ensnet_s and ensnet_n models, each trained on their corresponding BioDL training sets. When tested on the corresponding interaction-specific datasets BioDL_P_TE, BioDL_S_TE and BioDL_N_TE, the specific models consistently obtain performances higher than the interaction-generic ensnet_a, as one may expect.
Table 3.
Performance of ensnet_a, trained on the generic BioDL_A_TR dataset, compared with the ensnet p, s and n models trained on type-specific datasets containing protein, small molecule or nucleotide interaction interfaces and scored performance on the interaction-specific test sets as indicated
| Model | Test set | ACC | SPEC | F1 | MCC | AP | AUC |
|---|---|---|---|---|---|---|---|
| ensnet_a | BioDL_P_TE | 0.828 | 0.902 | 0.289 | 0.192 | 0.248 | 0.733 |
| ensnet_p | 0.840 | 0.909 | 0.339 | 0.249 | 0.302 | 0.755 | |
| ensnet_a | BioDL_S_TE | 0.937 | 0.967 | 0.339 | 0.306 | 0.289 | 0.826 |
| ensnet_s | 0.944 | 0.970 | 0.413 | 0.384 | 0.388 | 0.864 | |
| ensnet_a | BioDL_N_TE | 0.901 | 0.947 | 0.272 | 0.219 | 0.238 | 0.835 |
| ensnet_n | 0.921 | 0.957 | 0.418 | 0.376 | 0.399 | 0.894 |
Note: Highest AUC per metric per test set indicated in bold (P < 1e−6).
3.5 Network architecture matters
We compared the performance of all seven of our models (six separate architectures and an ensemble) trained on HHC_TR and the four BioDL_*_TR, yielding 35 trained predictors. Each was applied to their corresponding test sets: three for HHC_TR-trained models (Homo_TE, Hetero_TE and combined HHC_TE), and two each for the BioDL_*_TR (BioDL_*_TE and ZK448_*_TE). See Supplementary Tables S4, S5, S6, S7 and S8, for HHC, BioDL_A, _P, _S and _N, respectively, for details. The ensemble ensnet predictors perform best on all test sets, as we already saw for the BioDL_P_TR models on BioDL_P_TE in Table 2.
We further compared our ensnet_hhc, ensnet_a, ensnet_s and ensnet_n predictors with other published and available state-of-the-art sequence-based predictors on the same datasets. The published predictors use various methods and architectures including Random Forest, Logistic Regression, Support Vector Machine and Neural Networks. Table 4 shows comparisons with SeRenDIP and other PPI predictors benchmarked by Zhang and Kurgan (2019) on the PPI datasets, and SCRIBER and DRNApred on the ZK448 small molecule and nucleotide (DNA/RNA) datasets, respectively. All predictors mentioned here in Table 4 use similar input features, such as protein length, ASA, RSA, PSSM and secondary structure predicted from sequence.
Table 4.
Performance comparison of our ensnet models and other state-of-the-art sequence-based interaction prediction methods on applicable test sets
| Model | Test set | ACC | SPEC | F1 | MCC | AP | AUC |
|---|---|---|---|---|---|---|---|
| Protein–protein interaction (PPI) | |||||||
| ensnet_hhc | Homo_TE | 0.767 | 0.849 | 0.485 | 0.335 | 0.491 | 0.769 ** |
| SeRenDIP | 0.277 | 0.724 | |||||
| ensnet_hhc | Hetero_TE | 0.849 | 0.916 | 0.197 | 0.114 | 0.155 | 0.661 * |
| SeRenDIP | 0.122 | 0.636 | |||||
| ensnet_a | ZK448_A_TE | 0.785 | 0.870 | 0.385 | 0.254 | 0.357 | 0.729 ** |
| SCRIBERa | n.a. | 0.896 | 0.333 | 0.230 | 0.287 | 0.715 | |
| SSWRFa | n.a. | 0.891 | 0.287 | 0.178 | 0.256 | 0.687 | |
| CRFPPIa | n.a. | 0.887 | 0.266 | 0.154 | 0.238 | 0.681 | |
| LORISa | n.a. | 0.887 | 0.263 | 0.151 | 0.228 | 0.656 | |
| SPRINGSa | n.a. | 0.882 | 0.229 | 0.111 | 0.201 | 0.625 | |
| PSIVERa | n.a. | 0.874 | 0.191 | 0.066 | 0.170 | 0.581 | |
| SPRINTa | n.a. | 0.873 | 0.183 | 0.057 | 0.167 | 0.570 | |
| SPPIDERa | n.a. | 0.870 | 0.198 | 0.071 | 0.159 | 0.517 | |
| Protein–small molecule interaction | |||||||
| ensnet_s | ZK448_S_TE | 0.899 | 0.945 | 0.419 | 0.364 | 0.409 | 0.849 ** |
| SCRIBER | 0.874 | 0.931 | 0.278 | 0.209 | 0.259 | 0.706 | |
| Protein–DNA/RNA interaction | |||||||
| ensnet_n | ZK448_N_TE | 0.871 | 0.927 | 0.469 | 0.396 | 0.460 | 0.823 ** |
| DRNApred (DNA) | 0.830 | 0.903 | 0.294 | 0.198 | 0.240 | 0.609 | |
| DRNApred (RNA) | 0.814 | 0.894 | 0.230 | 0.124 | 0.248 | 0.547 | |
Note: Highest scores per metric per test set indicated in bold; confidence for difference in AUC-ROC with runner-up.
Metrics according to Zhang and Kurgan (2019) on their ZK448 test set.
P < 0.05.
P < 0.005.
For comparing ensnet_hhc with SeRenDIP, we used exactly the same test sets as used by SeRenDIP. For comparing ensnet_a with the predictors as published in Zhang and Kurgan (2019), we exactly followed their testing approach: we calculated average of metrics over 10 subsets of randomly selected 50% of ZK448_A_TE, and we only considered PPIs as to be predicted interactions and all other types as non-interacting. For comparing ensnet_s with SCRIBER, we randomly selected 10 proteins (UniProt IDs in Supplementary Table S12) from ZK448_S_TE and calculated the comparison metrics based on the protein–small molecule binding propensities returned by their webserver. For comparing ensnet_n with DRNApred, we used all proteins in ZK448_N_TE (38 proteins) and calculated the comparison metrics based on the nucleotide interaction propensities returned by their webserver. The cutoff points were selected such that the number of false positives (FPs) and false negatives (FNs) are equal; this affects ACC, SPEC, F1 and MCC. As can be seen from Table 4, our ensnet_hhc, ensnet_a, ensnet_s and ensnet_n predictors perform better than the corresponding state-of-the-art methods, on virtually all considered metrics.
3.6 Feature importance
Figure 4 shows the top 15 ranking of the importance of the features, as measured by SHAP, for one of our models (ann_p), estimated from 5000 randomly selected amino acids from ZK448_P. As seen previously (Hou et al., 2017, 2021), protein sequence length is by far the most important feature. High values of length (red dots; residues of longer proteins), in general, have lower SHAP values, i.e. lower likelihood of a residue being predicted to be part of an interface. Also, the high importance of secondary structure, particularly coil (WP_PC) and α-helix (WP_PA) is consistent with our previous work. High probability of coil across the windows (red WM_PC) also has notable impact on the predictions, as can also be seen from the correlations in Supplementary Figure S14.
Fig. 4.
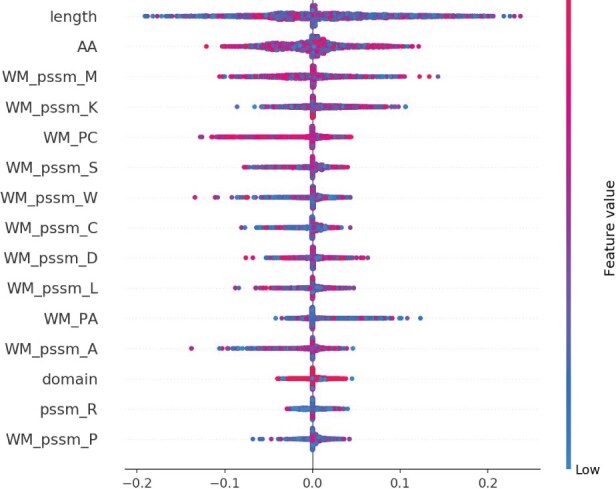
The top 15 ranking of the importance of the features based on the SHAP values for 5000 randomly selected amino acids in ZK448_P, indicating the contribution of a feature to a residue’s interface prediction. Colors represent the input values of a feature: blue for low and red for high values. The width of the distribution of a feature’s SHAP values shows its relative importance across the sampled residues. For aggregated features only the sum is shown, e.g. WM_PC is the sum of 3_wm_PC, 5_wm_PC, 7_wm_PC and 9_wm_PC
4 Discussion and conclusion
This work presents an in-depth and systematic comparison of multiple DL architectures for sequence-based prediction of protein interface residues. We include a series of neural nets, PIPENN, whose ensemble method performs well on generic and type-specific interface prediction tasks, including PPI, small molecule and nucleotide (DNA/RNA) interface prediction from sequence.
We explored multiple combinations of DL architecture building blocks, such as spatial forms, encoding schemes, network initializations, loss and activation functions and regularization mechanisms. Selected combinations resulted in six models and an ensemble, which we trained on existing and newly constructed training datasets. Performance was benchmarked on several independent test sets, facilitating fair comparison. Comparing the performance of our models to that of other published and available state-of-the-art sequence-based predictors on the same test sets, shows that our ensemble predictors obtain most accurate predictions on all interface types.
It is worth noting that the different prediction tasks are not equally difficult. We reproduce an earlier observation that homomeric PPI interface residues can be better predicted than heteromeric interfaces, as noted in Hou et al. (2017, 2019). Moreover, all architectures predict protein–small molecule and protein–nucleotide interface residues more accurately than protein–protein interface residues. This might be explained by differences in the size, specificity and structural heterogeneity of interfaces involved with these respective types of interaction: nucleotide interfaces are generally much smaller than PPI interfaces, affecting their variety and relative sequence locality; small molecule interactions typically require very specific chemical properties, leading to equally specific interface compositions; and the structural similarity between two random strands of DNA is much larger than that between two random proteins, so it stands to reason the similarity between (and consequently, predictability of) two protein–DNA interfaces is also greater than the similarity between two protein–protein interfaces.
One issue that likely affects our observations, and those of related studies as well, is that we necessarily operate under a ‘closed world’ assumption: not all interfaces are known, yet we assume that residues which were never observed to be part of an interface, truly are not part of one. Consequently, future experimental data are likely to reveal some of the residues that we label as negatives (not interacting) should in fact have been labeled positive.
Recent advances in protein structure prediction mean that structures are now available for increasing amounts of proteins (e.g. Su et al., 2021; Tunyasuvunakool et al., 2021), which opens up new types of features to be included in DL methods for interface prediction (e.g. Dai and Bailey-Kellogg, 2021; Xie and Xu, 2021). These are really exciting developments, and e.g. Dai and Bailey-Kellogg (2021) report for their ‘Unbound PInet (Aug 50)’ AUC-ROC and F1 of around 0.6 for PPI interface prediction on the DBD3 and DBD5 test sets. Some of the underlying methodology for structure prediction may also be directly applied to interface contact prediction, as recently reviewed by Cui et al. (2021).
In summary, we contribute the following: (i) BioDL, a dataset of protein sequences annotated with residue-level and type-specific interface annotation of sufficient size to perform DL; (ii) systematic characterization of different combinations of architectural building blocks, and their impact on the predictive performance of resulting neural nets; (iii) the PIPENN suite of neural net models, whose ensemble method outperforms state-of-the-art sequence-based models when it comes to predicting various types of protein interface. This conclusively demonstrates DL can contribute much to current efforts in computational protein interface prediction from sequence. We provide a public repository containing source code, datasets and pretrained models: https://github.com/ibivu/pipenn/.
Supplementary Material
Acknowledgements
We kindly acknowledge sponsoring by the VU HPC Council and the IT for Research (ITvO) BAZIS Linux computational cluster at the VU University Amsterdam.
Author contributions
R.H., B.S., K.A.F. and S.A. designed the experiments. H.d.F., K.A.F., B.S. and R.H. collected the datasets. R.H. implemented the methods and performed the experiments. R.H., B.S. and K.A.F. analyzed and interpreted the results. R.H., B.S., S.A., J.H. and K.A.F. wrote the article text. All authors revised the article and approved the final version for publication.
Financial Support: none declared.
Conflict of Interest: none declared.
Contributor Information
Bas Stringer, Department of Computer Science, IBIVU—Center for Integrative Bioinformatics, Vrije Universiteit, 1081HV Amsterdam, The Netherlands.
Hans de Ferrante, Department of Computer Science, IBIVU—Center for Integrative Bioinformatics, Vrije Universiteit, 1081HV Amsterdam, The Netherlands.
Sanne Abeln, Department of Computer Science, IBIVU—Center for Integrative Bioinformatics, Vrije Universiteit, 1081HV Amsterdam, The Netherlands.
Jaap Heringa, Department of Computer Science, IBIVU—Center for Integrative Bioinformatics, Vrije Universiteit, 1081HV Amsterdam, The Netherlands.
K Anton Feenstra, Department of Computer Science, IBIVU—Center for Integrative Bioinformatics, Vrije Universiteit, 1081HV Amsterdam, The Netherlands.
Reza Haydarlou, Department of Computer Science, IBIVU—Center for Integrative Bioinformatics, Vrije Universiteit, 1081HV Amsterdam, The Netherlands.
References
- Altschul S.F. et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asgari E., Mofrad M.R.K. (2015) Continuous distributed representation of biological sequences for deep proteomics and genomics. PLoS One, 10, e0141287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H.M. et al. (2000) The Protein Data Bank. Nucleic Acids Res., 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C.W. et al. (2008) Predicting RNA-binding sites of proteins using support vector machines and evolutionary information. BMC Bioinform., 9, S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho K. et al. (2014) On the properties of neural machine translation: encoder–decoder approaches. In: Proceedings of SSST-8, Eighth Workshop on Syntax, Semantics and Structure in Statistical Translation. Association for Computational Linguistics, Doha, Qatar, pp. 103–111.
- Chung J. et al. (2014) Empirical evaluation of gated recurrent neural networks on sequence modeling. In: NIPS 2014 Workshop on Deep Learning, December 2014, Montreal, Canada.
- Cui F. et al. (2021) Sequence representation approaches for sequence-based protein prediction tasks that use deep learning. Brief. Funct. Genomics, 20, 61–73. [DOI] [PubMed] [Google Scholar]
- Cui Y. et al. (2019) Predicting protein-ligand binding residues with deep convolutional neural networks. BMC Bioinform., 20, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai B., Bailey-Kellogg C. (2021) Protein interaction interface region prediction by geometric deep learning. Bioinformatics, 37, 2580–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumoulin V., Visin F. (2016) A guide to convolution arithmetic for deep learning. arXiv:1603.07285.
- Glorot X., Bengio Y. (2010) Understanding the difficulty of training deep feedforward neural networks. J. Mach. Learn. Res., 9, 249–256. [Google Scholar]
- Gupta A., Rush A.M. (2017) Dilated convolutions for modeling long-distance genomic dependencies. bioRxiv. 10.1101/200857. [DOI]
- Hanley J.A., McNeil B.J. (1982) The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology, 143, 29–36. [DOI] [PubMed] [Google Scholar]
- Hanson J. et al. (2018) Accurate prediction of protein contact maps by coupling residual two-dimensional bidirectional long short-term memory with convolutional neural networks. Bioinformatics, 34, 4039–4045. [DOI] [PubMed] [Google Scholar]
- He K. et al. (2015) Delving deep into rectifiers: Surpassing human-level performance on imagenet classification. In: IEEE International Conference on Computer Vision (ICCV 2015), Santiago, Chile, Vol. 1502. [Google Scholar]
- He K. et al. (2016a) Deep residual learning for image recognition. In: 2016 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Las Vegas, NV, USA, pp. 770–778.
- He K. et al. (2016b) Identity mappings in deep residual networks. In: Computer Vision—ECCV 2016. Springer International Publishing, Cham, Amsterdam, The Netherlands, pp. 630–645. [Google Scholar]
- Ho D., Lin Q. (2018) Person segmentation using convolutional neural networks with dilated convolutions. Electron. Imaging, 2018, 455–4557. [Google Scholar]
- Hochreiter S., Schmidhuber J. (1997) Long short-term memory. Neural Comput., 9, 1735–1780. [DOI] [PubMed] [Google Scholar]
- Hou Q. et al. (2015) Sequence specificity between interacting and non-interacting homologs identifies interface residues—a homodimer and monomer use case. BMC Bioinform., 16, 325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Q. et al. (2017) Seeing the trees through the forest: sequence-based homo- and heteromeric protein-protein interaction sites prediction using random forest. Bioinformatics, 33, 1479–1487. [DOI] [PubMed] [Google Scholar]
- Hou Q. et al. (2019) SeRenDIP: Sequential RemasteriNg to DerIve profiles for fast and accurate predictions of PPI interface positions. Bioinformatics, 35, 4794–4796. [DOI] [PubMed] [Google Scholar]
- Hou Q. et al. (2021) SeRenDIP-CE: sequence-based interface prediction for conformational epitopes. Bioinformatics, 37, 3421–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S., Thornton J.M. (1996) Principles of protein-protein interactions. Proc. Natl. Acad. Sci. USA, 93, 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley D. et al. (2018) Sequential regulatory activity prediction across chromosomes with convolutional neural networks. Genome Res., 28, 739–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg S.M., Lee S.-I. (2017). A unified approach to interpreting model predictions. In: Guyon I. et al. (eds.), Advances in Neural Information Processing Systems, Vol. 30. Curran Associates, Inc., Red Hook, NY, pp. 4765–4774. [Google Scholar]
- Mistry J. et al. (2021) Pfam: the protein families database in 2021. Nucleic Acids Res., 49, D412–D419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen B. et al. (2009) A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC Struct. Biol., 9, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quang D., Xie X. (2016) DanQ: a hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences. Nucleic Acids Res., 44, e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronneberger O. et al. (2015) U-net: convolutional networks for biomedical image segmentation. Lecture Notes Comput. Sci., 9351, 234–241. [Google Scholar]
- Shi Q. et al. (2021) Deep learning for mining protein data. Brief. Bioinform., 22, 194–218. [DOI] [PubMed] [Google Scholar]
- Sperandio O. (2012) Editorial: toward the design of drugs on protein-protein interactions. Curr. Pharm. Des., 18, 4585. [DOI] [PubMed] [Google Scholar]
- Su H. et al. (2021) Improved protein structure prediction using a new multi-scale network and homologous templates. Adv. Sci., 8, 2102592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunyasuvunakool K. et al. (2021) Highly accurate protein structure prediction for the human proteome. Nature, 596, 590–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velankar S. et al. (2013) SIFTS: Structure Integration with Function, Taxonomy and Sequences resource. Nucleic Acids Res., 41, D483–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S. et al. (2017) Accurate de novo prediction of protein contact map by ultra-deep learning model. PLoS Comput. Biol., 13, e1005324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. et al. (2019) SmoPSI: analysis and prediction of small molecule binding sites based on protein sequence information. Comput. Math. Methods Med., 2019, 1926156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z., Xu J. (2021) Deep graph learning of inter-protein contacts. Bioinformatics, 38, 947–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J. et al. (2013) BioLiP: a semi-manually curated database for biologically relevant ligand–protein interactions. Nucleic Acids Res., 41, D1096–D1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F., Koltun V. (2016) Multi-scale context aggregation by dilated convolutions. 4th International Conference on Learning Representations (ICLR) 2016, San Juan, Puerto Rico, May 2-4, 2016, Conference Track Proceedings
- Zhang J., Kurgan L. (2018) Review and comparative assessment of sequence-based predictors of protein-binding residues. Brief. Bioinform., 19, 821–837. [DOI] [PubMed] [Google Scholar]
- Zhang J., Kurgan L. (2019) SCRIBER: accurate and partner type-specific prediction of protein-binding residues from proteins sequences. Bioinformatics, 35, i343–i353. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.