

Abstract
Bone loss during skeletal unloading, whether due to neurotrauma resulting in paralysis or prolonged immobilization due to a variety of medical illnesses, accelerates bone loss. In this review the evidence that skeletal unloading leads to bone loss, at least in part, due to disrupted insulin like growth factor (IGF) signaling, resulting in reduced osteoblast proliferation and differentiation, will be examined. The mechanism underlying this disruption in IGF signaling appears to involve integrins, the expression of which is reduced during skeletal unloading. Integrins play an important, albeit not well defined, role in facilitating signaling not only by IGF but also by other growth factors. However, the interaction between selected integrins such as αυβ3 and β1 integrins and the IGF receptor are of especial importance with respect to the ability of bone to respond to mechanical load. Disruption of this interaction blocks IGF signaling and results in bone loss.
Keywords: Bone, IGF, Integrin, Mechanical load, Osteoblast, Osteoclast
Impact of mechanical load on bone
Mechanical loading has a profound influence on bone modeling and remodeling [1]. Bone formation occurs in those regions undergoing increased stress. For example when the ulna undergoes cyclic compression along its axis, because of its natural curvature, the medial surface is put under the greatest stress. This is the site of greatest bone formation following this procedure [2]. Similar results are found in other models of mechanical load. Both frequency and strain rate of the loading stimulus are important [3, 4]. The osteocyte is generally thought to be the major cell type in bone responding to mechanical load. These cells form a syncytium in bone, with cellular processes connecting each other and to the lining cells and osteoblasts on the surface of bone via gap junctions. The osteocyte lacunae may serve as stress risers to amplify the overall strain stimulus in the vicinity of the osteocyte enhancing the fluid shear stress thought to be key for mechanotransduction [5]. This initial signal involves calcium influx through stretch activated and L type channels [6, 7], ATP release which acts through its purinergic receptors to further increase intracellular calcium concentrations, prostaglandin E2 (PGE2) production [8, 9], and nitric oxide (NO) release [10]. These cellular responses are found in vitro in studies of both osteocytes and osteoblasts, so osteoblasts as well as osteocytes may respond directly to stress in vivo. Of particular relevance for this review is that IGF-I and integrin expression are also increased by mechanical load in osteocytes and osteoblasts in vivo and in vitro [11, 12], and the IGF-I receptor can be activated by fluid sheer stress in osteoblasts in vitro [13]. As will be emphasized in this review, the combined signaling from selected integrins and IGF-I are critical for the skeletal response to mechanical load.
Skeletal unloading, on the other hand, as occurs during prolonged bed rest and immobilization resulting from spinal injuries, amputations, fractures, and arthritic conditions, leads to bone loss. The most extreme example is the microgravity environment of space flight, which results in the near cessation of bone formation, as evidenced by the appearance of an extensive arrest line in the periosteum of cortical bone in both the tibia and humerus [14, 15]. Skeletal unloading leads to a decrease in osteoblast number and activity [16–23], likely due to a decrease in proliferation of osteoprogenitor cells [24]. The hindlimb elevation (tail suspension) model was developed to simulate skeletal unloading without the trauma associated with other manipulations such as nerve resection. This procedure is well tolerated by the animals with minimal evidence of stress as indicated by continued weight gain [25] and normal levels and circadian rhythms of corticosterone [26]. Skeletal unloading by hindlimb elevation results in decreased bone formation, mineralization, and maturation [25, 27–31], decreased osteoblast numbers [32], reduced serum and skeletal osteocalcin levels [33], lowered ash content of bone [25, 27, 28], and decreased bone strength [28, 34]. When bone marrow stromal cells (BMSC) from the bones of the unloaded limbs are cultured in vitro, there are fewer osteoprogenitors, and they proliferate more slowly [35], indicating that skeletal unloading causes a persistent change in cell function that can be assessed in vitro. In contrast to the unloaded bones of the hindlimbs, no significant change in bone mass or bone formation is observed in the humeri, mandible, and cervical vertebrae during hindlimb elevation [25]. The lack of effect of hindlimb elevation on these normally loaded bones indicates that local factors rather than systemic factors dominate the response of bone to skeletal unloading. To seek a local factor that could explain these changes, we first examined the role of insulin like growth factors (IGF).
IGF signaling pathways
IGF-I and its homologous family member IGF-II are both made by bone cells. IGF-I is the dominant IGF in postnatal murine bone, whereas IGF-II dominates in human bone. Although IGF-II has its own receptor (IGF-IIR or mannose-6 phosphate receptor), the IGF-I receptor (IGF-IR) is the major means by which these growth factors regulate cell growth and differentiation in that the IGF-IIR has no known signaling function. Since much of the data I will discuss have been generated in rats and mice, I will discuss primarily the results with IGF-I and its receptor. The IGF-I receptor is comprised of two alpha and two beta subunits (Fig. 1) (review in [36]). IGF-I binding to the receptor results in activation of its intrinsic tyrosine kinase. The kinase domain resides within amino acids 956–1256; activation entails the sequential tyrosine phosphorylation of residues Y1135, Y1131, and Y1136 which alters the structure of the β chain enabling its kinase activity to be expressed [37]. Mutation of these tyrosines to phenylalanine impairs the ability of IGF-IR to complex with other signaling molecules, including specific integrins [38]. These and subsequent phosphorylations create multiple docking sites for a variety of endogenous substrates including members of the insulin receptor substrate (IRS) family which associate with IGF-IR via phosphotyrosine binding (PTB) and src homology 2 (SH2) domains, growth receptor binding protein-2 (Grb2), which binds to specific motifs in the IGF-I receptor as well as in IRS, and the p85 subunit of phosphatidyl inositol 3 kinase (PI3K), which binds to other specific motifs within IRS. Src homology collagen (Shc), when tyrosine phosphorylated in response to IGF-I, binds to the SH2 domain of Grb2, which in turn forms a complex with son of sevenless (Sos), a guanine nucleotide exchange factor (GEF) that mediates GDP/GTP exchange in ras and thus activates it. Ras then activates Raf (MAPKKK), which phosphorylates and activates MEK (MAPKK), which in turn phosphorylates and activates extracellular signal-regulated kinase (ERK1/2 or MAPK). These are serine/threonine phosphorylations. Activated ERK enters the nucleus to phosphorylate and so activate transcription factors (e.g., elk-1 and c-jun) leading to increased cyclin D1 and reduced p21cip and p27kip expression. The increased levels of cyclin D1 and reduced levels of the cell cycle inhibitors p21cip and p27kip stimulate cell cycle progression from G1 to S, thus completing the pathway by which IGF-I and other growth factors promote proliferation. Activation of PI3K sets up a different pathway. PI3K phosphorylates phosphatidyl inositol bisphosphate (PIP2) to phosphatidyl inositol trisphosphate (PIP3) in the membrane, recruiting protein kinase B (PKB or Akt) to the membrane where it is phosphorylated and activated by phosphoinositide dependent kinase 1/2 (PDK1/2). The activated Akt then phosphorylates and inactivates Bad, a proapoptotic member of the bcl-2 family. This pathway blocks apoptosis. However, PI3K and Akt can enter the nucleus and by phosphorylating critical transcription factors also lead to increased cyclin D1 levels.
Fig. 1.
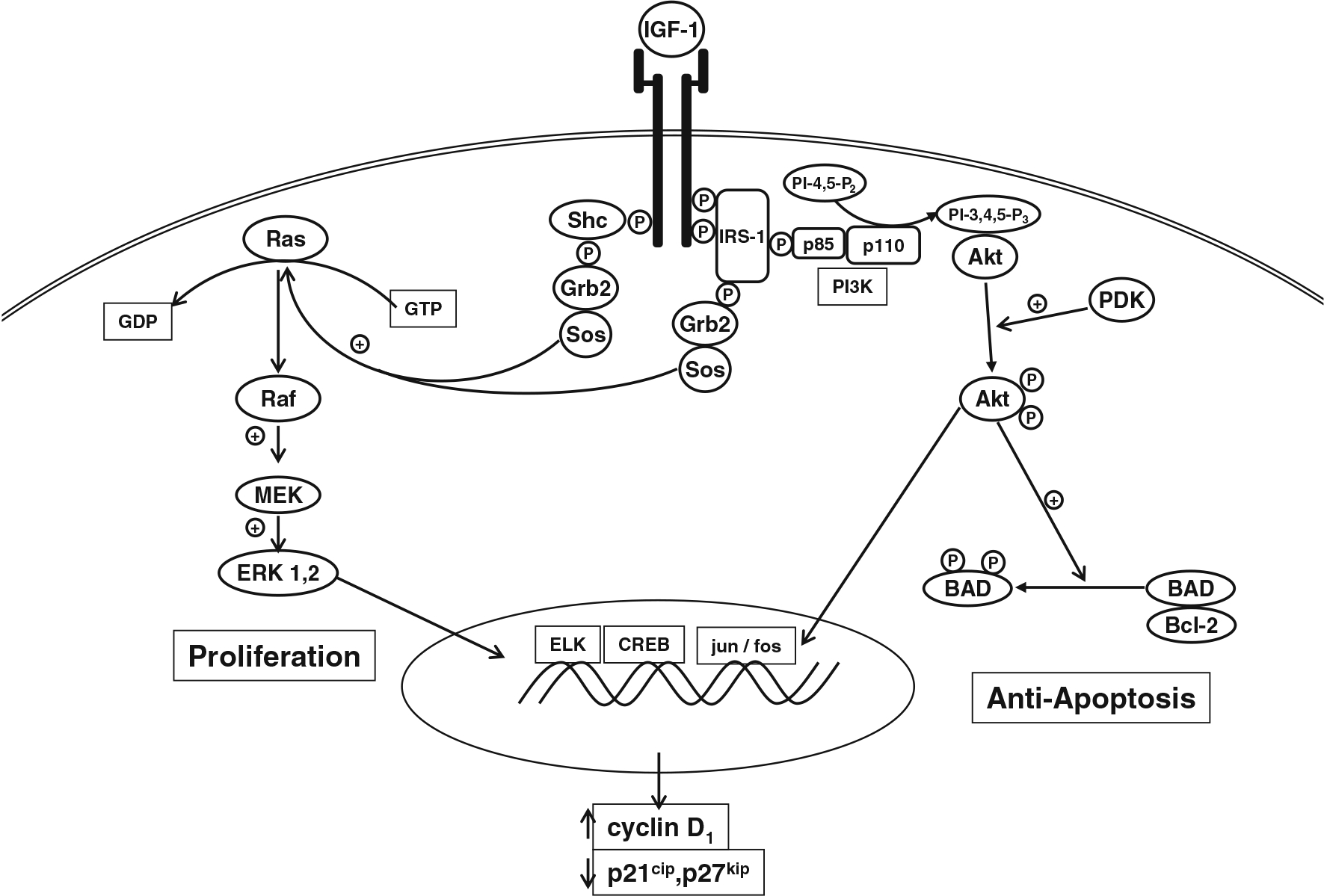
IGF signaling pathway. IGF I or II binds to the IGF-I receptor, a heterotetramer comprised of two alpha and two beta subunits. Upon binding of IGF, the receptor undergoes autophosphorylation of critical tyrosine residues, and two major pathways are activated. The first pathway leads to activation of Ras and the MAPK pathway eventuating in the activation of ERK1/2, which can enter the nucleus to induce genes important for proliferation. The second pathway leads to the activation of PI3K resulting in activation of Akt, which exerts anti-apoptotic actions by phosphorylating and so inactivating Bad, a proapoptotic regulator of Bcl-2
IGF-I signaling in bone
We have focused on IGF-I because it is abundantly produced by murine bone [39], is a well studied regulator of osteoblast proliferation and differentiation [40], and when deleted by genetic engineering results in animals with retarded bone growth [41, 42] and bone formation [43]. These abnormalities are readily reversed with exogenous IGF-I [43]. Overexpression of IGF-I in bone under the osteocalcin [44] or collagen 1 [45] promoter increases bone formation. Deletion of the IGF-I receptor (IGF-IR) from mature osteoblasts results in mice with poorly mineralized bone [46]. Although the number of colony forming units in BMSC cultures from such mice is normal, the colonies fail to mineralize [47], indicating an important role for IGF-I signaling in osteoblast differentiation as well as proliferation. Similarly, IGF-1 is required for normal osteoclastogenesis in that the IGF-I null mouse has increased trabecular bone, fewer and smaller osteoclasts, and defective bone resorption [48]. On the other hand overexpression of IGF-I in osteoblasts increases osteoclast number in vivo [45], and addition of IGF-I to co-cultures of osteoblasts and osteoclast precursors promotes osteoclast formation in vitro [48]. These data indicate that IGF-I signaling in both osteoblasts and osteoclasts is required for all aspects of bone remodeling.
IGF signaling in mechanotransduction
As previously mentioned, IGF-I production is increased by mechanical loading, and fluid flow is synergistic with IGF-I in the activation of the IGF-IR [13]. On the other hand, skeletal unloading results in resistance to IGF-I with respect to its anabolic actions [39, 49–52]. When IGF-I is infused into unloaded growing rats, their unloaded bones (tibiae) do not increase in size as assessed by changes in fat-free weight as do the bones of normally loaded animals [39]. Furthermore, IGF-I fails to stimulate bone formation (BFR) in the unloaded bones (tibiae), although stimulation of BFR in the humerus is equivalent to that seen in the normally loaded rats [51, 52]. Proliferation of osteoblasts in vivo is depressed whereas apoptosis is increased in the unloaded bones, and neither respond to IGF-I infusion, unlike the situation in normally loaded bones [52]. BMSC from unloaded bones have normal levels of the IGF-I receptor and normal binding of IGF-I to this receptor, but fail to respond to IGF-I with receptor activation as assessed by phosphorylation [52, 53]. The downstream pathways are likewise impacted in that ras is not activated, and ERK1/2 are not phosphorylated in response to IGF-I in BMSC from unloaded bones, in contrast to that from normally loaded animals, and IGF-I stimulated phosphorylation of Akt is reduced [52]. These results indicate that the resistance to IGF-I in unloaded bone is primarily due to a failure of IGF-I to activate its own receptor. This is not a problem with binding of IGF-I to the receptor or receptor levels, which are not altered by skeletal unloading [52]. When the animals are reloaded after a period of unloading, they show an accentuated response to IGF-I resulting in bone formation rates above the normally loaded controls [49]. The resistance to IGF-I caused by skeletal unloading persists when the bone cells (BMSC) are studied in vitro. BMSC from the tibiae of hindlimb-elevated rats form fewer colony forming units in vitro and fail to respond to IGF-I administration with an increase in proliferation [52, 53]. Thus, mechanical loading (or unloading) of bone profoundly alters IGF signaling. The question is why. Based on our own work and that of others, we hypothesize that the key to understanding the impact of mechanical load on the skeletal response to IGF-I lies in integrin activation.
Integrin signaling
Integrins are comprised of an alpha and beta subunit [54–56]. There are at least 18 α and 8 β genes so far identified in the human genome, several of which produce multiple transcripts by alternative splicing. These combine to form 24 different functional integrins in mammals. Each subunit can have multiple partners (Fig. 2). Seven of the human α subunits contain an I (interactive or inserted) domain (α1,α2,α10,α11,αL,αM, αX,αD, and αE) between the β propeller repeats 2 and 3 in the extracellular portion of the molecule which is involved in ligand binding and intercellular adhesion. Non-I domain containing α subunits include α3, α4, α5, α6, α7, α8, α9, αυ, and αIIb. All human integrin β subunits contain an I-like domain along with 4 EGF-like repeats in their extracellular domains. The RGD-binding site is located at the interface between the β–propeller domain of the α subunit and the I-like domain of the β subunit (Fig. 3). Different integrins bind preferentially to different matrix proteins, although both the integrins and the matrix proteins are promiscuous. Thus vitronectin binds αυβ5 or 3, osteopontin binds αυβ3, collagen binds α1 or 2β1, and fibronectin binds α3,5, or 8β1 and αυβ3, although the strength of binding varies. Limited crystallographic data indicate that ligand binding induces major conformational changes in the integrin subunits. In general the cytoplasmic tails of the integrin subunits are short (less than 75 amino acids, β4 is an exception). The α subunits show little homology in their cytoplasmic tails except for a GFFKR motif near the transmembrane domain important for association with the β subunit. The tails of the β subunits are more homologous. Most β subunit tails have NPxY/F motifs as part of PTB (phosphotyrosine-binding) domains. The cytoplasmic tails of the integrin subunits recruit a number of proteins, the profile of which varies from integrin to integrin. β1and β3 bind talin, β1 binds α actinin, β1,2,7 bind filamin, α4 binds paxillin, and a number of integrins bind focal adhesion kinase (FAK). Such proteins link the integrins to the cytoskeleton, which is important for many of the functions of the integrins. In addition a select number of integrins such as αυβ3 bind to caveolin-1 in the membrane. Some integrins are found in only a limited number of cell types or tissues such as αIIbβ3 in platelets or α6β4 in keratinocytes, but other integrins such as αυβ3 are widely distributed. Thus it is not surprising that gene deletions of the different integrins produce many different phenotypes, a number of which are embryonic lethal [57].
Fig. 2.
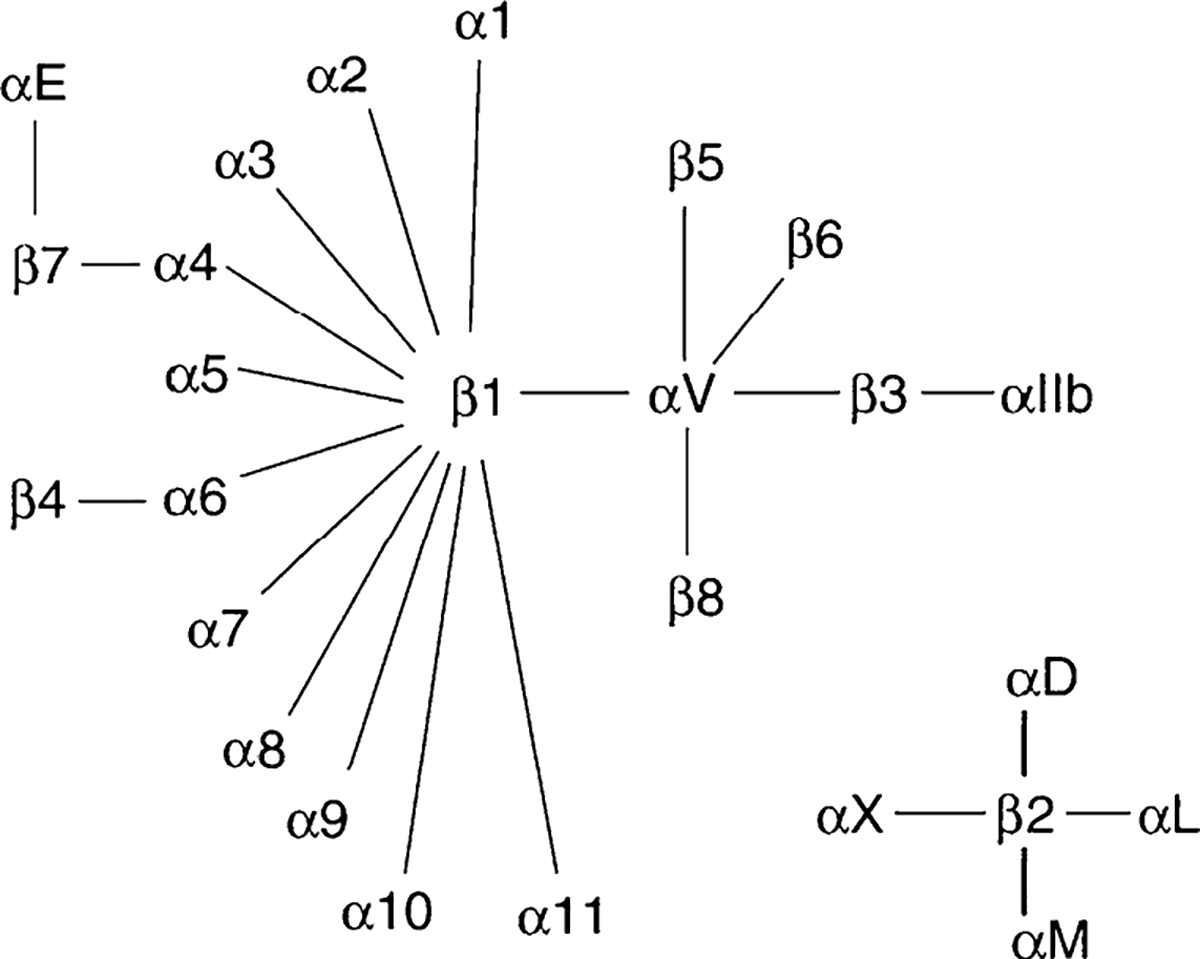
The members of the human integrin superfamily and how they combine to form heterodimeric integrins. At least 18 α subunits and 8 β subunits have been identified in humans, which combine to generate 24 different integrins. Integrin subunits that can bind to each other to form a heterodimer are connected by solid lines
Fig. 3.
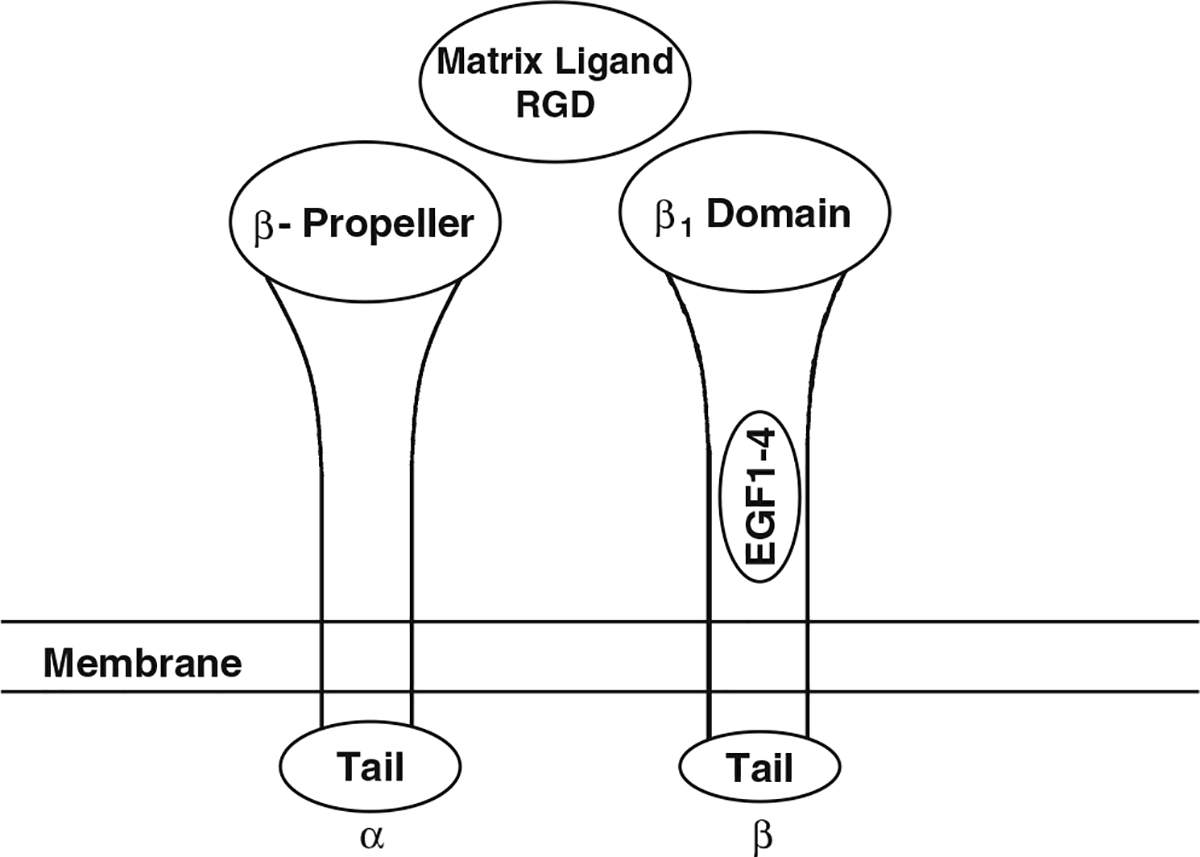
A model of an integrin lacking the I domain in the α subunit (the typical integrin involved with IGF signaling in bone). The β-propeller domain of the α subunit and the β1 domain of the β subunit form the binding domain for the extracellular ligand, in this case a matrix RGD containing protein such as osteopontin, vitronectin, or fibronectin. Binding of the ligand causes a conformational change in the integrin subunits resulting in separation of the intracellular portions of the tails, enabling binding to a number of proteins to those tails such as FAK and talin that mediate integrin signaling
Binding of integrins to their extracellular matrix proteins leads to clustering of the integrins with reorganization of the actin cytoskeleton and translocation of a number of proteins including signaling proteins to the focal adhesion plaque [58]. Focal adhesion kinase (FAK) or its homolog phosphotyrosine kinase 2 (Pyk2) plays an important role, acting as both a scaffolding protein to bring a number of proteins to the focal adhesion plaque including growth factor receptors, PI3K, Shc, Grb2 and src family kinases [59]. The src kinases phosphorylate tyrosines on FAK to provide docking sites for these proteins. Grb2 and Shc can be activated by integrins through FAK, and by binding Sos can then activate the ras/MAPK pathway. Activation of this pathway can also take place by a FAK independent mechanism in which the activated integrin binds to caveolin-1, which serves as the scaffolding protein to bring the src family kinases and growth factor receptors to the complex. As for the src family kinases recruited to FAK, the src kinases recruited to the caveolin-1 complex may activate the ras/MAPK pathway by recruiting and activating Shc/Grb2/Sos. This pathway appears to be limited to certain integrins (e.g., α1β1, α5β1, αυβ3), and these integrins appear to be the ones most supportive of growth factor stimulation of proliferation [60, 61].
Integrins in bone
Osteoblasts and BMSC express a number of integrins, including αυ, 1, 2, 3, 5, 6, 8 and β1, 3, 5 [62–66]. BMP-2 increases a number of these integrins, increasing the adhesion of osteoblasts to vitronectin and osteopontin [67]. Blocking antibodies to αυβ prevent BMP-2 from stimulating osteoblasts differentiation [67]. Immunoprecipitation methods and clustering analysis following adhesion to different substrates indicate that favored αβ combinations include αυ, 1, 2, 3, 5β1, αυβ1, 3, 5. Fibronectin, a ligand for several integrins including α4β1, α5β1, α8β1, αυβ1, and αυβ3 promotes bone cell adhesion and stimulation of osteoblast proliferation [68–70]. Addition of antibodies to fibronectin or soluble fibronectin fragments to the culture of differentiated osteoblasts results in apoptosis [70]. Within 60min of plating the osteoblasts on fibronectin or collagen, phosphorylation of FAK and ERK is observed as is a rise in the mRNA levels of fos and c-jun [71]. The Damsky laboratory [72] created a transgenic model overexpressing a dominant negative form of β1 under the osteocalcin promoter such that it was expressed only in mature osteoblasts. They found decreased bone mass and increased cortical porosity in the long bones and thinner flat bones of the skull, decreased bone formation rates in cortical bone, and failure of terminal differentiation of osteoblasts in vitro suggesting an important role for β1 in osteoblast function. The β2 null mouse also shows a reduction in trabecular bone with normal osteoclast numbers [73]. Bone marrow stromal cells from these mice show reduced osteogenic potential in vivo, and decreased mineralization in vitro. Overexpression of this integrin leads to increased bone formation [73].
The major integrin in the mature osteoclast is αυβ3, although osteoclast precursors express αυβ5 [74]. αυβ3 is critical for osteoclast function, but not for their formation. In particular, αυβ3 is required for the formation of the sealing ring necessary for the ability of osteoclasts to resorb bone. Mice null for β3 grow normally, but have increased bone mass and poorly functioning albeit abundant osteoclasts. In contrast, mice null for β5 show increased osteoclastogenesis especially after ovariectomy [75], suggesting a reciprocal interaction with β3. In the osteoclast when αυβ3 is activated, c-src binds to the β-subunit tail, recruits and activates another non-receptor tyrosine kinase syk, which mediates the activation of Vav3, a guanine nucleotide exchange factor for RhoA required for the formation of the sealing ring [76, 77]. The role of αυβ3 in regulating osteoclast function has been exploited clinically. An αυβ3 antagonist, L-000845704, was found to decrease bone turnover markers (resorption markers faster than formation markers) and increase lumbar spine BMD in post-menopausal osteoporotic women [78]. Whether such a drug would be useful to prevent or reverse bone loss accompanying skeletal unloading remains to be tested.
Integrins as sensors for mechanical load in bone
Integrins form an important link between the extracellular matrix and the cytoskeleton, and thus are in a position to transduce mechanical signals imposed on bone to responses by the bone cells. During skeletal unloading by hindlimb elevation we found a reduction in integrin expression [52]. This result has recently been confirmed by others [79]. Reloading these animals restores integrin expression (Long et al. unpublished). Fluid shear stress or mechanical deformation of bone cells increases and/or activates selected integrins [80, 81], whereas culturing mesenchymal stem cells (presumed osteoblast precursors) in a rotary cell culture system decreases downstream integrin signaling (decreased FAK and pyk2 phosphorylation and RhoA activation) [82, 83]. As mentioned previously, osteocytes are thought to be central to the ability of bone to sense mechanical load. These cells show attachments between their canalicular processes and the lining of the canalicular wall, attachments which contain αυβ3 [84], 3 is involved in both the osteoblastic and osteoclastic response to mechanical load. As mentioned above mice overexpressing a dominant negative β1 integrin subunit have been developed [72]. Although these mice showed a reduction in trabecular bone and loss of bone strength, their response to skeletal unloading (hindlimb elevation) did not differ significantly from wildtype controls [89]. However, they were not tested for their response to mechanical loading.
Role of integrins in regulating IGF-I responsiveness in bone
As previously noted, activation of integrins by their extracellular ligands leads to clustering into focal adhesion plaques. Receptors for growth factors, including IGF-IR, may be included in these clusters [90]. Although this section is focused on IGF-IR/integrin interactions in osteoblasts, the c-fms (m-CSF receptor) and/or IGF-IR may have similar interactions with integrins in the osteoclast. The importance of IGF-IR/integrin interactions with respect to IGF signaling is supported by observations that the matrix on which bone cells are plated makes a difference with respect to the strength of IGF-I signaling. Vitronectin and fibronectin increase the binding of the β3 integrin subunit to IGF-IR and enhance the ability of IGF-I to activate its receptor.
The role of integrins in regulating IGF-I responsiveness has been demonstrated in a number of tissues. Perhaps the most extensive examination emanates from the Clemmons laboratory which has studied this phenomenon in aortic smooth muscle cells [91–93]. The laboratory found that echistatin (a disintegrin) or blocking antibodies to the integrin αυβ3 blocked IGF-I stimulated proliferation, IGF-I receptor autophosphorylation, IRS-1 phosphorylation, and binding of the p85 subunit of PI3K to IRS-1. Their proposed mechanism is that integrin activation recruits the src homology 2 domain containing protein tyrosine phosphatase-2 (SHP-2) to the β3 integrin subunit. IGF-I in turn via the IGF-IR phosphorylates and so activates the transmembrane protein src homology domain containing protein tyrosine phosphatase substrate-1 (SHPS-1), which recruits SHP-2 to SHPS-1 for an as yet undefined role in further signaling. When αυβ3 activation is blocked, SHP-2 instead is recruited to the IGF-IR, where it dephosphorylates and so terminates the activation of the IGF-IR. Our results in bone cells indicate a different mechanism, however. We [52] have shown that although echistatin blocks IGF-I activation of the IGF-IR, neither skeletal unloading nor echistatin alters the recruitment of SHP-2 to the IGF-IR nor the timing of IGF-IR phosphorylation and dephosphorylation. Instead, the IGF-IR is just not phosphorylated in response to IGF-I in BMSC from unloaded bone or normal cells treated with echistatin. IGF-I responsiveness in bone cells may require direct binding of αυβ3 to the IGF-IR [94]. Indeed, IGF-I stimulates the phosphorylation of the β3 integrin subunit, and so activates it [94].
The matrix protein vitronectin is a potent stimulator of β3 integrin subunit phosphorylation, and potentiates the ability of IGF-I to do likewise [93, 95]. Mutation of the tyrosines involved (aa 773, 785) in the β3 integrin subunit blocks IGF-I signaling [93, 95]. Knocking down either the β1 or β3 integrin subunit in osteoblasts blocks IGF-I signaling, and the combined knockdown is especially inhibitory. Activation of the β3 integrin subunit may be the mechanism by which IGF-I and mechanical loading also activate (by phosphorylation) FAK and Pyk2 [96]. Skeletal unloading results in decreased expression of αυβ3 [52] as well as several other integrins including the β1 subunit, and decreased activation of FAK.
Our hypothesis is that mechanical load stimulates formation of an IGF-IR/integrin complex in osteoblasts. This process is maximized by activating the integrin with its matrix substrate (e.g., vitronectin) and the IGF-IR with IGF-I. We further hypothesize that the IGF-IR/integrin complex recruits non-receptor tyrosine kinases to the complex which are required for IGF-IR phosphorylation/activation in response to IGF-I. Skeletal unloading disrupts this mechanism by reducing integrin levels. Reloading restores integrin expression and enhances the formation of the integrin/IGF-IR complex accentuating the IGF-I responsiveness. Recent results from Kapur et al. [13] as well as our own laboratory support this hypothesis by demonstrating that fluid shear stress on bone cells stimulates IGF-IR phosphorylation independent of IGF-I, that this response is potentiated by fibronectin and vitronectin, and that the response is blocked by echistatin and knockdown of β1 and β3 integrin subunits. This model is shown in Fig. 4. This model need not exclude contributions from other mechanisms such as wnt signaling.
Fig. 4.
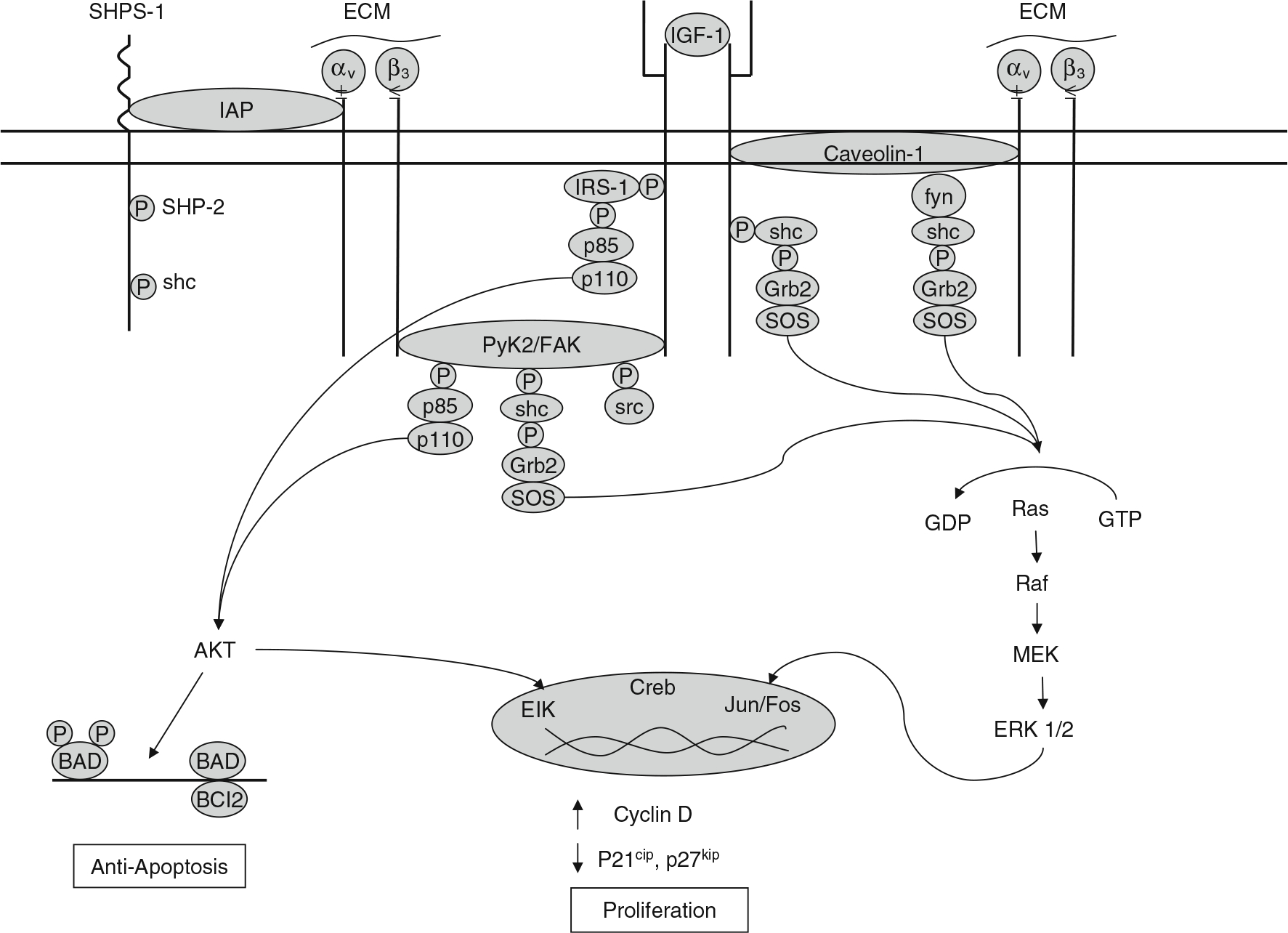
Model for IGF/integrin signaling interactions in regulating the skeletal response to load. The IGF-IR forms a complex with αυβ3 integrin that is required for IGF-I activation of the IGF-IR. This complex may be formed via the scaffolding function of FAK/Pyk2 or that of caveolin-1. SHPS-1 may play a role by regulating access of the phosphatase SHP-2 to this complex. Mechanical load increases whereas unloading decreases formation of this complex and so regulates IGF-I responsiveness. Formation of the integrin/IGF-IR complex brings to the IGF-IR non-receptor kinases such as FAK and src family kinases which may activate the IGF-IR independently and/or synergistically with IGF-I. The source of IGF-I may originate from osteocytes and osteoblasts. Production of IGF-I as well as expression and activation of integrins in these cells are stimulated by mechanical load
Clinical implications
Disuse osteoporosis is a major health problem contributing to substantial morbidity through increased risk of fractures especially in the elderly. The bone lost during extended bed rest or immobilization is generally not regained in the elderly. The mechanisms underlying the imbalance between bone formation and resorption during skeletal unloading are not well understood. We are proposing that the skeletal response to mechanical load requires complex formation between selected integrins and IGF-IR. Disruption of this complex during skeletal unloading results in a relative reduction in bone formation leading to osteoporosis, but this can be restored by reloading. If this hypothesis turns out to be valid, we anticipate that drugs that selectively regulate the integrin signaling pathway will prove efficacious in modulating the skeletal response to IGF-I and in so doing prevent or reverse the loss of bone during disuse.
Acknowledgements
The author has drawn from the work of his current and former postdoctoral fellows Drs. Paul Kostenuik, Takashi Sakata, Shigeki Nishida, Yongmei Wang, and Roger Long as well as his long standing collaboration with Dr. Bernard Halloran. The work has been and is supported by a Veterans Affairs Research Enhancement Award Program, a grant from the National Aeronautics and Space Administration (NNA04CC67G), a grant from the National Institutes of Health (RO1 DK54793) to the author, and a National Space Biomedical Research Institute Postdoctoral Fellowship to Dr. Long.
Abbreviations
- BMSC
Bone marrow stromal cells
- FAK
Focal adhesion kinase
- grb2
Growth receptor binding protein-2
- GEF
Guanine nucleotide exchange factor
- IGF-R
Insulin like growth factor (IGF) and its receptor
- IRS
Insulin receptor substrate
- MAPK
Mitogen activated protein kinase
- NO
Nitric oxide
- PI3K
Phosphatidyl inositol 3 kinase
- PIP2 and PIP3
Phosphatidyl inositol bis- and tris-phosphate
- PDK
Phosphoinositide dependent kinase
- PTB
Phosphotyrosine binding protein
- Pyk
Phosphotyrosine kinase
- PGE2
Prostaglandin E2
- PKB/Akt
Protein kinase B
- SOS
Son of sevenless
- SH2
src homology 2
- SHPS
SH2 domain containing protein tyrosine phosphatase (SHP) and its substrate
Footnotes
Conflict of interest statement No conflict of interest.
The skeletal response to mechanical load is critical for maintenance of skeletal integrity. This review will assess the interacting roles that insulin like growth factor I (IGF-I) signaling and selected integrins play in this response. Skeletal unloading results in decreased integrin expression, resistance to the anabolic actions of IGF-I, and bone loss.
References
- 1.Robling AG, Castillo AB, Turner CH (2006) Biomechanical and molecular regulation of bone remodeling. Ann Rev Biomed Eng 8:455–498 [DOI] [PubMed] [Google Scholar]
- 2.Robling AG, Hinant FM, Burr DB, Turner CH (2002) Improved bone structure and strength after long-term mechanical loading is greatest if loading is separated into short bouts. J Bone Miner Res 17:1545–1554 [DOI] [PubMed] [Google Scholar]
- 3.Lanyon LE, Rubin CT (1984) Static vs dynamic loads as an influence on bone remodelling. J Biomech 17:897–905 [DOI] [PubMed] [Google Scholar]
- 4.Turner CH, Owan I, Takano Y (1995) Mechanotransduction in bone: role of strain rate. Am J Physiol 269:E438–E442 [DOI] [PubMed] [Google Scholar]
- 5.Weinbaum S, Cowin SC, Zeng Y (1994) A model for the excitation of osteocytes by mechanical loading-induced bone fluid shear stresses. J Biomech 27:339–360 [DOI] [PubMed] [Google Scholar]
- 6.Li J, Duncan RL, Burr DB, Gattone VH, Turner CH (2003) Parathyroid hormone enhances mechanically induced bone formation, possibly involving L-type voltage-sensitive calcium channels. Endocrinology 144:1226–1233 [DOI] [PubMed] [Google Scholar]
- 7.Rawlinson SC, Pitsillides AA, Lanyon LE (1996) Involvement of different ion channels in osteoblasts’ and osteocytes’ early responses to mechanical strain. Bone 19:609–614 [DOI] [PubMed] [Google Scholar]
- 8.Genetos DC, Geist DJ, Liu D, Donahue HJ, Duncan RL (2005) Fluid shear-induced ATP secretion mediates prostaglandin release in MC3T3-E1 osteoblasts. J Bone Miner Res 20:41–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jorgensen NR, Geist ST, Civitelli R, Steinberg TH (1997) ATP- and gap junction-dependent intercellular calcium signaling in osteoblastic cells. J Cell Biol 139:497–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bakker AD, Soejima K, Klein-Nulend J, Burger EH (2001) The production of nitric oxide and prostaglandin E(2) by primary bone cells is shear stress dependent. J Biomech 34:671–677 [DOI] [PubMed] [Google Scholar]
- 11.Lean JM, Jagger CJ, Chambers TJ, Chow JW (1995) Increased insulin-like growth factor I mRNA expression in rat osteocytes in response to mechanical stimulation. Am J Physiol 268:E318–E327 [DOI] [PubMed] [Google Scholar]
- 12.Reijnders CM, Bravenboer N, Tromp AM, Blankenstein MA, Lips P (2007) Effect of mechanical loading on insulin-like growth factor-I gene expression in rat tibia. J Endocrinol 192:131–140 [DOI] [PubMed] [Google Scholar]
- 13.Kapur S, Mohan S, Baylink DJ, Lau KH (2005) Fluid shear stress synergizes with insulin-like growth factor-I (IGF-I) on osteoblast proliferation through integrin-dependent activation of IGF-I mitogenic signaling pathway. J Biol Chem 280:20163–20170 [DOI] [PubMed] [Google Scholar]
- 14.Morey ER, Baylink DJ (1978) Inhibition of bone formation during space flight. Science 201:1138–1141 [DOI] [PubMed] [Google Scholar]
- 15.Wronski TJ, Morey ER (1983) Effect of spaceflight on periosteal bone formation in rats. Am J Physiol 244:R305–R309 [DOI] [PubMed] [Google Scholar]
- 16.Vico L, Novikov VE, Very JM, Alexandre C (1991) Bone histomorphometric comparison of rat tibial metaphysis after 7-day tail suspension vs. 7-day spaceflight. Aviat Space Environ Med 62:26–31 [PubMed] [Google Scholar]
- 17.Oganov VS, Rakhmanov AS, Novikov VE, Zatsepin ST, Rodionova SS, Cann C (1991) The state of human bone tissue during space flight. Acta Astronaut 23:129–133 [DOI] [PubMed] [Google Scholar]
- 18.Vico L, Alexandre C (1992) Microgravity and bone adaptation at the tissue level. J Bone Miner Res 7(Suppl 2):S445–S447 [DOI] [PubMed] [Google Scholar]
- 19.Zerath E, Holy X, Malouvier A, Caissard JC, Nogues C (1991) Rat and monkey bone study in the Biocosmos 2044 space experiment. Physiologist 34:S194–S195 [PubMed] [Google Scholar]
- 20.Jee WS, Wronski TJ, Morey ER, Kimmel DB (1983) Effects of spaceflight on trabecular bone in rats. Am J Physiol 244:R310–R314 [DOI] [PubMed] [Google Scholar]
- 21.Yagodovsky VS, Triftanidi LA, Gorokhova GP (1976) Spaceflight effects on skeletal bones of rats (light and electron microscopic examination). Aviat Space Environ Med 47:734–738 [PubMed] [Google Scholar]
- 22.Vico L, Chappard D, Palle S, Bakulin AV, Novikov VE,Alexandre C (1988) Trabecular bone remodeling after seven days of weightlessness exposure (BIOCOSMOS 1667). Am J Physiol 255:R243–R247 [DOI] [PubMed] [Google Scholar]
- 23.Turner RT, Evans GL, Wakley GK (1995) Spaceflight results in depressed cancellous bone formation in rat humeri. Aviat Space Environ Med 66:770–774 [PubMed] [Google Scholar]
- 24.Garetto LP, Gonsalves MR, Morey ER, Durnova G, Roberts WE(1990) Preosteoblast production 55 hours after a 12.5-day spaceflight on Cosmos 1887. Faseb J 4:24–28 [DOI] [PubMed] [Google Scholar]
- 25.Globus RK, Bikle DD, Morey-Holton E (1984) Effects of simulated weightlessness on bone mineral metabolism. Endocrinology 114:2264–2270 [DOI] [PubMed] [Google Scholar]
- 26.Halloran BP, Bikle DD, Cone CM, Morey-Holton E (1988) Glucocorticoids and inhibition of bone formation induced by skeletal unloading. Am J Physiol 255:E875–E879 [DOI] [PubMed] [Google Scholar]
- 27.Globus RK, Bikle DD, Morey-Holton E (1986) The temporal response of bone to unloading. Endocrinology 118:733–742 [DOI] [PubMed] [Google Scholar]
- 28.Abram AC, Keller TS, Spengler DM (1988) The effects of simulated weightlessness on bone biomechanical and biochemical properties in the maturing rat. J Biomech 21:755–767 [DOI] [PubMed] [Google Scholar]
- 29.Bikle DD, Halloran BP, Cone CM, Globus RK, Morey-Holton E(1987) The effects of simulated weightlessness on bone maturation. Endocrinology 120:678–684 [DOI] [PubMed] [Google Scholar]
- 30.Kidder LS, Klein GL, Stuart CA, Lee TC, Gundberg CM, Alcock N, Cooper CW, Simmons DJ (1990) Skeletal effects of sodium fluoride during hypokinesia. Bone Miner 11:305–318 [DOI] [PubMed] [Google Scholar]
- 31.LeBlanc A, Marsh C, Evans H, Johnson P, Schneider V, Jhingran S (1985) Bone and muscle atrophy with suspension of the rat. J Appl Physiol 58:1669–1675 [DOI] [PubMed] [Google Scholar]
- 32.Halloran BP, Bikle DD, Wronski TJ, Globus RK, Levens MJ, Morey-Holton E (1986) The role of 1,25-dihydroxyvitamin D in the inhibition of bone formation induced by skeletal unloading. Endocrinology 118:948–954 [DOI] [PubMed] [Google Scholar]
- 33.Patterson-Buckendahl P, Globus RK, Bikle DD, Cann CE, Morey-Holton E (1989) Effects of simulated weightlessness on rat osteocalcin and bone calcium. Am J Physiol 257:R1103–R1109 [DOI] [PubMed] [Google Scholar]
- 34.Shaw SR, Zernicke RF, Vailas AC, DeLuna D, Thomason DB, Baldwin KM (1987) Mechanical, morphological and biochemical adaptations of bone and muscle to hindlimb suspension and exercise. J Biomech 20:225–234 [DOI] [PubMed] [Google Scholar]
- 35.Machwate M, Zerath E, Holy X, Hott M, Modrowski D, Malouvier A, Marie PJ (1993) Skeletal unloading in rat decreases proliferation of rat bone and marrow-derived osteoblastic cells. Am J Physiol 264:E790–E799 [DOI] [PubMed] [Google Scholar]
- 36.Le Roith D, Bondy C, Yakar S, Liu JL, Butler A (2001) The somatomedin hypothesis: 2001. Endocr Rev 22:53–74 [DOI] [PubMed] [Google Scholar]
- 37.Favelyukis S, Till JH, Hubbard SR, Miller WT (2001) Structure and autoregulation of the insulin-like growth factor 1 receptor kinase. Nat Struct Biol 8:1058–1063 [DOI] [PubMed] [Google Scholar]
- 38.Kiely PA, Leahy M, O’ Gorman D, O’ Connor R (2005) RACK1-mediated integration of adhesion and insulin-like growth factor I (IGF-I) signaling and cell migration are defective in cells expressing an IGF-I receptor mutated at tyrosines 1250 and 1251. J Biol Chem 280:7624–7633 [DOI] [PubMed] [Google Scholar]
- 39.Bikle DD, Harris J, Halloran BP, Morey-Holton E (1994) Altered skeletal pattern of gene expression in response to spaceflight and hindlimb elevation. Am J Physiol 267:E822–E827 [DOI] [PubMed] [Google Scholar]
- 40.Canalis E (1985) Effect of growth factors on bone cell replication and differentiation. Clin Orthop 193:246–263 [PubMed] [Google Scholar]
- 41.Baker J, Liu JP, Robertson EJ, Efstratiadis A (1993) Role of insulin-like growth factors in embryonic and postnatal growth. Cell 75:73–82 [PubMed] [Google Scholar]
- 42.Powell-Braxton L, Hollingshead P, Warburton C, Dowd M, Pitts-Meek S, Dalton D, Gillett N, Stewart TA (1993) IGF-I is required for normal embryonic growth in mice. Genes Dev 7:2609–2617 [DOI] [PubMed] [Google Scholar]
- 43.Bikle D, Majumdar S, Laib A, Powell-Braxton L, Rosen C, Beamer W, Nauman E, Leary C, Halloran B (2001) The skeletal structure of insulin-like growth factor I-deficient mice. J Bone Miner Res 16:2320–2329 [DOI] [PubMed] [Google Scholar]
- 44.Zhao G, Monier-Faugere MC, Langub MC, Geng Z, Nakayama T, Pike JW, Chernausek SD, Rosen CJ, Donahue LR, Malluche HH, Fagin JA, Clemens TL (2000) Targeted overexpression of insulin-like growth factor I to osteoblasts of transgenic mice: increased trabecular bone volume without increased osteoblast proliferation. Endocrinology 141:2674–2682 [DOI] [PubMed] [Google Scholar]
- 45.Jiang J, Lichtler AC, Gronowicz GA, Adams DJ, Clark SH, Rosen CJ, Kream BE (2006) Transgenic mice with osteoblast-targeted insulin-like growth factor-I show increased bone remodeling. Bone 39:494–504 [DOI] [PubMed] [Google Scholar]
- 46.Zhang M, Xuan S, Bouxsein ML, von Stechow D, Akeno N, Faugere MC, Malluche H, Zhao G, Rosen CJ, Efstratiadis A, Clemens TL (2002) Osteoblast-specific knockout of the insulin-like growth factor (IGF) receptor gene reveals an essential role of IGF signaling in bone matrix mineralization. J Biol Chem 277:44005–44012 [DOI] [PubMed] [Google Scholar]
- 47.Wang Y, Nishida S, Boudignon BM, Burghardt A, Elalieh HZ, Hamilton MM, Majumdar S, Halloran BP, Clemens TL, Bikle DD (2007) IGF-I receptor is required for the anabolic actions of parathyroid hormone on bone. J Bone Miner Res 22:1329–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Y, Nishida S, Elalieh HZ, Long RK, Halloran BP, Bikle DD(2006) Role of IGF-I signaling in regulating osteoclastogenesis. J Bone Miner Res 21:1350–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boudignon BM, Bikle DD, Kurimoto P, Elalieh H, Nishida S, Wang Y, Burghardt A, Majumdar S, Orwoll BE, Rosen C, Halloran BP (2007) Insulin-like Growth Factor-I Stimulates Recovery of Bone Lost after a Period of Skeletal Unloading. J Appl Physiol 103:125–131 [DOI] [PubMed] [Google Scholar]
- 50.Kostenuik PJ, Halloran BP, Morey-Holton ER, Bikle DD (1997) Skeletal unloading inhibits the in vitro proliferation and differentiation of rat osteoprogenitor cells. Am J Physiol 273:E1133–E1139 [DOI] [PubMed] [Google Scholar]
- 51.Sakata T, Halloran BP, Elalieh HZ, Munson SJ, Rudner L, VentonL, Ginzinger D, Rosen CJ, Bikle DD (2003) Skeletal unloading induces resistance to insulin-like growth factor I on bone formation. Bone 32:669–680 [DOI] [PubMed] [Google Scholar]
- 52.Sakata T, Wang Y, Halloran BP, Elalieh HZ, Cao J, Bikle DD(2004) Skeletal unloading induces resistance to insulin-like growth factor-I (IGF-I) by inhibiting activation of the IGF-I signaling pathways. J Bone Miner Res 19:436–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kostenuik PJ, Harris J, Halloran BP, Turner RT, Morey-Holton ER, Bikle DD (1999) Skeletal unloading causes resistance of osteoprogenitor cells to parathyroid hormone and to insulin-like growth factor-I. J Bone Miner Res 14:21–31 [DOI] [PubMed] [Google Scholar]
- 54.Giancotti FG, Ruoslahti E (1999) Integrin signaling. Science 285:1028–1032 [DOI] [PubMed] [Google Scholar]
- 55.Schwartz MA (2001) Integrin signaling revisited. Trends Cell Biol 11:466–470 [DOI] [PubMed] [Google Scholar]
- 56.Takada Y, Ye X, Simon S (2007) The integrins. Genome biology 8:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sheppard D (2000) In vivo functions of integrins: lessons from null mutations in mice. Matrix Biol 19:203–209 [DOI] [PubMed] [Google Scholar]
- 58.Asthagiri AR, Nelson CM, Horwitz AF, Lauffenburger DA (1999) Quantitative relationship among integrin-ligand binding, adhesion, and signaling via focal adhesion kinase and extracellular signal-regulated kinase 2. J Biol Chem 274:27119–27127 [DOI] [PubMed] [Google Scholar]
- 59.Horne WC, Sanjay A, Bruzzaniti A, Baron R (2005) The role(s) of Src kinase and Cbl proteins in the regulation of osteoclast differentiation and function. Immunol Rev 208:106–125 [DOI] [PubMed] [Google Scholar]
- 60.Wary KK, Mainiero F, Isakoff SJ, Marcantonio EE, Giancotti FG(1996) The adaptor protein Shc couples a class of integrins to the control of cell cycle progression. Cell 87:733–743 [DOI] [PubMed] [Google Scholar]
- 61.Wary KK, Mariotti A, Zurzolo C, Giancotti FG (1998) A requirement for caveolin-1 and associated kinase Fyn in integrin signaling and anchorage-dependent cell growth. Cell 94:625–634 [DOI] [PubMed] [Google Scholar]
- 62.Kaiser E, Sato M, Onyia JE, Chandrasekhar S (2001) Parathyroid hormone (1–34) regulates integrin expression in vivo in rat osteoblasts. J Cell Biochem 83:617–630 [DOI] [PubMed] [Google Scholar]
- 63.Lai CF, Chaudhary L, Fausto A, Halstead LR, Ory DS, Avioli LV, Cheng SL (2001) Erk is essential for growth, differentiation, integrin expression, and cell function in human osteoblastic cells. J Biol Chem 276:14443–14450 [DOI] [PubMed] [Google Scholar]
- 64.Gronthos S, Simmons PJ, Graves SE, Robey PG (2001) Integrin-mediated interactions between human bone marrow stromal precursor cells and the extracellular matrix. Bone 28:174–181 [DOI] [PubMed] [Google Scholar]
- 65.Moursi AM, Globus RK, Damsky CH (1997) Interactions between integrin receptors and fibronectin are required for calvarial osteoblast differentiation in vitro. J Cell Sci 110:2187–2196 [DOI] [PubMed] [Google Scholar]
- 66.Pistone M, Sanguineti C, Federici A, Sanguineti F, Defilippi P, Santolini F, Querze G, Marchisio PC, Manduca P (1996) Integrin synthesis and utilization in cultured human osteoblasts. Cell Biol Int 20:471–479 [DOI] [PubMed] [Google Scholar]
- 67.Lai CF, Cheng SL (2005) Alphavbeta integrins play an essential role in BMP-2 induction of osteoblast differentiation. J Bone Miner Res 20:330–340 [DOI] [PubMed] [Google Scholar]
- 68.Cowles EA, Brailey LL, Gronowicz GA (2000) Integrin-mediated signaling regulates AP-1 transcription factors and proliferation in osteoblasts. J Biomed Mater Res 52:725–737 [DOI] [PubMed] [Google Scholar]
- 69.Gronthos S, Stewart K, Graves SE, Hay S, Simmons PJ (1997) Integrin expression and function on human osteoblast-like cells. J Bone Miner Res 12:1189–1197 [DOI] [PubMed] [Google Scholar]
- 70.Globus RK, Doty SB, Lull JC, Holmuhamedov E, Humphries MJ, Damsky CH (1998) Fibronectin is a survival factor for differentiated osteoblasts. J Cell Sci 111:1385–1393 [DOI] [PubMed] [Google Scholar]
- 71.Mizuno M, Fujisawa R, Kuboki Y (2000) Type I collagen-induced osteoblastic differentiation of bone-marrow cells mediated by collagen-alpha2beta1 integrin interaction. J Cell Physiol 184:207–213 [DOI] [PubMed] [Google Scholar]
- 72.Zimmerman D, Jin F, Leboy P, Hardy S, Damsky C (2000) Impaired bone formation in transgenic mice resulting from altered integrin function in osteoblasts. Dev Biol 220:2–15 [DOI] [PubMed] [Google Scholar]
- 73.Miura Y, Miura M, Gronthos S, Allen MR, Cao C, Uveges TE, Bi Y, Ehirchiou D, Kortesidis A, Shi S, Zhang L (2005) Defective osteogenesis of the stromal stem cells predisposes CD18-null mice to osteoporosis. Proc Natl Acad Sci USA 102:14022–14027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ross FP, Teitelbaum SL (2005) alphavbeta3 and macrophage colony-stimulating factor: partners in osteoclast biology. Immunol Rev 208:88–105 [DOI] [PubMed] [Google Scholar]
- 75.Lane NE, Yao W, Nakamura MC, Humphrey MB, Kimmel D, Huang X, Sheppard D, Ross FP, Teitelbaum SL (2005) Mice lacking the integrin beta5 subunit have accelerated osteoclast maturation and increased activity in the estrogen-deficient state. J Bone Miner Res 20:58–66 [DOI] [PubMed] [Google Scholar]
- 76.Faccio R, Teitelbaum SL, Fujikawa K, Chappel J, Zallone A, Tybulewicz VL, Ross FP, Swat W (2005) Vav3 regulates osteoclast function and bone mass. Nat Med 11:284–290 [DOI] [PubMed] [Google Scholar]
- 77.Zou W, Kitaura H, Reeve J, Long F, Tybulewicz VL, Shattil SJ, Ginsberg MH, Ross FP, Teitelbaum SL (2007) Syk, c-Src, the alphavbeta3 integrin, and ITAM immunoreceptors, in concert, regulate osteoclastic bone resorption. J Cell Biol 176:877–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Murphy MG, Cerchio K, Stoch SA, Gottesdiener K, Wu M, Recker R (2005) Effect of L-000845704, an alphaVbeta3 integrin antagonist, on markers of bone turnover and bone mineral density in postmenopausal osteoporotic women. J Clin Endocrinol Metab 90:2022–2028 [DOI] [PubMed] [Google Scholar]
- 79.Dufour C, Holy X, Marie PJ (2007) Skeletal unloading induces osteoblast apoptosis and targets alpha5beta1-PI3K-Bcl-2 signaling in rat bone. Exp Cell Res 313:394–403 [DOI] [PubMed] [Google Scholar]
- 80.Liedert A, Kaspar D, Blakytny R, Claes L, Ignatius A (2006) Signal transduction pathways involved in mechanotransduction in bone cells. Biochem Biophys Res Commun 349:1–5 [DOI] [PubMed] [Google Scholar]
- 81.Pavalko FM, Norvell SM, Burr DB, Turner CH, Duncan RL, Bidwell JP (2003) A model for mechanotransduction in bone cells: the load-bearing mechanosomes. J Cell Biochem 88:104–112 [DOI] [PubMed] [Google Scholar]
- 82.Meyers VE, Zayzafoon M, Douglas JT, McDonald JM (2005) RhoA and cytoskeletal disruption mediate reduced osteoblastogenesis and enhanced adipogenesis of human mesenchymal stem cells in modeled microgravity. J Bone Miner Res 20:1858–1866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Meyers VE, Zayzafoon M, Gonda SR, Gathings WE, McDonald JM (2004) Modeled microgravity disrupts collagen I/integrin signaling during osteoblastic differentiation of human mesenchymal stem cells. J Cell Biochem 93:697–707 [DOI] [PubMed] [Google Scholar]
- 84.Wang Y, McNamara LM, Schaffler MB, Weinbaum S (2007) A model for the role of integrins in flow induced mechanotransduction in osteocytes. Proc Natl Acad Sci USA 104:15941–15946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Carvalho RS, Bumann A, Schaffer JL, Gerstenfeld LC (2002) Predominant integrin ligands expressed by osteoblasts show preferential regulation in response to both cell adhesion and mechanical perturbation. J Cell Biochem 84:497–508 [PubMed] [Google Scholar]
- 86.Morinobu M, Ishijima M, Rittling SR, Tsuji K, Yamamoto H, Nifuji A, Denhardt DT, Noda M (2003) Osteopontin expression in osteoblasts and osteocytes during bone formation under mechanical stress in the calvarial suture in vivo. J Bone Miner Res 18:1706–1715 [DOI] [PubMed] [Google Scholar]
- 87.Fujihara S, Yokozeki M, Oba Y, Higashibata Y, Nomura S, Moriyama K (2006) Function and regulation of osteopontin in response to mechanical stress. J Bone Miner Res 21:956–964 [DOI] [PubMed] [Google Scholar]
- 88.Ishijima M, Tsuji K, Rittling SR, Yamashita T, Kurosawa H, Denhardt DT, Nifuji A, Noda M (2002) Resistance to unloading-induced three-dimensional bone loss in osteopontin-deficient mice. J Bone Miner Res 17:661–667 [DOI] [PubMed] [Google Scholar]
- 89.Iwaniec UT, Wronski TJ, Amblard D, Nishimura Y, van der Meulen MC, Wade CE, Bourgeois MA, Damsky CD, Globus RK (2005) Effects of disrupted beta1-integrin function on the skeletal response to short-term hindlimb unloading in mice. J Appl Physiol 98:690–696 [DOI] [PubMed] [Google Scholar]
- 90.Borges E, Jan Y, Ruoslahti E (2000) Platelet-derived growth factor receptor beta and vascular endothelial growth factor receptor 2 bind to the beta 3 integrin through its extracellular domain. J Biol Chem 275:39867–39873 [DOI] [PubMed] [Google Scholar]
- 91.Maile LA, Badley-Clarke J, Clemmons DR (2001) Structural analysis of the role of the beta 3 subunit of the alpha V beta 3 integrin in IGF-I signaling. J Cell Sci 114:1417–1425 [DOI] [PubMed] [Google Scholar]
- 92.Zheng B, Clemmons DR (1998) Blocking ligand occupancy of thealphaVbeta3 integrin inhibits insulin-like growth factor I signaling in vascular smooth muscle cells. Proc Natl Acad Sci USA 95:11217–11222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Clemmons DR, Maile LA (2005) Interaction between insulin-like growth factor-I receptor and alphaVbeta3 integrin linked signaling pathways: cellular responses to changes in multiple signaling inputs. Mol Endocrinol 19:1–11 [DOI] [PubMed] [Google Scholar]
- 94.Long RK, Nishida S, Bikle DD (2005) IGF-1 stimulated b3 integrin recruitment is associated with potentiation of the IGF-1 receptor. J Bone Min Res 20:S250 [Google Scholar]
- 95.Maile LA, Busby WH, Sitko K, Capps BE, Sergent T, Badley-Clarke J, Clemmons DR (2006) Insulin-like growth factor-I signaling in smooth muscle cells is regulated by ligand binding to the 177CYDMKTTC184 sequence of the beta3-subunit of alphaVbeta3. Mol Endocrinol 20:405–413 [DOI] [PubMed] [Google Scholar]
- 96.Boutahar N, Guignandon A, Vico L, Lafage-Proust MH (2004) Mechanical strain on osteoblasts activates autophosphorylation of focal adhesion kinase and proline-rich tyrosine kinase 2 tyrosine sites involaved in ERK activation. J Biol Chem 279:30588–30599 [DOI] [PubMed] [Google Scholar]