

Abstract
We report the development of a robust user-friendly Escherichia coli (E. coli) expression system, derived from the BL21(DE3) strain, for site-specifically incorporating unnatural amino acids (UAAs) into proteins using engineered E. coli tryptophanyl-tRNA synthetase (EcTrpRS)-tRNATrp pairs. This was made possible by functionally replacing the endogenous EcTrpRS-tRNATrp pair in BL21(DE3) E. coli with an orthogonal counterpart from Saccharomyces cerevisiae, and reintroducing it into the resulting altered translational machinery tryptophanyl (ATMW-BL21) E. coli strain as an orthogonal nonsense suppressor. The resulting expression system benefits from the favorable characteristics of BL21(DE3) as an expression host, and is compatible with the broadly used T7-driven recombinant expression system. Furthermore, the vector expressing the nonsense-suppressing engineered EcTrpRS-tRNATrp pair was systematically optimized to significantly enhance the incorporation efficiency of various tryptophan analogs. Together, the improved strain and the optimized suppressor plasmids enable efficient UAA incorporation (up to 65% of wild-type levels) into several different proteins. This robust and user-friendly platform will significantly expand the scope of the genetically encoded tryptophan-derived UAAs.
Keywords: BL21(DE3), unnatural amino acid, tryptophanyl-tRNA synthetase, genetic code expansion
Graphical Abstract
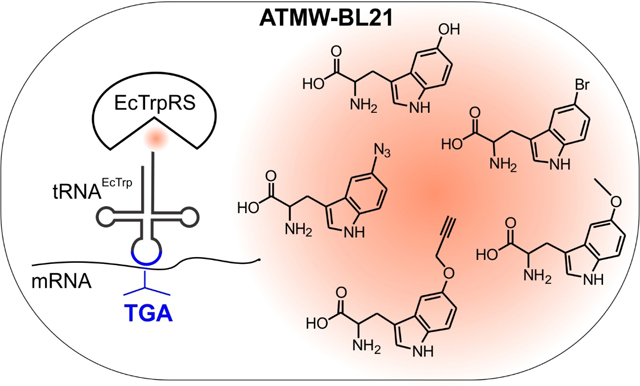
Introduction
Genetic code expansion (GCE) technology enables the study and manipulation of protein structure-function via site-specific incorporation of unnatural amino acids (UAAs).[1–5] A successful GCE platform relies on three components: 1) a reassigned codon that encodes the UAA, 2) an orthogonal aminoacyl-tRNA synthetase (aaRS)/transfer RNA (tRNA) pair, and 3) the desired UAA supplemented to the growth media. The amber nonsense codon, UAG, is used most frequently as the reassigned codon for GCE.[5] Orthogonal aaRS/tRNA pairs for GCE is traditionally borrowed from a different domain of life; in bacteria, the orthogonal pairs for GCE are imported from eukaryotes and archaea, while in eukaryotes, bacteria-derived pairs are typically used. Nearly the entire UAA toolbox available for prokaryotic protein expression is built using two archaea-derived aaRS/tRNA pairs: M. jannaschii tyrosyl and the Methanosarcina-derived pyrrolysyl pair.[1–5] Although numerous UAAs have been genetically encoded using these platforms, there is a continued demand for new pairs that can be engineered to genetically encode distinct structural classes of UAAs. Additionally, new pairs can be combined with the existing ones to facilitate site-specific incorporation of multiple, distinct UAAs into target proteins.[6]
Recently, we reported the ability to expand the genetic code of E. coli using its endogenous tryptophanyl-tRNA synthetase (EcTrpRS)/tRNA pair.[7] This was achieved by first functionally substituting the endogenous tryptophanyl pair of E. coli with a yeast counterpart, followed by reintroducing this ‘liberated’ pair into the resulting altered translational machinery tryptophanyl (ATMW1) strain of E. coli as an orthogonal nonsense suppressor.[7,8] Subsequently, the EcTrpRS was evolved in the ATMW1 strain to charge several tryptophan-derived UAAs with high fidelity and efficiency, including 5-hydroxytryptophan (5HTP), 5-azidotryptophan (5AzW), 5-propargyloxytryptophan (5PrW), 5-aminotryptophan (5AmW), 5-methoxytryptophan (5MTP), and 5-bromotryptophan (5BrW). We further demonstrated that the EcTrpRS/tRNA pair is orthogonal in mammalian cells and its engineered variants can be used to site-specifically incorporate the aforementioned tryptophan analogs into proteins in these cells. This new toolbox has enabled several novel applications since its development.[6,7,9–12] For example, novel chemoselective bioconjugation reactions have been developed to label 5-hydroxytryptophan, one of the UAAs genetically encoded using this pair.[9–12] Furthermore, a biosynthetic pathway has been developed to generate 5HTP in cells, obviating the need for its exogenous supply.[13] In addition, the EcTrpRS/tRNA pair was shown to be orthogonal to the archaea-derived tyrosyl and pyrrolysyl pairs, and these three pairs were used together to demonstrate the first example of site-specific incorporation of three different UAAs into one protein.[6] 5HTP-targeted bioconjugation reactions were shown to be orthogonal to existing bioorthogonal conjugation reactions, such as strain-promoted azide-alkyne cycloaddition and inverse electron demand Diels-Alder reactions between tetrazines and strained alkenes, enabling their concurrent use to label proteins at up to three distinct sites with different entities.[6] Further engineering of this pair holds the promise to genetically encode additional enabling UAAs.
However, the scope of this toolbox in our first-generation E. coli expression system has been somewhat limited by a few significant drawbacks. The ATMW1 strain, derived from the E. coli MG1655 strain, is not ideally suited for recombinant protein expression. In addition to the presence of detrimental proteases, this strain is not compatible with the popular T7 RNAP driven recombinant protein expression systems that are widely used in the scientific community. Finally, the first-generation pEVOL suppression vector, expressing the engineered EcTrpRS/tRNA pair, affords modest efficiency of UAA incorporation relative to other optimized UAA mutagenesis platforms.[14,15] Here we overcome these limitations by first developing an ATMW E. coli strain from BL21(DE3), a widely adopted expression host, to allow efficient, T7-compatible protein expression for incorporating our tryptophan analogs. Additionally, we systematically optimized our suppression vector to significantly enhance their efficiency. Together, our work provides a robust and user-friendly prokaryotic protein expression system for incorporating tryptophan-like UAAs using the EcTrpRS/tRNA pair.
Results
A BL21(DE3) derived ATMW E. coli strain
The first generation ATMW1 platform[7] was generated from the EcNR1 E. coli strain,[16] a derivative of the MG1655 strain that was optimized for efficient genome engineering. Although the facile recombination-mediated genome manipulation in EcNR1 was helpful for developing the ATMW1 strain, it is a suboptimal host for recombinant protein expression. In particular, the MG1655 strain encodes several proteases detrimental for recombinant protein expression, and does not encode the T7 RNA polymerase (T7RP), thereby precluding the use of the popular T7 expression systems. When designing the second generation ATMW strain, we selected BL21(DE3) as the progenitor, which is widely used for robust recombinant protein expression.[17] BL21(DE3) lacks the Lon and OmpT proteases, which enhances the integrity of recombinant proteins, and contains the lambda prophage containing T7RP under the IPTG inducible lacUV promoter, thereby enabling T7 driven protein expression.[18,19]
Generation of the ATMW strain involves knocking out the trpS and trpT genes from the E. coli genome, encoding EcTrpRS and tRNAEcTrp, respectively, in the presence of a plasmid expressing the yeast TrpRS/tRNA pair. Unlike the EcNR1 progenitor strain used for developing ATMW1, the BL21(DE3) strain does not encode temperature-inducible lambda Red genes (Exo, Beta, and Gam)[20] needed for recombination-mediated genome engineering. Instead, we used the pKD46 plasmid, which expresses these lambda Red genes from an arabinose-inducible promoter.[21] After performing the necessary recombinations, pKD46 can be removed from the strain simply by culturing the strain at elevated temperatures, thanks to its temperature-sensitive origin of replication. To generate the ATMW strain from BL21(DE3) – named ATMW-BL21 – we performed the steps described previously.[7] Briefly, the BL21(DE3) strain was transformed with the plasmid pUltraG_ScW40CCA, expressing the yeast TrpRS/tRNA pair, and pKD46. Next, the EcTrpRS and tRNATrp were removed from the genome by sequential recombination with a zeocin and a gentamycin resistance cassette, respectively (Figure 1a). Each recombination step was confirmed by diagnostic genomic PCR analysis as described before.[7] Finally, pKD46 was removed by culturing the strain at 37 °C, affording the final ATMW-BL21 strain with the genotype BL21(DE3) pUltraG_ScW40cca trpS::ZeoR trpT::GentR (Figure 1a).
Figure 1.

(a) The scheme demonstrating the strategy to develop ATMW-BL21 strain from BL21(DE3). In the presence of the pUltra plasmid expressing the yeast tryptophanyl pair, EcTrpRS and tRNAEcTrp were replaced with zeocin and gentamycin resistance genes, respectively, by recombination. The plasmid encoding the recombination machinery, pKD46, was removed at the end by heat induction. (b) ATMW-BL21 exhibits a growth rate comparable to its progenitor BL21(DE3) strain, with or without the pUltraG complementation plasmid. OD600, optical density measured at 600 nm.
Evaluating ATMW-BL21 for viability and protein expression
It is essential that a robust expression platform does not suffer from a significant growth defect. Replacing an endogenous aaRS/tRNA pair with a heterologous counterpart poses the risk of compromising the viability of the strain, if the latter pair performs suboptimally in the new host cell. We compared the growth profile of ATMW-BL21 to its progenitor BL21(DE3), with or without the pUltraG complementing plasmid, to show that it has no growth defect (Figure 1b). ATMW-BL21 and its progenitor grew virtually identically during the exponential phase, reaching similar final density (Figure 1b). It also exhibited similar growth characteristics relative to the previously reported ATMW1 strain (Supplementary Figure 1). The absence of a growth defect suggests that the complementing yeast TrpRS/tRNA pair efficiently substitutes the function of the endogenous EcTrpRS/tRNA pair.
Next, we tested the performance of the ATMW-BL21 strain for recombinant protein expression and compared it to its progenitor, as well as the first-generation ATMW1 strain. First, we expressed a wild-type green superfolder fluorescent protein (sfGFP) from a T5-lac promoter that recognizes the endogenous RNA polymerase and can be expressed in both MG1655 and BL21(DE3) derived E. coli strains. Both BL21(DE3) and ATMW-BL21 expressed this reporter over two-fold more efficiently than MG1655-derived EcNR1 and ATMW1, corroborating the superiority of BL21(DE3) as a protein expression host (Figure 2a). Next, we used a wild-type sfGFP reporter expressed from a T7 promoter and, as expected, observed robust protein expression only in the BL21(DE3) and ATMW-BL21 strains but not in ATMW1 (Figure 2b). These observations confirm that ATMW-BL21 retains the beneficial properties of its progenitor BL21(DE3) strain as a robust expression host, and enables robust T7 promoter-driven recombinant protein expression.
Figure 2.

Evaluating protein expression in ATMW-BL21, ATMW1, and their progenitor strains. (a) Expression of wild-type sfGFP from a T5-lac promoter in different strains, measured as the normalized fluorescence of the reporter in resuspended cells. (b) Expression of a wild-type sfGFP from a T7 promoter compared in various strains, EcNR1Z and BL21(DE3), measured as the normalized fluorescence of the reporter in resuspended cells. (c) 5HTP-dependent expression of sfGFP-151-TGA in ATMW1 and ATMW-BL21 using the “unoptimized” pEVOL expression system: pEVOL-tacI EcTrpRS-14 proK. Expression of full-length sfGFP was measured in resuspended cells using its characteristic fluorescence.
Incorporation of tryptophan analogs using EcTrpRS/tRNAEcTrpUCA pair in ATMW-BL21
After confirming its ability to serve as a robust expression host, we evaluated the efficiency of UAA mutagenesis in the ATMW-BL21 strain using the engineered EcTrpRS/tRNAEcTrpUCA pair in response to the TGA stop codon. A sfGFP reporter harboring a TGA codon at the 151 position was used as the reporter and expressed from the T5-lac promoter. The previously described pEVOL suppressor plasmid was used to express the engineered EcTrpRS-h14 [7] from a tacI promoter and the tRNAEcTrpUCA from the proK promoter. As expected, in both ATMW1 and ATMW-BL21 strains, expression of the sfGFP-151-TGA reporter was observed in the presence of 5HTP, while little expression was found in its absence (Figure 2c). However, the expression level was significantly lower relative to a wild-type sfGFP reporter control, which does not encode a premature nonsense codon (Figure 2c). Although ATMW-BL21 facilitated higher expression levels of the sfGFP-TGA reporter relative to ATMW1, the efficiency was only about 10% relative to wild-type sfGFP. In contrast, other optimized UAA mutagenesis systems have been reported to provide >50% suppression efficiency,[14,15] indicating there is room for optimizing our suppression vector design.
Optimization of the suppressor plasmid
To optimize the first generation pEVOL [15] suppressor vector (Figure 3a), we generated several different variants by systematically altering the origin of replication (ori), and the promoters driving EcTrpRS and tRNAEcTrpUCA (Figure 3b). The pEVOL plasmid has a p15a ori that is compatible with the complementing pUltraG plasmid in ATMW-BL21, harboring a CloDF13 ori,[22] and typical plasmids for recombinant protein expression that contain oris from the pBR322 compatibility group. First, we replaced the p15a ori of pEVOL with RSF [23] and ColA [24] ori, which satisfy the necessary compatibility requirements mentioned above, but would be maintained in the cell at different copy numbers. Efficiency of these vectors were tested using the aforementioned sfGFP-151-TGA reporter (Figure 3c). While the RSF and ColA variants of pEVOL were functional, the original p15a afforded the most efficient 5HTP incorporation (Figure 3c). Consequently, in the subsequent designs of the suppressor plasmid, we retained the p15a ori.
Figure 3.

Development of an optimized pEVOL suppressor plasmid. a) Vector map of the first-generation pEVOL vector (full sequence provided in SI). (b) A scheme showing the components of the vector which were altered for optimization: p15A, RSF, or ColA ori (light blue); tacI or glnS promoter (navy blue); EcTrpRS-h13 or -h14 (gray); proK, lpp, or leuV promoter (navy blue). (c) Expression of sfGFP-151-TGA using pEVOL vectors (proK-tRNA and tacI-TrpRS) with varying oris in ATMW-BL21 in the presence or absence of 5HTP. (d) Expression of sfGFP-151-TGA using pEVOL vectors containing either an inducible tacI or constitutive glnS promoter on the EcTrpRS (tRNA expressed from proK). (e) Expression of sfGFP-151-TGA using pEVOL vectors with different combinations of EcTrpRS and tRNAEcTrpUCA promoters (tacI/glnS = EcTrpRS promoter; leuV/lpp = tRNAEcTrpUCA promoter). (f) MS analysis of the sfGFP-151-TGA reporter protein confirming 5HTP incorporation using pEVOL-tacI-EcTrpRS-h13-leuV-tRNA. Full MS spectrum is provided in Supplementary Figure 2. (g) SDS-PAGE analysis of purified sfGFP-151– 5HTP and sfGFP-WT. Full SDS-PAGE gel is provided in Supplementary Figure 3. For all sfGFP reporter expression analyses, its characteristic fluorescence was measured in resuspended cells and normalized to OD600.
Another important factor driving UAA incorporation efficiency are the promoters driving EcTrpRS and tRNAEcTrpUCA expression. For EcTrpRS expression, we explored the weakly active constitutive glnS promoter, and the strong but inducible tacI promoter. For expressing tRNAEcTrpUCA, three constitutively active promoters were explored: proK, lpp,[25] and leuV,[26] with increasing promoter activity.[25,27] First, we compared the performance of pEVOL variants using a tacI or glnS promoter to drive the expression of EcTrpRS-h14, while the tRNA was still expressed from the proK promoter, to reveal that the tacI-driven system was significantly more efficient (Figure 3d). Then, we tested pEVOL variants that use leuV and lpp promoters to express tRNAEcTrpUCA, and for each case, we also expressed the EcTrpRS either from tacI or glnS (Figure 3e). These variants showed significantly higher suppression efficiency (>60% of wild-type GFP for tacI/leuV variant), which was much improved relative to the original suppressor plasmid. Additionally, we incorporated the previously reported EcTrpRS mutant h13 in the tacI/leuV vector, which was previously shown to have similar substrate scope as h14, but showed a slightly higher activity.[7] Using the pEVOL-tacI-EcTrpRS-h13-leuV-tRNA suppression system, we were able to express and purify sfGFP-151–5HTP with a yield of 129 mg/L (wild-type sfGFP yield was 164 mg/L), and confirmed the incorporation of 5HTP by MS analysis (Figure 3f and Supplementary Figure 2) and purity by SDS-PAGE analysis (Figure 3g and Supplementary Figure 3).
Incorporation of additional tryptophan analogs using the polyspecificity of EcTrpRS-h13
We previously showed that EcTrpRS-h13 and –h14 can charge a number of structurally similar UAAs, while still discriminating against the 20 canonical amino acids.[7] Such polyspecific aaRS mutants are valuable, since these enable rapid expansion of the UAA toolbox without having to develop a distinct mutant for each new UAA. We tested the expression of the sfGFP-151-TGA reporter using our optimized pEVOL plasmids in the presence of additional 5-substituted tryptophan analogs (Figure 4), which are established substrates for the engineered EcTrpRS mutants.[7] The suppressor plasmid using the most active promoters (tacI/leuV) yielded the highest expression levels, but was associated with higher reporter expression in the absence of an added UAA (Figure 4a). Replacing the strong tacI promoter for weaker glnS promoter driving the EcTrpRS-h13 led to lower background suppression, but also weaker expression overall (Figure 4a). Characterization of the analogous suppressor plasmids with a lpp promoter driving the tRNA also yielded similar results (Supplementary Figure 4).
Figure 4.

a) Evaluating the incorporation of different tryptophan analogs into sfGFP-151-TGA in ATMW-BL21 using three different pEVOL vectors. EcTrpRS-h13 and –h14 show slightly different substrate preference. The use of the stronger leuV promoter to express the tRNAEcTrpUCA and tacI promoter to express the EcTrpRS provides substantial increase in suppression efficiency. (b) Structures of the tryptophan analog UAAs used in the study.
Relative to the first-generation suppression plasmids, the use of the optimized tacI/leuV plasmid was associated with significantly higher levels of nonsense suppression in the absence of a substrate UAA, likely due to elevated expression of the suppressor pair. We have previously shown that this engineered EcTrpRS selectively charges the UAAs shown in Figure 4b when they are supplemented in the media, even though substantial suppression activity is observed in the absence of any UAA.[7] As described above (Figure 3f and Supplementary Figure 2), the MS analysis of the sfGFP-151-TGA reporter protein expressed using the optimized suppressor plasmid in the presence of 5HTP showed clean incorporation of this UAA. To further confirm the high fidelity of UAA incorporation with our optimized vector, we purified the sfGFP-151-TGA reporter expressed in the presence of each of the five UAAs shown in Figure 4 band performed ESI-MS analysis to confirm exclusive UAA-dependent incorporation at the target site (Supplementary Figure 5). Each UAA was used at 1 mM final concentration in the growth medium, which is standard for most UAA incorporation experiments.
Application of ATMW-BL21 to other recombinant proteins
After developing an efficient UAA incorporation system using the EcTrpRS/tRNAEcTrpUCA pair, we sought to further demonstrate its utility using proteins beyond GFP. First, we attempted the expression of a T7-promoted ketosteroid isomerase (KSI), which would further corroborate the ability of the ATMW-BL21 strain to support T7-driven recombinant protein expression. Two different TGA mutants of KSI, at positions 7 or 78, were expressed in ATMW-BL21 coexpressing the optimized suppressor plasmid and in the presence of 1 mM 5HTP in the expression medium.[14] Both mutants were isolated by immobilized metal-ion chromatography with good yields (54 mg/L and 36 mg/L for 7-TGA and 78-TGA KSI proteins, respectively). Whole-protein ESI-MS analysis confirmed the incorporation of 5HTP at the desired site (Supplementary Figure 6) and SDS-PAGE analysis confirmed their purity (Figure 5b and Supplementary Figure 7).
Figure 5.

ATMW-BL21 expression of other recombinant proteins. (a) Structure of KSI showing incorporation with 5HTP (PDB: 1OCV). (b) SDS-PAGE analysis of KSI-7–5HTP and KSI-78–5HTP. Full SDS-PAGE gel is provided in Supplementary Figure 7. (c) Cartoon structure of Fab with 5HTP incorporated in the heavy chain. (d) SDS-PAGE analysis of Fab WT and 169–5HTP mutants purified using ATMW-BL21 and ATMW1, with yields shown below. Full-length SDS-PAGE gel is provided in Supplementary Figure 8.
We further attempted to use our optimized platform to express the antigen binding fragment (Fab) of the anti HER2 (human epidermal growth factor 2) antibody (αHer2 Fab). Antibodies and their conjugates are frequently used as research reagents for analyses such as ELISA, immunofluorescence, flow cytometry, etc., as well as biotherapeutics. UAA mutagenesis provides a versatile way to create homogeneous, site-specific antibody conjugates.[6,9,28–30] We have previously shown that 5HTP-targeted bioconjugation chemistries can be used to site-specifically label antibody fragments.[6,9] The αHer2 Fab reporter was expressed from arabinose-inducible promoters encoded in a pBK vector and encoded a TGA at position 169 of the heavy chain. Using our optimized pEVOL suppression vector (tacI-h13-leuV), we were able to express and purify the Fab-169–5HTP mutant (Figure 5d and Supplementary Figure 8), and compared the isolated yield between ATMW1 and ATMW-BL21. We found that ATMW-BL21 afforded a two-fold increase in the yield of Fab-169–5HTP relative to ATMW1, further validating the utility of the optimized system.
Discussion
The genetic code expansion technology promises exciting new ways to probe and manipulate biology. To fulfill that promise, and to enable facile adoption by the broader scientific community, it is essential that the technology is optimized to offer robust performance, and is compatible with broadly used protein expression systems. Indeed, the development of optimized suppressor plasmids such as pEVOL and pUltra[14,15] in E. coli significantly facilitated broader adoption of the GCE technology using the archaea-derived tyrosyl and the pyrrolysyl pairs. Our recently reported use of the EcTrpRS/tRNAEcTrpUCA pair has introduced new UAAs to this toolbox. However, their use in the E. coli expression system was restricted solely to the ATMW1 strain, a suboptimal expression host, diminishing the potential utility of this toolbox. To overcome this limitation, we developed the BL21(DE3)-derived ATMW-BL21 strain (Figure 1a) that is compatible with the T7 expression systems and provide enhanced expression levels (Figure 2–3). The functional replacement of the endogenous E. coli tryptophanyl pair with its yeast counterpart was seamless, evidenced by a lack of any significant growth defect (Figure 1b). As expected, ATMW-BL21 retained the beneficial properties of its progenitor and facilitated robust T7-driven protein expression at comparable levels.
However, the UAA incorporation efficiency in this strain using the first-generation suppressor plasmid was found to be low (Figure 2c). We designed and tested several variants of the original suppressor plasmid, by systematically altering the origin of replication (ori), and the promoters expressing the engineered EcTrpRS and the tRNAEcTrpUCA, to determine the factor(s) limiting expression. Although the original p15a ori was found to be the optimal choice, the use of stronger promoters to drive the expression of the engineered pair significantly enhanced UAA incorporation efficiency (Figure 3d–e). In particular, expressing the tRNA from stronger promoters leuV and lpp provided improved efficiency, even when a weaker promoter was used to drive the expression of engineered EcTrpRS mutants (Figure 3e). We also employed a slightly different engineered EcTrpRS variant h13 [7] that offer higher suppression efficiency, affording reporter expression at approximately 65% of wild-type levels.
The optimized suppressor plasmids also led to a significant increase in reporter expression in the absence of added UAA. We have previously shown that the engineered EcTrpRS variants do charge tryptophan in the absence of the UAA at low levels,[7] even though the cognate UAAs are exclusively incorporated when they are present (Supplementary Figure 5). It is likely that our optimized suppression plasmid amplifies this background activity. We used MS analysis to show that the optimized suppression plasmid exclusively incorporated the full set of tryptophanyl UAAS, 5HTP, 5MTP, 5Brw, 5PrW, and 5AzW, by our polyspecific EcTrpRS-h13 when these are supplemented in the growth medium (Supplementary Figure 5). If a lower suppression level in the absence of the UAA is desired for certain applications, the variant suppressor plasmid employing the weaker promoter combination can be used.
In summary, we developed an optimized E. coli expression system for efficient incorporation of tryptophan analogs, comprising the ATMW-BL21 expression host that offer several advantages relative to its predecessor, as well as highly efficient suppression plasmids encoding the engineered EcTrpRS/tRNAEcTrpUCA pair. We demonstrated the utility of this new system by expressing sfGFP, KSI, and the Fab fragment of Her2 site-specifically incorporating tryptophan analogs with high fidelity and efficiency. The UAAs encoded using this pair offer diverse potential applications, such as bioconjugation, biophysical probing of protein structure and function, and multi-UAA incorporation, and the platform reported here is expected to broaden the utility of this toolbox, owing to its improved robustness and user-friendly nature.
Materials and Methods
All cloning and plasmid propagation was done in DH10B E. coli cells, with the exception of pEVOL vectors containing tRNAEcTrpUCA, which were propagated using the ATMW-BL21 strain followed by gel extraction. Restriction enzymes were purchased from New England Biolabs (NEB). Polymerase chain reactions (PCR) were carried out using the Phusion High-Fidelity DNA Polymerase from Thermo Scientific. T4 DNA Ligase was purchased from Enzymatics. For DNA purification of PCR products and plasmids, spin columns from Epoch Life Science and Macherey-Nagel Binding Buffer NTI were used. Primers were purchased from GENEWIZ and Integrated DNA Technologies (IDT), and Sanger sequencing was done by GENEWIZ and Eton Biosciences. Primer sequences used in this work can be found in Supplementary Table 1. Antibiotics and isopropyl β-D-1- thiogalactopyranoside (IPTG) were purchased from Sigma-Aldrich or Fisher Scientific. Media components were purchased from Fisher Scientific. For LB plates and liquid cultures, the following antibiotic concentrations were used, unless otherwise mentioned: 95 μg/ml spectinomycin, 20 μg/ml chloramphenicol, 100 μg/ml ampicillin, 15 μg/ml zeocin, and 10 μg/ml gentamycin.
Bacterial strains
ATMW1 was previously generated in the Chatterjee lab.[7] The EcNR1 strain was a gift from Prof. G.M Church. The EcNR1Z strain was generated in the Chatterjee lab and has the genotype EcNR1 lambda Red::ZeoR.[8]
Building ATMW-BL21 using Lambda Red recombination
ATMW-BL21 was developed following a strategy similar to make ATMW1,[7] except: A) the recombination machinery was provided by the pKD46 plasmid (instead of a genomically encoded copy), and B) in the final step, to remove the recombination machinery, after the genotype BL21(DE3) pUltraG_ScW40cca trpS::ZeoR trpT::GentR was achieved, the resulting strain was grown at 37 °C to remove pKD46.
BL21(DE3) cells containing pKD46 and the complementation plasmid, pUltra ScW40, were grown overnight at 30 °C from a single colony with shaking at 250 rpm. The next day, the cells were diluted 1:100 into 2 mL LB containing ampicillin and spectinomycin and allowed to grow at 30 °C with shaking at 250 rpm. Upon reaching an OD600 of 0.1, L-arabinose was added to a final concentration of 10 mM to induce lambda Red expression from the pKD46 plasmid. Cultures were grown to an OD600 of 0.4 and chilled on ice-water for 10 minutes and centrifuged at 4000 × g, 4 °C for 10 minutes. The supernatant was decanted from each culture and pellets were resuspended in 1 mL ice-cold, sterile deionized water. The centrifugation and wash was repeated once, with the final pellet being resuspended in 50 μL sterile deionized water, affording one transformation per 2 mL LB culture. 50 μL cell aliquots were electroporated with 50 ng double-stranded DNA (dsDNA) containing the necessary recombination cassette. After electroporation, cells were recovered in 1 mL sterile LB in a culture tube for 120 minutes, shaking at 250 rpm before plating on LB agar with selection conditions. The first recombination step (trpS::ZeoR) was recovered, plated, and grown at 30 °C to maintain the pKD46 plasmid, and the second and final recombination (trpT::GentR) was recovered, plated, and grown at 37 °C to remove pKD46, confirmed by a lack of growth in the presence of ampicillin. Colonies from selection plates were re-streaked on selection conditions and single colonies were subjected to colony PCR using KAPA-2G polymerase (Kapa Biosystems) as described previously.[7]
Growth analysis
5 mL starter cultures of ATMW1, BL21(DE3), BL21(DE3) + pUltraG_ScW40cca, and ATMW-BL21 with appropriate antibiotics were grown overnight in LB at 37 °C. Each starter culture was diluted to a OD600 of 0.03 in triplicate, in 15 mL LB with respective antibiotics in sterile 50 mL Erlenmeyer flasks, with shaking (250 r.p.m.) at 37 °C. Growth was monitored every hour by measuring OD600 in a 10-mm cuvette. Error bars on graph represent s.d.
Construction of pEVOL suppression plasmids
RSF ori:
The RSF ori was PCR amplified out of pRSF-KpDcr1 (Addgene #32022) with the primers RSF-NheI-F and RSF-HindIII-R, digested with NheI/HindIII, and inserted into the pEVOL vector backbone. ColA ori: The ColA ori was PCR amplified from pCOLA-T7-WspR:D70E (Addgene #79164) with the primers ColA-HindIII-F and ColA-NheI-R, digested with NheI/HindIII, and inserted into the pEVOL vector backbone. EcTrpRS-h13/h14: The EcTrpRS-h13 or h14 gene was PCR amplified with the primers H13 NotI-F and H13 NotI-R, digested with NotI, and inserted into the pEVOL-tac vector backbone. glnS promoter: A pEVOL plasmid containing GlnS and EcTrpRS-h14 already existed[6], so to make a EcTrpRS-h13 variant the gene was PCR amplified with the primers GlnS-NdeI-F and pEvol-SacI-R, digested with NdeI/SacI, and inserted in the pEVOL-glnS vector backbone. lpp promoter: An overlap extension PCR method was used to replace the proK promoter in pEVOL-tac with lpp. The first piece of the lpp promoter was PCR amplified with the primers lpp-PstI-F and lpp-EcTrptRNA-iR, and the second piece EcTrp-tRNA-F and pEvol-Mj-R, with a 20-nt overlap between primers lpp-EcTrptRNA-iR and EcTrp-tRNA-F. The two pieces were joined together using the terminal primers lpp-PstI-F and pEvol-Mj-R, digested with PstI/NcoI, and inserted into the pEVOL-tac vector backbone. To swap EcTrpRS-h13 in the place of h14, or vice versa, the same strategy as the glnS promoter section was used. To make a pEVOL-glnS plasmid with the lpp promoter, glnS was amplified with primers pUII-RS-sqF and pEvol-SacI-R, digested with NotI/SacI, and inserted into the pEVOL-lpp backbone. leuV promoter: the leuV promoter and tRNAEcTrpUCA gene was purchased as a gBlock® from IDT, PCR amplified with the primers leuV-PstI-F and PylCas-AvrII-R, digested with PstI/XhoI and inserted into the pEVOL-tac or glnS vector backbone.
sfGFP-151TGA reporter expression and fluorescence analysis
ATMW-BL21 cells were co-transformed with a pET22b-sfGFP-151TGA reporter plasmid and the desired pEVOL suppressor plasmid. For wild-type reporter expression analysis, a pET22b-sfGFP-WT plasmid was used alone. A 5 mL overnight culture containing the appropriate antibiotics was used to inoculate 1:100 a 20 mL LB culture (in a 50 mL sterile conical tube) supplemented with antibiotics. Cultures were grown to an OD600 of 0.5–0.6, followed by induction with a final concentration of 1 mM IPTG and +/− 1 mM UAA. The cultures were expressed at 30 °C with shaking at 250 r.p.m for 16 hours. The cultures were spun down (5,000 × g, 10 minutes, 4 °C), the supernatant was removed, and cells were resuspended in 1 mL PBS. Cells were diluted 100-fold in a 96-well plate (Thermo Scientific, 96 Well Black/Clear Bottom Plate) in PBS and fluorescence readings and OD600 were collected in a SpectraMAX M5 (Molecular Devices), ex = 488 nm for sfGFP. The fluorescence of each sample was normalized relative to its optical density. Error bars represent the standard deviation of the mean of two independent experiments.
KSI-7/78TGA reporter expression and analysis
ATMW-BL21 cells were co-transformed with a pET22b-L-lac-KSI-7TGA or pET22b-L-lac-KSI-78TGA reporter plasmid and the pEVOL-tac EcTrpRS-h14 proK suppressor plasmid. A 5 mL overnight culture, containing the appropriate antibiotics, was used to inoculate 1:100 a 20 mL LB culture (in a 50 mL sterile conical tube) supplemented with antibiotics. Cultures were grown to 0.5–0.6 OD600, followed by induction with a final concentration of 1 mM IPTG and +/− 1 mM UAA. The cultures were expressed at 30 °C with shaking at 250 r.p.m for 16 hours. The cultures were spun down (5,000 × g, 10 minutes, 4 °C), the supernatant was removed, and prepared for protein purification.
αHer2-Fab reporter expression and analysis
ATMW-BL21 and ATMW1 cells were co-transformed with a pBK-aher2-FAB-169TGA reporter plasmid and the pEVOL-tac EcTrpRS-h13 leuV suppressor plasmid. A 5 mL overnight culture, containing the appropriate antibiotics, was used to inoculate 1:100 a 250 mL LB culture (in a 1 L sterile Erlenmeyer flask). Cultures were grown to 0.5–0.6 OD600, followed by induction with a final concentration of 1 mM IPTG, 0.02% arabinose, and +/− 1 mM 5-HTP. The cultures were expressed at room temperature shaking at 250 r.p.m for 16 hours, harvested, then prepared for purification.
Protein purification of sfGFP and KSI
The cell pellets were resuspended in a ratio of 1 mL lysis buffer to 10 mL expression culture volume. Lysis buffer: B-PER Bacterial Protein Extraction Reagent (Thermo Scientific), 1× Halt Protease Inhibitor Cocktail (Thermo Scientific), 0.01% Pierce Universal Nuclease (Thermo Scientific). Cells were nutated for 30 minutes at room temperature, then clarified by centrifuging at 18,000 × g for 10 minutes at 4 °C. Both reporter proteins contain a C-terminal hexahistidine tag and were purified using a HisPur Ni-NTA resin (Thermo Scientific) following the manufacturer’s protocol. Protein purity was confirmed by SDS-PAGE analysis, and protein molecular mass was characterized using ESI-MS (Agilent Technologies, 1260 Infinity ESI–TOF).
Protein purification of αHer2-Fab
The cell pellets were resuspended in a periplasmic lysis buffer at a ratio of 150 mL to 2 L expression culture. Periplasmic lysis buffer: 20% sucrose, 30 mM Tris-HCl pH 8.0, 1 mM EDTA, 0.2 mg/mL lysozyme, 1x Halt Protease Inhibitor Cocktail. Cells were incubated for 30 minutes at 37 °C, then lysate was diluted 1:1 with binding buffer (50 mM NaOAc, pH 5.2) and clarified by centrifugation at 17,000 × g for 30 minutes at 4 °C. The supernatant was then purified using Pierce Protein G agarose (Thermo Scientific) following the manufacturer’s protocol. After purification, the protein was exchanged into 100 mM phosphate buffer, pH 7.0, using a 10 kDa Millipore-Sigma Amicon ultra centrifugal filter unit.
Unnatural amino acids
5HTP, 5BrW, and 5AmW were purchased from Chem-Impex International (Wood Dale, IL). 5MTP was purchased from Fisher Scientic. 5AzW was synthesized in the lab using a previously reported method,[31] and 5PrW was synthesized as discussed previously.[7]
Supplementary Material
Highlights.
The endogenous tryptophanyl-tRNA synthetase/tRNA pair of BL21(DE3) E. coli strain was functionally substituted with a yeast counterpart
The resulting altered translational machinery tryptophan (ATMW-BL21) strain enables robust T7-driven recombinant protein expression site-specifically incorporating various tryptophan analogs
Optimized suppression vectors expressing engineered E. coli tryptophanyl-tRNA synthetase/tRNA pairs resulted in improved efficiency of unnatural amino acid incorporation
Acknowledgments
The EcNR1 strain was a gift from G.M. Church (Harvard). This work was supported by NIGMS (R35GM136437 to AC).
Abbreviations used
- ATM
altered translational machinery
- aaRS
aminoacyl-tRNA synthetase
- sfGFP
superfolder GFP
- 5HTP
5-hydroxytryptophan
- tRNAUCA
opal suppressor tRNA
- T7RP
T7 RNA polymerase
Footnotes
Elise D. Ficaretta: Conceptualization, investigation, methodology, formal analysis, writing original draft, data curation. Chester J.J. Wrobel: Investigation (ATMW-BL21 cell-line generation). Soumya J.S. Roy: Investigation (chemical synthesis). Sarah B. Erickson: Investigation (antibody expression). James S. Italia: Investigation (plasmid development). Abhishek Chatterjee: Conceptualization, writing review and editing, supervision, project administration, funding acquisition
Declaration of interests
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
A patent application has been submitted regarding the work described in this manuscript. Dr. Chatterjee owns stock at BrickBio, Inc., and a member of its scientific advisory board.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Chin JW, Expanding and reprogramming the genetic code, Nature. 550 (2017). 10.1038/nature24031. [DOI] [PubMed] [Google Scholar]
- [2].Dumas A, Lercher L, Spicer CD, Davis BG, Designing logical codon reassignment-Expanding the chemistry in biology †, Chem. Sci. 6 (2015) 50–69. 10.1039/c4sc01534g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Italia JS, Zheng Y, Kelemen RE, Erickson SB, Addy PS, Chatterjee A, Expanding the genetic code of mammalian cells, Biochem. Soc. Trans. 45 (2017) 555–562. 10.1042/BST20160336. [DOI] [PubMed] [Google Scholar]
- [4].Mukai T, Lajoie MJ, Englert M, Söll D, Rewriting the Genetic Code, Annu. Rev. Microbiol. 71 (2017) 557–577. 10.1146/annurev-micro-090816-093247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Young DD, Schultz PG, Playing with the Molecules of Life, ACS Chem. Biol. 13 (2018) 854–870. 10.1021/acschembio.7b00974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Italia JS, Addy PS, Erickson SB, Peeler JC, Weerapana E, Chatterjee A, Mutually Orthogonal Nonsense-Suppression Systems and Conjugation Chemistries for Precise Protein Labeling at up to Three Distinct Sites, J. Am. Chem. Soc. (2019). 10.1021/jacs.8b12954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Italia JS, Addy PS, Wrobel CJJ, Crawford LA, Lajoie MJ, Zheng Y, Chatterjee A, An orthogonalized platform for genetic code expansion in both bacteria and eukaryotes, Nat. Chem. Biol. 13 (2017) 446–450. 10.1038/nchembio.2312. [DOI] [PubMed] [Google Scholar]
- [8].Italia JS, Latour C, Wrobel CJJ, Chatterjee A, Resurrecting the Bacterial Tyrosyl-tRNA Synthetase/tRNA Pair for Expanding the Genetic Code of Both E. coli and Eukaryotes, Cell Chem. Biol. 25 (2018) 1304–1312. 10.1016/j.chembiol.2018.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Addy PS, Erickson SB, Italia JS, Chatterjee A, A Chemoselective Rapid Azo-Coupling Reaction (CRACR) for Unclickable Bioconjugation, JACS. (2017). 10.1021/jacs.7b05125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Addy PS, Italia JS, Chatterjee A, An Oxidative Bioconjugation Strategy Targeted to a Genetically Encoded 5-Hydroxytryptophan, ChemBioChem. 19 (2018) 1375–1378. 10.1002/cbic.201800111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Addy PS, Zheng Y, Italia JS, Chatterjee A, A “Quenchergenic” Chemoselective Protein Labeling Strategy, ChemBioChem. 20 (2019) 1659–1663. 10.1002/cbic.201800817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Addy PS, Erickson SB, Italia JS, Chatterjee A, Labeling Proteins at Site-Specifically Incorporated 5-Hydroxytryptophan Residues Using a Chemoselective Rapid Azo-Coupling Reaction, Methods Mol. Biol. 2033 (2019) 239–251. [DOI] [PubMed] [Google Scholar]
- [13].Chen Y, Tang J, Wang L, Tian Z, Cardenas A, Fang X, Chatterjee A, Xiao H, Creation of Bacterial Cells with 5-Hydroxytryptophan as a 21st Amino Acid Building Block, Chem. 6 (2020) 2717–2727. 10.1016/j.chempr.2020.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chatterjee A, Sun SB, Furman JL, Xiao H, Schultz PG, A Versatile Platform for Single-and Multiple-Unnatural Amino Acid Mutagenesis in Escherichia coli, Biochemistry. 52 (2013) 1828–1837. 10.1021/bi4000244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Young TS, Ahmad I, Yin JA, Schultz PG, An Enhanced System for Unnatural Amino Acid Mutagenesis in E. coli, J. Mol. Biol. 395 (2010) 361–374. 10.1016/j.jmb.2009.10.030. [DOI] [PubMed] [Google Scholar]
- [16].Wang HH, Isaacs FJ, Carr PA, Sun ZZ, Xu G, Forest CR, Church GM, Programming cells by multiplex genome engineering and accelerated evolution, Nature. 460 (2009) 894–898. 10.1038/nature08187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Studier FW, Moffatt BA, Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes, J. Mol. Biol. 189 (1986) 113–130. 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- [18].Rosano GL, Ceccarelli EA, Neubauer P, Bruno-Barcena JM, Schweder T, Recombinant protein expression in Escherichia coli: advances and challenges, Front. Microbiol. 5 (2014) 1–17. 10.3389/fmicb.2014.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Rosano GL, Morales ES, Ceccarelli EA, New tools for recombinant protein production in Escherichia coli: A 5-year update, Protein Sci. 28 (2019) 1412–1422. 10.1002/pro.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Mosberg JA, Lajoie MJ, Church GM, Lambda Red Recombineering in Escherichia coli Occurs Through a Fully Single-Stranded Intermediate, Genetics. (2010). 10.1534/genetics.110.120782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Datsenko KA, Wanner BL, One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products, Proc. Natl. Acad. Sci. U. S. A. 97 (2000) 6640–6645. 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].E.V.& Antoine HJJN Stuitje R, Cornelis E. Spelt, Identification of mutations affecting replication control of plasmid Clo DF13, Nature. 290 (1981) 264–267. [DOI] [PubMed] [Google Scholar]
- [23].Weinberg DE, Nakanishi K, Patel DJ, Bartel DP, The inside-out mechanism of dicers from budding yeasts, Cell. 146 (2011) 262–276. 10.1016/j.cell.2011.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wang XC, Wilson SC, Hammond MC, Next-generation RNA-based fluorescent biosensors enable anaerobic detection of cyclic di-GMP, Nucleic Acids Res. 44 (2016). 10.1093/nar/gkw580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Inouye S, Inouye M, Up-promoter mutations in the Ipp gene of Escherichia coli, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hughes RA, Ellington AD, Rational design of an orthogonal tryptophanyl nonsense suppressor tRNA, Nucleic Acids Res. 38 (2010) 6813–6830. 10.1093/nar/gkq521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ross W, Salomon J, Holmes WM, Gourse RL, Activation of Escherichia coli leuV Transcription by FIS, J. Bacteriol. 181 (1999) 3864–3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hallam TJ, Wold E, Wahl A, Smider VV, Antibody conjugates with unnatural amino acids, Mol. Pharm. 12 (2015) 1848–1862. 10.1021/acs.molpharmaceut.5b00082. [DOI] [PubMed] [Google Scholar]
- [29].Kim CH, Axup JY, Schultz PG, Protein conjugation with genetically encoded unnatural amino acids, Curr. Opin. Chem. Biol. 17 (2013) 412–419. 10.1016/j.cbpa.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sun SB, Schultz PG, Kim CH, Therapeutic applications of an expanded genetic code, ChemBioChem. 15 (2014) 1721–1729. 10.1002/cbic.201402154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].M.L. and Johnson ME An Efficient Synthesis of 5-Azidotryptophan, Tetrahedron Lett. 35 (1994) 6255–6258. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.