

Graphical abstract
Keywords: Saccharomyces cerevisiae, Yhr202w, Smn1, AnalogYeast, metabolomics
Highlights
-
•
A quarter of the Saccharomyces cerevisiae genome is not functionally annotated.
-
•
We created a database of sensitive sequence similarity predictions for yeast proteins.
-
•
We use our database to identify candidate enzymes for non-annotated reactions.
-
•
We demonstrate a new function for the previously uncharacterized enzyme Yhr202w.
-
•
Our methodology allows uncovering new functions of uncharacterized proteins.
Abstract
Despite decades of research and the availability of the full genomic sequence of the baker’s yeast Saccharomyces cerevisiae, still a large fraction of its genome is not functionally annotated. This hinders our ability to fully understand cellular activity and suggests that many additional processes await discovery. The recent years have shown an explosion of high-quality genomic and structural data from multiple organisms, ranging from bacteria to mammals. New computational methods now allow us to integrate these data and extract meaningful insights into the functional identity of uncharacterized proteins in yeast. Here, we created a database of sensitive sequence similarity predictions for all yeast proteins. We use this information to identify candidate enzymes for known biochemical reactions whose enzymes are unidentified, and show how this provides a powerful basis for experimental validation. Using one pathway as a test case we pair a new function for the previously uncharacterized enzyme Yhr202w, as an extra-cellular AMP hydrolase in the NAD degradation pathway. Yhr202w, which we now term Smn1 for Scavenger MonoNucleotidase 1, is a highly conserved protein that is similar to the human protein E5NT/CD73, which is associated with multiple cancers. Hence, our new methodology provides a paradigm, that can be adopted to other organisms, for uncovering new enzymatic functions of uncharacterized proteins.
Introduction
One of the biggest revolutions in cell biology of eukaryotes came with the complete sequencing of the first eukaryotic genome, that of the yeast Saccharomyces cerevisiae.1 Computational analysis of the linear sequence uncovered a genome holding under 6000 genes that are mostly devoid of introns,2, 3 and lacking alternative splicing. This gives rise to a simple eukaryotic cell that can function with under 6000 protein products. Despite the simplicity of the yeast genome, a large portion of yeast proteins have not been functionally characterized. This hinders not only our understanding of cell biology but also of any biological process that relies on cells such as development and disease states.
One class of proteins that was intensively studied before the genome sequencing era is enzymes. Enzymes are molecular machines that can reduce the activation energy for metabolic reactions to occur4 and hence to understand cellular metabolism it is essential to map enzyme substrates and products. The specific enzymatic step that enzymes perform was historically discovered either through biochemical purification step based on activity or through genetic screens coupled with biochemical follow-ups.5, 6, 7, 8, 9, 10 These approaches required a clear growth phenotype or testable biochemical activity. While this made it feasible to uncover the major players and pathways, it was not an optimal approach to identify and characterize enzymes that provide non-essential metabolites, that have isoenzymes, or other parallel pathways. The complete genome sequence of yeast provided an ability to identify the presence of new proteins and as such new enzymes, however many remained without a functional annotation.
In recent years an attempt to uncover new enzymatic functions was undertaken by utilizing untargeted metabolomics approaches. Metabolomic profiling provides a map of all metabolic changes that occur in the absence of specific genes.11, 12, 13, 14 Despite the ability of such methods to uncover enzymatic functions in some cases,15, 14 as a general rule, the immense metabolic rewiring that occurs in each strain has made it difficult to extrapolate exact gene functions from such data without a guiding hypothesis.
One way by which hypotheses could be formed for gene functions is by relying on the rich information that has been accumulating in multiple organisms at the biochemical, genomic and structural levels. To date, these data have not yet been fully tapped to create hypotheses as to protein functions and hence support the discovery of new enzyme functions.
Here, we derived sensitive sequence similarity predictions (SSSP) for all yeast proteins using the HHSearch platform.16 These predictions, now easily accessible through our website (https://www.weizmann.ac.il/molgen/AnalogYeast/) uncover new mammalian proteins that are similar to yeast proteins not previously found by simple sequence comparisons alone. Together with the simple sequence-based predictions, this suggests that a larger fraction of the yeast proteome is conserved to humans than previously appreciated.
Utilizing the knowledge that accumulated in other organisms, from bacteria through plants, invertebrates and vertebrates, we derive functional predictions for yeast proteins. We focused on the fraction of the proteome that has unknown function and specifically, on the likely enzymes in this group. By crossing the list of uncharacterized enzymes with a map of missing ones in known pathways, we provide a new methodology for predicting enzymatic functions of uncharacterized proteins. Using a test case of the protein product of YHR202W, we show how such predictions can be rapidly validated using metabolomic pipelines. These allow us to support the role of Yhr202w (which we now name, Smn1 for Scavenger MonoNucleotidase 1) as a newly-identified AMP hydrolase.
Results
Sensitive sequence similarity predictions expand the known degree of similarity between yeast and human proteomes
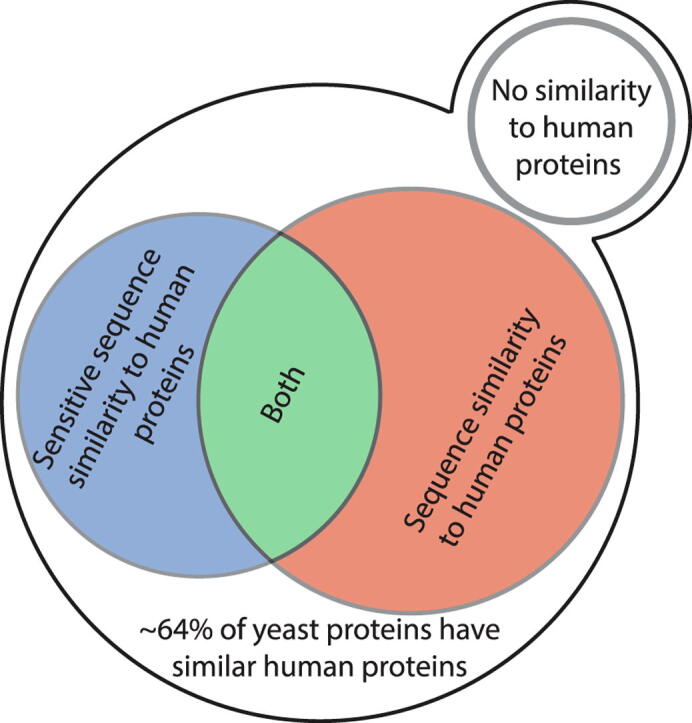
Ever since the establishment of the baker’s yeast as a leading model for eukaryotic cells, there has been an effort to characterize the exact extent of similarity between this simple unicellular organism and human cells. Previous efforts started even before the complete compilation of the yeast or human genome sequences17 and suggested that as little as 25% of genes in yeast will have human homologs. These efforts were simplified as whole genome sequences became available18 leading to a higher degree of predicted homology (31%). A compendium of many sequence-based prediction algorithms gathered by the “Quest for orthologs” database19 presented the most comprehensive comparison to date and suggested that ∼55% of yeast gene products have homologs in humans based on sequence (Figure 1, Supplementary Table 1).
Figure 1.

Sensitive sequence similarity predictions increase the percent of yeast proteins for which similar human proteins can be found. A Venn diagram showing similarity levels between S. cerevisiae and human proteomes. Shown are the percentage of yeast proteins with similar human proteins uncovered by sensitive sequence similarity predictions (HHSearch), homologs uncovered by sequence alone (Quest for Orthologs), or the combination of both.
However, 1 billion years of evolution have created proteins that are similar in function and in structure but have little similarity at the sequence level. Such proteins require identification using more sensitive approaches. Here, we took advantage of such a tool, HHSearch16 that was optimized to predict similarity based on amino acid sequence and secondary structure predicted from the amino acid sequence. Importantly, while most of these similarities result from divergent evolution, others might be the result of convergent evolution hence we use the term similarity and not homology.
We extracted such SSSPs using HHSearch on all yeast proteins and found that over 40% of yeast proteins had predicted similar proteins in the human proteome (Figure 1) (Supplementary Table 2). Since the available databases for this method are based on the limited available protein structures and protein domains, it is not surprising that the number of similar proteins that we found based on this strategy is less than those found based on the sequence comparison alone. Importantly, the SSSP did not simply constitute a subset of the 55% of homologs based on simple sequence comparisons. In fact, we found similar proteins for an additional 10% of the yeast proteome bringing the overall predicted similarity to humans to be about two thirds of the yeast proteome (Figure 1). This stresses, again, the validity of using yeast as a representative model for understanding conserved cellular functions.
A large fraction of the yeast proteome remains uncharacterized
To truly understand cellular function, it is essential to know the activity of the constituting proteins. However, many yeast proteins have remained uncharacterized despite decades of efforts. To uncover the exact fraction of proteins for which no defined molecular function has been identified, we took two complementary approaches. First, we used text mining in the relevant fields to identify entries in the Saccharomyces Genome Database (SGD)20 that suggest a lack of known function (Figure 2(A)). In parallel, we manually curated all SGD descriptions of protein functions and generated a list of those that did not have sufficient support for a molecular function (Figure 2(A), Supplementary Table 3). Each approach in as of itself showed that ∼25% of yeast genes are yet to be characterized, and the combination of these approaches had a similar magnitude (Figure 2(A)). Hence, we decided to continue working with the list that we manually curated.
Figure 2.

A large fraction of uncharacterized yeast proteins have similar proteins in other organisms. (A) A bar plot of the number of yeast proteins that are still functionally uncharacterized. Shown are the numbers based on either text mining of the SGD database, or by manual curation as well as the union of both. (B) Pie chart showing the percent of functionally characterized vs uncharacterized yeast proteins out of those that have at least one paralog (center). Out of each group shown is the percentage that have at least one paralog that is also uncharacterized. (C) Bar plots presenting the percent of yeast proteins that have similar proteins in other model organisms – divided by either all proteins or only the uncharacterized ones. Left – separation to discrete model species. Right – the contribution of each model species to the overall similarity level.
How have so many proteins evaded functional discovery despite decades of research? One hypothesis is that these proteins may be part of paralogous pairs or protein families. Having such similar proteins in the proteome may hinder functional annotation since backup by a paralog may reduce the phenotypes of losing a gene making the likelihood of capturing it by genetic methodologies smaller. To assay this hypothesis, we took a list of all yeast paralogous proteins21 and divided them into characterized and uncharacterized ones (from the curated list). We then asked what is their probability to have a paralog that is itself uncharacterized. We found that indeed proteins that are uncharacterized have a much bigger probability of having a paralog that is also not functionally characterized, supporting the back-up hypothesis (Figure 2(B)).
Another reason why these proteins may have been understudied is because they are yeast specific. To explore this hypothesis, we expanded our search for similar proteins to create SSSP of yeast proteins to all organisms as well as defined domains (Supplementary Table 2). To make these similarity predictions accessible to the yeast community, we organized them into an easily searchable database (https://www.weizmann.ac.il/molgen/AnalogYeast/). In this database (Using the term Analogy that includes both divergent and convergent evolution,22) for each yeast protein we present all predicted proteins based on SSSP as well as their description from UniProt,23 Pfam24 and links to the respective databases. In addition, for relevant cases we added information regarding their involvement in human diseases25 and/or their enzymatic activity.26
Using these predictions, we found that the uncharacterized proteins are half as likely, relative to the whole proteome, to have similar proteins identified by SSSP in other model organisms (Figure 2(C)). This suggests that indeed conservation has both promoted research as well as, maybe, incentivized it. However, nearly 800 yeast proteins with unknown functions do have similar proteins in some species or have conserved domains. For these, advanced approaches can, and should, be used to gain a better understanding of function (Figure 2(C)). Importantly, 18% are conserved in humans. Hence, uncovering their function becomes critical not only for a better understanding of yeast cells (for biotechnological applications or as drug targets for antifungals) but also as a basis for better understanding conserved cellular functions.
Many uncharacterized proteins show similarity to enzymes in other organisms
Proteins can be assigned into functional categories such as structural proteins that define the building blocks of cellular architecture, regulatory and chaperoning proteins that function through binding of other biomolecules (proteins, DNA or RNA), and enzymes. The last century saw a huge burst of enzyme discovery through the fields of genetics and biochemistry, enabling the mapping of enzymes carrying out the majority of central metabolism pathways.27, 28, 29 However, in recent years it is becoming clear that many additional enzymatic activities, that contribute to peripheral metabolism, signaling and stress responses, exist. Since such reactions are not essential, at least not under standard growth conditions, the identification of the enzymes carrying them out has been lagging behind.
To test whether some of the uncharacterized proteins that we defined have the potential to be enzymes we used the predictions from the SSSP algorithms to look for those proteins that show similarity to proteins with annotated enzymatic functions in any other organism. Importantly, since our analysis defines proteins as similar even if only a small part of the sequence is similar, sometimes not even in the active site of the enzyme, this type of analysis obviously gives rise to potential false positives and negatives. Taking that into account, our analysis uncovered that ∼20% of the uncharacterized proteins may be enzymes (Figure 3). This is a slightly smaller number than the fraction of enzymes in characterized proteins (∼28%) (Figure 3). This suggests that either proteins that are enzymes have been, historically, more likely to be identified, or rather that some enzymes are part of protein families that have not been structurally characterized in any organism.
Figure 3.

A substantial fraction of uncharacterized proteins have similarity to enzymes in other species. Pie charts displaying the percent of proteins (either the functionally characterized yeast proteins or the uncharacterized ones) with predicted similarity to enzymes. From the Uncharacterized proteins an additional pie chart demonstrates the magnitude of novel detections enabled by the SSSP searches.
Of the potential enzymes in our uncharacterized group, ∼20% were already annotated in SGD as potential enzymes based on manual curation (Figure 3). Regardless, our analysis reveals a rough estimate of over 200 proteins that may be yeast enzymes not previously characterized (Supplementary Table 4). Moreover, these proteins show similarity to defined enzymes in other organisms, some of which have been studied and whose substrates or products are known. This suggests that we could directly use these similarities to predict the enzymatic functions of uncharacterized yeast proteins.
Metabolic gaps in known pathways can be filled by predicting enzyme functions based on similarity
Over the years there has been an effort to map metabolic pathways and the enzymes carrying out each step in multiple organisms.30, 28 In yeast, this has led to the curation of over 572 reactions in 144 pathways consisting of 1417 enzymes. However, about 10% of the reactions (52) (Supplementary Table 5) still remain unaccounted for – meaning that it is clear that the step occurs but the enzyme carrying it out has not been identified or proven. These gaps of metabolic knowledge have been termed “pathway holes” and have awaited exploration.
We focused on these 52 pathway holes and probed whether any of our uncharacterized proteins for which we could find analogous enzymes, may account for the exact (or similar) reaction that occurs (Supplementary Table 5, Figure 4). We used Enzymatic Commission (EC) numbers, which define the exact enzyme function by a four-position hierarchical decision tree, to find proteins that can fit into the pathway “holes”. Generally, the first position of an EC number defines the general type of reaction, the second position defines a more specific reaction type, the third position defines the active enzymatic subclass and the fourth position defines the substrate.31 We chose to focus on enzymes that are either identical in their function and substrate to the defined hole (the EC number is identical to the “hole” in all four positions) or that are largely similar in their enzymatic functions (the EC number is identical up to the third position).
Figure 4.

Matching predicted enzymes with metabolic pathway holes. A plot depicting all pathways in yeast that have an enzymatic step for which an enzyme has not yet been identified (hole). Plotted are the number of proteins with matching enzymatic-annotation (EC number) uncovered by SSSP. Green represents a full (4- positions) match. Blue represents a partial (3- positions) match. Highlighted in orange is the pathway on which we focus (Figure 5).
We found that 15 out of the pathway holes have at least one candidate that fits in all four positions and 42 out of the holes have candidates that fit three-positions (Figure 4). Despite the fact that a four-position match should be the identical enzymatic reaction, some holes had multiple four-position candidates. This suggests that either this step has a large enzymatic redundancy or that additional pathways with more refined substrates or distinct cellular locals should be characterized in the future.
Assigning an enzyme for a pathway hole in the periplasmic NAD degradation pathway
To test case our predictions we chose to focus on an important activity that has evaded discovery using previous approaches. The pathway is a nucleoside salvage pathway converting NAD + to AMP and adenosine (Figure 5(A)). Interestingly, the enzyme carrying out the second step, converting AMP to adenosine, was suggested to reside in the periplasm32 and its activity is described by the EC number 3.1.3.5, yet was never identified in yeast (Figure 5(A)). Our data suggested that two candidates are a fit for this “pathway hole” (highlighted in Figure 4) - the uncharacterized proteins Yhr202w and Ydl024c. Incidentally, both proteins have a predicted signal peptide and are soluble,33, 34 (for a schematic of Yhr202w see Figure 5(B)), suggesting that they could be periplasmic. However, ydl024c was already annotated to an additional EC number (3.1.3.2) and was only matched to EC 3.1.3.5 based on similarity to a single human protein. In comparison, Yhr202w was not annotated to any other EC number and shows similarity to multiple enzymes of this function in several organisms: multiple bacterial species, Chinese cobras and humans. This made Yhr202w a more likely candidate. Moreover, Δyhr202w was shown to enable resistance to sodium selenite whose uptake is linked to that of phosphate, supporting a role for Yhr202w as a phosphatase as well as its extra-cellular localization.35, 36 We therefore first assayed whether Yhr202w is indeed a secreted enzyme by tagging the protein on its C’ with a Green Fluorescence Protein (GFP) and replacing its promotor with an inducible promotor (GALpr) to allow a strong inducible expression. To catch the intracellular phase by microscopy, before it is all secreted and cannot be imaged, we performed time-lapse microscopy (Supplementary Video) and could indeed capture a short phase where punctate structures, that could be secretory vesicles, appear (Figure 5(C)). At longer induction times very little intracellular signal was seen as would be expected from a secreted enzyme (Supplementary Video). To follow potential secretion, we performed a protein secretion assay whereby protein levels are tracked both intra-cellularly as well as in the medium (Figure 5(D)). As a control, we followed the levels of cytosolically expressed mCherry which should not be secreted and can serve to identify events of leakage of intracellular proteins to the medium. Indeed, Yhr202w-GFP could be found in both the media and the cellular fraction whereas mCherry could mostly be found in the cellular fraction supporting the notion that Yhr202w is a secreted enzyme.
Figure 5.

Assigning an enzyme for a pathway hole in the periplasmic NAD degradation pathway. (A) The periplasmic NAD degradation pathway, consisting of two reactions, one with an EC number 3.6.1.22 giving rise to AMP from NAD + and one, for which no enzyme has yet been described, giving rise to adenosine from AMP. (B) Yhr202w is a soluble protein containing a 21 amino acid signal sequence according to TOPCONS topology prediction. (C) Fluorescent microscopy image taken out of a time-lapse experiment showing the localization of Yhr202w-GFP under control of a GALpr. Images are shown 3.5 h after activation of the GAL inducible promoter by transfer to growth in galactose containing medium. Scale bar = 5 µm. (D) Yhr202w secretion analyzed by western blot. Yhr202w can be found in both the secreted and in the cellular fraction, while a soluble mCherry expressed under a constitutive promoter can be found mainly in the cellular fraction. (E) A bar plot showing the fold change of metabolites uncovered by metabolomics on strains in which Yhr202w/Smn1 was either deleted or overexpressed. The results depict fold change relative to genetically matched controls, with error-bars indicating the standard error of means. Fold-changes are marked with stars if they have a P-value < 0.005 in a T-test with a Benjamini-Hochberg correction.
Since Yhr202w is indeed secreted and has the correct enzymatic domains, we decided to directly assay its effect on the pathway substrates and products utilizing a metabolomics approach. We analyzed deletion and overexpression strains of Yhr202w and compared them to control strains by untargeted metabolomics on whole cell lysates using instrumentation that will allow us to focus on bases and other small polar metabolites (For a full list of metabolite changes see Supplementary Table 6). Using this we could confirm that indeed overexpression of Yhr202w causes an accumulation of adenosine in cells coupled with a reduction in AMP, ADP and ATP (Figure 5(E)). Conversely, the deletion mutant causes a reduction in adenosine with only a very small increase in ADP and ATP which are most likely highly regulated in their cellular levels (Figure 5(E)). As expected, overexpression of Yhr202w resulted in reduced NAD + levels explained by the increased flux through the pathway (and suggesting that the limiting step of this pathway was Yhr202w) while the deletion resulted in an order of magnitude increase in NAD + and NADH. This striking effect can be explained by the presence of a feedback loop completely blocking NAD + degradation in the periplasm when Yhr202w is absent. We therefore decided to name Yhr202w Smn1 (Scavenger MonoNucleotidase 1).
Discussion
One of the big frontiers in modern cell biology is to map the “dark matter” of life hidden in the large percent of cellular proteins whose function is unknown. Our work shows that at least one quarter of proteins in the most highly studied eukaryotic model cell, yeast, are still uncharacterized. This highlights how fragmented our picture of cellular activity still is.
One of the places where this is clearest is yeast metabolism. While decades of biochemical studies have highlighted the central pathways and focused on core, essential metabolites, about 10% of these core pathways still have holes and more research is required to finalize their annotation. Moreover, it is now clear that yeast metabolism is more complex than previously thought. Our work suggests that ∼20% of uncharacterized proteins are enzymes, highlighting at least 200 different reactions and pathways that could occur inside a yeast cell that have not yet been annotated. This may be especially relevant for metabolites that comprise single reactions or are stress induced.
Importantly, now is the time to uncover these new metabolic reactions and the enzymes mediating them. First, large metabolomic datasets showing changes in hundreds of metabolites for deletions of every cellular gene have already been published.37 However, by themselves these datasets can only provide limited answers to what specific enzymes are doing. This is because each perturbation causes a rewiring of cellular metabolism, resulting in myriads of changes in known, and uncharacterized, metabolites. Hence reaching a clear hypothesis as to the primary function of a single protein from the metabolic signature of its mutant has been challenging.
On the other hand, similarity predictions are now also emerging as a powerful tool due to the availability of multiple sequenced genomes, accumulation of structural data and powerful algorithms for comparing sequences and structures. However, such predictions are also, in as of themselves, often not enough. Our analysis shows this by examples of how multiple enzymes could potentially fulfill the requirement for a single specific pathway hole. Conversely, other holes have no candidate enzymes. Moreover, it is now clear that multiple enzymes have paralogs or isoenzymes that may perform an identical function but in a different compartment, slowing down correct assignment.
Luckily the last few years have brought about extensive mapping of protein localizations in yeast.38, 39, 40 Hence, the true power is now to use emerging computational approaches for integrating the large amounts of available genomic data coupled with the availability of large scale metabolomic data41 and proteomic information to integrate them into predictive models.42, 43, 44, 45 Indeed, many such efforts have been undertaken (from bacteria, through invertebrates to humans) to utilize the abundant genomic and proteomic data to uncover a function for uncharacterized proteins relying on computational approaches.46, 47, 48
However, any such predictions must still be tested to reach a more certain assignment of enzyme function. Our work shows that a powerful way to test these predictions is by measuring both deletion as well as over-expression versions of the enzymes. Using both mutants is important since deletions alone may not show a strong phenotype either due to rewiring of metabolism or because of buffering by enzyme redundancy. For example, Yhr202w has three similar proteins and there exist multiple other pathways to eliminate NAD + or form AMP. Indeed, overexpression often gives a much stronger signature. However, the most powerful approach is to find metabolites that change in inverse directions in the two, inverse, mutants. While many metabolites change for each genetic manipulation, very few will display this unique characteristic signature, narrowing down the search range.
Generally, and regardless of the approach used, identifying a function for a yeast protein holds many advantages, even if its similar proteins were already studied in other organisms. First, uncovering functions for uncharacterized proteins helps to “catalog” the yeast proteome – essential for reaching at true understanding of this simple cell. In addition, yeast can serve as an evolutionary bridge between the various branches of organisms. This is especially useful when trying to uncover similarity between bacteria and mammals that have 3 billion years of evolution between them. In this case yeast can serve as a “springboard” – being only ∼1 billion years distant from mammals. Finally, yeast can serve as an excellent model for functional studies as well as drug screening. For example, we identified Smn1 as a protein similar to human E5NT/CD73. While the human E5NT/CD73 was already shown to degrade AMP to adenosine,49, 50 it was difficult to use for uncovering drug targets. Since it was shown to have a role in cancer progression and cyclic AMP (cAMP) signaling,51, 52, 50, 53 uncovering such drugs may be beneficial. Interestingly we can also see changes in cAMP levels in the overexpression strains of Smn1 suggesting that yeast can serve as a model for E5NT/CD73 cellular activities. Now, having the yeast protein may provide a powerful platform for screening of novel inhibitors of this periplasmic protein – circumventing the need for drugs to enter cells.
More globally what is most surprising from our analysis is that yeast is much more similar to mammalian cells than previously thought. While previous assessments on similarity suggested that around half of the yeast proteome is conserved to mammals, the contribution of advanced computational approaches now suggests that it may be up to ∼64% that are, in fact, conserved. This stratifies the belief that yeast is a superb model for human cellular activity and supports the need to continue and uncover the function of all of its proteins.
Materials and Methods
Computational analysis
Quest
The Quest for Orthologs database19 was downloaded on January 18th 2020, containing all homology hits between yeast and other organisms based on a combination of different homology algorithms and their benchmark criteria.
HHsearch
Protein sequences for all saccharomyces cerevisiae genes were obtained from SGD20 and rearranged to individual FASTA file formats using a homemade script. The individual FASTA files were submitted to a standalone HHSearch (from hhsuite3)16 and searched against Pdb70,54 PfamA V34,24 scop70-1.75 and scop40.55, 56 All proteins with the word “dubious” in their description were discarded, as well as hits with similarity score below 95 (out of 100). The result files were combined to a single .csv file using a homemade script. If the match was through a PDB structure, the host organism was added from the PDB description. Further information for each protein was added from UniProt,23 including the indicated EC numbers.31 Additionally, the involvement of each human protein with specific diseases was added based on a MalaCards search25 conducted on GeneCards57 version V4.13 on February 26th, 2020. Further analyses were performed on this assembled database, from here on termed, AnalogYeast, using homemade scripts (https://github.com/Maya-Schuldiner-lab/AnalogYeast). Raw prediction results can also be found on our lab webpage: https://mayaschuldiner.wixsite.com/schuldinerlab/analogyeast
Curation of genes whose proteins have unknown functions
Using the SGD database,20 we assembled three lists containing proteins of unknown function. The first is a text mining list, containing all protein entries that included the keywords “uncharacterized”, “unknown”, “predicted”, “associates with”, “possibly”, “presumed” and “putative”, under the categories “Brief Summary” and “Function Summary”. The second list was manually curated based on SGD descriptions. The third list is a union of the two preceding lists.
Metabolic pathway holes
The list of pathway holes was acquired from the SGD YeastPathways database28 on June 11 2021.
Yeast strains, strain construction and culturing conditions
All strains in this study are based on the BY4741 laboratory strain.58 All information on strains, plasmids and primers can be found in Supplementary Table 7.
For metabolomics analysis all yeast strains were grown in standard synthetic dextrose (SD) medium (6.7 g/L yeast nitrogen base with ammonium sulfate, 2% glucose, and all necessary amino acids) from OD600 of ∼0.1 overnight in a 30 °C incubator. Then the cultures were diluted back to OD600 of ∼0.1 and incubated again overnight. Next the cultures were back diluted a last time to OD600 of ∼0.1 and left to grow until OD600 of ∼0.5 in 40 ml culture breathable tubes (LIFEGENE).
Secretion assay
Yeast strains were grown in standard synthetic dextrose (SD) medium (6.7 g/L yeast nitrogen base with ammonium sulfate, 2% galactose) from OD600 of ∼0.1 overnight at 30 °C. Next, the cultures were back diluted to OD600 of ∼0.1 and let to grow until OD600 of ∼0.5. The media fraction was separated from the cells using centrifugation (3000g for 3 min) and cells were washed with DDW. Both media and cell fractions were precipitated using 10% Trichloroacetic acid (TCA) (Sigma) for 20 min on ice, centrifuged for 15 min at 14,000g at 4 °C, the supernatant was aspirated, pellet was washed in cold acetone, dried at room temperature for 30 min and resuspended in urea lysis buffer (8 M urea in 50 mM tris pH 7.5 and oComplete Protease Inhibitor (Roche)). The cells were beaten with 100 µl of glass beads (scientific industries) for 10 min at 4 °C. Then 0.1% SDS and 50 mM DTT were added to both the cells and the media fractions and boiled at 95 °C for 5 min. Glass beads and cell debris were removed and the samples were resolved on 4–20% precast polyacrylamide gel (Bio-Rad), transferred to nitrocellulose membrane (PALL), and probed with a monoclonal mouse α-cherry (ab125096, Abcam) and a polyclonal rabbit α-GFP (ab290, Abcam). Secondary antibodies were alexa680 α-rabbit (ab175773, Abcam) and alexa790 α-mouse (ab186695, Abcam) that enable scanning using an Odyssey imaging system (LI-COR Biosciences).
Microscopy of yeast strains
Imaging of yeast strains was performed using a VisiScope Confocal Cell Explorer system (VisView), composed of a Yokogawa spinning disk scanning unit (CSU-W1) coupled with an inverted microscope (IX83; ×60 oil objective; Olympus) at an excitation wavelength of 488 nm for GFP. Images were taken by a connected PCO-Edge sCMOS camera (PCO) controlled by VisView software.
Metabolomics
Sample preparation
Each culture was filtered using a filtration apparatus onto a 25 mm nylon membrane (GVS), washed once with 5 ml of DDW and filtered again. The filter was transferred to 5 ml cold 50% acetonitrile in DDW, vortexed and snapped frozen in liquid nitrogen.
Sample mass spec analysis
Sample analysis was carried out by MS-Omics using a Thermo Scientific Vanquish LC coupled to Thermo Q Exactive HF MS. An electrospray ionization interface was used as ionization source. Analysis was performed in negative and positive ionization mode. The Ultra-performance liquid chromatography was performed using a slightly modified version of a previously described protocol.59 Peak areas were extracted using Compound Discoverer 3.1 (Thermo Scientific). Identification of compounds was performed at four levels; Level 1: identification by retention times (compared against in-house authentic standards), accurate mass (with an accepted deviation of 3 ppm), and MS/MS spectra, Level 2a: identification by retention times (compared against in-house authentic standards), accurate mass (with an accepted deviation of 3 ppm). Level 2b: identification by accurate mass (with an accepted deviation of 3 ppm), and MS/MS spectra, Level 3: identification by accurate mass alone (with an accepted deviation of 3 ppm).
CRediT authorship contribution statement
Nir Cohen: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Supervision, Validation, Visualization, Writing – original draft. Amit Kahana: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Visualization, Writing – original draft. Maya Schuldiner: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing – original draft.
Acknowledgments
Acknowledgements
We would like to thank Dr. Simon Fishilevich for providing the MalaCards source data. We thank Maayan Maron and Anastasia Zarankin from the Weizmann Institute of Science IT division for creating the AnalogYeast web site. We thank Dr. Adi Millman from the laboratory of Prof. Rotem Sorek for fruitful discussions. We thank Dr. Emma Fenech for sharing the list of all yeast paralogs. We thank Hadar Meyer for technical assistance. We would like to acknowledge the professional work by MS-Omics in metabolomics data acquisition. We thank Dr. Marton Megyeri, Dr. Eden Yifrach and Dr. Einat Zalckvar for critical reading of our manuscript.
Funding
This work in this manuscript is part of a project that has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (EU-H2020-ERC-CoG; grant name OnTarget, grant number 864068) and from a grant of the Israeli Science Foundation (ISF) (grant number 760/17). MS is an incumbent of the Dr. Gilbert Omenn and Martha Darling Professorial Chair in Molecular Genetics.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Edited by Michael Sternberg
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jmb.2022.167478.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Goffeau A., Barrell B.G., Bussey H., Davis R.W., Dujon B., Feldmann H., Galibert F., Hoheisel J.D., et al. Life with 6000 genes. Science. 1996;274:546. doi: 10.1126/science.274.5287.546. [DOI] [PubMed] [Google Scholar]
- 2.Dujon B. Yeasts illustrate the molecular mechanisms of eukaryotic genome evolution. Trends Genet. 2006;22:375–387. doi: 10.1016/j.tig.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 3.Ingolia N.T., Ghaemmaghami S., Newman J.R.S., Weissman J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Förster J., Famili I., Fu P., Palsson B.Ø., Nielsen J. Genome-scale reconstruction of the Saccharomyces cerevisiae metabolic network. Genome Res. 2003;13:244–253. doi: 10.1101/gr.234503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duntze W., Neumann D., Gancedo J.M., Atzpodien W., Holzer H. Studies on the regulation and localization of the glyoxylate cycle enzymes in Saccharomyces cerevisiae. Eur. J. Biochem. 1969;10:83–89. doi: 10.1111/j.1432-1033.1969.tb00658.x. [DOI] [PubMed] [Google Scholar]
- 6.Knobling A., Schiffmann D., Sickinger H.D., Schweizer E. Malonyl and palmityl transferase-less mutants of the yeast fatty-acid-synthetase complex. Eur. J. Biochem. 1975;56:359–367. doi: 10.1111/j.1432-1033.1975.tb02241.x. [DOI] [PubMed] [Google Scholar]
- 7.Korch C.T., Snow R. Allelic complementation in the first gene for histidine biosynthesis in SACCHAROMYCES CEREVISIAE. I. characteristics of mutants and genetic mapping of alleles. Genetics. 1973;74:287–305. doi: 10.1093/genetics/74.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lynen, F. (1969). [3] Yeast fatty acid synthase. In Lipids, (Elsevier), pp. 17–33.
- 9.Masselot M., de Robichon-Szulmajster H. Methionine biosynthesis in Saccharomyces cerevisiae. Molec. Gen. Genet. 1975;139:121–132. doi: 10.1007/BF00264692. [DOI] [PubMed] [Google Scholar]
- 10.Schweizer M., Roberts L.M., Höltke H.J., Takabayashi K., Höllerer E., Hoffmann B., Müller G., Köttig H., et al. The pentafunctional FAS1 gene of yeast: its nucleotide sequence and order of the catalytic domains. Mol. Gen. Genet. 1986;203:479–486. doi: 10.1007/BF00422073. [DOI] [PubMed] [Google Scholar]
- 11.Fuhrer T., Zampieri M., Sévin D.C., Sauer U., Zamboni N. Genomewide landscape of gene-metabolome associations in Escherichia coli. Mol. Syst. Biol. 2017;13:907. doi: 10.15252/msb.20167150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oliveira A.P., Ludwig C., Picotti P., Kogadeeva M., Aebersold R., Sauer U. Regulation of yeast central metabolism by enzyme phosphorylation. Mol. Syst. Biol. 2012;8:623. doi: 10.1038/msb.2012.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sauer U. Metabolic networks in motion: 13C-based flux analysis. Mol. Syst. Biol. 2006;2:62. doi: 10.1038/msb4100109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sévin D.C., Fuhrer T., Zamboni N., Sauer U. Nontargeted in vitro metabolomics for high-throughput identification of novel enzymes in Escherichia coli. Nature Methods. 2017;14:187–194. doi: 10.1038/nmeth.4103. [DOI] [PubMed] [Google Scholar]
- 15.Clasquin M.F., Melamud E., Singer A., Gooding J.R., Xu X., Dong A., Cui H., Campagna S.R., et al. Riboneogenesis in yeast. Cell. 2011;145:969–980. doi: 10.1016/j.cell.2011.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steinegger M., Meier M., Mirdita M., Vöhringer H., Haunsberger S.J., Söding J. HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinformatics. 2019;20:473. doi: 10.1186/s12859-019-3019-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Botstein D., Fink G.R. Yeast: an experimental organism for modern biology. Science. 1988;240:1439–1443. doi: 10.1126/science.3287619. [DOI] [PubMed] [Google Scholar]
- 18.Botstein D., Chervitz S.A., Cherry J.M. Yeast as a model organism. Science. 1997;277:1259–1260. doi: 10.1126/science.277.5330.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Altenhoff A.M., Garrayo-Ventas J., Cosentino S., Emms D., Glover N.M., Hernández-Plaza A., Nevers Y., Sundesha V., et al. The Quest for Orthologs benchmark service and consensus calls in 2020. Nucleic Acids Res. 2020;48:W538–W545. doi: 10.1093/nar/gkaa308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cherry J.M., Hong E.L., Amundsen C., Balakrishnan R., Binkley G., Chan E.T., Christie K.R., Costanzo M.C., et al. Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic Acids Res. 2012;40:D700–5. doi: 10.1093/nar/gkr1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fenech E.J., Ben-Dor S., Schuldiner M. Double the fun, double the trouble: paralogs and homologs functioning in the endoplasmic reticulum. Annu. Rev. Biochem. 2020;89:637–666. doi: 10.1146/annurev-biochem-011520-104831. [DOI] [PubMed] [Google Scholar]
- 22.Fitch W.M. Distinguishing Homologous from Analogous Proteins. Syst. Zool. 1975;19:99–113. [PubMed] [Google Scholar]
- 23.UniProt Consortium UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2021;49:D480–D489. doi: 10.1093/nar/gkaa1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mistry J., Chuguransky S., Williams L., Qureshi M., Salazar G.A., Sonnhammer E.L.L., Tosatto S.C.E., Paladin L., et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021;49:D412–D419. doi: 10.1093/nar/gkaa913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rappaport N., Twik M., Plaschkes I., Nudel R., Iny Stein T., Levitt J., Gershoni M., Morrey C.P., et al. MalaCards: an amalgamated human disease compendium with diverse clinical and genetic annotation and structured search. Nucleic Acids Res. 2017;45:D877–D887. doi: 10.1093/nar/gkw1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang A., Jeske L., Ulbrich S., Hofmann J., Koblitz J., Schomburg I., Neumann-Schaal M., Jahn D., et al. BRENDA, the ELIXIR core data resource in 2021: new developments and updates. Nucleic Acids Res. 2021;49:D498–D508. doi: 10.1093/nar/gkaa1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caspi R., Billington R., Ferrer L., Foerster H., Fulcher C.A., Keseler I.M., Kothari A., Krummenacker M., et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2016;44:D471–80. doi: 10.1093/nar/gkv1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karp P.D., Midford P.E., Billington R., Kothari A., Krummenacker M., Latendresse M., Ong W.K., Subhraveti P., et al. Pathway Tools version 23.0 update: software for pathway/genome informatics and systems biology. Brief. Bioinformatics. 2021;22:109–126. doi: 10.1093/bib/bbz104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu H., Li F., Sánchez B.J., Zhu Z., Li G., Domenzain I., Marcišauskas S., Anton P.M., et al. A consensus S. cerevisiae metabolic model Yeast8 and its ecosystem for comprehensively probing cellular metabolism. Nature Commun. 2019;10:3586. doi: 10.1038/s41467-019-11581-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karp P.D., Billington R., Caspi R., Fulcher C.A., Latendresse M., Kothari A., Keseler I.M., Krummenacker M., et al. The BioCyc collection of microbial genomes and metabolic pathways. Brief. Bioinformatics. 2019;20:1085–1093. doi: 10.1093/bib/bbx085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McDonald, A.G., Boyce, S., & Tipton, K.F. (2001). Enzyme classification and nomenclature. In eLS, John Wiley & Sons Ltd, ed. (Chichester, UK: John Wiley & Sons, Ltd), pp. 1–11.
- 32.Bogan K.L., Brenner C. 5′-Nucleotidases and their new roles in NAD+ and phosphate metabolism. New J. Chem. 2010;34:845. [Google Scholar]
- 33.Bernsel A., Viklund H., Hennerdal A., Elofsson A. TOPCONS: consensus prediction of membrane protein topology. Nucleic Acids Res. 2009;37:W465–8. doi: 10.1093/nar/gkp363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weill U., Cohen N., Fadel A., Ben-Dor S., Schuldiner M. Protein Topology Prediction Algorithms Systematically Investigated in the Yeast Saccharomyces cerevisiae. Bioessays. 2019;41 doi: 10.1002/bies.201800252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lazard M., Blanquet S., Fisicaro P., Labarraque G., Plateau P. Uptake of selenite by Saccharomyces cerevisiae involves the high and low affinity orthophosphate transporters. J. Biol. Chem. 2010;285:32029–32037. doi: 10.1074/jbc.M110.139865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pinson B., Merle M., Franconi J.-M., Daignan-Fornier B. Low affinity orthophosphate carriers regulate PHO gene expression independently of internal orthophosphate concentration in Saccharomyces cerevisiae. J. Biol. Chem. 2004;279:35273–35280. doi: 10.1074/jbc.M405398200. [DOI] [PubMed] [Google Scholar]
- 37.Mülleder M., Calvani E., Alam M.T., Wang R.K., Eckerstorfer F., Zelezniak A., Ralser M. Functional metabolomics describes the yeast biosynthetic regulome. Cell. 2016;167:553–565.e12. doi: 10.1016/j.cell.2016.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Breker M., Gymrek M., Moldavski O., Schuldiner M. LoQAtE–Localization and Quantitation ATlas of the yeast proteomE. A new tool for multiparametric dissection of single-protein behavior in response to biological perturbations in yeast. Nucleic Acids Res. 2014;42:D726–30. doi: 10.1093/nar/gkt933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weill U., Yofe I., Sass E., Stynen B., Davidi D., Natarajan J., Ben-Menachem R., Avihou Z., et al. Genome-wide SWAp-Tag yeast libraries for proteome exploration. Nature Methods. 2018;15:617–622. doi: 10.1038/s41592-018-0044-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yofe I., Weill U., Meurer M., Chuartzman S., Zalckvar E., Goldman O., Ben-Dor S., Schütze C., et al. One library to make them all: streamlining the creation of yeast libraries via a SWAp-Tag strategy. Nature Methods. 2016;13:371–378. doi: 10.1038/nmeth.3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ramirez-Gaona M., Marcu A., Pon A., Guo A.C., Sajed T., Wishart N.A., Karu N., Djoumbou Feunang Y., et al. YMDB 2.0: a significantly expanded version of the yeast metabolome database. Nucleic Acids Res. 2017;45:D440–D445. doi: 10.1093/nar/gkw1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Amantonico A., Oh J.Y., Sobek J., Heinemann M., Zenobi R. Mass spectrometric method for analyzing metabolites in yeast with single cell sensitivity. Angew. Chem. Int. Ed. Engl. 2008;47:5382–5385. doi: 10.1002/anie.200705923. [DOI] [PubMed] [Google Scholar]
- 43.Ibáñez A.J., Fagerer S.R., Schmidt A.M., Urban P.L., Jefimovs K., Geiger P., Dechant R., Heinemann M., et al. Mass spectrometry-based metabolomics of single yeast cells. Proc. Natl. Acad. Sci. USA. 2013;110:8790–8794. doi: 10.1073/pnas.1209302110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nobata C., Dobson P.D., Iqbal S.A., Mendes P., Tsujii J., Kell D.B., Ananiadou S. Mining metabolites: extracting the yeast metabolome from the literature. Metabolomics. 2011;7:94–101. doi: 10.1007/s11306-010-0251-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Urban P.L., Schmidt A.M., Fagerer S.R., Amantonico A., Ibañez A., Jefimovs K., Heinemann M., Zenobi R. Carbon-13 labelling strategy for studying the ATP metabolism in individual yeast cells by micro-arrays for mass spectrometry. Mol. Biosyst. 2011;7:2837–2840. doi: 10.1039/c1mb05248a. [DOI] [PubMed] [Google Scholar]
- 46.Kacsoh B.Z., Barton S., Jiang Y., Zhou N., Mooney S.D., Friedberg I., Radivojac P., Greene C.S., et al. New Drosophila Long-Term Memory Genes Revealed by Assessing Computational Function Prediction Methods. G3 (Bethesda) 2019;9:251–267. doi: 10.1534/g3.118.200867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garcia D.C., Cheng X., Land M.L., Standaert R.F., Morrell-Falvey J.L., Doktycz M.J. Computationally Guided Discovery and Experimental Validation of Indole-3-acetic Acid Synthesis Pathways. ACS Chem. Biol. 2019;14:2867–2875. doi: 10.1021/acschembio.9b00725. [DOI] [PubMed] [Google Scholar]
- 48.Zhang C., Wei X., Omenn G.S., Zhang Y. Structure and Protein Interaction-Based Gene Ontology Annotations Reveal Likely Functions of Uncharacterized Proteins on Human Chromosome 17. J. Proteome Res. 2018;17:4186–4196. doi: 10.1021/acs.jproteome.8b00453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jeffrey J.L., Lawson K.V., Powers J.P. Targeting metabolism of extracellular nucleotides via inhibition of ectonucleotidases CD73 and CD39. J. Med. Chem. 2020;63:13444–13465. doi: 10.1021/acs.jmedchem.0c01044. [DOI] [PubMed] [Google Scholar]
- 50.Narravula S., Lennon P.F., Mueller B.U., Colgan S.P. Regulation of endothelial CD73 by adenosine: paracrine pathway for enhanced endothelial barrier function. J. Immunol. 2000;165:5262–5268. doi: 10.4049/jimmunol.165.9.5262. [DOI] [PubMed] [Google Scholar]
- 51.Clayton A., Al-Taei S., Webber J., Mason M.D., Tabi Z. Cancer exosomes express CD39 and CD73, which suppress T cells through adenosine production. J. Immunol. 2011;187:676–683. doi: 10.4049/jimmunol.1003884. [DOI] [PubMed] [Google Scholar]
- 52.Gödecke A. cAMP: fuel for extracellular adenosine formation? Br. J. Pharmacol. 2008;153:1087–1089. doi: 10.1038/bjp.2008.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sciaraffia E., Riccomi A., Lindstedt R., Gesa V., Cirelli E., Patrizio M., De Magistris M.T., Vendetti S. Human monocytes respond to extracellular cAMP through A2A and A2B adenosine receptors. J. Leukoc. Biol. 2014;96:113–122. doi: 10.1189/jlb.3A0513-302RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Andreeva A., Howorth D., Chothia C., Kulesha E., Murzin A.G. SCOP2 prototype: a new approach to protein structure mining. Nucleic Acids Res. 2014;42:D310–4. doi: 10.1093/nar/gkt1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Andreeva A., Kulesha E., Gough J., Murzin A.G. The SCOP database in 2020: expanded classification of representative family and superfamily domains of known protein structures. Nucleic Acids Res. 2020;48:D376–D382. doi: 10.1093/nar/gkz1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stelzer G., Rosen N., Plaschkes I., Zimmerman S., Twik M., Fishilevich S., Stein T.I., Nudel R., et al. The genecards suite: from gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinformatics. 2016;54 doi: 10.1002/cpbi.5. 1.30.1–1.30.33. [DOI] [PubMed] [Google Scholar]
- 58.Brachmann C.B., Davies A., Cost G.J., Caputo E., Li J., Hieter P., Boeke J.D. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115–132. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 59.Hsiao J.J., Potter O.G., Chu T.-W., Yin H. Improved LC/MS Methods for the Analysis of Metal-Sensitive Analytes Using Medronic Acid as a Mobile Phase Additive. Anal. Chem. 2018;90:9457–9464. doi: 10.1021/acs.analchem.8b02100. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.