

Abstract
Objective
The aim of this study was to analyze the phenotypic spectrum, treatment, and prognosis of 72 Chinese children with SCN2A variants.
Methods
The SCN2A variants were detected by next-generation sequencing. All patients were followed up at a pediatric neurology clinic in our hospital or by telephone.
Results
In 72 patients with SCN2A variants, the seizure onset age ranged from the first day of life to 2 years and 6 months. The epilepsy phenotypes included febrile seizures (plus) (n = 2), benign (familial) infantile epilepsy (n = 9), benign familial neonatal-infantile epilepsy (n = 3), benign neonatal epilepsy (n = 1), West syndrome (n = 16), Ohtahara syndrome (n = 15), epilepsy of infancy with migrating focal seizures (n = 2), Dravet syndrome (n = 1), early infantile epileptic encephalopathy (n = 15), and unclassifiable developmental and epileptic encephalopathy (n = 8). Approximately 79.2% (57/72) patients had varying degrees of developmental delay. All patients had abnormal MRI findings with developmental delay. 91.7% (55/60) patients with de novo SCN2A variants had development delay, while only 16.7% (2/12) patients with inherited SCN2A variants had abnormal development. 83.9% (26/31) SCN2A variants that were located in transmembrane regions of the protein were detected in patients with development delay. Approximately 69.2% (9/13) SCN2A variants detected in patients with normal development were located in the non-transmembrane regions. Approximately 54.2% (39/72) patients were seizure-free at a median age of 8 months. Oxcarbazepine has been used by 38 patients, and seizure-free was observed in 11 of them (11/38, 28.9%), while 6 patients had seizure worsening by oxcarbazepine. All 3 patients used oxcarbazepine and with seizure onset age > 1 year presented seizure exacerbation after taking oxcarbazepine. Valproate has been used by 53 patients, seizure-free was observed in 22.6% (12/53) of them.
Conclusion
The phenotypic spectrum of SCN2A-related epilepsy was broad, ranging from benign epilepsy in neonate and infancy to severe epileptic encephalopathy. Oxcarbazepine and valproate were the most effective drugs in epilepsy patients with SCN2A variants. Sodium channel blockers often worsen seizures in patients with seizure onset beyond 1 year of age. Abnormal brain MRI findings and de novo variations were often related to poor prognosis. Most SCN2A variants located in transmembrane regions were related to patients with developmental delay.
Keywords: epilepsy, SCN2A gene, variant, phenotype, treatment
Introduction
The etiology of epilepsy is a major determinant of clinical course and prognosis. Six etiologic groups of epilepsy include structural, metabolic, genetic, infectious, and immune, as well as an unknown group (Scheffer et al., 2017). As genetic testing is broadly used in pediatric neurology, more than half of epilepsy children are thought to have a genetic cause (Reif et al., 2017). At present, voltage-gated sodium channel genes such as SCN1A, SCN2A, SCN3A, and SCN8A were reported to be causative genes of epilepsy (Ademuwagun et al., 2021), among them SCN2A has been reported to be the second most common, next only to SCN1A, the first reported causative gene for epilepsy (Heyne et al., 2019). Epilepsy caused by SCN2A variants mostly starts in early childhood and has a wide phenotypic spectrum, ranging from self-limited epilepsy with a favorable outcome to developmental and epileptic encephalopathy, and most of them respond well to sodium channel blockers (SCBs) (Grinton et al., 2015; Trump et al., 2016; Dilena et al., 2017; Flor-Hirsch et al., 2018; Kim et al., 2020; Melikishvili et al., 2020; Miao et al., 2020; Penkl et al., 2021). China has a large population and a large number of epilepsy children. However, the epilepsy phenotypes and prognosis caused by SCN2A variation in Chinese children have not yet been studied in a large sample. In this study, the phenotypic spectrum, treatment, and prognosis of epilepsy children with SCN2A variants were studied in a Chinese cohort from two pediatric clinical centers.
Materials and Methods
Participants
In this study, epilepsy children who were suspected of genetic etiology and identified with SCN2A variants by next-generation sequencing were enrolled in Peking University First Hospital and Shenzhen Children’s hospital from September 2006 to January 2021. All epilepsy patients fulfilled the following criteria: (1) no identifiable immediate or remote cause and (2) no metabolic or mitochondrial disorders. Clinical information includes the age of seizure onset, seizure types, developmental milestones, neurologic status, electroencephalogram (EEG), brain MRI, and treatment data of the patients and their relatives were collected using a pre-test questionnaire completed by the recruiting clinician by telephone or from medical records. Patients were followed up at a pediatric neurology clinic at our hospital or by telephone. The effect of anti-seizure medication (ASM) therapy were retrospectively assessed and classified according to the judgment of the treating physicians into seizure freedom, seizure reduction (reduction in seizure frequency > 50%), no effect or seizure worsening. This study was approved by the Ethics Committee of Peking University First Hospital and Shenzhen Children’s hospital, respectively. The written informed consent for the analysis and publication of clinical and genetic details was obtained from the patients or their parents.
Genetic Analysis
Blood samples were obtained from these probands and their family members when possible. Genomic DNA was extracted from peripheral blood by a standard method. All patients were screened for pathogenic variants either through a custom-designed gene panel in which candidate genes associated with epilepsy including SCN2A was selected as the genes of interest or by whole-exome sequencing. The potential pathogenic variations suggested by the targeted next-generation sequencing were validated using Sanger sequencing.
Results
SCN2A Variants
A total of 72 unrelated epilepsy patients with heterozygous SCN2A variants were collected. Among them, patients 1–8 have been reported in a previous study of benign familial epilepsy (Zeng et al., 2018). Fifty-nine SCN2A variants were identified, including 54 missense variants (91.5%, 54/59), 2 frameshift variants, 2 in-frame deletion variants, and 1 non-sense variant. A total of 22 SCN2A variants were novel. The SCN2A variants were scattered in different regions of the gene, and there were no obvious hot spot variants (see Figure 1). V261M, R853Q, H1853R, E999K, E1211K, R1319Q, A1500T, R1629H, and P1658S were recurrent variants, each was identified in two or three patients (see Table l). A total of 12 (12/72, 16.7%) patients had inherited variants, and the other 60 (60/72, 83.3%) patients had de novo variants. All 12 patients with inherited variants had a family history of epilepsy or febrile seizures. All of the affected parents had heterozygous variants as their children, except the mother of patient 48. She carries the same SCN2A variant with a ratio of about 21.5% in the peripheral blood by next-generation sequencing.
FIGURE 1.

The developmental outcome and treatment effects to oxcarbazepine of epilepsy patients with SCN2A variants. A total of 59 SCN2A variants was included. The shapes of variation sites represent different variation types (circle = missense variation; triangle = nonsense variation; square = frameshift variation; rhombu = in-frame deletion variation). The colors of the shape represent different developmental outcome (red = developmental delay; green = normal development; orange = both patients with normal development and developmental delay were observed). The colors of the variants represent the treatment effects of oxcarbazepine (green = seizure freedom; blue = seizure reduction; orange = no effect; red = seizure worsening; black = never used). The variation underlined indicates fever-sensitivity. Pentagons indicate one patient with the variants died.
TABLE 1.
The genetic testing results and clinical features of 72 patients with SCN2A variants.
| Patient | Gender | Variation | Reported/ novel |
Inheritance | Seizure onset age | Age at last follow-up | EEG features | Seizure types | MRI | Psychomotor development | Phenotype | Age at seizure free | Seizure free therapy | Other conditions |
| 1 | Male | c.2627A > G(p.N876S) | Reported | Maternal | 2 months | 5 years 6 months | Normal | FS | Normal | Normal | BFNIE | 4 months | OXC | |
| 2 | Female | c.2674G > A(p.V892I) | Reported | Maternal | 2 male | 6 years 11 months | FS | FS | Normal | Normal | BFNIE | 6 months | LEV, TPM | |
| 3 | Female | c.2872A > G(p.M958V) | Reported | Paternal | 3 months | 6 years 1 months | FD | FS | Normal | Normal | BFNIE | 4 months | VPA | |
| 4 | Male | c.668G > A(p.R223Q) | Reported | Paternal | 4 months | 11 years | Normal | FS | Normal | Normal | BFIE | 7 months | VPA | |
| 5 | Male | c.752T > C(p.V251A) | Reported | Maternal | 3 months | 9 years | FD | FS | Normal | Normal | BFIE | 4 months | PB | |
| 6 | Male | c.1307T > C(p.L436S) | Reported | Maternal | 3 months | 10 years | Normal | FS | Normal | Normal | BFIE | 8 months | VPA | |
| 7 | Female | c.1737C > G(p.S579R) | Reported | Paternal | 1 year 2 months | 6 years 2 months | Normal | FS | Normal | Normal | BFIE | 1 year 3 months | Self-limited | |
| 8 | Male | c.4835C > G(p.A1612G) | Reported | Maternal | 3 months | 8 years 10 months | Normal | FS | Normal | Normal | BFIE | 4 months | Self-limited | |
| 9 | Male | c.1523_1528 delAGAAAC (p.509_510del KQ) |
Novel | Paternal | 11 months | 10 years | GD | GTCS, AtS | Normal | Normal | FSP | 2 years | VPA | Fever sensetivity |
| 10 | Male | c.4988T > C(p.I1663T) | Novel | Maternal | 6 months | 7 years | GD, FD, MS | GTCS, MS, FS | Normal | Delay, walk at 3 years 6 months, speak at 1 year 3 months | DS | 5 years | VPA, CLZ, OXC | Fever sensetivity |
| 11 | Male | c.1108T > C(p.F370L) | Novel | De novo | 8 days | 2 years 7 months | BS, FS, SS | TSS, SS, FS | Normal | Delay, cannot control head and speak | OS | 8 months | VGB, TPM | Died at 2 years 7 months |
| 12 | Male | c.781G > A(p.V261M) | Reported | De novo | 4 days | 5 years | MD, FS | SS, FS | Normal | Delay, head control at 2 years 6 months, sit alone at 4 years | EIEE | 8 months | VPA, TPM | |
| 13 | Female | c.466A > C(p.K156Q) | Novel | De novo | 3 days | 1 year 4 months | GD, TS | TS | DWMM | Delay, cannot control head and speak | EIEE | Non-remission | ||
| 14 | Female | c.1261T > G(p.L421V) | Reported | De novo | 10 days | 4 years 3 months | BS, HPS, TSS, FS, SS | TSS, SS, FS | ACC, DWMM, DFTL, ELV | Delay, cannot control head and speak | OS → WS | Non-remission | Died at 4 years 3 months | |
| 15 | Male | c.5558A > G(p.H1853R) | Reported | De novo | 2 days | 2 years 9 months | BS, HPS, FS, SS | TSS, SS, FS | Normal | Delay, cannot control head and speak | OS → WS | 8 months | TPM | Died at 2 years 9 months |
| 16 | Male | c.4223T > C(p.V1408A) | Reported | De novo | 2 days | 8 years 1 months | FD | FS | Normal | Delay, walk at 1 year 8 months, normal speech | EIEE | 5 months | OXC, LEV | |
| 17 | Male | c.2995G > A(p.E999K) | Reported | De novo | 2 days | 3 years 10 months | HPS, MD | SS, FS | Normal | Delay, cannot control head and speak | WS | 3 months | VPA, LEV, TPM | |
| 18 | Male | c.4364T > A(p.I1455N) | Reported | De novo | 2 days | 7 years | HPS, FS, SS | SS, FS | Normal | Delay, cannot control head and speak | WS | 2 years 5 months | TPM, VPA | |
| 19 | Male | c.4454G > A(p. G1485D) | Novel | De novo | 3 months | 7 years 6 months | Normal | FS | Normal | Delay, walk at 1 years 4 months, poor school performance | EIEE | 2 years | VPA | |
| 20 | Male | c.1271T > C(p. V424A) | Reported | De novo | 1 days | 8 years 2 months | GD, FS | FS, GTCS | DFTL, ELV | Delay, cannot control head and speak | EIEE | Non-remission | Fever sensetivity | |
| 21 | Female | c.5144G > T(p. G1715V) | Reported | De novo | 8 months | 8 years 3 months | HPS, FD, MD, AS | SS, AS | Normal | Delay, walk at 2 years 1 months, can only speak a few words | WS | 3 years 1 months | VPA, LTG | |
| 22 | Male | c.3631G > A(p E1211K) | Reported | De novo | 4 months | 6 years 11 months | HPS, FD, SS, FS | SS, FS | ELV | Delay, control head at 1 years 4 months, cannot sit alone and speak | WS | Non-remission | ||
| 23 | Male | c.4498G > A(p.A1500T) | Reported | De novo | 2 days | 1 years 6 months | HPS, FD, SS, FS | SS, FS, TSS | Normal | Delay, cannot control head and speak | WS | Non-remission | ||
| 24 | Male | c.5196delC (p.P1733Lfs*36) |
Novel | De novo | 11 months | 5 years | HPS, SS | SS | DFTL | Delay, cannot control head and speak | WS | Non-remission | ||
| 25 | Male | c.4933G > A(p.G1645R) | Novel | De novo | 2 years | 5 years 11 months | GD | GTCS, FS | Normal | Delay, poor speech and school performance | DEE | 5 years 4 months | LEV, VPA | Fever sensetivity |
| 26 | Female | c.4399C > G(p.L1467V) | Novel | De novo | 10 months | 6 years 7 months | FD | FS | ELV | Delay before seizure onset, cannot walk and speak | DEE | 1 years 11 months | VPA, LEV, TPM | |
| 27 | Male | c.1128_1130de lCTT(p.377del L) |
Novel | De novo | 2 years 3 months | 6 years 4 months | FD | FS | Normal | Delay before seizure onset, walk at 1 years 6 months, cannot speak | DEE | 2 years 10 months | VPA, LEV | |
| 28 | Male | c.4303C > T(p.R1435*) | Reported | De novo | 2 years 6 months | 7 years 3 months | FD | FS | Normal | Delay before seizure onset, walk at 2 years, cannot speak | DEE | 3 years 4 months | VPA, LEV, TPM | ASD |
| 29 | Female | c.4015A > G(p.N1339D) | Reported | De novo | 14 days | 2 years 8 months | BS, HPS, FS, SS, MS | TSS, FS, MS | ELV, ACC, DFTL | Delay, cannot control head and speak | OS → WS | Non-remission | Died at 2 years 8 months | |
| 30 | Male | c.781G > A(p.V261M) | Reported | De novo | 3 days | 4 years 1 months | MD, FS | FS | Normal | Delay, walk at 1 year 2 months, slightly poor language performance | EIEE | Non-remission | ||
| 31 | Male | c.5558A > G(p.H1853R) | Reported | De novo | 2 days | 5 years 8 months | FD, FS | FS | Normal | Delay, walk at 1 year 6 months, poor language performance | EIEE | 1 month | PB | |
| 32 | Male | c.605C > T(p.A202V) | Reported | De novo | 3 days | 6 years 2 months | MD, FS | FS | Normal | Delay, walk at 1 year 2 months, slightly poor language performance | EIEE | 3 months | PB | |
| 33 | Male | c.5317G > A(p.A1773T) | Reported | De novo | 8 months | 13 years | HPS, GD, FD, SS, MS, AS | FS, SS, MS, AS | Normal | Delay, walk at 4 years, cannot speak | WS | Non-remission | ||
| 34 | Male | c.3631G > A(p.E1211K) | Reported | De novo | 1 year | 5 years | HPS, MD, SS | SS | DFL, ELV | Delay before seizure onset, walk at 4 years 8 months, cannot speak | WS | Non-remission | ASD | |
| 35 | Male | c.3956G > T(p.R1319L) | Reported | De novo | 2 days | 6 years 1 month | MD, FD, FS, SS, AS | FS, SS, AS | Normal | Delay, walk with help at 6 years, cannot speak | EIEE | Non-remission | ||
| 36 | Male | c.4712T > C(p.I1571T) | Reported | De novo | 2 days | 1 year 3 months | BS, HPS, MD, FS, SS | TSS, FS, S | Normal | Delay, cannot control head and speak | OS → WS | Non-remission | ||
| 37 | Female | c.4523A > T(p.K1508I) | Novel | De novo | 1 day | 2 years 9 months | BS, HPS, MD, FS, SS, TS | TS, SS, FS | DFTL, DWMM | Delay, cannot control head and speak | OS → WS | Non-remission | ||
| 38 | Female | c.4498G > A(p.A1500T | Reported | De novo | 2 days | 3 years 9 months | BS, HPS, FD, FS, SS | TSS, SS, FS | Normal | Delay, cannot control head and speak | OS → WS | Non-remission | ||
| 39 | Male | c.4025T > C(p.L1342P) | Reported | De novo | 6 months | 4 years 5 months | HPS, MD | SS | ACC, DWMM, ELV | Delay, cannot control head and speak | WS | 1 year 6 months | VPA, OXC | |
| 40 | Male | c.4036A > G(p.I1346V) | Reported | De novo | 1 days | 3 years 11 months | BS, HPS, FS, SS, TSS | FS, SS, TSS | ACC, DFTL | Delay, cannot control head and speak | OS → WS | Non-remission | ||
| 41 | Male | c.807G > T(p.L269F) | Novel | De novo | 1 days | 2 year 5 months | HPS, MD, FS, SS | FS, SS | DFTL | Delay, cannot control head and speak | WS | 3 months | LEV | |
| 42 | Male | c.4972C > T(p.P1658S) | Reported | De novo | 1 days | 3 years 3 months | MD, FD | FS | DFTL, HA | Delay, cannot control head and speak | EIEE | Non-remission | ||
| 43 | Male | c.4948C > A(p.L1650I) | Novel | De novo | 2 days | 1 year | BS, HPS, FS, SS | FS, SS, TSS | Normal | Delay, cannot control head and speak | OS → WS | Non-remission | ||
| 44 | Male | c.5237G > A(p.C1746Y) | Novel | De novo | 1 year 3 months | 6 years 9 months | GD, MD, FD, SS | FS, SS | ELV | Delay before seizure onset, cannot walk and speak | EIEE | 1 year 10 months | LEV, VPA, TPM | |
| 45 | Female | c.2657T > C(p.L886S) | Reported | De novo | 1 days | 5 years 1 months | BS, FD, FS | TSS, FS | DWMM | Delay, cannot control head and speak | OS | Non-remission | ||
| 46 | Female | c.4432C > A(p.Q1478K) | Novel | De novo | 1 month (30 days) | 3 years 3 months | FS | FS | Normal | Normal | BIE | 3 months | OXC | |
| 47 | Female | c.3579_3580 delCT > (p.W1194Vfs*9) |
Reported | De novo | 1 year 5 months | 5 year | GD, MD, FD, SS | SS | HA | Delay before seizure onset, cannot walk and speak | DEE | Non-remission | ||
| 48 | Female | c.2558G > A(p.R853Q) | Reported | Maternal | 10 months | 1 year 2 months | HPS, GD, MD, FS | FS, SS | DFTL,ACC | Delay before seizure onset, cannot sit and speak | WS | Non-remission | ||
| 49 | Male | c.781G > A(p.V261M) | Reported | De novo | 2 days | 2 years 3 months | Normal | FS | Normal | Normal | BNE | 3 months | OXC | |
| 50 | Male | c.640T > C(p.S214P) | Reported | De novo | 2 months | 2 years 11 months | HPS, FD, SS | FS, SS | ACC | Delay, cannot control head and speak | WS | Non-remission | Fever sensetivity | |
| 51 | Female | c.3936G > T(p.R1312S) | Novel | De novo | 4 months | 1 year 6 months | MD, FS, SS | FS, SS | DFTL | Delay before seizure onset, cannot control head and speak | DEE | Non-remission | ||
| 52 | Male | c.5558A > G(p.H1853R) | Reported | De novo | 9 days | 1 year 8 months | BS, HPS, FD, FS, SS, TSS | TSS, SS, FS | ACC, DWMM, ELV | Delay, cannot control head and speak | OS → WS | Non-remission | ||
| 53 | Male | c.5640A > C(p.E1880D) | Novel | De novo | 1 days | 1 year 4 months | HPS, ED, MD, SS, MS, TS | TS, FS, SS, MS | DFTL | Delay, cannot control head and speak | WS | Non-remission | ||
| 54 | Female | c.1253A > T(p.N418I) | Novel | De novo | 1 year 5 months | 2 years 3 months | MD, SS, MS | FS, MS, SS | Normal | Delay before seizure onset, cannot walk and speak | DEE | Non-remission | ||
| 55 | Female | c.3043G > A(p.D1015N) | Reported | De novo | 1 year | 13 years | Normal | FS | Normal | Normal | FSs | 11 years | VPA | Fever sensetivity |
| 56 | Female | c.4886G > A(p.R1629H) | Reported | De novo | 1 days | 5 years 2 months | BS, MD | TSS, FS | Normal | Delay, walk at 2 years, can speak only a few words | OS | 4 m | OXC, KD | |
| 57 | Male | c.3956G > A(p.R1319Q) | Reported | De novo | 4 months | 2 years 2 months | MD, FS | FS | Normal | Normal | BIE | 5 months | OXC | |
| 58 | Male | c.2558G > A(p.R853Q) | Reported | De novo | 11 months | 1 years | HPS, FD, SS | SS, FS | ELV, DFTL | Delay before seizure onset, 6 months control head | WS | Non-remission | ||
| 59 | Male | c.4886G > A(p.R1629H) | Reported | De novo | 4 days | 5 years 11 months | BS, MD, FD | FS | Normal | Delay, slightly poor language performance | EIEE | 1 month | OXC | |
| 60 | Female | c.4972C > T(p.P1658S) | Reported | De novo | 2 months | 7 years 2 months | FD | FS | Normal | Delay, walk at 2 years 8 months, speak at 9 months | EIEE | Non-remission | ||
| 61 | Male | c.3956G > A(p.R1319Q) | Reported | De novo | 2 months | 6 years 3 months | FD, FS | FS | DFL | Delay, normal motor development, cannot speak | DEE | 5 months | OXC | ASD |
| 62 | Male | c.2870C > A(p.T957N) | Novel | De novo | 2 days | 4 years 4 months | FD | FS | Normal | Normal | BIE | 3 months | OXC | |
| 63 | Male | c.2558G > A(p.R853Q) | Reported | De novo | 8 months | 1 year 4 months | HPS | FS, SS | ACC | Delay, cannot control head and speak | WS | Non-remission | ||
| 64 | Male | c.2995G > A(p.E999K) | Reported | De novo | 1 day | 1 year 3 months | BS, HPS, MFS, SS | FS, SS | Normal | Delay, cannot control head and speak | EIMFS | 1 year | CBZ | |
| 65 | Male | c.4901G > T(p.G1634V) | Reported | De novo | 1 days | 3 months | BS, MFS, SE | FS, SS | ACC, ELV | Delay, cannot control head | EIMFS | Non-remission | Died at 3 months | |
| 66 | Female | c.3631G > A(p.E1211K) | Reported | De novo | 10 months | 4 years 2 months | HPS, FD, SS, NCSE | FS, SS | Normal | Delay before seizure onset, cannot walk and speak | WS | 2 years 2 months | VPA | |
| 67 | Female | c.4969C > T(p.L1657F) | Novel | De novo | 6 days | 2 years 8 months | HPS, GD, MD, FD, SS, FS | FS, SS | ACC, ELV, DFTL | Delay, cannot control head and speak | EIEE | Non-remission | ||
| 68 | Male | c.4391C > T(p.T1464I) | Novel | De novo | 2 days | 2 years 2 months | FS, GD, MD, TS | FS, SS, TS | DWMM | Delay, control head at 2 years, cannot sit and speak | EIEE | 1 year 3 months | VGB, PRP | |
| 69 | Female | c.1288G > A(p.E430K) | Novel | De novo | 8 days | 1 year 8 months | BS, HPS, MD, FS, SS | FS, SS, TSS | ACC, DWMM, DFTL, ELV |
Delay, cannot control head and speak | OS → WS | Non-remission | ||
| 70 | Male | c.707C > G(p.T236S) | Reported | De novo | 20 days | 6 months | BS, HPS, FS, TSS, SS | FS, SS, TSS | ACC | Delay, cannot control head and speak | OS → WS | Non-remission | ||
| 71 | Male | c.5645G > A(p.R1882Q) | Reported | De novo | 2 days | 9 months | BS, HPS, GD, FS, SE, SS | FS, TSS | Normal | Delay, cannot control head and speak | OS → WS | Non-remission | ||
| 72 | Male | c.4610T > C(p.I1537T) | Novel | Paternal | 3 months | 1 year 6 months | MD | FS | Normal | Normal | BFIE | 4 months | LEV |
MS, myoclonic seizure; FS, focal seizure; GTCS, generalized tonic clonic seizure; AtS, atonic seizure; SS, spasm; TSS, tonic spasm seizure; TS, tonic seizure; AS, absence seizure; FD, focal discharges; MD, multifocal discharges; BS, burst suppression; GD, generalized discharges; HPS, hypsarrhythmia; SE, status epilepticus; NCSE, nonconvulsive status epilepticus; DWMM, delayed white matter myelination; ELV, Enlargement of lateral ventricles; ACC, agenesis of corpus callosum; DFTL, dysplasia of frontotemporal lobes; DFL, dysplasia of frontal lobes; HA, hippocampal atrophy; BFNIE, benign familial neonatal-infantile epilepsy; BNE, benign neonatal epilepsy; BIE, benign infantile epilepsy; WS, West syndrome; EIEE, early infantile epileptic encephalopathy; OS, Ohtahara syndrome; DEE, developmental and epileptic encephalopathy. EIMFS, epilepsy of infancy with migrating focal seizures; FSs, febrile seizures; FSP, febrile seizures plus; DS, Dravet syndrome; KD, ketogenic diet; OXC, oxcarbazepine; LEV, levetiracetam; TPM, topiramate; VPA, valproate; PB, phenobarbital; CLZ, clonazepam; LTG, lamotrigine; KD, ketogenic diet; CBZ, carbamazepine; PRP, perampanel; VGB, vigabatrin; ASD, autistic spectrum disorder.
Clinical Phenotypes of Patients With SCN2A Variants
Among 72 patients with SCN2A variants, 50 are men, 22 are women. The seizure onset age was ranged from the first day of life to 2 years and 6 months. A total of 36 patients had seizure onset in neonates (50.0%, 36/72). A total of 18 patients had seizure onset between 1 and 6 months of age (25.0%, 18/72). A total of 11 patients had seizure onset between 7 month and 1 year of age (15.3%, 11/72). Seizure onset age was beyond 1 year in 7 patients (9.7%, 7/72). The seizure onset age of 5 patients with non-missense variants was between 11 months to 2 years and 6 months.
Focal seizures were observed in 65 patients(90.3%, 65/72), epileptic spasms in 38(52.8%, 38/72), tonic spasms in 15, myoclonic seizures in 5, tonic seizures in 4, generalized tonic-clonic seizures in 4, absence seizures in 3, and atonic seizures in 1, respectively. A total of 41 (41/72, 56.9%) patients presented 2 or more seizure types. Seizures manifested fever-sensitivity in 6 (8.3%, 6/72) patients (patient 9, 10, 20, 25, 50, and 55).
A total of 72 patients with SCN2A variants underwent video EEG. Interictal EEG abnormalities were heterogeneous, such as focal or multifocal epileptic discharges in 45 patients, hypsarrhythmia in 30, burst suppression in 18, and generalized discharges in 13, respectively. A total of 10 patients had normal interictal EEG. Seizures were recorded in 48 patients, such as focal seizures in 34 patients, epileptic spasms in 31, tonic spasms in 4, myoclonic seizures in 5, tonic seizures in 4, and absence seizures in 3, respectively.
Brain MRI was performed in all 72 patients with SCN2A variants, which revealed abnormalities in 29 (29/72, 40.3%) patients. The abnormalities included dysplasia of frontal or frontotemporal lobes in 16, enlargement of the unilateral or bilateral lateral ventricle in 13, agenesis of the corpus callosum in 12, delayed white matter myelination in 8, and hippocampal atrophy in 2 patients, respectively (see Figure 2). The other 43 probands had normal brain MRI.
FIGURE 2.

Abnormal brain MRI of 3 patients with SCN2A variants. Brain MRI of Patient 14 at the age of 22 months. (a,b). Axial and coronal images (T1WI) showing agenesis of corpus callosum, delayed white matter myelination, dysplasia of frontotemporal lobes and enlargement of lateral ventricles. Brain MRI of Patient 29 at the age of 7 months. (c,f) Sagittal images (T1WI) showing enlargement of lateral ventricles, agenesis of corpus callosum and dysplasia of frontotemporal lobes. Brain MRI of Patient 39 at the age of 12 months. (d,e) Axial and coronal images (T2WI) showing agenesis of corpus callosum, delayed white matter myelination and enlargement of lateral ventricles. The arrow points to the lesion.
Among 72 patients with SCN2A variants, 57 patients (57/72, 79.2%) had varying degrees of developmental delay, and the other 15 patients had normal development. The 28 of 57 (28/57, 49.1%) patients with developmental delay cannot control head at last follow-up (median age: 2 years and 7 months; range: 3 months to 8 years and 2 months). All patients had epileptic spasms, burst suppression and hypsarrhythmia, abnormal MRI findings had developmental delay. Autism spectrum disorder (ASD) was diagnosed in 3 patients. All affected parents of the proband had normal development.
In 72 patients, the phenotypes were diagnosed febrile seizures (plus) (n = 2), benign (familial) infantile epilepsy (n = 9), benign familial neonatal-infantile epilepsy (n = 3), benign neonatal epilepsy (n = 1), West syndrome (n = 16), Ohtahara syndrome (n = 15), epilepsy of infancy with migrating focal seizures (EIMFS) (n = 2), Dravet syndrome (n = 1), early infantile epileptic encephalopathy (EIEE) (n = 15), and unclassifiable developmental and epileptic encephalopathy (DEE) (n = 8) (Figure 3). A total of 16 (16/72, 22.2%) patients were initially diagnosed with West syndrome. A total of 15 patients (15/72, 20.8%) were diagnosed with Ohtahara syndrome at first, but 12 (12/15, 80%) of them evolved into West syndrome afterward. Both Patient 9 and Patient 10 were probands of generalized epilepsy with febrile seizures plus (GEFS+) families. Several paternal family members of Patient 9 had histories of febrile seizures in childhood. The Patient 10 was detected with maternal SCN2A variation I1663T which was inherited from his grandmother. His mother had 2 febrile seizures in early childhood. However, his maternal grandmother had no history of seizures. The Patient 48 was diagnosed with intractable West syndrome, recurrent SCN2A variation R853Q was detected. His mosaic mother had several seizures before 1 year of age. Her seizures were self-limited without using any ASM therapy.
FIGURE 3.

Distribution of 72 Chinese epilepsy patients with SCN2A variants according to phenotypes [n (%)].
Genotype–Phenotype Correlation
Of the 59 SCN2A variants, 31 were located in transmembrane regions, while the other 28 were in non-transmembrane regions. Carriers of 2 recurrent variants (R1319Q and V261M) included both patients with normal development (Patient 57 and Patient 49) and patients with developmental delay (Patient 61, Patient 12, and Patient 30). A total of 13 variants were only detected in patients with normal development, 9 (9/13, 69.2%) of them were located in non-transmembrane regions, and the remaining 4 were located in transmembrane regions which accounting for 12.9% (4/31) of all transmembrane region variations. A total of 44 variants were only detected in patients with developmental delay, 26 (26/44, 59.1%) were located in transmembrane regions, accounting for 83.9% (26/31) of transmembrane region variants, and 18 were located in non-transmembrane regions (see Figure 1). The 10 of 12 (83.3%) patients with inherited SCN2A variants had normal intelligence; however, the other 2 (2/12, 16.7%) patients had a developmental delay (Patient 10 and 48). Among 60 patients with de novo SCN2A variants, 55 had development delay (91.7%, 55/60), and the remaining 5 patients had normal development.
Seizure Treatment and Prognosis
At the last follow-up (median age: 4 years and 4 months; range: 3 months to 13 years), 39 (54.2%, 39/72) patients were seizure-free at a median age of 8 months (range: 1 month to 5 years 4 months of age), the remaining 33 patients still had refractory seizures (median age: 2 years 8 months; range: 3 months to 13 years). Among 39 patients with seizure freedom, 2 patients who were diagnosed with benign familial infantile epilepsy did not use any ASM therapy, 21 (21/39, 53.8%) patients used monotherapy, 11 used two-drug treatment, and 5 used polytherapy. All 33 patients with uncontrolled seizures have tried at least 2 ASM therapies. All 15 patients had normal development were seizure-free. Of the 29 children with abnormal brain MRI, 23 (23/29, 79.3%) patients still had seizures at the last follow-up, and only 6 had seizure freedom.
At least one patient in the study experienced seizure control after treatment with SCBs such as oxcarbazepine, carbamazepine, lamotrigine, and other ASM therapy like valproate, topiramate, levetiracetam, phenobarbital, ACTH, vigabatrin, and perampanel. The effect of these ASM therapies is shown in Figure 4. No patient experienced seizure control after using phenytoin, zonisamide, lacosamide, clonazepam, nitrazepam, clobazam, cannabidiol, ketogenic diet, and vagus nerve stimulation.
FIGURE 4.
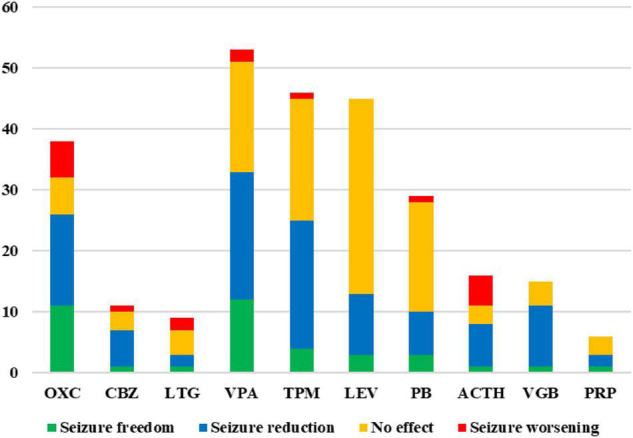
Treatment effects of anti-seizure medication (ASM) therapies in epilepsy patients with SCN2A variants. Number of treated patients and their seizure outcome (green = seizure freedom; blue = seizure reduction; orange = no effect; red = seizure worsening) that have been treated with different ASM therapies. Only effects of ASM therapies that at least one patient achieved seizure free are shown. OXC, oxcarbazepine; CBZ, carbamazepine; LTG, lamotrigine; VPA, valproate; TPM, topiramate; LEV, levetiracetam; PB, phenobarbital; VGB, vigabatrin; PRP, perampanel.
Oxcarbazepine has been used in 38 patients, seizure freedom, seizure reduction, no effect, and seizure worsening were observed in 11 (11/38, 28.9%), 15 (15/38, 39.5%), 6 (6/38, 15.8%), and 6 (6/38, 15.8%) patients, respectively. Among those 38 patients, 35 patients had seizure onset age < 3 months, 6 patients had seizure onset age between 4 months and 1 year of age, and the other 3 patients had seizure onset age > 1 year. For 29 patients with seizure onset age < 3 months, seizure freedom, seizure reduction, no effect, and seizure worsening were observed in 8 (8/29, 27.6%), 13 (13/29, 44.8%), 5 (5/29, 17.2%), and 3 (3/29, 10.3%) patients (patients 20, 25, and 37), respectively. For 6 patients with seizure onset age between 4 months and 1 year of age, seizure freedom, seizure reduction, and no effect were observed in 3, 2, and 1 patient, respectively, no patients experienced seizure worsening. All 3 patients with seizure onset age > 1 year had seizure exacerbation caused by oxcarbazepine (Patients 27, 47, and 50). The Patient 10 was diagnosed with Dravet syndrome and his seizure was controlled after the addition of oxcarbazepine at the age of 5 years old and had no relapse for nearly 2 years. The effects of oxcarbazepine in patients with different SCN2A variants have been presented in Figure 1. Carbamazepine has been used in 11 patients, seizure freedom, seizure reduction, no effect, and seizure worsening were observed in 1 (1/11, 9.1%), 6 (6/11, 54.5%), 3 (3/11, 27.3%), and 1 (1/11, 9.1%) patients, respectively. Lamotrigine has been used in 9 patients, seizure freedom, seizure reduction, no effect, and seizure worsening were observed in 1 (1/9, 11.1%), 2(2/9, 22.2%), 4(4/9, 44.4%), and 2 (2/9, 22.2%) patients, respectively.
Valproate has been used in 53 patients with SCN2A variants, seizure freedom, seizure reduction, no effect, and seizure worsening were observed in 12 (12/53, 22.6%), 21 (21/53, 39.6%), 18 (18/53, 34.0%), and 2 (2/53, 3.8%) patients, respectively. Seizures were controlled by topiramate, levetiracetam, and phenobarbital in 4 (4/46, 8.7%), 3 (3/45, 6.7%), and 3 (3/29, 10.3%) patients, respectively. One patient was seizure free after taking ACTH, vigabatrin, and perampanel, respectively. No patient had seizure exacerbation caused by levetiracetam, vigabatrin, and perampanel. Seizure worsening caused by ACTH was observed in 5 (5/16, 31.3%) patients.
Five (5/72, 6.9%) patients died at the age of 3 months to 4 years and 3 months (Patients 11, 14, 15, 29, and 65). All those 5 patients started seizures in neonate. Four patients were initially diagnosed with Ohtahara syndrome, and the other patient diagnosed with EIMFS. Three of them (Patients 14, 29, and 65) manifested intractable seizures with no effect to multiple ASM therapies. The causes of those 3 patients were unknown. Both Patients 11 and 15 were seizure free at the age of 8 months, and suffered possible sudden unexpected death in epilepsy (SUDEP).
Discussion
SCN2A gene is located on chromosome 2q24.3. The gene which contains 26 exons encodes the α2 subunit of the voltage-gated sodium channel (Nav1.2). Nav1.2 is mainly expressed in the initial part of excitatory neuron axons and unmyelinated axons. The protein is widely distributed in the cortex, hippocampus, striatum, and midbrain. Variations in SCN2A gene are associated with a spectrum of neurodevelopmental and epileptic disorders, such as epilepsy, intellectual disability, ASD, schizophrenia, and periodic ataxia, presenting an autosomal dominant inheritance (Carroll et al., 2016; Yokoi et al., 2018; Long et al., 2019; Schwarz et al., 2019; Suddaby et al., 2019; Epifanio et al., 2021). In recent years, a lot of SCN2A variants have been reported. The variation types include missense variation, in-frame deletion or insertion variation, non-sense variation, frameshift variation, and splice site variation. It has been reported that missense variation was the most common variation type of SCN2A variants (Wolff et al., 2017). In this study, we have found 59 SCN2A variants in 72 Chinese epilepsy patients, and 22 of them are novel variants. The SCN2A variants detected in our study show no hotspot and more than 90% of the variants are missense variants. Other variation types such as in-frame deletion or insertion variant, non-sense variant, and frameshift variant were also presented in our study, but the percentage is small. In our cohort, more than 80% of patients had de novo SCN2A variants.
Sugawara et al. (2001) firstly reported SCN2A missense variant R187W in a Japanese family with GEFS+. The affected patients in this family showed febrile seizures and focal epilepsy. Heron et al. (2002) reported that SCN2A gene was the major causative gene of benign familial neonatal-infantile epilepsy. At first, some researchers believed that missense variations tend to result in benign epilepsy, whereas truncation variations lead to severe and intractable epilepsy (Yamakawa, 2006). With the wide application of next-generation sequencing in clinical practice, SCN2A variants have been reported in severe early onset epileptic encephalopathy and most of them were de novo variants.
Most patients with SCN2A variants start seizures in early childhood. Wolff et al. (2017) reported that about half of patients with SCN2A variants had seizure onset in the neonate. In our study, half of the patients started seizures during the neonatal period. Nearly 80% of patients started seizures within 6 months of age. It suggests that seizures caused by SCN2A variants tend to start in early infancy. The seizure types of patients with SCN2A variants are varied. In our cohort, the most common seizure types were focal seizures and epileptic spasms, observed in about 90% and 50% of patients, respectively. Other seizure types, such as tonic spasm, myoclonic seizures, atonic seizures, tonic seizures, clonic seizures, generalized tonic-clonic seizures, and absence seizures were also observed in some patients. Those seizure types were relatively rare but all of them have been reported in the literature. In our study, the abnormal interictal EEG of epilepsy patients with SCN2A variants such as focal or multifocal epileptic discharges, hypsarrhythmia, burst suppression, and generalized discharges. About 40% of patients in this study had brain MRI abnormalities, such as dysplasia of the frontal or frontotemporal lobes, enlargement of unilateral or bilateral lateral ventricle, agenesis of the corpus callosum, delayed white matter myelination, and hippocampal atrophy. The dysplasia of frontal or frontotemporal lobes is a common defect in our patients with brain MRI abnormalities. The abnormal neuroimages mainly indicated cerebral dysplasia. Nearly 80% of patients had developmental delays, and about half of them cannot control their heads at a median age of 2 years and 7 months.
Wolff et al. (2017) reported 66 families or sporadic cases with SCN2A variants which were collected by a multicenter study that participated by 74 clinical or research institutions. The phenotypes reported in the multicenter study included benign (familial) neonatal/infantile epilepsy, Ohtahara syndrome, EIMFS, encephalopathy with early infantile-onset epilepsy, West syndrome, myoclonic-atonic epilepsy, Lennox–Gastaut syndrome, epileptic encephalopathy with infantile/childhood-onset epilepsy, intellectual disability, and/or autism without epilepsy. Most of the patients were diagnosed with epilepsy. In this study, we analyzed the epilepsy phenotypes of patients with SCN2A variants. The oldest child in our group at the last follow-up was 13 years old. Some of the patients in this study were diagnosed with epilepsy at our clinical center at an early stage of life and were confirmed with SCN2A variants in recent years. The common epilepsy phenotypes of patients with SCN2A variants include benign epilepsy in the first year of life, Ohtahara syndrome, West syndrome, and EIEE. Those epilepsy phenotypes account for more than 80% of this cohort. Most of the patients who were initially diagnosed with Ohtahara syndrome evolved into infantile spasms. Other rare epilepsy phenotypes include febrile seizures (plus), EIMFS, and Dravet syndrome in our study. Wolff et al. (2017) did not report any patient with Dravet syndrome, febrile seizures, or febrile seizures plus. In addition, only 6 patients (8.3%, 6/72) in our study had fever-sensitive seizures, indicating that fever sensitivity is a rare feature of epilepsy patients with SCN2A variants. All those 8 patients diagnosed with unclassifiable DEE had a developmental delay before seizure onset and developmental regression after seizure onset and cannot be diagnosed with any known epilepsy syndrome. In this study, some patients with developmental delay showed little improvement after seizure control, which further suggests that the variation itself has a significant impact on brain development. The mother of Patient 48 carried a mosaic SCN2A variant R853Q with a ratio of about 20% in the peripheral blood. R853Q is a recurrent variation that has been reported repeatedly in the literature (Ganguly et al., 2021). Both the Patient 48 and two other patients with the same variant in our study, as well as patients with the same variant reported in the literature, were diagnosed with West syndrome. However, the mother of Patient 48 had self-limited seizures before 1 year of age and normal development. It indicates that the phenotype severity caused by SCN2A variants is related to the dose of variation.
At the last follow-up, about half of the patients (54.2%, 39/72) were seizure-free at the median age of 8 months. Few of them were self-limited. Liao et al. (2010) found that SCN2A had high expression in the initial segment of the axon of hippocampal neuron of a mouse during 5–15 days after birth, and the function was gradually replaced by the protein encoded by SCN8A. It speculated that this may be the reason why benign epilepsy due to SCN2A variants could be self-limited with age. About half of the patients with seizure control were treated with monotherapy, while the rest were treated with 2 or 3 drugs. The seizures were not controlled in nearly half of the patients, and the median age of these patients at the last follow-up was 2 years and 8 months, with the oldest being 13 years. All those patients presented with refractory epilepsy. Seizures were not controlled in all patients with brain MRI abnormalities in this study.
It has been suggested that SCBs are effective drugs in the treatment of epilepsy of patients with SCN2A variation (Reif et al., 2017). More than half of the patients in our study had used SCBs. Probably because it comes in liquid form, oxcarbazepine was the most commonly used SCBs in our cohort which has been used in 39 patients. Although oxcarbazepine was indeed the most effective ASM therapy in our study, the rate of seizure control was still less than 30%. It has been reported (Wolff et al., 2017) that, patients with seizure onset age less than 3 months always carry SCN2A variants that cause the gain of function and SCBs were often effective for seizures. However, those patients had a seizure onset age later than 3 months, SCN2A variation often causes loss of function and SCBs worsen the seizure. Brunklaus et al. (2020) also reported that individuals with gain-of-function SCN2A/3A/8A most frequently present with early-onset epilepsy (<3 months), and have a good response to SCBs, which is not completely consistent with our results. In this study, about 27% of patients with seizure onset age <3 months had seizures controlled by oxcarbazepine, but another 3 patients had seizure exacerbation. Half of the patients with onset age from 4 months to 1 year had seizure control after administration of oxcarbazepine and no patient presented seizure exacerbation. All 3 patients with onset age > 1 year had seizure exacerbation due to oxcarbazepine. SCN2A gain-of-function has recently been recognized as a cause of early infantile-onset epileptic encephalopathies, whereas loss-of-function SCN2A variations often cause ASD or intellectual disability with later-onset mild epilepsy or without epilepsy (Yamakawa, 2006; Ben-Shalom et al., 2017; Wolff et al., 2017). Based on the results of our study and those in the literature, SCBs are not recommended for patients with seizure onset age > 1 year, while SCBs can be tried for patients with seizure onset age < 1 year, but the possibility of seizure exacerbation still needs to be warned. Phenotypes caused by SCN2A variation are associated with underlying functional changes caused by the variants. Although the rate of seizure control was low, in this study, seizure control was observed in one patient by carbamazepine and lamotrigine, respectively. Since a few cases of Dravet syndrome caused by SCN2A variants has been reported (Wang et al., 2012), the effect of SCBs for those patients has not been reported. In our study, Patient 10 was diagnosed with Dravet syndrome, he was seizure free after adding oxcarbazepine at the age of 5 years old, suggesting that Dravet syndrome is caused by SCN2A variant. It is different from that Dravet syndrome caused by SCN1A variation is mostly not responsive to or might even be exacerbated by SCBs. Although SCN2A and SCN1A are both sodium channel genes, the underlying pathogenesis of Dravet syndrome caused by SCN2A variation may be different from that of SCN1A.
Besides of SCBs, valproate was also the effective ASM therapy in this study, with a seizure control rate of about 22%, slightly lower than oxcarbazepine. In addition, the proportion of exacerbations caused by valproate was lower than that caused by oxcarbazepine. In this study, a large number of patients had used topiramate, levetiracetam, and phenobarbital, and seizure control was achieved in some patients by each of those drugs, although the rates of seizure control were relatively low. In addition, although there were fewer patients who had used ACTH, vigabatrin, and perampanel, there was one case of seizure control for each drug, respectively. ACTH is the preferred drug for the treatment of West syndrome, but nearly one-third of the patients in this study experienced increased seizure frequency after the use of ACTH. The specific mechanism of exacerbation of seizures needs to be further studied. No seizure exacerbation occurred after taking levetiracetam, perampanel, or vigabatrin.
In this study, 5 children died, accounting for about 7% (5/72) of the cases in this group. All 5 patients started seizures during the neonatal period and were diagnosed with severe epileptic syndromes such as Ohtahara syndrome or EIMFS. Two of the patients were seizure-free and the possible cause of death was SUDEP, while the other 3 patients still had frequent seizures and the exact cause of death was unknown. The underlying pathophysiology of SUDEP remains unclear. SUDEP cannot be predicted in advance, because the complete underlying pathophysiology of the phenomenon is likely multifactorial and prognostic biomarkers were still not found (Goldman et al., 2016; Sahly et al., 2022). Our study indicates that frequent seizures and SCN2A variations themselves may be important factors leading to death in these patients.
Phenotypes caused by SCN2A variants are heterogeneous. It has been reported that phenotypic variability from benign infantile epilepsy to Ohtahara syndrome was observed in 3 affected individuals of a family with SCN2A variation (Syrbe et al., 2016). In our study, both benign epilepsy with normal development and epileptic encephalopathy with developmental delay were observed in unrelated patients carrying variants R1319Q and V261M. This was also confirmed by the different phenotypes in the family of Patient 48, ranging from normal to Dravet syndrome in the carriers of the same SCN2A variant.
More than 80% of patients with inherited SCN2A variants had a benign outcome in our study. However, more than 90% of patients with de novo SCN2A variants showed developmental delay, which was identical to the reported studies (Wolff et al., 2017). In this study, the seizure onset age of patients with non-missense SCN2A variants was late, and the seizure onset age was after 11 months, which was consistent with the literature reports (Lauxmann et al., 2018). It may be most non-missense variations will lead to loss-of-function, and the early manifestations of such functional changes are mostly developmental delay or autism, while epilepsy often begins in late infancy or early childhood (Begemann et al., 2019). The number of SCN2A variants located in transmembrane regions was similar to that in non-transmembrane regions in our study. Only 13 SCN2A variants merely in patients with normal development, and about 70% of them were located in non-transmembrane regions. However, more than 80% of variants located in transmembrane regions were related to patients with developmental delay. This may be due to SCN2A variants in transmembrane regions having a greater effect on protein function than those in non-transmembrane regions.
SCN2A is a common causative gene of genetic epilepsy in children. Patients with SCN2A de novo variation usually have a poor prognosis lacking precise treatment. In this study, epilepsy patients with SCN2A variants from 2 pediatric clinical centers in China were studied. Multi-center, large-sample, and prospective studies are needed to further analyze the genotype–phenotype correlations of SCN2A-related epilepsy for precise medicine.
Data Availability Statement
The data presented in this study are available through Clinvar (http://www.clinvar.com/), with the following accession numbers SCV002099454 – SCV002099517. Further inquiry can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Peking University First Hospital and Shenzhen Children’s hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
YZ and JXL contributed to the design and implementation of the research. ZY and YJ were involved in supervised the work. JD and QZ were responsible for assessing the pathogenicity of variants. XN, YC, DW, JZ, JC, XY, and JLL were responsible for follow-up of the patients. QZ and YY contributed to the analysis of the results and to the writing of the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Funding
This work was supported by the Key Research Project of the Ministry of Science and Technology of China (Grant Nos. 2016YFC0904400 and 2016YFC0904401) and Sanming Project of Medicine in Shenzhen (Grant No. SZSM201812005).
References
- Ademuwagun I. A., Rotimi S. O., Syrbe S., Ajamma Y. U., Adebiyi E. (2021). Voltage Gated Sodium Channel Genes in Epilepsy: mutations, Functional Studies, and Treatment Dimensions. Front. Neurol. 12:600050. 10.3389/fneur.2021.600050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begemann A., Acuña M. A., Zweier M., Vincent M., Steindl K., Bachmann-Gagescu R., et al. (2019). Further corroboration of distinct functional features in SCN2A variants causing intellectual disability or epileptic phenotypes. Mol. Med. 25:6. 10.1186/s10020-019-0073-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shalom R., Keeshen C. M., Berrios K. N., An J. Y., Sanders S. J., Bender K. J. (2017). Opposing effects on Nav1.2 function underlie differences between SCN2A variants observed in individuals with autism spectrum disorder or infantile seizures. Biol. Psychiatry 82 224–232. 10.1016/j.biopsych.2017.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunklaus A., Du J., Steckler F., Ghanty I. I., Johannesen K. M., Fenger C. D., et al. (2020). Biological concepts in human sodium channel epilepsies and their relevance in clinical practice. Epilepsia 61 387–399. 10.1111/epi.16438 [DOI] [PubMed] [Google Scholar]
- Carroll L. S., Woolf R., Ibrahim Y., Williams H. J., Dwyer S., Walters J., et al. (2016). Mutation screening of SCN2A in schizophrenia and identification of a novel loss-of-function mutation. Psychiatr. Genet. 26 60–65. 10.1097/YPG.0000000000000110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilena R., Striano P., Gennaro E., Bassi L., Olivotto S., Tadini L., et al. (2017). Efficacy of sodium channel blockers in SCN2A early infantile epileptic encephalopathy. Brain Dev. 39 345–348. 10.1016/j.braindev.2016.10.015 [DOI] [PubMed] [Google Scholar]
- Epifanio R., Giorda R., Merlano M. C., Zanotta N., Romaniello R., Marelli S., et al. (2021). SCN2A Pathogenic Variants and Epilepsy: heterogeneous Clinical, Genetic and Diagnostic Features. Brain Sci. 12:18. 10.3390/brainsci12010018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flor-Hirsch H., Heyman E., Livneh A., Reish O., Watemberg N., Litmanovits I., et al. (2018). Lacosamide for SCN2A-related intractable neonatal and infantile seizures. Epileptic Disord. 20 440–446. 10.1684/epd.2018.1001 [DOI] [PubMed] [Google Scholar]
- Ganguly S., Thompson C. H., George A. L., Jr. (2021). Enhanced slow inactivation contributes to dysfunction of a recurrent SCN2A mutation associated with developmental and epileptic encephalopathy. J. Physiol. 599 4375–4388. 10.1113/JP281834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman A. M., Behr E. R., Semsarian C., Bagnall R. D., Sisodiya S., Cooper P. N. (2016). Sudden unexpected death in epilepsy genetics: molecular diagnostics and prevention. Epilepsia 57 17–25. 10.1111/epi.13232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinton B. E., Heron S. E., Pelekanos J. T., Zuberi S. M., Kivity S., Afawi Z., et al. (2015). Familial neonatal seizures in 36 families: clinical and genetic features correlate with outcome. Epilepsia 56 1071–1080. 10.1111/epi.13020 [DOI] [PubMed] [Google Scholar]
- Heron S. E., Crossland K. M., Andermann E., Phillips H. A., Hall A. J., Bleasel A., et al. (2002). Sodium-channel defects in benign familial neonatal-infantile seizures. Lancet 360 851–852. 10.1016/S0140-6736(02)09968-3 [DOI] [PubMed] [Google Scholar]
- Heyne H. O., Artomov M., Battke F., Bianchini C., Smith D. R., Liebmann N., et al. (2019). Targeted gene sequencing in 6994 individuals with neurodevelopmental disorder with epilepsy. Genet. Med. 21 2496–2503. 10.1038/s41436-019-0531-0 [DOI] [PubMed] [Google Scholar]
- Kim H. J., Yang D., Kim S. H., Kim B., Kim H. D., Lee J. S., et al. (2020). The phenotype and treatment of SCN2A-related developmental and epileptic encephalopathy. Epileptic Disord. 22 563–570. 10.1684/epd.2020.1199 [DOI] [PubMed] [Google Scholar]
- Lauxmann S., Verbeek N. E., Liu Y., Zaichuk M., Müller S., Lemke J. R., et al. (2018). Relationship of electrophysiological dysfunction and clinical severity in SCN2A-related epilepsies. Hum. Mutat. 39 1942–1956. 10.1002/humu.23619 [DOI] [PubMed] [Google Scholar]
- Liao Y., Deprez L., Maljevic S., Pitsch J., Claes L., Hristova D., et al. (2010). Molecular correlates of age-dependent seizures in an inherited neonatal-infantile epilepsy. Brain 133 1403–1414. 10.1093/brain/awq057 [DOI] [PubMed] [Google Scholar]
- Long S., Zhou H., Li S., Wang T., Ma Y., Li C., et al. (2019). The Clinical and Genetic Features of Co-occurring Epilepsy and Autism Spectrum Disorder in Chinese Children. Front. Neurol. 10:505. 10.3389/fneur.2019.00505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikishvili G., Dulac O., Gataullina S. (2020). Neonatal SCN2A encephalopathy: a peculiar recognizable electroclinical sequence. Epilepsy Behav. 111:107187. 10.1016/j.yebeh.2020.107187 [DOI] [PubMed] [Google Scholar]
- Miao P., Tang S., Ye J., Wang J., Lou Y., Zhang B., et al. (2020). Electrophysiological features: the next precise step for SCN2A developmental epileptic encephalopathy. Mol. Genet. Genom. Med. 8:e1250. 10.1002/mgg3.1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penkl A., Reunert J., Debus O. M., Homann A., Och U., Rust S., et al. (2021). A mutation in the neonatal isoform of SCN2A causes neonatal-onset epilepsy. Am. J. Med. Genet. A. 188 941–947. 10.1002/ajmg.a.62581 [DOI] [PubMed] [Google Scholar]
- Reif P. S., Tsai M. H., Helbig I., Rosenow F., Klein K. M. (2017). Precision medicine in genetic epilepsies: break of dawn? Expert Rev. Neurother. 17 381–392. 10.1080/14737175.2017.1253476 [DOI] [PubMed] [Google Scholar]
- Sahly A. N., Shevell M., Sadleir L. G., Myers K. A. (2022). SUDEP risk and autonomic dysfunction in genetic epilepsies. Auton. Neurosci. 237:102907. 10.1016/j.autneu.2021.102907 [DOI] [PubMed] [Google Scholar]
- Scheffer I. E., Berkovic S., Capovilla G., Connolly M. B., French J., Guilhoto L., et al. (2017). ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia 58 512–521. 10.1111/epi.13709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz N., Bast T., Gaily E., Golla G., Gorman K. M., Griffiths L. R., et al. (2019). Clinical and genetic spectrum of SCN2A-associated episodic ataxia SCN2A. Eur. J. Paediatr. Neurol. 23 438–447. 10.1016/j.ejpn.2019.03.001 [DOI] [PubMed] [Google Scholar]
- Suddaby J. S., Silver J., So J. (2019). Understanding the schizophrenia phenotype in the first patient with the full SCN2A phenotypic spectrum. Psychiatr. Genet. 29 91–94. 10.1097/YPG.0000000000000219 [DOI] [PubMed] [Google Scholar]
- Sugawara T., Tsurubuchi Y., Agarwala K. L., Ito M., Fukuma G., Mazaki-Miyazaki E., et al. (2001). A missense variant of the Na+ channel alpha II subunit gene Na(v)1.2 in a patient with febrile and afebrile seizures causes channel dysfunction. Proc. Natl. Acad. Sci. U.S.A. 98 6384–6389. 10.1073/pnas.111065098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syrbe S., Zhorov B. S., Bertsche A., Bernhard M. K., Hornemann F., Mütze U., et al. (2016). Phenotypic Variability from Benign Infantile Epilepsy to Ohtahara Syndrome Associated with a Novel Mutation in SCN2A. Mol. Syndromol. 7 182–188. 10.1159/000447526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trump N., McTague A., Brittain H., Papandreou A., Meyer E., Ngoh A., et al. (2016). Improving diagnosis and broadening the phenotypes in early-onset seizure and severe developmental delay disorders through gene panel analysis. J. Med. Genet. 53 310–317. 10.1136/jmedgenet-2015-103263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. W., Shi X. Y., Kurahashi H., Hwang S. K., Ishii A., Higurashi N., et al. (2012). Prevalence of SCN1A mutations in children with suspected Dravet syndrome and intractable childhood epilepsy. Epilepsy Res. 102 195–200. 10.1016/j.eplepsyres.2012.06.006 [DOI] [PubMed] [Google Scholar]
- Wolff M., Johannesen K. M., Hedrich U. B. S., Masnada S., Rubboli G., Gardella E., et al. (2017). Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain 140 1316–1336. 10.1093/brain/awx054 [DOI] [PubMed] [Google Scholar]
- Yamakawa K. (2006). Na channel gene mutations in epilepsy–the functional consequences. Rev. Epilepsy Res. 70 S218–S222. 10.1016/j.eplepsyres.2005.11.025 [DOI] [PubMed] [Google Scholar]
- Yokoi T., Enomoto Y., Tsurusaki Y., Naruto T., Kurosawa K. (2018). Nonsyndromic intellectual disability with novel heterozygous SCN2A mutation and epilepsy. Hum. Genome Var. 5:20. 10.1038/s41439-018-0019-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Q., Yang X., Zhang J., Liu A., Yang Z., Liu X., et al. (2018). Genetic analysis of benign familial epilepsies in the first year of life in a Chinese cohort. J. Hum. Genet. 63 9–18. 10.1038/s10038-017-0359-x [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data presented in this study are available through Clinvar (http://www.clinvar.com/), with the following accession numbers SCV002099454 – SCV002099517. Further inquiry can be directed to the corresponding author.