

Abstract
Kidney organoids generated from hPSCs have provided an unlimited source of renal tissue. Human kidney organoids are an invaluable tool for studying kidney disease and injury, developing cell-based therapies, and testing new therapeutics. For such applications, large numbers of uniform organoids and highly reproducible assays are needed. We have built upon our previously published kidney organoid protocol to improve the overall health of the organoids. This simple, robust 3D protocol involves the formation of uniform embryoid bodies in minimum component medium containing lipids, insulin-transferrin-selenium-ethanolamine supplement and polyvinyl alcohol with GSK3 inhibitor (CHIR99021) for 3 days, followed by culture in knock-out serum replacement (KOSR)-containing medium. In addition, agitating assays allows for reduction in clumping of the embryoid bodies and maintaining a uniform size, which is important for reducing variability between organoids. Overall, the protocol provides a fast, efficient, and cost-effective method for generating large quantities of kidney organoids.
Introduction
In recent years, a number of protocols to differentiate human pluripotent stem cells into kidney organoids have been developed1, 2, 3, 4, 5. Kidney organoids have provided an important tool to aid research into new regenerative medicine approaches, model kidney-related diseases, perform toxicity studies and therapeutic drug development. Despite their wide applicability, kidney organoids have limitations such as lack of maturation, limited long-term culture capacity in vitro, and a paucity of several cell types found in the human kidney6, 7, 8. Recent work has focused on improving the level of organoid maturation, extending the culture periods and expanding the complexity of kidney cell populations by modifying the existing protocols9, 10, 11, 12. In this present iteration of our established protocol5, 13, we have modified the medium components in the first stage of the protocol to a serum-free base medium supplemented with insulin-transferrin-selenium-ethanolamine (ITSE), lipids, polyvinyl alcohol (E5-ILP) and CHIR99021 (Figure 1). These changes provide a fully-defined, serum-free, low-protein medium, with less components than our previous medium composition5, 13 and without additional growth factors. As a result, the first stage medium is less labor-intensive to prepare than our previously published version, and may reduce batch-to-batch variability5. Previous studies have shown that both insulin and transferrin are important in serum-free culture14, 15, however, high levels of insulin can be inhibitory to mesoderm differentiation16. We have maintained the low insulin levels as provided in the original protocol, and further reduced levels of KOSR (containing insulin) in second stage of the assay. In line with other protocols for kidney organoid formation, lower levels of KOSR are beneficial to maintaining a balance between proliferation and differentiation of the kidney tissue17. In addition, we have lowered the glucose concentration in our Stage II medium13.
Figure 1: Protocol overview.
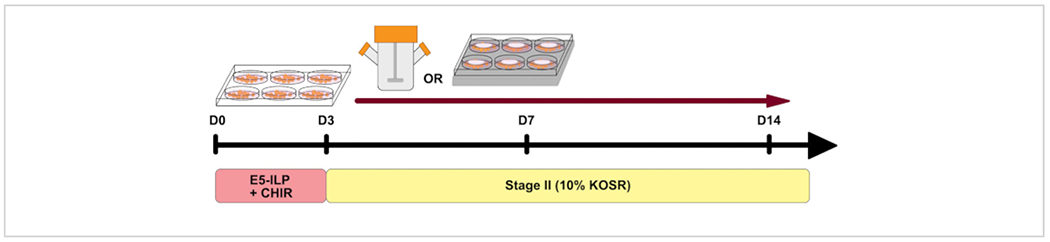
Schematic overview of the protocol showing timing of the two stages and use of spinner flasks and 6MSP. Please click here to view a larger version of this figure.
Our method describes a setup for suspension assay of kidney organoids, yielding up to ~1,000 organoids from an initial ~60% confluent hPSC 100 mm culture plate as described in the original publication5, 13. This protocol can be easily scaled up to starting with multiple 100 mm or 150 mm plates to further increase the organoid numbers.
Protocol
All experiments using hPSCs were performed in compliance with institutional guidelines, and were carried out in a Class II biosafety hood with appropriate personal protective equipment. All reagents are cell culture-grade unless stated otherwise. All cultures are incubated at 37 °C, 5% CO2 air atmosphere. At all stages of the assay, embryoid bodies or kidney organoids can be collected, and fixed or prepared for analysis. The hPSC lines used to generate this data have been fully characterized and published18.
1. Preparing culture plates
NOTE: Approximately 1 h prior to splitting hPSCs, coat 2 x 100 mm tissue culture plates with a stem cell qualified basement membrane matrix extract (BME). One may pre-coat the plates, seal them with a paraffin film and store at 4 °C according to manufacturers’ instructions.
Prepare 2 x 100 mm tissue culture-treated plates (1 for kidney organoid assay, 1 to maintain the cell line) and a 15 mL conical tube in the Class II biosafety hood.
Aliquot 8 mL of cold, serum-free Dulbecco’s Modified Eagle Medium (DMEM) into a 15 mL conical tube and ~4 mL into each of the 100 mm plates, enough to cover the bottom of each plate with medium.
-
Take a 100 μL aliquot of BME out of the freezer (−20 °C). Using a 2 mL serological pipette, take ~1 mL of cold DMEM from the 15 mL conical tube. Slowly thaw the BME aliquot by gently pipetting up and down with the cold DMEM, avoiding making bubbles.
NOTE: Do not let BME aliquot sit at room temperature. Use immediately.
Transfer the thawed DMEM/BME back into the 15 mL conical tube with the remaining DMEM. With a 10 mL serological pipette, gently mix the diluted BME by pipetting up and down at least 8 times to evenly disperse the BME, avoiding making bubbles.
-
Transfer 4 mL of the diluted BME into each plate with DMEM and gently swirl the plate so that the BME is evenly distributed. Incubate the coated plate for 1 h at room temperature or 30 min at 37 °C.
NOTE: Use 50 μL of BME per 100 mm plate. Use of other hPSC culture media and cell lines may require different concentrations of BME.
2. Passaging hPSCs
NOTE: For routine hPSC culture, passage cell lines at 70-80% confluency.
Aspirate the culture medium from the hPSC plate to be passaged. Add ~ 8 mL of Dulbecco’s phosphate-buffered saline (DPBS) to the hPSC plate and gently swirl to wash the cells.
-
Aspirate DBPS and add 2 mL of gentle cell dissociation reagent (GCDR) to the 100 mm plate, drop by drop on top to cover the cells.
NOTE: Other dissociation reagents may also be used. Adjust accordingly.
-
Incubate at room temperature for ~6-8 min until the colonies are breaking up and cells are refractive under phase contrast (Figure 2A).
NOTE: The timing may vary between cell lines. Adjust accordingly.
While incubating, prepare a 50 mL conical tube. Add 16 mL of hPSC medium (8 mL per 100 mm plate) and add Rho-associated kinase inhibitor (ROCKi) to a final concentration of 5 μM.
Aspirate DMEM from the BME-coated plates, and add 8 mL of hPSC medium plus ROCKi to each plate.
-
When the cells are ready (as described in point 2.3, Figure 2A), aspirate the GCDR and tilt the plate ~45° towards the experimenter and scrape the cells with a cell lifter.
NOTE: If cells are detatching, omit aspirating GCDR and proceed.
Turn the plate ~90° and scrape again to lift the remaining cells. Keep the plate ~45° and wash the cells down with 3 mL of hPSC medium using a 10 mL serological pipette.
-
Gently pipette up and down to break up large clumps (no more than 2-3 times) and seed the cells at the appropriate ratio for the cell line of interest onto the prepared plates. Place the plate with the cells in the incubator and move the plate gently in figure eight motions to distribute the cells evenly.
NOTE: In this experiment hPSC lines were split at a ratio of 1:5, this may vary for other cell lines and conditions. Leave the plate undisturbed over night.
After ~ 24 h, examine the cells for attachment. Look for small individual colonies attached. Aspirate the spent medium and replenish with 8 mL of fresh hPSC medium (no ROCKi added).
-
Continue observing and feeding daily until the cells reach ~60% confluency to start the kidney organoid assay (usually reached 48 to 72 h post passaging). The colonies will ideally be discrete and not merging (Figure 2B).
NOTE: It is very important to limit the cells to no more than 80% confluency in order to maintain their pluripotency state. Confluent cultures, rough handling or higher passages may lead to unwanted spontaneous differentiation or low efficiency of kidney organoid formation.
Figure 2: Stages of the protocol.

(A) Bright-field image of hPSC colony treated with GCDR. (B) Optimal confluency, and colony size to begin a kidney organoid assay. (C) hPSCs treated with dispase for 6 minutes. Red arrows point to edges of the colonies curling up. (D) Organoid assays on an orbital shaker. (E) Use of 200 μm cell strainer to sieve out large embryoid bodies. (F) Embryoid bodies at day 3 (D3) before transferring to Stage II medium. (G) Emergence of tubule formation can be observed at day 8 (D8) and (H) optimal timepoint for organoid harvesting and treatment at day 14 (D14). (I) Spinner flask used for bulk culture on a multi-position magnetic plate. (J) Assay on a multi-well magnetic stir plate. Scale bars, 200 μm. Please click here to view a larger version of this figure.
3. Day 0 - Setting up the kidney organoid assay
-
Before starting, prepare both the E5-ILP and Stage II media as per formulations (Table 1 and Table 2).
NOTE: The media can be stored for up to 14 days at 4 °C.
For one kidney organoid assay (one 100 mm culture plate is needed for one 6-well plate), prepare complete E5-ILP medium in a 50 mL conical tube: 18 mL of E5-ILP supplemented with 8 μM CHIR99021 (14.4 μL), 3.3 μM ROCKi (6 μL), 0.1 mM beta-mercaptoethanol (32.7 μL).
Place 2 mL of complete E5-ILP medium into each well of a 6 well ultra-low attachment plate.
-
Wash hPSCs at ~60 % confluency (Figure 2B) twice with ~ 8 mL of DPBS. Aspirate DPBS then add 2 mL of dispase per 100 mm plate, drop by drop to cover the cells and incubate for 6 min at 37 °C.
NOTE: After 6 min, the edges of the colonies will start to curl up (Figure 2C, red arrows) while the rest of the colony remains attached. If this is not obtained after 6 min, place the cells back into the incubator for additional 30 s. Other hPSC media and matrix may not be compatible with this timing. Laminin based BME coating is not compatible with dispase. If laminin based BME are the standard hPSC matrix, coat one of the plates in section 1 with the BME described in this method to be used for the kidney organoid assay.
-
Wash cells 3x with ~10 mL of DPBS. Aspirate DPBS then tilt the plate ~45° and scrape down with a cell lifter.
NOTE: Dispase is not deactivated, hence it needs to be washed out thoroughly. Do not reduce the number of washes.
Wash the colonies down from the top with 6 mL of complete E5-ILP medium using a 10 mL serological pipette. Pipette up and down gently to break up any large colonies (2 or 3 times is usually enough).
-
Distribute the colony clusters evenly by adding 1 mL per well into the 6-well plate. Place the plate on an orbital shaker (settings: orbital = 30, reciprocal = 330°, vibration = 5° - 2 s) that is placed in the 37 °C incubator (Figure 2D).
NOTE: The vibration feature is important for adequate distribution of organoids and to prevent clumping.
Table 1: E5-ILP medium composition.
Pipette all the reagents except the chemically defined lipids and anti-mycoplasma reagent directly into the upper chamber of a 0.22 μm Stericup filtration unit. After filtration, add the lipids and anti-mycoplasma reagent. Store at 4 °C for up to two weeks.
| Reagent | Stock conc. | Working conc. | Amount per 250 mL |
|---|---|---|---|
| TeSR-E5 | n/a | n/a | 238.48 mL |
| PVA | 10% | 0.25% | 6.25 mL |
| Pen-Strep | 100x | 1x | 2.5 mL |
| ITSE | 100x | 0.1x | 250 μL |
| Chemically defined Lipids | 100x | 1x | 2.5 mL |
| Plasmocin | 25 mg/mL | 2.5 μg/mL | 25 μL |
Table 2: Stage II medium composition.
Pipette all the reagents except and anti-mycoplasma reagent directly into the upper chamber of a 0.22 μm Stericup filtration unit. Once filtered, add anti-mycoplasma reagent. Store at 4 °C for up to two weeks.
| Reagent | Stock conc. | Working conc. | Amount per 500 mL |
|---|---|---|---|
| DMEM (Low Glucose) | n/a | n/a | 417.5 mL |
| KOSR | n/a | 10% | 50 mL |
| PVA | 10% | 0.25% | 12.5 mL |
| Pen-Strep | 100x | 1x | 5 mL |
| MEM-NEAA | 100x | 1x | 5 mL |
| GlutaMAX | 100x | 1x | 5 mL |
| HEPES | 100x | 1x | 5 mL |
| Plasmocin | 25 mg/mL | 2.5 μg/mL | 50 μL |
4. Day 2 - Feeding by half-medium change
NOTE: Within the 48 h, colony clusters will form embryoid bodies.
-
Prepare the complete medium: For one 6-well plate prepare 12 mL of E5-ILP medium + 8 μM CHIR99021 in a 15 mL conical tube.
NOTE: Beta-mercaptoethanol and ROCKi are not required.
-
Let the embryoid bodies settle at the bottom of the plate, tilt the plate ~45° then aspirate the medium slowly from the top, leave ~1 mL per well.
NOTE: Embryoid bodies at this stage clump rapidly. Do not leave them to settle for > 5 min.
Add 2 mL of prepared complete medium (section 4.1) per well. Return the plate back onto the shaker.
5. Day 3 - Transfer of embryoid bodies to Stage II medium
Prepare a 50 mL conical tube and DMEM (low glucose). Let the embryoid bodies settle at the bottom of the plate. Tilt the plate ~45° and aspirate the medium from the top slowly, leave ~1 mL per well.
Collect all the embryoid bodies carefully from each well using a 10 mL serological pipette and transfer them to the 50 mL conical tube.
Wash each well to collect any remaining embryoid bodies with ~ 1 mL of DMEM (low glucose) and add them to the same 50 mL conical tube.
-
Leave the embryoid bodies to settle to the bottom of the tube, ~5 min. While waiting, add 2 mL of Stage II medium to each well of the 6-well plate. Seive out large embryoid bodies (>300 μm) using a 200 μm cell strainer (Figure 2E).
Use a new 50 mL conical tube and place the 200 μm cell strainer on top. Pipette all of the embryoid bodies using a 10 mL serological pipette carefully over the cell strainer.
Rinse the cell strainer with an additional ~5 mL of DMEM (low glucose) to collect any embryoid bodies stuck in the cell strainer. Allow the embryoid bodies to settle to bottom of the conical tube.
When the embryoid bodies are settled, aspirate the supernatant and wash with ~10 mL of DMEM (low glucose).
Aspirate DMEM and re-suspend the embryoid bodies in 6 mL of Stage II medium.
Transfer the embryoid bodies back into the 6 well ultra-low attachment plate, distributing them evenly among the 6 wells.
-
Carry out half medium changes as described in steps 4.2 and 4.3 every other day.
NOTE: From day 3 onwards, the embryoid bodies will have a ‘golden’ and smooth, spherical appearance (Figure 2F). From ~ day 6, tubule formation in individual embryoid bodies will become apparent, with increasing numbers over the following days reaching optimum numbers and growth by day 14 (Figure 2G,H). To eliminate occasional clumping forming, upon visually observing the kidney organoids, or very small embryoid bodies without tubules, sieve out the <200 and large >500 μm organoids with a 500 and 200 μm cell strainers as described in steps 5.4.1 and 5.4.2.
6. Transfer to spinner flask and feeding
NOTE: A spinner flask may be used anytime from day 3 onwards for experiments that require large numbers of organoids. Routine transfer of organoids happens in our lab between days 6-8. Please see the Discussion section for alternatives if equipment is not available.
Transfer embryoid bodies into a 125 mL spinner flask with 45 mL of Stage II medium. Set magnetic stirrer speed to 120 rpm and place into the incubator (Figure 2I).
To feed embryoid bodies or kidney organoids, let the kidney organoids settle briefly to the bottom of the spinner flask. Lift the lid from one side arm of the flask and place the aspirating pipette inside, with the tip touching the opposite inside wall.
Slowly angle the aspirating pipette down and aspirate approximately half of the medium. Replenish with 20 mL of fresh Stage II medium by pipetting it through the same opening.
7. Setting up 6-well magnetic stir plate (6MSP)
NOTE: The 6MSP format may be used in place of spinner flasks if multiple conditions need to be tested. Use the 6MSP for compound or nephrotoxin treatments. This saves the amount of medium used in the second stage while maintaining nutrient availability through diffusion.
Clean the oval magnetic stir bars in a 50 mL conical tube by washing in a tissue culture suitable detergent briefly (if never used) or soak for > 1 h if previously used.
Briefly wash 3x in sterile DPBS.
Wash 1x for 5 min in 70% ethanol, 1x in sterile DPBS.
Rinse with anti-adherence solution and wash 1x in sterile DPBS and aspirate.
Carefully, using long sterile forceps place one magnetic stir bar into each well of the 6-well plate with embryoid bodies or kidney organoids.
-
Place the plate onto the 6MSP and set the speed to 120 rpm (Figure 2J). Maintain kidney organoids with half medium change as per section 4.2 and 4.3.
NOTE: In order for the magnetic stir bars to snap into position and start spinning, you may need to first put the power level to 100 briefly, then once they are all spinning, bring the power level down to 25.
Representative Results
In this most recent version of our protocol, kidney organoid differentiation is initiated in a defined, low protein medium. The assays are performed entirely in suspension and rely on the innate ability of hPSCs differentiation and organization for initiation of tubulogenesis. A single assay originating from a 100 mm ~60% confluent hPSC culture plate routinely yields 500-1,000 kidney organoids, as shown in our previous publication5. Due to such high numbers of organoids generated, this protocol is well suited for compound testing. We routinely use a 6-well format for compound testing however, this protocol can easily be scaled in the second stage (day 3 onwards) to other multi-well formats for higher-throughput compound testing. Immunofluorescence of paraffin sections shows presence of nephron segments in the organoids, i.e. renal tubules expressing Hepatocyte Nuclear Factor-1 beta (HNF1B) and Lotus Tetragonolobus Lectin (LTL) (Figure 3A - HNF1B, LTL), and podocyte clusters expressing V-maf Musculoaponeurotic Fibrosarcoma oncogene homolog B (MAFB) and nephrin (NPHS1) (Figure 3A - MAFB, Figure 3B - NPHS1). Furthermore, the modifications in this protocol can support expansion of endothelial cells as seen in Figure 3B showing staining with Platelet and Endothelial Cell Adhesion Molecule 1 (PECAM1) at day 26 of culture.
Figure 3: Expected results.
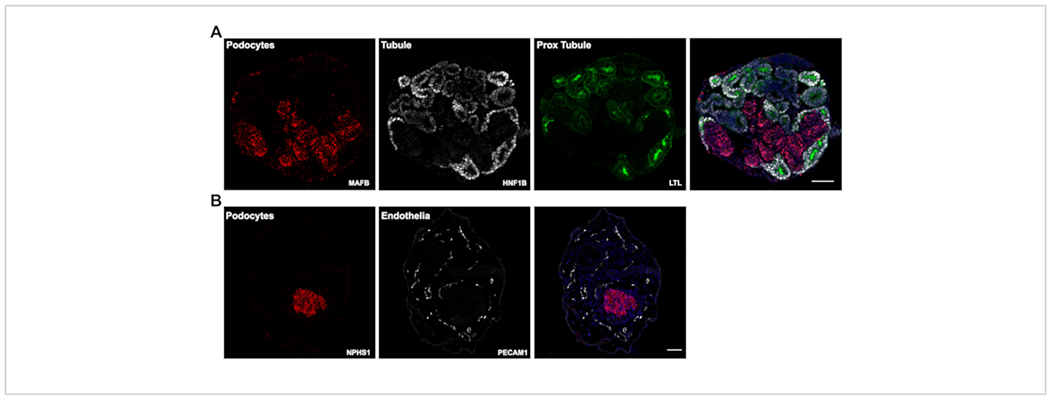
(A) Representative confocal images of immunofluorescently labeled paraffin sections of day 14 kidney organoids showing positive staining for tubule epithelia (HNF1B and LTL) and podocyte clusters (MAFB). (B) Day 26 kidney organoid sections labeled for podocyte clusters (NPHS1) and endothelial cells (PECAM1). Scale bars, 100 μm (A); 200 μm (B). Please click here to view a larger version of this figure.
Discussion
Previous studies have shown that the initial protocol steps are critical for intermediate mesoderm differentiation5, 19, 20 and, therefore, it is essential to implement a stringent medium composition at this stage. Removing undefined components such as serum, albumin, protein free hybridoma medium II from the first stage of the protocol may help to improve consistent differentiation efficiency between assays21.
The metabolic state of kidney cells is critical to their function, and glucose changes can lead to altered metabolic state22. Previous studies have described that high levels of glucose (up to 25 mM) can induce endothelial cell dysfunction and alter growth and oxidant capacity of kidney cells22, 23, 24. High levels of glucose have also been described to alter mitochondrial function24, which may be unfavorable when investigating kidney disease and nephrotoxicity or performing drug discovery using kidney organoids. We have, therefore, reduced the level of glucose in our protocol to promote a more in vivo-like metabolic state of the organoid kidney cells. As a result, the modifications to the kidney organoid assay provide a consistent, robust protocol, while maintaining its simplicity.
Kidney organoids are immature, and extended culture (>20 days) may lead to incidence of pro-fibrotic and non-renal cell types as previously described5, 25, leaving organoids less representative of healthy human kidney tissue. Based on our experience, the optimal treatment window, where the kidney organoids are at their healthiest is between days 14-18. Use of spinner flasks and multi-well magnetic stirrers as described above will enhance uniform nutrient availability as opposed to static culture21, 26. If the equipment for suspension culture such as the shaker or magnetic stirrers are not available, this protocol can still be carried out completely in the ultra-low attachment plates in static culture. There may however be increased incidence of embryoid bodies/organoid merging, leading to large specimens with necrotic cores due to hypoxia. Any organoids larger than 500 μm can be removed by using the cell strainers described. To reduce the chance of merging of the organoids in those cases, we suggest not seeding more than 100 organoids per 6-well. In addition, following feeding, the organoids should be evenly distributed by performing figure eight motions with the plate.
Low efficiency (<50%) of organoid formation may be observed. This usually occurs when the hPSC cultures have reached high confluency (>80%) during standard passaging. It is critical that hPSC maintenance is consistent and cells are not left to become over-confluent. High confluency and inconsistent passaging technique may also lead to spontaneous differentiation and increased cell death. If differentiation is present in the hPSC culture, we recommend removing the differentiated areas by aspirating with a fine pipette tip if it does not exceed 5% of the cell population, prior to starting the assay. If the differentiation areas exceed 5%, we recommend that a new batch of hPSCs is thawed and split at least once before starting a new assay.
We have observed that some hPSC lines are more prone to form non-renal cell types, such as cardiac or neural tissue. If this occurs, size filtration using the cell strainers may help to remove those organoids that contain non-renal outgrowths. Alternatively, changing the hPSC medium and/or matrix may help to reduce the non-renal outgrowths. From our experience, alternative hPSC media containing minimum components, and BME such as vitronectin, provide a more stringent pluripotent niche and thus help generate more homogeneous hPSC cultures.
Acknowledgments
This research was funded by the National Institutes of Health R01 DK069403, UC2 DK126122 and P30-DK079307 and ASN Foundation for Kidney Research Ben J. Lipps Research Fellowship Program to AP.
Footnotes
A complete version of this article that includes the video component is available at http://dx.doi.org/10.3791/62452.
Disclosures
Authors have nothing to disclose.
References
- 1.Takasato M et al. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature. 526 (7574), 564–568, (2015). [DOI] [PubMed] [Google Scholar]
- 2.Freedman BS et al. Modelling kidney disease with CRISPR-mutant kidney organoids derived from human pluripotent epiblast spheroids. Nature Communications. 6, 8715 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morizane R et al. Nephron organoids derived from human pluripotent stem cells model kidney development and injury. Nature Biotechnology. 33 (11), 1193–1200 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taguchi A et al. Redefining the in vivo origin of metanephric nephron progenitors enables generation of complex kidney structures from pluripotent stem cells. Cell Stem Cell. 14 (1), 53–67 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Przepiorski A et al. A simple bioreactor-based method to generate kidney organoids from pluripotent stem cells. Stem Cell Reports. 11 (2), 470–484 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Freedman BS et al. Modelling kidney disease with CRISPR-mutant kidney organoids derived from human pluripotentepiblast spheroids. Nature Communication. 6, 8715 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morizane R et al. Nephron organoids derived from human pluripotent stem cells model kidney development and injury. Nature Biotechnology. 33 (11), 1193–1200 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takasato M et al. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature. 526 (7574), 564–568 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Taguchi A, Nishinakamura R Higher-order kidney organogenesis from pluripotent stem cells. Cell Stem Cell. 21 (6), 730–746 e736 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Uchimura K, Wu H, Yoshimura Y, Humphreys BD Human pluripotent stem cell-derived kidney organoids with improved collecting duct maturation and injury modeling. Cell Reports. 33 (11), 108514 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Howden SE, Little MH Generating kidney organoids from human pluripotent stem cells using defined conditions. Methods in Molecular Biology. 2155, 183–192 (2020). [DOI] [PubMed] [Google Scholar]
- 12.Tanigawa S et al. Activin is superior to BMP7 for efficient maintenance of human iPSC-derived nephron progenitors. Stem Cell Reports. 13 (2), 322–337 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sander V et al. Protocol for large-scale production of kidney organoids from human pluripotent stem cells. STAR Protocols. 1 (3), 100150 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ekblom P, Thesleff I, Miettinen A, Saxen L Organogenesis in a defined medium supplemented with transferrin. Cell Differentiation. 10 (5), 281–288 (1981). [DOI] [PubMed] [Google Scholar]
- 15.Thesleff I, Ekblom P Role of transferrin in branching morphogenesis, growth and differentiation of the embryonic kidney. Journal of Embryology and Experimental Morphology. 82, 147–161 (1984). [PubMed] [Google Scholar]
- 16.Freund C et al. Insulin redirects differentiation from cardiogenic mesoderm and endoderm to neuroectoderm in differentiating human embryonic stem cells. Stem Cells. 26 (3), 724–733 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Nishikawa M et al. An optimal serum-free defined condition for in vitro culture of kidney organoids. Biochemistry and Biophysics Research Communication. 501 (4), 996–1002 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Oh JK et al. Derivation of induced pluripotent stem cell lines from New Zealand donors. Journal of the Royal Society of New Zealand. 1–14, (2020). [Google Scholar]
- 19.Takasato M et al. Directing human embryonic stem cell differentiation towards a renal lineage generates a self-organizing kidney. Nature Cell Biology. 16 (1), 118–126 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Lam AQ et al. Rapid and efficient differentiation of human pluripotent stem cells into intermediate mesoderm that forms tubules expressing kidney proximal tubular markers. Journal of American Society of Nephrology. 25 (6), 1211–1225 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bratt-Leal AM, Carpenedo RL, McDevitt TC Engineering the embryoid body microenvironment to direct embryonic stem cell differentiation. Biotechnology Progress. 25 (1), 43–51 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Imasawa T et al. High glucose repatterns human podocyte energy metabolism during differentiation and diabetic nephropathy. FASEB Journal. 31 (1), 294–307 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim KA et al. High glucose condition induces autophagy in endothelial progenitor cells contributing to angiogenic impairment. Biological and Pharmaceutical Bulletin. 37 (7), 1248–1252 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Piwkowska A, Rogacka D, Audzeyenka I, Jankowski M, Angielski S High glucose concentration affects the oxidant-antioxidant balance in cultured mouse podocytes. Journal of Cellular Biochemistry. 112 (6), 1661–1672 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Wu H et al. Comparative analysis and refinement of human PSC-derived kidney organoid differentiation with single-cell transcriptomics. Cell Stem Cell. 23 (6), 869–881 e868 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lei X, Deng Z, Duan E Uniform embryoid body production and enhanced mesendoderm differentiation with murine embryonic stem cells in a rotary suspension bioreactor. Methods in Molecular Biology, Clifton, N.J(2016). [DOI] [PubMed] [Google Scholar]