

Abstract
Objective:
The astroglial enzyme glutamine synthetase (GS) is deficient in small loci in the brain in adult patients with different types of focal epilepsy; however, the role of this deficiency in the pathogenesis of epilepsy has been difficult to assess due to a lack of sufficiently sensitive and specific animal models. The aim of this study was to develop an in vivo approach for precise and specific deletions of the GS gene in the postnatal brain.
Methods:
We stereotaxically injected various adeno-associated virus–Cre recombinase (AAV-Cre) constructs into the hippocampal formation and neocortex in 22- to 70-week-old GSflox/flox mice to knock out the GS gene in a specific and focal manner. The mice were subjected to seizure threshold determination, continuous video-EEG recordings, advanced in vivo neuroimaging, and immunocytochemistry for GS.
Results:
The construct AAV8-Glial Fibrillary Acidic Protein-Green Fluorescent Protein-Cre (AAV8-GFAP-GFP-Cre) eliminated GS in >99% of astrocytes in the injection center with a gradual return to full GS expression toward the periphery. Such focal GS deletion reduced seizure threshold, caused spontaneous recurrent seizures, and diminished functional connectivity.
Significance:
These results suggest that small loci of GS deficiency in the postnatal brain are sufficient to cause epilepsy and impaired functional connectivity. Additionally, given the high specificity and precise spatial resolution of our GS knockdown approach, we anticipate that this model will be extremely useful for rigorous in vivo and ex vivo studies of astroglial GS function at the brain-region and single-cell levels.
Keywords: Epilepsy, glutamate-glutamine cycle, metabolism, neurotransmission
Introduction
The enzyme glutamine synthetase (GS) is abundantly expressed in astrocytes and catalyzes the conversion of ammonia and glutamate to glutamine1, 2. GS may eliminate or reduce the toxic effects of glutamate and ammonia in the central nervous system by metabolizing these compounds. Moreover, multiple physiological processes, including glutamate and gamma-aminobutyric acid (GABA) synthesis, protein synthesis, and osmoregulation, rely on a steady supply of glutamine3. Hence, changes in the expression and activity of GS are expected to significantly impact central nervous system physiology.
There is growing evidence that diminished GS activity is implicated in the pathogenesis of several types of epilepsy. The most striking evidence comes from Haberle and colleagues, who described a small number of infants with loss-of-function mutations of the GS gene throughout the body. All the infants suffered from severe epileptic encephalopathy, and many died shortly after birth4–6. Additionally, several studies have identified GS deficiencies in small loci in the brain in adults with mesial temporal lobe epilepsy7, 8, neocortical epilepsy9, and glioblastoma-associated epilepsy10. These deficiencies are restricted to small regions of the hippocampal formation7, 8, the amygdala9, and the neoplastic tissue10, respectively, and they are likely acquired at some point during postnatal life. While a genetic loss of GS activity throughout the body clearly causes epilepsy4–6, the consequences of focal GS deficiencies in the adult brain remain incompletely understood.
The current approach for studying focal GS deficiencies in the postnatal central nervous system has been to microinject GS inhibitors, particularly methionine sulfoximine (MSO)11, into the brain of rodents. Using this approach, we found that inhibiting GS in the entorhinal-hippocampal region of rats results in recurrent seizures and neuropathological features that in many cases resemble human mesial temporal lobe epilepsy12–16. However, even though MSO is a potent inhibitor of GS, the chemical has several other effects, such as lowering tissue glutathione levels17, increasing astroglial glutamine content18, 19, and exciting neurons via a GS-independent mechanism20. Additionally, MSO diffuses throughout the injected tissue making it difficult to control and precisely confine the region of GS inhibition. This is concerning because the GS deficiency in human epilepsy is often restricted to minute brain regions such as the CA1, CA3, and the dentate hilus of the hippocampal formation7, 8.
To overcome these limitations, we developed a novel in vivo model of precisely targeted GS deletions that can be induced at any postnatal stage. We microinjected various adeno-associated virus – Cre recombinase (AAV-Cre) constructs into the postnatal brain of transgenic GSflox/flox mice and then assessed GS knockout efficiency, seizure threshold, spontaneous seizure activity, cerebrovascular reactivity, structural changes, and functional connectivity. Our working hypothesis was twofold: first, that this deletion strategy would effectively eliminate GS in precisely targeted regions of the postnatal brain; second, that GS deletion in parts of the hippocampus and neocortex would produce decreased seizure threshold, spontaneous recurrent seizures, and impaired functional connectivity, consistent with the idea that small loci of GS deficiency in the postnatal brain are sufficient to cause epilepsy. If our hypothesis is correct, then manipulations of astroglial GS activity may represent a novel antiepileptogenic or disease-modifying therapy. This is significant because no such therapies are available for human use.
Materials and methods
Animals
All animal care and use procedures were approved by the Institutional Animal Care and Use Committee of Yale University, and experiments were performed in accordance with current guidelines. Mice were housed on a 12-hour light/dark cycle in individually ventilated cages at constant temperature (22 ± 0.7 °C) and humidity (56 ± 6%) with unlimited access to food and water ad libitum. Biopsies from ear or tail were collected and genotyped for floxed GS as described in 21. The creation of floxed C57BL/6J GS mice (GSflox/flox) is detailed in 21.
Viral vectors, antibodies, and chemicals
Site specific recombination of the LoxP sites was achieved by using one of several viruses expressing Cre recombinase and green fluorescent protein (GFP) under various promoters in two separate expression cassettes. The following Cre-expressing adeno associated viruses (AAVs), at concentrations ranging from 4.1 × 1012 viral genome/mL to 8.1 × 1012 viral genome/ml were used: (1) AAV5-CMV-GFP-Cre Legacy (SignaGen Laboratories, Rockville, MD), (2) AAV5-CMV-GFP-Cre [Gene Therapy Center Vector Core at the University of North Carolina (Chapel Hill, NC)], (3) AAV8-CMV-GFP-Cre (Gene Therapy Center Vector Core), and (4) AAV8-GFAP-GFP-Cre (Gene Therapy Center Vector Core). In addition, a corresponding control virus was ordered for each of the four experimental viruses. Each control virus was procured from the same source and was of identical construction to the corresponding experimental virus other than lacking the Cre expression cassette. When possible, all viruses were injected without prior dilution. However, if the dilutions of a corresponding Cre and non-Cre (control) virus did not match, one of the viruses was diluted with Dulbecco’s phosphate buffered saline (PBS) to achieve concentration match.
Viral microinjection
Mice (22 – 70 weeks old) were induced into anesthesia with 5% isoflurane (Baxter, Deerfield, IL) in O2 and placed in a stereotaxic frame (David Kopf Instruments, Tujunga, CA). For the duration of the surgery, isoflurane was maintained at 1.5 – 3% and titrated based on respiratory status and response to intermittent foot pinch. Burr holes were made into the skull bilaterally, and virus was injected into the brain parenchyma using the following previously validated coordinates with bregma as the reference: dentate gyrus: AP −3.2 mm, ML ±3.1 mm, DV −3.5 mm; subiculum: AP −3.7 mm, ML ±3.5 mm, DV −3.44 mm; and entorhinal cortex: AP −4.1 mm, ML ±3.8 mm, DV −3.5 mm. Virus was delivered stereotaxically using a 5 μL 26-gauge syringe (Hamilton, Reno, NV). At each of the six injection sites per animal, 0.5 μL of virus was injected at a rate of 0.05 μL per 30 seconds.
Immunohistochemistry
The primary antibodies included chicken anti-GFP (GFP-1010, Aves Labs, Tigard, OR) and rabbit anti-GS (G2781Sigma, St. Louis, MO). The secondary antibodies were Alexa Fluor 488 goat anti-chicken IgG (A11039) and Alexa Fluor 555 goat anti-rabbit IgG (A21428), and Alexa Fluor 647 goat anti-mouse (A21235), all Life Technologies (Eugene, OR). All other reagents, unless otherwise noted, were obtained from Sigma Chemical Co. (St. Louis, MO). For histology and immunocytochemistry, mice were fully anesthetized with isoflurane and perfused transcardially with 0.9% NaCl followed by 4% formaldehyde in 0.1 M phosphate buffer, pH 7.4 (PB) for 5 minutes. The brains were rapidly removed from the skull and postfixed in the same fixative overnight at 4 °C and then sectioned on a Vibratome at 50 μm. The details on microscopy are given in supplementary material.
Seizure threshold study
Ten weeks after viral injection, seizure precipitation studies were conducted by measuring the time to first clonic twitch and time to generalized convulsions following an intraperitoneal injection of the convulsant agent pentylenetetrazol (PTZ, Sigma Aldrich, St. Louis, MO) at a dose of 90 mg/kg (diluted to 9 mg/mL in normal saline). Ten weeks post injection was chosen because AAVs may take several weeks to replicate and fully express their genes in living tissue 22. Administration of PTZ is one of several well-established tests used to determine the seizure threshold in various experimental paradigms and to evaluate the efficacy of anticonvulsive drugs 23, 24. Following induction of convulsive seizures, animals (n = 15) were fully anesthetized using isoflurane and euthanized as described below. For this study, experimenters were blinded to the viral vector used in each animal when recording time to seizure events.
Electrode implantation and video-intracranial EEG monitoring
In the video-EEG study, 11 animals were injected with virus as described above, split equally between Cre-expressing and control viruses only of serotype AAV8 GFAP (based on findings of the viral comparison study; see Results section). About four weeks after viral injection surgery, recording electrodes were implanted. To evaluate and maximize the survival of the animals the electrodes were implanted in two different arrangements. One subset was implanted with unilateral screw electrodes over the parietal cortex and a reference screw electrode in the skull (n = 5). Another subset was implanted with a single recording depth electrode in the hippocampus, and a reference depth electrode in the white matter of the cerebellum (n = 6).
Four to 6 weeks after viral injection, the mice were subjected to continuous video-EEG monitoring using the CEEGraph Vision LTM (Natus/Bio-Logic Systems Corp) monitoring system, as detailed in Dhaher et al 15. This time was chosen because AAVs may take several weeks to replicate and fully express their genes in living tissue 22. Seizures were identified by visual inspection of the EEG record. As detailed in 25 seizures were defined by EEG characteristics and not just by the duration of the discharge. The video record was examined to stage the seizures, using a modification of Racine’s criteria 26, as follows: subclinical, no remarkable behavior; stage 1, immobilization, eye blinking, twitching of vibrissae and mouth movements; stage 2, head nodding, often accompanied by facial clonus; stage 3, forelimb clonus; stage 4, rearing; stage 5, rearing, falling and generalized convulsions. Subclinical seizures were excluded from the analysis.
Animals were monitored daily for signs of distress and were immediately removed from the recordings is such signs appeared (i.e., immobility, poor grooming, ruffled fur, did not drink or eat). Animals were also removed from the recordings in the rare event that the headcap detached. Because of these issues, the total EEG monitoring time varied from 4 to 12 days.
In vivo brain imaging
To evaluate bilateral effects of unilateral GS – inhibition the Cre-expressing and control viruses only of serotype AAV8 GFAP were injected unilaterally in the right hippocampus. For each animal, 18 total injections (each injection: 0.5 μL of virus was injected at a rate of 0.05 μL per 30 seconds) were performed in 6 locations [(AP: −1.4 mm, ML: 0.8 mm), (AP: 1.8 mm, ML: 0.8 mm), (AP: −2,45 mm, ML: 0.8 mm), (AP: −1.4 mm, ML: 1.3 mm), ( AP: −1.8 mm, ML: 1.7 mm), (AP: −2.45 mm, ML: 1.7mm)] and at 3 depths in each location (DV: 1.0 mm, 1.5 mm, 2.0 mm), resulting in injection sites in the cortex, the radiatum and oriens layers of the hippocampus, and the molecular layer of the dentate gyrus. This cohort consisted of 22 animals (11 experimental and 11 controls). All animals underwent conventional magnetic resonance imaging (MRI) for anatomical assessments and functional MRI (fMRI) for cerebrovascular reactivity and connectivity assessments.
We used functional connectivity density (FCD) maps to evaluate global changes in functional connectivity measured from resting state fMRI 27. The FCD of a voxel or region of interest (ROI) represents the total number of functional connections to other voxels above a certain threshold. As previously described 28, FCD maps were calculated using the in-house script written in MATLAB (The MathWorks, Inc., Natick, MA). Each image in an fMRI series was cropped and masked to remove non-brain signals. The time series in each voxel was then bandpass filtered (0.001 to 0.1 Hz) to exclude slow drift of the signal. Pearson’s correlation coefficients were calculated between every voxel of the brain to every other voxel, where the number of voxels correlated above a threshold of 0.6 was used to generate FCD maps for each fMRI series. Short-and long-range FCD maps were calculated using a radius of 2 mm surrounding a given voxel.
Statistics
Unless otherwise stated, statistical analyses and graph plotting were conducted in Igor Pro 8.02 (WaveMetrics, Portland, OR). Statistical significance was assessed by a Student’s t-test for most of the comparisons and by a Welch’s t-test for the seizure count comparison (the latter test was chosen because of unequal variance between the groups) 29. Differences between groups were judged to be significant when p-values were smaller than 0.05. For multiple comparisons, a two-tailed t-test with a random field theory approach was used to achieve an adjusted significance threshold of p < 0.05 29. Error bars indicate the mean ± standard error of the mean (SEM) except when stated. No samples or animals were excluded from the analyses.
Blinding
During the PTZ seizure precipitation studies, experimenters were blinded to the viral vector used in each animal when recording time to seizure events. During grading of the continuous intracranial EEG data and accompanying video recordings, experimenters were blinded to the viral vector used in each animal (Cre-expressing vs. control).
See Supplementary Methods for additional details.
Results
Visualizing the injected virus suspension in vivo
To develop our injection approach, we injected two mice with intracerebral injections of USPIO nanoparticles. After 4 days, MRI showed a region of decreased T2 relaxation time of up to 21.2 mm3 around the injection sites, including local spread in the hippocampus and surrounding neocortex (Fig. 1A), suggesting that the virus likely reached these regions using our injection approach.
Figure 1. Spatial distribution and astroglial specificity of the knockdown approach.

A. Virus suspension with added superparamagnetic iron oxide (USPIO) nanoparticles was injected into the right hippocampus as a single 0.5 μL bolus, followed by Perfusion Weighted (PW) Dynamic Susceptibility Contrast (DSC) MRI, 4 days later in 2 normal mice. The regions of darker blue coloring in the T2 images (arrows) indicate the presence of USPIO nanoparticles in the parenchyma, reflecting the local spread of the infusion. B. Representative horizonal section of the hippocampal formation of a GSflox/flox mouse injected with AAV8-GFAP-GFP-Cre virus. Immunofluorescence for green fluorescent protein (GFP, green color) represents virus-transfected cells, and immunofluorescence for glutamine synthetase (GS, red color) represents GS-expressing astrocytes. This shows effective deletion of GS by absent GS-immunoreactivity in the virus-transfected (green) cells. C. Area of GS knockout for each virus at various magnifications. AAV5 CMV and AAV8 GFAP outperformed the AAV5 CMV Legacy and AAV8 CMV viruses. D. Example of region-ofinterest (ROI) selection in a horizontal hippocampal section transfected with AAV5 CMV. ROIs 1–5 are placed in areas of knockout whereas ROIs 6–7 are used as control regions to scale intensity measures. Red stained areas are not infected with virus and express GS normally. E. Knockdown efficiency by each of the 4 viruses as assessed by densitometry of GS in the injected brain region. The efficiency is expressed as 1 – relative GS intensity (*p < 0.05 and **p < 0.001). F. Representative field from the hippocampus of a GSflox/flox mouse injected with AAV8-GFAP-GFP-Cre virus and immunostained for green fluorescent protein. Note the characteristic astrocyte morphology of nearly all the transfected cells. G. The proportion of green fluorescent protein-positive cells show astrocyte morphology after transfection with each of the 4 virus types. Scale bars: B, D = 0.2 mm, F = 25 μm. Abbreviations: CA1, CA3, Cornu Ammonis subfields of the hippocampus; DH, dentate hilus; EC, entorhinal cortex; Gr, granule layer of the dentate gyrus; Mol, molecular layer of the dentate gyrus; SUB, subiculum.
AAV8-GFAP-GFP-Cre injections can eliminate astrocytic GS in discrete brain regions
To evaluate the transfection efficiency of each of the viruses, we first stained injected brain sections for the GFP reporter molecule. GFP immunostaining was visible in the dentate gyrus, subiculum, and entorhinal cortex in all virus injected animals (Supplementary Fig. S1A). Despite maintaining consistent delivery volumes (0.5 μL) and similar virus concentrations (4.1 – 8.1 × 1012 viral genome/mL) at each injected site, the density and extent of GFP-positive cells varied substantially among the viruses (Supplementary Fig. S1B and Fig. S2A–D). This result suggests that different virus types have unique capabilities to diffuse through tissue or transfect cells in regions of lower viral concentration (i.e., at the periphery of the injection field). Our most striking observation was that the AAV8-GFAP construct transfected a substantially larger area than the AAV5-CMV and AAV8-CMV constructs (approximately 2.5 × 105 pixels vs. 1.2 × 105, 1.4 × 105, and 7.4 × 104 pixels, respectively). Additionally, AAV8 GFAP was highly selective for astrocytes (Supplementary Fig. S2D) whereas the other vectors infected both neurons and astrocytes (Supplementary Fig. S2A – C).
Next, we determined the efficiency of GS-deletion at the cell and tissue levels using each Cre-expressing virus. Regardless of the type of virus tested, virtually none (< 1%) of the transfected (GFP-positive) cells were positive for GS (Fig. 1B), meaning that once a cell was transfected at levels to detect GFP-positivity, GS remained effectively knocked out. When assessing the knockout efficiency at the regional level, the smallest areas of GS elimination was with the AAV5-CMV Legacy and AAV8-CMV constructs, while the largest areas occurred with the AAV5-CMV and AAV8-GFAP (Fig. 1B, C). The difference in knockout efficiency between the first two and last two viruses was statistically significant (respectively, 95% CIs 47.8% to 78.1% and 78.8% to 87.5%, p = 0.0003 and 0.000007). In addition, AAV8-GFAP yielded a greater decrease in fluorescence intensity than AAV5-CMV (p = 0.018, Fig. 1D, E). Fig. 1E shows that the knockdown efficiency was not statistically significant for AAV5 CMV Legacy (95% CI −14.5% to 2.4%, p=0.12) or AAV8 CMV (95% CI 10.7% to 2.9%, p=0.19). Both the AAV5 CMV virus and the AAV8 GFAP virus significantly knocked down GS (respectively, 95% CIs 47.8% to 78.1% and 78.8% to 87.5%, p = 0.0003 and 0.000007). In addition, the increased knock-down of AAV8 GFAP over AAV5 CMV was statistically significant (p = 0.018).
Finally, we assessed the astrocyte selectivity of each virus. As expected, AAV8-GFAP exhibited near perfect astrocyte specificity (98.7% of GFP-positive cells were astrocytes based on morphological criteria, Fig. 1F). In contrast, AAV5-CMV Legacy, AAV5-CMV, and AAV8-CMV targeted mostly neurons with varying degrees of astrocyte labeling detected (10.0%, 21.3%, and 12.7% astrocyte specificity, respectively, Fig. 1G)
Bilateral cortico-entorhinal-hippocampal GS deletion lowers the threshold for acute seizures
Because inhibition or deficiency in GS may be a causative factor for seizures and epilepsy, we assessed the sensitivity of the GS deleted mice to provoked seizures using the intraperitoneal pentylenetetrazole (PTZ) approach. 23, 24We injected fifteen mice in both entorhinal-hippocampal regions with one of the four experimental viruses (2 AAV8-GFAP; 2 AAV8-CMV; 2 AAV5-CMV; 2 AAV5-CMV Legacy; n = 9) or four control viruses (3 AAV8-GFAP; 1 AAV8-CMV; 1 AAV5-CMV; 1 AAV5-CMV Legacy; n = 6), and then administered PTZ eight weeks later. Mice injected with Cre-expressing AAVs exhibited the first clonic twitch after approximately 41.2 ± 3.2 seconds (mean ± SEM), which was significantly less than animals injected with control virus (average 65.83 ± 12.9 seconds, p=0.044, Fig. 2A). However, the time to the first convulsive seizure was not statistically significant, though there was a nonsignificant trend for AAV-Cre injected animals to convulse faster (74.2 ± 9.4 seconds vs. 100.0 ± 18.0 seconds, p = 0.20, Fig. 2A). We did not have a sufficiently large number of animals to detect statistically significant differences in seizure threshold among the 4 experimental viruses.
Figure 2. Seizure threshold and spontaneous seizures.
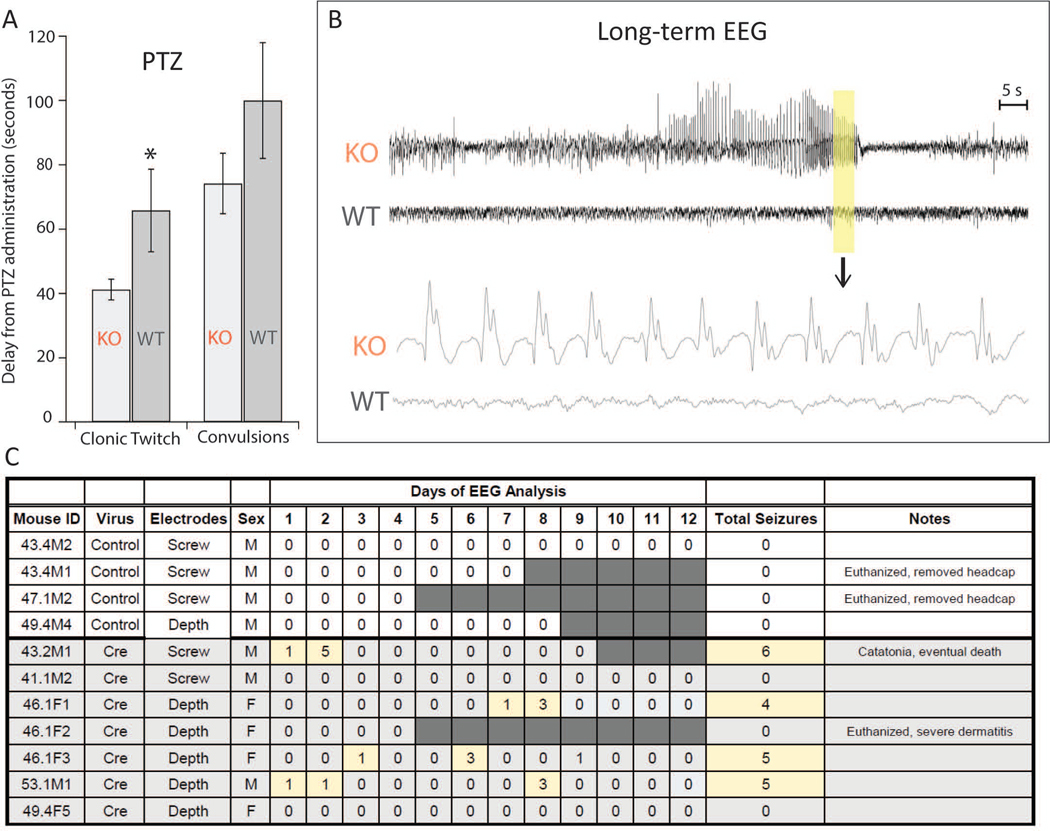
A. Pentylenetetrazole (PTZ)-induced seizures. GS knockout mice (KO, n = 9) demonstrate increased susceptibility to provoked seizures, as they have a shorter time to clonic twitch compared to controls (WT, n = 6, p < 0.05). There was a similar trend in time to convulsion, though this effect was not significant (p = 0.20). B. Representative examples of intracranial EEG recordings during a Racine stage 3 seizure in a GS-knockout mouse (KO). The two upper traces are from a KO mouse and control virus injected “wild type” (WT) mouse. The highlighted area is shown in the expanded traces below. Note the presence of rhythmic seizure activity in the KO mouse and normal EEG activity in the WT mouse. The Racine stage was determined from the concurrent video recording. C. Daily number of electrographic seizures encountered in control (n = 4) and Cre-injected (n = 7) mice over a continuous 4- to 12-day-period starting 4–6 weeks after infusion of AAV8-GFAP-GFP (control) or AAV8-GFAP-GFP-Cre (Cre) into the dentate gyrus of both hippocampi. The days with seizures are highlighted in yellow and days without EEG recording are highlighted in dark grey.
Bilateral entorhinal-hippocampal GS deletion causes spontaneous recurrent seizures
We next used continuous video-intracranial EEG recordings to determine whether deleting GS in bilateral entorhinal-hippocampal cortices would induce spontaneous recurrent seizures. We recorded from 6 animals injected with Cre expressing AAV8-GFAP and 4 animals injected with non-Cre expressing AAV8-GFAP for a continuous period of 4 to 12 days (Fig. 2B, C). The remaining animals did not survive the electrode implantation surgery. Spontaneous seizures occurred in 4/7 (57%) of the animals injected with Cre-expressing virus and in 0/4 (0%) of the animals injected with control virus (Fig. 2B, C). The seizures predominantly occurred in clusters, as evidenced by 11 of the 19 seizures occurring within 2-hours of another seizure in the same animal. Moreover, a large fraction of seizures (14/19, 74%) occurred between midnight and 8:00 in the morning. Common seizure behaviors included facial automatisms, tail extension, lordosis, rearing, and uncontrolled jumping.
Unilateral cortico-entorhinal-hippocampal GS deletion reduced functional connectivity
Previously, we demonstrated that embryonic deletion of cortical GS caused impaired cerebrovascular reactivity21. In addition, chemical inhibition of hippocampal GS in rats led to significant changes in fractional anisotropy throughout the brain30, and functional magnetic resonance imaging (fMRI) studies of focal epilepsy patients and animal models often show decreased functional connectivity31, 32. Here we sought to determine whether deleting GS in one hippocampus in adult mice would produce similar findings. Using blood oxygen level dependent (BOLD) imaging of GS-deficient (AAV8-GFAP-Cre-injected) vs. GS-intact brains, we found significant decreases in global functional connectivity across various regions of the brain in GS-deficient mice (Fig. 3A–C). We then created cerebrovascular reactivity maps by comparing the CO2-induced increase in the BOLD signal relative to baseline BOLD. We found no significant differences in cerebrovascular reactivity between GS-deficient and GS-intact brains (Fig. 3D–F). Finally, we found no significant differences in structural connectivity (maps of fractional anisotropy and mean diffusivity) between the two groups (Fig. 3G, H).
Figure 3. Functional connectivity, cerebrovascular reactivity, and structural imaging.

A. Resting-state functional magnetic resonance imaging (fMRI) showing group average parametric maps of functional connectivity density (FCD, percentage of brain voxels correlated at Pearson r ≥ 0.3) for control (left, WT) and Cre-positive (center, KO) groups. B, C. A voxel-level t-test shows clusters where FCD is significantly higher in the control group (WT) after correction for multiple comparison. Specific parametric maps show the density of connectivity with voxels within 1.5 mm (B) and beyond 1.5mm (C). D - F. Cerebrovascular reactivity. D. Group average parametric maps representing area under the curve of the fMRI signal (arbitrary units) during a hypercapnic challenge are shown for the control (top, WT) and Cre-positive (bottom, KO) groups. A representative fMRI signal curve for each group is shown for regions of interest located in the sensorimotor cortex (E) and the dorsal thalamus (F). After correction for multiple comparison, no significant group effect was found for cerebrovascular reactivity. G, H. Diffusion tensor imaging (DTI). Mean diffusivity (G) and fractional anisotropy (H) are shown as group average parametric maps overlaid on a coronal structural brain template for the control group (left, WT) and the Cre-injected (right, KO) group. After correction for multiple comparison, no significant group effect was found for either parameter.
Discussion
We established that stereotaxic injections of AAV8-GFAP-Cre in GSflox/flox mice can effectively delete GS in precisely targeted regions of the postnatal brain. Using this method, we discovered that these deletions produce a reduced seizure threshold, spontaneous recurrent seizures, and decreased functional brain connectivity. The significance of these findings will be discussed in detail below.
Advantages and limitations of the model
Microinjections of AAV8-GFAP-Cre into the brain of floxed C57BL/6J GS mice (GSflox/flox) at 22 to 70 weeks of age effectively deletes astroglial GS, as evidenced by immunohistochemistry at 4 to 6 weeks post-injection. Nearly all astrocytes in the center of the injection site completely lacked GS, and we observed a gradual return to full GS expression toward the periphery. While the USPIO nanoparticle imaging indicated that the injected virus diffused several hundreds of micrometers within the brain parenchyma, cells infected by the virus were limited to a smaller region. These results suggest that the virus gets progressively diluted toward the periphery and that a threshold virus concentration is needed to infect astrocytes and delete GS.
By adjusting the concentration and volume of the injected virus and the physical distance between injections, we propose the possibility of deleting GS in precisely targeted regions ranging in diameter of a few tens of micrometers to several hundreds of micrometers. Moreover, the zone at the injection periphery contains a mixture of GS-deleted and GS-intact astrocytes and possesses the ideal conditions for “internally controlled” comparison studies of GS-positive and GS-negative astrocytic domains within the same animal and brain region. The inclusion of GSflox/flox animals injected with the same strain of non-Cre expressing virus serves as an internal control for any effects related to the virus itself, such as tissue inflammation. Thus, our model is ideal for highly specific in vivo studies of postnatal GS function at the single cell (i.e., astrocyte domain) level, as well as at the brain regional (tens to hundreds of micrometers) level.
Our model has limitations. First, the stereotaxic procedure is relatively low throughput, as the procedure is performed on an individual basis and each site of injection requires about 5–10 minutes to complete. For example, while a small hypothalamic nucleus may only require a few injections, a larger thalamic nucleus or neocortical region may require multiple injections, thereby increasing the total surgery time. Second, precise stereotaxic injections may be challenging to perform in very young animals due to the small size and fragility of the skull. Third, unlike MSO, which rapidly deactivates GS, the virus-induced knockout occurs gradually and reaches maximum levels several weeks after injection. Fourth, the use of a tethered EEG monitoring system made it difficult to monitor the mice for a long, continuous period, like we were able to do in our prior rat studies. Mice do not tolerate the weight of the cables for a very long time and needed to be removed from the recordings after 4 to 12 days. Because of the short recording times, our study likely underestimates the true prevalence of seizures in both experimental and control animals, and our study is not suitable for accurate determination of seizure counts. Longer recordings, which may be possible with wireless systems, will be necessary to obtain such numbers. Nevertheless, the EEG data from the present study, along with data from mice and rats with focal brain GS inhibition or deletion12, 21, all support the concept that focal brain GS deficiency causes epileptic seizures.
GS deficiency and seizures
There is accumulating evidence from human and animal studies that GS dysfunction is implicated in the pathogenesis of some epilepsies. First, GS is deficient in discrete regions of the brain in patients with mesial temporal lobe epilepsy 7, 8, neocortical epilepsies 9, and glioblastoma-associated epilepsy 33. Second, infants with systemic GS deficiency due to mutations in the GS gene exhibit severe epileptic encephalopathy 4, 5. Third, systemic or intrahippocampal administration of the GS inhibitor MSO in rats results in acute or recurrent seizures, respectively 12, 19, and embryonic deletions of GS in the neocortex and hippocampus of mice causes spontaneous seizures 21. Here we show that deleting GS in very small brain regions is sufficient to reduce the seizure threshold and produce spontaneous recurrent seizures in some animals. This finding is clinically important, because the regions of GS deficiency in human patients with mesial temporal lobe epilepsy and neocortical epilepsies are quite small, typically involving the CA1, the CA3 and the dentate gyrus of the hippocampal formation in mesial temporal lobe epilepsy 7, 8, the amygdala in neocortical epilepsies 9, and the neoplastic tissue in glioblastoma-associated epilepsies 10. Collectively, these observations suggest that impaired GS activity in discrete regions of the postnatal brain may be linked to the pathogenesis of some focal epilepsies.
It should be noted that only 3 of the 5 GS deficient mice (60%) had recurrent seizures, whereas > 95% of rats infused with the GS inhibitor MSO into the hippocampal formation typically exhibit recurrent seizures. The difference in seizure prevalence between the two models can be explained by: (a) the much longer EEG monitoring period in MSO-infused rats (21 days) vs. GS knockout mice (4 – 12 days), (b) that MSO is likely to inhibit GS in a much larger area of the brain than the knockout approach, i.e. entire ventral entorhinal-hippocampal territory vs. multiple small regions within the hippocampal formation and neocortex, and (c) that the GS-floxed mice were generated in C57BL/6J mice, which are known to resistant to seizures34. The increased seizure prevalence in MSO-treated animals may also be related to the facts that MSO depletes brain glutathione levels and causes accumulation of astroglial glycogen 17–19. Reduced glutathione (GSH) is important for neutralizing reactive oxygen species in tissues, and studies have shown that oxidative stress increases seizure frequency and neuroinflammation in animal models of epilepsy 35, 36. Moreover, patients with MTLE and Lafora Disease have high glycogen levels in the hippocampal seizure focus 37 and throughout the brain 38, respectively, suggesting that increased astroglial glycogen is linked to seizures.
The seizure threshold study using PTZ showed a considerable difference of 23 seconds in time to twitch between experimental and control animals, but no significant difference in the time to convulsion. The lack of a difference in time to convulsion is not clear but may be related to the specific seizure triggering mechanism of the PTZ test and that animals with brain GS perturbations may respond differently than other animals to this test, as suggested in the study by Cloix et al. 39.
GS deficiency and multimodal imaging findings
Our findings of decreased short- and long-range connectivity by fMRI is consistent with clinical reports of epilepsy and other animal models of the disease31, 32. The default mode network in epilepsy, for example, shows a decrease in connectivity between the two main nodes of the default mode network, though preserved connectivity within each node 40–42. Changes in functional connectivity during the epileptogenic process remains unclear. A recent study of the rodent intrahippocampal kainic acid model of epilepsy demonstrated connectivity alterations with increased global connectivity and both decreased and increased local connectivity in epileptic animals43. This stands in contrast to reports that found decreases in global measures of connectivity directly related to seizure frequency32, 44. A recent human study also found decreased global functional connectivity in patients with mesial temporal lobe epilepsy, where the reduction is suggested to bear a direct relationship to neuro-cognitive deficits31. An interesting feature of our model is that the reduced functional connectivity is not accompanied by structural imaging changes. This is different from other commonly used models of mesial temporal lobe epilepsy, such as the kainic acid and pilocarpine models, which both show marked anatomical changes by structural brain imaging45, 46.
Previously, we demonstrated that mice with global embryonic deletion of GS in the cortex showed an abnormal vascular histology and a markedly blunted vasodilatory response to CO2 inhalation 21. So, the near normal cerebrovascular reactivity in the viral GS knockout suggests that global GS deletion may be required during embryonic development to compromise cerebrovascular reactivity; however, additional investigations will resolve this issue.
Finally, the lack of significant findings by structural imaging in the viral GS knockout remains surprising, as diffusion tensor imaging studies of rats infused with MSO into one hippocampal formation revealed significant changes in fractional anisotropy in multiple brain regions 30. These findings suggested that the changes may be dependent on the total seizure load. The differences between the two models could arise from the presumed larger region of GS deficiency caused by MSO than the virus, and the notion that chemically inhibited rats have a higher seizure load than virus injected mice.
Conclusions
We have developed a powerful and highly specific in vivo model of astroglial GS deficiency that can precisely target any region in the postnatal brain. Our model has several advantages over current approaches which use chemical inhibitors or embryonic knockouts targeting large portions of the brain. We found that deletion of GS in cortical regions causes spontaneous seizures and impaired functional connectivity – consistent with the postulated role of astroglial GS in the pathogenesis of some adult-onset focal epilepsies. Additionally, we expect that our model will be useful for many studies of astroglial GS function at the brain-regional and cellular levels, especially those studies whose completion was hindered by a lack of specific and anatomically targeted approaches.
Supplementary Material
Key points box.
We developed a mouse model for precise, genetic deletions of astroglial glutamine synthetase (GS) in small loci of the postnatal brain.
GS deletions in small regions of the hippocampus and neocortex reduced seizure threshold and induced spontaneous recurrent seizures.
Such deletions reduced functional connectivity by blood oxygen level dependent (BOLD) brain imaging.
These results suggest that small loci of GS deficiency in the postnatal brain are sufficient to cause epilepsy.
This model can also be used for rigorous studies of astroglial GS function at the brain-region and single-cell levels.
Acknowledgements
This work was supported by the Swebilius Foundation and by grants from the National Institutes of Health (NIH, NS058674, NS070824, NS109062, and NS109734) and the National Center for Advancing Translational Sciences (NCATS, a component of the NIH) (RR024139) and the Norwegian Research Council (Grant number 240844). The contents of the publication are solely the responsibility of the authors and do not necessarily represent the official view of NCATS or NIH. The authors have no conflicts of interest to declare. We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines. We thank Life Science Editors for editing assistance. The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.Krebs HA. Metabolism of amino-acids: The synthesis of glutamine from glutamic acid and ammonia, and the enzymic hydrolysis of glutamine in animal tissues Biochem J. 1935. Aug;29:1951–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martinez-Hernandez A, Bell KP, Norenberg MD. Glutamine synthetase: glial localization in brain Science. 1977. Mar 25;195:1356–1358. [DOI] [PubMed] [Google Scholar]
- 3.Norenberg MD, Jayakumar AR, Rama Rao KV, Panickar KS. New concepts in the mechanism of ammonia-induced astrocyte swelling Metab Brain Dis. 2007. Dec;22:219–234. [DOI] [PubMed] [Google Scholar]
- 4.Haberle J, Gorg B, Rutsch F, Schmidt E, Toutain A, Benoist JF, et al. Congenital glutamine deficiency with glutamine synthetase mutations N Engl J Med. 2005. Nov 3;353:1926–1933. [DOI] [PubMed] [Google Scholar]
- 5.Häberle J, Shahbeck N, Ibrahim K, Hoffmann GF, Ben-Omran T. Natural course of glutamine synthetase deficiency in a 3 year old patient Mol Genet Metab. 2011. May;103:89–91. [DOI] [PubMed] [Google Scholar]
- 6.Spodenkiewicz M, Diez-Fernandez C, Rufenacht V, Gemperle-Britschgi C, Haberle J. Minireview on Glutamine Synthetase Deficiency, an Ultra-Rare Inborn Error of Amino Acid Biosynthesis Biology (Basel). 2016. Oct 19;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eid T, Thomas MJ, Spencer DD, Runden-Pran E, Lai JC, Malthankar GV, et al. Loss of glutamine synthetase in the human epileptogenic hippocampus: possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy Lancet. 2004. Jan 3;363:28–37. [DOI] [PubMed] [Google Scholar]
- 8.van der Hel WS, Notenboom RG, Bos IW, van Rijen PC, van Veelen CW, de Graan PN. Reduced glutamine synthetase in hippocampal areas with neuron loss in temporal lobe epilepsy Neurology. 2005. Jan 25;64:326–333. [DOI] [PubMed] [Google Scholar]
- 9.Steffens M, Huppertz HJ, Zentner J, Chauzit E, Feuerstein TJ. Unchanged glutamine synthetase activity and increased NMDA receptor density in epileptic human neocortex: implications for the pathophysiology of epilepsy Neurochem Int. 2005. Nov;47:379–384. [DOI] [PubMed] [Google Scholar]
- 10.Rosati A, Marconi S, Pollo B, Tomassini A, Lovato L, Maderna E, et al. Epilepsy in glioblastoma multiforme: correlation with glutamine synthetase levels J Neurooncol. 2009. Jul;93:319–324. [DOI] [PubMed] [Google Scholar]
- 11.Roth JS, Wase A, Eichel HJ. Studies with methionine sulfoximine J Biol Chem. 1953. Feb;200:647–656. [PubMed] [Google Scholar]
- 12.Eid T, Ghosh A, Wang Y, Beckström H, Zaveri HP, Lee TS, et al. Recurrent seizures and brain pathology after inhibition of glutamine synthetase in the hippocampus in rats Brain. 2008. Aug;131:2061–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Y, Zaveri HP, Lee TS, Eid T. The development of recurrent seizures after continuous intrahippocampal infusion of methionine sulfoximine in rats: a video-intracranial electroencephalographic study Exp Neurol. 2009. Dec;220:293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lauritzen F, Perez EL, Melillo ER, Roh JM, Zaveri HP, Lee TS, et al. Altered expression of brain monocarboxylate transporter 1 in models of temporal lobe epilepsy Neurobiology of disease. 2012. Aug 10;45:165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dhaher R, Wang H, Gruenbaum SE, Tu N, Lee TS, Zaveri HP, et al. Effects of site-specific infusions of methionine sulfoximine on the temporal progression of seizures in a rat model of mesial temporal lobe epilepsy Epilepsy Res. 2015. Sep;115:45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gruenbaum SE, Wang H, Zaveri HP, Tang AB, Lee TS, Eid T, et al. Inhibition of glutamine synthetase in the central nucleus of the amygdala induces anhedonic behavior and recurrent seizures in a rat model of mesial temporal lobe epilepsy Epilepsy Behav. 2015. Oct;51:96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shaw CA, Bains JS. Synergistic versus antagonistic actions of glutamate and glutathione: the role of excitotoxicity and oxidative stress in neuronal disease Cell Mol Biol (Noisy-le-grand). 2002. Mar;48:127–136. [PubMed] [Google Scholar]
- 18.Bernard-Helary K, Ardourel MY, Hevor T, Cloix JF. In vivo and in vitro glycogenic effects of methionine sulfoximine are different in two inbred strains of mice Brain Res. 2002. Mar 8;929:147–155. [DOI] [PubMed] [Google Scholar]
- 19.Folbergrova J, Passonneau JV, Lowry OH, Schulz DW. Glycogen, ammonia and related metabolities in the brain during seizures evoked by methionine sulphoximine J Neurochem. 1969. Feb;16:191–203. [DOI] [PubMed] [Google Scholar]
- 20.Kam K, Nicoll R. Excitatory synaptic transmission persists independently of the glutamate-glutamine cycle J Neurosci. 2007. Aug 22;27:9192–9200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou Y, Dhaher R, Parent M, Hu QX, Hassel B, Yee SP, et al. Selective deletion of glutamine synthetase in the mouse cerebral cortex induces glial dysfunction and vascular impairment that precede epilepsy and neurodegeneration Neurochem Int. 2019. Feb;123:22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferrari FK, Samulski T, Shenk T, Samulski RJ. Second-strand synthesis is a ratelimiting step for efficient transduction by recombinant adeno-associated virus vectors J Virol. 1996. May;70:3227–3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mandhane SN, Aavula K, Rajamannar T. Timed pentylenetetrazol infusion test: a comparative analysis with s.c.PTZ and MES models of anticonvulsant screening in mice Seizure. 2007. Oct;16:636–644. [DOI] [PubMed] [Google Scholar]
- 24.Tollner K, Twele F, Loscher W. Evaluation of the pentylenetetrazole seizure threshold test in epileptic mice as surrogate model for drug testing against pharmacoresistant seizures Epilepsy Behav. 2016. Apr;57:95–104. [DOI] [PubMed] [Google Scholar]
- 25.Avoli M, Gloor P. Pathophysiology of focal and generalized convulsive seizures versus that of generalized non-convulsive seizures. In: Wolf P, editor. Epileptic Seizures and Syndromes. Montrouge, France: John Libby Eurotext Ltd; 1994. p. 547–561. [Google Scholar]
- 26.Racine RJ, Burnham WM, Gartner JG, Levitan D. Rates of motor seizure development in rats subjected to electrical brain stimulation: strain and inter-stimulation interval effects Electroencephalography and clinical neurophysiology. 1973;35:553–556. [DOI] [PubMed] [Google Scholar]
- 27.Tomasi D, Volkow ND. Functional connectivity density mapping Proc Natl Acad Sci U S A. 2010. May 25;107:9885–9890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaneko G, Sanganahalli BG, Groman SM, Wang H, Coman D, Rao J, et al. Hypofrontality and Posterior Hyperactivity in Early Schizophrenia: Imaging and Behavior in a Preclinical Model Biol Psychiatry. 2017. Mar 15;81:503–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Worsley KJ, Marrett S, Neelin P, Vandal AC, Friston KJ, Evans AC. A unified statistical approach for determining significant signals in images of cerebral activation Human brain mapping. 1996;4:58–73. [DOI] [PubMed] [Google Scholar]
- 30.Wang H, Huang Y, Coman D, Munbodh R, Dhaher R, Zaveri HP, et al. Network evolution in mesial temporal lobe epilepsy revealed by diffusion tensor imaging Epilepsia. 2017. May;58:824–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Struck AF, Boly M, Hwang G, Nair V, Mathis J, Nencka A, et al. Regional and global resting-state functional MR connectivity in temporal lobe epilepsy: Results from the Epilepsy Connectome Project Epilepsy Behav. 2021. Feb 18;117:107841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Christiaen E, Goossens MG, Raedt R, Descamps B, Larsen LE, Craey E, et al. Alterations in the functional brain network in a rat model of epileptogenesis: A longitudinal resting state fMRI study Neuroimage. 2019. Nov 15;202:116144. [DOI] [PubMed] [Google Scholar]
- 33.Rosati A, Poliani PL, Todeschini A, Cominelli M, Medicina D, Cenzato M, et al. Glutamine synthetase expression as a valuable marker of epilepsy and longer survival in newly diagnosed glioblastoma multiforme Neuro Oncol. 2013. May;15:618–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McKhann GM 2nd, Wenzel HJ, Robbins CA, Sosunov AA, Schwartzkroin PA. Mouse strain differences in kainic acid sensitivity, seizure behavior, mortality, and hippocampal pathology Neuroscience. 2003;122:551–561. [DOI] [PubMed] [Google Scholar]
- 35.Geronzi U, Lotti F, Grosso S. Oxidative stress in epilepsy Expert review of neurotherapeutics. 2018. May;18:427–434. [DOI] [PubMed] [Google Scholar]
- 36.Sharma S, Carlson S, Gregory-Flores A, Hinojo-Perez A, Olson A, Thippeswamy T. Mechanisms of disease-modifying effect of saracatinib (AZD0530), a Src/Fyn tyrosine kinase inhibitor, in the rat kainate model of temporal lobe epilepsy Neurobiology of disease. 2021. Aug;156:105410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dalsgaard MK, Madsen FF, Secher NH, Laursen H, Quistorff B. High glycogen levels in the hippocampus of patients with epilepsy J Cereb Blood Flow Metab. 2007. Jun;27:1137–1141. [DOI] [PubMed] [Google Scholar]
- 38.Duran J, Gruart A, Lopez-Ramos JC, Delgado-Garcia JM, Guinovart JJ. Glycogen in Astrocytes and Neurons: Physiological and Pathological Aspects Adv Neurobiol. 2019;23:311–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cloix JF, Tahi Z, Martin B, Hevor T. Selection of two lines of mice based on latency to onset of methionine sulfoximine seizures Epilepsia. 2010. Jan;51:118–128. [DOI] [PubMed] [Google Scholar]
- 40.Pittau F, Grova C, Moeller F, Dubeau F, Gotman J. Patterns of altered functional connectivity in mesial temporal lobe epilepsy Epilepsia. 2012. Jun;53:1013–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cataldi M, Avoli M, de Villers-Sidani E. Resting state networks in temporal lobe epilepsy Epilepsia. 2013. Dec;54:2048–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haneef Z, Lenartowicz A, Yeh HJ, Engel J Jr., Stern JM. Effect of lateralized temporal lobe epilepsy on the default mode network Epilepsy Behav. 2012. Nov;25:350–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li L, He L, Harris N, Zhou Y, Engel J Jr., Bragin A. Topographical reorganization of brain functional connectivity during an early period of epileptogenesis Epilepsia. 2021. Mar 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Christiaen E, Goossens MG, Descamps B, Larsen LE, Boon P, Raedt R, et al. Dynamic functional connectivity and graph theory metrics in a rat model of temporal lobe epilepsy reveal a preference for brain states with a lower functional connectivity, segregation and integration Neurobiology of disease. 2020. Jun;139:104808. [DOI] [PubMed] [Google Scholar]
- 45.Luna-Munguia H, Marquez-Bravo L, Concha L. Longitudinal changes in gray and white matter microstructure during epileptogenesis in pilocarpine-induced epileptic rats Seizure. 2021. Feb 13. [DOI] [PubMed] [Google Scholar]
- 46.Salo RA, Miettinen T, Laitinen T, Grohn O, Sierra A. Diffusion tensor MRI shows progressive changes in the hippocampus and dentate gyrus after status epilepticus in rat - histological validation with Fourier-based analysis Neuroimage. 2017. May 15;152:221–236. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.