

Abstract
Cells perform various functions by proteins via protein complexes. Characterization of protein complexes is critical to understanding their biological and clinical significances and has been one of the major efforts of functional proteomics. To date, most protein complexes are characterized by in vitro system from protein extracts after cells or tissues are lyzed, and it has been challenging to determine which of these protein complexes are formed in intact cells. Herein, we report an approach to preserve protein complexes using in vivo cross-linking followed by size exclusion chromatography and data independent acquisition mass spectrometry. This approach enables the characterization of in vivo protein complexes from cells or tissues, which allows the determination of protein complexes in clinical research. More importantly, the described approach can identify protein complexes that are not detected by in vitro system, which provide unique protein function information.
Graphical Abstract
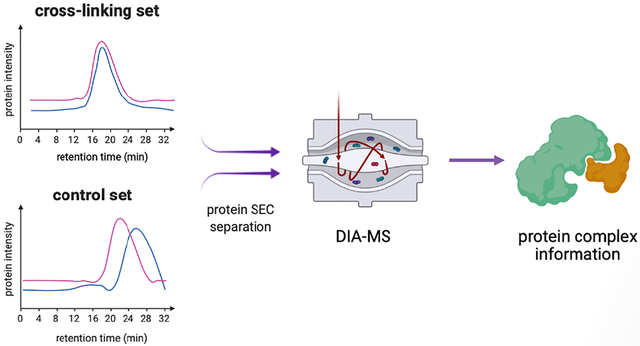
Every organism has a limited number of genes. Proteins as gene products carry out dynamic and diverse biological functions depending on their associated complexes, which play central roles in physiological and pathological processes1. The protein complexes dynamically assemble and dissemble based on the expression of different proteins and provide mechanistic insights into the organization of biological system. Characterization of protein complexes facilitates the understanding of diseases such as cancer and infectious disease related pathways2,3. Toward this end, affinity purification mass spectrometry (AP-MS) has become a powerful approach for large-scale analysis of protein complexes of target proteins4–6. However, AP-MS relies on accessibility and availability of antibody to each target protein for successful antibody purification or introducing accessible AP tags by genetic engineering, making them less applicable to clinically acquired specimens. Currently, large-scale characterizations of protein complexes using AP-MS remain difficult.
Protein co-fractionation using size exclusion chromatography (SEC) coupled with mass spectrometry (Co-Frac-MS) provides antibody and genetic engineering-independent technology for comprehensive characterization of protein complexes7–9. To date, Co-Frac-MS approaches have been successfully used in cell or tissue lysates of protein extracts. Such interactome studies include the analysis of cell samples in different human cell lines5, metazoan embryonic cells10 and tissue suspension10. These studies have revealed a large number of protein complexes that were either preserved or newly formed in vitro when proteins are released from different cellular components or from different cells in tissues. However, it is challenging to determine which of the identified protein complexes is formed in vivo in cellular compartments prior to lysing the cells or tissues.
Cross-linking mass spectrometry (XL-MS) is a powerful technology for studying in vivo protein complexes, due to its ability to stabilize protein interactions in their native environment prior to cell lysis and thus preventing loss and/or reorganization of protein interactions during biochemical manipulations11. In vivo chemical cross-linking has been coupled with affinity purification to identify protein complexes from living systems12–15. Recent advancement in XL-MS technologies has enabled effective proteome-wide in vivo analysis to define protein interaction landscapes of human cells16 and tissues17. While these studies are successful in constructing protein network topologies based on cross-linked sites, the definition of individual protein complexes cannot be easily assessed at the systems-level without prior knowledge.
Here, we developed an integrated approach, namely X-Co-Frac-MS (in vivo cross-linking (X) assisted Co-Fractionation MS) for global characterization of in vivo protein complexes. The platform used chemical cross-linking to preserve in vivo protein complexes through covalent bond formation prior to lysing samples in denaturing conditions to prevent in vitro protein complex formation. Cross-linked protein complexes were separated by SEC, digested, and analyzed by data independent acquisition mass spectrometry (DIA-MS). To identify genuine protein complexes, uncross-linked cells were used as the control and analyzed in the same way. In comparison to the control, the co-eluted protein complexes that were cross-linked and preserved in SEC were identified based on SEC retention time shift.
EXPERIMENTAL SECTION
Materials and reagents.
Chemicals, human IgG, and Protein A from Staphylococcus aureus were purchased for Sigma-Aldrich (St. Louis, MO), and HPLC-grade reagents were purchased from Fisher Scientific™ (Waltham, MA). Lys-C was purchased from FUJIFILM Wako Chemicals (Richmond, VA). HEK293 cell line was purchased from American Type Culture Collection (ATCC, Manassas, VA). Yarra-SEC-4000 column (300 × 7.8 mm, pore size 500 Å, particle size 3 μm) was purchased from Phenomenex® (Torrance, CA). Empore™ Extraction Disks (3M C18) were purchased from VWR (Radnor, PA). iRT Kit was purchased from Biognosys Inc. (Boston, MA).
Cell culture.
HEK293 cells were cultured in DMEM medium with 10% FBS and 1.5 % penicillin-streptomycin in a humidified air incubator with 5% CO2 at 37 °C.
Preparation of chemical cross-linking and SEC separation samples for MS analysis.
Live samples were washed with 1 × PBS for six times and treated with 1 % HCHO 1 × PBS solution for 10 min at 37 °C, and then HCHO was quenched by ammonium bicarbonate with a final concentration 1 M. The chemical cross-linked samples were lysed by 8 M urea lysis buffer (8 M urea in 1 × PBS solution with pH 7.4). Control samples were also treated in the same condition except for not treated with HCHO. Lysates were sonicated for 30 s three times at 15 % power in ice water and clarified by centrifugation at 16,000 × g for 12 min at 4 °C. Protein concentration from supernatant was measured by BCA kits and equal amount of proteins (0.5 mg) were directly fractionated on a Yarra-SEC-4000 column, and fractioned at 0.5 mL/min flow rate with 8 M urea in 0.1 M phosphate (pH 6.8) SEC buffer. Each fraction was collected at 0.33 min from 9 to 24 min post-injection in a 96-well plate, and fractions 6–45 (40 fractions) were analyzed by DIA-MS. The proteins in each fraction were reduced with 6 mM dithiothreitol (DTT) for 1h at 37 °C and alkylated with 12 mM iodoacetamide (IAA) for 45 min at room temperature in dark. The proteins were proteolyzed to peptides with Lys-C at 1mAU:10 μg enzyme to substrate ratio for 2h. Peptides were acidified with 50 % formic acid to 2 % final concentration with pH 3. The digested peptides were desalted on reverse-phase Empore™ packed C18 stage tips. Each fraction was dried on Speed-Vac (Thermo Scientific), then resuspended in 3 % acetonitrile with 0.1 % formic acid and supplemented with equal amounts of iRT Kit peptides to calibrate the internal retention time.
Quantitative proteomic analysis using DIA-MS.
Samples were analyzed in a Q Exactive™ Hybrid Quadrupole-Orbitrap mass spectrometer (Thermo Scientific). The digested peptides in each fraction were injected with equal proportional amounts with 1μg of maximin peptides in certain fraction. Peptides were separated on an ultimate 3000 RSLC system (Dionex) via reverse phase 75μm × 50 cm C18PepMap RSLC column (Thermo Scientific) with protection of 5 mm guard column C18 nano column. The column was heated to 50 °C using a column heater. The flow rate was 0.300 μL/min with 0.1 % formic acid and 3 % acetonitrile in water (A) and 0.1% formic acid, 80% acetonitrile (B). The peptides were separated with a 7%–30% B gradient in 60 mins. The parameters were as follows: MS1, AGC Target 3 × 106, Max IT – 200 ms, charge state include 2–6, isolation window 20.0 m/z, scan range 400–900 m/z; MS2, AGC Target – 3 × 106, Max IT – 100 ms.
Protein database building and quantification of DIA searching.
Raw DIA data were processed using Spectronaut (Biognosys), and the library was built using directDIA™ based on each set of 40 SEC fractions. The established library was used to match the set of DIA data from each SEC fraction. The protein abundance for each protein in all the fractions is provided in Table S1–S4.
Protein abundance quantification in each fraction.
The abundance of each protein in each fraction was relatively quantified in the chemical cross-linking and control set, such that the maximum intensity had an intensity of 1.0.
RESULTS AND DISCUSSION
Description of the method.
The X-Co-Frac-MS method involved five steps (Fig. 1a). (i) Cross-linking: Formaldehyde is used to cross-link protein complexes from intact cells or tissues to preserve in vivo protein complexes (Fig. 1b)18. (ii) Denaturing lysis: Proteins from cross-linked cells are denatured in 8M urea buffer to prevent in vitro protein complex formation. (iii) SEC separation: Denatured proteins and cross-linked protein complexes are separated by SEC. (iv) DIA-MS: Proteins collected from each SEC fraction are digested to peptides and analyzed by DIA-MS for protein identification and quantitation. (v) Data analysis: The retention time of each protein is compared between the control and cross-linked fractions to determine in vivo protein complexes based on protein retention time shift. The co-eluted proteins specially detected after in vivo cross-linking to resolve as protein complexes in reported databases19.
Figure 1.
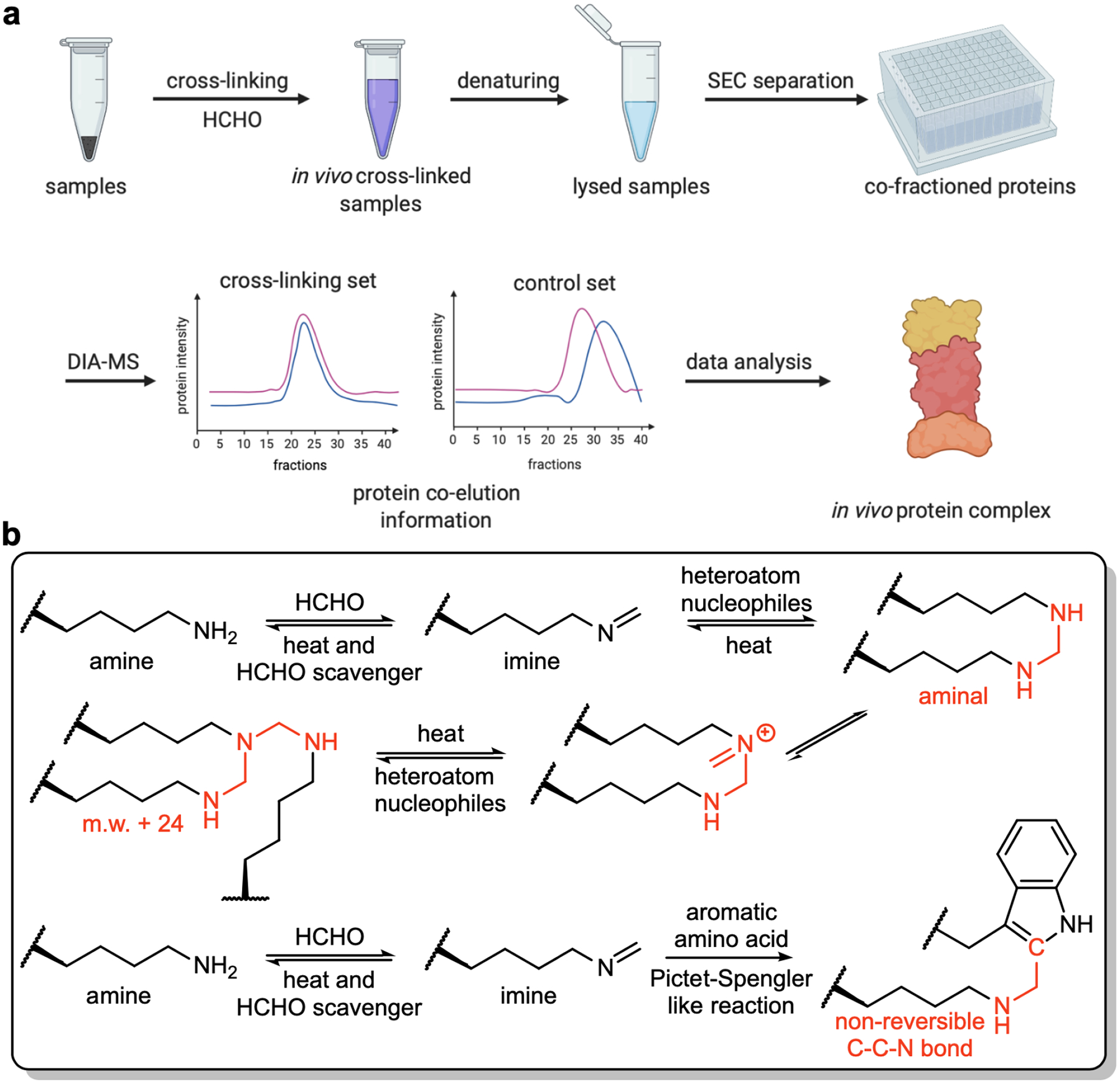
Workflow and mechanism of X-Co-Frac. (a) General workflow of in vivo protein complex detection. (b) Mechanism of in vivo chemical cross-linking.
Proof of principle study.
To evaluate the workflow and illustrate the feasibility for protein complex detection, we chose the protein complex of IgG and an IgG binding protein (protein A from Staphylococcus aureus (SpA)) as the model system20. We investigated whether cross-linked protein complex can be analyzed by SEC in denatured condition. First, we investigated whether X-Co-Frac could shift the SEC retention time for each protein in denaturing condition. IgG with and without cross-linking were separated by SEC in 8 M urea (Fig. 2a). There was no elution peak shift between IgG without cross-linking and IgG with cross-linking under 8 M urea condition. SpA with and without cross-linking also showed a similar retention time (Fig. 2b). Then, we tested the formation of the SpA and IgG complex after cross-linking (Fig. 2c). We treated the SpA-IgG complex with formaldehyde cross-linking under non-denatured condition. Then the cross-linked sample was denatured in 8 M urea followed by SEC separation using 8 M urea elution buffer. We found that cross-linking maintained the SpA-IgG complex, which was observed as a new peak in SEC trace (Fig. 2c). In comparison, SEC separation of the denatured IgG-SpA complex without cross-linking did not yield the same peak (red line in Fig. 2c). These results demonstrate that cross-linking enables the preservation of the SpA-IgG complex under denaturing condition.
Figure 2.
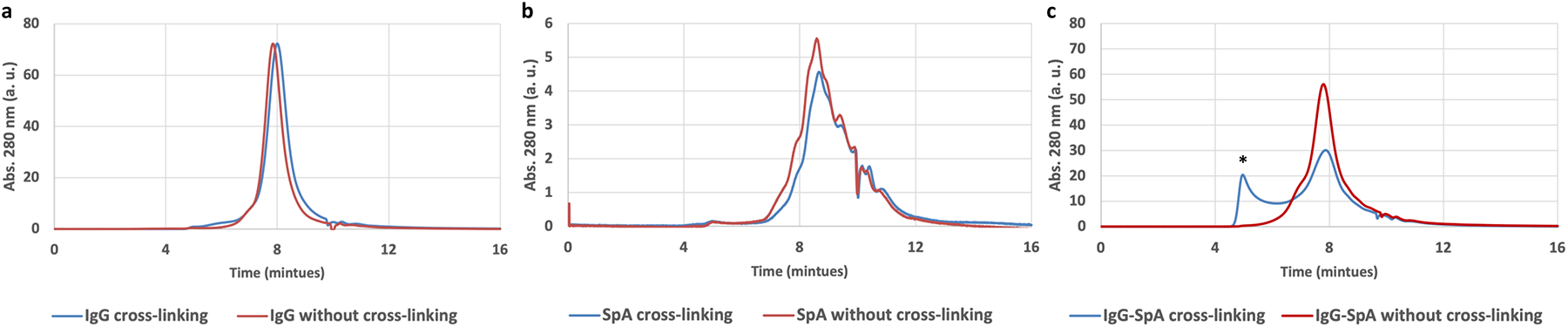
Identification of protein complex by X-Co-Frac separation in 8M urea denatured condition. (a) IgG with and without cross-linking separation by SEC. (b) SpA with and without cross-linking separation by SEC. (c) IgG-SpA complex with and without cross-linking separation by SEC (new peak *).
Characterization of protein complexes in HEK293 cells.
To explore the potential of in vivo cross-linking for global characterization of protein complexes, we applied the X-Co-Frac-MS method to HEK293 cells. This cell line has been widely used in human cell biology studies21, especially in protein-protein interaction study7,8. Here, HEK293 cells were treated with 1% HCHO, and then quenched by ammonium bicarbonate. The cross-linked (Set 1) and non-cross-linked control cells (Set 2) were lysed in denaturing buffer containing 8 M urea. The two sets were separated by SEC with a denaturing elution buffer. The proteins were digested directly with Lys-C in 8 M urea elution buffer, followed by DIA-MS to identify and quantify the proteins in each fraction. Forty DIA-MS maps constituted from each set, and the protein complexes were analyzed based on the changed proteins retention time in the Set 1 compared with the Set 2. Based on the shifted protein SEC retention time, in vivo cross-linked and co-eluted protein complexes were characterized using a protein complex database19. The following principles were applied for whole protein complex profiling. First, the proteins in each complex were identified in both sets. Second, the co-eluted protein complexes in the Set 1 were selected, and then proteins with a retention time shift compared to the Set 2 were chosen as candidates. Third, the selected candidates would be accounted for identification of protein complexes if co-eluted protein peaks were only detected from Set 1 comparing to those from Set 2.
We then applied X-Co-Frac-MS to the protein complex identification of HEK293 cells with CORUM database. We identified 272 protein complexes in our study. One of the identified complexes was the 26S proteasome (CORUM complex 181), a broadly studied protein complex8,22,23 that is responsible for more than 80% of intracellular protein degradation24. Selecting 26S proteasome regulatory subcomplex as an example25, in the Set 1, six proteins of regulatory subcomplex, PRS4, PRS7, PRS6A, PRS6B, PRS8, and PRS10, were co-eluted at fraction 12 and fraction 16 after in vivo cross-linking (Fig. 3a). In the Set 2, these proteins were mainly eluted at later fraction, suggesting that these proteins from 26S proteasome regulatory subunits were covalently tethered together via in vivo cross-linking before the cells were lyzed. We used X-Co-Frac-MS to successfully identify the 26S proteasome related complex. From the Fig. 3a, we also knew that proteins appeared as a monomer as well as in complexes. The monomers showed among fractions 25–30 in set 2. Meanwhile, the proteins in set 1 mainly co-eluted at fractions 12 and 16 marked as protein complex and appeared as monomers with different tiny elution peaks near fraction 25. These proteins also showed in other protein complexes in CORUM database such as CORUM complex 32 and 193.
Figure 3.

Protein complexes identified by X-Co-Frac method for HEK293 cells. (a) SEC elution data sets of 26S proteasome regulatory subunits. (b) SEC elution data sets of Succinyl-CoA synthetase GDP-forming complex. (c) SEC elution data sets of identified three proteins in polycomb repressive complex 2.
We also found some protein complexes identified by X-Co-Frac-MS that were not shown in previous Co-Frac-MS studies8. For example, the succinyl-CoA synthetase GDP-forming complex (CORUM complex 393) (Fig. 3b), the two subunits, SUCB2 and SUCA, were co-eluted at fraction 26 only in Set 1. In Set 2, protein SUCA was eluted at fraction 32 and protein SUCB2 eluted at fraction 29, indicating that the two proteins did not co-elute in the absence of cross-linking. This example also proved that the X-Co-Frac-MS method is effective in analyzing native protein complexes.
On the other hand, we observed some protein complexes identified by Co-Frac-MS approach that were not observed in our X-Co-Frac-MS workflow. We listed an example of polycomb repressive complex 2 (CORUM complex 105), which showed in Fig. 3c. This complex contains five proteins in total and we consistently identified three of them in both cross-linking and control sets. For the cross-linking set, two histone binding proteins (RBBP4 and RBBP7) were mainly eluted at fraction 26 and fraction 27; the rest polycomb protein (SUZ12) was eluted at earlier fraction. The control set had the similar elution pattern of these three proteins, which suggested there is no co-elution for this Co-Frac-MS approach reported protein complex. This example suggests that some protein complexes detected from in vitro system may not exist in vivo.
The protein complexes identified by X-Co-Frac-MS were further compared to protein complexes previously reported from HEK293 cell extracts using the Co-Frac-MS without cross-linking8 (Fig. 4a). Here, X-Co-Frac-MS approach identified 272 protein complexes from HEK293 cells. While 180 of them were observed by previous report without cross-linking8, 92 protein complexes were only identified using our X-Co-Frac-MS approach, and 454 protein complexes were uniquely identified from in vitro cell lysate without cross-linking.
Figure 4.
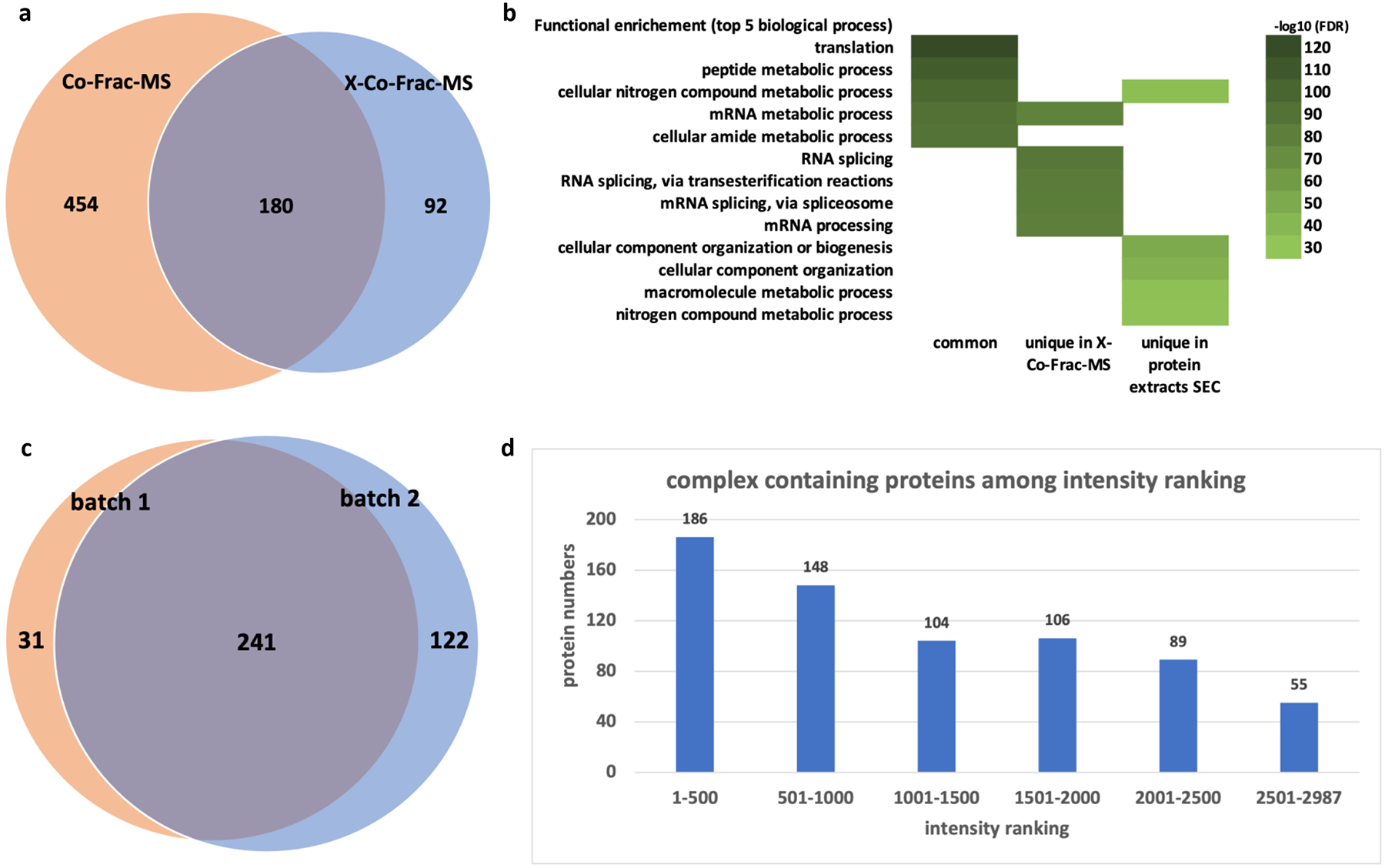
Protein complexes characterized by X-Co-Frac-MS in HEK293 cells. Comparison of X-Co-Frac-MS and Co-Frac-MS approaches, (a) Venn diagram of identified protein complex, (b) gene ontology for functional enrichment of identified complex proteins. (c) Venn diagram of protein complex overlap for the reproducibility of X-Co-Frac-MS approach. (d)Protein abundance ranking (batch 1) based on the total intensities of the identified proteins.
Next, we compared the gene ontology for functional enrichment of proteins identified from the protein complexes by in vivo, in vitro, and in common for biological process8. The top biological processes of proteins from commonly identified protein complexes were translation and metabolic process related according to the STRING database search result (Fig. 4b)26. Complexes identified by the X-Co-Frac method were enriched in the biological processes of RNA-related splicing and processing (Fig. 4b). Protein in complexes identified from Co-Frac approach showed enriched in the biological processes of cellular component organization and nitrogen compound metabolic process. (Fig. 4b). Further, we investigated the reproducibility of the X-Co-Frac workflow. Two batches of HEK293 cell sample were prepared and analyzed half-year apart by the same protocol (Fig. 4c). Common 241 protein complexes were identified from the two biological replicates. Batch 2 uniquely identified 122 protein complexes due to a higher number of proteins identified in batch 2 (Table S1–S4). Last, we found that proteins identified from complexes (batch 1) using X-Co-Frac method had wide ranging protein intensities (Fig. 4d). Among the 2,987 proteins identified in Set 1, 55 proteins were identified from protein complexes among the lowest abundant protein group. In the protein intensity ranking of 1001 to 2500, the number of complex-containing proteins was nearly the same on three rankings. This finding indicates that the protein complexes detected by our method were not protein intensity dependent.
CONCLUSION
We described a reproducible chemical cross-linking workflow named X-Co-Frac-MS for probing in vivo protein complexes. The workflow identifies protein complexes via in vivo cross-linking followed by SEC separation in denatured condition and quantitative proteomic analysis using DIA-MS. We tested the workflow using proteins IgG and SpA, and successfully applied to characterize protein complexes from HEK293 cells. Compared with published in vitro protein complexes identified from HEK293 cells using Co-Frac-MS8, our results identified previously uncharacterized protein complexes. We used three criteria to identify protein complexes. First, proteins were co-eluted in the same SEC fraction only after cross-linking. Second, the co-eluted proteins in databases showed as protein complexes. Third, the retention time of complex involved proteins were shifted and identified together in the cross-linking sample compared with the control. If proteins involved in known protein complexes meet the three criteria, they were identified as in vivo protein complexes. This allowed to reduce the false positive identification of protein complexes. However, it is possible that some of the protein complexes reported in this study or in the current protein complex databases are not formed by direct protein-protein interactions. Other molecules such as by DNAs27, RNAs28–30, carbohydrates31, lipids32, or small molecules33,34 can mediate protein complex formation. To determine if a protein complex is formed directly through protein-protein interaction, XL-MS can be employed to identify protein inter-connectivity at the residue level to distinguish direct from indirect interactions of protein complexes10.
For the X-Co-Frac method, in vivo protein complex profiling could be achieved by formaldehyde cross-linking prior to cell lysis. We chose formaldehyde for in vivo cross-linking over other cross-linking reagents due to its small size, fast reaction, excellent cell and tissue permeability35,36, broad utility in preserving tissues37, as well as applicability in studying interaction networks of protein complexes12–15.
As protein complexes in current databases are mainly identified from in vitro system, the approach developed here will provide unique information on authentic protein complexes as they occur in living systems, complementing existing complex-centric strategies. Importantly, this method can be generalized for characterizing in vivo protein complexes from various sample origins including tissues, for both research and clinical applications in the future.
Supplementary Material
Table S1. First batch of cross-linking set data matrix.
Table S2. First batch of control set data matrix.
Table S3. Second batch of cross-linking set data matrix.
Table S4. Second batch of control set data matrix.
ACKNOWLEDGMENT
This work was supported by Funding: National Cancer Institute, the Clinical Proteomic Tumor Analysis Consortium (CPTAC, U24CA210985) to H.Z. and R01GM074830 to L.H.
Footnotes
CONFLICT OF INTEREST DISCLOSURE
The authors declare no conflict of interest.
REFERENCES
- (1).Hartwell LH; Hopfield JJ; Leibler S; Murray AW Nature 1999, 402, C47–C52. [DOI] [PubMed] [Google Scholar]
- (2).Vidal M; Cusick ME; Barabási A Cell 2011, 144, 986–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Wang X; Wei X; Thijssen B; Das J; Lipkin SM; Yu H Nature Biotech. 2012, 30, 159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Malovanaya A; Lanz RB; Jung SY; Bulynko Y; Le NT; Chan DW; Ding C; Shi Y; Yucer N; Krenciute G; Kim B; Li C; Chen R; Li W; Wang Y; O’Malley BW; Qin J Cell 2011, 145, 787–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Huttlin EL et al. Nature 2017, 545, 505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Hein MY Cell 2015, 163, 712–723. [DOI] [PubMed] [Google Scholar]
- (7).Havugimana PC et al. Cell 2012, 150, 1068–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Heusel M; Bludau I; Rosenberger G; Hafen R; Frank M; Banaei-Esfahani A; van Drogen A; Collins BC; Gstaiger M; Aebersold R Mol. Syst. Biol 2019, 15, e8438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Smits AH; Vermeulen M Trends in Biotech. 2016, 34, 825–834. [DOI] [PubMed] [Google Scholar]
- (10).Yu C; Huang L Anal. Chem 2018, 90, 144–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Wan C et al. Nature 2015, 525, 339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Vasilescu J; Guo X; Kast J Proteomics 2004, 4, 3845–3854. [DOI] [PubMed] [Google Scholar]
- (13).Schmitt-Ulms G; Hansen K; Liu J; Cowdrey C; Yang J; DeArmond SJ; Cohen FE; Prusiner SB; Baldwin MA Nat. Biotechnol 2004, 22, 724–731. [DOI] [PubMed] [Google Scholar]
- (14).Guerrero C; Tagwerker C; Kaiser P; Huang L Mol. Cell Proteomics 2006, 5, 366–378. [DOI] [PubMed] [Google Scholar]
- (15).Yu C; Yang Y; Wang X; Guan S; Fang L; Liu F; Walters KJ; Kaiser P; Huang L Mol. Cell Proteomics 2016, 15, 2279–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Wheat A; Yu C; Wang X; Burke AM; Chemmama IE; Kaake RM; Baker P; Rychnovsky SD; Yang J; Huang L Proc. Natl. Acad. Sci. USA 2021, 118, e2023360118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Chavez JD; Lee CF; Caudal A; Keller A; Tian R; Bruce JE Cell Syst. 2018, 24, 136–141.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Tayri-Wilk T; Slavin M; Zamel J; Blass A; Cohen S; Motzik A; Sun X; Shalev DE; Ram O; Kalisman N Nat. Commun 2020, 11, 3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Giurgiu M; Reinhard J; Brauner B; Dunger-Kaltenbach I; Fobo G; Frishman G; Montrone C; Ruepp A Nucleic Acids Research 2019, 47, D559–D563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Deisenhofer J Biochemistry 1981, 20, 2361–2370. [PubMed] [Google Scholar]
- (21).Graham FL Smiley J; Russell WC; Nairn R J. Gen. Virol 1977, 36, 59–74 [DOI] [PubMed] [Google Scholar]
- (22).Wang X; Guerrero G; Kaiser P; Huang L Expert Rev. Proteomics 2007, 4, 649–665. [DOI] [PubMed] [Google Scholar]
- (23).Wang X et al. Molecular & Cellular Proteomics 2017, 16, 840–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Collins GA; Goldberg AL Cell 2017, 169, 792–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Dong Y Zhang S; Wu Z; Li X; Wang WL; Zhu Y; Stoilova-McPhie S; Lu Y; Finley D; Mao Y Nature 2019, 565, 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).https://string-db.org The latest version of the database is Szklarczyk D; Gable AL; Lyon D; Junge A; Wyder S; Huerta-Cepas J; Simonovic M; Doncheva NT; Morris JH; Bork P; Jensen LJ; von Mering C Nucleic Acids Res. 2019, 47, D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Ranish JA, Yi EC, Leslie DM; Purvine SO; Good-lett DR; Eng J; Aebersold R Nat. Genet 2003, 33, 349–355. [DOI] [PubMed] [Google Scholar]
- (28).Rissland OS Wiley Interdiscip. Rev. RNA 2017, 8, e1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Berg JM, Tymoczko JL, and Stryer L Aminoacyl-transfer RNA synthetases read the genetic code. In Biochemistry. (5th ed., section 29.2). New York, NY: W. H. Freeman, 2002. [Google Scholar]
- (30).Draper DE; Reynaldo LP Nucleic Acids Research 1999, 27, 381–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Toonstra C; Hu Y; Zhang HJ Proteome Res. 2019, 18, 2467–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).El-Fakharany E; Redwan EM RSC Adv, 2019,9, 36890–36906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Thompson AD; Dugan A; Gestwicki JE; Mapp AK ACS Chem. Biol 2012, 7, 1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Schreiber SL Cell 2021, 184, 3–9. [DOI] [PubMed] [Google Scholar]
- (35).Solomon MJ; Varshavsky A Proc. Natl. Acad. Sci. U.S.A 1985, 82, 6470–6474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Larance M; Kirkwood KJ; Tinti M; Murillo AB; Ferguson MA; Lamond AI Molecular & Cellular Proteomics 2016, 15, 2476–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Hoffman EA; Frey BL; Smith LM; Auble DT J. Biol. Chem 2015, 290, 26404–26411. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. First batch of cross-linking set data matrix.
Table S2. First batch of control set data matrix.
Table S3. Second batch of cross-linking set data matrix.
Table S4. Second batch of control set data matrix.