

Abstract
Imbalanced mitochondrial dynamics including inhibited mitochondrial fusion is associated with cardiac dysfunction as well as tumorigenesis. This study sought to explore the effects of promoting mitochondrial fusion on doxorubicin(Dox)-induced cardiotoxicity and its antitumor efficacy, with a focus on the underlying metabolic mechanisms. Herein, the inhibition of Mfn2-mediated mitochondrial fusion was identified as a key phenotype in Dox-induced cardiotoxicity. Restoration of Mfn2-mediated mitochondrial fusion enhanced mitochondrial oxidative metabolism, reduced cellular injury/apoptosis and inhibited mitochondria-derived oxidative stress in the Dox-treated cardiomyocytes. Application of lentivirus expressing Drp1 (mitochondrial fusion inhibitor) or Rote/Anti A (mitochondrial complex I/III inhibitors) blunted the above protective effects of Mfn2. Cardiac-specific Mfn2 transgenic mice showed preserved mitochondrial fusion and attenuated myocardial injury upon Dox exposure in vivo. The suppression of Mfn2-mediated mitochondrial fusion was induced by Dox-elicited upregulation of FoxO1, which inhibited the transcription of Mfn2 by binding to its promoter sites. In the B16 melanoma, Mfn2 upregulation not only attenuated tumor growth alone but also further delayed tumor growth in the presence of Dox. Mechanistically, Mfn2 synergized with the inhibitory action of Dox on glycolysis metabolism in the tumor cells. One common feature in both cardiomyocytes and tumor cells was that Mfn2 increased the ratio of oxygen consumption rate to extracellular acidification rate, suggesting Mfn2 triggered a shift from aerobic glycolysis to mitochondrial oxidative metabolism. In conclusion, targeting Mfn2-mediated mitochondrial fusion may provide a dual therapeutic advantage in Dox-based chemotherapy by simultaneously defending against Dox-induced cardiotoxicity and boosting its antitumor potency via metabolic shift.
Keywords: Doxorubicin cardiotoxicity, Mitochondrial fusion, Mfn2, FoxO1, Metabolism
Abbreviations: Ad, Adenoviruses; ChIP, Chromatin immunoprecipitation; cTnT, Cardiac troponin T; Dox, Doxorubicin; ECAR, Extracellular acidification rate; EF, Ejection fraction; GEO, Gene expression omnibus; LDH, Lactate dehydrogenase; LVESV, Left ventricular end-systolic volume; MDA, Malondialdehyde; MMP, Mitochondrial membrane potential; MOI, Multiplicities of infection; NRVMs, Neonatal rat ventricle myocytes; NTg, Non-transgenic; OCR, Oxygen consumption rate; OXPHOS, Mitochondrial oxidative phosphorylation; SOD, Superoxide dismutase; Tg, Transgenic; TUNEL, Terminal-deoxynucleoitidyl transferase mediated nick end labeling
Graphical abstract
Highlights
-
•
Dox exposure inhibits Mfn2-mediated mitochondrial fusion in the hearts.
-
•
The reduction of Mfn2 is mediated by FoxO1 in a transcriptional manner.
-
•
Mfn2 overexpression reduces Dox-induced injury and enhances its anticancer action.
-
•
Mfn2 induces a shift from aerobic glycolysis to mitochondrial oxidative metabolism.
1. Introduction
Since the late 1960s, anthracyclines like doxorubicin (Dox) have been widely used in cancer chemotherapy, and they continue to be mainstays in cancer treatment when combined with new-generation targeted therapies. The cardiotoxic effects of anthracyclines, however, severely restrict their therapeutic utility [1]. The modification or even termination of the anticancer treatments is often required. Cardiotoxicity tends to be more common during the first year following the end of therapy, thus increasing cancer survivors' premature morbidity and mortality [2].
Although clinical assessment allows for the early diagnosis of cardiotoxicity, primary interventions remain a therapeutic need that has yet to be addressed. Small clinical studies have revealed that conventional heart failure medication has a relatively moderate impact [2]. Dexrazoxane is the sole FDA-approved cardiac protectant, while the higher risk of developing subsequent malignancies has limited its use [3,4]. Ideally, optimal cardioprotective therapies should not only reduce the cardiotoxicity of chemotherapy, but should also retain or even augment chemotherapy's anticancer activity.
Mitochondrial fusion/fission dynamics has been recognized as a key regulator of cardiac function as well as tumorigenesis. Excessive mitochondrial fission is typically found in the heart under stress circumstances, resulting in mitochondrial damage and cardiac dysfunction. Accordingly, inhibiting this excessive mitochondrial fission provides beneficial effects for multiple cardiac diseases such as diabetic cardiomyopathy and ischemia-reperfusion injury [[5], [6], [7]]. Nonetheless, inhibition of mitochondrial fission may have a negative effect on cardiac mitochondrial quality and performance in the basal state [8]. In parallel with mitochondrial fission, mitochondrial fusion is also necessary for mitochondrial health maintenance. Promotion of mitochondrial fusion by upregulating fusion-related proteins reduces myocardial damage induced by ischemia or diabetes [9,10], and does not impair mitochondrial quality in the heart in the basal state [8], making it a safer strategy than mitochondrial fission inhibition. Interestingly, inhibited mitochondrial fusion is associated with tumor proliferation and invasion [11,12]. Despite this knowledge, there is still limited information about the effects of mitochondrial fusion intervention on Dox-induced cardiotoxicity and its antitumor efficacy.
Herein, we identify the suppression of Mfn2-mediated mitochondrial fusion as a key determinant of Dox-induced cardiotoxicity. In addition, it is determined that the inhibitory effect on Mfn2 is mediated by FoxO1 in a transcriptional manner. In the hearts, reconstitution of mitochondrial fusion through upregulation of Mfn2 alleviates Dox-induced myocardial injury by enhancing mitochondrial oxidative metabolism. In the tumor tissue, Mfn2-mediated mitochondrial fusion delays tumor growth and strengthens antitumor therapy of Dox mainly by inhibiting aerobic glycolysis metabolism. We demonstrate that promoting Mfn2-mediated mitochondrial fusion is a unique approach of reducing Dox-induced cardiotoxicity while enhancing its anticancer potency through metabolic regulation.
2. Materials and methods
2.1. Animals and treatments
All animal experimental procedures were carried out in compliance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals (8th Edition, 2011) and the study was authorized by the Fourth Military Medical University Ethics Committee. Dox (5 mg/kg/week, MedChem Express) was administered intraperitoneally to male C57BL/6 mice aged 8-10-weeks for consecutive 3 weeks [13,14]. The total cumulative Dox dosage was 15 mg/kg. Age-matched C57BL/6 mice were given the same volume of saline vehicles as the control animals.
Cardiac-specific Mfn2 Transgenic mice (Tg-Mfn2) were generated by crossing Rosa26-CAG-Loxp-Stop-Loxp-Mfn2 knock-in mice with αMHC-Mer-Cre-Mer (tamoxifen-inducible heart-specific Cre) mice on C57BL/6 genetic background, both of which were obtained from Shanghai Model Organisms Center. A 5-day intraperitoneal tamoxifen (20 mg/kg/d, MedChem Express) injection was used to elicit gene recombination as described previously [15] in 10-week-old mice followed by an additional 2 weeks to allow the clearance of tamoxifen. Age-matched male littermates (Rosa26-CAG-Loxp-Stop-Loxp-Mfn2) treated with comparable tamoxifen were served as non-transgenic controls (NTg). The information of the primers used in genotyping was provided in Supplementary Table 1.
2.2. Cardiomyocyte culture and adenoviral transfection
Primary neonatal cardiomyocytes were obtained from newborn Sprague-Dawley rats aged 1–2 days. Dox was applied to the cardiomyocytes for 24 h at the given dosage. To increase the expression of Mfn2 or FoxO1, the cardiomyocytes were transfected with adenoviruses encoding the Mfn2 gene (Ad-Mfn2), the FoxO1 gene (Ad-FoxO1), or an empty vector (Ad-EV) (Hanbio Technology, Shanghai, China) at varying multiplicities of infection (MOIs). NRVMs were transfected with FoxO1 siRNA (sense: GCAGACACCUUGCUAUUCATT; antisense: UGAAUAGCAAGGUGUCUGCTT) or negative control siRNA (#sc-37007, Santa Cruz Biotechnology) utilizing Lipofectamine RNAiMAX reagent to knock down FoxO1 (Invitrogen, Carlsbad, USA). The cardiomyocytes were exposed to Dox for additional 24 h after 48 h of adenoviral or siRNA transfection.
2.3. Cell viability, cTnT, and LDH measurement
Cell Counting Kit-8 test kits (Dojindo, Japan) were used to determine the vitality of cardiomyocytes according to the manufacturer's guidelines. Animal serum and cell supernatant were obtained to examine the release of cardiac injury indicators including lactate dehydrogenase (LDH) and cardiac troponin T (cTnT). The levels of LDH and cTnT were determined respectively using LDH assay kits from Beyotime Institute of Biotechnology (Nantong, China) and cTnT ELISA kits from Nanjing Jiancheng Bioengineering Institute (Nanjing, China).
2.4. Analysis of mitochondrial morphology
Tissue samples collected from the anterior wall of LV were preserved with 2.5% glutaraldehyde for 24 h at 4 °C and handled as previously reported [16]. Images were taken using a transmission electron microscope (TEM) (JEM-1230, JEOL Ltd., Tokyo, Japan) at 300 kV. Image-Pro Plus 6.0 software was used to determine the number and size of mitochondria. The morphometric analysis includes at least 300 mitochondria from at least eight pictures per heart. The proportion of mitochondria in a specific area categorized into three size groups (< 0.6 μm2, within 0.6–1.0 μm2, >1.0 μm2) was counted as previously described [6,17]. MitoTrackerRed CMXRos probe (100 nmol/L, Invitrogen, Carlsbad, USA, 30 min at 37 °C) was used to label mitochondria in the cells. Nikon A1R MP + confocal laser-scanning microscope was used to take the images. The number of mitochondria per cell and mitochondrial size were counted and measured as we previously described [10].
2.5. Quantification of apoptosis and ROS production
TUNEL labeling (Roche, Shanghai, China) and caspase-3 activity colorimetric assay kits (KeyGEN Biotech, Jiangsu, China) were used to determine cellular apoptosis as described previously [18]. The apoptosis index was obtained by dividing the number of TUNEL-positive apoptotic cells by the total number of nucleated cells stained with 4′,6-diamino-2-phenylindole (DAPI). The caspase 3 colorimetric assay kit is based on the hydrolysis of acetyl-Asp-Glu-Val-Asp p-nitroanilide (Ac-DEVD-pNA) by caspase 3, resulting in the release of the p-nitroaniline (pNA) moiety. The p-NA produces a yellow color and can be quantified using a spectrophotometer or a microtiter plate reader reading absorbance at 405 nm. DCFH-DA staining (Beyotime Biotechnology, Nantong, China) and MitoSOX staining (Invitrogen, Carlsbad, USA) were used to measure cellular and mitochondrial ROS levels in the cardiomyocytes. DCFH was added to the cardiac homogenate to evaluate ROS production, and the fluorescence was measured using a fluorometer [19]. Total superoxide dismutase (SOD), cellular CuZnSOD, mitochondrial MnSOD, and malondialdehyde (MDA) levels were determined utilizing standard assay kits (Beyotime Biotechnology, Nantong, China) following the manufacturer's recommendations.
2.6. Assessment of mitochondrial function
Mitochondrial membrane potential (MMP) was measured utilizing JC-1 MMP assay kits (Beyotime Biotechnology, Nantong, China) as directed by the manufacturer's instructions. High mitochondrial membrane potential is shown by red fluorescence, while low mitochondrial membrane potential is indicated by green fluorescence. The relative level of MMP was calculated by dividing the intensity of red fluorescence by the intensity of green fluorescence.
Oxygen consumption rate (OCR) was determined using an XF24 Extracellular Flux Analyzer (Agilent SeaHorse Bioscience, USA) [20]. The cells were stimulated with 1 μM oligomycin, 0.5 μM FCCP and 1 μM antimycin A + Rotenone sequentially to determine ATP-related OCR, maximal OCR and non-mitochondrial OCR. The concentrations of intracellular proteins were determined to normalize all the OCR data. Mitochondrial respiratory chain complex activity assay kits (Solarbio, Beijing, China) were used to assess the activities of mitochondrial complex I-IV as directed by the manufacturer's instructions.
2.7. Assessment of glycolytic capacity
Extracellular acidification rate (ECAR) was evaluated as an indication of glycolytic capacity using an XF24 Extracellular Flux Analyzer (Agilent SeaHorse Bioscience, USA) [10]. The cells were stimulated with 10 mM glucose, 1 μM oligomycin and 50 mM 2-deoxy-d-glucose (2-DG) sequentially to assess glucose metabolism capacity, maximal glycolytic capacity and non-glycolytic capacity respectively. The concentrations of intracellular proteins were determined to normalize all the ECAR data.
2.8. Measurement of cardiac function
A VEVO 3100 ultrasound equipment (Visual Sonics, Toronto, Canada) was used to perform echocardiography on anesthetized mice at the appropriate time as described previously [21]. Isoflurane was used to anesthetize the animals for the assessment. The images in M-mode were acquired. Left ventricular end-systolic volume (LVESV) and ejection fraction (EF) were measured and calculated by the system.
2.9. Histological analysis
Fluorescein isothiocyanate (FITC)-labeled wheat germ agglutinin (WGA) staining (Servicebio, Wuhan, China) was used to measure the cross-sectional area of the cardiomyocytes. Masson's trichrome staining (Servicebio, Wuhan, China) was used to assess myocardial interstitial fibrosis. The ratio of fibrotic area to total area was used to determine the collagen volume fraction.
2.10. Tumor studies
Dox-sensitive mouse melanoma (B16), human hepatocellular carcinoma (SUN-368), mouse hepatocellular carcinoma (Hepa 1–6) and mouse mammary carcinoma (4T1) cell lines were kindly provided by Associate Professor Xiang Zhang (Department of Biochemistry and Molecular Biology, Fourth Military Medical University) and cultured in DMEM medium. To explore the influence of Mfn2 on Dox's anticancer potential, tumor cells were transfected with adenoviruses harboring Mfn2 (Ad-Mfn2) for 48 h and then subjected to Dox challenge at the indicated dose for another 24 h. In addition, to assess the effect of Mfn2 on Dox chemotherapeutic capability in vivo, lentivirus encoding Mfn2 (LV-Mfn2) was employed to achieve stable expression of Mfn2 in B16 melanoma. Male C57BL/6 mice aged 8–10 weeks were injected subcutaneously with 1 × 106 B16 melanoma cells at the left back flank. The mice were given intraperitoneal Dox injections (7.5 mg/kg/week for consecutive 2 weeks) one week after cell injection. The total cumulative Dox dosage was 15 mg/kg. Tumor size was determined every week using the following formula: volume = 1/2 × length × width × height.
2.11. Quantitative real-time PCR and Western blot analysis
Quantitative real-time PCR (qRT-PCR) was used to determine the relative mRNA levels. Total RNA was taken from tissues or cells and reverse transcribed to cDNA. The experiments were carried out using a Bio-Rad Real-Time PCR System. DNA template, forward primers, reverse primers and SYBR Green PCR master mix were included in each reaction. The mRNA levels were standardized to 18S levels using the 2−ΔΔCt method. Supplementary Table 1 contained a list of all the primer sequences. Total proteins were obtained from the tissues or cells using RIPA buffer. The classic Western blotting procedure was used as previously indicated [25]. Supplementary Table 2 contained a list of all the antibodies used.
2.12. Chromatin immunoprecipitation analysis
The SimpleChIP Plus Enzymatic Chromatin IP kit (#9005, Cell Signaling Technology) was used to perform chromatin immunoprecipitation (ChIP) according to the manufacturer's instructions [22]. Briefly, 1% formaldehyde was used to cross-link the primary cardiomyocytes, which was then homogenized in the lysis solution. The enzymatically sheared chromatins were incubated with protein G magnetic beads and CHIP-grade antibodies against FoxO1 (#2880, Cell Signaling Technology). DNA was extracted from the precipitation and subsequently analyzed by real-time PCR. Supplementary Table 1 contained the sequences of the Mfn2 promoter region primers. IgG was employed as the negative control.
2.13. Luciferase activity assay
The luciferase activity assay was carried out using the Dual-Luciferase Reporter Assay System (Promega) as directed by the manufacturer's instructions. HEK-293T cells were transfected with plasmids coding for pGL-Mfn2-promoter [−2000/+0 bp] or pGL-Basic using lipofectamine 2000 (Invitrogen), and then infected with adenovirus containing FoxO1 (Ad-FoxO1) or an empty vector (Ad-EV). Luciferase activity was evaluated 48 h after infection.
2.14. Statistical analysis
All of the data were expressed as the means ± SEM (standard error of the means). The two-tailed unpaired Student's t-test was used for statistical analysis of two groups. One-way analysis of variance (ANOVA) or two-way ANOVA was used to compare more than two groups followed by Turkey or Bonferroni's multiple comparison test where appropriate. P values of less than 0.05 were considered statistically significant.
3. Results
3.1. Dox exposure inhibited Mfn2-mediated mitochondrial fusion in the hearts and cardiomyocytes
The effect of Dox on mitochondrial fusion/fission dynamics was studied both in vivo and in vitro. Increased serum cTnT levels in the Dox-treated mice indicated the model of Dox-induced cardiotoxicity was successfully constructed in vivo (Fig. 1A). The numbers of mitochondria per μm2 did not change significantly between the control and Dox treatments (Fig. 1B and C). Compared to control hearts, Dox-treated hearts showed lower mean mitochondrial size (Fig. 1B and D), an increased proportion of mitochondria with a size less than 0.6 μm2 and decreased proportion of mitochondria with a size between 0.6 and 1 μm2 or bigger than 1 μm2 (Fig. 1E). Among the mitochondrial fission proteins (Drp1 and Fis1) and fusion proteins (Mfn1, Mfn2 and Opa1), only the expression of Mfn2 was significantly reduced in the Dox-treated hearts (Fig. 1F and G). For in in vitro experiments, the primary cardiomyocytes were treated with different doses of Dox for 24 h. As shown in Supplementary Fig. S1, Dox reduced the cardiomyocyte viability in a concentration-dependent manner. The dose of 3 μM Dox caused moderate cardiomyocyte injury, when cell viability was reduced by about 40%. Thus 3 μM Dox was used to imitate the Dox injury model in vitro according to this preliminary result and the previous study [23]. The control primary cardiomyocytes mainly had highly elongated and interconnected mitochondria (Fig. 1H). When compared to control cardiomyocytes, Dox-exposed cardiomyocytes possessed smaller and shorter mitochondria. There was a decrease in mitochondrial size and an increase in the number of mitochondria per cell (Fig. 1H–J) as well as a decrease in Mfn2 expression (Fig. 1K) in Dox-treated cardiomyocytes. All of the findings suggested that Mfn2-mediated mitochondrial fusion was suppressed in both the Dox-treated hearts and cardiomyocytes.
Fig. 1.

Mfn2-mediated mitochondrial fusion was inhibited in the hearts and cardiomyocytes upon Dox exposure. (A) Serum cTnT level measured by ELISA. n = 6 mice in each group. Dox (doxorubicin) was administered to the mice intraperitoneally at the dosage of 5 mg/kg/week for consecutive 3 weeks. (B) Representative mitochondrial images obtained by transmission electron microscope. (Quantitative data are presented in C-E). Original magnification × 15000. Representative mitochondria on the bottom images (local magnifications of top images) were marked with light green pseudo-color to highlight the changes of the mitochondria. (C) Quantitative data of mitochondrial number per μm2. (D) Quantitative data of mean mitochondrial size. (E) The proportion of mitochondria in a specific area categorized into three size groups (< 0.6 μm2, within 0.6–1.0 μm2, >1.0 μm2) was counted. (F-G) The mitochondrial fission/fusion-related proteins' representative blots and quantitative data. (H) MitoTracker Red-stained mitochondrial morphology images. Original magnification × 600. Dox (doxorubicin) was applied to the cardiomyocytes for 24 h at the dosage of 3 μM. (I) Quantitative data of mean mitochondrial size in the cardiomyocytes. (J) Quantitative data of mitochondrial number per cell. (K) Representative blots and quantitative data of Mfn2 in the cardiomyocytes. n = 6 independent experiments per group in Figure A–E and Figure H–J. The blotting experiments were conducted 3 times independently per group in Figure F–G and Figure K. Two-tailed unpaired Student's t-test was used. **P < 0.01, ***P < 0.001. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.2. Mfn2 overexpression restored mitochondrial fusion and inhibited mitochondria-dependent apoptosis in the dox-treated cardiomyocytes
To investigate the role of Mfn2-mediated mitochondrial fusion in Dox-induced cardiotoxicity, adenovirus encoding Mfn2 (Ad-Mfn2) were transfected into the primary cardiomyocytes. As shown in Supplementary Fig. S2, the expression of Mfn2 was significantly increased after Ad-Mfn2 transfection in a dose-dependent manner. Ad-Mfn2 at an MOI of 25 resulted in a nearly 2.5-fold increase in Mfn2 expression, which may bring Mfn2 back to normal levels under the Dox conditions (Mfn2 levels was reduce to approximately 50%). This MOI was then chosen for subsequent experiments. As expected, the reduction of Mfn2 upon Dox insult was rescued to normal levels by Ad-Mfn2 (Fig. 2A). Ad-Mfn2 significantly increased mitochondrial size and reduced mitochondrial number per cell in the presence of Dox (Fig. 2B and C), indicating that Mfn2 overexpression restored mitochondrial fusion in the Dox-treated primary cardiomyocytes. Upregulation of Mfn2 reduced cellular injury and apoptosis in the Dox-treated cardiomyocytes, as shown by enhanced cell viability (Fig. 2D), decreased LDH release, caspase-3 activity and TUNEL apoptotic index (Fig. 2E–H). Moreover, Ad-Mfn2 substantially raised mitochondrial membrane potential (Fig. 2I and J) and prevented mitochondrial cytochrome c release (Fig. 2K and L). These data indicated that restoration of Mfn2-mediated mitochondrial fusion protected the cardiomyocytes against mitochondria-dependent apoptosis induced by Dox.
Fig. 2.

Mfn2 overexpression restored mitochondrial fusion and inhibited mitochondria-dependent apoptosis in the Dox-treated cardiomyocytes. (A) Representative blots and quantitative data of Mfn2. (B) MitoTracker Red-stained mitochondrial morphology images and quantitative data of mean mitochondrial size. Original magnification × 600. (C) Quantitative data of mitochondrial number per cell. (D) Relative cell viability. (E) Lactate dehydrogenase (LDH) release determined in cell supernatant. (F) Relative activity of caspase 3 expressed as a fold change compared with Con + Ad-EV. (G) Representative images of TUNEL and DAPI staining. (Quantitative data are presented in H). Original magnification × 400. (H) Quantitative data of the apoptotic index. (I) Representative images of mitochondrial membrane potential stained by JC-1. (Quantitative data are presented in J). Original magnification × 200. (J) Quantification of mitochondrial membrane potential. (K-L) Representative blots and quantitative data of cytosolic cytochrome c (cyt-c). n = 6 independent experiments per group in Figure B–J. The blotting experiments were conducted 3 times independently per group in Figure A and Figure K–L. One-way ANOVA with Turkey's multiple comparison test was used. *P < 0.05, **P < 0.01, ***P < 0.001. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.3. Mfn2 overexpression alleviated mitochondrial oxidative stress and improved mitochondrial oxidative phosphorylation (OXPHOS) capacity in the dox-treated cardiomyocytes
Mitochondrial ROS (red fluorescence) and cellular ROS (green fluorescence) were significantly increased in the Ad-EV-transfected cardiomyocytes upon Dox exposure (Fig. 3A–C). The co-staining results (merged yellow fluorescence) revealed that the majority of cellular ROS originated from mitochondria (Fig. 3A). Mfn2 overexpression inhibited both mitochondrial and cellular ROS production in Dox-treated cardiomyocytes (Fig. 3A–C). Dox suppressed mitochondrial oxidative phosphorylation (OXPHOS) capacity in the Ad-EV-transfected cardiomyocytes, including basal respiration, ATP-related respiration and maximal respiration capacity (Fig. 3D and E). Mfn2 overexpression significantly enhanced mitochondrial basal respiration and maximal respiration capacity in the Dox-treated cardiomyocytes (Fig. 3D and E). Extracellular acidification rate (ECAR) was determined as a glycolysis indicator. Dox exposure also inhibited extracellular glycolytic capacity (ECAR) in the Ad-EV-transfected cardiomyocytes, including basal glycolytic, glucose metabolism, maximal glycolytic and spare glycolytic capacity (Fig. 3F–G). Mfn2 overexpression had no significant effects on all glycolytic parameters in the Dox-treated cardiomyocytes (Fig. 3F–G). In addition, Dox exposure suppressed the activities of mitochondrial complex I, III (Fig. 3H) and nearly inhibited all the mRNA expressions of representative metabolic enzymes involved in OXPHOS and glycolysis (Fig. 3I). Mfn2 overexpression prevented the reduction of mitochondrial complex I, III activities (Fig. 3H) and restored the expression of several OXPHOS enzymes including IDH2, FH and MDH2 in the Dox-treated cardiomyocytes (Fig. 3I). The ratio of OCR/ECAR was eventually analyzed as a reflection of the relative magnitude of OXPHOS versus glycolysis. As shown in Fig. 3J, Dox exposure suppressed basal OCR/ECAR ratio, while Mfn2 overexpression significantly increased basal and maximal OCR/ECAR ratio in the Dox-treated cardiomyocytes. These results together suggested that Mfn2 overexpression prevented Dox-induced elevation of mitochondrial oxidative stress and inhibition of OXPHOS.
Fig. 3.

Mfn2 overexpression inhibited mitochondrial ROS and enhanced mitochondrial oxidative metabolism in the Dox-treated cardiomyocytes. (A) Representative images of MitoSOX-stained mitochondrial ROS (red fluorescence) and DCFH-DA-stained cellular ROS production (green fluorescence). Original magnification × 600. (B) Quantitative data of relative whole-cell ROS fluorescence density expressed as a fold change compared with Con + Ad-EV. (C) Quantitative data of relative mitochondrial ROS fluorescence density expressed as a fold change compared with Con + Ad-EV. (D-E) Oxygen consumption rate (OCR) and quantitative data of OCR. (F-G) Extracellular acidification rate (ECAR) and quantitative data of ECAR. (H) Quantitative data of mitochondrial complex I to IV (CI to CIV) activity expressed as a fold change compared with Con + Ad-EV. n = 6 independent experiments per group in Figure A–C and Figure H. (I) Quantitative mRNA expression of representative metabolic enzymes involved in mitochondrial oxidative phosphorylation and glycolysis. PDHX, pyruvate dehydrogenase complex component X; CS, citrate synthase; IDH, isocitrate dehydrogenase; FH, fumarate hydratase; MDH, malate dehydrogenase; HK, hexokinase; GPI, glucose-6-phosphate isomerase; PFKFB2, 6-phosphofructo-2-kinase; ALDOA, fructose bisphosphate aldolase A; PGAM2, phosphoglycerate mutase 2; ENO1, enolase 1; PKM, Pyruvate kinase M; LDH, lactate dehydrogenase. (J) The ratio of oxygen consumption rate (OCR) relative to extracellular acidification rate (ECAR). n = 3 independent experiments per group in Figure D–G and FigureI-J. One-way ANOVA with Turkey's multiple comparison test was used. *P < 0.05, **P < 0.01, ***P < 0.001. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.4. Mfn2-mediated mitochondrial fusion exerted protective effects via enhancing mitochondrial oxidative metabolism
To further explore the role of elevated mitochondrial fusion and enhanced mitochondrial complex I, III activities in Mfn2-mediated protective effects, lentivirus expressing Drp1 (LV-Drp1) was used to inhibit mitochondrial fusion in the cardiomyocytes. Rotenone (a mitochondrial complex I inhibitor) and antimycin A (a mitochondrial complex III inhibitor) were employed to block mitochondrial oxidative metabolism. No significant loss of cell viability was observed when the cardiomyocytes were treated with LV-Drp1 ranging from 10 to 40 MOI (Supplementary Fig. S3A), rotenone ranging from 0.03 to 0.1 μM (Supplementary Fig. S3B) or antimycin A ranging from 0.03 to 1 μM (Supplementary Fig. S3C). Rotenone combined with antimycin A (Rote 0.1 μM + Anti A 1 μM) also had no obvious influence on cell viability, and effectively inhibited the activities of both mitochondrial complexes I and III (Supplementary Figs. S3D–F). Drp1 expression was considerably upregulated in cells transfected with LV-Drp1 at a MOI of 40 (Supplementary Fig. S3G). These optimal non-toxic concentrations were then used for the subsequent experiments. As shown in Fig. 4A–E, application of LV-Drp1 (40 MOI, 48 h before Ad-Mfn2 transfection) or Rote/Anti A (Rote 0.1 μM + Anti A 1 μM, 30 min before Ad-Mfn2 transfection) blocked the inhibitory effects of Mfn2 on cellular injury/apoptosis and mitochondrial ROS in the Dox-treated cardiomyocytes. Moreover, the upregulating effects of Mfn2 on mitochondrial fusion and mitochondrial complex I/III activities were synchronously blunted by either LV-Drp1 or Rote/Anti A (Fig. 4E–I). These results above indicated that Mfn2 protected the cardiomyocytes against Dox-induced injury via enhancing mitochondrial fusion and oxidative metabolism.
Fig. 4.

Mfn2 protected the cardiomyocytes against Dox-induced injury via enhancing mitochondrial fusion and oxidative metabolism. The cardiomyocytes were treated with lentivirus expressing Drp1 (LV-Drp1) or Rote/Anti A (Rote 0.1 μM + Anti A 1 μM) before the transfection of adenoviruses encoding the Mfn2 gene (Ad-Mfn2) under Dox condition. (A) Relative cell viability. (B) Lactate dehydrogenase (LDH) release in cell supernatant. (C) Relative activity of caspase 3 expressed as a fold change compared with Dox. (D) Quantitative data of relative mitochondrial ROS fluorescence density. (E) Representative images of mitochondria-derived superoxide production stained with MitoSOX (upper, quantitative data are presented in D) and MitoTracker Red-stained mitochondrial morphology (bottom, quantitative data are presented in F–G). Original magnification × 600. (F) Quantitative data of mean mitochondrial size in the cardiomyocytes. (G) Quantitative data of mitochondrial number per cell. (H–I) Quantitative data of mitochondrial complex I and III activity. n = 6 independent experiments per group in Figure A–I. One-way ANOVA with Turkey's multiple comparison test was used. ***P < 0.001. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.5. Cardiac-specific overexpression of Mfn2 promoted mitochondrial fusion and attenuated dox-induced cardiotoxicity in vivo
After demonstrating that Mfn2-mediated mitochondrial fusion protected cardiomyocytes against Dox-induced injury in vitro, the protective effects of Mfn2 overexpression were then validated in vivo by using cardiac-specific Mfn2 Transgenic mice (Tg-Mfn2), which were generated by crossing Rosa26-CAG-Loxp-Stop-Loxp-Mfn2 knock-in mice with αMHC-Mer-Cre-Mer mice (Supplementary Figs. S4A and 4B). Mfn2 expression was significantly elevated in cardiac tissues of Tg-Mfn2 mice. In other tissues including liver, muscle and brain, the expression of Mfn2 was comparable between non-transgenic controls (NTg) and Tg-Mfn2 mice (Supplementary Fig. S4C). Compared to NTg animals, Tg-Mfn2 mice showed significantly increased Mfn2 expression in the Dox-treated hearts (Fig. 5A). Mfn2 overexpression markedly preserved cardiac function and attenuated myocardial injury upon Dox exposure, as shown by the increased ejection fraction (EF), decreased left ventricular end-systolic volume (LVESV) and reduced serum levels of cTnT (Fig. 5B–E). Moreover, Mfn2 overexpression promoted mitochondrial fusion in the Dox-treated hearts, as reflected by increased mean mitochondrial size, decreased proportion of mitochondria with a size less than 0.6 μm2 and increased proportion of mitochondria with a size between 0.6 and 1 μm2 or bigger than 1 μm2 (Fig. 5F–H). The number of mitochondria per μm2 did not differ significantly across the experimental groups (Fig. 5I).
Fig. 5.

Cardiac-Specific overexpression of Mfn2 promoted mitochondrial fusion and improved cardiac function in the Dox-treated mice. (A) Representative blots and quantitative data of Mfn2. The blotting experiments were conducted 3 times independently per group in Figure A. (B) Representative images of M-mode echocardiography (Quantitative data are presented in C-D). (C) EF, ejection fraction. (D) LVESV, left ventricular end-systolic volume. (E) Serum cTnT level measured by ELISA. (F) Representative mitochondrial images obtained by transmission electron microscope (Quantitative data are presented in G-I). Original magnification × 15000. Representative mitochondria on the bottom images (local magnifications of top images) were marked with light green pseudo-color to highlight the changes of the mitochondria. (G) The proportion of mitochondria in a specific area categorized into three size groups (< 0.6 μm2, within 0.6–1.0 μm2, >1.0 μm2) was counted. (H) Quantitative data of mean mitochondrial size. (I) Quantitative data of mitochondrial number per μm2. n = 6 independent experiments per group in Figure B–I. One-way ANOVA with Turkey's multiple comparison test was used. **P < 0.01, ***P < 0.001. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Abnormal cardiac structure is an important characteristic change in Dox-induced cardiotoxicity [24]. Compared with the vehicle-treated NTg mice, NTg mice treated with Dox showed cardiac atrophy and fibrosis, as shown by decreased cardiomyocyte cross-sectional area and increased collagen volume fraction (Fig. 6A–C). Mfn2 overexpression prevented cardiac atrophy and fibrosis in the Dox-treated mice (Fig. 6A–C). Oxidative stress and cardiomyocyte apoptosis have been reported to be the major contributors to Dox-induced cardiotoxicity [25]. As expected, there was an increase in cardiomyocyte apoptosis (as measured by the apoptosis index and caspase 3 activity in Fig. 6D–F) and oxidative stress (indicated by elevated ROS level, increased MDA content, decreased activity of total superoxide dismutase (SOD), cellular CuZnSOD and mitochondrial MnSOD in Fig. 6G–I) in the Dox-treated NTg hearts as compared to the vehicle-treated NTg hearts. Mfn2 overexpression significantly reduced cardiac apoptosis and prevented ROS production in the Dox-treated hearts (Fig. 6D–I). Together, these data indicated that Mfn2 overexpression restored mitochondrial fusion and alleviated Dox-induced cardiotoxicity in the animals, as manifested by suppressed myocardial apoptosis/oxidative stress and improved cardiac structure/function.
Fig. 6.

Cardiac-Specific overexpression of Mfn2 improved cardiac structure and inhibited myocardial apoptosis/oxidative stress in the Dox-treated mice. (A) Upper: Representative photomicrographs of wheat germ agglutinin-stained cardiomyocyte size (Quantitative data are presented in B). Original magnification × 800. Lower: Representative photomicrographs of Masson's trichrome-stained interstitial fibrosis (Quantitative data are presented in C). Original magnification × 400. (B) Quantitative data of the cross-sectional area of cardiomyocytes. (C) Quantitative data of the collagen volume fraction. (D) Representative images of TUNEL and DAPI staining. (Quantitative data are presented in E). Original magnification × 400. (E) Quantification of the apoptotic index. (F) Relative activity of caspase 3 expressed as a fold change compared with NTg + Vehicle. (G) Relative level of ROS determined by a fluorometer with fluorescent probe DCFH. DCFH was added to the cardiac homogenate to evaluate ROS production, and the fluorescence was measured using a fluorometer. (H) Malondialdehyde (MDA) level. (I) The activity of total superoxide dismutase (SOD), cellular CuZnSOD and mitochondrial MnSOD. n = 6 independent experiments per group in Figure A–I. One-way ANOVA with Turkey's multiple comparison test was used. **P < 0.01, ***P < 0.001.
3.6. The suppression of Mfn2-mediated mitochondrial fusion in dox-treated hearts was transcriptionally regulated by FoxO1
How Dox exposure inhibited Mfn2-mediated mitochondrial fusion in the hearts was further explored. Mfn2 mRNA expression was shown to be decreased in Dox-treated hearts and cardiomyocytes (Supplementary Figs. S5A–B), suggesting that Dox exposure may inhibit Mfn2 expression at the transcriptional level. Public cardiac mRNA expression profiling derived from the untreated or Dox-treated mice was downloaded from the Gene Expression Omnibus (GEO) database (GSE157904) to screen for potential transcription factors. The expression pattern of key transcription factors involved in this database was displayed in heatmap form in Fig. 7A. Statistical analysis of the expression pattern indicated that only FoxO1 mRNA was significantly increased in the Dox-treated cardiomyocytes (Fig. 7B). Consistently, we found that the protein expressions of total FoxO1 and nuclear FoxO1 were upregulated in the Dox-treated cardiomyocytes as compared to the control cardiomyocytes (Fig. 7C and D). Knockdown of FoxO1 with FoxO1 RNAi significantly increased Mfn2 mRNA and protein expression (Fig. 7E–H), restored mitochondrial fusion (Fig. 7I–K) and increased cell viability (Fig. 7L) in the Dox-treated cardiomyocytes in vitro. Since the protein expression of total FoxO1 was also upregulated in the Dox-treated hearts in vivo (Fig. 8A), the mice were then treated with FoxO1 inhibitor AS1842856 (HY-100596, MedChemExpress) at the dosage of 100 mg/kg by oral gavage twice daily for two days immediately following each Dox injection according to the previous study [26]. FoxO1 inhibitor upregulated the expression of myocardial Mfn2 mRNA and protein (Fig. 8B and C), improved cardiac performance (Fig. 8D–F) and restored mitochondrial fusion (Fig. 8G–I) in the Dox-treated mice.
Fig. 7.

Knockdown of FoxO1 restored Mfn2-mediated mitochondrial fusion and increased cell viability in the Dox-treated cardiomyocytes. (A) Heatmap showing the expression pattern of key transcription factors involved in the Gene Expression Omnibus (GEO) database (GSE157904). (B) Quantitative mRNA expression of FoxO1 in GSE157904. (C) Representative blots and quantitative data of total FoxO1 protein level. (D) Representative blots and quantitative data of nuclear FoxO1 protein level. Two-tailed unpaired Student's t-test was used to compare the two groups in Figure B–D. (E-G) Representative blots and quantitative data of FoxO1 and Mfn2 protein levels. The blotting experiments were conducted 3 times independently per group in Figure C–G. (H) Quantitative mRNA expression of Mfn2. n = 3 independent experiments per group in Figure B and Figure H. (I) Representative MitoTracker Red-stained mitochondrial morphology images. (Quantitative data are presented in J–K). Original magnification × 600. (J) Quantitative data of mean mitochondrial size in the cardiomyocytes. (K) Quantitative data of mitochondrial number per cell. (L) Relative cell viability. n = 6 independent experiments per group in FigureI-L. One-way ANOVA with Turkey's multiple comparison test was used to compare the groups in Figure F–L. *P < 0.05, **P < 0.01, ***P < 0.001. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Fig. 8.

FoxO1 inhibitor restored Mfn2-mediated mitochondrial fusion and enhanced cardiac function in the Dox-treated mice. (A) Representative blots and quantitative data of total FoxO1 protein level in the heart tissues. (B) Representative blots and quantitative data of Mfn2 protein level. The blotting experiments were conducted 6 times independently per group in Figure A–B. (C) Quantitative data of Mfn2 mRNA expression. (D) Representative M-mode echocardiography images. (Quantitative data are presented in E-F) (E) EF, ejection fraction. (F) LVESV, left ventricular end-systolic volume. (G) Quantitative data of mean mitochondrial size. (H) Representative mitochondrial images obtained by transmission electron microscope (Quantitative data are presented in G and I). Original magnification × 15000. Representative mitochondria on the bottom images (local magnifications of top images) were marked with light green pseudo-color to highlight the changes of the mitochondria. (I) The proportion of mitochondria in a specific area categorized into three size groups (< 0.6 μm2, within 0.6–1.0 μm2, >1.0 μm2) was counted. n = 6 independent experiments per group in Figure C–I. One-way ANOVA with Turkey's multiple comparison test was used. *P < 0.05, **P < 0.01, ***P < 0.001. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
On the other hand, using Ad-FoxO1 transfection, FoxO1 was upregulated to mimic the effect of Dox. As shown in Fig. 9A–D, Ad-FoxO1 ranged from 10 to 90 MOI reduced Mfn2 mRNA and protein expression in a dose-dependent manner. Ad-FoxO1 at an MOI of 30 significantly reduced Mfn2 expression and inhibited mitochondrial fusion in the cardiomyocytes (Fig. 9A–G). ChIP and PCR assay revealed that FoxO1 was predominantly bound to position 552–963 bp upstream of the transcriptional start site of the Mfn2 gene (PCR3) (Fig. 9H and I). Moreover, the Mfn2 promoter that bound to FoxO1 was significantly increased under the Dox condition (Fig. 9J and K). Luciferase reporter assay showed that upregulation of FoxO1 significantly inhibited the luciferase activity of the Mfn2 promoter reporter (Fig. 9L). Together, these results above indicated that Dox-elicited upregulation of FoxO1 inhibited Mfn2-mediated mitochondrial fusion at the transcriptional level.
Fig. 9.

FoxO1 inhibited the transcription of Mfn2 by binding to its promoter sites. Ad-FoxO1 reduced Mfn2 mRNA and protein expression in a dose-dependent manner. (A-C) Representative blots and quantitative data of FoxO1 and Mfn2 protein levels. The blotting experiments were conducted 3 times independently per group in Figure A–C. (D) Quantitative data of Mfn2 mRNA expression. n = 3 independent experiments per group in Figure D. One-way ANOVA with Bonferroni's multiple comparison test was used to compare the groups in Figure B–D. (E) Representative MitoTracker Red-stained mitochondrial morphology images (Quantitative data are presented in F-G). Original magnification × 600. (F) Quantitative data of mean mitochondrial size in the cardiomyocytes. (G) Quantitative data of mitochondrial number per cell. n = 6 independent experiments per group in Figure E–G. Two-tailed unpaired Student's t-test was used to compare the groups in Figure F–G. (H) ChIP analysis of FoxO1 at five positions of Mfn2 promoter in the cardiomyocytes. One-way ANOVA with Turkey's multiple comparison test was used. (I) Allocation of five primers of Mfn2 promoter. (J-K) ChIP analysis for FoxO1 binding to Mfn2 promoter using prime 3 in the cardiomyocytes subjected to different treatments. (L) FoxO1 inhibits the luciferase activity of the Mfn2 promoter in HEK-293T cells. n = 3 independent experiments per group in Figure H–L. Two-way ANOVA with Bonferroni's multiple comparison test was used to compare the groups in Figure K–L. *P < 0.05, **P < 0.01, ***P < 0.001; ###P < 0.001 vs. other primers. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.7. Mfn2 potentiated the anticancer activity of dox via inhibiting aerobic glycolysis metabolism
Finally, the effect of Mfn2 overexpression on Dox's anticancer efficacy was investigated. Since 3 μM Dox has been applied to the cardiomyocytes in our previous experiments, this dosage was then chosen for subsequent tumor cell experiments in vitro. As shown in Fig. 10A–C and Supplementary Fig. S6A-L, in various models of Dox-sensitive tumors including mouse melanoma (B16), human hepatocellular carcinoma (SUN-368), mouse hepatocellular carcinoma (Hepa 1–6) and mouse mammary carcinoma (4T1), Mfn2 overexpression via Ad-Mfn2 alone could induce cell death (a reduction in cell viability and an increase in LDH release) as well as enhance the effects of Dox on antagonizing tumor cells. Ad-Mfn2 promoted mitochondrial fusion (increased mitochondrial size and reduced mitochondrial number per cell) in the B16 tumor cells whether upon vehicle control (Con) or Dox exposure (Fig. 10D–F). To further explore the potential effect of Mfn2 on the antitumor action of Dox in vivo, B16 cell lines with stable overexpression of Mfn2 (LV-Mfn2) were established by using lentiviral transfection (Fig. 10G). In the mice with subcutaneous B16 melanoma xenografts, Mfn2 overexpression (LV-Mfn2) not only attenuated tumor growth alone but also further delayed tumor growth in the presence of Dox, resulting in considerably smaller tumors in Dox + LV-Mfn2 than in Dox + LV-EV group (Fig. 10H and I).
Fig. 10.

Mfn2 boosted the antitumor action of Dox in mouse B16 melanoma. (A) Representative blots and quantitative data of Mfn2 in the B16 tumor cells. (B) Relative cell viability in the B16 tumor cells. n = 6 per group. (C) Lactate dehydrogenase (LDH) release in cell supernatant. n = 6 per group. (D) Representative MitoTracker Red-stained mitochondrial morphology images (Quantitative data are presented in E-F). Original magnification × 600. (E) Quantitative data of mean mitochondrial size in the B16 tumor cells. (F) Quantitative data of mitochondrial number per cell. (G) B16 cell lines with stable overexpression of Mfn2 (LV-Mfn2) were established by using lentiviral transfection. The blotting experiments were conducted 3 times independently per group in Figure A and Figure G. (H) Tumor growth curve post B16 tumor cell injection. (I) Representative images of B16 melanoma and Quantitative data of tumor weight at 3 weeks post B16 tumor cell injection. n = 6 independent experiments per group in Figure B–F and Figure H–I. One-way ANOVA with Turkey's multiple comparison test was used. *P < 0.05, **P < 0.01, ***P < 0.001. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
To clarify how Mfn2 overexpression synergizes with Dox, the Seahorse platform was used to monitor the metabolism state of B16 cells. Under the control (Con) condition, Ad-Mfn2 enhanced mitochondrial oxidative function (increased mitochondrial complex I/III activities, Fig. 11A–C) and inhibited glycolytic capacity (decreased ECAR, Fig. 11D–E) in the B16 cells. Dox inhibited both mitochondrial oxidative function (decreased OCR and mitochondrial complex I activity, Fig. 11A–C) and glycolytic capacity (decreased ECAR, Fig. 11D–E) in the Ad-EV-transfected B16 cells. Ad-Mfn2 further inhibited glycolytic capacity in the Dox-treated B16 cells (Fig. 11D–E). The ratio of OCR/ECAR was also analyzed as a reflection of the relative magnitude of OXPHOS versus glycolysis in the B16 cells. Consequently, Dox exposure significantly suppressed basal and maximal OCR/ECAR ratio, while Mfn2 overexpression increased the maximal OCR/ECAR ratio in the presence or absence of Dox (Fig. 11F). In addition, under the control condition, Ad-Mfn2 increased the expression of one OXPHOS enzyme IDH1 and inhibited the expression of one glycolysis-related enzyme LDH2 (Fig. 11G). Dox exposure reduced the expression of several representative metabolic enzymes involved in OXPHOS and glycolysis, including IDH1, IDH2, ENO1, LDH1 and LDH2 (Fig. 11G). Under Dox-treated conditions, Mfn2 overexpression further reduced the expression of glycolysis-related enzyme LDH2 (Fig. 11G). To further ascertain whether the inhibition of glycolysis is required for the enhancing effect of Mfn2 on the anticancer action of Dox, 2-Deoxy-d-glucose (2-DG, 16 mM) was used to block aerobic glycolysis in B16 tumor cells according to the previous study [27]. Pretreatment with 2-DG increased tumor cell death in the Dox-treated B16 cells, while Ad-Mfn2 did not further enhance the tumor-killing effects of Dox following pretreatment with 2-DG (Supplementary Fig. S7). Taken together, these results suggested that Mfn2 overexpression synergized with the antitumor efficacy of Dox through inhibiting glycolysis metabolism.
Fig. 11.

Mfn2 synergized the inhibitory effects of Dox on glycolysis metabolism in the B16 tumor cells. (A-B) Oxygen consumption rate (OCR) and quantitative data of OCR. (C) Quantitative data of mitochondrial complex I to IV (CI to CIV) activity. n = 6 independent experiments per group in Figure C. (D-E) Extracellular acidification rate (ECAR) and quantitative data of ECAR. (F) The ratio of oxygen consumption rate (OCR) relative to extracellular acidification rate (ECAR). (G) Quantitative mRNA expression of representative metabolic enzymes involved in mitochondrial oxidative phosphorylation and glycolysis. PDHX, pyruvate dehydrogenase complex component X; CS, citrate synthase; IDH, isocitrate dehydrogenase; FH, fumarate hydratase; MDH, malate dehydrogenase; HK, hexokinase; GPI, glucose-6-phosphate isomerase; PFKFB2, 6-phosphofructo-2-kinase; ALDOA, fructose bisphosphate aldolase A; PGAM2, phosphoglycerate mutase 2; ENO1, enolase 1; PKM, Pyruvate kinase M; LDH, lactate dehydrogenase. n = 3 independent experiments per group in Figure A–B and Figure D–G. One-way ANOVA with Turkey's multiple comparison test was used. *P < 0.05, **P < 0.01, ***P < 0.001.
4. Discussion
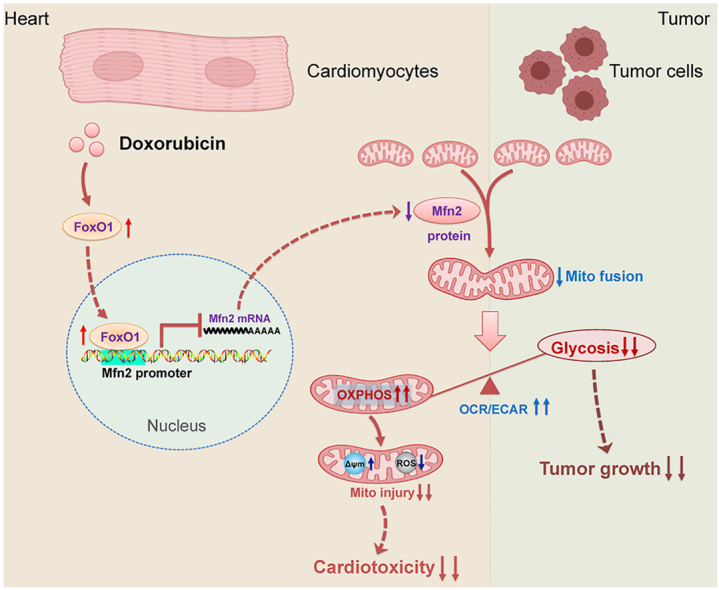
There has been substantial evidence indicating that anticancer Dox therapy may result in functional and structural impairment of the heart, which significantly restricts its therapeutic use. Up to now, the therapeutic strategies against Dox-induced cardiotoxicity without affecting its antitumor efficacy are still limited. In this study, we provide one important finding that promotion of Mfn2-mediated mitochondrial fusion protects against Dox-induced myocardial injury and enhances its anticancer activity, in which aerobic glycolysis is switched to mitochondrial oxidative metabolism (as evidenced by increased OCR-to-ECAR ratio in both cardiomyocytes and tumor cells, Fig. 12). Therefore, promoting mitochondrial fusion by upregulating Mfn2 might be a promising therapeutic strategy in Dox-based chemotherapy since it has the prospects of “killing two birds with one stone”, that is, reducing Dox-induced cardiac adverse effects while increasing its anti-cancer properties.
Fig. 12.

Schematic figure illustrating that Mfn2-mediated mitochondrial fusion alleviates Dox-induced cardiotoxicity and enhances its anticancer efficacy via metabolic shift from glycolysis to mitochondrial oxidative phosphorylation (OXPHOS). In the cardiomyocytes, Dox-elicited upregulation of FoxO1 negatively inhibits the transcriptional expression of Mfn2 directly by binding to its promoter. Reconstitution of mitochondrial fusion through upregulation of Mfn2 alleviates Dox-induced myocardial injury by enhancing OXPHOS. In the tumor cells, Mfn2-mediated mitochondrial fusion delays tumor growth and strengthens antitumor therapy of Dox mainly by inhibiting aerobic glycolysis metabolism. OCR/ECAR, the ratio of oxygen consumption rate to extracellular acidification rate. Δψm, mitochondrial membrane potential.
Cardiomyocytes continually perform the contractile function and are highly reliant upon energy supply through mitochondrial oxidative phosphorylation. Relative to cardiomyocytes, tumor cells are characterized by unlimited proliferation and are largely dependent on energy supply through aerobic glycolysis even under the normal oxygen condition, which is known as the Warburg effect [28]. Mitochondrial fusion and fission change the morphology of mitochondria dynamically and thus make energetic adjustments in accordance with the metabolic cellular needs [29]. As indicated in our study, the mitochondria in the cardiomyocytes are mainly elongated and interconnected (Con in Fig. 1H), while the mitochondria of the B16 tumor cells are mainly fragmented under the basal conditions (Con in Fig. 10D). It is meaningful that the distinct mitochondrial dynamics in cardiomyocytes and tumor cells are compatible with their metabolic characteristics, since mitochondrial fusion is often associated with enhanced OXPHOS in cardiomyocytes [5,30] while mitochondrial fission exhibited by tumor cells contributes to inhibited OXPHOS and enhanced aerobic glycolysis [31,32]. Therefore, although promoting mitochondrial fusion induces a similar metabolic shift from glycolysis to OXPHOS in both cardiomyocytes and tumor cells, the distinguishing metabolic patterns ultimately result in different cell fates, as shown in our study that mitochondrial fusion promotes cardiomyocyte survival in the setting of Dox cardiotoxicity while inducing tumor cell death in the present or absent of Dox. Our study suggests that more interventions targeting different metabolic features between myocardium and tumor may be novel therapeutic strategies to reduce Dox-induced cardiotoxicity with boosting its antitumor potential.
It is well-known that Dox inhibits mitochondrial respiration in the cardiomyocytes [33,34], while the effects of Dox on mitochondrial respiration in tumor cells are still largely unclear. Our study found that Dox significantly suppresses mitochondrial oxidative phosphorylation (OXPHOS) capacity, inhibits mitochondrial complex activity and several OXPHOS enzymes in both cardiomyocytes and B16 tumor cells. Along with this, Dox exposure also inhibits aerobic glycolytic capacity and several glycolysis enzymes in both cardiomyocytes and tumor cells. To sum up, a decrease in the OCR-to-ECAR ratio indicates that Dox has a greater effect on mitochondrial oxidative phosphorylation relative to glycolysis. This may be due to that Dox predominantly accumulates in mitochondria and nucleus [35], with the former interacting directly with Complex I and other oxidative phosphorylation proteins [24,36,37]. The accumulation of Dox in the nucleus binds to DNA and inhibits the activity of topoisomerase 2β, resulting in downstream suppression of mitochondrial gene expression and secondary impairment of oxidative phosphorylation [24,38].
Several mitochondrial fission and fusion proteins are involved in the delicate regulation of mitochondrial dynamic balance. The expression profiles of mitochondrial dynamics-related proteins are often altered in the setting of disrupted mitochondrial dynamics in cardiac diseases. In the present study, suppressed mitochondrial fusion in Dox-treated hearts or cardiomyocytes is induced by the decreased expression of Mfn2. Although previous studies have shown that Dox differently alters the level of fission/fusion proteins under various experimental conditions, the reduction of Mfn2 expression appears to be the most prominent phenotype among these studies [[39], [40], [41]]. Up to now, there are limited studies that target mitochondrial fusion modulation as an intervention in doxorubicin-induced cardiotoxicity, especially in vivo. Herein, using cellular and transgenic animal models in vitro and in vivo, we have demonstrated that reconstitution of Mfn2 restores mitochondrial fusion and subsequently alleviates Dox-induced mitochondrial dysfunction and cardiotoxicity. These findings further demonstrate the feasibility of targeting Mfn2-mediated mitochondrial fusion to prevent mitochondrial injury as occurred in Dox cardiotoxicity.
Besides being identified as a mitochondrial fusion protein, Mfn2 has been shown to have other non-fusion pleiotropic functions including mitophagy induction and mitochondria–endoplasmic reticulum (ER) tether [42,43]. Drp1 is a well-known mitochondrial fission protein, while it is not involved in mitochondria–ER contact sites [44] and has a similar function with Mfn2 in the aspect of accelerating mitophagy flux [45]. These characteristics make Drp1 overexpression an ideal fusion inhibitor without antagonizing the non-fusion function of Mfn2. In our study, the inhibitory effects of Mfn2 on Dox-induced cardiac injury are largely blunted by LV-Drp1, suggesting that enhanced mitochondrial fusion induced by Mfn2 could be the main contributor to its protective effects. Nevertheless, other unknown non-fusion functions besides Mfn2 could not be fully ruled out when regarding its protective effects. In addition, it is found that fission activator LV-Drp1 blunts the enhancing effects of Mfn2 on mitochondrial complex I/III activities in Dox-treated cardiomyocytes. In turn, mitochondrial complex I/III inhibitors Rote/Anti A also blunts the enhancing effects of Mfn2 on mitochondrial fusion (Fig. 4E–I). These findings imply that mitochondrial dynamics and mitochondrial complex activities may be causally linked under the Dox condition, further supporting the notion that mitochondrial morphology and function reciprocally regulate each other in response to the environment [46].
Another important finding in our study is that the molecular mechanism of how Dox inhibited Mfn2-mediated mitochondrial fusion has been revealed. We have demonstrated that the activation of FoxO1 was responsible for the inhibition of Mfn2 and mitochondrial fusion under Dox conditions. FoxO1 belongs to the Forkhead transcription factor family and is a key regulator of myocardial homeostasis. FoxO1 activation contributes to higher sensitivity to ischemic damage in diabetic hearts through inducing nitrosative stress and endoplasmic reticulum stress [47]. Furthermore, prolonged activation of cardiac FoxO1 induced by metabolic stress causes diabetic cardiomyopathy and heart failure via insulin receptor substrate downregulation [48]. It has been proposed that FoxO1 modulates mitochondrial dynamics indirectly via the ROCK1-Drp1 pathway. FoxO1 increases the transcription of ROCK1, which then phosphorylates Drp1 at Ser600 to activate Drp1-mediated mitochondrial fission [49]. Here we identify Mfn2 as a transcriptional target of FoxO1, revealing a previously unidentified direct link between FoxO1 and mitochondrial dynamics. Since the protein and mRNA expressions of FoxO1 were both increased in the Dox-treated cardiomyocytes (Fig. 7B–C), it is speculated that Dox exposure may elevate FoxO1 expression at the transcriptional level. There might be some transcription factors involved in the transcriptional activation of FoxO1, which needs further investigation.
It needs to be mentioned that the current study still has certain limitations. First, the boosting impacts of Mfn2 on the antitumor potency of Dox were mainly validated in the B16 melanoma in vivo and hepatocellular carcinoma as well as mammary carcinoma in vitro. Considering the heterogeneity and complexity of tumors, it is hard for us to evaluate the effects of Mfn2 in all the Dox-sensitive tumor models. As Mfn2 is generally considered to be a potential tumor suppressor gene [50,51], it is speculated that Mfn2 at least does not inhibit the antitumor therapy of Dox. Second, the analysis of potential transcription factors was based on the public GEO database but not our high-throughput mRNA sequencing. In addition to FoxO1, other omitted transcription factors might be implicated in the regulation of Mfn2 transcriptional repression under the Dox condition. Third, it is difficult to understand the complex mechanism of how Mfn2 affects the mRNA expressions of various OXPHOS and glycolysis-related enzymes at present. This may be addressed in future research with the development of advanced technology. Despite these limitations, it is believed that our study has yielded valuable novel information for identifying the reconstitution of Mfn2-mediated mitochondrial fusion as a dual protective strategy under the Dox condition.
In summary, we herein reveal that Mfn2-mediated mitochondrial fusion not only defends the heart against Dox-induced toxicity but also enhances the antitumor effects of Dox via metabolic shift. These findings suggest that targeting Mfn2-mediated mitochondrial fusion might be a novel potential option for cancer patients undergoing Dox-based chemotherapy.
Fundings
This study was supported by the grants from National Natural Science Foundation of China (No. 81970316, No. 82070387, No. 82070051, No. 81670354, No. 82111530058), Key Research and Development Plan of Shaanxi (No. 2021SF-141, 2022KWZ-18) and the Fundamental Research Funds for the Central Universities (No. xzy012019115).
Authors' contributions
Mingge Ding, Jianming Pei and Feng Fu conceived and designed the study. Mingge Ding, Rui Shi, Xiaoming Gu and Shumiao Zhang performed the animal experiments. Mingge Ding, Rui Shi, Shuli Cheng and Man Li carried out the cellular experiments. Mingge Ding, Rui Shi, Shuli Cheng, Man Li, Dema De, Chaoyang Liu, Juan Li and Min Jia performed the molecular biology experiments. Mingge Ding and Rui Shi analyzed the data. Mingge Ding drafted the manuscript. Mingge Ding, Rong Fan, Jianming Pei and Feng Fu revised and edited the manuscript. All authors have read and approved the final manuscript.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2022.102311.
Contributor Information
Mingge Ding, Email: dingmingge@xjtu.edu.cn.
Jianming Pei, Email: jmpei8@fmmu.edu.cn.
Feng Fu, Email: fufeng048@126.com.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Hulst M.B., Grocholski T., Neefjes J.J.C., van Wezel G.P., Metsa-Ketela M. Anthracyclines: biosynthesis, engineering and clinical applications. Nat. Prod. Rep. 2021 doi: 10.1039/d1np00059d. [DOI] [PubMed] [Google Scholar]
- 2.Cardinale D., Colombo A., Bacchiani G., Tedeschi I., Meroni C.A., Veglia F., Civelli M., Lamantia G., Colombo N., Curigliano G., Fiorentini C., Cipolla C.M. Early detection of anthracycline cardiotoxicity and improvement with heart failure therapy. Circulation. 2015;131(22):1981–1988. doi: 10.1161/CIRCULATIONAHA.114.013777. [DOI] [PubMed] [Google Scholar]
- 3.Shaikh F., Dupuis L.L., Alexander S., Gupta A., Mertens L., Nathan P.C. Cardioprotection and second malignant neoplasms associated with dexrazoxane in children receiving anthracycline chemotherapy: a systematic review and meta-analysis. J. Natl. Cancer Inst. 2016;108(4) doi: 10.1093/jnci/djv357. [DOI] [PubMed] [Google Scholar]
- 4.Tebbi C.K., London W.B., Friedman D., Villaluna D., De Alarcon P.A., Constine L.S., Mendenhall N.P., Sposto R., Chauvenet A., Schwartz C.L. Dexrazoxane-associated risk for acute myeloid leukemia/myelodysplastic syndrome and other secondary malignancies in pediatric Hodgkin's disease. J. Clin. Oncol. 2007;25(5):493–500. doi: 10.1200/JCO.2005.02.3879. [DOI] [PubMed] [Google Scholar]
- 5.Hernandez-Resendiz S., Prunier F., Girao H., Dorn G., Hausenloy D.J., Action E.-C.C. Targeting mitochondrial fusion and fission proteins for cardioprotection. J. Cell Mol. Med. 2020;24(12):6571–6585. doi: 10.1111/jcmm.15384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ding M., Feng N., Tang D., Feng J., Li Z., Jia M., Liu Z., Gu X., Wang Y., Fu F., Pei J. Melatonin prevents Drp1-mediated mitochondrial fission in diabetic hearts through SIRT1-PGC1α pathway. J. Pineal Res. 2018;65(2) doi: 10.1111/jpi.12491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Forte M., Schirone L., Ameri P., Basso C., Catalucci D., Modica J., Chimenti C., Crotti L., Frati G., Rubattu S., Schiattarella G.G., Torella D., Perrino C., Indolfi C., Sciarretta S., Italian C. Society of Cardiology Working group on, H. Molecular Biology of the, the role of mitochondrial dynamics in cardiovascular diseases. Br. J. Pharmacol. 2021;178(10):2060–2076. doi: 10.1111/bph.15068. [DOI] [PubMed] [Google Scholar]
- 8.Qin Y., Li A., Liu B., Jiang W., Gao M., Tian X., Gong G. Mitochondrial fusion mediated by fusion promotion and fission inhibition directs adult mouse heart function toward a different direction. Faseb. J. 2020;34(1):663–675. doi: 10.1096/fj.201901671R. [DOI] [PubMed] [Google Scholar]
- 9.Ong S.B., Subrayan S., Lim S.Y., Yellon D.M., Davidson S.M., Hausenloy D.J. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 2010;121(18):2012–2022. doi: 10.1161/CIRCULATIONAHA.109.906610. [DOI] [PubMed] [Google Scholar]
- 10.Ding M., Liu C., Shi R., Yu M., Zeng K., Kang J., Fu F., Mi M. Mitochondrial fusion promoter restores mitochondrial dynamics balance and ameliorates diabetic cardiomyopathy in an optic atrophy 1-dependent way. Acta Physiol. 2020;229(1) doi: 10.1111/apha.13428. [DOI] [PubMed] [Google Scholar]
- 11.Si L., Fu J., Liu W., Hayashi T., Nie Y., Mizuno K., Hattori S., Fujisaki H., Onodera S., Ikejima T. Silibinin inhibits migration and invasion of breast cancer MDA-MB-231 cells through induction of mitochondrial fusion. Mol. Cell. Biochem. 2020;463(1–2):189–201. doi: 10.1007/s11010-019-03640-6. [DOI] [PubMed] [Google Scholar]
- 12.Zhao J., Zhang J., Yu M., Xie Y., Huang Y., Wolff D.W., Abel P.W., Tu Y. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene. 2013;32(40):4814–4824. doi: 10.1038/onc.2012.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang X., Hu C., Kong C.Y., Song P., Wu H.M., Xu S.C., Yuan Y.P., Deng W., Ma Z.G., Tang Q.Z. FNDC5 alleviates oxidative stress and cardiomyocyte apoptosis in doxorubicin-induced cardiotoxicity via activating AKT. Cell Death Differ. 2020;27(2):540–555. doi: 10.1038/s41418-019-0372-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fisher P.W., Salloum F., Das A., Hyder H., Kukreja R.C. Phosphodiesterase-5 inhibition with sildenafil attenuates cardiomyocyte apoptosis and left ventricular dysfunction in a chronic model of doxorubicin cardiotoxicity. Circulation. 2005;111(13):1601–1610. doi: 10.1161/01.CIR.0000160359.49478.C2. [DOI] [PubMed] [Google Scholar]
- 15.Sohal D.S., Nghiem M., Crackower M.A., Witt S.A., Kimball T.R., Tymitz K.M., Penninger J.M., Molkentin J.D. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ. Res. 2001;89(1):20–25. doi: 10.1161/hh1301.092687. [DOI] [PubMed] [Google Scholar]
- 16.Zepeda R., Kuzmicic J., Parra V., Troncoso R., Pennanen C., Riquelme J.A., Pedrozo Z., Chiong M., Sánchez G., Lavandero S. Drp1 loss-of-function reduces cardiomyocyte oxygen dependence protecting the heart from ischemia-reperfusion injury. J. Cardiovasc. Pharmacol. 2014;63(6):477–487. doi: 10.1097/FJC.0000000000000071. [DOI] [PubMed] [Google Scholar]
- 17.Shimizu Y., Lambert J.P., Nicholson C.K., Kim J.J., Wolfson D.W., Cho H.C., Husain A., Naqvi N., Chin L.S., Li L., Calvert J.W. DJ-1 protects the heart against ischemia-reperfusion injury by regulating mitochondrial fission. J. Mol. Cell. Cardiol. 2016;97:56–66. doi: 10.1016/j.yjmcc.2016.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ji L., Fu F., Zhang L., Liu W., Cai X., Zhang L., Zheng Q., Zhang H., Gao F. Insulin attenuates myocardial ischemia/reperfusion injury via reducing oxidative/nitrative stress. Am. J. Physiol. Endocrinol. Metab. 2010;298(4):E871–E880. doi: 10.1152/ajpendo.00623.2009. [DOI] [PubMed] [Google Scholar]
- 19.Fan G.C., Zhou X., Wang X., Song G., Qian J., Nicolaou P., Chen G., Ren X., Kranias E.G. Heat shock protein 20 interacting with phosphorylated Akt reduces doxorubicin-triggered oxidative stress and cardiotoxicity. Circ. Res. 2008;103(11):1270–1279. doi: 10.1161/CIRCRESAHA.108.182832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen L., Liu L., Li C., Hu C., Su F., Liu R., Zeng M., Zhao D., Liu J., Guo Y., Long J. A mix of apple pomace polysaccharide improves mitochondrial function and reduces oxidative stress in the liver of high-fat diet-induced obese mice. Mol. Nutr. Food Res. 2017;61(3) doi: 10.1002/mnfr.201600433. [DOI] [PubMed] [Google Scholar]
- 21.Fu F., Zhao K., Li J., Xu J., Zhang Y., Liu C., Yang W., Gao C., Li J., Zhang H., Li Y., Cui Q., Wang H., Tao L., Wang J., Quon M.J., Gao F. Direct evidence that myocardial insulin resistance following myocardial ischemia contributes to post-ischemic heart failure. Sci. Rep. 2015;5:17927. doi: 10.1038/srep17927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen W.X., Zhang Z.G., Ding Z.Y., Liang H.F., Song J., Tan X.L., Wu J.J., Li G.Z., Zeng Z., Zhang B.X., Chen X.P. MicroRNA-630 suppresses tumor metastasis through the TGF-β- miR-630-Slug signaling pathway and correlates inversely with poor prognosis in hepatocellular carcinoma. Oncotarget. 2016;7(16):22674–22686. doi: 10.18632/oncotarget.8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou L., Li R., Liu C., Sun T., Htet Aung L.H., Chen C., Gao J., Zhao Y., Wang K. Foxo3a inhibits mitochondrial fission and protects against doxorubicin-induced cardiotoxicity by suppressing MIEF2. Free Radic. Biol. Med. 2017;104:360–370. doi: 10.1016/j.freeradbiomed.2017.01.037. [DOI] [PubMed] [Google Scholar]
- 24.Wallace K.B., Sardao V.A., Oliveira P.J. Mitochondrial determinants of doxorubicin-induced cardiomyopathy. Circ. Res. 2020;126(7):926–941. doi: 10.1161/CIRCRESAHA.119.314681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wenningmann N., Knapp M., Ande A., Vaidya T.R., Ait-Oudhia S. Insights into doxorubicin-induced cardiotoxicity: molecular mechanisms, preventive strategies, and early monitoring. Mol. Pharmacol. 2019;96(2):219–232. doi: 10.1124/mol.119.115725. [DOI] [PubMed] [Google Scholar]
- 26.Xia P., Chen J., Liu Y., Fletcher M., Jensen B.C., Cheng Z. Doxorubicin induces cardiomyocyte apoptosis and atrophy through cyclin-dependent kinase 2-mediated activation of forkhead box O1. J. Biol. Chem. 2020;295(13):4265–4276. doi: 10.1074/jbc.RA119.011571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuznetsov A.V., Javadov S., Margreiter R., Grimm M., Hagenbuchner J., Ausserlechner M.J. Structural and functional remodeling of mitochondria as an adaptive response to energy deprivation. Biochim. Biophys. Acta Bioenerg. 2021;1862(6) doi: 10.1016/j.bbabio.2021.148393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lunt S.Y., Vander Heiden M.G. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011;27:441–464. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- 29.Rodrigues T., Ferraz L.S. Therapeutic potential of targeting mitochondrial dynamics in cancer. Biochem. Pharmacol. 2020;182 doi: 10.1016/j.bcp.2020.114282. [DOI] [PubMed] [Google Scholar]
- 30.Ong S.B., Hall A.R., Hausenloy D.J. Mitochondrial dynamics in cardiovascular health and disease. Antioxidants Redox Signal. 2013;19(4):400–414. doi: 10.1089/ars.2012.4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao T., Zhang X., Zhao J., Zhou F., Wang Y., Zhao Z., Xing J., Chen B., Li J., Liu S. SIK2 promotes reprogramming of glucose metabolism through PI3K/AKT/HIF-1alpha pathway and Drp1-mediated mitochondrial fission in ovarian cancer. Cancer Lett. 2020;469:89–101. doi: 10.1016/j.canlet.2019.10.029. [DOI] [PubMed] [Google Scholar]
- 32.Chen H., Chan D.C. Mitochondrial dynamics in regulating the unique phenotypes of cancer and stem cells. Cell Metabol. 2017;26(1):39–48. doi: 10.1016/j.cmet.2017.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Studneva I., Palkeeva M., Veselova O., Molokoedov A., Ovchinnikov M., Sidorova M., Pisarenko O. Protective effects of a novel agonist of galanin receptors against doxorubicin-induced cardiotoxicity in rats. Cardiovasc. Toxicol. 2019;19(2):136–146. doi: 10.1007/s12012-018-9483-x. [DOI] [PubMed] [Google Scholar]
- 34.Zhu S.G., Kukreja R.C., Das A., Chen Q., Lesnefsky E.J., Xi L. Dietary nitrate supplementation protects against Doxorubicin-induced cardiomyopathy by improving mitochondrial function. J. Am. Coll. Cardiol. 2011;57(21):2181–2189. doi: 10.1016/j.jacc.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cova D., De Angelis L., Monti E., Piccinini F. Subcellular distribution of two spin trapping agents in rat heart: possible explanation for their different protective effects against doxorubicin-induced cardiotoxicity. Free Radic. Res. Commun. 1992;15(6):353–360. doi: 10.3109/10715769209049151. [DOI] [PubMed] [Google Scholar]
- 36.Doroshow J.H., Davies K.J. Redox cycling of anthracyclines by cardiac mitochondria. II. Formation of superoxide anion, hydrogen peroxide, and hydroxyl radical. J. Biol. Chem. 1986;261(7):3068–3074. [PubMed] [Google Scholar]
- 37.Davies K.J., Doroshow J.H. Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J. Biol. Chem. 1986;261(7):3060–3067. [PubMed] [Google Scholar]
- 38.Zhang S., Liu X., Bawa-Khalfe T., Lu L.S., Lyu Y.L., Liu L.F., Yeh E.T. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012;18(11):1639–1642. doi: 10.1038/nm.2919. [DOI] [PubMed] [Google Scholar]
- 39.Catanzaro M.P., Weiner A., Kaminaris A., Li C., Cai F., Zhao F., Kobayashi S., Kobayashi T., Huang Y., Sesaki H., Liang Q. Doxorubicin-induced cardiomyocyte death is mediated by unchecked mitochondrial fission and mitophagy. Faseb. J. 2019 doi: 10.1096/fj.201802663R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang H., Tao A., Song J., Liu Q., Wang H., Rui T. Doxorubicin-induced cardiomyocyte apoptosis: role of mitofusin 2. Int. J. Biochem. Cell Biol. 2017;88:55–59. doi: 10.1016/j.biocel.2017.05.006. [DOI] [PubMed] [Google Scholar]
- 41.Li J., Li Y., Jiao J., Wang J., Li Y., Qin D., Li P. Mitofusin 1 is negatively regulated by microRNA 140 in cardiomyocyte apoptosis. Mol. Cell Biol. 2014;34(10):1788–1799. doi: 10.1128/MCB.00774-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dorn G.W. 2nd, Mitofusins as mitochondrial anchors and tethers. J. Mol. Cell. Cardiol. 2020;142:146–153. doi: 10.1016/j.yjmcc.2020.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen Y., Dorn G.W. 2nd, PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 2013;340(6131):471–475. doi: 10.1126/science.1231031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu S., Zou M.H. Mitochondria-associated endoplasmic reticulum membranes in the heart. Arch. Biochem. Biophys. 2019;662:201–212. doi: 10.1016/j.abb.2018.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tong M., Zablocki D., Sadoshima J. The role of Drp1 in mitophagy and cell death in the heart. J. Mol. Cell. Cardiol. 2020;142:138–145. doi: 10.1016/j.yjmcc.2020.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schrepfer E., Scorrano L. Mitofusins, from mitochondria to metabolism. Mol. Cell. 2016;61(5):683–694. doi: 10.1016/j.molcel.2016.02.022. [DOI] [PubMed] [Google Scholar]
- 47.Guo W., Jiang T., Lian C., Wang H., Zheng Q., Ma H. QKI deficiency promotes FoxO1 mediated nitrosative stress and endoplasmic reticulum stress contributing to increased vulnerability to ischemic injury in diabetic heart. J. Mol. Cell. Cardiol. 2014;75:131–140. doi: 10.1016/j.yjmcc.2014.07.010. [DOI] [PubMed] [Google Scholar]
- 48.Battiprolu P.K., Hojayev B., Jiang N., Wang Z.V., Luo X., Iglewski M., Shelton J.M., Gerard R.D., Rothermel B.A., Gillette T.G., Lavandero S., Hill J.A. Metabolic stress-induced activation of FoxO1 triggers diabetic cardiomyopathy in mice. J. Clin. Invest. 2012;122(3):1109–1118. doi: 10.1172/JCI60329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shi Y., Fan S., Wang D., Huyan T., Chen J., Chen J., Su J., Li X., Wang Z., Xie S., Yun C., Li X., Tie L. FOXO1 inhibition potentiates endothelial angiogenic functions in diabetes via suppression of ROCK1/Drp1-mediated mitochondrial fission. Biochim. Biophys. Acta (BBA) - Mol. Basis Dis. 2018;1864(7):2481–2494. doi: 10.1016/j.bbadis.2018.04.005. [DOI] [PubMed] [Google Scholar]
- 50.Filadi R., Pendin D., Pizzo P. Mitofusin 2: from functions to disease. Cell Death Dis. 2018;9(3):330. doi: 10.1038/s41419-017-0023-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Allegra A., Innao V., Allegra A.G., Musolino C. Relationship between mitofusin 2 and cancer. Adv. Protein Chem. Struct. Biol. 2019;116:209–236. doi: 10.1016/bs.apcsb.2018.11.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.