

Abstract
Immunosuppressive factors in the tumor microenvironment (TME) impair T cell function and limit the antitumor immune response. T cell surface receptors and surface proteins that influence interactions and function in the TME are proven targets for cancer immunotherapy. However, how the entire surface proteome remodels in primary human T cells in response to specific suppressive factors in the TME remains to be broadly and systematically characterized. Here, using a reductionist cell culture approach with primary human T cells and stable isotopic labeling with amino acids in cell culture–based quantitative cell surface capture glycoproteomics, we examined how two immunosuppressive TME factors, regulatory T cells (Tregs) and hypoxia, globally affect the activated CD8+ surface proteome (surfaceome). Surprisingly, coculturing primary CD8+ T cells with Tregs only modestly affected the CD8+ surfaceome but did partially reverse activation-induced surfaceomic changes. In contrast, hypoxia drastically altered the CD8+ surfaceome in a manner consistent with both metabolic reprogramming and induction of an immunosuppressed state. The CD4+ T cell surfaceome similarly responded to hypoxia, revealing a common hypoxia-induced surface receptor program. Our surfaceomics findings suggest that hypoxic environments create a challenge for T cell activation. These studies provide global insight into how Tregs and hypoxia remodel the T cell surfaceome and we believe represent a valuable resource to inform future therapeutic efforts to enhance T cell function.
Keywords: T cells, tumor microenvironment, hypoxia, cell surface proteomics
Abbreviations: CAR, chimeric antigen receptor; PBMC, peripheral blood mononuclear cell; SILAC, stable isotopic labeling with amino acids in cell culture; Teff, effector T cell; TME, tumor microenvironment; Treg, regulatory T cell
Graphical abstract
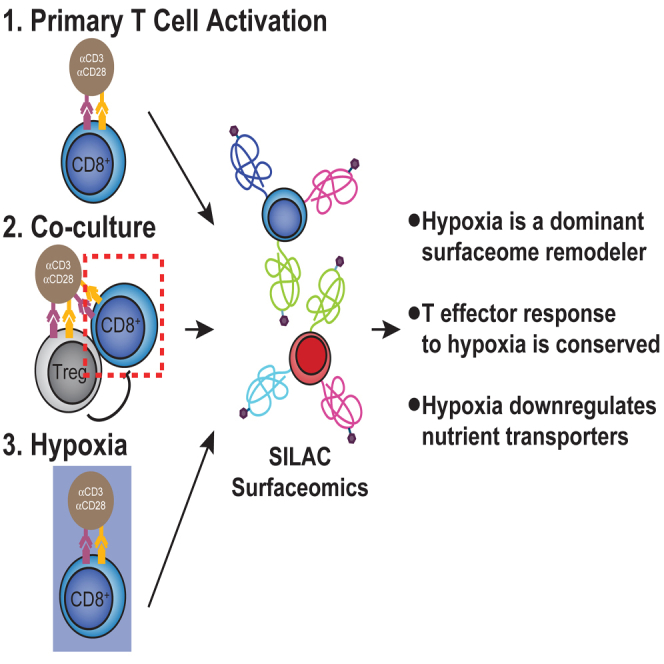
Highlights
-
•
Quantitative surface proteomics of primary human T cells
-
•
Activation, regulatory T cells, and hypoxia induce bidirectional surfaceome changes
-
•
Hypoxia dramatically remodels the primary T cell surface proteome
-
•
Both regulatory T cells and hypoxia downregulate nutrient transporter expression
In Brief
The tumor microenvironment (TME) features immunosuppressive regulatory T cells (Tregs) and areas of hypoxia. Given the importance of surface proteins in T cell antitumor function, we performed quantitative cell surface proteomics to determine how these factors affect the primary effector T cell surface proteome (surfaceome). We discovered that Treg coculture and hypoxia reduced expression of nutrient transporters, among other proteins implicated in T cell activation. Together, our findings reveal insights into how these immunosuppressive factors modulate the T cell surfaceome.
Cytotoxic CD8+ T cells promote tumor cell killing, and helper CD4+ T cells provide support for the antitumor immune response. In cancer, these cells function in a complex tumor microenvironment (TME) consisting of a mixture of tumor, somatic, and immune cells that create a unique environment in and around the tumor (1). Tumor-infiltrating effector T cells (CD8+ and CD4+, Teffs) also encounter immunosuppressive regulatory T cells (Tregs) in the TME (2, 3). Tregs express immune suppressors such as cytotoxic T-lymphocyte associated antigen 4 (CTLA-4) (4) and produce immunosuppressive adenosine (5) and cytokines such as transforming growth factor β (TGFβ), interleukin (IL)-10 (6), and IL-35 (7). Furthermore, Tregs and effector T cells compete for IL-2 for proliferation; high expression of the high-affinity IL-2 receptor on Tregs allows them to out-compete other T cells for available IL-2 (8). Tregs can also directly kill CD8+ T cells via the perforin pathway (9). Consistent with these many immunosuppressive activities, increased Treg tumor infiltration is associated with poor prognosis in a number of cancers, including non-small-cell lung, hepatocellular, renal cell, breast, cervical, ovarian, and gastric cancers, as well as melanoma (3, 10, 11).
Another hallmark of the TME is hypoxia due to poor and variable vascularization within the tumor (1, 12). Hypoxia is common in the core of tumors and induces dramatic transcriptional changes (1, 12, 13, 14, 15, 16, 17). Tumor-associated hypoxia strongly influences the function of numerous immune cells from both the myeloid (18) and lymphoid lineages (19, 20, 21), with both stimulatory and inhibitory effects reported (22). Hypoxia induces expression of ectonucleoside triphosphate diphosphohydrolase 1 (ENTPD1 or CD39) and 5′-nucleotidase (NT5E or CD73) that catalyze the production of immunosuppressive adenosine from ATP (23). Hypoxia also induces the Warburg effect, which leads to tumor acidification and subsequent decreased CD8+ T cell proliferation and cytotoxic activity (24). Although some studies examining the direct effect of hypoxic culture of CD8+ T cells showed hypoxia enhances cytotoxic activity (25), hypoxia also dramatically reduces cell proliferation (25, 26). Furthermore, hypoxia promotes recruitment of Tregs to the tumor (27) and CD8+ cells have been observed to be excluded from areas of tumor hypoxia (28). Recently, hypoxia was also linked to T cell exhaustion (29). Inhospitable, hypoxic regions in solid tumors may also limit the function of chimeric antigen receptor (CAR)-T cells (30), which could contribute to the limited success of targeting solid tumors with CAR-T cells. Collectively, although the response of CD8+ T cells to hypoxia is complex, hypoxia appears to have a net immunosuppressive effect on CD8+ T cell function (22).
The cell surface proteome, or surfaceome, mediates T cell interactions with the external environment, and the effect of external environmental factors on the T cell surfaceome has not yet been studied globally. Not only does the surfaceome help T cells sense and respond to the environmental conditions of the TME, but membrane proteins are useful surface markers and key regulators of the antitumor function of T cells. For example, proteins such as programmed death protein 1 (PD1) play crucial roles in the suppression of CD8+ cells (31). Consequently, many current immunotherapies target and modulate T cells through blockade or engagement of surface proteins (e.g., anti-PD1 or anti-CTLA-4 therapy, bispecific T cell engagers) (32). Therefore, profiling how the CD8+ T cell surfaceome changes in response to specific TME factors, such as Treg-mediated suppression or hypoxia, should expand our understanding of the basic biological response to these modulators.
We have taken a reductionist cell culture approach to understand how Tregs and hypoxia modulate the cell surface proteome of primary effector T cells. Using quantitative cell surface proteomics (33, 34) we identified bidirectional changes in the CD8+ T cell surfaceome following classic activation with agonistic antibodies to CD3 and CD28. We discovered that coculturing with Tregs, and especially hypoxic culture, significantly altered the activated CD8+ surfaceome in a manner consistent with reduced CD8+ activation. Hypoxia similarly induced dramatic changes in the CD4+CD25- conventional T cell surfaceome. Collectively, these in vitro findings provide a resource characterizing the effect of these immunosuppressive factors on the T cell surfaceome and identify surface proteins for selective targeting of suppressed effector T cells.
Experimental Procedures
Cell Isolation
Primary human T cells were isolated from leukoreduction chamber residuals following Trima Apheresis (Blood Centers of the Pacific) using established protocols (35). Briefly, peripheral blood mononuclear cells (PBMCs) were isolated using Ficoll separation in SepMate tubes (STEMCELL Technologies) in accordance with the manufacturer’s instructions. CD8+ T cells were isolated from PBMCs using either the EasySep Human CD8+ T cell Isolation Kit or the RosetteSep Human CD8+ T Cell Enrichment Cocktail (STEMCELL), following the manufacturer’s protocol. CD4+CD25- conventional T cells and CD4+CD127lowCD25+ Tregs were isolated from PBMCs with the EasySep Human CD4+CD127lowCD25+ Regulatory T Cell Isolation Kit (STEMCELL). Isolated cell populations were analyzed for purity by flow cytometry on a Beckman Coulter CytoFlex flow cytometer using a panel of antibodies (anti-CD3 [UCHT1], anti-CD4 [OKT4], anti-CD8a [RPA-T8], anti-CD25 [M-A251], anti-CD45RA [HI100], and anti-CD127 [A019D5], all from BioLegend). Pilot experiments comparing FACS sorting and magnetic bead separation showed similar results with respect to cell purity, so magnetic bead separation was used.
Cell Culture/SILAC Labeling
Following isolation, cells were adjusted to 1e6 cells/ml in RPMI 1640 Medium for stable isotopic labeling with amino acids in cell culture (SILAC) (Thermo Fisher) supplemented with 100 U/ml Penicillin/Streptomycin (Gemini Bio-Products), 10 mM HEPES (UCSF Cell Culture Facility), 100 μM non-essential amino acids (Lonza), 1 mM sodium pyruvate (VWR), 55 μM 2-mercaptoethanol (Gibco), 10 mM N-acetyl-cysteine (Sigma), and 10% dialyzed fetal bovine serum (Gemini). Media was also supplemented with either light L-[12C6,14N2] lysine/L-[12C6,14N4] arginine (Sigma) or heavy L-[13C6,15N2] lysine/L-[13C6,15N4] arginine (Cambridge Isotope Laboratories). CD8+ and CD4+ Teffs were stimulated by addition to tissue culture flasks coated with anti-CD3 (produced in-house, clone OKT3) and anti-CD28 ([D2Z4E], Cell Signaling) antibodies or with anti-CD3/anti-CD28 Dynabeads (Thermo) at a 1:1 bead:cell ratio in the presence of 50 U/ml recombinant human IL-2 (Thermo). Following 2 days of culture, the stimuli were removed and cells allowed to expand in heavy or light SILAC media with 50 U/ml IL-2 for 12 additional days. Tregs were stimulated using anti-CD3/anti-CD28 Dynabeads (Thermo) at a 1:1 bead:cell ratio and maintained in 300 U/ml IL-2. Following 2 days of culture, the stimuli were removed and cells allowed to expand in heavy or light SILAC media with 300 U/ml IL-2 for 9 days and then restimulated with more beads at 1:1 bead:cell ratio. At day 14, Tregs were counted and put into coculture with CD8 T cells at a 1:1 Treg:CD8+ T cell ratio. For flow cytometry examining the effect of expansion on surface marker expression, cells were stained with anti-CD69 [FN50], anti-CD71 (TFRC) [CY1G4], anti-SELL [DREG-56], and anti-IL7R [A019D5] antibodies, all from Biolegend. Flow cytometry data were acquired as above, analyzed using FlowJo (v10.7.1).
Baseline CD8+ Activation
CD8+ T cells were stimulated using anti-CD3/anti-CD28 Dynabeads (Thermo) at a 1:1 bead:cell ratio and maintained in 50 U/ml IL-2. Following 2 days of culture, the stimuli were removed and cells allowed to expand in heavy or light SILAC media with 50 U/ml IL-2 for 12 additional days, at which point CD8+ T cells were counted and restimulated 1:1 with anti-CD3/anti-CD28 Dynabeads for 3 days in the absence of IL-2. Isotopically labeled activated and resting cells were then combined 1:1 for downstream processing.
Coculture
On day 14, isotopically labeled CD8+ T cells were put into coculture with Tregs at a 1:1 Treg:CD8+ T cell ratio. In addition, at the time of initiation of coculture, anti-CD3/anti-CD28 Dynabeads were added at a 1:1 bead:CD8+ T cell ratio in the absence of IL-2. At the time of coculture, the CD8+ T cells grown in heavy SILAC media were cocultured with Tregs, while light-labeled CD8+ T cells were kept in monoculture and grown in light SILAC media. At the end of 3 days of coculture, CD8+ T cells were isolated from coculture using STEMCELL CD8+ enrichment kits and then combined in equal numbers with the CD8+ T cells grown in monoculture in the light SILAC media. This was done in the opposite combination of light/heavy SILAC media as well to ensure there was no bias in the experiment in assigning light or heavy SILAC media to those T cells grown in co- or monoculture.
Hypoxic T Cell Activation
To model activation in hypoxic conditions, expanded, SILAC-labeled CD8+, CD4+CD25-, or Treg cells were collected, resuspended in fresh SILAC media, and stimulated using anti-CD3/anti-CD28 Dynabeads at a 1:10 bead:cell ratio in the absence of IL-2. Cells were then either cultured at 37 °C, 5% CO2 in normoxic (20% O2) or hypoxic (1% O2) conditions for 3 days. Hypoxic culture was performed in a Coy Laboratory Products hypoxic cabinet using a nitrogen/5% CO2 balance blend. Cells were then separated from the Dynabeads and heavy and light cells mixed at a 1:1 ratio in both forward and reverse SILAC mode (CD8+, CD4+CD25- cells) before surface protein capture. For Western blotting and flow cytometry of IL-18R1 and CD70, cells were similarly isolated and activated as above. For Western blot, cells were harvested and lysed using commercial RIPA buffer (Millipore) supplemented with cOmplete Mini protease inhibitor cocktail (Sigma). Samples were normalized for protein content using BCA assay (Pierce) and run on 4 to 12% Novex Bolt Bis-Tris gels at 200V before transfer to PVDF membranes using an Invitrogen iBlot 2 system. Membranes were first stained for total protein content using Revert 700 stain (Li-Cor) in accordance with the manufacturer’s protocol. Membranes were then blocked with Li-Cor blocking buffer and stained overnight at 4 °C with 1:1000 diluted primary antibody in blocking buffer. Primary antibodies used included: 26407-1-AP (SLC5A6), 14195-1-AP (SLC7A1), and 22787-1-AP (SLC16A3), all from Proteintech. After staining with 800CW-conjugated anti-Rabbit IgG secondary antibody (Li-Cor 926–32211, 1:10,000 dilution), blots were visualized on a Li-Cor Odyssey CLx and images further processed using ImageJ (v2.1.0/1.53c). For flow cytometry, cells were stained with GHOST viability dye from Tonbo Biosciences and anti-IL18R1 [H44] and anti-CD70 [113–16] antibodies, both from Biolegend. Flow cytometry data were acquired as above, analyzed using FlowJo (v10.7.1), and statistically analyzed using a one-way ANOVA with a Tukey’s multiple comparisons test in GraphPad Prism (v8).
Cell Surface Capture
Cell surface glycoproteins were captured as previously described (36). Briefly, immediately after combining isotopically labeled cells, the cells were washed in PBS, pH 6.5, and glycoproteins oxidized with 1.6 mM NaIO4 (Sigma) in PBS, pH 6.5 for 20 min at 4 °C. Oxidized vicinal diols were subsequently biotinylated with 1 mM biocytin hydrazide (Biotium) in the presence of 10 mM aniline (Sigma) in PBS, pH 6.5 for 90 min at 4 °C. Cells were then flash frozen and stored at −80 °C before further preparation. To isolate glycoproteins for mass spectrometry, cell pellets were lysed with commercial RIPA buffer (VWR) supplemented with 1X Protease Inhibitor Cocktail (Sigma) and 1 mM EDTA (Sigma) for 30 min at 4 °C. Cells were further disrupted with probe sonication and biotinylated glycoproteins pulled down with NeutrAvidin-coated agarose beads (Thermo) for 1 h at 4 °C. Beads were transferred to Poly-Prep chromatography columns (Bio-Rad) and sequentially washed with RIPA (PBS pH 7.4 with 0.5% sodium deoxycholate [Thermo], 0.1% sodium dodecylsulfate [Fisher Scientific], 1% Nonidet P-40 substitute [VWR]), high-salt PBS (PBS pH 7.4, 2 M NaCl [Sigma]), and denaturing urea buffer (50 mM ammonium bicarbonate, 2 M Urea). After washing, beads were collected and glycoproteins reduced with 5 mM TCEP (Calbiochem) for 30 min at 37 °C and alkylated with 11 mM iodoacetamide (Sigma) for 30 min at room temperature. Beads were washed with urea buffer and trypsinized on-bead overnight at room temperature with 20 μg trypsin (Promega). The next day, the tryptic fraction was collected using Pierce Spin Columns before the beads were again transferred to PolyPrep columns and washed with RIPA, high-salt buffer, and urea buffer before a final wash with 50 mM ammonium bicarbonate. Beads were transferred to a fresh tube and glycopeptides liberated with 5000 U/ml PNGaseF for 3 h at 37 °C. This PNGaseF fraction was collected as above. Both tryptic and PNGaseF fractions were then desalted with SOLA HRP SPE columns (Thermo) following standard protocols, dried, and dissolved in 0.1% formic acid, 2% acetonitrile prior to LC-MS/MS analysis.
Mass Spectrometry
Mass spectrometry was performed as previously described (36), with some slight adjustments. All peptides were separated using an UltiMate 3000 UHPLC system (Thermo) with prepacked 0.75 mm × 150 mm Acclaim Pepmap C18 reversed phase columns (2 μm pore size, Thermo) and analyzed on a Q Exactive Plus (Thermo Fisher Scientific) mass spectrometer. For tryptic fractions, 1 μg of resuspended peptides was injected and separated using a linear gradient of 3 to 35% solvent B (solvent A: 0.1% formic acid, solvent B: 80% acetonitrile, 0.1% formic acid) over 230 min at 300 nl/min. Due to the low peptide yield of the PNGase fraction, the entire fraction was injected and subsequently separated using the same gradient over 170 min. Data-dependent acquisition was performed using a top 20 method (dynamic exclusion 35 s; selection of peptides with a charge of 2, 3, or 4). Full spectra with a resolution of 140,000 (at 200 m/z) were gathered in MS1 using an AGC target of 3e6, maximum injection time of 120 ms, and scan range of 400 to 1800 m/z. Centroided data from MS2 scans were collected at a resolution of 17,500 (at 200 m/z) with an AGC target of 5e4 and maximum injection time of 60 milliseconds. The normalized collision energy was set at 27 and an isolation window of 1.5 m/z with an isolation offset of 0.5 m/z was used.
Data Analysis/Statistics
SILAC proteomics data were analyzed as previously described (36). Briefly, each individual dataset was searched for peptides using ProteinProspector v6.2.2 against the human proteome (SwissProt database, August 3, 2017 release, 20,218 entries). Enzyme specificity was set to trypsin with up to two missed cleavages. Cysteine carbamidomethyl was set as the only fixed modification; methionine oxidation, N-terminal glutamate to pyroglutamate, and lysine/arginine SILAC labels were set as variable modifications. Asparagine deamidation was also listed as a variable modification for the PNGaseF fractions. During the search, the peptide mass tolerance was 6 ppm, fragment ion mass tolerance was 0.4 Da, and peptide identification was filtered by peptide score of 0.0005 in ProteinProspector, resulting in a false discovery rate (FDR) of <1% calculated using the number of decoy peptides in the SwissProt database. Skyline (UWashington) (37) software was used to perform quantitative analysis of SILAC ratios using an MS1 filtering function against a curated list of extracellular proteins generated via searches for “membrane” but not “mitochondrial” or “nuclear” using UniProt subcellular localization annotations, as previously described (5973 entries) (36). For datasets collected in forward and reverse SILAC mode, spectral libraries of experiments were analyzed simultaneously to allow MS1 peaks without an explicit peptide ID to be quantified using an aligned peptide retention time. The Skyline report was subsequently exported for ratiometric analysis using a previously reported custom R script (36). Briefly, low-quality identifications (isotope dot product <0.8) were removed. For the tryptic fraction, only proteins with two or more peptides were included in downstream analysis, whereas for the PNGase fraction, only peptides with N to D deamidation were included. Ratios derived from both fractions were then combined, centered on a mean of zero, and presented as median log2 enrichment values. Protein-level correlations of the PNGase and tryptic fractions can be found in the Supplemental Materials (supplemental Figs. S2, S7, S11, S14, and S16). Although combining both the tryptic and PNGase fractions reduces the specificity for cell surface proteins, coverage is dramatically enhanced, permitting expanded analysis of global changes induced by the factors tested in this work. Significance was then determined using a Mann–Whitney test of peptide ratios for all peptides for a given protein. Keratin 2, vimentin, and prothrombin showed dramatic enrichment in some light-labeled SILAC samples, suggesting these were contaminants and were therefore removed from downstream analysis. Heatmaps comparing expression levels between donors were generated using heatmapper.ca and other graphs were generated using GraphPad Prism (v8).
RNAseq data for naïve and activated CD8+ T cells were downloaded from the Database of Immune Cell eQTLs, Expression, and Epigenomics (DICE) (38). Expression data were gathered for all overlapping proteins found in the CD8+ activation surfaceomics dataset, and an average expression level was calculated from all available donors in the DICE database. The expression ratio between activated and naïve was then calculated and compared with enrichments observed in the surfaceomics data. STRING analysis was performed using the online STRING Database (v11.0) and visualized using Cytoscape (v3.7.2). Gene-set enrichment analysis was performed using R and the fgsea (v1.18.0) (39) package from Bioconductor as previously described (40). The biological process annotated gene set (c5) used was obtained from the MSigDB collection (v7.4.1). GO Cell Compartment terms were removed due to the surface enriched nature of the samples.
Experimental Design and Statistical Rationale
For all proteomics experiments cells from at least N = 2 to 3 donors were used, and where cells were not limiting, SILAC was performed with switched isotopic coding (forward and reverse mode). Tryptic and PNGase fractions resulting from glycoprotein enrichment were analyzed separately and combined at the final protein quantification stage. Additional details regarding data handling can be found above. For Western blot and flow cytometry experiments, cells from at least N = 2 to 3 donors were again used.
Results
Activation Dramatically Alters the CD8+ T Cell Surfaceome in a Bidirectional Fashion
The first step of our strategy was to study how the CD8+ cell surfaceome responds to activation, and how this response is altered by Treg suppression or hypoxia (Fig. 1A). Primary CD8+ cells were isolated from healthy donors and expanded 10 to 100-fold with anti-CD3 and anti-CD28 stimulation in the presence of IL-2. Cells were grown in medium containing light or heavy isotope-labeled lysine and arginine to quantitatively compare stimulation conditions using SILAC coupled with a glycoprotein cell surface capture technique and LC-MS/MS (33, 34, 36) (Fig. 1A, top). This led to quantitative and uniform labeling as assessed by isotope distribution on four abundant and constitutively expressed proteins (supplemental Fig. S1). We first compared the activation-induced changes in the CD8+ surfaceome before and after activation with anti-CD3/anti-CD28 for 3 days and then examined how the program was altered by the addition of primary Tregs or hypoxic culture (Fig. 1A, bottom).
Fig. 1.

Surface proteomics reveals both well-established and novel activation-induced changes in surface protein levels.A, top, schematic depicting expansion and SILAC labeling workflow. Primary human CD8+ T cells were isolated and expanded using anti-CD3/anti-CD28 stimulation in media supplemented with IL-2 and either heavy or light arginine and lysine. After expansion, cells were stimulated in varying conditions before surface protein enrichment and protein identification with LC-MS/MS. A, bottom, strategy for assessing the effect of immunosuppressive stimuli on the activated CD8+ T-cell surfaceome. First, the surfaceomic changes associated with CD8+ activation in monoculture under normoxic conditions were analyzed. These changes then served as a baseline for later experiments examining the surfaceomic consequences of activating CD8+ T cells in co-culture with primary Tregs or in hypoxic culture. B, Volcano plot of surface protein changes following stimulation of CD8+ T cells with anti-CD3/anti-CD28 beads. Data represent compiled results from N = 4 donors. Proteins with a −/+1.5-fold change and p < 0.05 were included in downstream analysis. Proteins significantly down- (blue) or upregulated (red) are indicated. C, Log2(Enrichment Ratio) of indicated proteins. Each dot represents data from an individual donor. Line represents the mean and error bars are standard deviation. SILAC, stable isotopic labeling with amino acids in cell culture.
Surfaceomic analysis of unstimulated and anti-CD3/anti-CD28-stimulated CD8+ T cells from four donors identified a total of 669 surface proteins (Fig. 1B, supplemental Table S1 and supplemental Fig. S2). Although there was donor-to-donor variation (supplemental Fig. S3), assessment of a compiled dataset including fold-change data from the four donors revealed that about 16% of these proteins (106/669) consistently showed significant (p < 0.05) 1.5-fold up- or downregulation. These significantly altered proteins showed strong correlation between most donors (supplemental Fig. S3). We observed changes in markers of T cell activation, including upregulation of two classic T cell activation markers (CD69 (41) and the transferrin receptor [TFRC] (42)), and downregulation of the IL-7 receptor (internalized upon activation) and vasoactive intestinal peptide receptor 1 (VIPR1) (43) (Fig. 1C). Although we found that the cell expansion necessary for SILAC labeling affected the basal expression of CD69, TFRC, and the IL-7 receptor (supplemental Fig. S4), our surfaceomics data demonstrate that these expanded cells were still sensitive to anticipated activation-induced changes.
Large-scale network analysis of the significantly altered proteins demonstrates the roughly symmetrical, bidirectional response of the T cell surfaceome to activation, with 66 proteins upregulated and 40 proteins downregulated (Figs. 1B and S5). In addition to CD69 and TFRC, numerous well-established T cell activation markers were also upregulated, including CD63 (41), CD83 (44), CD97 (45), and CD109 (46). Importantly, multiple solute carrier (SLC) transporters were also upregulated on activated CD8+ T cells, including the amino acid transporters SLC1A5 and SLC7A5 that have been previously implicated in supporting T cell activation (47). Of note, comparison of our proteomics data with RNAseq data from activated versus resting CD8+ cells in the DICE database revealed a loosely positive correlation (r = 0.25, p < 0.0001, supplemental Fig. S6A) (38). It is well known that protein and RNA levels show only mild correlations because of differences in stability and regulation. Assessment of only proteins that were significantly changed in our proteomics data revealed a stronger correlation (r = 0.53, p < 0.0001, supplemental Fig. S6B). Observed discrepancies could be due to differential activation conditions, but nonetheless underscore the importance of protein-level profiling to capture surface protein remodeling in immune cells.
More globally, pathway analysis of up- and downregulated cell surface proteins revealed the most significant enrichment for proteins implicated in immune function, with a slight trend toward upregulation of these proteins (supplemental Fig. S5A, proteins annotated for GO.0002376: immune system process are indicated with green borders). Collectively, these surfaceomic data identify classic (e.g., CD69) and some newly recognized (e.g., integrin αX [ITGAX], SLC39A14, bone marrow stromal antigen 2 [BST2]) markers for immune activation of primary CD8+ T cells.
Coculture With Tregs Modulates the Activated CD8+ Surfaceome
We next analyzed the effect that primary Tregs have upon the surfaceome of activated primary CD8+ T cells in a 1:1 ratio coculture after 3 days (Fig. 2A, supplemental Table S2 and supplemental Fig. S7). Relative to activation alone, Treg coculture had a mild global impact on the surfaceome of activated CD8+ cells, with significant (p < 0.05) up- or downregulation (−/+ 1.5-fold change) of only 34 out of 675 proteins detected (Fig. 2A). Changes in these proteins were again largely consistent between donors (supplemental Fig. S8). Of note, upregulation of TFRC and the proinflammatory cytokine receptor IL18R1 on the cell surface was blunted by the addition of Tregs (Fig. 2B). In the presence of Tregs, IL7R showed variable upregulation between donors, opposite of the trend seen with cell activation in monoculture (Figs. 1C and 2B). Similarly, L-selectin (SELL), a protein that is typically downregulated with T cell activation (48), was slightly upregulated on CD8+ T cells, again consistent with a suppressive effect of Tregs (Fig. 2B). Due to the smaller number of significantly changed proteins in this dataset, network analysis was not as striking as in the activation dataset (supplemental Fig. S9). However, in contrast to the CD8+ T cell activation dataset, which demonstrated an upward trend in proteins implicated in immune processes, there is a downward trend in immune-annotated proteins when CD8+ cells are activated in the presence of Tregs, consistent with an immunosuppressive effect. Furthermore, many proteins that were upregulated on activated CD8+ T-cells in monoculture (Figs. 1B and S5) were downregulated upon activation in the presence of Tregs (11 of 23 downregulated proteins, Figs. 2A and S9). Among these proteins are several SLCs, including SLC1A5 and SLC7A1. Collectively, these data suggest that the presence of Tregs reverses at least part, but not all, of the activation-induced surfaceomic response in CD8+ T cells.
Fig. 2.

Tregs and hypoxia induce surfaceomic changes representative of immunosuppression.A, CD8+ T cells were stimulated with anti-CD3/anti-CD28 beads either in the absence or presence of Tregs. Following culture, CD8+ cells were isolated for cell surface proteomics. Volcano plot shows compiled results from N = 4 donors. Proteins with a −/+1.5-fold change and p < 0.05 were included in downstream analysis. Proteins significantly down- (blue) or upregulated (red) are indicated. B, Log2(Enrichment Ratio) of indicated proteins. Each dot represents data from an individual donor. Line represents mean and error bars are standard deviation. C, CD8+ T cells were stimulated with anti-CD3/anti-CD28 beads in either normoxic (20% O2) or hypoxic (1% O2) conditions for 3 days. Volcano plot shows compiled results from N = 3 donors. Proteins with a −/+1.5-fold change and p < 0.05 were included in downstream analysis. Proteins significantly down- (blue) or upregulated (red) are indicated. D, Log2(Enrichment Ratio) of indicated proteins. Each dot represents data from an individual donor. Line represents mean and error bars are standard deviation. E, Top 20 enriched gene sets identified by GSEA of all proteins identified in the three datasets. Negative (downregulation) and positive (upregulation) normalized effect sizes are shown in blue and red, respectively. SILAC peptide ratio was used to prerank proteins, then GSEA was performed with the MySigDB C5 GO gene set collection. F, several proteins responsible for L-amino acid transport (GO:0015807) are upregulated in activated CD8+ T cells, but downregulated by both Tregs and hypoxia. Treg, regulatory T cell.
Hypoxia Triggers Large-Scale Surfaceomic Changes in Activated CD8+ T Cells Consistent With Immunosuppression and Anaerobic Reprogramming
We next wanted to test how hypoxia affects the surfaceome of activated CD8+ T cells. Over 3 days in culture, activated CD8+ T cells trended toward faster proliferation (∼1.3-fold) in normoxia (20% O2) compared with hypoxia (1% O2), and there were no substantial differences in cell viabilities (supplemental Fig. S10). Interestingly, surface proteomics of CD8+ T cells activated in normoxic or hypoxic conditions revealed substantial remodeling of the surface proteome. Of a total of 1064 proteins identified, 196 were significantly (p < 0.05) up- or downregulated (−/+ 1.5-fold change, Fig. 2C, supplemental Table S3 and supplemental Fig. S11) in hypoxia relative to normoxia. The fold changes observed for these significantly altered proteins showed higher correlation among donors (supplemental Fig. S12) than seen in the activation (supplemental Fig. S3) and Treg coculture (supplemental Fig. S8) datasets. The upregulation of the hypoxia-induced glucose transporter SLC2A3 (GLUT3) (49) we observed is consistent with a shift toward glycolysis. We also observed downregulation of activin receptor type-1 (ACVR1), which is sequestered in endosomes under hypoxic conditions (50) (Fig. 2D).
Network visualization of proteins significantly altered by hypoxia revealed multiple clusters of up- and downregulated proteins (supplemental Fig. S13A). Interestingly, gene ontology analysis did not identify significant enrichment for proteins involved in response to hypoxia (GO.0001666) or cellular response to hypoxia (GO.0071456). However, four of these significantly altered proteins are found in the “Hallmark Hypoxia” gene set (SDC4, SLC6A6, β-1,3-Galactosyltransferase 6 [B3GALT6], and SLC2A3) (51). SLC16A3, a transporter induced in hypoxia, is also upregulated in our culture conditions (Fig. 2C) (52). Among the 132 hypoxia-upregulated proteins we observed marked upregulation of proteins involved in protein glycosylation and glycoprotein metabolic processes (supplemental Fig. S13B). Tumor hypoxia is well established to cause glycan remodeling of tumor cell surface proteins (53), and our data suggest the same may be true for hypoxic T cells. In contrast, proteins implicated in immune function were strongly represented in the pool of 64 hypoxia-suppressed proteins. In addition to the late activation marker IL2RA (CD25), costimulatory receptors, such as CD80 and CD86, as well as receptors for interleukins-3, -12, and -18 were significantly downregulated in hypoxic conditions. Several SLCs implicated in T cell function, including SLC1A5 (47), and the proproliferative cytokines tumor necrosis factor ligand superfamily member 4 (TNFSF4, OX40 ligand) and TNFSF8 (CD30 ligand) were also downregulated. Of note, hypoxia led to downregulation of some immunosuppressive proteins, including CD70 (54) and T-cell immunoreceptor with Ig and ITIM domains (TIGIT) (55), but consistent upregulation of the checkpoint molecule V-type immunoglobulin domain-containing suppressor of T-cell activation (VISTA, C10orf54) (56) (Fig. 2D). Collectively, these data reveal that hypoxia induces dramatic remodeling of the activated CD8+ T cell surfaceome and markedly regulates numerous proteins important for T cell activation.
Analysis of All Datasets Reveals a Conserved Response to Immunosuppressive Stimuli
We next cross-referenced the observed surfaceomic changes associated with Treg coculture and hypoxia. Interestingly, gene set enrichment analysis (GSEA) comparing the expression level of all proteins identified in our datasets revealed striking downregulation of a number of biological processes in both the Treg coculture and hypoxia conditions (Fig. 2E), including L-amino acid transport (Fig. 2F). Consistent with this observation, analysis of proteins significantly upregulated with CD8+ activation but blunted due to hypoxia or Tregs identified three solute carriers (Fig. 3A, left). Of these, SLC1A5 and SLC7A1 have been previously implicated in supporting T cell function following activation. Western blotting for SLC5A6 and SLC7A1 confirmed these transporters are downregulated in hypoxia, in contrast to a known hypoxia-induced transporter, SLC16A3 (52), which is upregulated (Fig. 3B). Furthermore, the proinflammatory cytokine receptor IL18R1 and potentially inhibitory CD70 (54) that were upregulated with CD8+ activation exhibited blunted surface induction with hypoxia and Tregs. Flow cytometry for these surface markers using cells from seven donors revealed significant upregulation of IL18R1 with activation and downregulation of IL18R1 in hypoxia, consistent with our proteomics results (Fig. 3C). CD70 showed similar trends; however, these changes were not statistically significant (Fig. 3C). Strikingly, when examining proteins downregulated upon CD8+ activation in standard conditions but upregulated with hypoxia and Tregs, there are no commonly regulated proteins (Fig. 3A, right). This suggests that protein upregulation seen during activation in hypoxic conditions may be primarily a general response to hypoxia. Together, the downregulation of a small but common set of surface proteins by both Treg coculture and hypoxia may represent a conserved response of the T cell surface proteome to these two immunosuppressive stimuli.
Fig 3.

Analysis of all datasets reveals commonalities in the effects of Tregs and hypoxia on the activated CD8+T cell surfaceome.A, left, Venn diagram showing intersection of proteins that were upregulated upon CD8+ activation, but downregulated by Treg co-culture or hypoxia. The five proteins at the intersection of all three datasets are indicated. A, right, Venn diagram showing intersection of proteins downregulated upon CD8+ activation, but upregulated by Treg co-culture or hypoxia. B, Western blot for SLC5A6, SLC7A1, and SLC16A3. CD8+ T cells from three different donors were activated with anti-CD3/anti-CD28 beads in either normoxic (20% O2, “N”) or hypoxic (1% O2, “H”) conditions for 3 days before samples were collected for Western blot. A total protein stain is shown as a loading control. C, flow cytometry of cells from N = 7 donors were either left resting or stimulated in normoxic (20% O2) or hypoxic (1% O2) conditions for 3 days. Cells were then collected and stained for either IL18R1 or CD70. Mean fluorescence intensity (MFI) values are shown for each donor. Bars represent means and error bars show standard error of the mean. Statistical significance was determined using a one-way ANOVA with a Tukey’s multiple comparisons test (∗ indicates p < 0.05). Treg, regulatory T cell.
CD8+ and CD4+ T Cells Demonstrate Similar Hypoxia-Induced Surface Remodeling
Given the dramatic effect of hypoxic culture on the surface proteome of activated CD8+ T cells, we next determined how hypoxia modulates surface protein expression on another cell subset important for the antitumor immune response: CD4+CD25- conventional T cells. Analysis of CD4+CD25- T cells from the same donors as in Figure 2C again revealed dramatic surfaceomic remodeling when these cells were activated in hypoxic conditions (Fig. 4A, supplemental Table S4 and supplemental Fig. S14). Of the 1144 proteins identified, 992 were also identified in our CD8+ hypoxia dataset. The fold-change ratios of these commonly identified proteins showed strong correlation (R = 0.82, p < 0.0001) between CD8+ and CD4+CD25- cells (Fig. 4B). Interestingly, more proteins were significantly up- (306) or downregulated (204) in the CD4+CD25- dataset, and the magnitude of these changes was larger than those observed for the CD8+ cells. However, there was a large overlap in the sets of significantly altered proteins (Fig. 4, C and D). Consequently, functional enrichment for these overlapping proteins was similar to that observed when analyzing the CD8+ response alone, with significant downregulation of proteins involved in immune-related processes (e.g., CD70 and IL18R1) and upregulation of proteins involved in solute transport (e.g., SLC2A3, SLC16A3) and protein glycosylation. Similar experiments using Tregs also showed that hypoxia enhanced expression of SLC2A3 and SLC16A3 and reduced expression of IL18R1 (supplemental Table S5 and supplemental Figs. S15 and S16). Interestingly, hypoxia significantly changed 18% and 45% of detected surface proteins for CD8+ and CD4+CD25- cells, respectively, but only 9% of detected proteins were changed on Tregs (supplemental Fig. S15A). Furthermore, more than 75% of surface proteins significantly altered on the hypoxic CD8+ T cell surface were also altered on CD4+CD25- cells, but less than 50% of these proteins were altered on the Treg surface (supplemental Fig. S15B). Together, these data show there is substantial similarity in the surfaceomic response of CD8+ and CD4+ T cells to hypoxia, whereas the Treg surfaceome appears more resistant to hypoxia-induced surfaceomic changes.
Fig. 4.

CD8+and CD4+Teff surface proteomes respond similarly to hypoxia.A, CD4+CD25- T cells were stimulated with anti-CD3/anti-CD28 beads in either normoxic (20% O2) or hypoxic (1% O2) conditions for 3 days. Volcano plot shows compiled results from N = 3 donors. Proteins with a −/+1.5-fold change and p < 0.05 were included in downstream analysis. Proteins significantly down- (blue) or upregulated (red) are indicated. B, Spearman correlation comparing the log2(Enrichment Ratio) for both CD8+ and CD4+ T cells activated under hypoxic conditions. Venn diagrams showing proteins commonly down- (C) or upregulated (D) in hypoxia on both CD8+ and CD4+ cells. Below each Venn diagram are results from a GO biological process pathway analysis for commonly regulated proteins using the STRING database. The number of proteins identified and analysis FDR for each process are indicated.
Discussion
A major obstacle effector T cells encounter when mounting an antitumor immune response is the immunosuppressive TME. The TME is a complex environment, including a number of different cell types, stroma, and metabolic factors that cannot be fully replicated in simple cell culture. Nonetheless, several factors are known to affect effector T cell function, such as the presence of Tregs and intratumoral hypoxia. Our coculture and hypoxic culture experiments are intended to isolate the role of these important factors in remodeling the T cell surfaceome, and additional studies are needed to directly link our in vitro models to an actual TME. However, our studies illuminate the bidirectional surfaceomic changes on CD8+ T cells associated with cell activation and with immunosuppression following coculture with Tregs or hypoxic culture. The presence of Tregs partly reversed activation-induced changes in CD8+ T cells, consistent with suppressed cell activation. Perhaps surprisingly, the effect of hypoxia was much larger. Hypoxia triggered large-scale surfaceomic remodeling consistent with a general cellular response to the new metabolic demands of a low oxygen environment. Cross-referencing of the effects of both Treg coculture and hypoxia on the surface proteome of activated CD8+ T cells exposed a small, but intriguing list of common changes in the expression of proteins primarily involved in nutrient transport.
Our approach identified numerous well-established activation markers (e.g., CD69, TFRC, cytotoxic and regulatory T cell molecule [CRTAM]) that support T cell function. TNF receptor superfamily member 18 (TNFRSF or GITR), a known molecule vital for CD8+ antitumor function and agonism of which synergizes with anti-PD1 checkpoint therapy (57), was upregulated with activation, as was the exhaustion marker lymphocyte-activation gene 3 (LAG3). Interestingly, CD39, which facilitates conversion of extracellular ATP to ADP, was upregulated with activation, whereas NT5E, which converts ADP to immunosuppressive adenosine, was downregulated following activation. These opposing changes are consistent with the recent observation that following activation human T cells strongly upregulate CD39, whereas NT5E remains near baseline levels (58). One of the more striking observations in our activation dataset was the broad and significant upregulation of a number of solute transporters involved in transporting amino acids, vitamins, and other nutrients. Several of the upregulated transporters (SLC1A5, SLC29A1, SLC2A1, SLC7A1) have previously been implicated in supporting T cell function and are crucial to help T cells respond to the metabolic demands following activation (59, 60). A number of cell adhesion molecules also demonstrated divergent trends following T cell activation. Many were significantly upregulated, including CD84 (61), activated leukocyte cell adhesion molecule (ALCAM), which stabilizes the immunological synapse (62), and integrin αX, which is associated with improved migratory potential (63). However, neuronal cell adhesion molecule (NRCAM), intercellular adhesion molecule 1 (ICAM1), and integrin α4 (ITGA4), the latter of which supports T cell migration (64), were downregulated. The polarity of this response following activation may be representative of the complex interactions that an activated T cell must make with its environment to not only infiltrate an immunologically active zone, but also interact with target cells. Lastly, coculture with Tregs downregulated several classic markers upregulated in activation, TFRC, SLC1A5, and SLC7A1 (47). Of note, the exhaustion marker hepatitis A virus cellular receptor 2 (HAVCR2, or TIM-3) was also downregulated on CD8+ T cells activated in the presence of Tregs. This may be a consequence of diminished CD8+ activation in the presence of Tregs.
Multiple studies have specifically examined the direct effect of hypoxia on CD8+ T cells, either by modulating the activity of the canonical hypoxia-associated transcription factor hypoxia-inducible factor-1α (HIF-1α) or using hypoxic cell culture. Doedens et al. showed that knockdown of the negative HIF regulator von Hippel–Lindau disease tumor suppressor (VHL) enhanced the cytotoxic signature of CD8+ cells and led to sustained effector function (65). Similarly, Gropper et al. showed that CD8+ T cells cultured in 1% O2 exhibited enhanced cytolytic activity (25). However, over 5 days, these hypoxic CD8+ T cells proliferated half as quickly as cells cultured under normoxic conditions. This is consistent with other literature reports of reduced CD8+ or CD4+ Teff proliferation in hypoxic conditions (26, 66). Indeed, we too observed a trend toward reduced proliferation of CD8+ T cells after 3 days of hypoxic culture (supplemental Fig. S10), and this may have become more apparent after additional days in culture (25, 26). Taken with the results of other studies examining CD8+ function in hypoxia, low oxygen tension appears to exert a net immunosuppressive effect on the antitumor response (22). Our data add to these focused functional studies and revealed that hypoxia led to the most substantial surfaceomic changes of all conditions tested with respect to the number of proteins demonstrating significant change. Additionally, the response of CD4+CD25- and CD8+ T cells to hypoxia was more similar than that of Tregs, suggesting there may be divergent responses of Teffs and Tregs to hypoxia.
Analysis of the altered surface proteins in hypoxic culture (e.g., GLUT3 upregulation) reveals changes consistent with a metabolic change toward glycolysis. One protein that showed robust upregulation in both CD8+ and CD4+CD25- cells was SLC16A3, a hypoxia-induced lactate transporter that helps export lactate produced from glycolysis, which has previously been implicated in supporting tumor growth (52). Interestingly, the related SLC16A1 was recently shown to help intratumoral Tregs metabolically cope with high lactate levels in areas of tumor hypoxia (67). The role of SLC16A3 upregulation on hypoxic CD8+ and CD4+CD25- cells remains unclear and further functional follow-up is needed, but previous studies indicate inhibition of this transporter can enhance effector function (68). Interestingly, surfaceomic changes in response to both Treg coculture and hypoxia showed downregulation of transporters vital for fueling macromolecule production needed for both proliferation and effector function (Fig. 5). Whereas the glutamate transporter SLC1A5 and amino acid transporter SLC7A1 have been previously implicated in supporting T cell activation (47), SLC5A6 is a sodium-dependent multivitamin transporter that has not been previously implicated in T cell function. Together, these three transporters may represent a common set of proteins whose expression is modulated by immunosuppressive factors. Further mechanistic studies are needed to determine if downregulation of these transporters directly contributes to effector T cell suppression, but this premise is consistent with a recent study finding that hypoxia-induced metabolic stress promotes T cell exhaustion (29). It remains to be seen if these solute transporters are also downregulated on infiltrating T cells in vivo, but our findings help illuminate how Tregs and hypoxia may lead to the metabolic starvation of effector T cells.
Fig. 5.

Tregs and hypoxia limit nutrient transporter expression on CD8+T cells. Tregs and hypoxia suppress expression of nutrient transporters on CD8+ T cells, potentially limiting T cell nutrient uptake during activation, and subsequently, T cell proliferation. Treg, regulatory T cell.
Finally, one global observation from our hypoxic CD8+ data is that although a large number of proteins show significant change, the magnitude of these changes is moderate, with only 28/196 (14%) of significantly altered proteins showing a change of greater than twofold. This may suggest that the T cell response to hypoxia results in a distributed biology, where moderate changes in protein abundance collectively lead to a suppressed state. This complicates functional follow-up of proteins identified in this study, and additional studies are needed to relate our observations with TME-induced surfaceomic changes in vivo.
In conclusion, our cell surface proteomics data revealed upregulation of well-established activation markers following CD8+ stimulation, but also identified many additional upregulated proteins that may augment T cell effector function and help T cells cope with increased metabolic demands. Both immunosuppressive factors tested caused significant changes to the CD8+ surfaceome consistent with diminished T cell function. Interestingly, however, these factors converged on mediating downregulation of transporters crucial for linking the T cell activation response and the metabolic shift that follows stimulation (47). Furthermore, the response of both CD8+ and CD4+ effector T cells to hypoxia was remarkably conserved. Collectively, our findings provide important insight into the plasticity of the T cell surfaceome and lay the foundation for future efforts to not only further characterize, but also potentially therapeutically engage, T cell surface proteins within the TME.
Data Availability
The raw proteomics data, peaklists, ProteinProspector results, and Skyline quantification results have been deposited to the ProteomeXchange Consortium via the PRIDE (69) partner repository with the dataset identifier PXD024789. The R script used can be accessed on Github at https://github.com/byrn1855/T_Cell_SILAC.git. This article contains supplemental data. Full outputs from SILAC analysis can be found in supplemental Tables S1–S5. Complete peptide lists for each raw file analyzed can be found in supplemental Tables S6–S10. Flow cytometry data and all other data presented are available upon reasonable request.
Supplemental data
This article contains supplemental data.
Conflict of interest
A. A. is a cofounder of Tango Therapeutics, Azkarra Therapeutics, Ovibio Corporation; a consultant for SPARC, Bluestar, ProLynx, Earli, Cura, GenVivo and GSK; a member of the Scientific Advisory Board of Genentech, GLAdiator, Circle and Cambridge Science Corporation; receives grant/research support from SPARC and AstraZeneca; holds patents on the use of PARP inhibitors held jointly with AstraZeneca, which he has benefitted financially (and may do so in the future). A. M. is cofounder, member of the Boards of Directors and member of Scientific Advisory Boards of Spotlight Therapeutics and Arsenal Biosciences. A. M. has served as an advisor to Juno Therapeutics, was a member of the Scientific Advisory Board at PACT Pharma and was an advisor to Trizell. A. M. has received honoraria from Merck and Vertex, a consulting fee from AlphaSights, and is an investor in and informal advisor to Offline Ventures. A. M. owns stock in Arsenal Biosciences, Spotlight Therapeutics and PACT Pharma. The Marson lab has received research support from Juno Therapeutics, Epinomics, Sanofi, GlaxoSmithKline, Gilead and Anthem. J. A. W. is cofounder of Soteria Therapeutics, is on the Scientific Advisory Board of Jnana Therapeutics, Inception Therapeutics, IgGenix Inc, Red Tree Capital, Spotlight Therapeutics, Inzen Therapeutics, and receives research support from Bristol-Myers-Squibb, TRex Bio and Merck, Inc.
Acknowledgments
We thank Kevin Leung for valuable insight and discussions and the other members of the Wells Lab for their support.
Funding and additional information
J. A. W. is grateful for funding from the Harry and Dianna Hind Endowed Professorship in Pharmaceutical Sciences, NIH R35GM122451; J. A. W., A. A. and A. M. were supported on a common grant from the Parker Institute for Cancer Immunotherapy. Postdoctoral Fellowship support included a National Institutes of Health National Cancer Institute F32 (5F32CA239417 to J. R. B.). A. M. holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund, is an investigator at the Chan Zuckerberg Biohub and is a recipient of The Cancer Research Institute (CRI) Lloyd J. Old STAR grant. The Marson lab has received additional funds from the Innovative Genomics Institute (IGI), and the Simons Foundation.
Author contributions
J. B., A. W., E. S., J. C., A. A., A. M., and J. W. conceptualization; J. B. data curation; J. B. and A. W. formal analysis; A. M. and J. W. funding acquisition; J. B., A. W., E. S., and J. C. investigation; J. B., A. W., E. S., J. C., and L. K. methodology; A. M. and J. W. resources; A. A., A. W., and J. W. supervision; J. B. validation; J. B. visualization; J. B. and J. W. writing—original draft; J. B., A. W., E. S., J. C., L. K., A. A., A. M., and J. W. writing—reviewing and editing.
Footnotes
Present address for Amy M. Weeks: Department of Biochemistry, University of Wisconsin-Madison, Madison, WI 53706.
Supplemental Data
References
- 1.Quail D.F., Joyce J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013;19:1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zheng Y., Josefowicz S.Z., Kas A., Chu T.T., Gavin M.A., Rudensky A.Y. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature. 2007;445:936–940. doi: 10.1038/nature05563. [DOI] [PubMed] [Google Scholar]
- 3.Togashi Y., Shitara K., Nishikawa H. Regulatory T cells in cancer immunosuppression — implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019;16:356–371. doi: 10.1038/s41571-019-0175-7. [DOI] [PubMed] [Google Scholar]
- 4.Wing K., Onishi Y., Prieto-Martin P., Yamaguchi T., Miyara M., Fehervari Z., Nomura T., Sakaguchi S. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322:271–275. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 5.Deaglio S., Dwyer K.M., Gao W., Friedman D., Usheva A., Erat A., Chen J.F., Enjyoji K., Linden J., Oukka M., Kuchroo V.K., Strom T.B., Robson S.C. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 2007;204:1257–1265. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jarnicki A.G., Lysaght J., Todryk S., Mills K.H.G. Suppression of antitumor immunity by IL-10 and TGF-beta-producing T cells infiltrating the growing tumor: Influence of tumor environment on the induction of CD4+ and CD8+ regulatory T cells. J. Immunol. 2006;177:896–904. doi: 10.4049/jimmunol.177.2.896. [DOI] [PubMed] [Google Scholar]
- 7.Collison L.W., Workman C.J., Kuo T.T., Boyd K., Wang Y., Vignali K.M., Cross R., Sehy D., Blumberg R.S., Vignali D.A. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007;450:566–569. doi: 10.1038/nature06306. [DOI] [PubMed] [Google Scholar]
- 8.Thornton A.M., Shevach E.M. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J. Exp. Med. 1998;188:287–296. doi: 10.1084/jem.188.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grossman W.J., Verbsky J.W., Barchet W., Colonna M., Atkinson J.P., Ley T.J. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity. 2004;21:589–601. doi: 10.1016/j.immuni.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 10.Sato E., Olson S.H., Ahn J., Bundy B., Nishikawa H., Qian F., Jungbluth A.A., Frosina D., Gnjatic S., Ambrosone C., Kepner J., Odunsi T., Ritter G., Lele S., Chen Y.T., et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. U. S. A. 2005;102:18538–18543. doi: 10.1073/pnas.0509182102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Togashi Y., Nishikawa H. In: Emerging Concepts Targeting Immune Checkpoints in Cancer and Autoimmunity. Yoshimura A., editor. Springer International Publishing; New York City, NY: 2017. Regulatory T cells: Molecular and cellular basis for immunoregulation; pp. 3–27. [Google Scholar]
- 12.Vaupel P., Mayer A. In: Oxygen Transport to Tissue XXXVI. Swartz H.M., Harrison D.K., Bruley D.F., editors. Springer; New York City, NY: 2014. Hypoxia in tumors: Pathogenesis-related classification, characterization of hypoxia subtypes, and associated biological and clinical implications; pp. 19–24. [DOI] [PubMed] [Google Scholar]
- 13.Muz B., de la Puente P., Azab F., Azab A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (Auckl) 2015;3:83–92. doi: 10.2147/HP.S93413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Madsen C.D., Pedersen J.T., Venning F.A., Singh L.B., Moeendarbary E., Charras G., Cox T.R., Sahai E., Erler J.T. Hypoxia and loss of PHD2 inactivate stromal fibroblasts to decrease tumour stiffness and metastasis. EMBO Rep. 2015;16:1394–1408. doi: 10.15252/embr.201540107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Branco-Price C., Evans C.E., Johnson R.S. Endothelial hypoxic metabolism in carcinogenesis and dissemination: HIF-A isoforms are a NO metastatic phenomenon. Oncotarget. 2013;4:2567–2576. doi: 10.18632/oncotarget.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eales K.L., Hollinshead K.E.R., Tennant D.A. Hypoxia and metabolic adaptation of cancer cells. Oncogenesis. 2016;5 doi: 10.1038/oncsis.2015.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu K.H., Eichorst J.P., McGinnis C.S., Patterson D.M., Chow E.D., Kersten K., Jameson S.C., Gartner Z.J., Rao A.A., Krummel M.F. ZipSeq: Barcoding for real-time mapping of single cell transcriptomes. Nat. Methods. 2020;17:833–843. doi: 10.1038/s41592-020-0880-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lewis C., Murdoch C. Macrophage responses to hypoxia: Implications for tumor progression and anti-cancer Therapies. Am. J. Pathol. 2005;167:627–635. doi: 10.1016/S0002-9440(10)62038-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y., Ertl H.C.J. Starved and asphyxiated: How can CD8(+) T cells within a tumor microenvironment prevent tumor progression. Front. Immunol. 2016;7:32. doi: 10.3389/fimmu.2016.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dang E.V., Barbi J., Yang H.Y., Jinasena D., Yu H., Zheng Y., Bordman Z., Fu J., Kim Y., Yen H.R., Luo W., Zeller K., Shimoda L., Topalian S.L., Semenza G.L., et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. 2011;146:772–784. doi: 10.1016/j.cell.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Westendorf A.M., Skibbe K., Adamczyk A., Buer J., Geffers R., Hansen W., Pastille E., Jendrossek V. Hypoxia enhances immunosuppression by inhibiting CD4+ effector T cell function and promoting Treg activity. Cell. Physiol. Biochem. 2017;41:1271–1284. doi: 10.1159/000464429. [DOI] [PubMed] [Google Scholar]
- 22.Vuillefroy de Silly R., Dietrich P.-Y., Walker P.R. Hypoxia and antitumor CD8+ T cells: An incompatible alliance? Oncoimmunology. 2016;5 doi: 10.1080/2162402X.2016.1232236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hatfield S.M., Kjaergaard J., Lukashev D., Belikoff B., Schreiber T.H., Sethumadhavan S., Abbott R., Philbrook P., Thayer M., Shujia D., Rodig S., Kutok J.L., Ren J., Ohta A., Podack E.R., et al. Systemic oxygenation weakens the hypoxia and hypoxia inducible factor 1α-dependent and extracellular adenosine-mediated tumor protection. J. Mol. Med. 2014;92:1283–1292. doi: 10.1007/s00109-014-1189-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fischer K., Hoffmann P., Voelkl S., Meidenbauer N., Ammer J., Edinger M., Gottfried E., Schwarz S., Rothe G., Hoves S., Renner K., Timischl B., Mackensen A., Kunz-Schughart L., Krause S.W., et al. Inhibitory effect of tumor cell–derived lactic acid on human T cells. Blood. 2007;109:3812–3819. doi: 10.1182/blood-2006-07-035972. [DOI] [PubMed] [Google Scholar]
- 25.Gropper Y., Feferman T., Shalit T., Salame T.M., Porat Z., Shakhar G. Culturing CTLs under hypoxic conditions enhances their cytolysis and improves their anti-tumor function. Cell Rep. 2017;20:2547–2555. doi: 10.1016/j.celrep.2017.08.071. [DOI] [PubMed] [Google Scholar]
- 26.Vuillefroy de Silly R., Ducimetière L., Yacoub Maroun C., Dietrich P.Y., Derouazi M., Walker P.R. Phenotypic switch of CD8(+) T cells reactivated under hypoxia toward IL-10 secreting, poorly proliferative effector cells. Eur. J. Immunol. 2015;45:2263–2275. doi: 10.1002/eji.201445284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Facciabene A., Peng X., Hagemann I.S., Balint K., Barchetti A., Wang L.P., Gimotty P.A., Gilks C.B., Lal P., Zhang L., Coukos G. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T reg cells. Nature. 2011;475:226–230. doi: 10.1038/nature10169. [DOI] [PubMed] [Google Scholar]
- 28.Hatfield S.M., Kjaergaard J., Lukashev D., Schreiber T.H., Belikoff B., Abbott R., Sethumadhavan S., Philbrook P., Ko K., Cannici R., Thayer M., Rodig S., Kutok J.L., Jackson E.K., Karger B., et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci. Transl. Med. 2015;7:277ra30. doi: 10.1126/scitranslmed.aaa1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scharping N.E., Rivadeneira D.B., Menk A.V., Vignali P.D.A., Ford B.R., Rittenhouse N.L., Peralta R., Wang Y., Wang Y., DePeaux K., Poholek A.C., Delgoffe G.M. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat. Immunol. 2021;22:205–215. doi: 10.1038/s41590-020-00834-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodriguez-Garcia A., Palazon A., Noguera-Ortega E., Powell D.J.J., Guedan S. CAR-T cells hit the tumor microenvironment: Strategies to overcome tumor escape. Front. Immunol. 2020;11:1109. doi: 10.3389/fimmu.2020.01109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahmadzadeh M., Johnson L.A., Heemskerk B., Wunderlich J.R., Dudley M.E., White D.E., Rosenberg S.A. Tumor antigen–specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. 2009;114:1537–1544. doi: 10.1182/blood-2008-12-195792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ribas A., Wolchok J.D. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350–1355. doi: 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wollscheid B., Bausch-Fluck D., Henderson C., O'Brien R., Bibel M., Schiess R., Aebersold R., Watts J.D. Mass-spectrometric identification and relative quantification of N-linked cell surface glycoproteins. Nat. Biotechnol. 2009;27:378–386. doi: 10.1038/nbt.1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martinko A.J., Truillet C., Julien O., Diaz J.E., Horlbeck M.A., Whiteley G., Blonder J., Weissman J.S., Bandyopadhyay S., Evans M.J., Wells J.A. Targeting RAS-driven human cancer cells with antibodies to upregulated and essential cell-surface proteins. eLife. 2018;7 doi: 10.7554/eLife.31098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roth T.L., Puig-Saus C., Yu R., Shifrut E., Carnevale J., Li P.J., Hiatt J., Saco J., Krystofinski P., Li H., Tobin V., Nguyen D.N., Lee M.R., Putnam A.L., Ferris A.L., et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature. 2018;559:405–409. doi: 10.1038/s41586-018-0326-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leung K.K., Nguyen A., Shi T., Tang L., Ni X., Escoubet L., MacBeth K.J., DiMartino J., Wells J.A. Multiomics of azacitidine-treated AML cells reveals variable and convergent targets that remodel the cell-surface proteome. Proc. Natl. Acad. Sci. U. S. A. 2019;116:695–700. doi: 10.1073/pnas.1813666116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pino L.K., Searle B.C., Bollinger J.G., Nunn B., MacLean B., MacCoss M.J. The Skyline ecosystem: Informatics for quantitative mass spectrometry proteomics. Mass Spectrom. Rev. 2020;39:229–244. doi: 10.1002/mas.21540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmiedel B.J., Singh D., Madrigal A., Valdovino-Gonzalez A.G., White B.M., Zapardiel-Gonzalo J., Ha B., Altay G., Greenbaum J.A., McVicker G., Seumois G., Rao A., Kronenberg M., Peters B., Vijayanand P. Impact of genetic polymorphisms on human immune cell gene expression. Cell. 2018;175:1701–1715.e16. doi: 10.1016/j.cell.2018.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Korotkevich G., Sukhov V., Budin N., Shpak B., Artyomov M.N., Sergushichev A. Fast gene set enrichment analysis. bioRxiv. 2021 doi: 10.1101/060012. [preprint] [DOI] [Google Scholar]
- 40.Leung K.K., Wilson G.M., Kirkemo L.L., Riley N.M., Coon J.J., Wells J.A. Broad and thematic remodeling of the surfaceome and glycoproteome on isogenic cells transformed with driving proliferative oncogenes. Proc. Natl. Acad. Sci. U. S. A. 2020;117:7764–7775. doi: 10.1073/pnas.1917947117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pfistershammer K., Majdic O., Stöckl J., Zlabinger G., Kirchberger S., Steinberger P., Knapp W. CD63 as an activation-linked T cell costimulatory element. J. Immunol. 2004;173:6000–6008. doi: 10.4049/jimmunol.173.10.6000. [DOI] [PubMed] [Google Scholar]
- 42.Bayer A.L., Baliga P., Woodward J.E. Transferrin receptor in T cell activation and transplantation. J. Leukoc. Biol. 1998;64:19–24. doi: 10.1002/jlb.64.1.19. [DOI] [PubMed] [Google Scholar]
- 43.Vomhof-DeKrey E.E., Haring J.S., Dorsam G.P. Vasoactive intestinal peptide receptor 1 is downregulated during expansion of antigen-specific CD8 T cells following primary and secondary listeria monocytogenes infections. J. Neuroimmunol. 2011;234:40–48. doi: 10.1016/j.jneuroim.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aerts-Toegaert C., Heirman C., Tuyaerts S., Corthals J., Aerts J.L., Bonehill A., Thielemans K., Breckpot K. CD83 expression on dendritic cells and T cells: Correlation with effective immune responses. Eur. J. Immunol. 2007;37:686–695. doi: 10.1002/eji.200636535. [DOI] [PubMed] [Google Scholar]
- 45.Spendlove I., Sutavani R. The role of CD97 in regulating adaptive T-cell responses. Adv. Exp. Med. Biol. 2010;706:138–148. doi: 10.1007/978-1-4419-7913-1_12. [DOI] [PubMed] [Google Scholar]
- 46.Lin M., Sutherland D.R., Horsfall W., Totty N., Yeo E., Nayar R., Wu X.F., Schuh A.C. Cell surface antigen CD109 is a novel member of the α2 macroglobulin/C3, C4, C5 family of thioester-containing proteins. Blood. 2002;99:1683–1691. doi: 10.1182/blood.v99.5.1683. [DOI] [PubMed] [Google Scholar]
- 47.Ren W., Liu G., Yin J., Tan B., Wu G., Bazer F.W., Peng Y., Yin Y. Amino-acid transporters in T-cell activation and differentiation. Cell Death Dis. 2017;8 doi: 10.1038/cddis.2017.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ivetic A., Hoskins Green H.L., Hart S.J. L-Selectin: A major regulator of leukocyte adhesion, migration and signaling. Front. Immunol. 2019;10:1068. doi: 10.3389/fimmu.2019.01068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lauer V., Grampp S., Platt J., Lafleur V., Lombardi O., Choudhry H., Kranz F., Hartmann A., Wullich B., Yamamoto A., Coleman M.L., Ratcliffe P.J., Mole D.R., Schödel J. Hypoxia drives glucose transporter 3 expression through HIF-mediated induction of the long non-coding RNA NICI. J. Biol. Chem. 2019;295:4065–4078. doi: 10.1074/jbc.RA119.009827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang H., Lindborg C., Lounev V., Kim J.H., McCarrick-Walmsley R., Xu M., Mangiavini L., Groppe J.C., Shore E.M., Schipani E., Kaplan F.S., Pignolo R.J. Cellular hypoxia promotes heterotopic ossification by amplifying BMP signaling. J. Bone Miner Res. 2016;31:1652–1665. doi: 10.1002/jbmr.2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J.P., Tamayo P. The molecular signatures database hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Choi S.-H., Kim M.Y., Yoon Y.S., Koh D.I., Kim M.K., Cho S.Y., Kim K.S., Hur M.W. Hypoxia-induced RelA/p65 derepresses SLC16A3 (MCT4) by downregulating ZBTB7A. Biochim. Biophys. Acta (Bba) - Gene Regul. Mech. 2019;1862:771–785. doi: 10.1016/j.bbagrm.2019.06.004. [DOI] [PubMed] [Google Scholar]
- 53.Munkley J., Elliott D.J. Hallmarks of glycosylation in cancer. Oncotarget. 2016;7:35478–35489. doi: 10.18632/oncotarget.8155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.O’Neill R.E., Du W., Mohammadpour H., Alqassim E., Qiu J., Chen G., McCarthy P.L., Lee K.P., Cao X. T cell-derived CD70 delivers an immune checkpoint function in inflammatory T cell responses. J. Immunol. 2017;199:3700–3710. doi: 10.4049/jimmunol.1700380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Manieri N.A., Chiang E.Y., Grogan J.L. Tigit: A key inhibitor of the cancer immunity cycle. Trends Immunol. 2017;38:20–28. doi: 10.1016/j.it.2016.10.002. [DOI] [PubMed] [Google Scholar]
- 56.Deng J., Li J., Sarde A., Lines J.L., Lee Y.C., Qian D.C., Pechenick D.A., Manivanh R., Le Mercier I., Lowrey C.H., Varn F.S., Cheng C., Leib D.A., Noelle R.J., Mabaera R. Hypoxia-induced VISTA promotes the suppressive function of myeloid- derived suppressor cells in the tumor microenvironment. Cancer Immunol. Res. 2019;7:1079–1090. doi: 10.1158/2326-6066.CIR-18-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang B., Zhang W., Jankovic V., Golubov J., Poon P., Oswald E.M., Gurer C., Wei J., Ramos I., Wu Q., Waite J., Ni M., Adler C., Wei Y., Macdonald L., et al. Combination cancer immunotherapy targeting PD-1 and GITR can rescue CD8+ T cell dysfunction and maintain memory phenotype. Sci. Immunol. 2018;3 doi: 10.1126/sciimmunol.aat7061. [DOI] [PubMed] [Google Scholar]
- 58.Raczkowski F., Rissiek A., Ricklefs I., Heiss K., Schumacher V., Wundenberg K., Haag F., Koch-Nolte F., Tolosa E., Mittrücker H.W. CD39 is upregulated during activation of mouse and human T cells and attenuates the immune response to Listeria monocytogenes. PLoS One. 2018;13 doi: 10.1371/journal.pone.0197151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wei C.-W., Lee C.Y., Lee D.J., Chu C.F., Wang J.C., Wang T.C., Jane W.N., Chang Z.F., Leu C.M., Dzhagalov I.L., Hsu C.L. Equilibrative nucleoside transporter 3 regulates T cell homeostasis by coordinating lysosomal function with nucleoside availability. Cell Rep. 2018;23:2330–2341. doi: 10.1016/j.celrep.2018.04.077. [DOI] [PubMed] [Google Scholar]
- 60.Macintyre A.N., Gerriets V.A., Nichols A.G., Michalek R.D., Rudolph M.C., Deoliveira D., Anderson S.M., Abel E.D., Chen B.J., Hale L.P., Rathmell J.C. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014;20:61–72. doi: 10.1016/j.cmet.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Martin M., Romero X., de la Fuente M.A., Tovar V., Zapater N., Esplugues E., Pizcueta P., Bosch J., Engel P. CD84 functions as a homophilic adhesion molecule and enhances IFN-γ secretion: Adhesion is mediated by ig-like domain 1. J. Immunol. 2001;167:3668–3676. doi: 10.4049/jimmunol.167.7.3668. [DOI] [PubMed] [Google Scholar]
- 62.Kim M.N., Hong J.Y., Shim D.H., Sol I.S., Kim Y.S., Lee J.H., Kim K.W., Lee J.M., Sohn M.H. Activated leukocyte cell adhesion molecule stimulates the T-cell response in allergic asthma. Am. J. Respir. Crit. Care Med. 2018;197:994–1008. doi: 10.1164/rccm.201703-0532OC. [DOI] [PubMed] [Google Scholar]
- 63.Qualai J., Li L.X., Cantero J., Tarrats A., Fernández M.A., Sumoy L., Rodolosse A., McSorley S.J., Genescà M. Expression of CD11c is associated with unconventional activated T cell subsets with high migratory potential. PLoS One. 2016;11 doi: 10.1371/journal.pone.0154253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bettelli E., Glatigny S., Duhen R., Arbelaez C., Kumari S., Oukka M., Bettelli E. Integrin alpha 4 differentially affect the migration of effector and regulatory T cells (P4113) J. Immunol. 2013;190:133.10. [Google Scholar]
- 65.Doedens A.L., Phan A.T., Stradner M.H., Fujimoto J.K., Nguyen J.V., Yang E., Johnson R.S., Goldrath A.W. Hypoxia-inducible factors enhance the effector responses of CD8+ T cells to persistent antigen. Nat. Immunol. 2013;14:1173–1182. doi: 10.1038/ni.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Clambey E.T., McNamee E.N., Westrich J.A., Glover L.E., Campbell E.L., Jedlicka P., de Zoeten E.F., Cambier J.C., Stenmark K.R., Colgan S.P., Eltzschig H.K. Hypoxia-inducible factor-1 alpha–dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc. Natl. Acad. Sci. U. S. A. 2012;109:E2784–E2793. doi: 10.1073/pnas.1202366109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Watson M.J., Vignali P.D.A., Mullett S.J., Overacre-Delgoffe A.E., Peralta R.M., Grebinoski S., Menk A.V., Rittenhouse N.L., DePeaux K., Whetstone R.D., Vignali D.A.A., Hand T.W., Poholek A.C., Morrison B.M., Rothstein J.D., et al. Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature. 2021;591:645–651. doi: 10.1038/s41586-020-03045-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Renner K., Bruss C., Schnell A., Koehl G., Becker H.M., Fante M., Menevse A.N., Kauer N., Blazquez R., Hacker L., Decking S.M., Bohn T., Faerber S., Evert K., Aigle L., et al. Restricting glycolysis preserves T cell effector functions and augments checkpoint therapy. Cell Rep. 2019;29:135–150.e9. doi: 10.1016/j.celrep.2019.08.068. [DOI] [PubMed] [Google Scholar]
- 69.Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D.J., Inuganti A., Griss J., Mayer G., Eisenacher M., Pérez E., Uszkoreit J., Pfeuffer J., Sachsenberg T., Yilmaz S., et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019;47:D442–D450. doi: 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw proteomics data, peaklists, ProteinProspector results, and Skyline quantification results have been deposited to the ProteomeXchange Consortium via the PRIDE (69) partner repository with the dataset identifier PXD024789. The R script used can be accessed on Github at https://github.com/byrn1855/T_Cell_SILAC.git. This article contains supplemental data. Full outputs from SILAC analysis can be found in supplemental Tables S1–S5. Complete peptide lists for each raw file analyzed can be found in supplemental Tables S6–S10. Flow cytometry data and all other data presented are available upon reasonable request.