

Abstract
In the wake of the COVID-19 pandemic, drug repurposing has been highlighted for rapid introduction of therapeutics. Proposed drugs with activity against SARS-CoV-2 include compounds with positive charges at physiologic pH, making them potential targets for the organic cation secretory transporters of kidney and liver, i.e., the basolateral organic cation transporters, OCT1 and OCT2; and the apical multidrug and toxin extruders, MATE1 and MATE2-K. We selected several compounds proposed to have in vitro activity against SARS-CoV-2 (chloroquine, hydroxychloroquine, quinacrine, tilorone, pyronaridine, cetylpyridinium, and miramistin) to test their interaction with OCT and MATE transporters. We used Bayesian machine learning models to generate predictions for each molecule with each transporter and also experimentally determined IC50 values for each compound against labeled substrate transport into CHO cells that stably expressed OCT2, MATE1, or MATE2-K using three structurally distinct substrates (atenolol, metformin and 1-methyl-4-phenylpyridinium) to assess the impact of substrate structure on inhibitory efficacy. For the OCTs substrate identity influenced IC50 values, although the effect was larger and more systematic for OCT2. In contrast, inhibition of MATE1-mediated transport was largely insensitive to substrate identity. Unlike MATE1, inhibition of MATE2-K was influenced, albeit modestly, by substrate identity. Maximum unbound plasma concentration/IC50 ratios were used to identify potential clinical DDI recommendations; all the compounds interacted with the OCT/MATE secretory pathway, most with sufficient avidity to represent potential DDI issues for secretion of cationic drugs. This should be considered when proposing cationic agents as repurposed antivirals.
SIGNIFICANCE STATEMENT
Drugs proposed as potential COVID-19 therapeutics based on in vitro activity data against SARS-CoV-2 include compounds with positive charges at physiological pH, making them potential interactors with the OCT/MATE renal secretory pathway. We tested seven such molecules as inhibitors of OCT1/2 and MATE1/2-K. All the compounds blocked transport activity regardless of substrate used to monitor activity. Suggesting that plasma concentrations achieved by normal clinical application of the test agents could be expected to influence the pharmacokinetics of selected cationic drugs.
Introduction
In the wake of the COVID-19 pandemic, drug repurposing has been highlighted as a possible way to accelerate production of effective therapeutics (Yang et al., 2020). Candidates for repurposing include a number of compounds that possess positive charge at physiologic pH [e.g., hydroxychloroquine and tilorone (Puhl et al., 2020; Yang et al., 2020)], raising the prospect of adverse drug-drug interactions (DDIs) or toxicity through the interaction of such compounds with pathways associated with the clearance of cationic drugs [about 40% of all prescribed drugs (Neuhoff et al., 2003)] from the body.
The kidney is the preferential site of excretion of small, hydrophilic organic cations (Hagenbuch, 2010). In the kidney, the first step in the pathway for organic cation (OC) secretion (i.e., entry of substrate from the blood into renal proximal tubule cells) is dominated by activity of the organic cation transporter (OCT)2 (Motohashi et al., 2002) followed by the extrusion from inside the cell into the filtrate by the apically expressed multidrug and toxin extrusion proteins, MATE1 and MATE2-K (Tanihara et al., 2007). Inhibition of either the basolateral or apical transporters can lead to higher concentrations of pharmaceuticals in the blood, thereby altering their pharmacokinetics (Koepsell, 2013), whereas inhibition of the apical MATEs can result in drug accumulation and a resulting nephrotoxicity (Yokoo et al., 2007). Because of the presence within the kidney of this common pathway for the secretion of OCs, and the comparatively broad selectivity of both sets of processes (Bednarczyk et al., 2003; Martinez-Guerrero et al., 2016; Sandoval et al., 2018), the stage is set for DDIs (Lepist and Ray, 2012). Although the kidney is likely to play the predominant role in clearance of most cationic substrates, the liver activity of OCT1 (sinusoidal membrane) and MATE1 (canalicular membrane) likely mediates the biliary secretion of at least some organic cations (Koepsell, 2020). Understanding the influence of OCT and MATE transporters is critical to ensure safe use of any cationic drug that is planned on being administered to patients. The inhibitory potential of a potential “perpetrator” compound can be anticipated from comparison of its maximum (unbound) concentration of the in the blood (Cu,max) to its in vitro IC50 value against transport of a “victim” drug [Cu,max/IC50 (Giacomini et al., 2010; FDA, 2012; FDA, 2020)].
There is also increasing awareness that the profile of inhibition a potential perpetrator can exert on its target can be influenced by the identity of the substrate used as a gauge as transport activity (Belzer et al., 2013; Yin et al., 2016; Sandoval et al., 2018; Koepsell, 2019). Inhibition of OCT2 activity is, in fact, significantly influenced by the choice of substrate used to assess transport (Belzer et al., 2013; Sandoval et al., 2018). Although the interaction of inhibitory ligands with MATE1 does not seem to be systematically influenced by the choice of substrate used to assess transport activity (Martinez-Guerrero et al., 2016), whether the same is true for hMATE2-K has not yet been elucidated. Consequently, the use of several substrates must be considered when planning IC50 studies for the profile of inhibition of OC (Koepsell, 2019; FDA, 2020).
In the current study, we examined two sets of cationic compounds that have been recently proposed as potential COVID-19 therapeutics based on either in vitro activity against SARS-CoV-2 or published activity against other related viruses (Baker et al., 2020; Gawriljuk et al., 2020; Puhl et al., 2020; Yang et al., 2020) as potential inhibitors of the primary transporters involved in the renal and hepatic secretion of organic cations, i.e., OCT1, OCT2, MATE1, and MATE2-K. The first set consisted of five antiparasitic/antimalarial compounds proposed to exhibit antiviral activity: chloroquine, hydroxychloroquine, and quinacrine [proposed to inhibit SARS-CoV-2 activity (Uzunova et al., 2020; Pineda et al., 2021)]; and tilorone and pyronaridine [proposed to inhibit SARS-CoV-2 activity and investigated against activity of other viral infections, including ebola, Chikungunya virus, and other coronavirus-caused diseases such as Middle Eastern respiratory syndrome coronavirus MERS (Bae et al., 2020; Ekins and Madrid, 2020; Lane and Ekins, 2020; Puhl et al., 2020)]. The second set of molecules consisted of the widely accessible, over the counter antiseptic molecules cetylpyridinium (frequently used in mouthwash) and miramistin (which is available in Russia), both of which display broad-spectrum antiviral activities that have garnered attention for their potential use in COVID-19 treatment (Mukherjee et al., 2017; Baker et al., 2020; Osmanov et al., 2020; Vergara-Buenaventura and Castro-Ruiz, 2020). We found that all these compounds are effective in vitro inhibitors of both OCT1/2 and MATE1/2-K.
Materials and Methods
Chemicals
[3H]1-Methyl-4-phenylpyridinium (MPP) [specific activity (S.A.) 81.3 Ci/mmol] and [3H]atenolol [S.A. 15 Ci/mmol] were purchased from American Radiolabeled Chemicals, Inc, MO. [14C]Metformin [S.A. 107 mCi/mmol] was purchased from Moravek Biochemicals (Brea, CA). MPP, chloroquine, hydroxychloroquine, Ham’s F-12 Kaighn’s modified medium, and Dulbecco’s modified Eagle’s medium were obtained from Sigma-Aldrich Co. Other reagents were of analytical grade and obtained through standard commercial sources.
Cell Culture
CHO cells containing a single integrated Flp Recombination Target site were obtained from Invitrogen and were used for stable expression of OCT1, OCT2, MATE1, and MATE2-K, as previously described (Pelis et al., 2007; Astorga et al., 2012). Expression of transporters was maintained through hygromycin (100 µg/ml; Invitrogen, Carlsbad, CA) selective pressure; wild-type (non-OCT2-expressing) CHO cells were maintained through Zeocin (100 µg/ml; Invitrogen) selective pressure. These cells were cultured under 5% CO2/95% air with subculture every 3 to 4 days. When seeded into 96-well plates (Greiner; VWR Intl., Arlington Heights, IL) for transport assays, cells were grown to confluence in antibiotic-free medium.
Uptake Experiments with Cultured Cells that Stably Expressed OCT2, MATE1, and MATE2-K
For uptake experiments, CHO cells expressing a target transporter were seeded in 96-well cell culture plates (Greiner; VWR Intl., Arlington Heights, IL) at densities sufficient for the cells to reach confluence within 24 (150,000 cells per well) or 48 hours (75,000 cells per well). Plates containing culture media were placed in an automatic fluid aspirator/dispenser (model 406, BioTek, Winooski, VT) and rinsed/aspirated two times with room temperature Waymouth buffer (pH 7.4), after which substrate/inhibitor-containing transport buffer (50 μl) was automatically introduced into each well. After the experimental incubation, the transport reaction was stopped by the rapid addition (and simultaneous aspiration) of cold (4°C) Waymouth buffer. After final aspiration of the cold stop, 200 μl of scintillation cocktail (Microscint 20, Perkin-Elmer, Waltham, MA) was added to each well, and the plates were sealed (Topseal-A; Perkin-Elmer) and allowed to sit for at least 2 hours before radioactivity was assessed in a 6 channel, multiwell scintillation counter (Wallac Trilux 1450 Microbeta, Perkin-Elmer). In studies measuring MATE1- and MATE2-K-mediated transport, the cells were first preincubated for 20 minutes in a buffer containing 20 mM NH4Cl. Transport was initiated by aspirating this medium and replacing it with an NH4Cl-free medium, thereby rapidly establishing an outwardly directed H+ gradient (∼pH 6 in the cytoplasm vs. 7.4 in the medium) that persisted for several minutes (data not shown).
OCT2 and MATE1 Bayesian Model Predictions
We previously described the development of individual substrate-specific Bayesian machine learning models for OCT2 and MATE1 with commercial software available to us (Martinez-Guerrero et al., 2016; Sandoval et al., 2018). We used these previously published datasets to generate Bayesian classification models with our proprietary Assay Central software (Sandoval et al., 2018) and used the resulting models for prediction of the compounds used in this study (where a score greater than 0.5 is classed as active) as potential inhibitors alongside the in vitro data generated. Molecular similarity analysis of substrate probes was performed using Discovery Studio (Biovia, San Diego, CA) to calculate using MDL fingerprints to generate Tanimoto similarities.
Data Analysis
Results are presented as means ± S.D. Statistical analyses were performed using ANOVA (Tukey’s post-test), with P < 0.05 considered statistically significant (Prism, 8 GraphPad Software, Inc, San Diego, CA).
Results
Characterization of OCT1/2 and MATE1/2-K Transport Activity
Selection of Substrates and Inhibitors
Three known substrates of OCTs and MATEs, [3H]MPP, [3H]atenolol, and [14C]metformin (Fig. 1) were selected for study because their structures differed substantially from one another, (see Supplemental Table 3) as are the kinetic characteristics of their transport (Martinez-Guerrero et al., 2016; Yin et al., 2016; Sandoval et al., 2019). MPP is a prototypic substrate for OC transport research (Lazaruk and Wright, 1990), and atenolol and metformin, both of which are secreted by the renal/hepatic OCT1/2-MATE1/2-K pathways (Nies et al., 2011), are therapeutic agents in wide use in the United States and other countries. The test inhibitors (Fig. 1) included five amines with known antiparasitic and/or antiviral activity (chloroquine, hydroxychloroquine, pyronaridine, tilorone, and quinacrine), all of which are dibasic at physiologic pH; and two monovalent quaternary ammonium compounds (cetylpyridinium and miramistin) with strong antiseptic properties. All seven of these compounds have been proposed for use or tested in vitro against SARS-CoV-2 (Baker et al., 2020; Gawriljuk et al., 2020; Puhl et al., 2020; Uzunova et al., 2020).
Fig. 1.

Structures of the three test substrates (atenolol, metformin, and MPP) and seven test inhibitors (chloroquine, hydroxychloroquine, quinacrine, tilorone, pyronaridine, cetylpyridinium, and miramistin).
Time Dependence of Transport Activity
The functional expression of OCT2, MATE1, or MATE2-K was assessed by measuring the uptake of [3H]atenolol (∼200 nM), [14C]metformin (∼10 µM), and [3H]MPP (∼15 nM) into CHO cells that stably expressed each transporter. Figure 2 shows representative time courses of net uptake (reported in terms of substrate clearance to facilitate comparisons of transport efficiency) of the three radiolabeled substrates by OCT2 (Fig. 2, A–C), MATE1 (Fig. 2, D–F), and MATE2-K (Fig. 2, G–I). Of the three test substrates, rates of mediated atenolol transport (Fig. 2, A, D, and G) were generally substantially lower; 60-second rates ranged from ∼2% to 20% of those for MPP and metformin. The comparatively low rate of MATE2-K-mediated metformin transport was an exception; mediated metformin transport was only about 10% of atenolol transport (Fig. 2, G and H). For OCT2- and MATE1-mediated transport, the time courses of net uptake of the three test substrates were nearly linear for 60 seconds, so 1-minute uptakes were used in subsequent experiments to estimate the initial rate of substrate transport; 2-minute uptakes were used for MATE1 and MATE2-K.
Fig. 2.

Time course uptake experiments of ∼200 nM [3H]atenolol, ∼10 µM [14C]metformin, and ∼15nM [3H]MPP by CHO-OCT2 (A, B, and C), CHO-MATE1 (D, E, and F), or CHO-MATE2-K (G, H, and I). Uptake is expressed as clearance (µl cm−2). Each point is the mean of triplicate measures of uptake determined in a single representative experiment. Experiments with MATE1- and MATE2-K-expressing CHO cells were measured in the presence (triangles) or absence (circles) of 1 mM unlabeled MPP. Experiments with OCT2-expressing cells also included parallel measurements with wild-type CHO cells (triangles).
Inhibition of Transport Activity
All of the test inhibitors blocked net uptake of the three test substrates (atenolol, metformin, and MPP) by OCT2, MATE1, and MATE2-K (Table 1), and the kinetics of inhibition were adequately described by the following relationship (Groves et al., 1994):
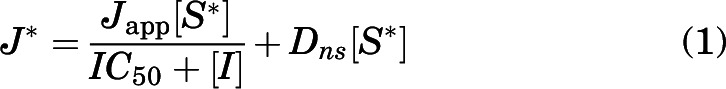 |
where J* is the total rate of carrier-mediated transport of labeled substrate from a concentration of substrate equal to [S*] (which for all three substrates was generally at least 50 times less than the apparent Michaelis constant, Ktapp, for transport of that substrate); IC50 is the concentration of inhibitor that reduced mediated (i.e., blockable) substrate transport by 50%; Japp is a constant comprised of the maximal rate of substrate transport times the ratio of the inhibitor IC50 and the Ktapp for transport of the labeled substrate; and Dns is a first order rate constant describing the nonblockable component of total substrate accumulation (consisting of diffusion, nonspecific binding, and residual substrate not eliminated by the rinse procedure).
TABLE 1.
IC50 values for inhibition of atenolol, metformin, and MPP transport into CHO cells stably transfected with hOCT1, hOCT2, hMATE1, or hMATE2-K. Each value is a mean IC50 (in μM; ±S.D.) of three or more separate experiments.
| Inhibitors | ||||||||
|---|---|---|---|---|---|---|---|---|
| IC50 | ||||||||
| Transporter | Substrates | Chloroquine | Hydroxychloroquine | Quinacrine | Tilorone | Pyronaridine | Cetylpyridinium | Miramistin |
| µM | ||||||||
| hOCT2 | [3H]Atenolol | 2.9 ± 0.02 | 5.4 ± 0.7 | 0.6 ± 0.16 | 1.11 ± 0.77 | 1.7 ± 0.5 | 22.5 ± 3.1 | 1.2 ± 0.05 |
| [14C]Metformin | 15.7 ± 1.6 | 15.3 ± 5.03 | 2.08 ± 0.14 | 4.4 ± 0.34 | 5.2 ± 0.7 | 69.3 ± 18.3 | 11.5 ± 0.6 | |
| [3H]MPP | 106.4 ± 19.3 | 103.7 ± 40.2 | 30.3 ± 3.9 | 60.4 ± 11.8 | 65.1 ± 17.7 | 161.8 ± 62.1 | 54.4 ± 4.8 | |
| hMATE1 | [3H]Atenolol | 1.7 ± 0.19 | 2.3 ± 0.6 | 16.9 ± 6.4 | 52.9 ± 21.5 | 56.1 ± 22.1 | 20.2 ± 2.8 | 5.7 ± 0.9 |
| [14C]Metformin | 2.4 ± 1.5 | 2.3 ± 0.3 | 17.13 ± 3.3 | 67.9 ± 19.4 | 84.7 ± 22.6 | 17.1 ± 6.1 | 11.98 ± 2.2 | |
| [3H]MPP | 2.9 ± 1.6 | 3.6 ± 0.19 | 22.9 ± 8.2 | 84.0 ± 35 | 81.7 ± 32.3 | 19.0 ± 11.2 | 12.4 ± 2.3 | |
| hMATE2K | [3H]Atenolol | 3.1 ± 2.5 | 5.5 ± 3.7 | 1.8 ± 0.8 | N/A | N/A | 23.4 ± 7.2 | 21.1 ± 6.8 |
| [14C]Metformin | 11.1 ± 18.2 | 3.4 ± 4.2 | 7.3 ± 8.3 | N/A | N/A | 7.6 ± 5.4 | 29.6 ± 27.9 | |
| [3H]MPP | 4.2 ± 2.4 | 6.9 ± 0.7 | 0.6 ± 0.3 | N/A | N/A | 36.3 ± 12.3 | 12.4 ± 2.8 | |
| hOCT1 | [3H]Atenolol | 18.1 ± 3.7 | 39.3 ± 13.9 | 52.6 ± 7.5 | 8.4 ± 0.4 | 10.8 ± 1.6 | 7.5 ± 2.2 | 16.5 ± 4.2 |
| [3H]MPP | 37.1 ± 21.4 | 46.92 ± 12.3 | 10.5 ± 1.8 | 18.3 ± 4.4 | 35.1 ± 11.2 | 30.1 ± 8.2 | 45.9 ± 4.5 | |
Figures 3, 4, and 5 show the profiles of inhibition of OCT2-, MATE1- and MATE2-K-mediated transport, respectively, for the three test substrates as produced by the battery of inhibitors [note: the nonblockable component of substrate transport (Dns[S*]) was subtracted from each profile]. Of particular note, inhibition of OCT2-mediated transport displayed a marked sensitivity to the substrate used to assess transport activity. Figure 3H compares IC50 values for inhibition produced by each test compound against OCT2-mediated transport of the three substrates. MPP transport (black bars) was consistently the least sensitive to inhibition, whereas atenolol (white bars) was most sensitive; inhibition of metformin transport (gray bars) was, for all the test compounds, intermediate in sensitivity. This pattern of substrate-dependent inhibition of OCT2 transport is evident in the pairwise comparisons of IC50 values shown in Fig. 7 (panels A, B, and C). For the seven test compounds, IC50s for inhibition of OCT2-mediated MPP transport were, on average, 36-fold larger than those for atenolol, and 9-fold larger than those metformin. IC50 values for inhibition of metformin transport were also consistently larger (4.5-fold) than those for atenolol.
Fig. 3.

Inhibition of labeled substrate (atenolol, metformin, or MPP) uptake into OCT2-expressing CHO cells produced by chloroquine (A), hydroxychloroquine (B), quinacrine (C), tilorone (D), pyronaridine (E), cetylpyridinium (F), and miramistin (G). One min uptakes (pH 7.4) of ∼15 nM [3H]MPP, 150 nM [3H]atenolol, or 15 µM [14C]metformin were measured in the presence of increasing concentrations of each inhibitor. Each point is the mean (±S.D.) of results determined in two or three separate experiments (see Table 1 for a summary of results). The bar graph (H) compares inhibitor constants (IC50) generated against the three substrates (atenolol, metformin, or MPP) for each of the inhibitors. *Indicates cases where inhibition of the substrate was significantly different from the other substrates, # indicates MPP different from atenolol but not different from metformin at the level of P < 0.05.
Fig. 4.

Inhibition of labeled substrate (atenolol, metformin, or MPP) uptake in MATE1-expressing CHO cells by chloroquine (A), hydroxychloroquine (B), quinacrine (C), tilorone (D), pyronaridine (E), cetylpyridinium (F), and miramistin (G). Two min uptakes (pH 7.4) of ∼15 nM [3H]MPP, 150 nM [3H]atenolol, or 10 µM [14C]metformin were measured in the presence of increasing concentrations of each inhibitor. Each point is the mean (±S.D.) of results determined in three or more separate experiments (see Table 1 for a summary of results). The bar graph (H) compares inhibitor constants (IC50) generated against the three substrates (atenolol, metformin, or MPP) for each of the inhibitors. *Indicates the different value that was significant at the level of P < 0.05.
Fig. 5.

Inhibition of labeled substrate (atenolol, metformin, or MPP) uptake in hMATE2-K-expressing CHO cells by chloroquine (A), hydroxychloroquine (B), quinacrine (C), cetylpyridinium (D), and miramistin (E). Two min uptakes (pH 7.4) of ∼15 nM [3H]MPP, 150 nM [3H]atenolol, or 10 µM [14C]metformin were measured in the presence of increasing concentrations of each inhibitor . Each point is the mean (±S.D.) of results determined in three or more separate experiments (see Table 1 for a summary of results). The bar graph (H) compares inhibitor constants (IC50) generated against the three substrates (atenolol, metformin, or MPP) for each of the inhibitors. *Indicates a difference between MPP & atenolol significant at the level of P < 0.05.
Fig. 7.

Pairwise comparison of log IC50 values for inhibition of OCT2- (A, B, and C), MATE1- (D, E, and F), and MATE2-K- (G, H, and I) mediated transport of each substrate by the tested inhibitors. Dashed lines represent equivalent inhibition of the compared substrates; the solid line represents a simple linear regression of the data.
In contrast to the substrate-dependence of OCT2 inhibition, inhibition of MATE1-mediated transport (Fig. 4) was largely insensitive to the substrate used to assess transport activity. Figure 4H compares IC50 values for inhibition produced by each test compound against MATE1-mediated transport of the three substrates. Chloroquine and hydroxychloroquine proved to be very effective inhibitors, with IC50 values ranging from 1.7 µM to 3.6 µM (Table 1), but they were equally effective blocking all three test substrates. Similarly, the comparatively modest inhibition of transport produced by tilorone and pyronaridine (IC50s of between 50 and 85 µM; Table 1), was not influenced by substrate identity (nor was the moderately effective inhibition produce by the quaternary antiseptics, cetylpyridinium and miramistin; Table 1). The routine absence of substrate-dependent inhibition of MATE1-mediated transport is evident in the pairwise comparisons of IC50 values shown in Fig. 7 (panels D, E and F). For the seven test compounds, there were no significant differences between the IC50s for inhibition of MATE1-mediated transport of either atenolol, metformin, or MPP.
Assessing the influence of substrate structure on inhibition of MATE2-K-mediated transport was complicated by the comparatively low rates of metformin transport we observed (Fig. 2) that likely contributed to the larger variance evident in the inhibition profiles obtained for MATE2-K (Fig. 5 vs. Figs. 3 and 4). Figure 5H visually compares IC50 values for inhibition produced by each test compound against MATE2-K-mediated transport of the three substrates. Chloroquine, hydroxychloroquine, and quinacrine were all comparatively effective inhibitors of transport activity (IC50s of 0.6 to 11 µM). But whereas substrate had no evident influence on inhibition produced by chloroquine (IC50s of 3–11 µM), substrate identity was correlated with differences (albeit modest; Table 1) in IC50 values obtained for hydroxychloroquine (P < 0.05) as well as for cetylpyridinium (Fig. 5, B, D, and H). But the consistent ‘pattern’ of substrate influence observed for inhibition of OCT2 (i.e., inhibitory efficacy for all inhibitors tested running from atenolol > metformin > MPP; Fig. 3) was not evident for inhibition of MATE2-K transport; whereas metformin was most sensitive to inhibition by hydroxychloroquine, it was least sensitive to inhibition by quinacrine. Similarly, the sensitivity to cetylpyridinium and miramistin was reversed for metformin versus MPP.
Although OCT1 plays a comparatively minor role in clearance of cationic drugs (Hagenbuch, 2010), we determined the profiles of inhibition of OCT1-mediated transport of MPP and atenolol (Fig. 6). Like OCT2, the inhibition of OCT1-mediated transport displayed a sensitivity, albeit comparatively modest, to the substrate used to assess transport activity for some of the inhibitors tested. Figure 6H compares IC50 values for inhibition produced by each test compound against OCT1-mediated transport of MPP and atenolol (low rates of metformin transport precluded accurate estimation of IC50 values). Atenolol transport (white bars) was most sensitive to inhibition by pyronaridine, cetylpyridinium, and miramistin, whereas MPP transport (black bars) was most sensitive to quinacrine. Substrate identity did not influence inhibition by chloroquine, hydroxychloroquine, or tilorone.
Fig. 6.

Inhibition of labeled substrate (atenolol or MPP) uptake in hOCT1-expressing CHO cells by chloroquine (A), hydroxychloroquine (B), quinacrine (C), tilorone (D), pyronaridine (E), cetylpyridinium (F), and miramistin (G). Two min uptakes (pH 7.4) of ∼15 nM [3H]MPP or 200 nM [3H]atenolol were measured in the presence of increasing concentrations of each inhibitor. Each point is the mean (±S.D.) of results determined in two or more separate experiments. The bar graph (H) compares inhibitor constants (IC50) generated against the two substrates (atenolol or MPP) for each of the inhibitors. *Indicates the different value that was significant at the level of P < 0.05
Influence of Clinical Concentrations of Chloroquine and Hydroxychloroquine on Activity of OCT1, OCT2, MATE1, and MATE2-K
The combination of comparatively low IC50 values for inhibition of OCTs and MATEs, with the relatively high plasma concentrations associated with therapeutic use of both chloroquine and hydroxychloroquine (Table 2), suggested that use of these agents could result in potentially adverse DDIs. The latest FDA Guidance for Industry (FDA, 2020) suggests that an investigational drug should be considered to have the potential to inhibit these transporters in vivo if the Cu,max / IC50 value is ≥0.1. With this in mind, Fig. 8 shows for chloroquine and hydroxychloroquine the ratios for Cu,max and IC50 (Cu,max / IC50) for inhibition of transport of each test substrate by OCT2, MATE1, and MATE2-K (Table 2). This ‘interaction ratio’ exceeded 0.1 for atenolol and/or metformin for all three transporters.
TABLE 2.
Cu,max/IC50 ratio for chloroquine, hydroxychloroquine, quinacrine, tilorone, and pyronaridine. IC50 values were taken from Table 1. Cu,max values taken from the literature as indicated.
| Inhibitors | ||||||
|---|---|---|---|---|---|---|
| Chloroquine | Hydroxychloroquine | Quinacrine | Tilorone | Pyronaridine | ||
| Maximum Unbound Plasma Concentration [Cu,max (μM)] | 1.1a | 1.2a | 0.18b | 0.11c | 0.10d | |
| Transporter | Substrate | Cu,max/IC50 | ||||
| hOCT2 | [3H]Atenolol | 0.39* | 0.22* | 2.27* | 0.10* | 0.06 |
| [14C]Metformin | 0.07 | 0.08 | 0.65* | 0.024 | 0.018 | |
| [3H]MPP | 0.01 | 0.01 | 0.04 | 0.002 | 0.001 | |
| hMATE1 | [3H]Atenolol | 0.66* | 0.51* | 0.007 | 0.002 | 0.002 |
| [14C]Metformin | 0.47* | 0.51* | 0.008 | 0.002 | 0.001 | |
| [3H]MPP | 0.39* | 0.33* | 0.007 | 0.001 | 0.001 | |
| hMATE2K | [3H]Atenolol | 0.36* | 0.21* | 0.08 | N/D | N/D |
| [14C]Metformin | 0.10* | 0.35* | 0.019 | N/D | N/D | |
| [3H]MPP | 0.27* | 0.17* | 0.23* | N/D | N/D | |
aNicol et al., 2020.
bBjörkman et al., 1989.
cZhang et al., 2010.
dCroft et al., 2012.
*Indicates values above the 0.1 cut off for recommending a clinical DDI study.
Fig. 8.

Bar graph represents Cu,max/IC50 ratios and their relation to the 0.1 cutoff value (for recommending a clinical DDI study) for chloroquine (A), hydroxychloroquine (B), and quinacrine (C) generated against OCT2, MATE1, and MATE2-K. Data were taken from Table 2.
OCT2 and MATE1 Bayesian Model Predictions
We previously demonstrated how substrate-specific OCT2 and MATE1 data can be used for generating predictive computational models that can inform structure activity relationships (Martinez-Guerrero et al., 2016; Sandoval et al., 2018). Here we used the corresponding array of Bayesian models (Supplemental Table 1) to generate predictions for each of the compounds (Supplemental Table 2). Although we had not previously described atenolol as a substrate for either MATE1 or OCT2, we did have models for metformin and MPP. Both models for MATE1 indicated that hydroxychloroquine had the highest score, although this was borderline. For OCT2, the model for interaction with metformin performed well, correctly predicting quinacrine as the most potent inhibitor (with the highest score in the model). The MPP model did not perform as well, scoring 4 of the 7 compounds as inhibitors. Interestingly, the OCT2 Bayesian models for cimetidine and the fluorescent substrate, NBD-NTMA, appear to correspond very well with the inhibition data from this study and this could be due to their closer structural resemblance to atenolol (Supplemental Table 3).
Discussion
Here we determined the interaction of OCT1, OCT2, MATE1, and MATE2-K with a set of cationic compounds that have received attention for their potential treatment of COVID-19 (Baker et al., 2020; Gawriljuk et al., 2020; Puhl et al., 2020; Yang et al., 2020). It is worth emphasizing that, although there is some in vitro data for several of these molecules, to date, there is no clinical evidence that confirms the in vivo efficacy against COVID-19 [e.g., for hydroxychloroquine (Giri et al., 2020; Self et al., 2020; Ong et al., 2021)].
All the tested compounds proved to be effective inhibitors of these transporters. However, the degree of inhibition of OCT2 by the test compounds proved to be markedly influenced by the substrate used to assess transport activity. We previously reported a marked difference in IC50 values for inhibition of OCT2-mediated transport of MPP and metformin (Belzer et al., 2013), with MPP transport proving to be much less sensitive (about 10-fold) to inhibition than was metformin transport. This observation was confirmed and expanded upon in subsequent studies (Hacker et al., 2015; Yin et al., 2016). We recently determined the profiles of inhibition produced by 400+ test compounds against OCT2-mediated transport of six structurally distinct substrates (Sandoval et al., 2018). Again, MPP transport proved markedly less sensitive to transport of any of the other test substrates; although some systematic differences were noted between these other test substrates, IC50 values for inhibition generally differed by <3-fold). However, Yin et al. (2016) found that OCT2-mediated transport of atenolol was substantially more sensitive to inhibition than was transport of metformin. Consequently, in the current study we determined inhibition profiles for the test agents against transport of MPP, metformin, and atenolol. As before, MPP transport was least sensitive to inhibition, and as noted by Yin et al. (2016), atenolol transport proved to be most sensitive to inhibition. Inhibition of OCT1 also influenced by substrate identity (Fig. 6; Table 1). Although the size of the effect was more modest than that observed for OCT2 (as much as 50-fold), OCT1-mediated MPP transport was generally least sensitive to inhibition. Overall, the differences in IC50 values observed between OCT-mediated transport of MPP, metformin, and atenolol by us (Fig. 3) and others (Yin et al., 2016) underscore the importance, recently emphasized by Koepsell and formalized by the FDA, of using multiple substrates to test the inhibitory potential of novel molecular entities (NMEs) on activity of OCTs (Koepsell, 2019; FDA, 2020)
The systematic low sensitivity to inhibition of OCT-mediated MPP transport is likely influenced by its mechanistically complex interaction with OCTs. The surface of OCT1, for example, has multiple binding sites for MPP, including an allosteric inhibitory site (Keller et al., 2019). That this complexity may be a general property of the OCTs is supported by the deduced kinetic mechanism of MPP interaction with OCT2 that includes a unique inhibitory site for MPP [but not metformin (Sandoval et al., 2019)]. Although MPP was originally proposed as a test substrate for assessing OCT transport activity (Giacomini et al., 2010; Huang et al., 2010; FDA, 2012), its consistent low sensitivity to inhibition suggests that it should not be used for that purpose to avoid underestimating the inhibitory potential of the NME under study.
It is worth pointing out that there is some discrepancy in the literature with the values for chloroquine inhibition of OCT2. Whereas Zolk et al., 2009b reported an IC50 of >1 mM for chloroquine’s inhibition of MPP transport, Belzer et al., 2013 reported a value of 129 μM, in close agreement with the results presented here (Table 1). Recently, (Yee et al., 2021) reported an IC50 of ∼100 µM against OCT2-mediated metformin transport, compared with the 15 µM value we measured; they also reported an IC50 of 0.8 µM against MATE1-mediated metformin transport, similar to our value of 2.4 µM. Modest differences in affinity for substrate and inhibitors have been reported between two common sequence variants of OCT2, the preponderant wild-type sequence in which amino acid residue 270 is an alanine, and the p.270Ala > Ser (c.808G > T) variant that is expressed at a frequency of ∼10% in different ethnic groups (Leabman et al., 2002; Chen et al., 2009). These differences including a decrease in sensitivity to inhibition by propranolol, cimetidine, and metformin (Zolk et al., 2009a). However, the IC50s for chloroquine’s inhibition of OCT2-mediated MPP transport were quite similar: IC50 of 1087 µM for wild type versus 926 µM for p.270Ala > Ser (Zolk et al., 2009a). Nevertheless, we confirmed that the sequence of the transporter used in our studies was that of the preponderant p.Ala270Ser variant. We suggest the differences in IC50 values noted here versus those reported by Zolk et al. reflect an undefined, technical issue.
Substrate identity had little impact on the inhibition profiles for MATE1, an observation that was expected. We previously have showed that inhibition of MATE1-mediated transport produced by ∼400 test compounds was effectively the same for four structurally distinct substrates, including MPP and metformin (Martinez-Guerrero et al., 2016). Yin et al., 2016 did find modest differences (5-fold) in IC50 values for MATE1-mediated transport of metformin and atenolol, but in the present study, we found no significant differences in IC50 values produced by the test agents against the MATE1-mediated transport of either MPP, metformin, or atenolol. These observations suggest that, whereas ligand interaction (substrates and inhibitors) with OCT2 is not restricted to a common site, the kinetic profiles of ligand interaction with MATE1 are consistent with a single binding site (or set of mutually exclusive binding sites) where inhibitors and substrates interact.
The influence of substrate identity on inhibitor interaction with MATE2-K was rather ambiguous. Although there were modest differences in IC50 values observed for inhibition produced by hydroxychloroquine and cetylpyridinium, the differences were modest (2–5-fold) and lacked the consistent profile observed for inhibition of OCT2-mediated transport of different substrates. We suggest that the current data set is too small and variable to draw any conclusions about whether, unlike MATE1, inhibition of MATE2-K is systematically influenced by substrate identity.
Although the compounds tested blocked all three transporters to differing extents, chloroquine, hydroxychloroquine, and quinacrine were particularly effective inhibitors of OCT2, MATE1, and MATE2-K, with IC50 values generally on the order of 1 to 10 µM (Table 1). The most recent FDA Guidance for Industry (FDA, 2020) recommends that, when the maximum unbound plasma concentration of an NME (or repurposed drug) is ≥10% of its IC50 (Cu,max/IC50 ≥ 0.1), an in vivo (clinical) DDI study be conducted; the European Medicine Agency and Japan’s Pharmaceuticals and Medical Devices Agency have similar (albeit modestly different) recommendations (EMA, 2012; PMDA, 2016). Table 2 shows the Cu,max/IC50 ratios for several combinations of transporters, substrates, and inhibitors included in the present study. Of note, chloroquine and hydroxychloroquine inhibition of metformin transport by both MATE1 and MATE-2K displayed ratios > 0.1. Pyronaridine and tilorone, although both displaying high to moderate interactions with all four transporters, generally displayed ratios substantially lower than 0.1, due in part to their comparatively low Cu.max levels (Table 2). For OCT1, the relevant parameter for a drug concentration is the maximum hepatic inlet concentration (Iin,max,u) (Parkinson, 2019), for chloroquine Iin,max,u concentration can reach ∼20 uM (Alam et al., 2016) resulting in an (IC50/Iin,max,u) ratio of 0.77 against atenolol and 1.6 against MPP. If the maximum hepatic inlet concentration of a drug is consistently higher than those found in plasma, the IC50 values against OCT1 in this study (10–50 µM) could lead to DDI with OCT1.
Cetylpyridinium and miramistin have both generated recent interest because of their known antiseptic effectiveness against coronaviruses (Mukherjee et al., 2017; Baker et al., 2020; Osmanov et al., 2020). Cetylpyridinium has been proposed to have a potential role in reducing the number of viral particles in the oral cavity (Vergara-Buenaventura and Castro-Ruiz, 2020) and has been assessed in a recent controlled clinical trial (Seneviratne et al., 2021). The increasing attention these compounds have received increases the likelihood of oral ingestion of solutions that can contain high concentrations of these compounds [e.g., 20 mM (Rösing et al., 2017)]. Although human pharmacokinetic data are unavailable, a recent study on the pharmacokinetics of oral ingestion of drug inactive ingredients in rats (Pottel et al., 2020) reported a Cmax for cetylpyridinium of 0.26 µM, so the IC50 values observed for these quaternary ammonium compounds against both OCT1/2 (1 to 150 µM) and MATEs 1 and 2-K (6 to 35 µM) suggest a modest chance for generation of DDIs. For drugs that are rapidly metabolized [e.g., hydroxychloroquine (Giri et al., 2020)], the interaction of their metabolites with these transport proteins should be assessed to more precisely predict the overall risk of DDI.
To summarize, these data support the conclusion that the tested antivirals are effective inhibitors of the OCT- and MATE-mediated elements of the renal hepatic OC secretory pathways, supporting the view that dedicated DDI trials be conducted to assess risk of DDIs associated with their repurposed use against SARS-CoV-2.
Acknowledgments
We would like to Dr. Ana C. Puhl and Dr. Thomas R. Lane for discussions on SARS-CoV-2, Dr. Antony J. Williams for discussions on cetylpyridinium and miramistin, Dr. Alex Clark for software assistance, and Biovia for providing Discovery Studio.
Abbreviations
- Cu,max
maximum unbound plasma concentration
- DDI
drug-drug interaction
- FDA
Food and Drug Administration
- Iin,max,u
maximum unbound hepatic inlet concentration
- MATE
multidrug and toxin extruders
- MPP
1-methyl-4-phenylpyridinium
- NME
novel molecular entity
- OC
organic cation
- OCT
organic cation transporter
- S.A.
specific activity
Authorship Contributions
Participated in research design: Martinez-Guerrero, Ekins, Wright.
Conducted experiments: Martinez-Guerrero, Zhang, Zorn.
Contributed new reagents or analytic tools: Zorn, Ekins.
Performed data analysis: Martinez-Guerrero, Zhang, Zorn, Ekins, Wright.
Wrote or contributed to the writing of the manuscript: Martinez-Guerrero, Ekins, Wright.
Footnotes
This work was supported by National Institutes of Health National Institute of General Medical Sciences [Grants 5R01GM129777, 1R-41GM131433-01A1, R44-GM122196-02A1, 5P30-ES006694].
S.E. is owner and K.M.Z. is an employee of Collaborations Pharmaceuticals, Inc. All others have no actual or perceived conflict of interest with the contents of this article.
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Alam K, Pahwa S, Wang X, Zhang P, Ding K, Abuznait AH, Li L, Yue W (2016) Downregulation of organic anion transporting polypeptide (OATP) 1B1 transport function by lysosomotropic drug chloroquine: implication in OATP-mediated drug-drug interactions. Mol Pharm 13:839–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astorga B, Ekins S, Morales M, Wright SH (2012) Molecular determinants of ligand selectivity for the human multidrug and toxin extruder proteins MATE1 and MATE2-K. J Pharmacol Exp Ther 341:743–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae J-Y, Lee GE, Park H, Cho J, Kim Y-E, Lee J-Y, Ju C, Kim W-K, Kim JI, Park M-S(2020) Pyronaridine and artesunate are potential antiviral drugs against COVID-19 and influenza. bioRxiv:2020.2007.2028.225102. [Google Scholar]
- Baker N, Williams AJ, Tropsha A, Ekins S (2020) Repurposing quaternary ammonium compounds as potential treatments for COVID-19. Pharm Res 37:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednarczyk D, Ekins S, Wikel JH, Wright SH (2003) Influence of molecular structure on substrate binding to the human organic cation transporter, hOCT1. Mol Pharmacol 63:489–498. [DOI] [PubMed] [Google Scholar]
- Belzer M, Morales M, Jagadish B, Mash EA, Wright SH (2013) Substrate-dependent ligand inhibition of the human organic cation transporter OCT2. J Pharmacol Exp Ther 346:300–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björkman S, Elisson LO, Gabrielsson J (1989) Pharmacokinetics of quinacrine after intrapleural instillation in rabbits and man. J Pharm Pharmacol 41:160–163. [DOI] [PubMed] [Google Scholar]
- Chen YLi SBrown CCheatham SCastro RALeabman MKUrban TJChen LYee SWChoi JH, et al. (2009) Effect of genetic variation in the organic cation transporter 2 on the renal elimination of metformin. Pharmacogenet Genomics 19:497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft SL, Duparc S, Arbe-Barnes SJ, Craft JC, Shin C-S, Fleckenstein L, Borghini-Fuhrer I, Rim H-J (2012) Review of pyronaridine anti-malarial properties and product characteristics. Malar J 11:270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekins S, Madrid PB (2020) Tilorone, a broad-spectrum antiviral for emerging viruses. Antimicrob Agents Chemother 64:e00440–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EMA (2012) Guideline on the investigation of drug interactions, in (Agency EM ed). [Google Scholar]
- FDA (2012) Guidance for industry: drug interaction studies - study design, data analysis, implications for dosing, and labeling recommendations, in (U.S.Food D, Administration ed) pp 1–79. [Google Scholar]
- FDA (2020) In vitro drug interaction studies — Cytochrome P450 enzyme- and transporter-mediated drug interactions guidance for industry, in (U.S.Food D, Administration ed). [Google Scholar]
- Gawriljuk VOKyaw Zin PPFoil DHBernatchez JBeck SBeutler NRicketts JYang LRogers TPuhl AC, et al. (2020) Machine learning models identify inhibitors of SARS-CoV-2. bioRxiv:2020.2006.2016.154765. [Google Scholar]
- Giacomini KMHuang SMTweedie DJBenet LZBrouwer KLChu XDahlin AEvers RFischer VHillgren KM, et al. ; International Transporter Consortium (2010) Membrane transporters in drug development. Nat Rev Drug Discov 9:215–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri A, Das A, Sarkar AK, Giri AK (2020) Mutagenic, genotoxic and immunomodulatory effects of hydroxychloroquine and chloroquine: a review to evaluate its potential to use as a prophylactic drug against COVID-19. Genes Environ 42:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves CE, Evans KK, Dantzler WH, Wright SH (1994) Peritubular organic cation transport in isolated rabbit proximal tubules. Am J Physiol 266:F450–F458. [DOI] [PubMed] [Google Scholar]
- Hacker K, Maas R, Kornhuber J, Fromm MF, Zolk O (2015) Substrate-dependent inhibition of the human organic cation transporter OCT2: a comparison of metformin with experimental substrates. PLoS One 10:e0136451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagenbuch B (2010) Drug uptake systems in liver and kidney: a historic perspective. Clin Pharmacol Ther 87:39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SM, Zhang L, Giacomini KM (2010) The international transporter consortium: a collaborative group of scientists from academia, industry, and the FDA. Clin Pharmacol Ther 87:32–36. [DOI] [PubMed] [Google Scholar]
- Keller T, Gorboulev V, Mueller TD, Dötsch V, Bernhard F, Koepsell H (2019) Rat organic cation transporter 1 contains three binding sites for substrate 1-methyl-4-phenylpyridinium per monomer. Mol Pharmacol 95:169–182. [DOI] [PubMed] [Google Scholar]
- Koepsell H (2013) The SLC22 family with transporters of organic cations, anions and zwitterions. Mol Aspects Med 34:413–435. [DOI] [PubMed] [Google Scholar]
- Koepsell H (2019) Multiple binding sites in organic cation transporters require sophisticated procedures to identify interactions of novel drugs. Biol Chem 400:195–207. [DOI] [PubMed] [Google Scholar]
- Koepsell H (2020) Organic cation transporters in health and disease. Pharmacol Rev 72:253–319. [DOI] [PubMed] [Google Scholar]
- Lane TR, Ekins S (2020) Toward the target: tilorone, quinacrine, and pyronaridine bind to ebola virus glycoprotein. ACS Med Chem Lett 11:1653–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaruk KDA, Wright SH (1990) MPP+ is transported by the TEA(+)-H+ exchanger of renal brush-border membrane vesicles. Am J Physiol 258:F597–F605. [DOI] [PubMed] [Google Scholar]
- Leabman MKHuang CCKawamoto MJohns SJStryke DFerrin TEDeYoung JTaylor TClark AGHerskowitz I, et al. ; Pharmacogenetics of Membrane Transporters Investigators (2002) Polymorphisms in a human kidney xenobiotic transporter, OCT2, exhibit altered function. Pharmacogenetics 12:395–405. [DOI] [PubMed] [Google Scholar]
- Lepist EI, Ray AS (2012) Renal drug-drug interactions: what we have learned and where we are going. Expert Opin Drug Metab Toxicol 8:433–448. [DOI] [PubMed] [Google Scholar]
- Martinez-Guerrero LJ, Morales MH, Ekins S, Wright SH (2016) Lack of influence of substrate on ligand interaction with human MATE1. Mol Pharmacol 90: 254–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motohashi H, Sakurai Y, Saito H, Masuda S, Urakami Y, Goto M, Fukatsu A, Ogawa O, Inui K (2002) Gene expression levels and immunolocalization of organic ion transporters in the human kidney. Journal of the American Society of Nephrology 13:866–874. [DOI] [PubMed] [Google Scholar]
- Mukherjee PK, Esper F, Buchheit K, Arters K, Adkins I, Ghannoum MA, Salata RA (2017) Randomized, double-blind, placebo-controlled clinical trial to assess the safety and effectiveness of a novel dual-action oral topical formulation against upper respiratory infections. BMC Infect Dis 17:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhoff S, Ungell AL, Zamora I, Artursson P (2003) pH-dependent bidirectional transport of weakly basic drugs across Caco-2 monolayers: implications for drug-drug interactions. Pharm Res 20:1141–1148. [DOI] [PubMed] [Google Scholar]
- Nicol MR, Joshi A, Rizk ML, Sabato PE, Savic RM, Wesche D, Zheng JH, Cook J (2020) Pharmacokinetics and pharmacological properties of chloroquine and hydroxychloroquine in the context of COVID-19 infection. Clin Pharmacol Ther 108:1135–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nies AT, Koepsell H, Damme K, Schwab M (2011) Organic cation transporters (OCTs, MATEs), in vitro and in vivo evidence for the importance in drug therapy. Handb Exp Pharmacol 201:105–167. [DOI] [PubMed] [Google Scholar]
- Ong WY, Go ML, Wang DY, Cheah IK, Halliwell B (2021) Effects of antimalarial drugs on neuroinflammation-potential use for treatment of COVID-19-related neurologic complications. Mol Neurobiol 58:106–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osmanov A, Farooq Z, Richardson MD, Denning DW (2020) The antiseptic Miramistin: a review of its comparative in vitro and clinical activity. FEMS Microbiol Rev 44:399–417. [DOI] [PubMed] [Google Scholar]
- Parkinson A (2019) Regulatory recommendations for calculating the unbound maximum hepatic inlet concentration: a complicated story with a surprising and happy ending. Drug Metab Dispos 47:779–784. [DOI] [PubMed] [Google Scholar]
- Pelis RM, Dangprapai Y, Wunz TM, Wright SH (2007) Inorganic mercury interacts with cysteine residues (C451 and C474) of hOCT2 to reduce its transport activity. Am J Physiol Renal Physiol 292:F1583–F1591. [DOI] [PubMed] [Google Scholar]
- Pineda B, Pérez de la Cruz V, Hernández Pando R, Sotelo J (2021) Quinacrine as a potential treatment for COVID-19 virus infection. Eur Rev Med Pharmacol Sci 25:556–566. [DOI] [PubMed] [Google Scholar]
- PMDA (2016) Guideline of drug interaction studies for drug development and appropriate provision of information, in (Ministry of Health LaW, Japan. ed). [Google Scholar]
- Pottel JArmstrong DZou LFekete AHuang X-PTorosyan HBednarczyk DWhitebread SBhhatarai BLiang G, et al. (2020) The activities of drug inactive ingredients on biological targets. Science 369:403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puhl ACFritch EJLane TRTse LVYount BLSacramento CQTavella TACosta FTMWeston SLogue J, et al. (2020) Repurposing the ebola and marburg virus inhibitors tilorone, quinacrine and pyronaridine: in vitro activity against SARS-CoV-2 and potential mechanisms. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rösing CK, Cavagni J, Gaio EJ, Muniz FWMG, Ranzan N, Oballe HJR, Friedrich SA, Severo RM, Stewart B, Zhang YP (2017) Efficacy of two mouthwashes with cetylpyridinium chloride: a controlled randomized clinical trial. Braz Oral Res 31:e47. [DOI] [PubMed] [Google Scholar]
- Sandoval PJ, Morales M, Secomb TW, Wright SH (2019) Kinetic basis of metformin-MPP interactions with organic cation transporter OCT2. Am J Physiol Renal Physiol 317:F720–F734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval PJ, Zorn KM, Clark AM, Ekins S, Wright SH (2018) Assessment of substrate dependent ligand interactions at the organic cation transporter OCT2 using six model substrates. Mol Pharmacol 94:1057–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Self WHSemler MWLeither LMCasey JDAngus DCBrower RGChang SYCollins SPEppensteiner JCFilbin MR, et al. ; National Heart, Lung, and Blood Institute PETAL Clinical Trials Network (2020) Effect of hydroxychloroquine on clinical status at 14 days in hospitalized patients with COVID-19: a randomized clinical trial. JAMA 324:2165–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seneviratne CJBalan PKo KKKUdawatte NSLai DNg DHLVenkatachalam ILim KSLing MLOon L, et al. (2021) Efficacy of commercial mouth-rinses on SARS-CoV-2 viral load in saliva: randomized control trial in Singapore. Infection 49:305–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanihara Y, Masuda S, Sato T, Katsura T, Ogawa O, Inui K (2007) Substrate specificity of MATE1 and MATE2-K, human multidrug and toxin extrusions/H(+)-organic cation antiporters. Biochem Pharmacol 74:359–371. [DOI] [PubMed] [Google Scholar]
- Uzunova K, Filipova E, Pavlova V, Vekov T (2020) Insights into antiviral mechanisms of remdesivir, lopinavir/ritonavir and chloroquine/hydroxychloroquine affecting the new SARS-CoV-2. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie 131:110668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergara-Buenaventura A, Castro-Ruiz C (2020) Use of mouthwashes against COVID-19 in dentistry. Br J Oral Maxillofac Surg 58:924–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CWPeng TTHsu HYLee YZWu SHLin WHKe YYHsu TAYeh TKHuang WZ, et al. (2020) Repurposing old drugs as antiviral agents for coronaviruses. Biomed J 43:368–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee SW, Vora B, Oskotsky T, Zou L, Jakobsen S, Enogieru OJ, Koleske ML, Kosti I, Rödin M, Sirota M, Giacomini KM (2021) Drugs in COVID-19 clinical trials: predicting transporter-mediated drug-drug interactions using in vitro assays and real-world data. Clinical Pharmacology & Therapeutics [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J, Duan H, Wang J (2016) Impact of substrate-dependent inhibition on renal organic cation transporters hOCT2 and hMATE1/2-K-mediated drug transport and intracellular accumulation. J Pharmacol Exp Ther 359:401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoo S, Yonezawa A, Masuda S, Fukatsu A, Katsura T, Inui K (2007) Differential contribution of organic cation transporters, OCT2 and MATE1, in platinum agent-induced nephrotoxicity. Biochem Pharmacol 74:477–487. [DOI] [PubMed] [Google Scholar]
- Zhang X, Duan J, Zhai S, Yang Y, Yang L (2010) Performance of tiloronoxim and tilorone determination in human blood by HPLC-MS/MS: method validation, uncertainty assessment and its application to a pharmacokinetic study. J Chromatogr B Analyt Technol Biomed Life Sci 878:492–496. [DOI] [PubMed] [Google Scholar]
- Zolk O, Solbach TF, König J, Fromm MF (2009a) Functional characterization of the human organic cation transporter 2 variant p.270Ala>Ser. Drug Metab Dispos 37:1312–1318. [DOI] [PubMed] [Google Scholar]
- Zolk O, Solbach TF, König J, Fromm MF (2009b) Structural determinants of inhibitor interaction with the human organic cation transporter OCT2 (SLC22A2). Naunyn Schmiedebergs Arch Pharmacol 379:337–348. [DOI] [PubMed] [Google Scholar]