

ABSTRACT
Human immunodeficiency virus (HIV)-infected macrophages are long-lived cells that sustain persistent virus expression, which is both a barrier to viral eradication and contributor to neurological complications in patients despite antiretroviral therapy (ART). To better understand the regulation of HIV-1 in macrophages, we compared HIV-infected primary human monocyte-derived macrophages (MDM) to acutely infected primary CD4 T cells and Jurkat cells latently infected with HIV (JLAT 8.4). HIV genomes in MDM were actively transcribed despite enrichment with heterochromatin-associated H3K9me3 across the complete HIV genome in combination with elevated activation marks of H3K9ac and H3K27ac at the long terminal repeat (LTR). Macrophage patterns contrasted with JLAT cells, which showed conventional bivalent H3K4me3/H3K27me3, and acutely infected CD4 T cells, which showed an intermediate epigenotype. 5′-Methylcytosine (5mC) was enriched across the HIV genome in latently infected JLAT cells, while 5′-hydroxymethylcytosine (5hmC) was enriched in CD4 cells and MDMs. HIV infection induced multinucleation of MDMs along with DNA damage-associated p53 phosphorylation, as well as loss of TET2 and the nuclear redistribution of 5-hydoxymethylation. Taken together, our findings suggest that HIV induces a unique macrophage nuclear and transcriptional profile, and viral genomes are maintained in a noncanonical bivalent epigenetic state.
IMPORTANCE Macrophages serve as a reservoir for long-term persistence and chronic production of HIV. We found an atypical epigenetic control of HIV in macrophages marked by heterochromatic H3K9me3 despite active viral transcription. HIV infection induced changes in macrophage nuclear morphology and epigenetic regulatory factors. These findings may identify new mechanisms to control chronic HIV expression in infected macrophages.
KEYWORDS: HIV, latency, macrophage, microglia, histone, chromatin, DNA methylation, hydroxymethylation
INTRODUCTION
Human immunodeficiency virus (HIV) results in lifelong infection requiring continuous antiretroviral therapy (ART) to suppress viral replication and prevent immune deficiency (1). A major barrier to cure is the existence of long-lived infected cells that persist during ART. Multiple anatomic sites may contribute to viral persistence, including blood, lymphoid tissue, gut-associated lymphoid tissue, bone marrow, and brain (2–4). While most attention has focused on CD4-positive (CD4+) T cell reservoirs, myeloid cells are well established to harbor virus in the central nervous system (CNS), where macrophages and microglia are the principal infected cell type (5–7). Functional cure therefore requires attention not only to CD4+ T cell reservoirs but to other cells, including myeloid reservoirs.
Monocytes are generally resistant to HIV infection but become permissive as they mature into macrophages (8, 9). Macrophages are thus terminally differentiated nondividing cells, yet they are susceptible to robust HIV infection (10, 11). This feature differentiates them from T cells, which require activation and cell proliferation to become robustly susceptible in vitro (although limited low-level infection of resting T cells may be achieved in some models) (12–14). Thus, macrophage infection and establishment of integrated provirus occur in the context of a unique cellular microenvironment relative to T cells, particularly with regard to chromatin structure. Another distinguishing feature of macrophage infection is that they are long-lived cells yet resistant to HIV-induced killing, and productively infected macrophages persist for prolonged periods, unlike activated CD4+ T cells that are killed by active virus replication (8, 15, 16). This feature enables infected myeloid cells to serve as long-term reservoirs in vivo, particularly in the CNS (17–20). Finally, while the resting CD4+ T cell long-term reservoir is typically thought of as latent, low-level virus expression generally persists throughout the life span of infected macrophages (17, 21–23). Indeed, persistent low-level virus expression from long-lived infected brain macrophages is thought to be a driver of neurological complications that occur in infected people despite ART (24, 25). Thus, macrophages likely control HIV differently than CD4+ T cells, and studies of epigenetic control in T cell models may not be sufficient to understand persistent infection in differentiated primary macrophages.
Transcriptional and epigenetic regulation of HIV in T-cells has been studied extensively (26, 27). Transcriptional regulation occurs primarily at the 5′ long terminal repeat (LTR) and is mediated by various transcription factors that control the modification and positioning of nucleosomes that restrict transcription initiation and elongation (26, 28, 29). In contrast to T cells, less is known about epigenetic factors that control HIV infection in macrophages. Here, we investigate epigenetic features, including active and repressive histone marks and DNA methylation and hydroxymethylation status across the HIV genome in infected primary human monocyte-derived macrophages (MDMs) that regulate the HIV genome in postintegration latency. We compare MDMs to productively infected primary CD4+ T cells and also with an established latency model in Jurkat T cells using JLAT cells. Our results revealed surprising noncanonical bivalent chromatin structures of the HIV-1 genome in primary infected macrophages.
RESULTS
Epigenetic profiles of HIV genomes in primary macrophages, primary CD4 T cells, and latent cell line JLAT.
Primary MDMs and CD4 T cells were infected with the brain-derived macrophage-tropic HIV-1 primary isolate YU2 (30) using conditions to optimize stable integrated infections (Fig. 1A), while the JLAT 8.4 T cell line carries a single clonally integrated latent HIV genome derived from a prototype strain (31). The epigenetic modification of histones associated with HIV genomes in MDMs, CD4 T cells, or latently infected JLAT 8.4 cells was investigated by chromatin immunoprecipitation coupled with quantitative PCR (ChIP-qPCR) assay using primers sampling regions across the entire viral genome (Fig. 1B). We assayed for histone H3K4me3, H3K9me3, H3K9ac, H3K9me2, H3K27ac, and H3K27me3 (Fig. 1C). Total histone H3 and IgG occupancy were also analyzed by ChIP-qPCR (Fig. S1 in the supplemental material).
FIG 1.

Histone modifications across the HIV genome in MDMs, CD4 T cells, and JLAT 8.4 cells. (A) Timeline of experimental design. (B) Schematic of HIV genome and positions of primers used for ChIP and DIP. (C) ChIP-qPCR analysis of the HIV genome in MDMs, CD4 T cells, and JLAT 8.4 cells for H3K4me3, H3K9me3, H3K9ac, H3K9me2, H3K27ac, and H3K27me3 using primers spaced across the genome as indicated. Cellular control primers targeted the actin and GAPDH promoters and 10q CTCF and 10q TERRA transcript regions. Note that primer sites Nuc_0, Nuc_1, A, B, S, and T target sequences in both 5′ and 3′ LTRs but are depicted graphically with one or the other. Error bars are standard deviation for 3 technical replicates.
We observed significant differences in the pattern of histone modification between each of these HIV infection models. H3K4me3, a euchromatic mark associated with most transcriptional start sites, was enriched at site B in MDMs and T cells and in JLAT 8.4 cells at Nuc_0, site A, and site S within the nef region and LTR. Nucleosome positions have been well characterized for HIV LTR in JLAT cells (32, 33). H3K9me3, a mark associated with constitutive heterochromatin, was highly enriched across the entire HIV genome in MDMs (approaching 20% input), while relatively lower levels were observed in latently infected JLAT 8.4 cells and intermediate levels in CD4 T cells. H3K27me3, a facultative heterochromatic mark associated with polycomb-mediated repression, was enriched in JLAT 8.4 at Nuc_1 and site B, as well as within the body of the genome within the env gene (P and Q regions). H3K9ac was detected in Nuc_0 and Nuc_1 and site B in MDMs, with lower levels in JLAT 8.4 and CD4 T cells. Controls for H3K4me3 and H3K9ac were enriched at active cellular genes actin and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) as expected, and heterochromatic marks for H3K9me3 or H3K27me3 were enriched at telomeric sites (10q CTCF and TERRA) as expected (with the exception that JLAT 8.4 lacked H3K27me3 enrichment at telomeres). IgG levels were generally below threshold significance at all primer positions with the exception of Nuc_1 in all three cells and positions G, P, and Q in JLAT 8.4 cells, which showed a low background signal (Fig. S1). These findings indicate that HIV genomes in MDMs are subject to distinct histone modification patterns compared to acute and latent CD4 T cell infection models. We also note that some of these epigenetic marks are enriched in HIV regions outside the LTR.
Atypical bivalent histone modification of the MDM LTR.
To further explore the ChIP data, we reanalyzed the histone modifications, focusing only on the LTR and adjacent nucleosome 2 (site B) for each infection model (Fig. 2). We observed that the LTR in MDMs was highly enriched with histone H3K9me3 and to a lesser extent with H3K27ac. The LTR in CD4 T cells also showed a relatively high enrichment of H3K9me3, as well as H3K9me2, the highest enrichment in H3K4me3 at site B, and the lowest density of total H3 relative to the other cell types tested. JLAT 8.4 cells were highly enriched for H3K4me3 at Nuc_0 and had low levels of H3K9me2 and H3K9me3. H3K27me3 was enriched at Nuc_1 and site B in JLAT relative to the other cell types. The latter pattern of histone modifications has been reported previously for JLAT cells (34–36) and corresponds with bivalent (H3K4me3/K27me3) histone modification observed in some developmentally regulated genes and pluripotent stem cells (37). In contrast to JLAT cells, the H3K4me3 enrichment in CD4 peaked at site B, located close to nucleosome 2. The relatively high H3K9me3 seen in MDM and CD4 cells, combined with H3K4me3 in CD4 T cells and to a lesser extent in MDMs, is similar to that observed at lineage-specific regulatory elements, such as methylation pause sites in adipocytes (38). The combination of H3K9me3 and H3K27ac that we find in MDMs is similar to that observed for transposable elements in ES cells (39).
FIG 2.
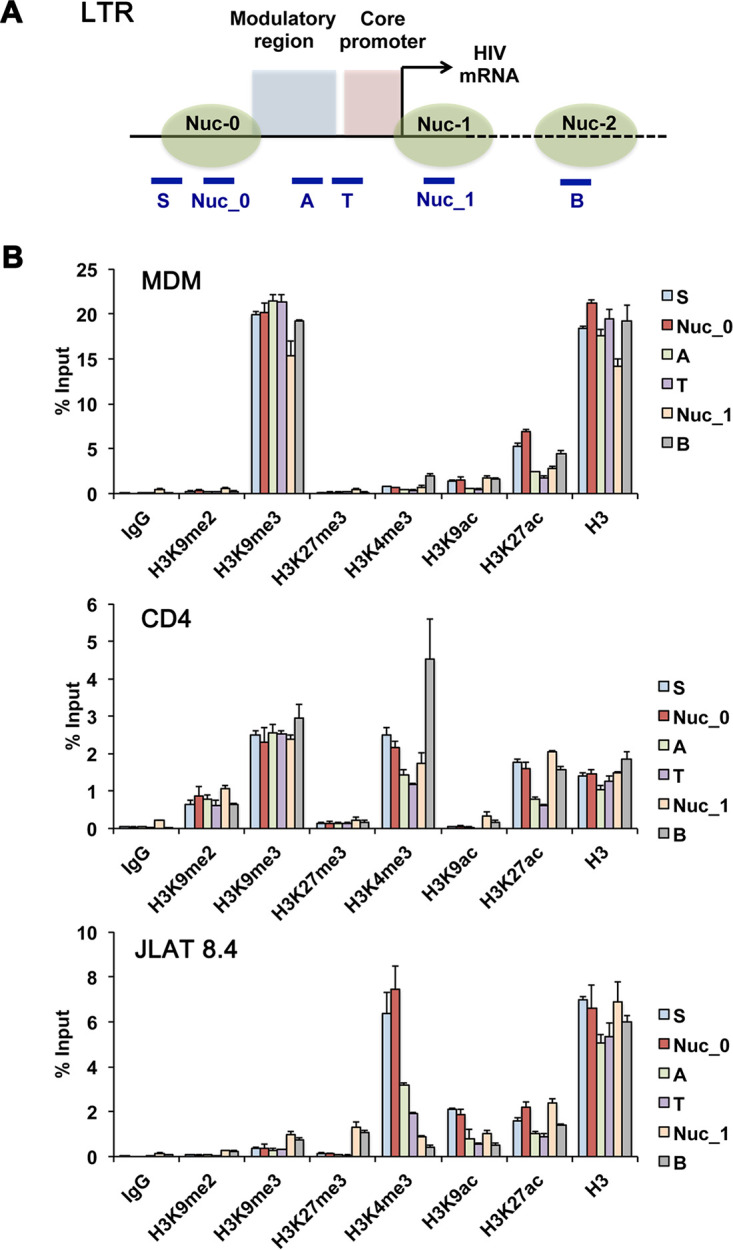
ChIP-qPCR focusing on HIV LTR. (A) Schematic of HIV 5′ LTR with adjacent nucleosome 2 and positions of primers used in panel B. Solid line represents LTR DNA, and dashed line represents HIV genomic region downstream of 5′ LTR. Primers that amplify sequences duplicated in 5′ and 3′ LTRs are listed. (B) ChIP-qPCR data from Fig. 1 was regraphed to directly compare enrichments of each histone modification across the LTR region and adjacent nucleosome 2 site for MDMs, CD4 cells, and JLAT8.4 cells. Error bars are standard deviation for 3 technical replicates.
Relative abundance of unintegrated and total vDNA.
Since the HIV-1 genome can exist as either integrated or unintegrated form, but the integrated form is responsible for viral expression, we assessed their relative abundances in YU2-infected macrophages. Total HIV copy number was quantified based on gag gene amplification, and episomal 2-LTR circles were quantified using outward-directed primers specific for this form of viral DNA (40). One week after infection, total viral copies exceeded 2-LTR circles by more than an order of magnitude (Table 1). Thus, while unintegrated viral-derived DNA is known to persist in MDMs longer than in replicating cells (41), this result suggests that our ChIP and DNA immunoprecipitation (DIP) analyses reflect predominantly integrated proviral DNA.
TABLE 1.
Quantification of HIV-1 total and 2-LTR circular episomal vDNA
| Target | No. of copies per ng DNA | No. of copies per cell |
|---|---|---|
| β-Globin | 75.3a | 1 |
| Gag | 84.6 | 1.12 |
| 2-LTR | 5.47 | 0.07 |
β-Globin is reported as equivalent cell number based on serial dilution of peripheral blood mononuclear cell (PBMC).
Antisense transcripts in infected MDMs.
Antisense transcripts are known to be a source of generating H3K9me3 (42), and antisense transcripts have been reported for HIV, particularly during latent infection (43, 44). Therefore, we assayed for antisense HIV transcripts using HIV-specific antisense reverse transcriptase (RT) primers at nucleotide positions 15 (AS RT-2), 2944 (AS RT-1), and 7431 (AS RT-3) in conjunction with qPCR primer pairs positioned at 351 to 461 (primer 2), 3161 to 3275 (primer 1), 7846 to 7733 (primer 3), and 8333 to 8426 (primer 4) based on the YU2 HIV genome (Fig. 3A). Antisense transcripts were detected at all positions in YU2-infected MDMs and CD4 T cells, but not in uninfected control cells (Fig. 3B) or in reaction mixtures lacking RT (Fig. 3C). The relative levels of antisense transcript in MDMs and CD4 T cells correlated with the relative ratio of sense transcripts for nef and tat (Fig. 3D). These findings indicated that both MDMs and CD4 T cells have measurable levels of antisense HIV and raise the possibility that antisense transcription contributes to H3K9me3 formation, as has been observed for many endogenous retroviruses (45).
FIG 3.

Antisense RNA transcripts of HIV in MDM and CD4 infection. (A) Schematic of HIV genome showing the position of three HIV-specific antisense reverse transcription primers (AS RT1, AS RT2, or AS RT3) and four qPCR primer pairs (1, 2, 3, and 4) used for antisense transcript quantification. (B) RT-qPCR of antisense transcripts in uninfected and YU2-infected CD4 T cells and MDMs, calculated relative to cellular GUSB sense transcript. (C) RT-qPCR of antisense transcripts in YU2-infected CD4 T cells and MDMs with or without addition of RT. (D) Sense transcription of nef or tat for the same infection and RNA samples as shown in panel B.
Cytosine methylation and hydroxymethylation of HIV genomes.
To investigate whether DNA modifications differed in MDMs and CD4 T cell infection, we assessed the levels of cytosine methylation (5mC) and hydroxymethylation (5hmC) of HIV DNA in MDMs, CD4 T cells, and JLAT 8.4 cells using methylcytosine DNA immunoprecipitation (MeDIP) or hydroxymethylcytosine DNA IP (hMeDIP) assays (Fig. 4). We detected elevated levels of 5mC on the JLAT 8.4 genome, including locations at site B, which was previously identified as a CpG island adjacent to the 5′ LTR in JLAT (34), and the 3′ regions within the env gene (P, Q, and R). Relatively low or undetectable levels of 5mC were found associated with HIV infection in MDMs and CD4 T cells. In contrast, both MDMs and CD4 T cells showed a broad pattern of hydroxymethylation (5hmC) across the HIV genomes, whereas this modification was mostly absent from JLAT 8.4 cells. Cellular controls for 5mC and 5hmC were highly enriched at cellular telomeric positions and absent in actively transcribed genes Actin and GAPDH. These findings indicate the 5mC is formed in long-term latently infected JLAT cells but is generally not formed during productive infection of primary MDMs or CD4 T cells, while 5hmC forms during infection of MDMs and CD4 T cells with actively transcribing HIV genomes.
FIG 4.

DNA immunoprecipitation (DIP) assay for 5mC or 5hmc across the HIV genome for infected MDMs, CD4 T cells, or JLAT 8.4 cells. MeDIP for 5mC (left panels, orange bars) or hMeDIP for 5hmC (right panels, green bars) or control IgG were analyzed using PCR primers across HIV genome, as indicated in Fig. 1A. Cellular gene control sites at 10q or XqYq TERRA or promoter regions for actin or GAPDH.
Differential expression of histone and nuclear viral response proteins in MDMs and CD4 T cells.
To assess whether these epigenetic variations were associated with cell type-specific differences in global histone modifications, we assayed total cellular histone modification levels with or without HIV-1 YU2 infection by Western blotting (Fig. 5A). We found that histone levels were generally less abundant in MDMs than CD4 cells (normalized to actin and total protein), although these modifications did not change significantly upon HIV infection. Among the histone modifications, H3K9me2 appeared depleted in MDMs relative to CD4 cells, and H3K9me3 showed a slight increase (∼1.47-fold) upon HIV infection in MDMs. We next assayed chromatin proteins that have been implicated in nuclear antiviral functions (Fig. 5B). We found that PML had a different distribution of slow-mobility isoforms in HIV-infected MDMs that were not detectable in CD4 T cells. These slower-mobility PML isoforms may reflect PML isoforms and posttranslational modifications induced by virus infections in the nucleus (46). Both DAXX and Lamin B1 were less abundant in MDMs than in CD4 T cells but were not affected by HIV infection. In contrast, the methylcytosine oxidase TET2 was downregulated in HIV-infected MDMs but not CD4 T cells. This is consistent with previous studies showing that TET2 is a target of ubiquitin-mediated degradation by HIV Vpr in macrophages (47, 48), and it suggests this effect is cell type specific. We also found that lamin A/C is expressed at much higher levels in MDMs than CD4, while the reverse occurs for lamin B1 expression. These differences may reflect the very different nuclear morphology and cell cycle properties of MDMs and CD4 T cells. HIV also induced p53 in MDMs, but not in CD4 T cells (Fig. 5C). Strikingly, we found that MDMs had near-undetectable levels of poly(ADP-ribose) polymerase (PARP1), although HIV induced total cellular levels of poly(ADP-ribose) (PAR), suggesting that other PARPs may be activated in MDMs in response to HIV infection. Taken together, these findings underscore substantial differences in nuclear protein biology in MDMs and CD4 T cells and their distinct responses to HIV infection.
FIG 5.

Western blot analysis of MDM and CD4 T cells with HIV-1 YU2 or mock infection. (A) Western blots were probed for a panel of histone modifications of H3K9me3, H3K9me2, H3K27me3, H3K4me3, H3ac, H3, H2B, and HP1γ. (B) Western blots probed for PML, Daxx, TET2, lamin B1, and lamin A/C. (C) Western blots probed for p53, PARP1, PAR, actin, and GAPDH.
HIV-induced changes in MDM nuclear organization.
MDMs form multinucleated and giant cells in response to activation signals and in response to HIV infection both in vitro and in the brain in vivo (8, 49–51). We examined the changes in MDM nuclear morphology before and after HIV infection, using immunofluorescence microscopy to detect nuclear proteins and viral p24 Gag antigen (Fig. 6). HIV infection led to a marked increase in multinucleated cells enriched with H3K4me3 (∼2.0-fold) and 5hmC (∼1.6-fold) (Fig. 6A, B, and E). We also observed an increase in punctate PML and DAXX colocalized nuclear bodies in each of the multiple nuclei in infected MDMs (Fig. 6C to E). We also observed that the 5hmC signal changed substantially upon HIV infection in MDMs (Fig. 6F). In the absence of infection, most 5hmC appeared perinuclear, while after HIV infection, 5hmC was strongly enriched in each of the nuclei of the multinucleated MDMs. These findings demonstrate that HIV infection remodels MDM nuclear morphology, induces an antiviral response increase in PML nuclear bodies, and induces a strong nuclear relocalization of 5hmC.
FIG 6.

Immunofluorescence microscopy of MDM with HIV-1 YU2 or mock infection. (A) YU2 or mock-infected MDMs stained with H3K4me3 (green) or HIV protein p24 (red) and merged with DAPI (blue). (B) 5hmC (green), 5mC (red), and merged with DAPI (blue). (C) PML (green), p24 (red), or merged with DAPI (blue). (D) PML (green), DAXX (red), and merged with DAPI (blue). The images were taken with 20× (panels A to C) or 100× (panel D) lenses. Scale bar, 10 μm. (E) Quantification of fluorescence intensity for images represented in panels A to D are provided for cells with n > 100 with distribution around mean intensity. P values were determined by two-tailed Student’s t test. ****, P < 0.0001. (F) Immunofluorescence for 5hmC in MDM mock (top panels) or YU2 infected (lower panels) with 5hmC (green) or DAPI (blue, merged). Images taken with 20× lens.
DISCUSSION
While much is known about HIV infection and latency in CD4 T cells, relatively less is known about the regulation of HIV in macrophages. Here, we examined the epigenetic features of HIV in primary human MDMs, employing a brain-derived HIV-1 primary isolate relevant to in vivo infection, and compared it with CD4+ T cells and the latently infected T cell line, JLAT 8.4. Our comparison suggests that MDMs use different mechanisms than CD4+ T cells to regulate HIV infection. We found HIV sequences in MDMs are enriched with H3K9me3 throughout the viral genome, with an atypical bivalent histone modification characterized by high H3K9me3 in combination with H3K27ac (Fig. 7). We also observed that 5′-hydroxymethylcytosine (5hmC) was enriched across the HIV genome in MDMs and CD4 cells, but not in JLAT cells, which were elevated in 5mC. These data suggest that the epigenetic regulatory features in MDMs are different than those observed in both productively infected CD4 T cells and latently infected JLAT cells and potentially distinct from previously characterized macrophage activation states.
FIG 7.
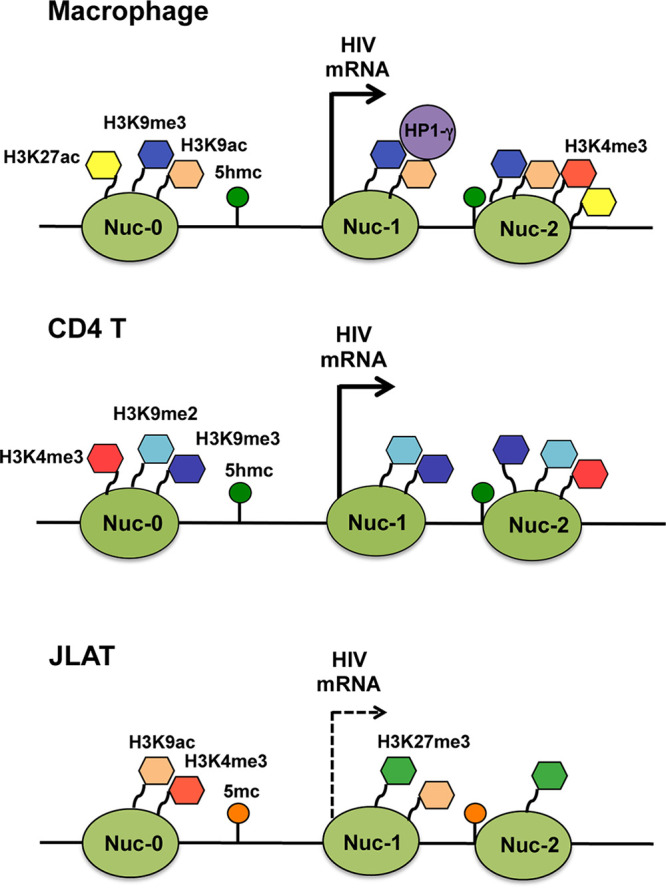
Model of HIV epigenetic regulation. Summary of histone tail modifications associated with LTR region in HIV-infected MDM, CD4 T cell, and JLAT 8.4 cells.
Our findings are similar to previous reports of H3K9me3-associated heterochromatin that was associated with silencing of HIV LTR postintegration (52, 53). However, in MDM infection where HIV is actively transcribing, the H3K9me3 does not appear to confer transcriptional silencing. In CD4 T cells, H3K9me3 formation was found to depend on the histone methyltransferase SUV39H1 or the SETDB1-associated HUSH complex (54). In microglial cells, transcription factor C/EBP alpha, corepressor CTIP2, and histone demethylase LSD1 have all been implicated in the formation of silent heterochromatin at the HIV LTR (53, 55, 56). COUP-TF and CTIP2 were found to recruit HP1alpha and H3K9me3 methylation in microglial cells (22, 57, 58). HIC1 and HMGA1 were also found to be chromatin-associated repressors of HIV transcription in microglial cells (59). Whether these same factors function to generate H3K9me3 in MDMs is unknown. Epigenetic control of HIV transcription and replication is also known to depend on the surrounding chromatin environment (60). We noted that HIV genomic regions outside the LTR, and especially within the regions encompassing the env open reading frame (ORF), have distinguishing epigenetic marks. In JLAT 8.4, this region was enriched for H3K27me3 and 5mC. JLAT 8.4 is a clonal isolate with a single HIV integration site (31) and is therefore likely to have a more homogeneous epigenetic landscape than HIV-infected primary cells. Thus, some of the epigenetic differences we observed between the MDMs, CD4, and JLAT are likely to reflect differences in integration sites, as well as the major differences in cell type and metabolic state of the infected cells.
DNA methylation is also known to contribute to HIV silencing of the 5′ LTR in JLAT and CD4+ T cells (34, 61). We found high levels of 5mC at the LTR in JLAT cells, as expected, but no significant 5mC in MDMs. On the other hand, 5hmC levels were enriched in MDMs across the entire HIV genome. To our knowledge, hydroxymethylcytosine has not yet been described in the regulation of HIV in macrophage or T cells. HIV Vpr has been shown to cause ubiquitin-mediated degradation of TET2 (47, 48), the major enzyme responsible for enzymatically converting methylcytosine to hydroxymethylcytosine in hematopoietic cells (62). Our data show that TET2 is selectively degraded in MDM infection, but not in CD4 T cells (Fig. 5). We also observed increased intensity and relocalization of 5hmC from the nuclear periphery to the nucleus in HIV-infected MDMs (Fig. 6). A similar localization of 5hmC to the nuclear periphery was observed in developing mouse retinal photoreceptor cells (63). How the HIV-dependent degradation of TET2 is balanced with the enrichment and redistribution of 5hmC on the HIV genome requires further investigation.
Total levels of histone proteins were lower in MDMs, as were most modified histones, relative to CD4+ T cells. This may reflect the postmitotic state of MDMs relative to cycling CD4+ T cells. However, the amount of H3K9me3 relative to other histone modifications appeared to be higher in MDMs. While HIV infection did not alter global levels of any histone modification, it did induce many other changes in MDM proteins. In addition to the loss of TET2, HIV induced several modifications associated with viral infection and DNA damage response, including modification of PML, induction of p53, and the generation of PAR. PARP1 has been found to form a complex with Vpr (64), but our findings suggest that PARP1 is undetectable prior to HIV infection in MDM cells. Other PARPs, such as PARP2 or tankyrase, may be responsible for PARylation in MDM cells.
Imaging studies revealed that HIV infection leads to a large increase in multinucleated giant cells, which is well described in vitro and in vivo. These form in response to macrophage activation due to direct interaction with pathogens or phagocytic substrates (65, 66). For HIV-infected cells, multinucleate giant cells may also result from Env-mediated cell-cell fusion. More recent studies suggest that macrophage multinucleation can arise from mitotic polyploidy and chromothripsis and not exclusively as a result of cellular fusions (67). Our findings are consistent with the induction of DNA damage based on the increase in phosphorylated p53 and PAR that correlates with the formation of the multinucleated macrophages after HIV infection. This is also consistent with other reports showing that HIV can activate p53 in human MDMs (hMDMs) and that p53 functions to restrict HIV replication (68).
HIV transcriptional regulation has been shown to be controlled primarily by factors that bind to the LTR and the TAR regions, including nucleosomes positioned in close proximity to the transcription start site. Recent studies suggested unintegrated HIV DNA, especially 2-LTR circles, were associated with repressive chromatin structure (69, 70) and that circular HIV DNA persists in macrophages since they are nondividing cells (71). Most of the assays in this study do not distinguish between episomal and integrated HIV provirus, but we found that the abundance of 2-LTR circles was only a fraction of that compared to total HIV genomes in infected cells. It is also known that most HIV genomes integrate into transcriptionally active loci in CD4 T cells (72). However, a recent study found that when HIV does integrate into H3K9me3-enriched chromatin regions, the provirus is transcriptionally silent and refractory to activation induced by phorbol myristate acetate (PMA)/ionomycin treatment (60). Our data suggest that epigenetic regulation of HIV genomes in postmitotic MDMs where HIV transcription is not silenced is different than that found for CD4 T cells. We found that HIV-1 genomes in MDMs were enriched in H3K9me3 but lacked DNA methylation and were transcriptionally active. Others have reported that H3K9me3 can be associated with transcriptional activation (73, 74) and mRNA elongation (75), especially when this histone modification paired with H3K9ac or H3K4me2 (76). H3K9me3 is also found coupled with H3K27ac at transposable elements that can be transcriptionally activated in some cell types and stress conditions (39). We provide evidence that HIV genomes have H3K9me3 distributed throughout the viral genome and that antisense transcription occurs that may initiate at the 3′ LTR. The detection of antisense transcription in MDMs is consistent with the model that antisense transcripts can recruit factors that generate H3K9me3 heterochromatin (34, 35). However, the elevated H3K9me3 in MDMs does not correspond to transcriptional repression, suggesting that H3K9me3 may not be sufficient to repress HIV genome transcription in macrophage. It is likely that additional repressive factors that restrict RNA polymerase II function may be not fully operational at the HIV LTR in MDM infection.
Our study has several caveats. We examined cells 10 days after infection under conditions where integration would be complete and the stable level of sustained virus production is typically established in macrophages (8), but it is possible that further epigenetic changes could occur at later time points, which will be important to assess in future studies. It is also plausible that unintegrated viral DNA may contribute to epigenetic profiles, although unintegrated 2-LTR circles were present at <10% of total HIV copy numbers, indicating it would contribute, at most, a small fraction. In the ChIP and DIP studies, transduction with Vpx was done to boost early steps of infection to levels sufficient for robust signals and data interpretation, and we cannot exclude the possibility that histone and methylation/hydroxymethylation profiles could be affected by Vpx. We studied only a single HIV-1 strain, although it is a highly relevant primary virus cloned directly from human brain. Finally, MDMs are a primary cell type and thus closer to the in vivo myeloid cells infected by HIV than transformed cell lines, but they may not necessarily reflect, in all respects, macrophages and microglia in the brain, the primary site for myeloid cell infection.
In summary, we report here using primary human monocyte-derived macrophages and a brain-derived HIV-1 primary isolate, evidence for noncanonical bivalent chromatin structures of the viral genome, which may contribute to distinct regulation of virus expression in myeloid cells.
MATERIALS AND METHODS
Cells and viral infection.
Monocytes were purified from healthy donors and cultured in Iscove’s modified Dulbecco’s medium (IMDM) containing 10% human AB serum with penicillin, streptomycin, and 1% glutamine. Monocytes were maintained in culture for 7 days to allow differentiation into monocyte-derived macrophages (MDMs) prior to HIV-1 infection. CD4 T cells were purified from healthy donors and cultured in RPMI 1640 containing 10% fetal calf serum (FCS) with penicillin, streptomycin, and 1% glutamine. T cells were stimulated with 5 μg/mL phytohemagglutinin (PHA) for 3 days prior to HIV-1 infection and then treated with 10 ng/mL interleukin-2 (IL-2). Cell purification used negative selection employing the RosetteSep platform (Stemcell Technologies) and was carried out by the Penn CFAR Immunology Core. All data represent a minimum of three independent experiments using cells from different donors.
The brain-derived HIV-1 pYU2 infectious molecular clone (IMC) with all accessory genes intact (30) was provided by B. Hahn (University of Pennsylvania), and infectious virus was generated by transfection of 293T cells. Virus was treated with DNase (to prevent inadvertent transfection of residual plasmid during infections) and quantified by HIV-1 p24 Gag antigen by enzyme-linked immunosorbent assay (ELISA). To enhance entry into CD4 T cells, the HIV-1 YU2 IMC was cotransfected with plasmid encoding vesicular stomatitis virus G protein (VSVg) to generate mixed pseudotypes, harvested, and quantified similarly (YU2/VSVg). To enhance infection of MDMs in the chromatin and DNA immunoprecipitation (ChIP and DIP, respectively) experiments, transduction with the Vpx protein of simian immunodeficiency virus (SIV) was carried out. Vpx-containing pseudotype virions were generated by cotransfecting 293T cells with plasmids encoding SIV Gag, the SIV Vpx gene, and VSVg (77) provided by J. Skowronski (Case Western Reserve University). Viral particles were harvested 3 days later and quantified by SIV Gag p27 antigen by ELISA, and 3 ng of p27 was used for transduction per 106 MDMs at the same day of HIV-1 infection. MDMs at 7 days postplating or CD4 T cells 3 days post-PHA stimulation were infected with HIV-1 YU2 using 7 ng of viral p24 Gag antigen per 106 MDM or 3.5 ng of viral p24 Gag antigen per 106 CD4 T cells. Cells were infected by spinoculation at 1,200 × g for 2 h at room temperature and then maintained in culture until they were analyzed.
For ChIP, MeDIP, and hMeDIP assays, 50 nM of the RT inhibitor efavirenz (EFV) was added to MDMs 6 days after infection to restrict further rounds of reinfection, and then cells were cultured for additional 4 days before harvest to maximize integration. For CD4 T cells, EFV was added 4 days postinfection and then cultured for an additional 4 days before harvest. For reverse transcriptase PCR (RT-PCR), EFV was added to MDM cells 7 days postinfection, and cells were then cultured for an additional 7 days before harvest; EFV was added to CD4 T cells 2 days postinfection, and then cells were cultured for an additional 2 days before harvest. For Western blot analysis, cells were harvested 7 days postinfection.
To assess the relative abundance of integrated versus unintegrated HIV-1 genomes, MDMs were infected with YU2/VSVg using 50 ng p24 Gag antigen per 2 × 106 cells, EFV was added 4 days later, and total DNA was harvested 3 days later. Total gag was quantified using primer/probe as described in reference 78, with a standard curve comprised of serial dilutions of ACH-2 cells that contain a single HIV genome per cell (79). To quantify 2-LTR circles, a primer/probe set using outward-directed primers in U5 and U3 specific for unintegrated circular viral DNA was used based on that described in reference 40 and modified to match the YU2 sequence. As a standard curve, serial dilutions were used of a plasmid constructed containing the 2-LTR junction generated from the YU2 IMC. Cellular β-globin gene copies were quantified using primers and probes as described in reference 80 with a standard curve generated from serial dilutions of peripheral blood mononuclear cells. Primer and probe sequences are shown in Table S1 in the supplemental material.
ChIP-qPCR assays.
ChIP-qPCR assays were performed as described previously (81). Quantification of precipitated DNA was determined using real-time PCR and the threshold cycle (ΔΔCT) method for relative quantitation (ABI 7900HT Fast real-time PCR system). Rabbit IgG (catalog no. 2729S; Cell Signaling), anti-H3K4me3 (catalog no. 07-473; MilliporeSigma), H3K9me2 (catalog no. C15410060; Diagenode), H3K9me3 (catalog no. C15410056; Diagenode), H3K9ac (catalog no. 07-352; MilliporeSigma), H3K27me3 (catalog no. C15410069; Diagenode), H3K27ac (catalog no. ab4729; Abcam), and pan-histone H3 (catalog no. 07-690; MilliporeSigma) were used in ChIP assays. Primers for ChIP and DIP assays are listed in Table S2.
MeDIP and hMeDIP assays.
Total genomic DNA was purified using Wizard genomic DNA purification kit (Promega; catalog no. A1120) and then subjected to methylcytosine DNA immunoprecipitation (MeDIP) or hydroxymethylcytosine DNA IP (hMeDIP) assays. The MeDIP or hMeDIP assays were performed using the MagMeDIP kit (Diagenode; catalog no. C02010021) or hMeDIP kit (Diagenode; catalog no. C02010031). Quantification of precipitated DNA was determined using real-time PCR and the ΔΔCT method for relative quantitation (ABI 7900HT Fast real-time PCR system).
Western blot assay.
Rabbit anti-H3K4me3, H3K9me2, H3K9me3, H3K27me3, and pan-histone H3 antibodies used in Western blotting were same as antibodies used in ChIP assays. Rabbit polyclonal anti-H3ac (catalog no. 06-599; MilliporeSigma), anti-H2B (catalog no. 07-371; Millipore Sigma), anti-lamin B1 (catalog no. 12586S; Cell Signaling), anti-TET2 (catalog no. 21207-1-AP; Proteintech), anti-PARP-1 (catalog no. ALX-210-302-R100; Alexis), anti-Daxx (catalog no. D7810; MilliporeSigma), anti-GAPDH (catalog no. 2118; Cell Signaling); mouse monoclonal anti-p53 (catalog no. OP43; MilliporeSigma), anti-PML (catalog no. ab96051; Abcam), anti-lamin A/C (MANLAC1; DSHB), anti-PAR (catalog no. 4335-MC-100; Trevigen), anti-HP1γ (catalog no. MAB3450; Chemicon), and actin-peroxidase antibody (catalog no. A3854; Sigma) were used.
Immunofluorescence.
On day 8 post-HIV infection, cells were washed with 1× phosphate-buffered saline (PBS) and fixed for 15 min with 2% paraformaldehyde (Electron Microscopy Sciences) in 1× PBS, then washed twice with 1× PBS, recovered with 70% ethanol, washed with 1× PBS, and permeabilized for 15 min with 0.3% Triton X-100 (Sigma) in PBS. Cells were then incubated in blocking solution (0.2% fish gelatin, 0.5% bovine serum albumin [BSA] in 1×PBS) for 30 min at room temperature. Primary antibodies were diluted in blocking solution and applied on the cells for 1 h at room temperature followed by 1× PBS washing. For 5hmC and 5mC staining, after the permeabilization and PBS washing steps, the cells were treated with 4 N HCl for 30 min at room temperature. Cells were then washed with 1× PBS three times, incubated with 5hmC or 5mC antibodies in blocking solution. Cells were further incubated with fluorescence-conjugated secondary antibodies in blocking solution for 1 h at room temperature, counterstained with DAPI (4′,6-diamidino-2-phenylindole), and mounted in Fluoromount-G medium (SouthernBiotech). Images were taken at Nikon upright microscope using 20× or 100× lens and processed with Adobe Photoshop CS6. Antibodies used in immunofluorescence include mouse anti-p24 (catalog no. ab9071; Abcam), rabbit anti-H3K4me3 (catalog no. 07-433; MilliporeSigma), rabbit anti-PML (catalog no. A301-167A; Bethyl), mouse anti-5mC (catalog no. C15200081-100; Diagenode), rabbit anti-5hmC (catalog no. 39769; Active Motif), goat anti-DAXX (catalog no. sc-167A; Santa Cruz), and Alexa Fluor 594 or Alexa Fluor 488 (Invitrogen). Original fluorescence images were captured with standardized acquisition parameters using a Nikon 80i upright microscope with ImagePro Plus software (Media Cybernetics). Fluorescence image intensity was quantified by gathering each set of images into a single multipoint, multichannel ND file using NIS-Elements AR software (Nikon Instruments) and analyzed together. For each individual image, the DAPI channel was used to define the size, shape, and location for each nucleus, and the outline was converted to a region of interest and applied to all available channels for that image. Mean intensity was then collected in each channel for each region of interest.
RNA extraction and quantitative RT-PCR.
RNA was isolated from 2 × 106 cells using RNeasy Plus minikit (Qiagen). Reverse transcription was performed with either random decamers or HIV antisense-specific primers. HIV antisense qPCR was carried out using 4 specific primer pairs and the antisense reverse transcription cDNA, while cellular β-glucuronidase (GUSB) and HIV tat and nef qPCR was done using random decamer cDNA. Real-time PCR was performed with SYBR green probe in an ABI Prism 7900 and the ΔΔCT method for relative quantitation. Primers for HIV antisense-specific reverse transcription and RT-qPCR are listed in Table S3.
ACKNOWLEDGMENTS
This work was supported by NIH grant R61/33 133696 (R.G.C., P.M.L.).
We thank J. Skowronski and B. Hahn for constructs. We acknowledge assistance from Andreas Wiedmer and the Wistar Cancer Center Core Facilities in Imaging and Flow Cytometry and the Immunology and Virology Cores of the Penn Center for AIDS Research (P30-AI045008).
Footnotes
Supplemental material is available online only.
Contributor Information
Ronald G. Collman, Email: collmanr@pennmedicine.upenn.edu.
Paul M. Lieberman, Email: Lieberman@wistar.org.
Guido Silvestri, Emory University.
REFERENCES
- 1.Pitman MC, Lau JSY, McMahon JH, Lewin SR. 2018. Barriers and strategies to achieve a cure for HIV. Lancet HIV 5:e317–e328. 10.1016/S2352-3018(18)30039-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rothenberger MK, Keele BF, Wietgrefe SW, Fletcher CV, Beilman GJ, Chipman JG, Khoruts A, Estes JD, Anderson J, Callisto SP, Schmidt TE, Thorkelson A, Reilly C, Perkey K, Reimann TG, Utay NS, Nganou Makamdop K, Stevenson M, Douek DC, Haase AT, Schacker TW. 2015. Large number of rebounding/founder HIV variants emerge from multifocal infection in lymphatic tissues after treatment interruption. Proc Natl Acad Sci USA 112:E1126-34. 10.1073/pnas.1414926112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lamers SL, Rose R, Maidji E, Agsalda-Garcia M, Nolan DJ, Fogel GB, Salemi M, Garcia DL, Bracci P, Yong W, Commins D, Said J, Khanlou N, Hinkin CH, Sueiras MV, Mathisen G, Donovan S, Shiramizu B, Stoddart CA, McGrath MS, Singer EJ. 2016. HIV DNA Is frequently present within pathologic tissues evaluated at autopsy from combined antiretroviral therapy-treated patients with undetectable viral loads. J Virol 90:8968–8983. 10.1128/JVI.00674-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eisele E, Siliciano RF. 2012. Redefining the viral reservoirs that prevent HIV-1 eradication. Immunity 37:377–388. 10.1016/j.immuni.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kruize Z, Kootstra NA. 2019. The role of macrophages in HIV-1 persistence and pathogenesis. Front Microbiol 10:2828. 10.3389/fmicb.2019.02828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wallet C, De Rovere M, Van Assche J, Daouad F, De Wit S, Gautier V, Mallon PWG, Marcello A, Van Lint C, Rohr O, Schwartz C. 2019. Microglial cells: the main HIV-1 reservoir in the brain. Front Cell Infect Microbiol 9:362. 10.3389/fcimb.2019.00362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Richman DD, Margolis DM, Delaney M, Greene WC, Hazuda D, Pomerantz RJ. 2009. The challenge of finding a cure for HIV infection. Science 323:1304–1307. 10.1126/science.1165706. [DOI] [PubMed] [Google Scholar]
- 8.Collman R, Hassan NF, Walker R, Godfrey B, Cutilli J, Hastings JC, Friedman H, Douglas SD, Nathanson N. 1989. Infection of monocyte-derived macrophages with human immunodeficiency virus type 1 (HIV-1). Monocyte-tropic and lymphocyte-tropic strains of HIV-1 show distinctive patterns of replication in a panel of cell types. J Exp Med 170:1149–1163. 10.1084/jem.170.4.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rich EA, Chen IS, Zack JA, Leonard ML, O'Brien WA. 1992. Increased susceptibility of differentiated mononuclear phagocytes to productive infection with human immunodeficiency virus-1 (HIV-1). J Clin Invest 89:176–183. 10.1172/JCI115559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weinberg JB, Matthews TJ, Cullen BR, Malim MH. 1991. Productive human immunodeficiency virus type 1 (HIV-1) infection of nonproliferating human monocytes. J Exp Med 174:1477–1482. 10.1084/jem.174.6.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stevenson M, Brichacek B, Heinzinger N, Swindells S, Pirruccello S, Janoff E, Emerman M. 1995. Molecular basis of cell cycle dependent HIV-1 replication. Implications for control of virus burden. Adv Exp Med Biol 374:33–45. 10.1007/978-1-4615-1995-9_4. [DOI] [PubMed] [Google Scholar]
- 12.Agosto LM, Yu JJ, Dai J, Kaletsky R, Monie D, O'Doherty U. 2007. HIV-1 integrates into resting CD4+ T cells even at low inoculums as demonstrated with an improved assay for HIV-1 integration. Virology 368:60–72. 10.1016/j.virol.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dai J, Agosto LM, Baytop C, Yu JJ, Pace MJ, Liszewski MK, O'Doherty U. 2009. Human immunodeficiency virus integrates directly into naive resting CD4+ T cells but enters naive cells less efficiently than memory cells. J Virol 83:4528–4537. 10.1128/JVI.01910-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pace MJ, Graf EH, Agosto LM, Mexas AM, Male F, Brady T, Bushman FD, O'Doherty U. 2012. Directly infected resting CD4+T cells can produce HIV Gag without spreading infection in a model of HIV latency. PLoS Pathog 8:e1002818. 10.1371/journal.ppat.1002818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gendelman HE, Orenstein JM, Baca LM, Weiser B, Burger H, Kalter DC, Meltzer MS. 1989. The macrophage in the persistence and pathogenesis of HIV infection. AIDS 3:475–495. 10.1097/00002030-198908000-00001. [DOI] [PubMed] [Google Scholar]
- 16.Sattentau QJ, Stevenson M. 2016. Macrophages and HIV-1: an unhealthy constellation. Cell Host Microbe 19:304–310. 10.1016/j.chom.2016.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abbas W, Tariq M, Iqbal M, Kumar A, Herbein G. 2015. Eradication of HIV-1 from the macrophage reservoir: an uncertain goal? Viruses 7:1578–1598. 10.3390/v7041578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Joseph SB, Arrildt KT, Sturdevant CB, Swanstrom R. 2015. HIV-1 target cells in the CNS. J Neurovirol 21:276–289. 10.1007/s13365-014-0287-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Churchill M, Nath A. 2013. Where does HIV hide? A focus on the central nervous system. Curr Opin HIV AIDS 8:165–169. 10.1097/COH.0b013e32835fc601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen MF, Gill AJ, Kolson DL. 2014. Neuropathogenesis of HIV-associated neurocognitive disorders: roles for immune activation, HIV blipping and viral tropism. Curr Opin HIV AIDS 9:559–564. 10.1097/COH.0000000000000105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumar A, Abbas W, Herbein G. 2014. HIV-1 latency in monocytes/macrophages. Viruses 6:1837–1860. 10.3390/v6041837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Redel L, Le Douce V, Cherrier T, Marban C, Janossy A, Aunis D, Van Lint C, Rohr O, Schwartz C. 2010. HIV-1 regulation of latency in the monocyte-macrophage lineage and in CD4+ T lymphocytes. J Leukoc Biol 87:575–588. 10.1189/jlb.0409264. [DOI] [PubMed] [Google Scholar]
- 23.Siliciano RF, Greene WC. 2011. HIV latency. Cold Spring Harb Perspect Med 1:a007096. 10.1101/cshperspect.a007096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Williams DW, Veenstra M, Gaskill PJ, Morgello S, Calderon TM, Berman JW. 2014. Monocytes mediate HIV neuropathogenesis: mechanisms that contribute to HIV associated neurocognitive disorders. Curr HIV Res 12:85–96. 10.2174/1570162x12666140526114526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yadav A, Collman RG. 2009. CNS inflammation and macrophage/microglial biology associated with HIV-1 infection. J Neuroimmune Pharmacol 4:430–447. 10.1007/s11481-009-9174-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mbonye U, Karn J. 2017. The molecular basis for human immunodeficiency virus latency. Annu Rev Virol 4:261–285. 10.1146/annurev-virology-101416-041646. [DOI] [PubMed] [Google Scholar]
- 27.Boehm D, Ott M. 2017. Host methyltransferases and demethylases: potential new epigenetic targets for HIV cure strategies and beyond. AIDS Res Hum Retroviruses 33:S8–S22. 10.1089/aid.2017.0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khoury G, Darcis G, Lee MY, Bouchat S, Van Driessche B, Purcell DFJ, Van Lint C. 2018. The molecular biology of HIV latency. Adv Exp Med Biol 1075:187–212. 10.1007/978-981-13-0484-2_8. [DOI] [PubMed] [Google Scholar]
- 29.Mbonye U, Karn J. 2014. Transcriptional control of HIV latency: cellular signaling pathways, epigenetics, happenstance and the hope for a cure. Virology 454–455:328–339. 10.1016/j.virol.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Y, Kappes JC, Conway JA, Price RW, Shaw GM, Hahn BH. 1991. Molecular characterization of human immunodeficiency virus type 1 cloned directly from uncultured human brain tissue: identification of replication-competent and -defective viral genomes. J Virol 65:3973–3985. 10.1128/JVI.65.8.3973-3985.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jordan A, Bisgrove D, Verdin E. 2003. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J 22:1868–1877. 10.1093/emboj/cdg188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rafati H, Parra M, Hakre S, Moshkin Y, Verdin E, Mahmoudi T. 2011. Repressive LTR nucleosome positioning by the BAF complex is required for HIV latency. PLoS Biol 9:e1001206. 10.1371/journal.pbio.1001206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Verdin E, Paras P, Jr., Van Lint C. 1993. Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J 12:3249–3259. 10.1002/j.1460-2075.1993.tb05994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kauder SE, Bosque A, Lindqvist A, Planelles V, Verdin E. 2009. Epigenetic regulation of HIV-1 latency by cytosine methylation. PLoS Pathog 5:e1000495. 10.1371/journal.ppat.1000495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Williams SA, Chen LF, Kwon H, Ruiz-Jarabo CM, Verdin E, Greene WC. 2006. NF-kappaB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. EMBO J 25:139–149. 10.1038/sj.emboj.7600900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Turner AW, Margolis DM. 2017. Chromatin regulation and the histone code in HIV latency. Yale J Biol Med 90:229–243. [PMC free article] [PubMed] [Google Scholar]
- 37.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, Jaenisch R, Wagschal A, Feil R, Schreiber SL, Lander ES. 2006. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125:315–326. 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 38.Matsumura Y, Nakaki R, Inagaki T, Yoshida A, Kano Y, Kimura H, Tanaka T, Tsutsumi S, Nakao M, Doi T, Fukami K, Osborne TF, Kodama T, Aburatani H, Sakai J. 2015. H3K4/H3K9me3 bivalent chromatin domains targeted by lineage-specific DNA methylation pauses adipocyte differentiation. Mol Cell 60:584–596. 10.1016/j.molcel.2015.10.025. [DOI] [PubMed] [Google Scholar]
- 39.He J, Fu X, Zhang M, He F, Li W, Abdul MM, Zhou J, Sun L, Chang C, Li Y, Liu H, Wu K, Babarinde IA, Zhuang Q, Loh YH, Chen J, Esteban MA, Hutchins AP. 2019. Transposable elements are regulated by context-specific patterns of chromatin marks in mouse embryonic stem cells. Nat Commun 10:34. 10.1038/s41467-018-08006-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Butler SL, Hansen MS, Bushman FD. 2001. A quantitative assay for HIV DNA integration in vivo. Nat Med 7:631–634. 10.1038/87979. [DOI] [PubMed] [Google Scholar]
- 41.Gillim-Ross L, Cara A, Klotman ME. 2005. HIV-1 extrachromosomal 2-LTR circular DNA is long-lived in human macrophages. Viral Immunol 18:190–196. 10.1089/vim.2005.18.190. [DOI] [PubMed] [Google Scholar]
- 42.Ninova M, Fejes TK, Aravin AA. 2019. The control of gene expression and cell identity by H3K9 trimethylation. Development 146:dev181180. 10.1242/dev.181180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kobayashi-Ishihara M, Terahara K, Martinez JP, Yamagishi M, Iwabuchi R, Brander C, Ato M, Watanabe T, Meyerhans A, Tsunetsugu-Yokota Y. 2018. HIV LTR-driven antisense RNA by itself has regulatory function and may curtail virus reactivation from latency. Front Microbiol 9:1066. 10.3389/fmicb.2018.01066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saayman S, Ackley A, Turner AW, Famiglietti M, Bosque A, Clemson M, Planelles V, Morris KV. 2014. An HIV-encoded antisense long noncoding RNA epigenetically regulates viral transcription. Mol Ther 22:1164–1175. 10.1038/mt.2014.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Groh S, Schotta G. 2017. Silencing of endogenous retroviruses by heterochromatin. Cell Mol Life Sci 74:2055–2065. 10.1007/s00018-017-2454-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tavalai N, Stamminger T. 2009. Interplay between herpesvirus infection and host defense by PML nuclear bodies. Viruses 1:1240–1264. 10.3390/v1031240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Q, Su L. 2019. Vpr enhances HIV-1 Env processing and virion infectivity in macrophages by modulating TET2-dependent IFITM3 expression. mBio 10:e01344-19. 10.1128/mBio.01344-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lv L, Wang Q, Xu Y, Tsao LC, Nakagawa T, Guo H, Su L, Xiong Y. 2018. Vpr targets TET2 for degradation by CRL4(VprBP) E3 ligase to sustain IL-6 expression and enhance HIV-1 replication. Mol Cell 70:961–970.e5. 10.1016/j.molcel.2018.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Herrtwich L, Nanda I, Evangelou K, Nikolova T, Horn V, Sagar Erny D, Stefanowski J, Rogell L, Klein C, Gharun K, Follo M, Seidl M, Kremer B, Munke N, Senges J, Fliegauf M, Aschman T, Pfeifer D, Sarrazin S, Sieweke MH, Wagner D, Dierks C, Haaf T, Ness T, Zaiss MM, Voll RE, Deshmukh SD, Prinz M, Goldmann T, Holscher C, Hauser AE, Lopez-Contreras AJ, Grun D, Gorgoulis V, Diefenbach A, Henneke P, Triantafyllopoulou A. 2016. DNA damage signaling instructs polyploid macrophage fate in granulomas. Cell 167:1264–1280.e18. 10.1016/j.cell.2016.09.054. [DOI] [PubMed] [Google Scholar]
- 50.Koenig S, Gendelman HE, Orenstein JM, Dal Canto MC, Pezeshkpour GH, Yungbluth M, Janotta F, Aksamit A, Martin MA, Fauci AS. 1986. Detection of AIDS virus in macrophages in brain tissue from AIDS patients with encephalopathy. Science 233:1089–1093. 10.1126/science.3016903. [DOI] [PubMed] [Google Scholar]
- 51.Budka H. 1986. Multinucleated giant cells in brain: a hallmark of the acquired immune deficiency syndrome (AIDS). Acta Neuropathol 69:253–258. 10.1007/BF00688301. [DOI] [PubMed] [Google Scholar]
- 52.Marban C, Suzanne S, Dequiedt F, de Walque S, Redel L, Van Lint C, Aunis D, Rohr O. 2007. Recruitment of chromatin-modifying enzymes by CTIP2 promotes HIV-1 transcriptional silencing. EMBO J 26:412–423. 10.1038/sj.emboj.7601516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Du Chene I, Basyuk E, Lin YL, Triboulet R, Knezevich A, Chable-Bessia C, Mettling C, Baillat V, Reynes J, Corbeau P, Bertrand E, Marcello A, Emiliani S, Kiernan R, Benkirane M. 2007. Suv39H1 and HP1gamma are responsible for chromatin-mediated HIV-1 transcriptional silencing and post-integration latency. EMBO J 26:424–435. 10.1038/sj.emboj.7601517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tchasovnikarova IA, Timms RT, Matheson NJ, Wals K, Antrobus R, Gottgens B, Dougan G, Dawson MA, Lehner PJ. 2015. Epigenetic silencing by the HUSH complex mediates position-effect variegation in human cells. Science 348:1481–1485. 10.1126/science.aaa7227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cherrier T, Le Douce V, Eilebrecht S, Riclet R, Marban C, Dequiedt F, Goumon Y, Paillart JC, Mericskay M, Parlakian A, Bausero P, Abbas W, Herbein G, Kurdistani SK, Grana X, Van Driessche B, Schwartz C, Candolfi E, Benecke AG, Van Lint C, Rohr O. 2013. CTIP2 is a negative regulator of P-TEFb. Proc Natl Acad Sci USA 110:12655–12660. 10.1073/pnas.1220136110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Le Douce V, Colin L, Redel L, Cherrier T, Herbein G, Aunis D, Rohr O, Van Lint C, Schwartz C. 2012. LSD1 cooperates with CTIP2 to promote HIV-1 transcriptional silencing. Nucleic Acids Res 40:1904–1915. 10.1093/nar/gkr857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rohr O, Marban C, Aunis D, Schaeffer E. 2003. Regulation of HIV-1 gene transcription: from lymphocytes to microglial cells. J Leukoc Biol 74:736–749. 10.1189/jlb.0403180. [DOI] [PubMed] [Google Scholar]
- 58.Cherrier T, Suzanne S, Redel L, Calao M, Marban C, Samah B, Mukerjee R, Schwartz C, Gras G, Sawaya BE, Zeichner SL, Aunis D, Van Lint C, Rohr O. 2009. p21(WAF1) gene promoter is epigenetically silenced by CTIP2 and SUV39H1. Oncogene 28:3380–3389. 10.1038/onc.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Le Douce V, Forouzanfar F, Eilebrecht S, Van Driessche B, Ait-Ammar A, Verdikt R, Kurashige Y, Marban C, Gautier V, Candolfi E, Benecke AG, Van Lint C, Rohr O, Schwartz C. 2016. HIC1 controls cellular- and HIV-1- gene transcription via interactions with CTIP2 and HMGA1. Sci Rep 6:34920. 10.1038/srep34920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Einkauf KB, Osborn MR, Gao C, Sun W, Sun X, Lian X, Parsons EM, Gladkov GT, Seiger KW, Blackmer JE, Jiang C, Yukl SA, Rosenberg ES, Yu XG, Lichterfeld M. 2022. Parallel analysis of transcription, integration, and sequence of single HIV-1 proviruses. Cell 185:266–282.e15. 10.1016/j.cell.2021.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Blazkova J, Trejbalova K, Gondois-Rey F, Halfon P, Philibert P, Guiguen A, Verdin E, Olive D, Van Lint C, Hejnar J, Hirsch I. 2009. CpG methylation controls reactivation of HIV from latency. PLoS Pathog 5:e1000554. 10.1371/journal.ppat.1000554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Meldi KM, Figueroa ME. 2015. Cytosine modifications in myeloid malignancies. Pharmacol Ther 152:42–53. 10.1016/j.pharmthera.2015.05.002. [DOI] [PubMed] [Google Scholar]
- 63.Singh RK, Diaz PE, Binette F, Nasonkin IO. 2018. Immunohistochemical detection of 5-methylcytosine and 5-hydroxymethylcytosine in developing and postmitotic mouse retina. J Vis Exp 58274. 10.3791/58274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Muthumani K, Choo AY, Zong WX, Madesh M, Hwang DS, Premkumar A, Thieu KP, Emmanuel J, Kumar S, Thompson CB, Weiner DB. 2006. The HIV-1 Vpr and glucocorticoid receptor complex is a gain-of-function interaction that prevents the nuclear localization of PARP-1. Nat Cell Biol 8:170–179. 10.1038/ncb1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brooks PJ, Glogauer M, McCulloch CA. 2019. An overview of the derivation and function of multinucleated giant cells and their role in pathologic processes. Am J Pathol 189:1145–1158. 10.1016/j.ajpath.2019.02.006. [DOI] [PubMed] [Google Scholar]
- 66.McNally AK, Anderson JM. 2011. Macrophage fusion and multinucleated giant cells of inflammation. Adv Exp Med Biol 713:97–111. 10.1007/978-94-007-0763-4_7. [DOI] [PubMed] [Google Scholar]
- 67.Herrtwich L, Nanda I, Evangelou K, Nikolova T, Horn V, Sagar Erny D, Stefanowski J, Rogell L, Klein C, Gharun K, Follo M, Seidl M, Kremer B, Munke N, Senges J, Fliegauf M, Aschman T, Pfeifer D, Sarrazin S, Sieweke MH, Wagner D, Dierks C, Haaf T, Ness T, Zaiss MM, Voll RE, Deshmukh SD, Prinz M, Goldmann T, Holscher C, Hauser AE, Lopez-Contreras AJ, Grun D, Gorgoulis V, Diefenbach A, Henneke P, Triantafyllopoulou A. 2018. DNA damage signaling instructs polyploid macrophage fate in granulomas. Cell 174:1325–1326. 10.1016/j.cell.2018.08.015. [DOI] [PubMed] [Google Scholar]
- 68.Shi B, Sharifi HJ, DiGrigoli S, Kinnetz M, Mellon K, Hu W, de Noronha CMC. 2018. Inhibition of HIV early replication by the p53 and its downstream gene p21. Virol J 15:53. 10.1186/s12985-018-0959-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Geis FK, Goff SP. 2019. Unintegrated HIV-1 DNAs are loaded with core and linker histones and transcriptionally silenced. Proc Natl Acad Sci USA 116:23735–23742. 10.1073/pnas.1912638116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Machida S, Depierre D, Chen HC, Thenin-Houssier S, Petitjean G, Doyen CM, Takaku M, Cuvier O, Benkirane M. 2020. Exploring histone loading on HIV DNA reveals a dynamic nucleosome positioning between unintegrated and integrated viral genome. Proc Natl Acad Sci USA 117:6822–6830. 10.1073/pnas.1913754117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hamid FB, Kim J, Shin CG. 2017. Distribution and fate of HIV-1 unintegrated DNA species: a comprehensive update. AIDS Res Ther 14:9. 10.1186/s12981-016-0127-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang GP, Ciuffi A, Leipzig J, Berry CC, Bushman FD. 2007. HIV integration site selection: analysis by massively parallel pyrosequencing reveals association with epigenetic modifications. Genome Res 17:1186–1194. 10.1101/gr.6286907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brinkman AB, Roelofsen T, Pennings SW, Martens JH, Jenuwein T, Stunnenberg HG. 2006. Histone modification patterns associated with the human X chromosome. EMBO Rep 7:628–634. 10.1038/sj.embor.7400686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim A, Kiefer CM, Dean A. 2007. Distinctive signatures of histone methylation in transcribed coding and noncoding human beta-globin sequences. Mol Cell Biol 27:1271–1279. 10.1128/MCB.01684-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vakoc CR, Mandat SA, Olenchock BA, Blobel GA. 2005. Histone H3 lysine 9 methylation and HP1gamma are associated with transcription elongation through mammalian chromatin. Mol Cell 19:381–391. 10.1016/j.molcel.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 76.Wiencke JK, Zheng S, Morrison Z, Yeh RF. 2008. Differentially expressed genes are marked by histone 3 lysine 9 trimethylation in human cancer cells. Oncogene 27:2412–2421. 10.1038/sj.onc.1210895. [DOI] [PubMed] [Google Scholar]
- 77.Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S, Florens L, Washburn MP, Skowronski J. 2011. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 474:658–661. 10.1038/nature10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Palmer S, Wiegand AP, Maldarelli F, Bazmi H, Mican JM, Polis M, Dewar RL, Planta A, Liu S, Metcalf JA, Mellors JW, Coffin JM. 2003. New real-time reverse transcriptase-initiated PCR assay with single-copy sensitivity for human immunodeficiency virus type 1 RNA in plasma. J Clin Microbiol 41:4531–4536. 10.1128/JCM.41.10.4531-4536.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hermankova M, Siliciano JD, Zhou Y, Monie D, Chadwick K, Margolick JB, Quinn TC, Siliciano RF. 2003. Analysis of human immunodeficiency virus type 1 gene expression in latently infected resting CD4+ T lymphocytes in vivo. J Virol 77:7383–7392. 10.1128/JVI.77.13.7383-7392.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.O'Doherty U, Swiggard WJ, Malim MH. 2000. Human immunodeficiency virus type 1 spinoculation enhances infection through virus binding. J Virol 74:10074–10080. 10.1128/jvi.74.21.10074-10080.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chau CM, Lieberman PM. 2004. Dynamic chromatin boundaries delineate a latency control region of Epstein-Barr virus. J Virol 78:12308–12319. 10.1128/JVI.78.22.12308-12319.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 and Tables S1 to S3. Download jvi.00162-22-s0001.pdf, PDF file, 0.2 MB (218.5KB, pdf)