

Abstract
The genome is hierarchically organized into multiple three-dimensional (3D) architectures including chromatin loops, domains, compartments, and regions associated with nuclear lamina and nucleoli. Changes in these architectures have been associated with normal development, aging, and a wide range of diseases. Despite its critical importance, understanding how the genome is spatially organized in single cells, how organization varies in different cell types in mammalian tissue, and how organization affects gene expression remains a major challenge. Previous approaches are limited by a lack of capacity to directly trace chromatin folding in 3D, and to simultaneously measure genomic organization in relation to other nuclear components and gene expression in the same, single cells. We developed an image-based 3D genomics technique termed chromatin tracing, which enables direct 3D tracing of chromatin folding along individual chromosomes in single cells. More recently, we also developed Multiplexed Imaging of Nucleome Architectures (MINA), which enables simultaneous measurements of multiscale chromatin folding, associations of genomic regions with nuclear lamina and nucleoli, and copy numbers of numerous RNA species in the same, single cells in mammalian tissue. Here we provide detailed protocols for chromatin tracing in cell lines and MINA in mammalian tissue, which take 3–4 days for experimental work and 2–3 days for data analysis. We expect these developments to be broadly applicable and to impact many lines of research on 3D genomics by depicting multiscale genomic architectures associated with gene expression, in different types of cells and tissue undergoing different biological processes.
Introduction
The mammalian genome is hierarchically organized into loops, domains, compartments and chromosome territories in the nucleus during the interphase of the cell cycle1–7. This three-dimensional (3D) genome organization affects fundamental genomic processes including transcriptional regulation, DNA replication, chromosomal translocation, somatic hypermutation, DNA damage, repair, and recombination8–13. Despite its critical importance, a precise map of the 3D genome organization in single cells is missing, due to technical limitations. The widely adopted high-throughput chromosome conformation capture techniques, including Hi-C14, measure contact frequencies between pairs of genomic loci to infer the genome organization. However, these sequencing-based methods do not directly reveal the chromatin folding trajectory in 3D, but depend on maximum likelihood computation to estimate the 3D folding, and often rely on population averaging to achieve high genomic resolution15,16. To overcome these challenges and directly trace 3D chromatin folding in single cells, we developed an imaging approach based on multiplexed sequential fluorescence in situ hybridization (FISH)17. This approach, termed chromatin tracing, sequentially images and pinpoints numerous genomic positions in 3D to reconstruct and super-resolve chromatin folding traces of individual chromosomes in single cells17. With chromatin tracing, we and others have visualized chromatin folding across a variety of length scales, from the A-B compartmentalization of individual chromosomes, to topologically associating domains (TADs) and TAD-like structures, and to chromatin loops such as promoter-enhancer loops17–25. Chromatin tracing has also been combined with immunofluorescence staining to reveal distinct chromatin folding in different epigenetic states, such as the folding organization of activated and inactivated X chromosomes in female human cells17,26–28.
The 3D genome architecture is known to be associated with gene expression and with other nuclear structures including nuclear lamina and nucleoli9,29–31. However, these features were profiled by different sequencing techniques, and there is no technology that can combine these parameters into a single method that can be applied within the same cell. To quantify the spatial expression patterns of hundreds to thousands of RNA species in single cells, we previously co-developed a multiplexed error-robust fluorescence in situ hybridization (MERFISH) method that uses combinatorial barcoding and sequential imaging to achieve highly multiplexed RNA imaging18,32–34. To study the single-cell multi-omic correlations among chromatin folding, gene expression, and the associations of chromatin regions with nuclear landmarks such as nuclear lamina and nucleoli, we integrated chromatin tracing, MERFISH, and multiplexed protein imaging into a single technology termed Multiplexed Imaging of Nucleome Architectures (MINA)18. Using MINA, we jointly profiled multiscale chromatin folding, expression patterns of numerous RNA species, and spatial proximities of numerous genomic regions to nucleoli and nuclear lamina in the same single cells in mammalian tissue sections. Compared with concurrently developed approaches including Hi-M and ORCA that studied fine-scale chromatin folding and single or multiple (up to 29) RNA species in Drosophila embryos22,23, the MINA method has the following improvements. First, it offers two additional layers of nucleomic information – the genomic profiles of lamina and nucleolar associations – in mammalian tissue. Second, it expands the scale of gene expression profiling from tens to hundreds of RNA species. Third, it extends the range of genomic length scales probed in the same chromatin tracing experiment to over four orders of magnitude. MINA can uniquely reveal multiple facets of nucleomic architectures and their associations with gene expression in different cell types and cell states in complex mammalian tissue.
In summary, chromatin tracing is an image-based 3D genomic method for tracing chromatin folding of individual chromosomes in single cells. MINA enables simultaneous measurements of multiscale chromatin folding, associations of genomic regions with nuclear lamina and nucleoli, and copy numbers of numerous RNA species in the same, single cells in mammalian tissue. Here we present detailed experimental setup and data analysis pipelines of the chromatin tracing and MINA techniques.
Development of the method
The chromatin tracing technique and MINA use a microarray-based DNA oligo pool design and synthesis strategy35–37. First, a template oligo pool is bioinformatically designed with our home-built ProbeDealer software38, and commercially synthesized. Primary probes are then synthesized from the template oligo pool via limited-cycle PCR, in vitro transcription, reverse transcription, alkaline hydrolysis and oligo purification17,18,32. To allow multiplexed imaging, each primary probe contains a primary targeting region that targets a genomic locus or RNA species, and one or more overhang secondary readout regions that allow binding of dye-labeled secondary DNA oligo probes. Primary probes targeting the same genomic/RNA target share the same secondary readout sequences. For the combinatorial barcoding of MERFISH in MINA implementations, we use a previously described Modified Hamming Distance 4 (MHD4) code for MERFISH probe design32. Primary probes for each RNA species contain a unique combination of 4 out of 16 readout sequences, forming a unique combinatorial 16-bit binary barcode for each RNA species with 4 bits being “1”s and 12 bits being “0”s. For chromatin tracing at the TAD-to-chromosome length scale, we typically design 1000 template oligos unique to the central 100-kilobase (kb) of each TAD. For finer-scale chromatin tracing, we typically design 150 template oligos targeting each 5-kb region. For MERFISH, we typically design 48 template oligos for each RNA species. After probe design and synthesis, all primary probes are hybridized to the genomic/RNA targets. Dye-labeled secondary probes are then sequentially introduced and hybridized to the secondary readout regions on the primary probes, producing a distinctively bright signal that indicates the target location. We repeatedly take 3D epifluorescence images via z-stepping, extinguish the fluorescence via photobleaching, and then introduce new secondary probes to image the next targets. After image acquisition, we map the 3D positions of targets, link them into chromatin traces, or decode them as different RNA species.
In the MINA implementation, to distinguish individual cells in tissue sections, we label the cell boundary with oligo-conjugated wheat germ agglutinin (WGA). We then perform immunofluorescence staining with anti-fibrillarin primary antibody and Alexa Fluor 647-conjugated secondary antibody to visualize the nucleoli. After post-fixation of the antibodies, we hybridize primary probes targeting chromatin regions and RNA species simultaneously in the same tissue section. We then repeat the process of secondary probe hybridization, image acquisition and photobleaching until all targets have been profiled. Finally, to approximate the nuclear lamina positions, we label the nuclei with DAPI stain. With the cell boundary labeling, we quantify the copy number of each RNA species in individual cells, and perform single-cell RNA profile clustering to identify the different cell types in the tissue. Meanwhile, we reconstruct chromatin traces, nuclear lamina, and nucleolar positions within single cells, which together with the RNA analyses allows us to analyze cell-type-specific nucleome architectures and their joint variations with gene expression in situ.
Applications of the methods
Our first demonstration of the chromatin tracing technique visualized the 3D chromatin folding of human chromosomes in cell culture17. It revealed a series of spatial folding principles of single chromosomes at the TAD-to-chromosome length scale, such as the polarized organization of A and B compartments and unique folding configurations of active and inactive X chromosomes17. Next, to study enhancer-promoter looping in transcription regulation, we recently increased the genomic resolution of chromatin tracing to the promoter-enhancer length scale in our MINA implementation18, and demonstrated chromatin tracing of the cis-regulatory region of gene Scd2 in single cells of mouse fetal liver tissue sections. We also simultaneously traced the entire mouse chromosome 19 (mChr19) at the TAD-to-chromosome length scale to demonstrate multi-scale chromatin tracing in the same tissue18.
In the MINA implementation, we further used MERFISH to profile the RNA copy numbers of cell type marker genes and genes from the traced chromatin regions18. We showed that different cell types identified by MERFISH have distinct chromatin folding at both the TAD-to-chromosome length scale and the promoter-enhancer length scale, and the folding changes are associated with gene expression changes from the same regions18. In addition, lamina and nucleolar association profiles of the TADs were analyzed in the same cells, and were shown to differ among cell types and to only partially correlate with chromatin folding patterns18. We showed that both intra-chromosomal self-associating interactions (e.g. through phase separation) and extra-chromosomal interactions (e.g. with nucleoli and nuclear lamina) are necessary to maintain the correct chromatin organization18.
We expect the chromatin tracing and MINA methods to be broadly applicable to study how 3D genome/nucleome organization regulates development, aging and diseases across virtually all biomedical fields and in a variety of organisms. Indeed, Sawh et al. have adapted TAD-to-chromosome scale and sub-TAD scale chromatin tracing to C. elegans embryos20. Mateo et al. and Cardozo Gizzi et al. have studied 2-kb to 20-kb resolution chromatin organizations in Drosophila embryos22,23. Our MINA method further profiled chromatin tracing at both TAD-to chromosome and 5-kb length scales in mammalian tissue18. We expect the protocols to facilitate future research on 3D genome/nucleome organization and gene expression regulation.
Limitations
One potential limitation of our chromatin tracing and MINA approaches is that the non-specific binding of FISH probes and autofluorescence in some mammalian tissue types may overwhelm the true FISH signals. For example, it is typically challenging to perform single molecule FISH in fresh frozen mouse pancreas tissues39. Previous reports suggest that non-specific binding of FISH probes to proteins can be a major source of background signals40. Additional development such as the integration of tissue clearing40 may be required to adapt our techniques to certain tissue types.
Another limitation is the high up-front cost required to achieve high genome coverage. Each chromatin tracing or MINA experiment typically maps tens of genomic loci. For example, we mapped 69 genomic loci along mouse chromosome 1918. The major limiting factor of genome coverage is the cost of oligos. For instance, it costs several thousand US dollars to purchase the template oligo pool used in the experiment above, although the same oligo pool can be repeatedly used in primary probe syntheses. As the oligo pool cost is largely proportional to the genome coverage, the cost is high for a chromatin tracing experiment targeting the entire mouse or human 3D genome.
A third limitation is the genomic resolution. DNA FISH targets are usually above several kb in genomic length. It is challenging to detect shorter targets with sufficient detection efficiency. Our chromatin tracing approach typically achieves above 80% detection efficiency at the TAD-to-chromosome length scale, whereas 5-kb resolution chromatin tracing has a detection efficiency of around 50% in mouse fetal liver. To enhance the detection efficiency, signal amplification approaches such as branched amplification41 may be combined with chromatin tracing in future developments.
Comparison with other methods
To study 3D genome organizations in single cells, several single-cell versions of the Hi-C (or Hi-C-like) technique have been developed42–44, including the most recent Dip-C technique44. Dip-C can obtain millions of chromatin contacts in single cells and reconstruct diploid human genome structures at 20-kb resolution44. In comparison to our image-based techniques, these sequencing-based methods are advantageous in terms of the higher genomic coverage. However, chromatin tracing and MINA have several complementary advantages: First, chromatin tracing and MINA preserve the real 3D positional information of targeted genomic loci and cell locations, whereas Hi-C studies need to infer chromatin folding patterns from contact events, and cell positions are lost during the sequencing procedure. Second, our image-based techniques can be applied to tissue sections to study 3D genome structures in situ, which has not been achieved with single-cell Hi-C techniques so far. Third, chromatin tracing can be combined with image-based RNA expression profiling and protein imaging in single cells, as demonstrated by our MINA development18. As it is challenging and costly to do Hi-C and RNA-seq in the same single cells45,46, MINA can be a useful and preferable alternative for analyzing co-variations of chromatin structure and gene expression across different cell types and cell states. Fourth, the co-imaging of nuclear landmarks in MINA allows us to profile nuclear lamina and nucleolar associations together with chromatin folding in single cells, which no single-cell sequencing technique has achieved so far. Fifth, our image-based technique has a demonstrated throughput of thousands of cells per experiment (from one or two days of imaging)18, significantly higher than the cell number throughput of high-resolution single-cell Hi-C techniques44.
In comparison to other concurrently developed, combined chromatin tracing and RNA imaging approaches including ORCA and Hi-M22,23, MINA has several differences. First, although all three implementations perform chromatin tracing, MINA simultaneously profiles nucleolar and lamina associations in addition to chromatin folding, enabling the analyses of spatial proximities between chromatin regions to nuclear landmarks. In other words, MINA expands the analysis from chromatin folding to more “nucleomic” (whole nucleome) architectures. Second, both ORCA and Hi-M focus on fine-scale chromatin tracing at 2-kb to 20-kb genomic resolutions, whereas MINA achieves multiscale chromatin tracing in the same sample across four orders of magnitude of genomic length, ranging from 5-kb to tens of megabases. Third, while ORCA and Hi-M profile expression patterns of 29 or one RNA species, respectively, MINA expands the scale of gene expression profiling to well over a hundred RNA species. Fourth, ORCA and Hi-M were developed in Drosophila embryos, whereas MINA has been established with mammalian tissue sections. Fifth, when performing cell segmentation for single-cell RNA profile analysis, ORCA does not visualize cell boundaries, but instead uses the probed genomic locus positions to approximate the cell centers for the segmentation algorithm. This procedure is sufficient when cells of the same type are largely grouped in space, and neighboring cells share roughly the same transcriptome. Hi-M does not implement cell segmentation. In contrast, MINA segments cells by visualizing cell boundaries with WGA labeling. This procedure is necessary when different cell types are intermixed in tissue and accurate segmentation of neighboring cells is required to quantify distinct single-cell RNA profiles.
Overview of the procedure
The protocol presented procedures for both chromatin tracing in cell lines and MINA in tissues (Fig.1). The procedure for chromatin tracing in cell lines consists of chromatin tracing probe design with ProbeDealer (steps 1–9), primary probe synthesis from template oligo pool (steps 14–22), cell culture preparation (step 23A), chromatin tracing primary probe hybridization (step 24A), sequential hybridization and imaging (steps 25–39), and data analysis (steps 40–48). The procedure for MINA in tissues consists of chromatin tracing probe design with ProbeDealer (steps 1–9), MERFISH probe design with ProbeDealer (steps 10–13), primary probe synthesis from template oligo pool (steps 14–22), WGA-oligo conjugation and tissue preparation (step 23B), MINA primary probe hybridization (step 24B), sequential hybridization and imaging (steps 25–39), and data analysis (steps 40–48).
Fig.1. Schematic diagram of the chromatin tracing and MINA protocols.
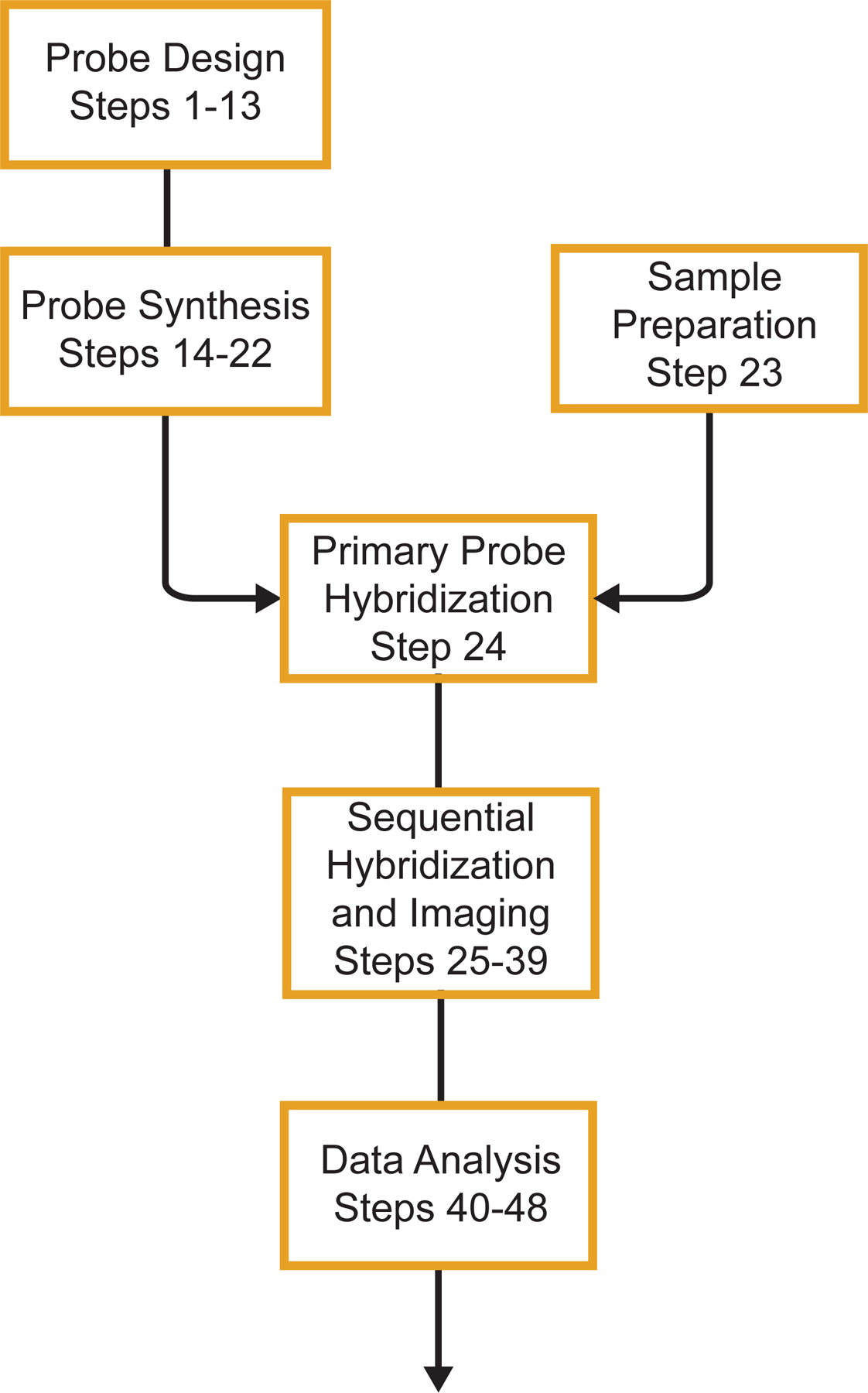
A schematic outline of the major steps in the chromatin tracing and MINA protocols. The major steps include chromatin tracing and MERFISH probe design, primary probe synthesis, sample preparation, primary probe hybridization, sequential hybridization and imaging, and data analysis.
Experimental design
Probe design and library amplification
We use a previously developed microarray-based oligo library synthesis strategy35–37 to synthesize a template oligo pool and generate primary probes from the template oligo pool with a high-yield enzymatic amplification approach32. For chromatin tracing, each template oligo for a primary probe contains 4 regions (from 5’ to 3’): i) a 20-nt forward priming region for oligo pool amplification, ii) a 30-nt readout region for dye-labeled secondary probe hybridization, iii) a 30-nt targeting region for binding to the genomic region of interest, and iv) a 20-nt reverse priming region for oligo pool amplification17,18. Alternatively, a second 30-nt readout region can be added between region iii and region iv to double the readout signal intensity18. For MERFISH probe design, each template oligo for a primary probe contains 6 regions (from 5’ to 3’): i) a 20-nt forward priming region, ii) a 20-nt readout region, iii) a 30-nt targeting region, iv) a second 20-nt readout region, v) a third 20-nt readout region, and vi) a 20-nt reverse priming region18,47. The sequences of the 20-nt priming regions are derived from random sequences that are screened to ensure the lack of homology with the mouse and human genomes and good performance in PCR priming32. The 30-nt readout regions for chromatin tracing are concatenations of 20-nt priming sequences and 10-nt halves created as above. The 20-nt readout sequences for MERFISH are introduced in a previous study47. The 30-nt targeting regions and sequences of the entire template oligo pool can be designed with our home-built MATLAB application ProbeDealer38. Users can provide genomic coordinates of target genomic loci for chromatin tracing probe design, or Ensembl transcript IDs for MERFISH probe design. ProbeDealer then generates the 30-nt targeting region sequences, BLASTs them against the genome and transcriptome to ensure oligo binding specificity, and concatenates additional readout and priming regions to assemble template oligos. Other software including OligoArray and OligoMiner are also useful alternatives for designing the template oligo pool48–50.
To synthesize primary probes from the template oligo pool, we follow a previously introduced high-yield enzymatic amplification procedure, which consists of limited-cycle PCR, in vitro transcription, reverse transcription, alkaline hydrolysis and oligo purification32 (Fig. 2a).
Fig.2. Primary probe synthesis.
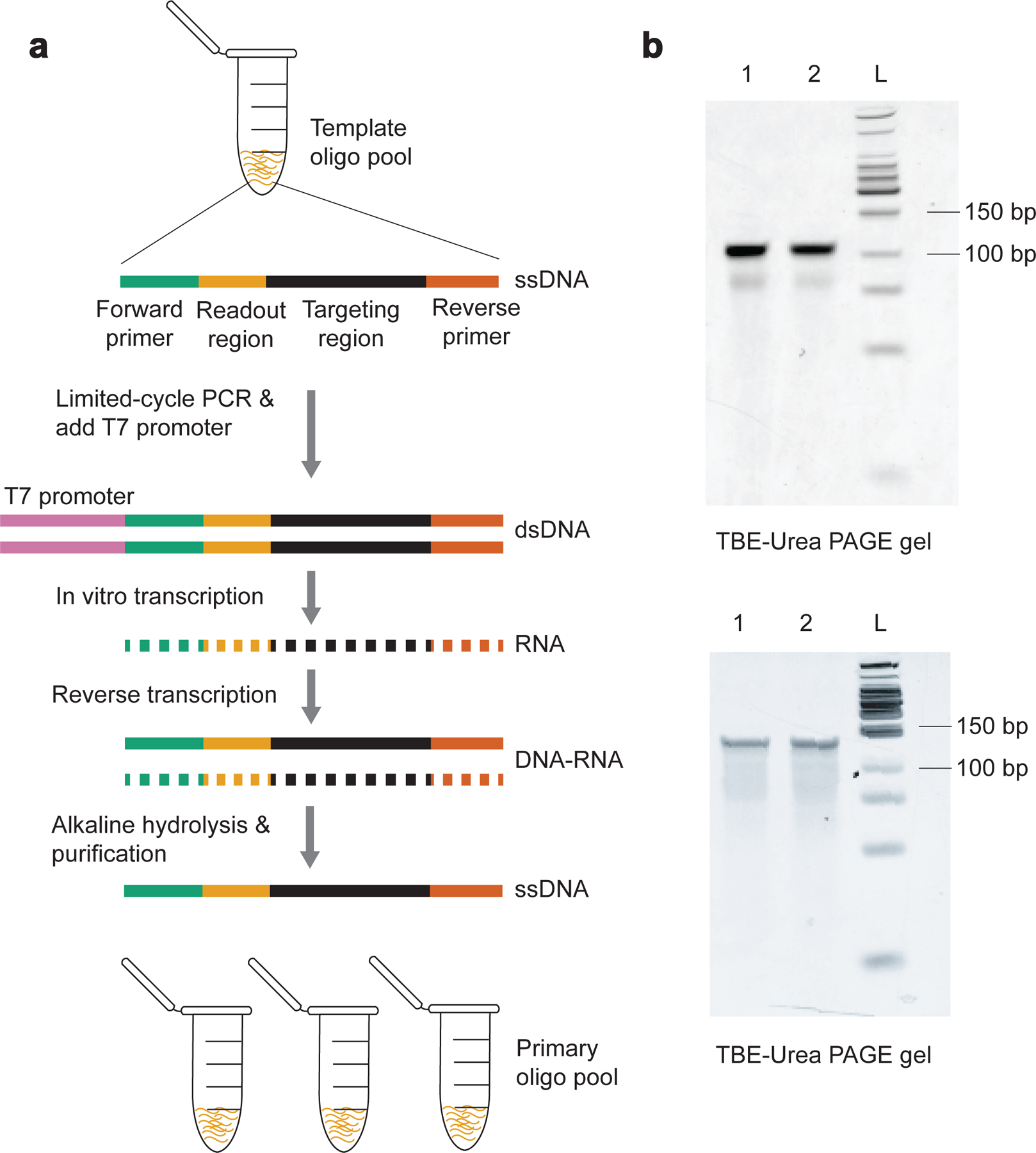
a, An outline of the primary probe synthesis procedure. It includes limited-cycle PCR, in vitro transcription, reverse transcription, alkaline hydrolysis, and oligo purification. Each template oligo in this example contains a forward primer, a readout region, a target region, and a reverse primer. b, Example results of TBE-Urea polyacrylamide gel electrophoresis for chromatin tracing and MINA experiments. (Upper panel) Lane 1 and 2 are 100-nt chromatin tracing primary probes for tracing mouse chromosome X. (Lower panel) Lane 1 and 2 are 130-nt MERFISH primary probes for mouse fetal liver tissue samples. L: Low molecular weight DNA ladder. Raw data are provided in SourceData_Fig2.
Sample preparation
Chromatin tracing and MINA can be performed in both cultured cells and tissue sections. Cells can be grown on 40-mm-diameter #1.5 glass coverslips. For tissue sample preparation, tissue samples are dissected, immediately embedded into optimal cutting temperature compounds and frozen into blocks. Tissue blocks can be stored at −80 °C until cryosectioning. Tissue blocks are routinely cryosectioned at a thickness of 10 μm onto poly-L-lysine treated coverslips. Tissue sections are then fixed in 4% (wt/vol) paraformaldehyde in DPBS for 20 min and incubated with oligo-conjugated WGA to label the cell membrane. After a brief post-fixation of oligo-conjugated WGA, we permeabilize the tissue sections with 0.5% (vol/vol) Triton X-100 in DPBS to facilitate probe penetration. We then store the tissue sections in 100% ethanol until probe hybridization. The integrity of DNA and RNA are both preserved in ethanol.
Probe hybridization
To implement chromatin tracing, live cells grown on coverslips are first fixed in 4% (wt/vol) paraformaldehyde in DPBS for 10 min, permeabilized with 0.5% (vol/vol) Triton X-100 in DPBS to expedite probe penetration, and washed with DPBS. The pre-fixed and pre-permeabilized tissue sections can be washed with DPBS directly. The cell line or tissue samples are then treated briefly with hydrochloric acid to denature the double-stranded genomic DNA. We further digest the samples with RNase A to reduce the non-specific binding of primary probes to RNA. Following a pre-hybridization blocking step, the samples are hybridized with primary probes, denatured at 86 °C for 3 min and incubated at 37 °C overnight. Stringent washing steps are then performed to reduce the non-specific binding of primary probes. We then apply 0.1-μm yellow-green beads as fiducial markers to correct for sample drift during sequential hybridizations. At this point, the samples are ready for secondary probe hybridization and sequential imaging.
In the MINA implementation, to visualize the nucleoli, we perform anti-fibrillarin immunofluorescence staining using the pre-fixed and pre-permeabilized sample washed with DPBS. A post-fixation step can protect the antibody signals from the harsh treatment conditions during FISH. We then briefly treat the sample with hydrochloric acid. As we hybridize primary probes targeting DNA and RNA simultaneously, all MINA procedures should be performed in an RNase-free setting. We thus omit the RNase treatment step and directly perform a short pre-hybridization blocking step. To avoid degradation of the RNase inhibitor in the primary probe hybridization buffer from heat denaturation, we denature the sample at 86 °C first without adding the primary probe solution, and follow immediately with chromatin tracing and MERFISH primary probe hybridization and overnight incubation at 37 °C. Stringency washing steps are performed to reduce non-specific probe binding. We then attach yellow-green fiducial beads to the coverslip to enable sample drift correction. Next, we continue with sequential secondary probe hybridization and imaging. An outline of the MINA method is illustrated in Fig. 3.
Fig.3. Outline of the MINA method.
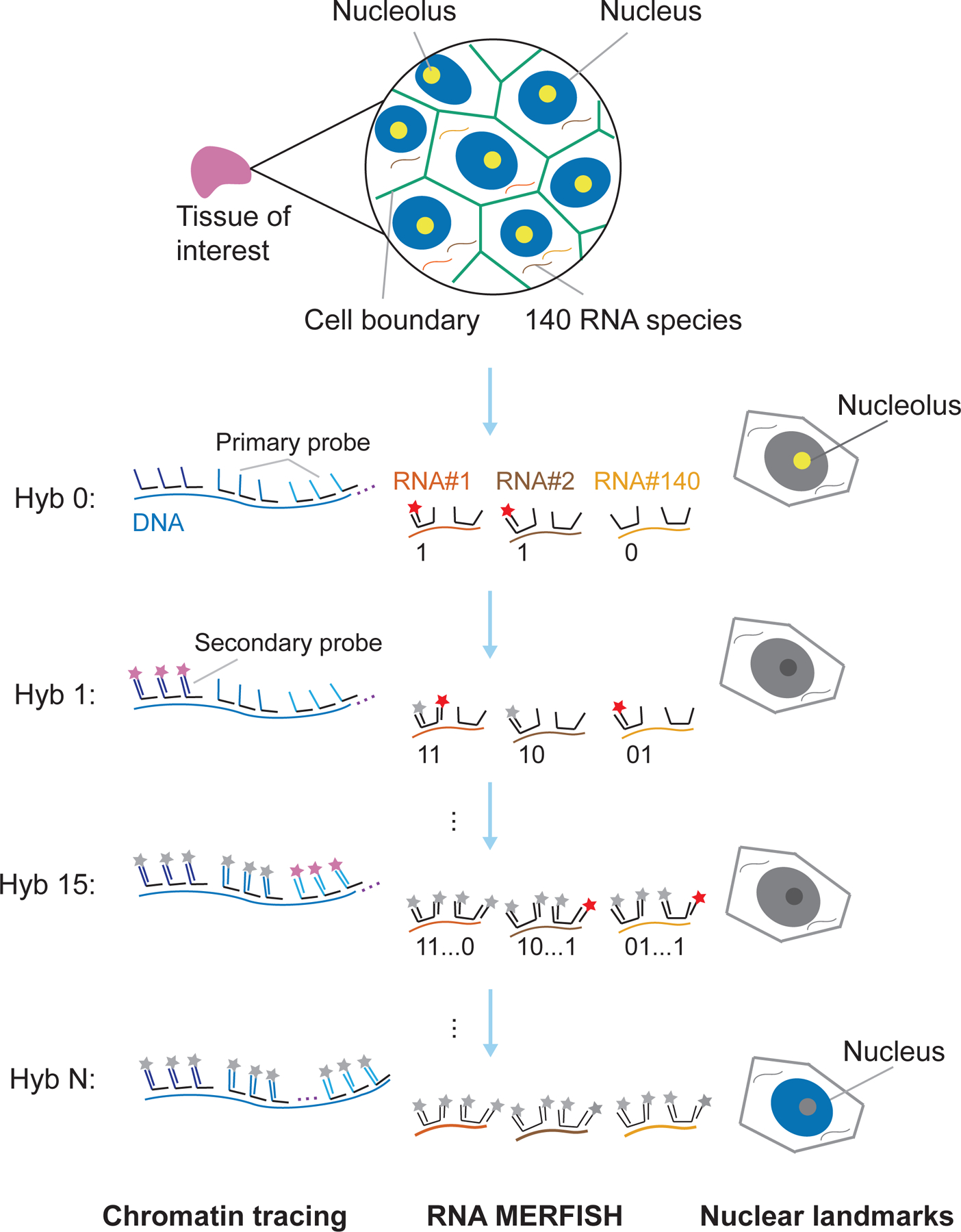
Primary probes for chromatin tracing and MERFISH are hybridized to the tissue of interest. Nucleolar and nuclear lamina landmarks are visualized with anti-fibrillarin immunofluorescence staining and approximated with DAPI staining respectively. During sequential hybridization and imaging, MERFISH bit 1 and anti-fibrillarin immunofluorescence staining are visualized in Hyb0. In subsequent hybridization and imaging rounds, target genomic loci (imaged in Hyb1 to Hyb N-1) and other MERFISH bits (imaged in Hyb1 to Hyb15) are sequentially imaged with dye-labeled secondary probes, separated by photobleaching. In the final round (Hyb N), DAPI staining images are acquired to approximate nuclear lamina positions. The figure is adapted with permission from the published MINA work18.
Sequential secondary hybridization and imaging
After primary probe hybridization, we assemble the sample into an FCS2 flow chamber. We then repeatedly perform secondary probe hybridization, imaging, and photobleaching. For chromatin tracing, we sequentially hybridize Alexa Fluor 647- and ATTO 565-conjugated 30-nt secondary probes to the readout regions on primary probes. For MERFISH, we use Alexa Fluor 750-labeled 20-nt secondary probes with sequences complementary to MERFISH primary probe readout regions. We employ an automated fluidics system32,51 to perform buffer exchange. When applicable, prior to sequential hybridization and imaging, we acquire fibrillarin immunofluorescence images by taking z-stack images with 647-nm laser illumination. We use an Alexa Fluor 647-conjugated secondary antibody for fibrillarin immunofluorescence staining. To correct for sample drift during sequential hybridization and imaging, we acquire yellow-green fiducial bead z-stack images with 488-nm laser illumination. In each round of secondary probe hybridization, we hybridize the secondary probes, wash away unbound probes, and flow oxygen-scavenging imaging buffer52 through the sample chamber. We then collect images from multiple fields of view (FOVs) using an automated microscope17,18. We take z-stack images with 750-nm, 647-nm, 560-nm and 488-nm laser illuminations at each FOV. Next, we photobleach the sample and continue with the next round of secondary probe hybridization and imaging.
We acquire WGA labeling patterns using ATTO 565-labeled secondary probes during an additional round of secondary probe hybridization and imaging. After the full tracing procedure, we flow DAPI stain through the sample chamber and take z-stack images with 405-nm laser illumination. The sequential hybridization and imaging pipeline is illustrated in Fig. 4.
Fig.4. Outline of MINA sequential hybridization and imaging.
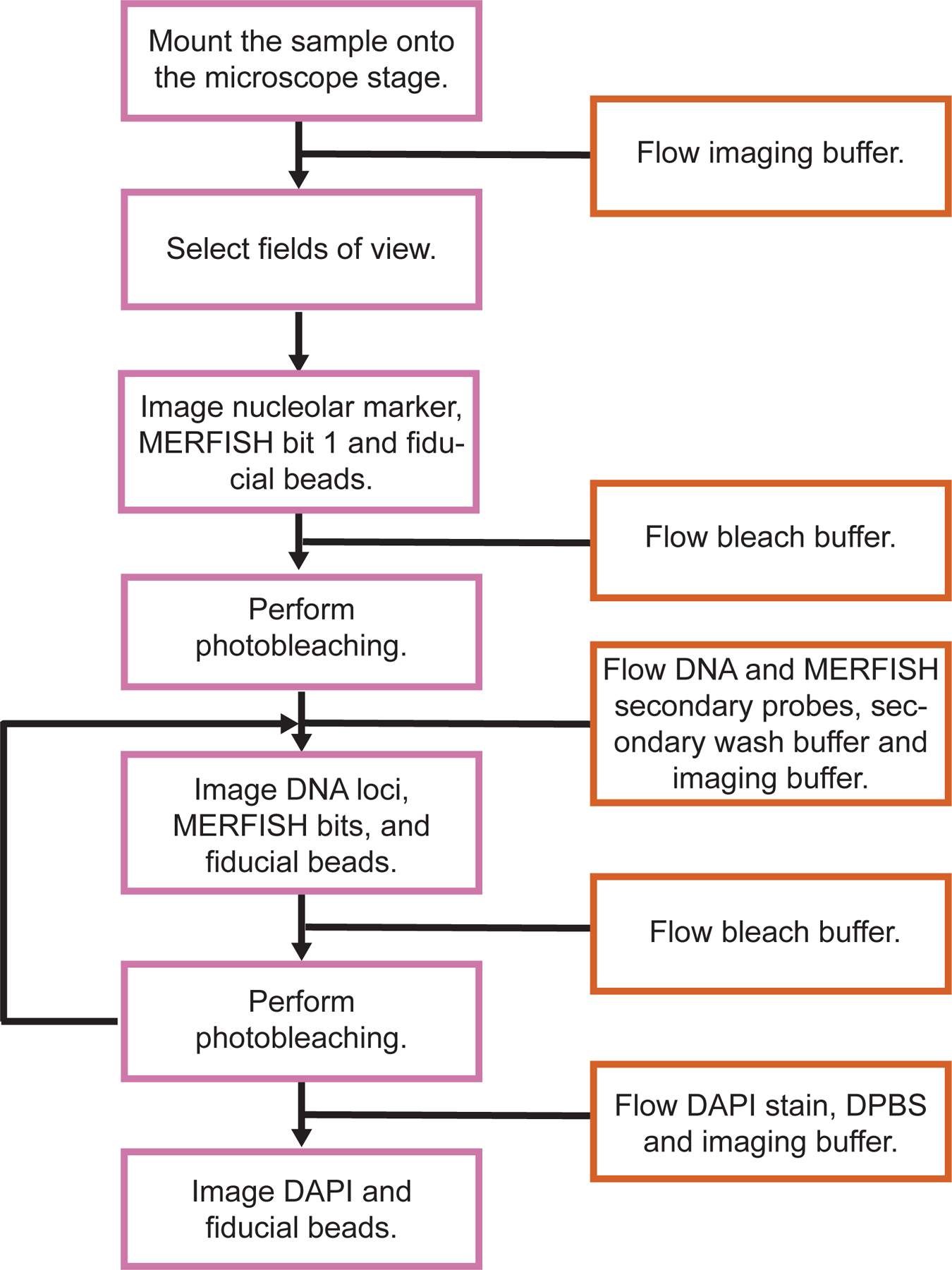
Schematic diagram of MINA sequential hybridization and imaging.
Image analysis
Prior to 3D chromatin folding analysis, we first correct chromatic aberrations between the two readout fluorescent channels (560-nm and 647-nm) for chromatin tracing with z-stepping images of TetraSpeck fluorescent beads on a coverslip, and correct sample drift with the yellow-green fiducial bead images. For each round of secondary hybridization and each FOV, we load the raw image stacks of both readout fluorescent channels, and fit the 3D positions of all potential target foci with Gaussian curve fitting. We then link the foci into chromatin traces based on their genomic identities and spatial clustering. After the chromatin trace reconstruction, one can analyze the pairwise distance/contact event between genomic loci (e.g. between promoter and enhancer), A and B compartmentalization of large scale chromatin traces, radius of gyration of each trace (which measures the compactness of the traced region), the scaling relationship between spatial distance and genomic distance (which measures the intermixing of chromatin regions), and the positioning of each labeled genomic locus relative to the entire traced region (e.g. the tendency of each TAD to localize to the surface of chromosome territory)17,18.
To perform image analysis for MERFISH, we first correct sample drift with the fiducial bead images. We then employ a pixel-based algorithm to decode the RNA species identities. For each hybridization round, we load the raw image stacks of the 750-nm channel, subtract local background, and retain regional maxima above an intensity threshold as potential RNA signals, which correspond to the “1”s in the binary barcodes, while the other pixels correspond to “0”s. After analyzing all hybridization rounds, the full barcode of each pixel is compared to the MERFISH codebook to identify the RNA identity of the pixel. Connected pixels with the same RNA identity are counted as the same RNA molecule. After decoding, we assign individual RNA molecules to single cells. To segment individual cells, we apply the watershed algorithm to the WGA-labeled cell boundary images. Following MERFISH and cell boundary image processing, one can identify cell types by clustering single-cell RNA expression profiles and analyze enrichment of gene expression in each cell type18. By combining chromatin tracing and MERFISH data, one can study chromatin folding organizations in different cell types and cell states and the co-variations between gene expression and chromatin structures (including all the chromatin folding metrics mentioned in the last paragraph).
To extract the 3D positions of nucleoli, we extract nucleolar positions with adaptive thresholding of the nucleolar images. To approximate nuclear lamina positions, we reconstruct a gradient image from the DAPI image to extract the edges of each nucleus. With the additional nucleolar and nuclear lamina position information, one can identify chromatin regions in spatial proximity to nucleoli/nuclear lamina and analyze the heterogeneity of such associations across different cell types, as well as the correlations of such associations with chromatin folding and gene expression18.
We developed a MATLAB application called MinaAnalyst for data analysis (Fig. 5). MinaAnalyst accepts raw images as inputs and performs data analysis for MERFISH, chromatin tracing, and MINA.
Fig.5. Data analysis with MinaAnalyst.
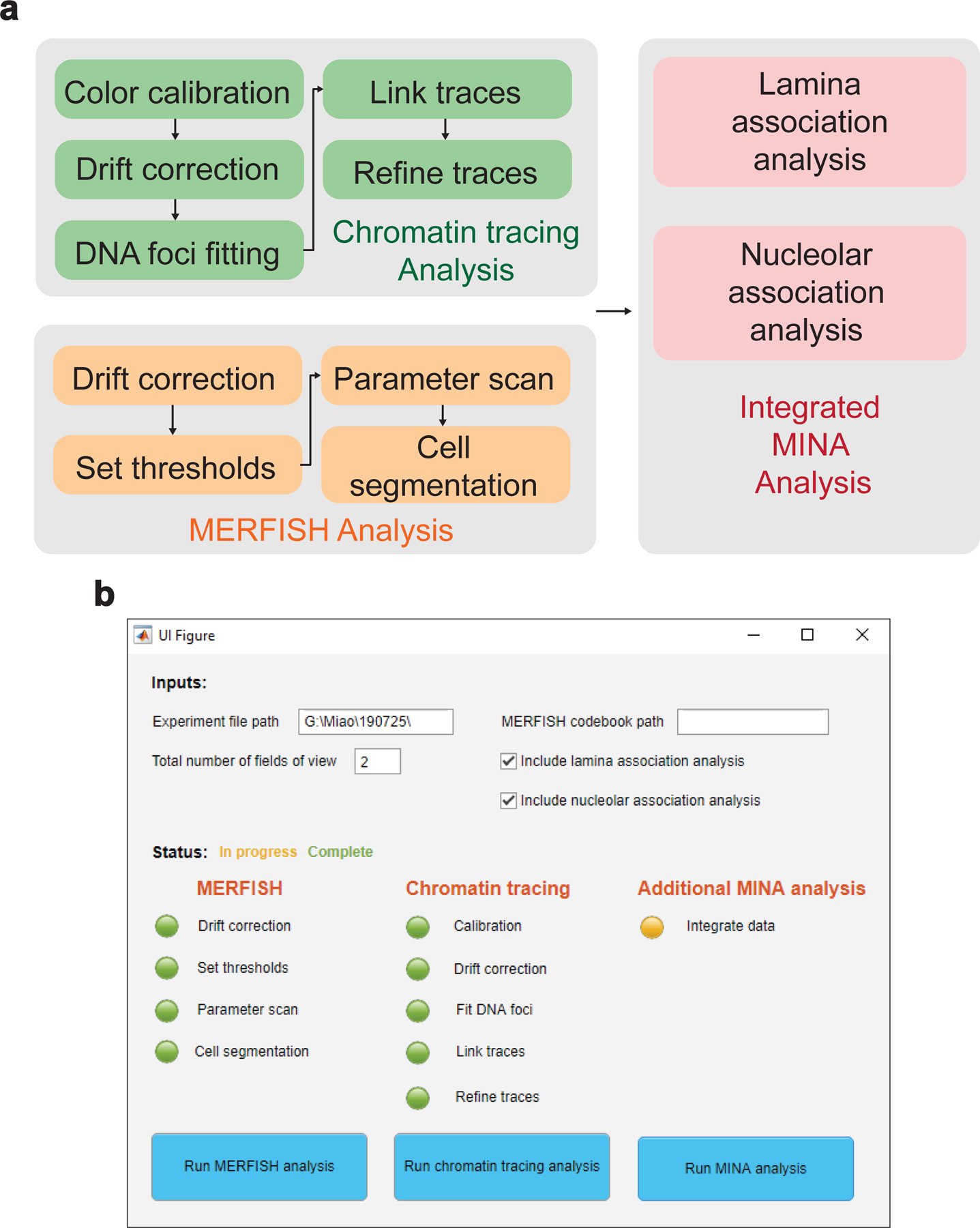
a, Data analysis pipeline with MinaAnalyst. Raw images are separately analyzed with MERFISH and chromatin tracing analyses, followed by integrated MINA analysis. b, MATLAB GUI for the MinaAnalyst application. Required inputs include experiment file path for raw images and total number of FOVs. Optional inputs include MERFISH codebook path and whether to include nuclear lamina and nucleolar association analyses. Users can choose different analysis options by clicking the corresponding buttons.
Controls
To validate the chromatin tracing approach, we routinely compare spatial distance measured by chromatin tracing with Hi-C contact frequency measured by sequencing between corresponding pairs of genomic regions17,18. A strong anti-correlation between the spatial distance and the Hi-C contact frequency at the population averaged level on a log-log plot indicates good chromatin tracing data quality. For TAD-to-chromosome scale tracing, we also analyze the correlations between A-B compartmentalization schemes measured from chromatin tracing and from Hi-C data17.
To validate the profiling of nucleolar/nuclear lamina associations of chromatin regions, we analyze the correlations of nucleolar/lamina association ratios with A-B compartment scores. Significant anti-correlations at the population averaged level are expected, since compartment B regions (with negative compartment scores) and nucleoli/lamina associated chromatin domains are all enriched with inactive chromatin29–31.
To validate the MERFISH measurements, we compare RNA copy numbers from MERFISH with bulk RNA-seq results. A strong, positive correlation between the probed genes’ RNA copy numbers from MERFISH and their FPKM values from bulk RNA-seq indicates that the MERFISH measurement is of high quality18,32.
Materials
Biological materials
-
Wild-type pregnant female C57BL/6 mice aged 8–15 weeks (The Jackson Laboratory) for MINA experiments
! CAUTION Experimental procedures involving animals should be performed according to the institutional and governmental regulations. All animal experiments described here are approved by the Institutional Animal Care and Use Committee (IACUC) of Yale University.
-
IMR-90 cell line (ATCC Cat# CCL-186, RRID:CVCL_0347) for chromatin tracing experiments
! CAUTION The cell lines used should be regularly checked to ensure they are authentic and are not infected with mycoplasma.
Reagents
Common reagents for chromatin tracing and MINA
Template oligo pool (CustomArray (now part of Genscript), custom order)
Forward and reverse primers for limited-cycle PCR (Integrated DNA Technologies, custom order). Sequences for the forward and reverse primers can be found in Supplementary Table 1.
20x EvaGreen dye (Biotium, cat. no. 31000-T)
Q5 Hot start high-fidelity 2x master mix (New England Biolabs, cat. no. M0494L)
DNA Clean & Concentrator kit (Zymo, cat. no. D4003)
Oligo binding buffer (Zymo, cat.no. D4060-1-40)
HiScribe T7 Quick High Yield RNA Synthesis Kit (New England Biolabs, cat. no. E2050S)
RNasin Ribonuclease Inhibitor (Promega, cat. no. N2611)
CTP solution (Thermo Scientific, cat. no. R0451)
dNTP solution mix (New England Biolabs, cat. no. N0447S)
Reverse transcription primer (Integrated DNA Technologies, custom order). Sequences of the reverse transcription primers can be found in Supplementary Table 1.
Maxima H Minus Reverse Transcriptase (Thermo Scientific, cat. no. EP0752)
Low molecular weight DNA ladder (New England Biolabs, cat. no. N3233S)
EDTA, 0.5 M solution, pH 8.0 (AmericanBio, cat. no. AB00502–01000)
Sodium hydroxide, 1 M solution (NaOH; Fisher Scientific, cat. no. S25549)
Zymo-spin V Columns w/ Reservior (Zymo, cat. no. C1016-25)
15% Mini-PROTEAN TBE-Urea Gel, 15 well, 15ul (Bio-Rad Laboratories, cat. no. 4566056)
Novex TBE-Urea sample buffer (2x; Invitrogen, cat. no. LC6876)
10× TBE buffer (AmericanBio, cat. no. AB14024–01000)
GelGreen nucleic acid gel stain (Biotium, cat. no.41004)
Dulbecco’s phosphate buffered saline (DPBS), 1× solution (Gibco, cat. no. 14190-144)
-
Paraformaldehyde, 16% (wt/vol) aqueous solution (Electron Microscopy Sciences, cat. no. 15710)
! CAUTION Paraformaldehyde is a toxic reagent. Perform all operations in a chemical fume hood with protective gloves.
Triton X-100 (Sigma, cat. no. T8787)
-
Hydrochloric acid (HCl, 36.5% – 38% (wt/wt); J.T.Baker, cat. no. 9535-00)
! CAUTION Hydrochloric acid is a corrosive and hazardous liquid. Handle it with protective gloves in a chemical fume hood.
UltraPure SSC, 20× solution (Invitrogen, cat. no. 15557044)
-
Formamide (AmericanBio, cat. no. AB00600-00500)
! CAUTION Formamide is toxic and a potential teratogen. Handle it with protective gloves in a chemical fume hood.
Tween-20 (AmericanBio, cat. no. AB02038-00500)
Dextran sulfate sodium salt from Leuconostoc spp. (Sigma-Aldrich, cat. no. D8906-50G)
-
Yellow-green fiducial beads (Invitrogen, cat. no. F8803)
▲CRITICAL The bead diameter should be smaller than the diffraction limited resolution of the imaging system to ensure localization precision.
Alexa Fluor 647-labeled chromatin tracing secondary probes (Integrated DNA Technologies, custom order). Sequences of the oligos can be found in Supplementary Table 2.
ATTO 565-labeled chromatin tracing secondary probes (Integrated DNA Technologies, custom order). Sequences of the oligos can be found in Supplementary Table 2.
-
TetraSpeck microspheres, 0.1 μm, fluorescent blue/green/orange/dark red (Invitrogen, cat. no. T7279)
▲CRITICAL The bead diameter should be smaller than the diffraction limited resolution of the imaging system to ensure localization precision.
Ethylene carbonate (Sigma-Aldrich, cat. no. E26258-500G)
Reagents specific to chromatin tracing
Eagle’s Minimum Essential Medium (ATCC, cat. no. 30–2003)
Premium Grade Fetal Bovine Serum (VWR, cat. no. 97068–085)
Penicillin-Streptomycin (10,000 U/mL; Gibco, cat. no. 15140–122)
-
Sodium borohydride (Sigma-Aldrich, cat. no. 480886–25G)
! CAUTION Sodium borohydride is a toxic and corrosive reagent. Handle it with protective gloves in a chemical fume hood. Sodium borohydride releases flammable gases when in contact with water. Never allow it to contact water during storage.
Ribonuclease A (AmericanBio, cat. no. AB12023–00100)
Reagents specific to MINA
-
DBCO-PEG5-NHS ester (Kerafast, cat. no. FCC310)
▲CRITICAL NHS esters are moisture-sensitive. Store the reagent under a desiccated environment and prepare stock solutions immediately before use.
Dimethyl sulfoxide (DMSO; Sigma-Aldrich, cat. no. D4540)
Unconjugated wheat germ agglutinin (WGA; Vector Laboratories Cat# L-1020, RRID:AB_2336868)
3’-azide-modified oligonucleotide (CGGTACGCACTTCCGTCGACGCAATAGCTC/3AzideN/, Integrated DNA Technologies, custom order).
Optimal cutting temperature compound (Tissue-Tek O.C.T.; VWR, cat. no. 25608–930)
0.01% (wt/vol) poly-L-lysine (Millipore, cat. no. A-005-C)
Ethyl alcohol, 200 proof (Sigma-Aldrich, cat. no. E7023–500ML)
Hanks’ balanced salt solution (HBSS, 1× solution; Gibco, cat. no. 14025092)
Bovine serum albumin (Sigma-Aldrich, cat. no. A9647-100G)
Glycine (AmericanBio, cat. no. 56-40-6)
Yeast tRNA (Invitrogen, cat. no. 15401011)
Anti-fibrillarin primary antibody (Abcam Cat# ab5821, RRID:AB_2105785)
Alexa Fluor 647-conjugated anti-rabbit secondary antibody (Thermo Fisher Scientific Cat# A-31573, RRID:AB_2536183)
Alexa Fluor 750-labeled MERFISH secondary probes (Integrated DNA Technologies, custom order). Sequences of the oligos can be found in Supplementary Table 2.
Tris-HCl, 1M solution, pH 8.0 (Sigma-Aldrich, cat. no. T2694–1L)
D-(+)-Glucose (Sigma-Aldrich, cat. no. G8270)
Glucose oxidase from Aspergillus niger (Sigma-Aldrich, cat. no. G2133–250KU)
Catalase from bovine liver (Sigma-Aldrich, cat. no. C30)
(±)-6-Hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid (TROLOX; Sigma-Aldrich, cat. no. 238813)
RNase inhibitor, Murine (New England BioLabs, cat. no. M0314L)
200 mM Ribonucleoside vanadyl complex (New England BioLabs, cat. no. S1402S)
-
DAPI, 1 mg/mL solution (Fisher Scientific, cat. no. EN62248)
! CAUTION DAPI is a known mutagen and should be handled with protective gloves in a chemical fume hood. The dye must be disposed of in accordance with local environmental health and safety regulations.
Equipment
Common equipment for chromatin tracing and MINA
PCR machine (thermal cycler; Bio-Rad, model no. T100)
DNA SpeedVac vacuum concentrator (Thermo Fisher Scientific, model no. DNA120-115)
Nanodrop spectrophotometer (Thermo Scientific, model no. ND-1000UV/Vis)
40-mm-diameter, #1.5 coverslips (Bioptechs, cat. no. 40-1313-0319)
Falcon 60 mm tissue culture dishes (Corning, cat. no. 353004)
CELLSTAR polypropylene tubes (15 mL and 50 mL, Greiner Bio-One, cat. no. 188261 and 227270)
Desktop centrifuge (Eppendorf, cat. no. 5424)
Digital dry bath, 120V (Bio-Rad)
Water bath (PolyScience, model no. WBE28)
blueBox Pro Transilluminator with imaging hood (miniPCR, model no. QP-1700–02)
Nikon Ti2-U inverted research microscope
Oil-immersion objective (60x, numerical aperture (NA) = 1.4; Nikon CFI Plan Apo Lambda)
sCMOS camera (Hamamatsu, model no. Orca Flash 4.0 V3)
750-nm laser (MPB Communications, cat. no. 2RU-VFL-P-500–750-B1R)
647-nm laser (MPB Communications, cat. no. 2RU-VFL-P-1000–647-B1R)
560-nm laser (MPB Communications, cat. no. 2RU-VFL-P-1000–560-B1R)
488-nm laser (MPB Communications, cat. no. 2RU-VFL-P-500–488-B1R)
405-nm laser (Coherent, model no. OBIS 405nm LX 50mW)
980-nm laser (Thorlabs, cat no. LP980-SF15)
Rotating filter wheel (Thorlabs, cat. no. FW102C)
Mechanical shutters (Vincent Associates, cat. no. LS3S2Z0)
Multi-band dichroic mirror (Chroma, cat. no. ZT405/488/561/647/752rpc-UF2)
Multi-band emission filter (Chroma, cat. no. ZET405/488/561/647–656/752m)
Position sensitive detector (Thorlabs, cat. no. PDP90A)
Piezo z positioner (Mad City Labs, cat. no. Nano-F100S)
950-nm short pass filter (Semrock, cat. no. FF01–950/SP-25)
Motorized x-y sample stage (Marzhauser, cat. no. SCAN IM 112×74)
8-channel modular valve positioners (Hamilton Company, cat. no. 36798 and 36766)
Flow chamber (Bioptechs, model no. FCS2)
Peristaltic pump (Gilson, cat. no. F155004)
Acquisition/analysis workstation (Intel Core i7-8700K CPU 3.70 GHz processor, 36.0 GB installed RAM)
Seagate 8 TB expansion desktop USB 3.0 external hard drive
Equipment specific to chromatin tracing
150 cm2 rectangular canted tissue culture flasks (Corning, cat. no. 355000)
Equipment specific to MINA
Amicon Ultra-0.5 Centrifugal Filter Unit, 10 KDa cutoff (Amicon Ultra, cat. no. UFC501024)
Cryostat (Leica, model no. CM1950)
25 mm × 20 mm × 5 mm Tissue-Tek Cryomold (VWR, cat. no. 25608–916)
Reagent setup
Common reagent setup for chromatin tracing and MINA
-
Alkaline hydrolysis buffer
Mix 500 μL 0.5 M EDTA and 500 μL 1 M NaOH. ▲CRITICAL Prepare fresh.
-
GelGreen nucleic acid gel staining solution
Mix 5 mL 10× TBE buffer and 45 mL Milli-Q H2O to make 1× TBE solution. Add 20 μL GelGreen nucleic acid gel stain to the solution and mix gently. ▲CRITICAL Prepare fresh.
-
4% (wt/vol) paraformaldehyde in DPBS
Mix 9 mL DPBS and 3 mL 16% (wt/vol) paraformaldehyde. ▲CRITICAL The solution can be stored in darkness at room temperature (22 °C) for a week.
-
0.5% (vol/vol) Triton X-100 in DPBS
Add 250 μL Triton X-100 to 50 mL DPBS. Vortex until the solution becomes homogenous. The solution can be stored at room temperature for several months.
-
0.1 M HCl solution
Add 45.8 mL Milli-Q H2O to a 50 mL Falcon tube. Add 4.2 mL 37% (wt/wt) HCl into Milli-Q H2O to make 1 M HCl. Mix 45 mL Milli-Q H2O and 5 mL 1 M HCl to make 0.1 M HCl solution. The solution can be stored at room temperature for several months.
! CAUTION Hydrochloric acid is a corrosive and hazardous liquid. Handle it with protective gloves in a chemical fume hood. Note that, if the order is reversed and Milli-Q H2O is wrongly added into 37% (wt/wt) HCl, the first drops of water may heat up, evaporate, and carry splashes of acid out of the tube onto people.
-
2x SSC buffer/bleach buffer
Mix 45 mL Milli-Q H2O and 5 mL 20x SSC to make the 2x SSC solution. The solution can be stored at room temperature for several months.
-
2x SSCT solution
Add 50 μL Tween-20 to 50 mL 2x SSC to make the 2x SSCT solution. The solution can be stored at room temperature for several months.
-
Secondary probe hybridization buffer/Secondary wash buffer
Warm up ethylene carbonate at 60 °C. Aliquot it into 25 mL stock solutions. The stock solutions can be stored at room temperature for several months. Warm up the 25 mL ethylene carbonate aliquots before use. Add 25 mL Milli-Q H2O to make 50% (vol/vol) ethylene carbonate solution. ▲CRITICAL The diluted 50% (vol/vol) ethylene carbonate solution can be stored at room temperature for one week.
To make the secondary probe hybridization buffer, mix 20 mL 50% (vol/vol) ethylene carbonate solution, 25 mL Milli-Q H2O and 5 mL 20x SSC. ▲CRITICAL Secondary probe hybridization buffer can be stored at room temperature for one week.
Reagent setup specific to chromatin tracing
-
1 mg/mL sodium borohydride in DPBS
Weigh 10 mg sodium borohydride in a 15 mL Falcon tube and dissolve it into 10 mL DPBS. ▲CRITICAL Prepare fresh and add the solution to the sample immediately after preparation.
! CAUTION Sodium borohydride is corrosive and toxic. It should be weighed with protective gloves.
-
0.1 mg/mL ribonuclease A solution
Dissolve ribonuclease A in 1 mL H2O to make the 100 mg/mL stock solution. ▲CRITICAL The stock solution can be stored at room temperature for months. Add 3 μL stock solution to 3 mL DPBS to make 0.1 mg/mL ribonuclease A solution. ▲CRITICAL Prepare the buffer fresh.
-
Chromatin tracing pre-hybridization buffer
Mix 500 μL 20x SSC, 2.5 mL formamide, 2 mL Milli-Q H2O and 5 μL Tween-20. Gently mix the solution until it is homogenous. ▲CRITICAL Prepare the buffer fresh.
-
Chromatin tracing hybridization buffer
Mix 10 μL 20x SSC, 50 μL formamide, and 40 μL 50% dextran sulfate. Gently tap the tube until the solution is homogenous. ▲CRITICAL Prepare the buffer fresh.
Reagent setup specific to MINA
-
2 mg/mL WGA in DPBS
Add 5 mL DPBS to the 10 mg WGA powder and mix gently. The solution can be stored at 4 °C for several months.
-
Blocking buffer
Weigh 0.5 g bovine serum albumin and 1.126 g glycine. Dissolve them into 49 mL DPBS. Add 50 μL Tween-20 into the solution. Mix them gently until the solution is homogenous. Blocking buffer can be stored at 4 °C for several weeks.
-
MINA pre-hybridization buffer
Mix 300 μL 20x SSC, 1.5 mL formamide and 1.2 mL Milli-Q H2O. Add 30 μL 200 mM ribonucleoside vanadyl complex to the solution. ▲CRITICAL Prepare the buffer fresh.
-
MINA hybridization buffer
Mix 50 μL formamide, 10 μL 1% yeast tRNA, 10 μL 20x SSC, 20 μL 50% dextran sulfate, 9 μL Milli-Q H2O, 1 μL murine RNase inhibitor. Mix them gently until the solution is homogenous. ▲CRITICAL Prepare the buffer fresh.
-
Imaging buffer
Mix 5 mL 20x SSC, 2.5 mL 1 M Tris-HCl and 38 mL Milli-Q H2O. Weigh 5 g glucose and add it to the solution. Weigh 25 mg TROLOX, dissolve it in 250 μL ethanol, and add the TROLOX solution to the imaging buffer. Add 25 μL murine RNase inhibitor. Add 20 mL H2O to dissolve the glucose oxidase powder into 50 mg/mL solution. Add 500 μL glucose oxidase solution and 50 μL catalase immediately before imaging. ▲CRITICAL Imaging buffer can be stored at 4 °C for one week.
-
1 μg/mL DAPI solution
Add 3 μL 1 mg/mL DAPI solution to 3 mL DPBS to make 1 μg/mL DAPI solution. ▲CRITICAL The 1 mg/ml DAPI solution should be stored at 4 °C. Prepare the 1 μg/mL DAPI solution fresh.
Equipment setup
Wide-field epifluorescence microscope
For imaging, we use a home-built microscope system (Fig. 6a) with a Nikon Ti2-U body equipped with a Nikon CFI Plan Apo Lambda 60x Oil (NA1.40) objective lens and a Hamamatsu Orca Flash 4.0 V3 camera. With this objective lens and camera combination, we routinely collect 1536×1536-pixel images with a pixel size of 108 nm/pixel, although it is possible to enlarge the FOV to 2048×2048 pixels. For epifluorescence illumination, we use a 750-nm laser to excite and image Alexa Fluor 750 on secondary probes, a 647-nm laser to excite and image Alexa Fluor 647 on secondary probes and on the anti-rabbit secondary antibody, a 560-nm laser to excite and image ATTO 565 on secondary probes, and a 488-nm laser to excite and image the yellow-green fiducial beads for drift correction. For these four lasers above, we use a filter wheel with neutral density filters to tune the laser intensities during imaging, and four mechanical shutters to control laser on-off. In addition, we use a 405-nm laser to excite and image the DAPI stain. The intensity and on-off of this 405-nm laser are controlled via USB communication to the laser console.
Fig.6. Schematic illustration of the home-built microscope and focus-lock system.
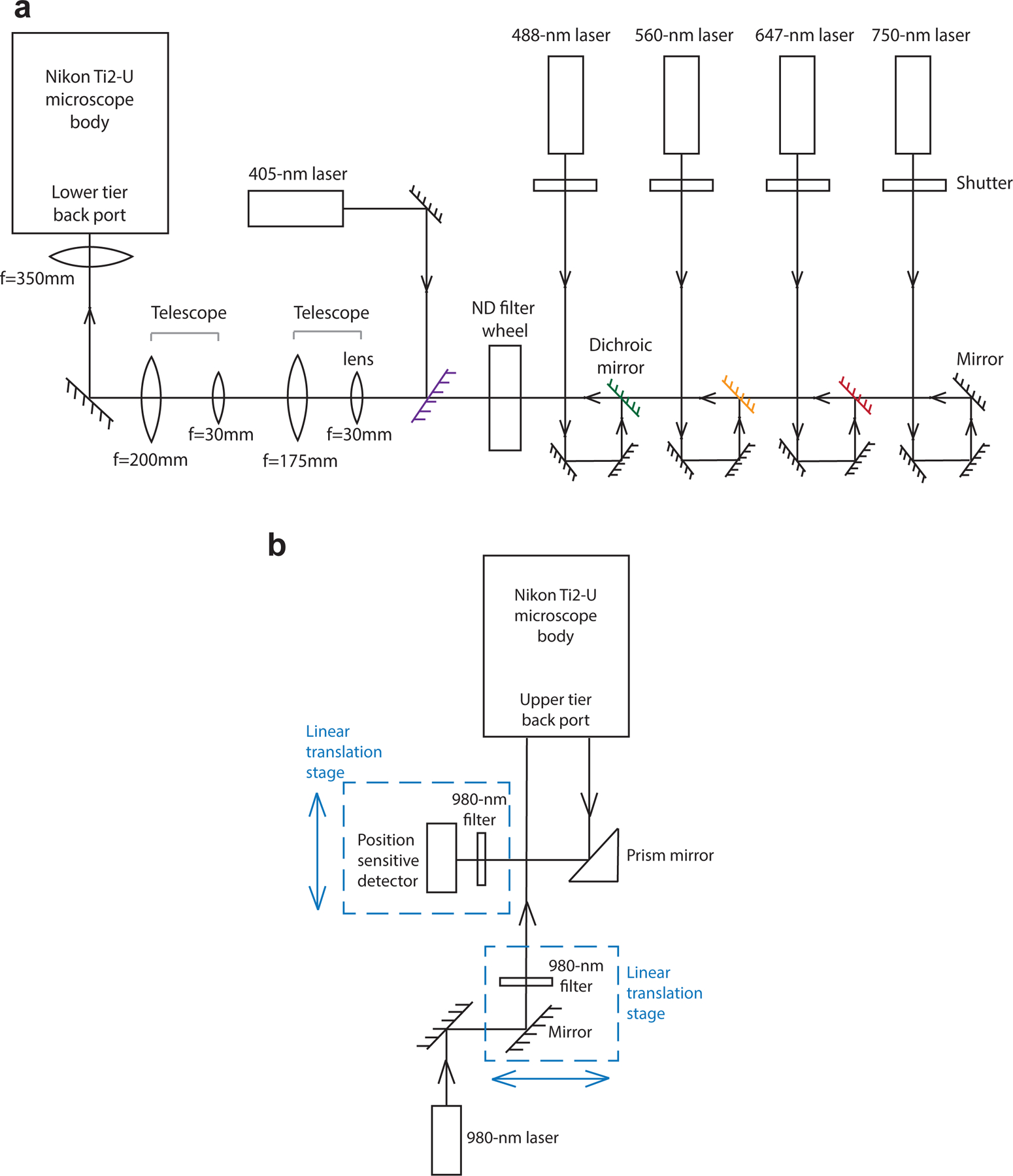
a, A schematic illustration of the home-built microscope setup. The 750-nm, 647-nm, 560-nm and 488-nm lasers are firstly directed through a neutral density (ND) filter wheel, which tunes laser intensities. The digitally controlled 405-nm laser and the four laser lines above are combined, pass through two telescopes for beam diameter expansion, and are directed into the lower tier back port of the Nikon Ti2-U microscope body. b, A schematic illustration of the home-built focus-lock system. A 980-nm laser passes through a 980-nm bandpass filter and is aligned to be parallel with the optical axis of the objective lens but translated away from the center axis. The back reflected 980-nm laser displaced to the other side of the center axis is captured by a prism mirror and is directed to a position sensitive detector.
Our Nikon Ti2-U microscope body has a two-tier dichroic fluorescence filter turret setup. In the lower tier turret, we direct the five laser lines above to the sample with a multi-band dichroic mirror, and filter fluorescent emission with a multi-band emission filter. The upper tier turret is dedicated to a home-built active focus-lock system (Fig. 6b). In this turret, we direct a 980-nm laser to the sample with a dichroic mirror. This laser is aligned parallel with the optical axis of the objective lens but displaced away from the center of the objective lens. As a result, the laser hits the glass-water interface of the sample chamber at an angle, and its reflected light travels down the objective lens also in parallel with the optical axis, but displaced away from the center, and away from the original light path. This reflected 980-nm light is directed to a position sensitive detector. A drift of the sample in the z direction leads to a displacement signal from the position sensitive detector, and this signal is fed back to a piezo z positioner below the objective lens to actively compensate for the drift and maintain focus. In addition, a 950-nm short pass filter is added to the lower tier turret to prevent reflected 980-nm light from reaching the camera.
To automatically scan and image multiple FOVs, we use a motorized x-y sample stage. To perform optical z sectioning, we use the same piezo z positioner mentioned above.
Fluidics setup
The design and instrumentation of the fluidics setup have been described in detail before51. In brief, three 8-channel modular valve positioners are connected in a daisy chain, leading to 22 input fluid channels (2 out of 24 channels are used for the daisy chain connections). Each input channel is connected to a tube with buffer for hybridization, wash, imaging, or bleaching. A single output fluid channel leaves the valve system (2 out of the 3 output channels are used for the daisy chain connections). This output channel is connected to a flow chamber that holds the sample. The flow chamber is then connected to a peristaltic pump that drives the liquid flow through the whole system.
User-microscope interface
Python-based image acquisition and flow control software (hal4000 for image acquisition, kilroy for flow control, and dave for coordinating hal4000 and kilroy in sequential hybridization and imaging) were developed by Xiaowei Zhuang lab and are downloaded from https://github.com/ZhuangLab/storm-control.
Procedure
Chromatin tracing probe design with ProbeDealer ● Timing 1 d
-
1
Download and install BLAST+ from NCBI.
▲CRITICAL STEP MAC users need to add the BLAST executable bin to the PATH environment variable.
-
2
Download and install MATLAB version R2018a or a more recent version following the manufacturer’s instructions. Install the MATLAB Bioinformatics Toolbox to use ProbeDealer38.
-
3
Download the ProbeDealer package from https://campuspress.yale.edu/wanglab/probedealer.
-
4
Install the ProbeDealer application. Double-click the “ProbeDealer.mLapp” file in the package to open the graphical user interface (GUI) of the application.
-
5
Download the hg38 human and mm10 mouse BLAST databases from https://campuspress.yale.edu/wanglab/probedealer. Input the folder that contains relevant genome BLAST databases.
▲CRITICAL STEP If performing MINA, users will also need to design MERFISH probes (steps 10–13) and should therefore input both the “TxShortHeader” and “Tx” subfolders.
-
6
Choose the “chromatin tracing” probe type from the selection panel.
▲CRITICAL STEP Users can choose to “only target antisense strand of genes”, to “avoid exon regions”, or choose both these options. The “only target antisense strand of genes” option is suitable for chromatin tracing experiments without RNase treatment. Choose both options when performing chromatin tracing and RNA detection simultaneously, such as the MINA method.
▲CRITICAL STEP The ProbeDealer application uses default parameters (probe length range, melting temperature range, GC content range, secondary structure formation melting temperature range, cross hybridization melting temperature range) for probe design. Users can customize the parameters based on their specific applications.
-
7
Input the genome target sequences in fasta or Excel spreadsheet formats. The Excel spreadsheet file should specify the chromosome ID and start and end positions of the target region.
-
8
Choose the output probe type and specify the output folder to store the output files. Final oligos will be generated in both fasta and Excel spreadsheet formats.
-
9
Purchase the template oligo pool from CustomArray, GenScript or Twist Bioscience.
(OPTIONAL) MERFISH probe design with ProbeDealer ● Timing 1 d
▲CRITICAL STEP If performing MINA, you will also need to design MERFISH probes.
-
10
Choose the “MERFISH” probe type in the selection panel of ProbeDealer.
▲CRITICAL STEP The ProbeDealer application uses default parameters (probe length range, melting temperature range, GC content range, secondary structure formation melting temperature range, cross hybridization melting temperature range) for probe design. Users can customize the parameters based on their specific applications.
-
11
Input target sequences in Excel spreadsheets. The spreadsheet should contain two columns. The first column specifies the Ensembl transcript IDs and the second column contains the gene FPKM values or other measures of relative gene expression.
▲CRITICAL STEP By default, ProbeDealer accepts up to 140 target transcripts for MERFISH probe design.
▲CRITICAL STEP ProbeDealer accepts Ensembl transcript IDs without version variants. For example, ProbeDealer accepts ENST00000000 instead of ENST00000000.0.
-
12
Specify the output folder to store the final oligos and the codebook. Final oligos are stored in fasta or Excel spreadsheet formats. The codebook contains the MHD4 codes assigned to each transcript.
-
13
Order the template oligo pool from CustomArray, GenScript or Twist Bioscience.
Primary probe synthesis from template oligo pool ● Timing 1.5 d
-
14Limited-cycle PCR to amplify the template oligo pool. Prepare a template-primer master mix of the template oligo pool and primers as follows.
Reagent Quantity (μL) Milli-Q H2O 189 Forward primer (100 μM) 5 Reverse primer (100 μM) 5 Template oligo pool (40–180 ng/μL) 1 Total 200 Set up the PCR system as indicated below.Reagent Quantity (μL) Milli-Q H2O 12.5 20x EvaGreen dye 2.5 Q5 Hot start high-fidelity 2x master mix 25 Template-primer master mix 10 Total 50 Check the melting temperature of the primers on the website https://tmcalculator.neb.com/#!/main. Set up the cycling conditions of the PCR machine as suggested below.Cycle Denaturation Annealing Extension 1 98 °C, 3 min 2-end 98 °C, 10 s Primer melting temperature (67 °C-72 °C), 10 s 72 °C, 15 s End the PCR reaction before the amplification curve reaches a plateau.
▲CRITICAL STEP End the PCR reaction before the amplification curve reaches a plateau to reduce non-specific amplifications.
▲CRITICAL STEP End the PCR reaction at the end of the elongation step of the current amplification cycle.
-
15
DNA purification. Use the DNA Clean & Concentrator kit from Zymo Research and elute purified DNA with 13 μL Milli-Q water.
■PAUSE POINT Purified DNA can be stored at −20 °C until further use.
-
16In vitro transcription. Set up the in vitro transcription system as indicated below. Use the following table when performing primary probe synthesis for chromatin tracing.
Reagent Quantity (μL) DEPC-treated ddH2O 7.5 NTP mix 10 T7 RNA polymerase mix 2 RNasin Ribonuclease Inhibitor 1 Purified DNA from step 15 10 Total 30.5 To accommodate the disproportionate use of cytosine in MERFISH probes, add additional CTP during in vitro transcription. Use the following table when synthesizing primary probes for MERFISH.Reagent Quantity (μL) DEPC-treated ddH2O 6.16 CTP mix 1.34 NTP mix 10 T7 RNA polymerase mix 2 RNasin Ribonuclease Inhibitor 1 Purified DNA from step 15 10 Total 30.5 -
17
Incubate the system in the PCR machine at 37 °C for 16 h.
-
18Reverse transcription. Set up the reverse transcription system as indicated below. Use the following system when performing primary probe synthesis for chromatin tracing. To synthesize dye-labeled primary probes, use fluorophore-conjugated reverse transcription primers. Fluorophores such as Alexa Fluor 647 can be conjugated to the 5’ end of reverse transcription primers.
Reagent Quantity (μL) dNTP mix (10 mM) 12 Reverse transcription primer (100 μM) 10 Maxima H Minus Reverse Transcriptase 1.5 5x reverse transcription buffer 15 RNasin Ribonuclease Inhibitor 1.5 Unpurified RNA product from step 16 30 Total 70 To account for the additional requirement for guanine in MERFISH probes, double the concentration of all dNTPs during reverse transcription. Use the following table when doing probe synthesis for MERFISH.Reagent Quantity (μL) dNTP mix (10 mM) 24 Reverse transcription primer (100 μM) 10 Maxima H Minus Reverse Transcriptase 1.5 5x reverse transcription buffer 15 RNasin Ribonuclease Inhibitor 1.5 Unpurified RNA product from step 16 30 Total 82 Incubate the system in the PCR machine at 50 °C for 60 min.
-
19
RNA degradation. Add 75 μL alkaline hydrolysis buffer to the system to hydrolyze synthesized RNA molecules. Incubate the system at 95 °C for 10 min.
-
20
Oligo purification. Add 300 μL oligo binding buffer to the unpurified oligos in step 19 and mix gently. Add 1200 μL ethanol and transfer the solution into Zymo-spin V Columns. Centrifuge at 12,000 xg for 30 s and discard the flow-through. Wash the columns once with 750 μL DNA wash buffer. Centrifuge at 12,000 xg for 30 s and discard the flow-through. Elute the oligos with 80 μL-100 μL DEPC-treated water. Centrifuge at 12,000 ×g for 30 s and collect the solution as the primary probe oligo pool solution.
-
21
Take a 2 μL aliquot to measure the oligo concentration using a Nanodrop spectrophotometer. The concentration is 300 ng/μL-500 ng/μL for chromatin tracing primary probes and 900 ng/μL-1100 ng/μL for MERFISH primary probes. Take a 1 μL aliquot for TBE-Urea polyacrylamide gel electrophoresis. Use a SpeedVac vacuum concentrator to dry the oligo pool solution. Store the dry oligos at −20 °C until further use.
■PAUSE POINT Dry oligos can be stored at −20 °C for years.
-
22
TBE-Urea polyacrylamide gel electrophoresis. Prepare a 15% Mini-PROTEAN TBE-Urea polyacrylamide gel and pre-run the gel in 1x TBE at 70 V for 1 h at 60 °C. Mix 1 μL purified oligos from step 20, 9 μL Milli-Q water, and 10 μL 2x TBE-Urea sample buffer. Heat the sample mixture at 95 °C for 10 min and then load the sample mixture into each well. Run the gel at 70 V at 60 °C for 50 min. For gel staining, incubate the gel with GelGreen nucleic acid gel staining solution for 30 min at 4 °C and image the gel with blueBox Pro Transilluminator with imaging hood (Fig. 2b).
? TROUBLESHOOTING
Sample preparation ● Timing 2–4 days (3–4 days for chromatin tracing in cell lines; 2 days for MINA in tissue sections)
-
23Follow option A for preparing cell lines for chromatin tracing and option B for preparing tissue samples for MINA.
- Culture of cell lines for chromatin tracing. TIMING 3–4 days
-
IMR-90 cell culture. Culture cells (we use IMR-90 cells) with Eagle’s Minimum Essential Medium containing 10% (vol/vol) fetal bovine serum and 1x Penicillin-Streptomycin in 150 cm2 cell culture flasks. Grow cells onto 40-mm-diameter, #1.5 coverslips in Falcon 60 mm tissue culture dishes.▲CRITICAL STEP We typically passage cells at a split ratio of 1:2–1:8. Passaging generations should be less than 20 to avoid accumulated mutations or structural variations.
-
Fix the IMR90 cells prepared in step 23A(i) in freshly made 4% (wt/vol) paraformaldehyde in DPBS for 10 min at room temperature. Wash with DPBS twice.■PAUSE POINT Fixed cells can be stored in DPBS at 4 °C for weeks.
-
- WGA-oligo conjugation and tissue preparation for MINA TIMING 2 days
-
DBCO solution preparation. Dissolve DBCO-PEG5-NHS ester in DMSO to a concentration of 10 mM.▲CRITICAL STEP NHS esters are moisture-sensitive. To avoid moisture condensation onto the product, equilibrate the vial to room temperature before opening.▲CRITICAL STEP Prepare the stock solution immediately before use. Avoid buffers containing primary amines and azides.? TROUBLESHOOTING
-
DBCO conjugation to WGA. Add 2.7 μL of the DBCO-PEG5-NHS ester solution to 100 μL of 2 mg/mL WGA in DPBS. Incubate the solution mixture at room temperature for 1 h so the free primary amine groups of WGA can be conjugated with DBCO.? TROUBLESHOOTING
-
DBCO-labeled WGA purification. Terminate the reaction and remove excess DBCO molecules with 10 KDa cutoff Amicon Ultra-0.5 centrifugal filter unit.? TROUBLESHOOTING
-
Oligo conjugation to WGA. Combine 20 μL of 100 μM 3’-azide-modified oligonucleotide with purified DBCO-conjugated WGA. Incubate the reaction at 4 °C for at least 12 h. The sample mixture can be directly used as oligo-conjugated WGA.? TROUBLESHOOTING
- Mouse maintenance. Maintain all mice under constant temperature (22 °C) and humidity (40%–60%).
-
Mouse fetal liver tissue collection. Sacrifice female mice on day 14.5 of pregnancy by isoflurane inhalation and cervical dislocation. Isolate embryos at embryonic day 14.5 from the uterus and immerse them in cold DPBS. Dissect fetal liver from the embryo and embed it with optimal cutting temperature compound in a 25-mm × 20-mm × 5-mm Tissue-Tek Cryomold. Freeze tissue blocks immediately with dry ice or liquid nitrogen.■PAUSE POINT Frozen tissue blocks can be stored at −80 °C for years until cryosectioning.
-
Mouse fetal liver tissue sectioning. Treat coverslips with 30 μL 0.01% (wt/vol) poly-L-lysine at room temperature for 15 min prior to tissue sectioning. Perform cryosectioning of frozen tissue blocks at a thickness of 10 μm at −15 °C in a cryostat. Attach tissue sections to the center of the coverslip.? TROUBLESHOOTING
-
Tissue fixation. After cryosectioning, fix tissue sections immediately in 4% (wt/vol) paraformaldehyde in DPBS for 20 min at room temperature, and wash with DPBS for 3 min twice.▲CRITICAL STEP Equilibrate the tissue section at room temperature for 15 s before adding 4% (wt/vol) paraformaldehyde in DPBS. Otherwise, the tissue section may disintegrate.■PAUSE POINT Tissue sections can be stored in ethanol at −20 °C for weeks after fixation.? TROUBLESHOOTING
-
Oligo-conjugated WGA staining. Wash tissue sections with Hanks’ balanced salt solution for 5 min, stain them with oligo-conjugated WGA prepared in step 23B(iv) at a concentration of 2–5 μg/mL in Hanks’ balanced salt solution with 2000× diluted murine RNase inhibitor for 20 min at 37 °C, and wash the sample with Hanks’ balanced salt solution 3 times.▲CRITICAL STEP It is critical to use calcium-containing Hanks’ balanced salt solution. WGA is a carbohydrate-binding lectin that requires calcium for its optimal binding activity.
- Post-fix tissue sections in 4% (wt/vol) paraformaldehyde in DPBS for 10 min. Wash with DPBS twice.
-
Permeabilize the tissue section with 0.5% (vol/vol) Triton X-100 in DPBS for 15 min at room temperature. Wash twice with DPBS.■PAUSE POINT After permeabilization, tissue sections can be stored at −20 °C in ethanol for weeks before MINA primary probe hybridization.
-
Primary probe hybridization ● Timing 1.5 d
-
24Follow option A for chromatin tracing in cell lines and option B for MINA in tissue sections.
- Chromatin tracing primary probe hybridization.
-
Treat the cells prepared in step 23A(ii) with freshly made 1 mg/mL sodium borohydride in DPBS for 10 min at room temperature. Wash twice with DPBS.! CAUTION Sodium borohydride is corrosive and toxic. It should be weighed with protective gloves.
- Permeabilize the cells with 0.5% (vol/vol) Triton X-100 in DPBS for 10 min at room temperature. Wash with DPBS for 2 min twice.
- Treat the cells with 0.1 M HCl for 5 min at room temperature. Wash with DPBS twice.
- Treat the cells with freshly prepared 0.1 mg/mL ribonuclease A solution for 45 min in a 37 °C incubator.
- Wash cells in 2x SSC twice. Incubate the cells at room temperature for 30 min in freshly made chromatin tracing pre-hybridization buffer.
- Dissolve the SpeedVac-dried chromatin tracing primary probes prepared in step 21 in 10 μL Milli-Q water and mix well. Add 1 μL dissolved chromatin tracing primary probes in 25 μL of chromatin tracing hybridization buffer. Final probe concentration is 6–20 μM. Mix the probes well with gentle tapping.
- Pipet the chromatin tracing hybridization buffer containing primary probes onto a glass slide. Flip the coverslip onto the glass slide so that the cells are in contact with the hybridization buffer.
- Put the coverslip-slide assembly on an 86 °C digital dry bath and perform heat denaturation for 3 min.
- Incubate the assembly overnight for 15–18 h at 37 °C in a humid chamber.
- Gently remove the coverslip from the coverslip-slide assembly and put the coverslip into a Falcon 60 mm tissue culture dish, with cells facing upwards. Wash with 2x SSCT at 60 °C in a water bath for 15 min twice.
- Wash with 2x SSCT at room temperature for 15 min. Then switch the buffer to 2x SSC.
-
Attach yellow-green fiducial beads onto the coverslip. Dilute yellow-green fiducial beads at a 1:100 ratio in water. Add 15 μL diluted bead solution to 3 mL 2x SSC. Immediately vortex the solution and pour it into the tissue culture dish containing samples.▲CRITICAL STEP Yellow-green fiducial beads become very sticky in salt solutions. Add beads immediately to the sample after diluting beads in 2x SSC.
- Assemble the coverslip into an FCS2 flow chamber. Proceed to step 25 for sequential hybridization and imaging.
-
- MINA primary probe hybridization.
- Immunofluorescence staining. Replenish processed mouse fetal liver tissue sections prepared in step 23B(xi) with DPBS. Wash twice for 3 min each with DPBS.
- Block the tissue section with blocking buffer for 30 min at room temperature.
- Incubate the sample with anti-fibrillarin primary antibody at a concentration of 1:100 in blocking buffer with 2000× diluted murine RNase inhibitor at 4 °C overnight. Wash with DPBS for 5 min for three times.
- Incubate the sample with Alexa Fluor 647-conjugated anti-rabbit secondary antibody at a concentration of 1:1000 in blocking buffer with 2000× diluted murine RNase inhibitor at room temperature for 1 h. Wash with DPBS for 5 min for three times.
-
Post-fixation. Post-fix the sample with 4% (wt/vol) paraformaldehyde in DPBS for 10 min and wash with DPBS for 3 min twice.▲CRITICAL STEP A post-fixation step is required to protect the antibody staining signals from the harsh FISH procedure.
- HCl treatment. Treat the sample with 0.1 M HCl for 5 min. Wash with DPBS for 3 min twice.
- Heat denaturation. Prior to probe hybridization, block the sample with MINA pre-hybridization buffer for 5 min at room temperature.
-
For heat denaturation, drop 20 μL MINA hybridization buffer onto a glass slide. Flip the coverslip so that the tissue section can be in contact with the MINA hybridization buffer. Place the coverslip-tissue-slide assembly on top of an 86 °C digital dry bath for 3 min.▲CRITICAL STEP No MINA primary probes are added in MINA hybridization buffer during heat denaturation. We apply heat denaturation first and then hybridize MINA primary probes. This strategy protects protein-based murine RNase inhibitors from denaturation. Otherwise, RNA FISH signals can be disrupted.
- Probe hybridization. After heat denaturation, gently remove the coverslip and wash it briefly with 2x SSC. For MINA primary probe hybridization to each sample, prepare 12.5 μL MINA hybridization buffer with 24–28 μM MERFISH primary probes and 4–8 μM chromatin tracing primary probes. Drop the buffer onto a piece of parafilm, flip the coverslip so that the tissue section can be in contact with the MINA hybridization buffer containing probes.
- Incubate the coverslip sample assembly at 37 °C in a humid chamber for 18–24 h.
- Remove the coverslip from the sample assembly carefully and put it in a tissue culture dish, with tissue sections facing upwards. Wash the sample with 2x SSCT at 60 °C in a water bath for 15 min twice. Wash the sample with 2x SSCT at room temperature for 15 min once.
- Pre-hybridize the first Alexa Fluor 750-labeled MERFISH secondary probe in secondary probe hybridization buffer for 20 min.
- Wash the sample twice with secondary wash buffer for 2 min each.
-
Dilute yellow-green fiducial beads at a 1:100 ratio in water. Add 15 μL diluted bead solution to 3 mL 2x SSC. Immediately vortex the solution and pour it into the sample dish. Proceed with sequential hybridization and imaging.▲CRITICAL STEP Yellow-green fiducial beads become very sticky in salt solutions. Add beads immediately to the sample after diluting beads in 2x SSC.
Sequential hybridization and imaging ● Timing ~1 h per round
-
25
Assemble the FCS2 microfluidic chamber. Put the upper white half with perfusion tubes on the bench.
-
26
Follow the manufacturer’s instruction and mount the pieces from bottom to top: upper white half; upper gasket, microaqueduct slide; singular lower gasket; 40-mm-diameter, #1.5 coverslips with cells facing the singular gasket; and self-locking base.
▲CRITICAL STEP Make sure that the side of the microaqueduct slide with grooves faces towards the cells.
-
27
Preparation of secondary probes. Dilute Alexa Fluor 647 and ATTO 565-labeled chromatin tracing secondary probes in secondary probe hybridization buffer at 7.5 nM concentration. Dilute Alexa Fluor 750-labeled MERFISH secondary probes in secondary probe hybridization buffer at 3.75 nM concentration.
-
28
Connect all tubes containing secondary probes, secondary wash buffer, imaging buffer and bleach buffer to the corresponding input channels of the three 8-channel modular valves.
-
29
Dilute TetraSpeck beads at a concentration of 1:200 in DPBS. Prepare a coverslip-slide assembly, with TetraSpeck bead solution filled in between.
-
30
Mount the TetraSpeck bead slide onto the microscope.
-
31
Focus onto the coverslip surface and take z-stack images of TetraSpeck beads in the 647-nm channel and 560-nm channel to correct the color shift for chromatin tracing. The z-stack image covers a range of about 5.5 μm, with a 200-nm step size and 0.4 s exposure time at each step.
▲CRITICAL STEP Adjust the focus lock and z-stack starting position accordingly so that the z-stack fully covers the TetraSpeck beads in the axial direction.
-
32
Unmount the TetraSpeck bead slide and mount the sample. Select FOVs. Save the FOV positions for sequential imaging of the same FOVs.
▲CRITICAL STEP For MINA implementations in tissue sections, select regions where tissues remain intact.
-
33For chromatin tracing in cell lines, follow option A. For MINA in tissue samples, follow option B.
- Chromatin tracing
-
Focus onto the coverslip surface and take z-stack images in the 647-nm and 560-nm channels for secondary probe hybridization imaging. Take z-stack images in the 488-nm channel for fiducial bead imaging. The z-stacks cover a range of about 7 μm, with a 200-nm step size and 0.4 s exposure time at each step. The laser power densities at the sample are 0.036 kW/cm2 at 488 nm, 0.20 kW/cm2 at 560 nm, and 0.31 kW/cm2 at 647 nm.▲CRITICAL STEP Yellow-green fiducial beads serve as fiducial markers to correct for sample drift during the sequential hybridizations.▲CRITICAL STEP Adjust the focus lock and z-stack starting position accordingly so that the z-stack fully covers the cell layer in the axial direction.▲CRITICAL STEP If the primary probes are conjugated with fluorophores, acquire images for the chromosome territories prior to secondary hybridizations. If the primary probes are non-conjugated, perform sequential secondary hybridizations directly.? TROUBLESHOOTING
-
To recapitulate the human chromosome 21 (hChr21) tracing results in the original chromatin tracing work17, image the first 17 TADs in the 647-nm channel from hyb1 to hyb17. Image the last 17 TADs in the 560-nm channel from hyb1 to hyb17.? TROUBLESHOOTING
-
- MINA
-
Image the pre-hybridized sample from step 24B(xiv). Focus onto the coverslip surface and take z-stack images in the 750-nm channel for MERFISH secondary probe hybridization imaging. Take z-stack images in the 647-nm channel for 3D fibrillarin imaging, and in the 488-nm channel for fiducial bead imaging. The z-stacks cover a range of about 7 μm, with a 200-nm step size and 0.4 s exposure time at each step. The laser power densities at the sample are 0.036 kW/cm2 at 488 nm, 0.20 kW/cm2 at 560 nm, 0.31 kW/cm2 at 647 nm, and 1.1 kW/cm2 at 750 nm. This step (denoted as Hyb0) acquires the first round of MERFISH images in the 750-nm channel and 3D fibrillarin staining images in the 647-nm channel.▲CRITICAL STEP Adjust the focus lock and z-stack starting position accordingly so that the z-stack fully covers the cell layer in the axial direction.? TROUBLESHOOTING
-
After image acquisition for the pre-hybridized sample (denoted as Hyb0), continue with 40 rounds of secondary hybridizations with the same image parameters as in step 33B(i). To recapitulate our recent MINA work18, image the 16 rounds of MERFISH readout hybridizations in the 750-nm channel from Hyb0 to Hyb15. Image the first 40 TADs of mChr19 in the 647-nm channel from Hyb1 to Hyb40. Image the last 10 TADs of mChr19 in the 560-nm channel from Hyb21 to Hyb30. Image the 19 consecutive loci upstream of Scd2 in the 560-nm channel from Hyb1 to Hyb19. Image the WGA labeling in the 560-nm channel in Hyb31.? TROUBLESHOOTING
-
-
34For each round of secondary probe hybridization, set the microfluidics steps as follows.
Solution Flow speed (mL/min) Volume (mL) Secondary probes 0.6 2.5 Secondary probe hybridization 0 Hybridize for 30 min. Secondary wash buffer 0.6 1.2 Imaging buffer 0.6 1.2 ▲CRITICAL STEP Keep the flow rate <0.6 mL/min. The cells can detach from the coverslip with high flow speed.
? TROUBLESHOOTING
-
35
To extinguish the fluorescence signals after each imaging round, flow 1.2 mL bleach buffer through the chamber to replace the imaging buffer. For photobleaching, the laser power densities at the sample are 1.1 kW/cm2 at 647 nm, 2.2 kW/cm2 at 560 nm and 1.1 kW/cm2 at 750 nm.
? TROUBLESHOOTING
-
36
Repeat steps 34–35 for all secondary hybridizations.
-
37
(OPTIONAL) At the end of MINA experiments only, flow 2.5 mL 1 μg/mL DAPI solution at a speed of 0.6 mL/min through the chamber and incubate for 10 min. The schematic diagram of the sequential hybridization and imaging procedure is illustrated in Fig. 4.
-
38
(OPTIONAL) Flow 1.2 mL DPBS at a speed of 0.6 mL/min through the chamber to wash off DAPI solution. Flow 1.2 mL imaging buffer at a speed of 0.6 mL/min through the chamber. Take z-stack images for fiducial beads in the 488-nm channel and for DAPI stain in the 405-nm channel for each FOV. The z-stacks cover a range of about 7 μm, with a 200-nm step size and 0.4 s exposure time at each step. The laser power densities at the sample are 0.036 kW/cm2 at 488 nm and 0.014 kW/cm2 at 405 nm.
? TROUBLESHOOTING
-
39
Transfer image data to the analysis workstation for image analysis.
▲CRITICAL STEP The images generated from chromatin tracing and MINA typically occupy 300 GB and 1.5 TB of hard drive space, respectively. Allocate sufficient hard drive space to store the data.
Data analysis with MinaAnalyst ● Timing 2–3 d
-
40
Install MinaAnalyst. Download and install MATLAB version R2018a or a more recent version following the manufacturer’s instructions.
-
41
Download the MinaAnalyst package from https://campuspress.yale.edu/wanglab/mina-analyst/.
-
42
Double-click the “MinaAnalyst.mLapp” file in the package to open the GUI of the application (Fig. 5b).
-
43Input the following files and parameters in the application GUI:
- Experiment file path (required): folder path of the imaging data on local computer. (Please refer to the “parameters_default.m” file introduced below and our example MINA dataset for the default naming conventions of image files.)
- Total number of FOVs (required): number of imaged areas in the dataset.
- (OPTIONAL) MERFISH codebook path: folder path for the MERFISH codebook. This input is only required for MERFISH and MINA analyses. When required, the folder should contain a file named “CodeBook.mat”, a file named “AllCodes.mat”, and a sub-folder named “CodeMatrices” containing files named “CodeMatrix1.mat”, “CodeMatrix2.mat”,…, “CodeMatrix140.mat”. These files and sub-folder comprise the codebook necessary for MERFISH analysis, and are generated by the ProbeDealer software during MERFISH probe design. An empty file indicates that the MinaAnalyst folder contains these files and sub-folder.
- (OPTIONAL) Include lamina association analysis for MINA analysis when the dataset includes nuclear (DAPI) imaging.
-
(OPTIONAL) Include nucleolar association analysis for MINA analysis when the dataset includes nucleolar imaging.▲CRITICAL STEP Additional default parameters and detailed annotations of the parameters are listed in the “parameters_default.m” file in the application package. Users may modify these parameters using the MATLAB script editor or a text editor. Please note that edits made in the MinaAnalyst GUI overwrite the default parameters in the “parameters_default.m” file.
-
44
Perform data analysis. To perform chromatin tracing analysis alone, click the “Run chromatin tracing analysis” button. To perform MERFISH analysis alone, click the “Run MERFISH analysis” button. To perform full MINA analysis (including chromatin tracing analysis, MERFISH analysis, and additional lamina and/or nucleolar analyses if selected), click the “Run MINA analysis” button. Analysis progress is indicated in the GUI panel. The MINA analysis should automatically perform both the chromatin tracing and MERFISH analyses, and generate all the output files listed below.
? TROUBLESHOOTING
-
45Outputs. When performing the chromatin tracing analysis, check that an output file named “ChrList.mat” has been generated that can be accessed through MATLAB. The file contains a structure array named “Chr”, each element of which corresponds to a chromatin trace. The fields of the structure should include:
- x: x coordinates (unit: μm) of each chromatin locus in the trace.
- y: y coordinates (unit: μm) of each chromatin locus in the trace.
- z: z coordinates (unit: μm) of each chromatin locus in the trace.
- r: an array of 1 or 0 values indicating whether each chromatin locus is present (1) or missing (0) in the trace. When a chromatin locus is missing, the corresponding x, y, and z coordinates are set to “NaN” (a “not a number” space holder in MATLAB).
- FOV: serial number of the imaging FOV where this chromatin trace is located.
-
46(OPTIONAL) If performing the MERFISH analysis, check that an output file named “SingleCellAnalysisResults.mat” has been generated and that this contains two structure arrays “CellList” and “MolList”. The “CellList” elements correspond to individual cells and should contain the following fields:
- CellID: serial number of the cell.
- FOV: serial number of the imaging FOV where the cell is located.
- PixelList: pixel indices (linear indices in MATLAB) of the cell area in the FOV.
- Center: center position of the cell in the FOV (unit: pixels).
- RNACopyNumber: an array of RNA copy numbers for each probed transcript species in the cell.
- TotalRNACopyNumber: total detected RNA copy number in the cell.
- OnEdge: a 1 or 0 value indicating whether the cell is overlapping the edge of the FOV (1) or not (0).
The “MolList” elements correspond to individual RNA molecules, and should contain the following fields:
FieldOfView: serial number of the imaging FOV where the molecule is located.
MoleculeX: x coordinate of the molecule (unit: pixels).
MoleculeY: y coordinate of the molecule (unit: pixels).
GeneID: ID number of the transcript species in the MERFISH codebook.
CellID: serial number of the cell that contains this molecule, which corresponds to the “CellID” field of the “CellList” structure.
CellCenterX: x coordinate of the center of the cell in the FOV (unit: pixels).
-
CellCenterY: y coordinate of the center of the cell in the FOV (unit: pixels).
? TROUBLESHOOTING
-
47(OPTIONAL) Check the chromatin traces are matched to individual cells, and the following new fields have been added to the “Chr” structure:
- CellID: serial number of the cell that contains this chromatin trace, which corresponds to the “CellID” of the “CellList” structure. If the matching fails (e.g. due to sample drift, chromatin traces on the edges of an imaging area may be out of the field when imaging cell boundaries), the “CellID” value of the chromatin trace is set to “NaN”.
- MeanXWGA: x coordinate of the center of the chromatin trace in the FOV using the coordinate system of the MERFISH analysis (unit: pixels). The drift correction procedure in the chromatin tracing analysis converts all chromatin locus coordinates to the coordinate system of the Hyb0 imaging round. The MERFISH analysis procedure converts all RNA molecule coordinates to the coordinate system of the cell boundary imaging round. This file (and similarly the next one) records the chromatin trace center position in the latter coordinate system.
- MeanYWGA: y coordinate of the center of the chromatin trace in the FOV using the coordinate system of the MERFISH analysis (unit: pixels).
-
48
(OPTIONAL) If the “Include lamina association analysis” option is selected during MINA analysis, add a field named “MinDisToLamina” to the “Chr” structure to record the distance from each chromatin locus to the nuclear lamina (unit: μm). If the “Include nucleolar association analysis” option is selected, add a field named “MinDisToNucleolar” to the “Chr” structure to record the minimum distance from each chromatin locus to nucleoli (unit: μm). When a chromatin locus is missing in the chromatin trace, the corresponding “MinDisToLamina” and/or “MinDisToNucleolar” value is set to “NaN”.
? TROUBLESHOOTING
Troubleshooting
Troubleshooting advice can be found in Table 1.
Table 1.
Troubleshooting guide
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| Step 22 | Non-specific band in the TBE-Urea polyacrylamide gel. | Non-specific amplifications during limited-cycle PCR. | Pause the limited-cycle PCR right before the amplification curve reaches plateau. Use the correct melting temperature of the primers for annealing in the PCR reaction. |
| Step 23B(vii) | Tissue sections detach from the coverslip. | Insufficient drying of tissue sections after cryosectioning. | Allow the tissue section to dry at room temperature for 15 s prior to tissue fixation. |
| The 0.01% (wt/vol) poly-L-lysine treatment is insufficient for tissue attachment during the experiment. | Increase the poly-L-lysine concentration and incubation time. | ||
| Step 33B(ii) | High background of oligo-conjugated WGA. (Fig. 8a) | The concentration of DBCO-PEG5-NHS ester is too high. | Perform a titration of the DBCO-PEG5-NHS ester to find the optimal concentration. |
| Insufficient purification of DBCO-conjugated WGA. | Perform spin column-based buffer exchange with DPBS 3 times to remove potential residual DBCO. | ||
| Step 33 | DNA FISH signals are too weak. (Fig. 8a) | Insufficient chromatin tracing primary probe concentration. | Increase the chromatin tracing primary probe concentration. Perform a titration of primary probes when working with new samples or probe sets. |
| Insufficient primary probe incubation time. | Prolong the primary probe incubation time (up to 48 hrs). | ||
| Insufficient tissue permeabilization. | Treat the sample with 0.5% (vol/vol) Triton X-100 for 30 min or longer. | ||
| Insufficient denaturation of the genomic DNA. | Increase the heat denaturation time from 3 min to 5 min. | ||
| Step 33 | DNA FISH background signals are too high. | Chromatin tracing primary probe concentration is too high. | Decrease the chromatin tracing primary probe concentration. Perform a titration of primary probes when working with new samples or probe sets. |
| Autofluorescence is too strong in the sample. | Perform 1 mg/mL sodium borohydride treatment to the sample for 10 min to reduce autofluorescence. | ||
| Step 33B | RNA FISH signals are too weak or no RNA FISH signals. (Fig. 8a) | The RNA species are degraded during the experiment. | Use RNase-free reagents and equipment during the experiment. Apply RNase inhibitors as required in the protocol. |
| Insufficient primary probe concentration. | Increase the primary probe concentration during tracing. Perform a titration of primary probes when working with new samples or probe sets. | ||
| Insufficient primary probe incubation. | Prolong the primary probe incubation time (up to 48 hrs) | ||
| Insufficient sample permeabilization. | Treat the sample with 0.5% (vol/vol) Triton X-100 for 30 min or longer during sample permeabilization. | ||
| Step 33B | RNA FISH background signals are too high. | Primary probe concentration is too high. | Decrease the primary probe concentration. Perform a titration of primary probes when working with new samples or probe sets. |
| Autofluorescence is too strong in the sample. | Perform 1 mg/mL sodium borohydride treatment to the sample for 10 min to reduce autofluorescence. | ||
| Step 33B | RNA single-molecule FISH signals are patchy without distinct single-molecule foci. | Certain/many RNA species with high expression levels are included in the MERFISH library or target RNA species are localized within spatial clusters. | Check the FPKM values and identities of the target RNA species when designing probes. |
| Step 33B(i) | Poor antibody staining quality for anti-fibrillarin antibody. (Fig. 8a) | Insufficient sample permeabilization. | Treat the sample with 0.5% (vol/vol) Triton X-100 for 30 min or longer during sample permeabilization. |
| Primary antibody concentration of the anti-fibrillarin antibody is too low. | Incubate the sample with higher anti-fibrillarin primary antibody concentration. A 1:100 dilution is typically used for immunofluorescence staining. | ||
| Step 38 | Poor DAPI staining quality. | Insufficient washing of DAPI stain. | Use 2x SSC to wash away non-specific DAPI stain. |
| Steps 34, 35 | Fiducial bead signals get weaker during sequential imaging. | Fiducial beads gradually get photobleached during the sequential hybridization procedure. | Increase the 488-nm channel laser intensity around hyb20 when performing more than 20 rounds of secondary probe hybridizations. |
| Steps 34, 35 | Sample drift is too large during the sequential hybridizations. | Flow speed is too high. | Decrease the flow speed. |
| Tissue sections dislodge from the coverslip. | Increase the poly-L-lysine concentration and incubation time for coverslip treatment prior to tissue section attachment. | ||
| Steps 34, 35 | Focus lock is lost during the sequential hybridizations. | Air bubble in the flow system. | Tighten the chamber before buffer flow. |
| The selected FOVs are too far away from each other. | All selected FOVs are recommended to be within a 2 mm × 5 mm area. | ||
| Step 44 | Detection efficiency is too low for chromatin tracing or MERFISH. | Start positions of the z-stack images are too low or too high. | Adjust the start positions of the z-stack images to make sure the z-range covers a layer of cells. |
| DNA FISH hybridization conditions are not ideal. | Adjust the DNA primary probe concentration, DNA primary probe incubation time, cell permeabilization, and genome heat denaturation conditions. | ||
| RNA FISH hybridization conditions are not ideal. | Adjust the RNA primary probe concentration, RNA primary probe incubation time, and cell permeabilization. Apply murine RNase inhibitors at required steps. | ||
| Steps 46, 48 | Poor cell segmentation or nuclei segmentation. | Poor labeling quality for the cell membrane (WGA). | Adjust the WGA-oligo conjugation conditions by titrating the WGA and DBCO-PEG5-NHS ester molar ratios and adjusting the purification conditions of DBCO-conjugated WGA. |
| Poor labeling quality for the nuclei (DAPI). | Adjust the DAPI incubation and washing conditions. | ||
| Default parameters for cell segmentation and nuclei segmentation are not ideal for the specific sample. | Adjust the parameters in the “parameters_default.m” file until the algorithms are ideal for the specific sample. |
Timing
Steps 1–9, chromatin tracing probe design with ProbeDealer: 1 d.
Steps 10–13, (OPTIONAL) MERFISH probe design with ProbeDealer: 1 d.
Steps 14–22, primary probe synthesis from template oligo pool: 1.5 d.
Step 23, sample preparation: 3–4 d.
Step 24, primary probe hybridization: 1.5 d.
Steps 25–39, sequential hybridization and imaging: ~1 h per round for 15 FOVs.
Steps 40–48, data analysis with MinaAnalyst: 2–3 d.
Anticipated results
This protocol describes a detailed procedure of template oligo pool probe design, primary probe synthesis, sample processing, primary probe hybridization, sequential hybridization and imaging, and image analysis for both chromatin tracing and MINA. Here we provide a typical set of results for MINA that includes a chromatin tracing component.
First, at the level of TAD-to-chromosome length scale, the chromatin tracing component of MINA reveals 3D folding of individual chromatin traces and generates spatial distance matrices of individual traces, with each matrix element representing the spatial distance between a pair of TADs (Fig. 7a). Population averaging yields a mean spatial distance matrix (Fig. 7b). Calculation of the Pearson correlation coefficient between the inverse Hi-C contact frequencies and mean spatial distances shows a strong correlation and validates the chromatin tracing data quality (Fig. 7c). Based on the mean spatial distance matrix, a normalization and compartmentalization analysis yields population-averaged A-B compartment scores of the target TADs17,18 (Fig. 7d). MINA further measures the positions of nucleoli and nuclear lamina and quantifies the probabilities of different TADs being in spatial proximity to the nucleoli and nuclear lamina. Analyses of the correlations between A-B compartment scores and nucleolar/lamina association ratios show the expected anti-correlations (Fig. 7e, 7f). In addition, the chromatin tracing component of MINA can also reveal the different tendencies of different chromatin regions to be localized to the chromosome surface (Fig. 7g).
Fig.7. Example results of single-copy and ensemble analyses of chromatin tracing.
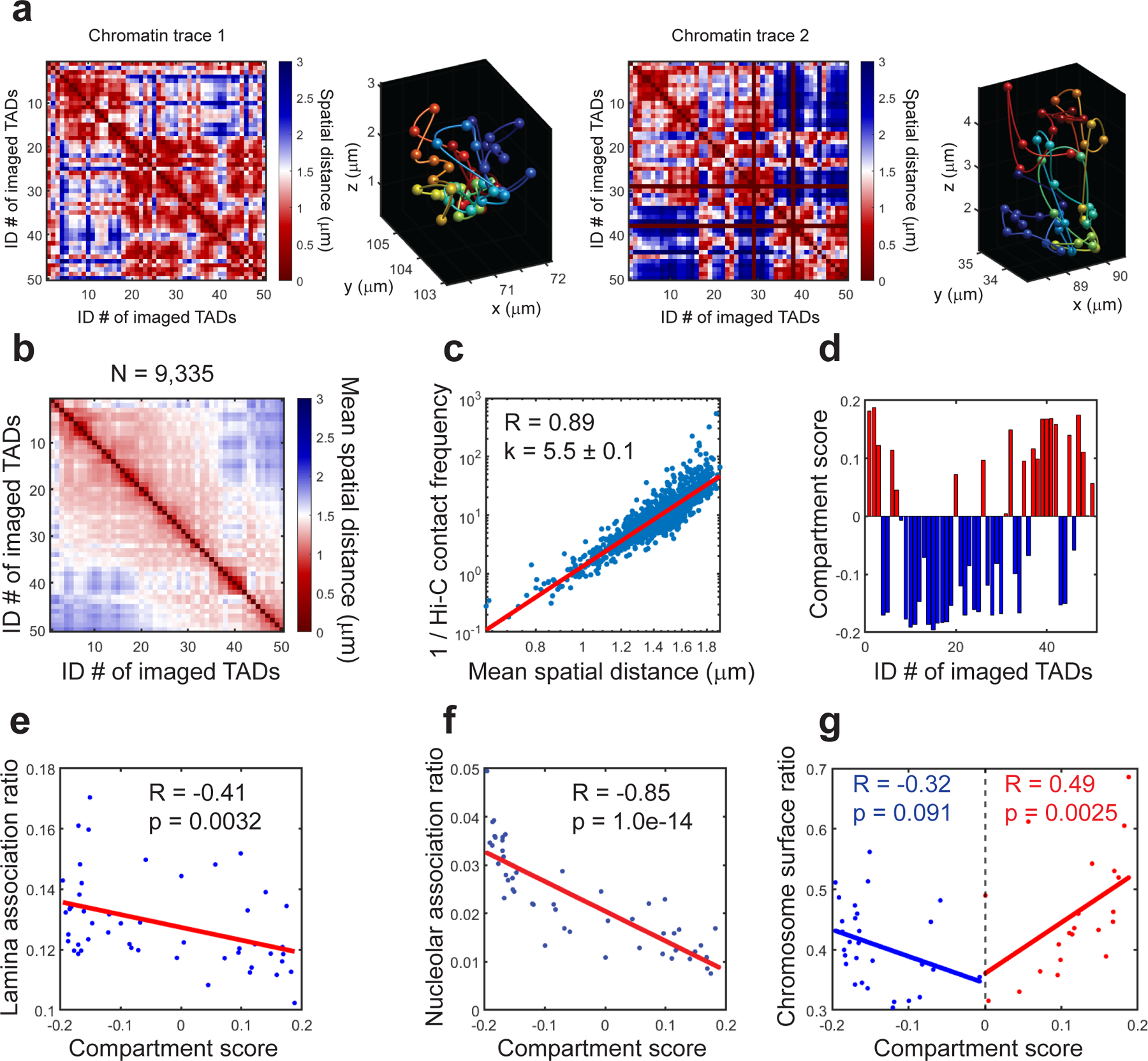
a, Two single-copy chromatin traces of mChr19 in mouse fetal liver. (Left panels) Spatial distance matrices of the two single-copy chromatin traces. Each matrix element represents the spatial distance between a pair of imaged TADs in the chromatin trace. (Right panels) The reconstructed chromatin traces in 3D, with TADs represented by pseudo-colored spheres connected by smooth curves to guide the eye. b, Mean spatial distance matrix for mChr19 TADs. Each element represents the mean spatial distance between a pair of imaged TADs. A total of 9,335 copies of mChr19 are analyzed. c, Correlations between the inverse Hi-C contact frequency and mean spatial distances. d, Compartment scores of imaged TADs. Red bars: compartment A TADs. Blue bars: compartment B TADs. e, Correlation between lamina association ratios and compartment scores of mChr19 TADs. f, Correlation between nucleolar association ratios and compartment scores of mChr19 TADs. g, Correlation between chromosome surface ratios and compartment scores of mChr19 TADs. Red and blue dots represent compartment A and B TADs, respectively. This figure is generated using one replicate from the published MINA work18. All experiments performed are approved by IACUC of Yale University. Raw data are provided in SourceData_Fig7.
Second, at the promoter-enhancer length scale with 5-kb genomic resolution, the chromatin tracing component of MINA similarly generates a mean spatial distance matrix, with each element showing the distance between a pair of target genomic loci. By comparing the mean distance matrices from different cell types, MINA can reveal cell-type-specific enhancer-promoter interactions between target loci (Fig. 8b).
Fig.8. Example results of gene expression and cell-type-specific genome architecture analyses.
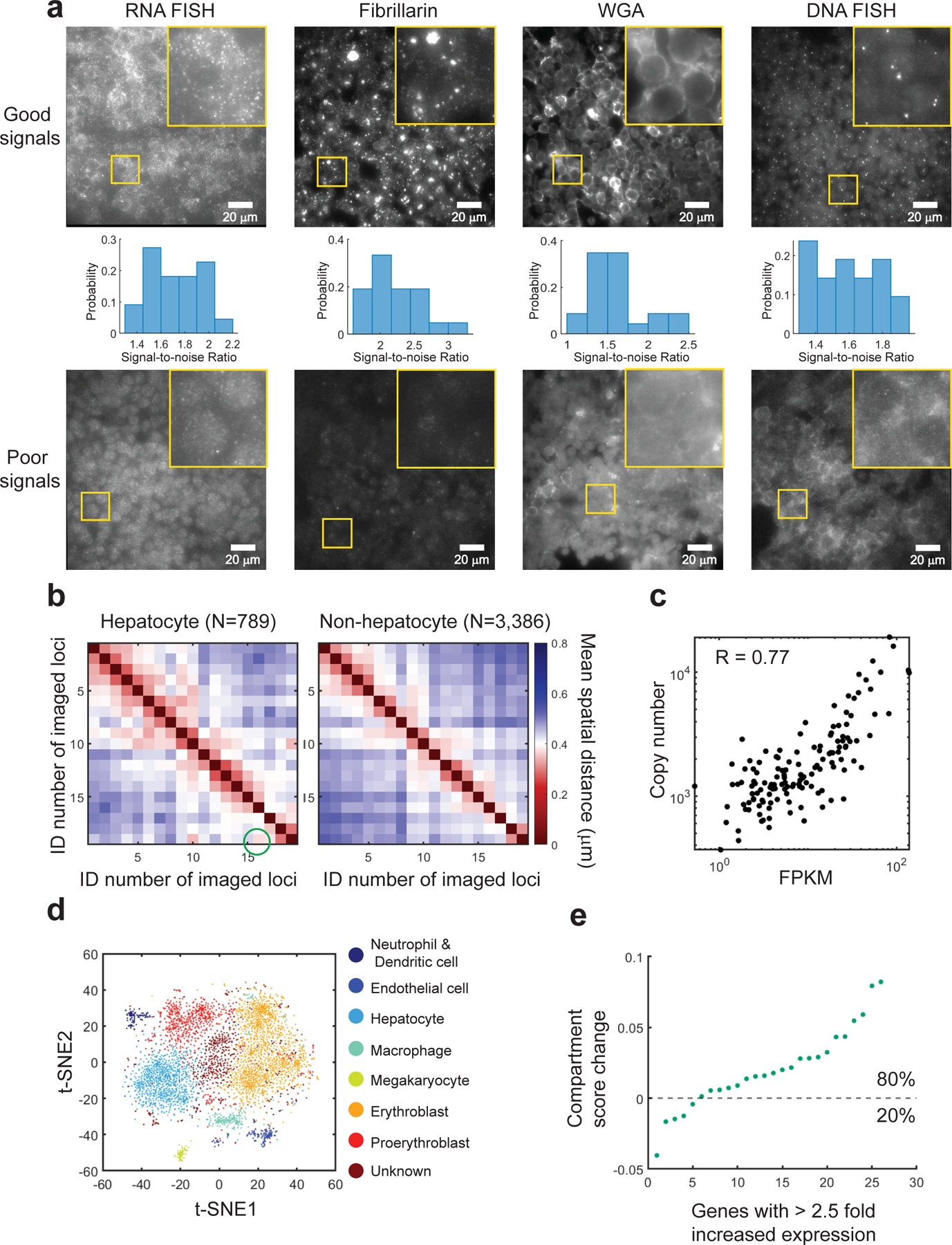
a, Example raw images of successful (upper panels) and poor (lower panels) RNA FISH signals, fibrillarin antibody staining signals, WGA staining signals and DNA FISH signals. Yellow boxes in the top-right corner contain 3.5x zoom-in images of the selected regions. The histograms in the middle panels show distributions of signal-to-noise ratios in raw images with good signals. b, Mean spatial distance matrices for the upstream cis-regulatory region of the gene Scd2 in mouse fetal liver hepatocytes and non-hepatocytes. Each element represents the mean spatial distance between a pair of imaged 5-kb loci. The green circle highlights a hepatocyte-specific enhancer-promoter interaction between locus 16 (a candidate enhancer) and locus 19 (the Scd2 promoter). A one-side t-test of the spatial distances between locus 16 and locus 19 in hepatocytes versus non-hepatocytes has a p-value of 0.019. A total of 789 copies and 3,386 copies of chromatin traces are analyzed for hepatocytes and non-hepatocytes, respectively. c, Correlation between total RNA copy numbers derived from MERFISH and FPKM values derived from bulk RNA sequencing. d, t-distributed stochastic neighbor embedding plot for the single-cell RNA expression profiles. Each pseudo-colored cluster represents a cell type in mouse fetal liver. Cell type identities are assigned based on cell type marker gene enrichment in each cluster. e, Change in compartment scores of the TADs that contain genes with more than 2.5 fold increase in expression when comparing one cell type to another. The genes are ranked according to the compartment score change. In most cases, the increased gene expression is associated with a positive change in compartment score. Panels b-e are generated using one replicate from the published MINA work18. All experiments performed are approved by IACUC of Yale University. Raw data are provided in SourceData_Fig8.
Third, in terms of RNA expression profiling, the MERFISH component of MINA quantifies the copy numbers of all targeted RNA species in single cells. The RNA copy numbers correlate with bulk RNA-sequencing results at the population level (Fig. 8c). The MERFISH results further allow cell type identification by clustering of single-cell gene expression profiles18 (Fig. 8d). The cell type identification allows all nucleome architectures above and the relationship between them to be analyzed in a cell-type specific fashion (e.g. Fig. 8b), and yields the co-variation of nucleome and transcriptome features (Fig. 8e).
Supplementary Material
Supplementary Table 1. Sequences of the forward primers, reverse primers and reverse transcription primers for primary probe synthesis.
Supplementary Table 2. Sequences of Alexa Fluor 750-labeled MERFISH secondary probes, Alexa Fluor 647-labeled chromatin tracing secondary probes, and ATTO 565-labeled chromatin tracing secondary probes.
Acknowledgements
S.W. is supported by NIH Director’s New Innovator Award 1DP2GM137414, NCI grant 1R33CA251037, NHGRI grant 1R01HG011245, and 4DN NCI grant 1U01CA260701. This work is partially supported by NHGRI grant 1R01HG011245. M.H. and Y.Cheng are supported by the China Scholarship Council (CSC) Grant. J.S.D.R. is supported by NIH Predoctoral Training Grant (2T32GM007499). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Code availability
The ProbeDealer package can be downloaded from https://campuspress.yale.edu/wanglab/probedealer. The MinaAnalyst package can be downloaded from https://campuspress.yale.edu/wanglab/mina-analyst/.
Competing interests
S.W. is one of the inventors on a patent applied for by Harvard University related to MERFISH. The other authors declare no competing interests.
Data availability
An example raw dataset of two imaging fields are downloadable from https://campuspress.yale.edu/wanglab/mina-analyst/. The full datasets used to generate Fig. 7 and Fig. 8 are not uploaded online due to the prohibitively large size. The full datasets are available from the corresponding author upon request.
References
- 1.Finn EH & Misteli T Molecular basis and biological function of variability in spatial genome organization. Science 365, eaaw9498 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gorkin DU, Leung D & Ren B The 3D Genome in Transcriptional Regulation and Pluripotency. Cell Stem Cell 14, 762–775 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levine M, Cattoglio C & Tjian R Looping Back to Leap Forward: Transcription Enters a New Era. Cell 157, 13–25 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dekker J & Mirny L The 3D Genome as Moderator of Chromosomal Communication. Cell 164, 1110–1121 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bickmore WA & van Steensel B Genome Architecture: Domain Organization of Interphase Chromosomes. Cell 152, 1270–1284 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Cremer T & Cremer M Chromosome territories. Cold Spring Harb. Perspect. Biol 2, a003889 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonora G, Plath K & Denholtz M A mechanistic link between gene regulation and genome architecture in mammalian development. Curr. Opin. Genet. Dev 27, 92–101 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pope BD et al. Topologically associating domains are stable units of replication-timing regulation. Nature 515, 402–405 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Steensel B & Furlong EEM The role of transcription in shaping the spatial organization of the genome. Nat. Rev. Mol. Cell Biol 20, 327–337 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y et al. Spatial Organization of the Mouse Genome and Its Role in Recurrent Chromosomal Translocations. Cell 148, 908–921 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Senigl F et al. Topologically Associated Domains Delineate Susceptibility to Somatic Hypermutation. Cell Rep 29, 3902–3915.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Misteli T & Soutoglou E The emerging role of nuclear architecture in DNA repair and genome maintenance. Nat. Rev. Mol. Cell Biol 10, 243–254 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akdemir KC et al. Disruption of chromatin folding domains by somatic genomic rearrangements in human cancer. Nat. Genet 52, 294–305 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lieberman-Aiden E et al. Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science 326, 289–293 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rao SSP et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 159, 1665–1680 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Q, Sun Q, Czajkowsky DM & Shao Z Sub-kb Hi-C in D. melanogaster reveals conserved characteristics of TADs between insect and mammalian cells. Nat. Commun 9, 188 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang S et al. Spatial organization of chromatin domains and compartments in single chromosomes. Science 353, 598–602 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu M et al. Multiplexed imaging of nucleome architectures in single cells of mammalian tissue. Nat. Commun 11, 2907 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bintu B et al. Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science 362, eaau1783 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sawh AN et al. Lamina-Dependent Stretching and Unconventional Chromosome Compartments in Early C. elegans Embryos. Mol. Cell 78, 96–111.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nir G et al. Walking along chromosomes with super-resolution imaging, contact maps, and integrative modeling. PLoS Genet 14, e1007872 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mateo LJ et al. Visualizing DNA folding and RNA in embryos at single-cell resolution. Nature 568, 49–54 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cardozo Gizzi AM et al. Microscopy-Based Chromosome Conformation Capture Enables Simultaneous Visualization of Genome Organization and Transcription in Intact Organisms. Mol. Cell 74, 212–222.e5 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Su J-H, Zheng P, Kinrot SS, Bintu B & Zhuang X Genome-Scale Imaging of the 3D Organization and Transcriptional Activity of Chromatin. Cell 182, 1641–1659.e26 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu M & Wang S Chromatin Tracing: Imaging 3D Genome and Nucleome. Trends Cell Biol 31, 5–8 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Plath K, Mlynarczyk-Evans S, Nusinow DA & Panning B Xist RNA and the Mechanism of X Chromosome Inactivation. Annu. Rev. Genet 36, 233–278 (2002). [DOI] [PubMed] [Google Scholar]
- 27.Galupa R & Heard E X-chromosome inactivation: new insights into cis and trans regulation. Curr. Opin. Genet. Dev 31, 57–66 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Lessing D, Anguera MC & Lee JT X chromosome inactivation and epigenetic responses to cellular reprogramming. Annu. Rev. Genomics Hum. Genet 14, 85–110 (2013). [DOI] [PubMed] [Google Scholar]
- 29.Guelen L et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 453, 948–951 (2008). [DOI] [PubMed] [Google Scholar]
- 30.Németh A et al. Initial Genomics of the Human Nucleolus. PLOS Genet 6, e1000889 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Koningsbruggen S et al. High-resolution whole-genome sequencing reveals that specific chromatin domains from most human chromosomes associate with nucleoli. Mol. Biol. Cell 21, 3735–3748 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen KH, Boettiger AN, Moffitt JR, Wang S & Zhuang X Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, aaa6090 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moffitt JR et al. Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science 362, eaau5324 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xia C, Fan J, Emanuel G, Hao J & Zhuang X Spatial transcriptome profiling by MERFISH reveals subcellular RNA compartmentalization and cell cycle-dependent gene expression. Proc. Natl. Acad. Sci 116, 19490–19499 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boyle S, Rodesch MJ, Halvensleben HA, Jeddeloh JA & Bickmore WA Fluorescence in situ hybridization with high-complexity repeat-free oligonucleotide probes generated by massively parallel synthesis. Chromosome Res 19, 901–909 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamada NA et al. Visualization of Fine-Scale Genomic Structure by Oligonucleotide-Based High-Resolution FISH. Cytogenet. Genome Res 132, 248–254 (2011). [DOI] [PubMed] [Google Scholar]
- 37.Beliveau BJ et al. Versatile design and synthesis platform for visualizing genomes with Oligopaint FISH probes. Proc. Natl. Acad. Sci 109, 21301–21306 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu M et al. ProbeDealer is a convenient tool for designing probes for highly multiplexed fluorescence in situ hybridization. Sci. Rep 10, 22031 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Farack L & Itzkovitz S Protocol for Single-Molecule Fluorescence In Situ Hybridization for Intact Pancreatic Tissue. STAR Protoc 1, 100007 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moffitt JR et al. High-performance multiplexed fluorescence in situ hybridization in culture and tissue with matrix imprinting and clearing. Proc. Natl. Acad. Sci 113, 14456–14461 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xia C, Babcock HP, Moffitt JR & Zhuang X Multiplexed detection of RNA using MERFISH and branched DNA amplification. Sci. Rep 9, 7721 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stevens TJ et al. 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature 544, 59–64 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nagano T et al. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 502, 59–64 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tan L, Xing D, Chang C-H, Li H & Xie XS Three-dimensional genome structures of single diploid human cells. Science 361, 924–928 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dey SS, Kester L, Spanjaard B, Bienko M & van Oudenaarden A Integrated genome and transcriptome sequencing of the same cell. Nat. Biotechnol 33, 285–289 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Macaulay IC et al. G&T-seq: parallel sequencing of single-cell genomes and transcriptomes. Nat. Methods 12, 519–522 (2015). [DOI] [PubMed] [Google Scholar]
- 47.Moffitt JR et al. High-throughput single-cell gene-expression profiling with multiplexed error-robust fluorescence in situ hybridization. Proc. Natl. Acad. Sci 113, 11046–11051 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rouillard J-M OligoArray 2.0: design of oligonucleotide probes for DNA microarrays using a thermodynamic approach. Nucleic Acids Res 31, 3057–3062 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beliveau BJ et al. OligoMiner provides a rapid, flexible environment for the design of genome-scale oligonucleotide in situ hybridization probes. Proc. Natl. Acad. Sci 115, E2183–E2192 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Passaro M et al. OligoMinerApp: a web-server application for the design of genome-scale oligonucleotide in situ hybridization probes through the flexible OligoMiner environment. Nucleic Acids Res 48, W332–W339 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moffitt JR & Zhuang X RNA Imaging with Multiplexed Error-Robust Fluorescence In Situ Hybridization (MERFISH). in Methods in Enzymology vol. 572 1–49 (Elsevier, 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rasnik I, McKinney SA & Ha T Nonblinking and long-lasting single-molecule fluorescence imaging. Nat. Methods 3, 891 (2006). [DOI] [PubMed] [Google Scholar]
Key references using this protocol
Wang, S. et al. Science 353, 598–602 (2016):
Liu, M. et al. Nat. Commun. 11, 2907 (2020):
https://doi.org/10.1038/s41467–020-16732–5
Hu, M. et al. Scientific Reports 10, 22031 (2020):
Key data used in this protocol
Liu, M. et al. Nat. Commun. 11, 2907 (2020):
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Sequences of the forward primers, reverse primers and reverse transcription primers for primary probe synthesis.
Supplementary Table 2. Sequences of Alexa Fluor 750-labeled MERFISH secondary probes, Alexa Fluor 647-labeled chromatin tracing secondary probes, and ATTO 565-labeled chromatin tracing secondary probes.
Data Availability Statement
An example raw dataset of two imaging fields are downloadable from https://campuspress.yale.edu/wanglab/mina-analyst/. The full datasets used to generate Fig. 7 and Fig. 8 are not uploaded online due to the prohibitively large size. The full datasets are available from the corresponding author upon request.