

Abstract
Inhibition of glutaminase-1 (GLS-1) hampers the proliferation of tumor cells reliant on glutamine. Known glutaminase inhibitors have potential limitations, and in vivo exposures are potentially limited due to poor physicochemical properties. We initiated a GLS-1 inhibitor discovery program focused on optimizing physicochemical and pharmacokinetic properties, and have developed a new selective inhibitor, Compound 27 (IPN60090), which is currently in phase 1 clinical trials. Compound 27 attains high oral exposures in preclinical species, with strong in vivo target engagement, and should robustly inhibit glutaminase in humans.
Graphical Abstract
INTRODUCTION
Glutaminase (GLS-1), an enzyme that converts glutamine to glutamate, is a promising therapeutic target in tumors dependent on glutamine.1,2,3,4 Inhibition of GLS-1 should affect the production of multiple biochemical intermediates required for cellular proliferation and maintenance, including glutathione (involved in cellular redox balance), α-ketoglutarate (tricarboxylic acid cycle intermediate), and ammonia (building block for protein synthesis) (Figure 1). In addition, by increasing the amount of glutamine in the tumor microenvironment, glutaminase inhibition may help support activation and proliferation of T-cells to combat tumor growth.5,6
Figure 1.
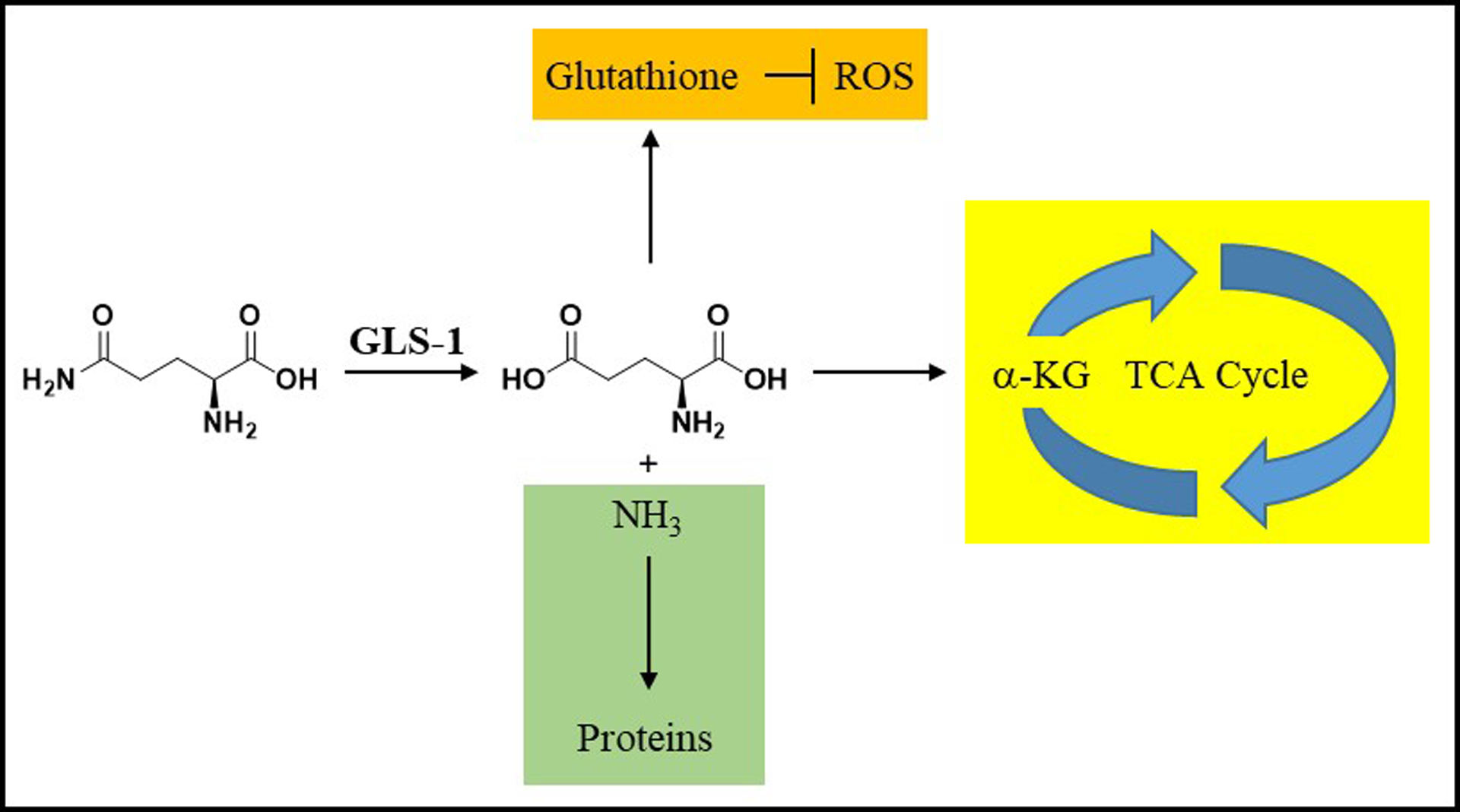
Glutamine metabolism through GLS-1. GLS-1 converts glutamine to glutamate and ammonia. The produced glutamate can be further converted by the cellular machinery to glutathione (which regulates reactive oxygen species (ROS)) or alpha-ketoglutarate (α-KG) (key building block in the tricarboxylic acid (TCA) cycle). The produced ammonia is a building block for cellular protein syntheses.
A growing pool of evidence suggests that tumors rewire metabolic pathways to sustain their needs for energy and cellular building blocks, and that these changes can increase reliance on GLS-1. For example, we and others have disclosed that certain tumors characterized by loss or mutations in KEAP and/or NRF2 genes, which encode for proteins that regulate glutathione production, are sensitive to GLS-1 inhibition.7,8 Additionally, there are multiple reports demonstrating additive or synergistic effects of GLS-1 inhibition in tumor cells when combined with a variety of agents in both in vitro and in vivo models.5,9,10,11,12,13,14 GLS-1 inhibition has also been reported to sensitize tumor cells to radiation.15,16
There are two known isoforms of glutaminase: GLS-1 (also called kidney-type or KGA), and GLS-2 (also called liver-type or LGA). GLS-2 expression appears limited primarily to the liver, while GLS-1 is ubiquitous. As mentioned above, GLS-1 has been validated as a cancer target, and our program focused on selective GLS-1 inhibitors.
GLS-1 can exist as two variants (KGA and GAC), both of which appear susceptible to the same classes of inhibitors. There are three main classes of GLS-1 inhibitors reported, represented by the irreversible and unselective active-site inhibitor 6-diazo-5-oxy-L-norleucine (DON, 1)17 and the GLS-1-selective allosteric inhibitors bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES, 2)18 and “compound 968” (3)19 (Figure 2).
Figure 2.
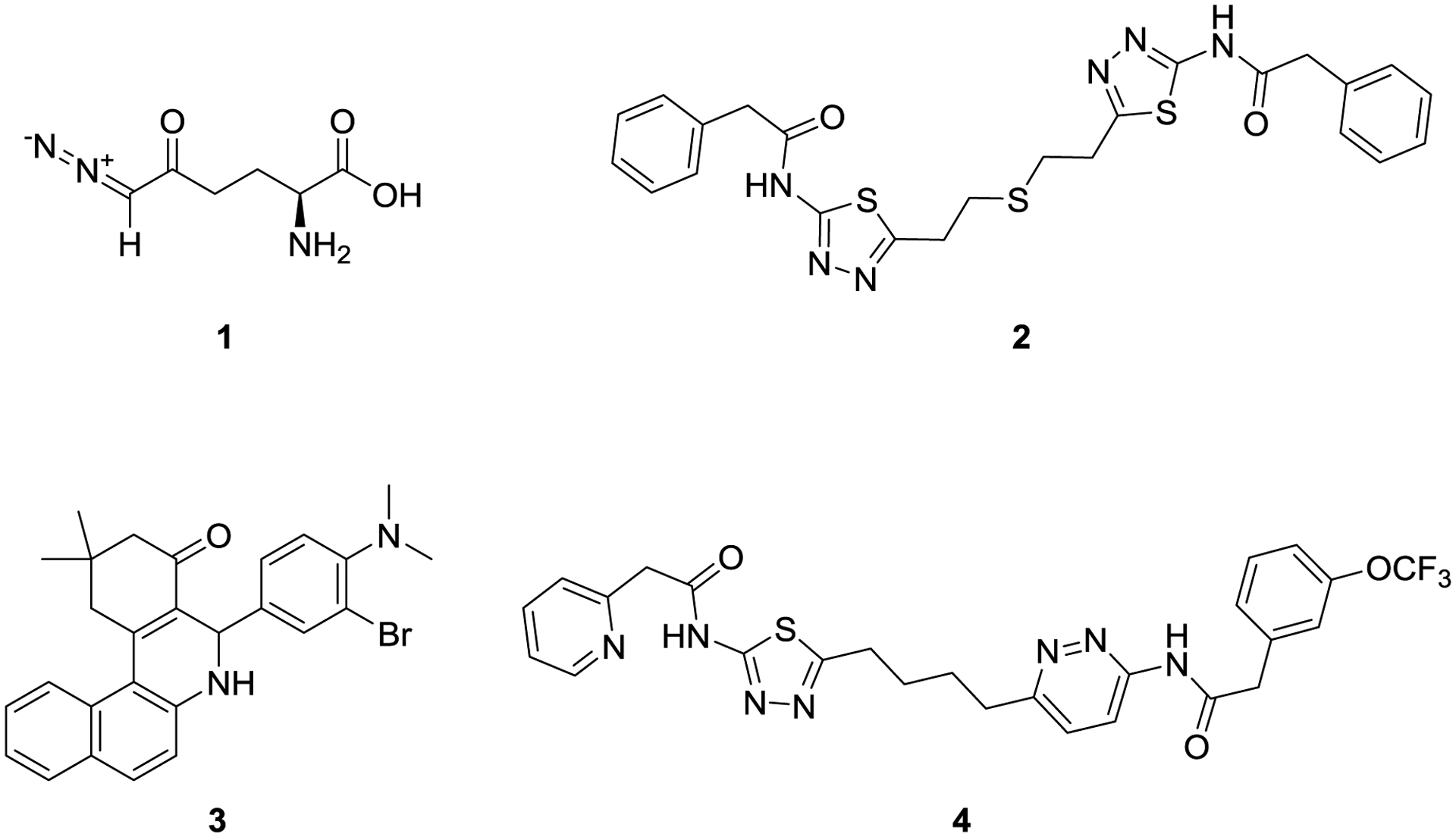
Structures of known GLS inhibitors DON (1), BPTES (2), “compound 968” (3) and CB-839 (4).
Compound 1, first reported to inhibit glutamine catabolism by Willis and Seegmiller17, inhibits multiple glutamine-utilizing enzymes including glutaminase, and its polypharmacology presumably underlies unacceptable toxicities observed in the clinic, although efforts are ongoing to potentially overcome these issues through the use of targeted prodrugs with low dosing regimens.20 Compound 3, reported by Cerione and co-workers, appears to operate through a unique allosteric mechanism; information on exact binding modes is currently limited but the compound has been reported to inhibit both GLS-1 and GLS-2.21,22,23
Compound 2, first disclosed in a patent from Elan18 and inhibition mechanism elucidated by Robinson and co-workers,24 also has a unique allosteric binding mode, which has been elucidated with protein crystallography.25 Compound 2 is reported to have high selectivity over GLS-2, and has served as a chemical starting point for multiple discovery efforts,26 including those of Johns Hopkins University27, Calithera Biosciences8, University of Pittsburgh28,29, Zhejiang University of Technology30, AstraZeneca31, Pfizer32, Agios33, and Rhizen34. Calithera’s efforts have progressed the furthest, with CB-839 (telaglenastat, 4) currently in pivotal clinical trials for multiple oncology indications.35,26 Compound 4 is a potent inhibitor of GLS-1, although its physicochemical and PK properties appear to require fairly high doses (recommended phase 2 doses of 600–800 mg, twice a day36,37,38). Both preclinical and clinical results for compound 4 are encouraging for the use of GLS-1 inhibitors in combination therapies against multiple cancer types.
We also selected compound 2 as an initial lead and focused on improving upon its metabolic stability and solubility profile. Our program prioritized properties in the hope that an eventual candidate would be able to achieve high sustained exposures relative to potencies in the clinic, and thus be positioned to test the clinical potential of maximally inhibiting glutaminase. We describe herein the efforts that culminated in the discovery of IPN60090, a compound with excellent physicochemical and pharmacokinetic properties, currently in phase 1 clinical trials for solid tumors.
RESULTS AND DISCUSSION
Lead Identification.
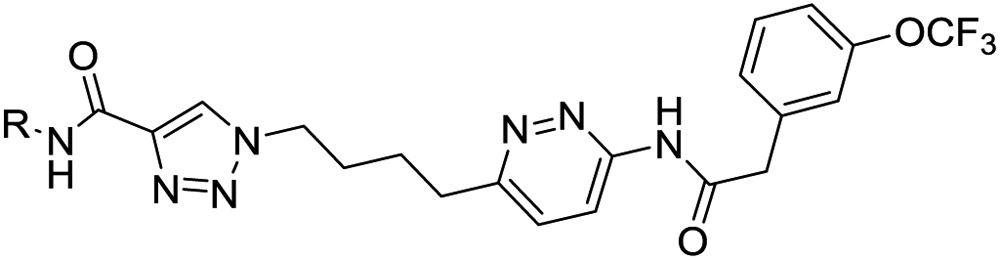
Our strategy started with consideration of a published crystal structure of compound 2 bound to full-length human GLS-1 (GAC isoform).39 Compound 2 binds at the interface of GLS-1 molecules in a 4:2 protein:inhibitor ratio, with the two aminothiadiazole moeities making key hydrogen-bonding interactions with the protein. We therefore embarked on a broad empirical exercise testing aminothiadiazole alternatives expected to present similar patterns of hydrogen bond donors and acceptors. We discovered a variety of unique GLS-1 inhibitors40; this manuscript focuses on the series that produced our clinical candidate, containing an amidotriazole linked to an acylaminopyridazine. Figure 3 shows our proposed chemical series and its hypothesized binding mode to the targeted site of GLS-1. For sake of discussion, we label the various portions of the scaffold as “cores”, “linker” and “wings”.
Figure 3.
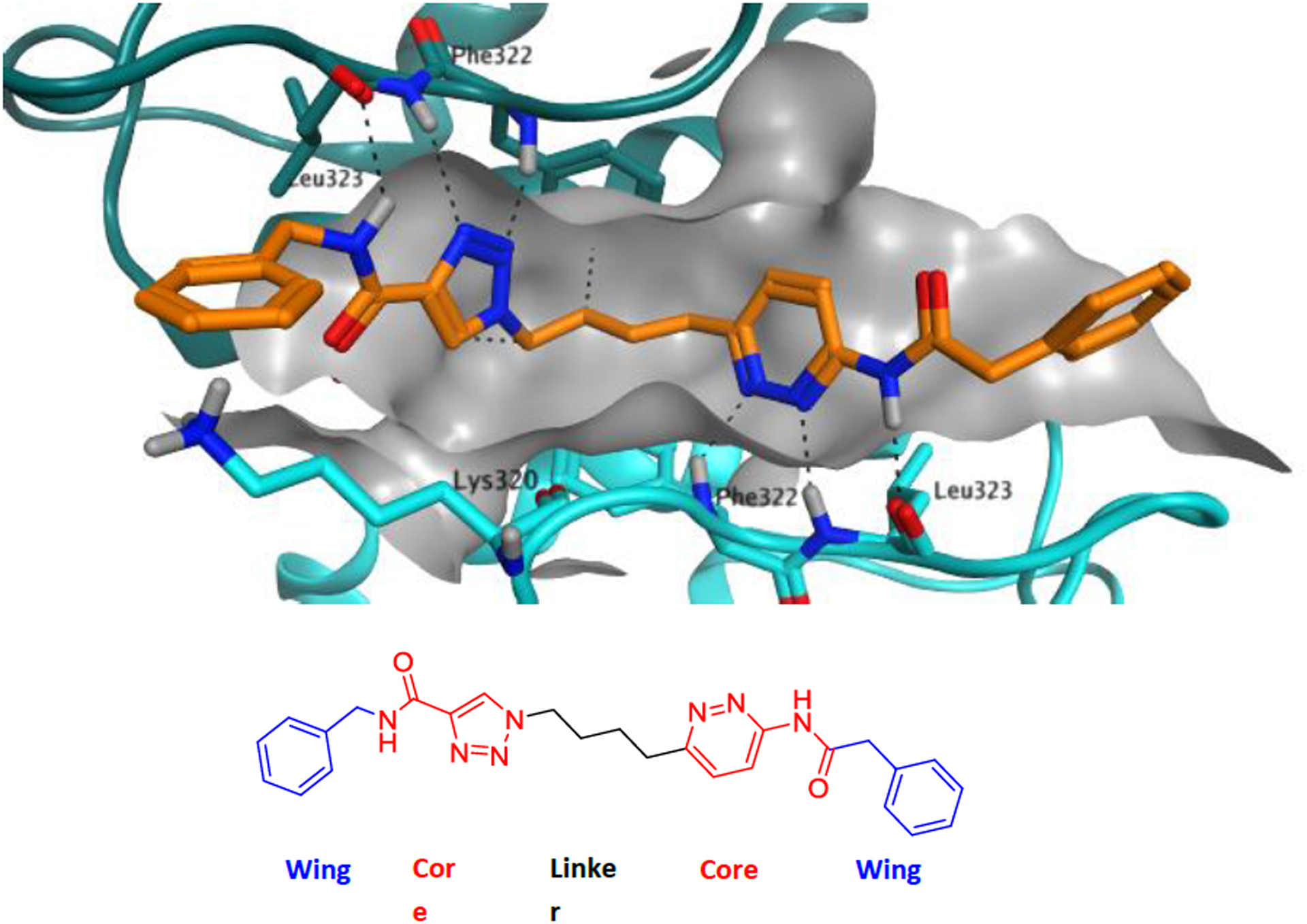
Proposed binding of series example 5 in the GLS-1 tetramer, modeled using PDB 3UO9.39
The acylaminopyridazine core is a known motif in GLS-1 inhibitors and is present in 4; pyridazine can be considered an isostere of thiadiazole, in the same way that benzene and thiophene are considered to be isosteres of each other.41 Amidotriazole cores are, to our knowledge, unprecedented in GLS-1 inhibitors. Comparison of amidotriazole and acylaminopyridazine fragments suggest a close overlay of most hydrogen bond donors and acceptors (Figure 4).
Figure 4.
The hydrogen bonding motifs of amidotriazoles overlay well onto those of aminopyridazines.
The four-carbon linker was first popularized by Thangavelu and co-workers as a substitute for the thioether linker present in 242, and was suggested by modeling and early empirical studies to be an optimal length for our series. We were able to further improve upon this linker, as will be described later in the manuscript. The wings are tolerant of a wide variety of modifications and were a major focus of our optimization efforts.
Scaffold Proof of Concept.
For proof of potency for this novel scaffold, we synthesized compounds 5, 6 and 7, which contain benzyl, or pyridylmethyl and trifluoromethoxybenzyl wings, similar to those present in compounds 2 and 4. Enzyme potencies were evaluated in a GLS-1 (GAC isoform) enzyme assay indirectly measuring conversion of glutamine to glutamate, and cellular potencies were evaluated in a cellular viability assay (A549 cell line). For comparison purposes, we include data from the same assays for compounds 2 and 4. All three of our new compounds demonstrate good potencies in enzyme and cell-based assays (Table 1), thus giving us confidence in exploring the scaffold further.
Table 1.
Scaffold Proof of Concept
| Compound | Structure | GLS IC50 (nM)a |
A549 IC50 (nM)b |
Microsomal Stability (mL/min/kg)c |
pH 7.4 Solubility (μM)d |
|
|---|---|---|---|---|---|---|
| Hum | Rat | |||||
| 5 |
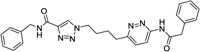
|
14 | 26 | 49 | 128 | 0.04 |
| 6 |
|
6 | 11 | 88 | 283 | 0.8 |
| 7 |
|
14 | 31 | 52 | 162 | 0.4 |
| Compound 2 | 25 | 276 | 87 | 530 | 0.2 | |
| Compound 4 | 6 | 6 | 62 | 203 | 0.3 | |
Inhibition of purified recombinant human GAC assessed via a dual-coupled enzyme assay. IC50 values are reported as the mean of at least three determinations, except for compound 5 (n=1). Standard deviations and counts are included in the supporting information.
Inhibition of proliferation of A549 cells. IC50 values are reported as the mean of at least three determinations, except for compound 5 (n=2). Standard deviations and counts are included in the supporting information.
Liver microsomal intrinsic clearance (mL/min/kg protein).
Kinetic aqueous solubility (μM) in phosphate buffer (100 mM) at pH 7.4; shaken for 1 hour at RT.
Further profiling of compounds 5-7 revealed two major liabilities, also issues for literature comparators 2 and 4, which were the primary foci of our lead optimization efforts (Table 1). Firstly, microsomal stabilities were low (most Clint’s ranked as high clearance by internal criteria, >83 mL/min/kg in rat and >58 mL/min/kg in human); secondly, solubilities were very low (<1 μM). The low solubilities, and possibly the low microsomal stabilities, are likely attributable to the large number of aromatic rings in the compounds, therefore our first optimization efforts focused on replacing one of the aromatic wings with smaller and/or less lipophilic groups. Based on the liabilities just described, the key data that we monitored at this early stage were potencies, microsomal stabilities and solubilities.
Triazole Wing Exploration.
Our scan of triazole wing pieces focused on replacing the heteroaryl motif of parent compound 7 (Table 2). All replacements investigated showed a loss in potency. However, the simple methylamide 8 still retained a reasonable amount of potency, with a higher ligand binding efficiency (0.30 compared to 0.27 for 7)43. More importantly, compound 8 showed much improved microsomal stabilities relative to the original compounds and literature comparators 2 and 4, especially human. We therefore favored the methylamide to keep constant as we probed other substituents on the scaffold. While the microsomal properties of compound 8 were promising, solubility remained low.
Table 2.
SAR on the Triazole Wing: Benzyl Replacements
| Compound | R | GLS IC50 (nM)a |
A549 IC50 (nM)b |
Microsomal Stability (mL/min/kg)c |
pH 7.4 Solubility (μM)d |
|
|---|---|---|---|---|---|---|
| Human | Rat | |||||
| 8 | Methyl | 32 | 103 | 0.3 | 28 | 0.4 |
| 9 |
|
100 | 527 | 9 | 64 | 0.02 |
| 10 |
|
14 | 57 | 35 | 47 | 0.15 |
| 11 |
|
88 | 471 | 11 | 30 | 37 |
| 12 |
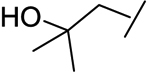
|
118 | 789 | 8 | 14 | 0.91 |
| 13 |
|
23 | 172 | 28 | 50 | 0.6 |
Inhibition of purified recombinant human GLS-1 (GAC isoform) assessed via a dual-coupled enzyme assay. IC50 values are reported as the mean of at least three determinations. Standard deviations and counts are included in the supporting information.
Inhibition of proliferation of A549 cells. IC50 values are reported as the mean of at least three determinations. Standard deviations and counts are included in the supporting information.
Liver microsomal intrinsic clearance (mL/min/kg protein).
Kinetic aqueous solubility (μM) in phosphate buffer (100 mM) at pH 7.4; shaken for 1 hour at RT.
Pyridazine Wing Exploration.
Keeping the triazole wing constant as a methyl amide, we explored pyridazine wings (Table 3). In the hope of improving solubility, we first replaced the trifluoromethylbenzyl group of compound 8 with simple pyridylmethyl sidechains (compounds 14 and 15), which did improve solubilities while retaining most of the potency of 8. The ortho-pyridine 14 was chosen for further optimization, because of its superior cell potency.
Table 3.
SAR of Pyridazine Wing: Pyridylmethyls
| Compound | R | GLS IC50 (nM)a |
A549 IC50 (nM)b |
Microsomal Stability (mL/min/kg)c |
pH 7.4 Solubility (μM)d |
|
|---|---|---|---|---|---|---|
| Hum | Rat | |||||
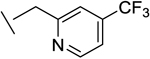
| 14 |
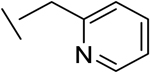
|
71 | 246 | 10 | 3 | 80 |
| 15 |
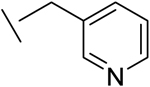
|
67 | 643 | 4 | 8 | 33 |
| 16 |
|
42 | 130 | 4 | 55 | 69 |
| 17 |
|
49 | 103 | 2 | 0.4 | 15 |
| 18 |
|
22 | 50 | 22 | 16 | 90 |
| 19 |
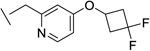
|
46 | 86 | 9 | 25 | 91 |
| 20 |
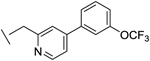
|
10 | 8 | 23 | 56 | 6 |
Inhibition of purified recombinant human GLS-1 (GAC isoform) assessed via a dual-coupled enzyme assay. IC50 values are reported as the mean of at least three determinations. Standard deviations and counts are included in the supporting information.
Inhibition of proliferation of A549 cells. IC50 values are reported as the mean of at least three determinations. Standard deviations and counts are included in the supporting information.
Liver microsomal intrinsic clearance (mL/min/kg protein).
Kinetic aqueous solubility (μM) in phosphate buffer (100 mM) at pH 7.4; shaken for 1 hour at RT.
The 4-trifluoromethyl variant (compound 16) was well tolerated, with only a slight loss of cellular potency compared to the trifluoromethoxybenzyl analog 8, and more importantly retained the improved solubility. We therefore embarked on a more extensive scan of 4-substituted-2-pyridylmethyl wingpieces, resulting in many analogs with good to excellent potencies, low microsomal clearances and reasonable solubilities. Particularly noteworthy were analogs 16 and 19, which showed promising overall profiles, and analog 20, which while less soluble was especially potent in our cellular assays.
Linker: Benefits of Fluorine.
We were interested in probing the effects of fluorine substitution on potencies and microsomal stabilities.44 Molecular modeling suggested that a fluorine substituent would be well tolerated on the linker, specifically on either of the two internal carbons.45 For synthetic reasons, we focused on fluorination of the internal carbon closer to the triazole core. Fluorination is not only tolerated, but consistently improved both cell potencies (albeit moderately) and microsomal stabilities (Table 4). Fluorination introduces a chiral center; we focused on the R-enantiomer because it often showed small potency advantages (e.g. compound 21 vs compound 22).
Table 4.
Linker Fluorination and Pyridine Methylation
| Compound | R | GLS IC50 (nM)a |
A549 IC50 (nM)b |
Microsomal Stability (mL/min/kg)c |
pH 7 Solubility (μM) |
|
|---|---|---|---|---|---|---|
| Hum | Rat | |||||
| 21 |
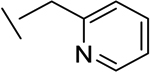
|
32 | 101 | 1 | 1 | 115d |
|
22 (enantiomer of 21) |
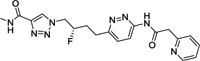
|
62 | 208 | 4 | 2 | ND |
| 23 |
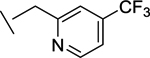
|
44 | 78 | 0.2 | 23 | 63e |
| 24 |
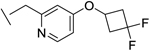
|
25 | 24 | 4 | 14 | 37e |
| 25 |
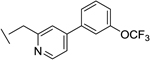
|
10 | 1 | 13 | 12 | 0.2e |
| 26 |
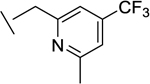
|
21 | 17 | 4 | 8 | 5e |
| 27 |
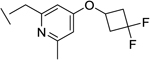
|
31 | 26 | 0.7 | 19 | 40e |
Inhibition of purified recombinant human GLS-1 (GAC isoform) assessed via a dual-coupled enzyme assay. IC50 values are reported as the mean of at least three determinations. Standard deviations and counts are included in the supporting information.
Inhibition of proliferation of A549 cells. IC50 values are reported as the mean of at least three determinations. Standard deviations and counts are included in the supporting information.
Liver microsomal intrinsic clearance (mL/min/kg protein).
Kinetic aqueous solubility (μM) in phosphate buffer (100 mM) at pH 7.4; shaken for 1 hour at RT.
Thermodynamic aqueous solubility (μM) in phosphate buffer (100 mM) at pH 7.0; shaken for 1 hour then equilibrated overnight at RT.
We also found that introduction of an ortho-methyl group to the pyridine could, in select cases, slightly improve potencies (compound 26 vs compound 23). While the ortho-methyl group of compound 27 did not improve potencies relative to the des-methyl analog 24, ultimately 27 showed superior in vivo properties.
Final Triaging.
Based primarily on physicochemical and metabolic properties coupled with cellular potencies, we selected compounds 24, 26 and 27 for further characterization. In addition, we profiled analog 25, on the rationale that this example, despite overall poorer properties, may have a lower bar on required exposures due to its excellent potencies. We triaged the set based on solubilities, permeabilities, microsomal stabilities, and in vivo PK (rat and dog). Tables 5 and 6 show summaries of in vitro and in vivo PK (rat and dog) data for these compounds.
Table 5.
In Vitro Properties of Select Compounds
| 24 | 25 | 26 | 27 | |
|---|---|---|---|---|
|
Thermodynamic
Solubilities (μM)a pH 7 FESSIF SGF |
37 80 9015 |
0.2 11 342 |
5 22 46 |
38b 19b 9446b |
| Caco2 AB/BA (cm/sec*10−6)c | 1.8/20 | 5.8/21 | 7.7/25 | 5.2/25 |
|
Microsomal Stability
Clint
(mL/min/kg)d Rat Dog Human |
14 2 4 |
22 77 13 |
8 0 4 |
15 5 0 |
|
Hepatocytes Stability
Clint
(mL/min/kg)e Rat Dog Human |
5 4 0 |
34 12 3 |
5 9 2 |
6 7 3 |
|
Protein Binding
(% bound)f Rat Dog Human |
97 72 97 |
99 98 99 |
95 90 96 |
99 98 98 |
Thermodynamic aqueous solubility (μM) in phosphate buffer (100 mM) at pH 7.0, FeSSIF (Fed State Simulated Intestinal Fluid) at pH 5.0, or SGF (fasted state Simulated Gastrointestinal Fluid) at pH 1.6; shaken for 1 hour then equilibrated overnight at RT.
Thermodynamic aqueous solubilities for the crystalline most stable polymorph of 27, identified during later development, in phosphate buffer (50 mM) at pH 6.8, FeSSIF or SGF, shaken for 24 hours at 25 °C (phosphate buffer) or 37 °C (FeSSIF and SGF).
Permeability in Caco-2 cells AB (apical to basolateral) and BA (basolateral to apical) movement of 10 μM test compound in 21 day cultured Caco-2 cells ((cm/s) × 10−6).
Liver microsomal intrinsic clearance (mL/min/kg protein).
Liver hepatocyte intrinsic clearance (mL/min/kg protein).
Plasma protein binding determined by equilibrium dialysis.
Table 6.
In Vivo Properties of Select Compoundsa
| 24b | 25b | 26 | 27c | |
|---|---|---|---|---|
| Rat d | ||||
| CL (mL/min/kg) | 3 | 8 | 1 | 0.3 |
| Vdss (L/kg) | 0.2 | 1 | 0.2 | 0.1 |
| t1/2 (h) | 1.2 | 2.7 | 3.3 | 5.3 |
| F % | 48 | 19 | 28 | 70 |
| Cmax (μM) | 5.8 | 0.35 | 4.4 | 32 |
| AUClast (h*μM) | 16 | 1.2 | 24 | 208 |
| Dog e | ||||
| CL (mL/min/kg) | 3 | 2.9 | 0.9 | 0.6 |
| Vdss (L/kg) | 0.5 | 0.5 | 0.2 | 0.2 |
| t1/2 (h) | 2.3 | 2.5 | 2.6 | 6.3 |
| F % | 34 | 8 | 21 | >100% |
| Cmax (μM) | 2.7 | 0.58 | 4.5 | 31 |
| AUClast (h*μM) | 10 | 2.5 | 23 | 250 |
Cmax, AUClast and F % were determined from the oral dose and CL, Vdss and t1/2 were determined from the IV dose.
Dosed as a hydrochloride salt.
Dosed as a bis-hydrochloride salt.
Male Sprague-Dawley rats. Doses were 0.3 mg/kg IV (intravenous) and 3 mpk PO (per os), except for compound 25 (0.3 mpk IV, 2 mpk PO).
Male beagle dogs. Doses were 0.3 mg/kg IV (intravenous) and 3 mpk PO (per os).
Compound 27 was chosen for further evaluation based on its overall profile, including significantly longer half-lives in rat and dog relative to the other leading compounds.. Importantly, while compound 25 is significantly more potent than compound 27, we believe its higher in vivo clearances and low oral exposures could present challenges at later stages of development, and therefore favored the less potent compound 27. Compound 27 has moderate solubility in pH 7 buffer and simulated intestinal fluid, but high solubility in simulated gastrointestinal fluid. These solubility results were attained for the crystalline freebase form that was carried into development. The compound shows mild efflux in a Caco-2 assay, used to assess intestinal permeability; this mild efflux potential has a minimal effect on oral absorption, based on the high bioavailabilities observed with oral dosing.
Deeper Profiling of Compound 27 (IPN60090).
A summary of potency, selectivity and pharmacokinetic properties of compound 27 is shown in Figure 5. Compound 27 is a highly selective inhibitor of GLS-1, with no activity observed against GLS-2. Testing in a cellular target engagement assay, which measured conversion of glutamine to glutamate, showed a similar IC50 to that seen in the cellular viability assay. As summarized in Figure 5, compound 27 showed no significant inhibition of the hERG channel or common cytochrome P450 enzymes. Compound 27 also showed no significant activities against an 80-member Eurofins CEREP panel of various ion channels and receptors, or against a 97-member Eurofins DiscoverX kinase panel. Specific results from the CEREP and DiscoverX panels are contained in the supporting information.
Figure 5.
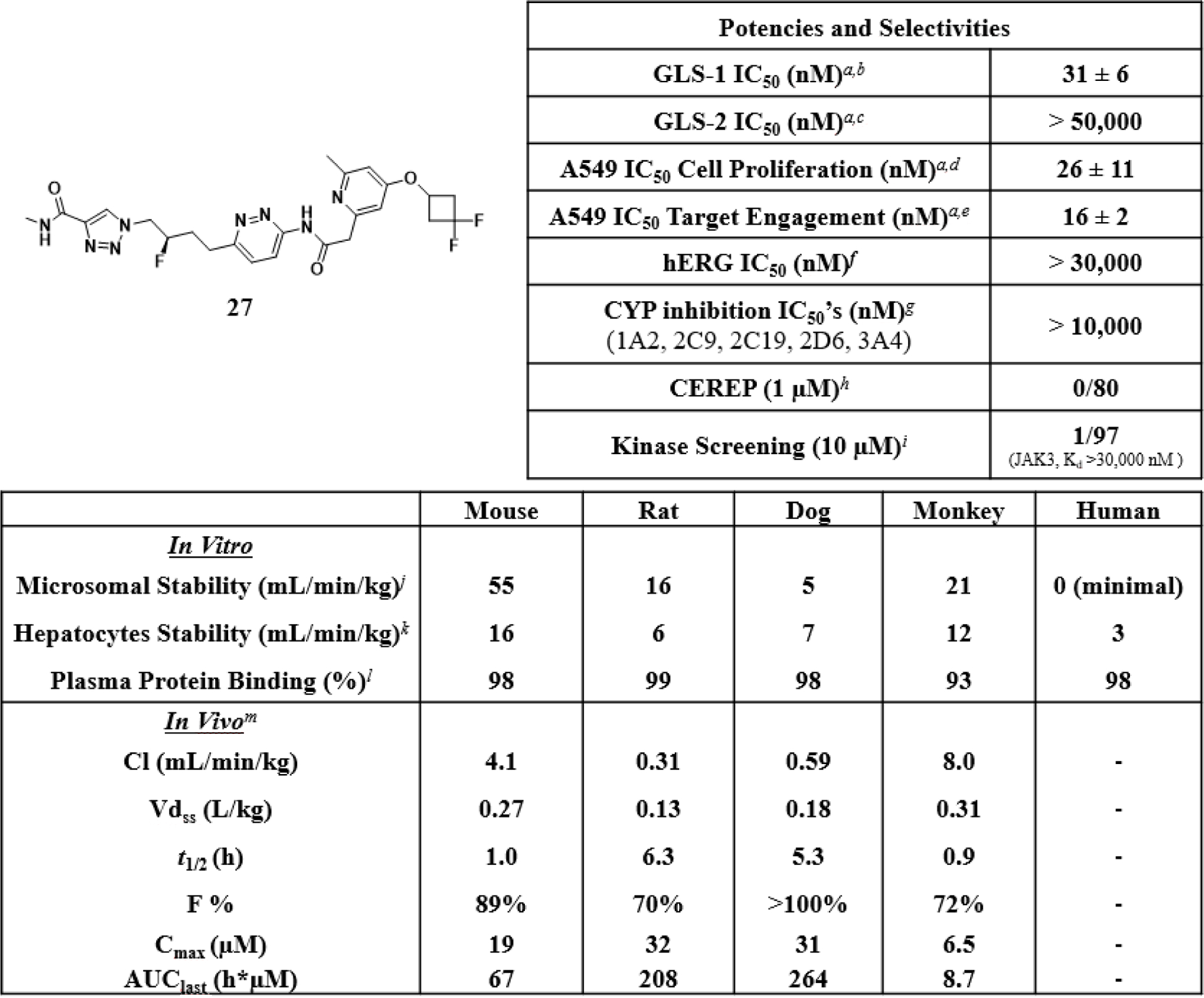
Summary of potencies, selectivities and pharmacokinetic properties of compound 27. aAll IC50 values are reported as the mean of at least two determinations. bInhibition of purified recombinant human GLS-1 (GAC isoform) assessed via a dual-coupled enzyme assay. IC50 value is the mean ± standard deviation for 36 determinations. cInhibition of purified recombinant human GLS-2 assessed via a dual-coupled enzyme assay. dInhibition of proliferation of A549 cells. IC50 value is the mean ± standard deviation for 36 determinations. eInhibition of cellular glutaminase assessed via direct measurement of glutamine and glutamate levels in the cellular media. IC50 value is the mean ± standard deviation for 4 determinations. fhERG QPatch assay, CHO cell line. gInhibition of metabolism of known substrates for the indicated cytochrome P450 enzymes. hEurofins CEREP profiling service; details in the supporting information. iEurofins DiscoverX KINOMEscan profiling service, scanEDGE product; details in the supporting information. jLiver microsomal intrinsic clearance (mL/min/kg protein). kLiver hepatocyte intrinsic clearance (mL/min/kg protein). lPlasma protein binding determined by equilibrium dialysis. mDosed as a bis-hydrochloride salt to female CD-1 mice, male Sprague-Dawley rats, male cynomolgus monkeys and male beagle dogs. Doses were 0.3 mg/kg IV (intravenous) and 3 mpk PO (per os), except for mouse (3 mpk IV, 10 mpk PO). Cmax, AUClast and F % were determined from the oral dose and CL, Vdss and t1/2 were determined from the IV dose.
The pharmacokinetic properties of compound 27 in rat and dog are superior to its properties in mouse and monkey, with rat and dog showing lower in vivo clearances and 5–6x longer half-lives. Because of the low volumes of distribution across species, half-lives are short unless clearances are extremely low, as is the case for rat and dog. Clearances across species are qualitatively in agreement with in vitro metabolic stabilities. Given that the in vitro clearances in human microsomes and hepatocytes are very low (similar to or lower than values for rat and dog), we expect low in vivo clearances and favorable half-lives in humans, more similar to rat and dog than to mouse and monkey.
Compound 27 was further tested in ascending single dose oral PK experiments in mouse, rat and dog (Figure 6). Across the dose ranges tested (up to 200 mg/kg in mouse, 100 mg/kg in rat and 10 mg/kg in dog), maximal concentrations and total exposures continually increased with dose, with high exposures achieved across species.
Figure 6.
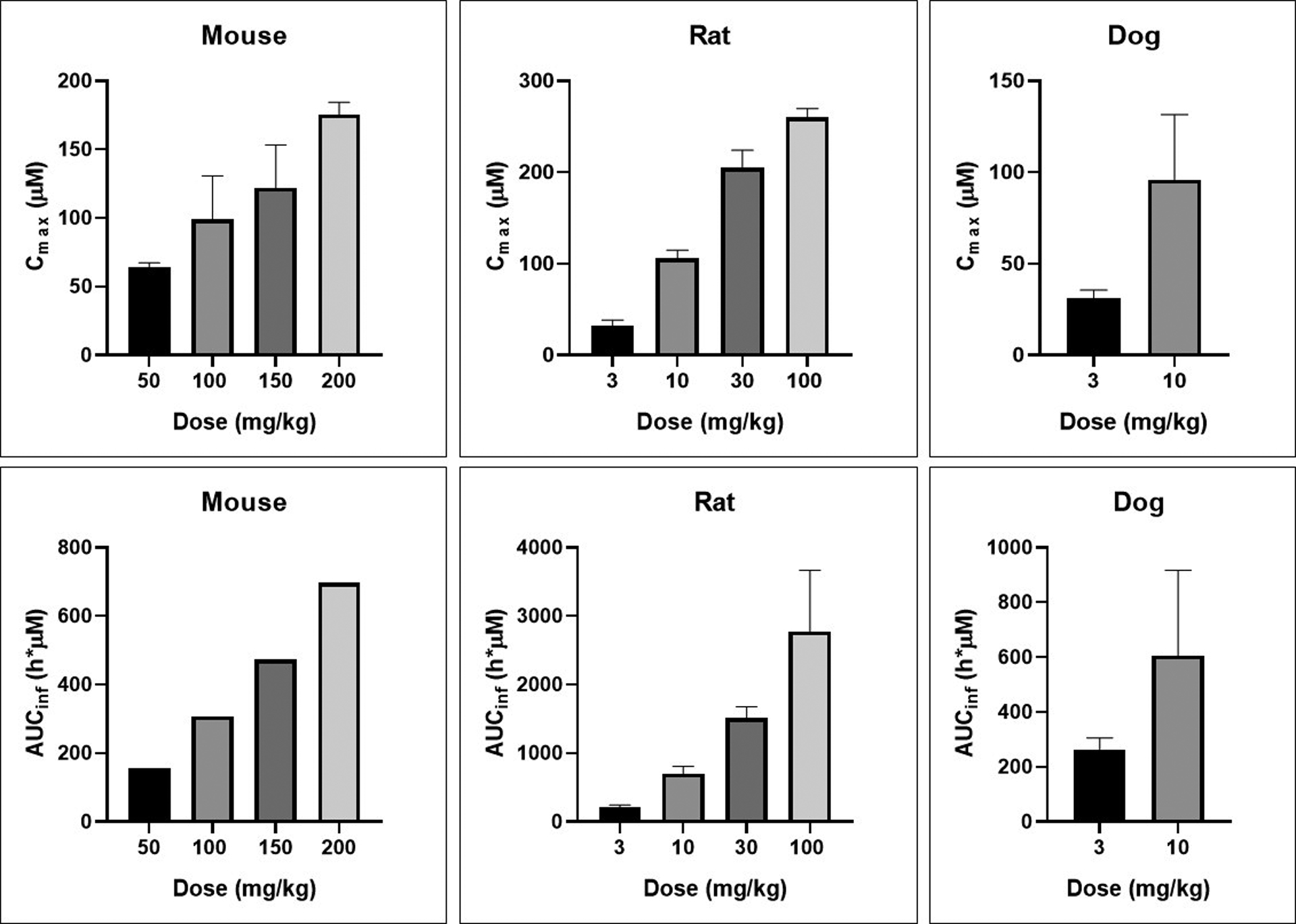
Ascending dose oral PK of compound 27 (bis-hydrochloride salt) in mouse, rat and dog dosed in 0.5% aqueous methylcellulose vehicle. Values are the means with standard deviation for three animals except for mouse AUC values, which are composite determinations as described in the experimental section.
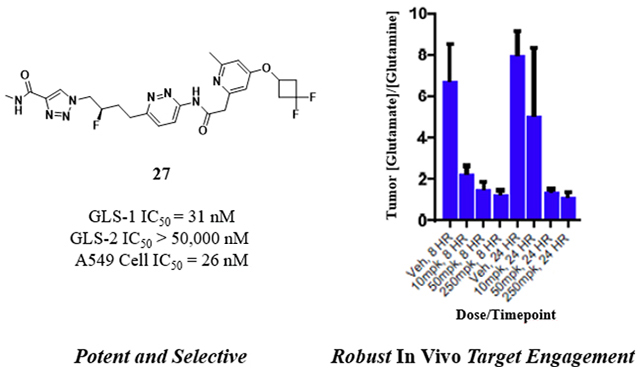
In Vivo Target Engagement of Compound 27.
To demonstrate that 27 could robustly inhibit glutaminolysis in vivo, and to determine plasma concentrations required for in vivo target engagement, compound was orally administered in an H460 non-small cell lung cancer xenograft model at doses of 10, 50 or 250 mg/kg (Figure 7). To determine target engagement, we analyzed tumor samples at the 8 and 24 hour time points for changes in glutamine and glutamate concentrations relative to vehicle control, on the expectation that inhibition of glutaminase should increase levels of glutamine relative to glutamate. Figure 7a shows the ratio of glutamate to glutamine in tumors when animals were treated with ascending doses of compound 27. The expected changes were indeed observed, with the effect at 50 mg/kg similar to that at 250 mg/kg despite an increase in plasma concentrations, suggesting that we are maximally inhibiting target at doses of 50–250 mg/kg. The average free plasma concentrations observed at these doses and timepoints were 0.058–0.316 μM (Figure 7b).
Figure 7.
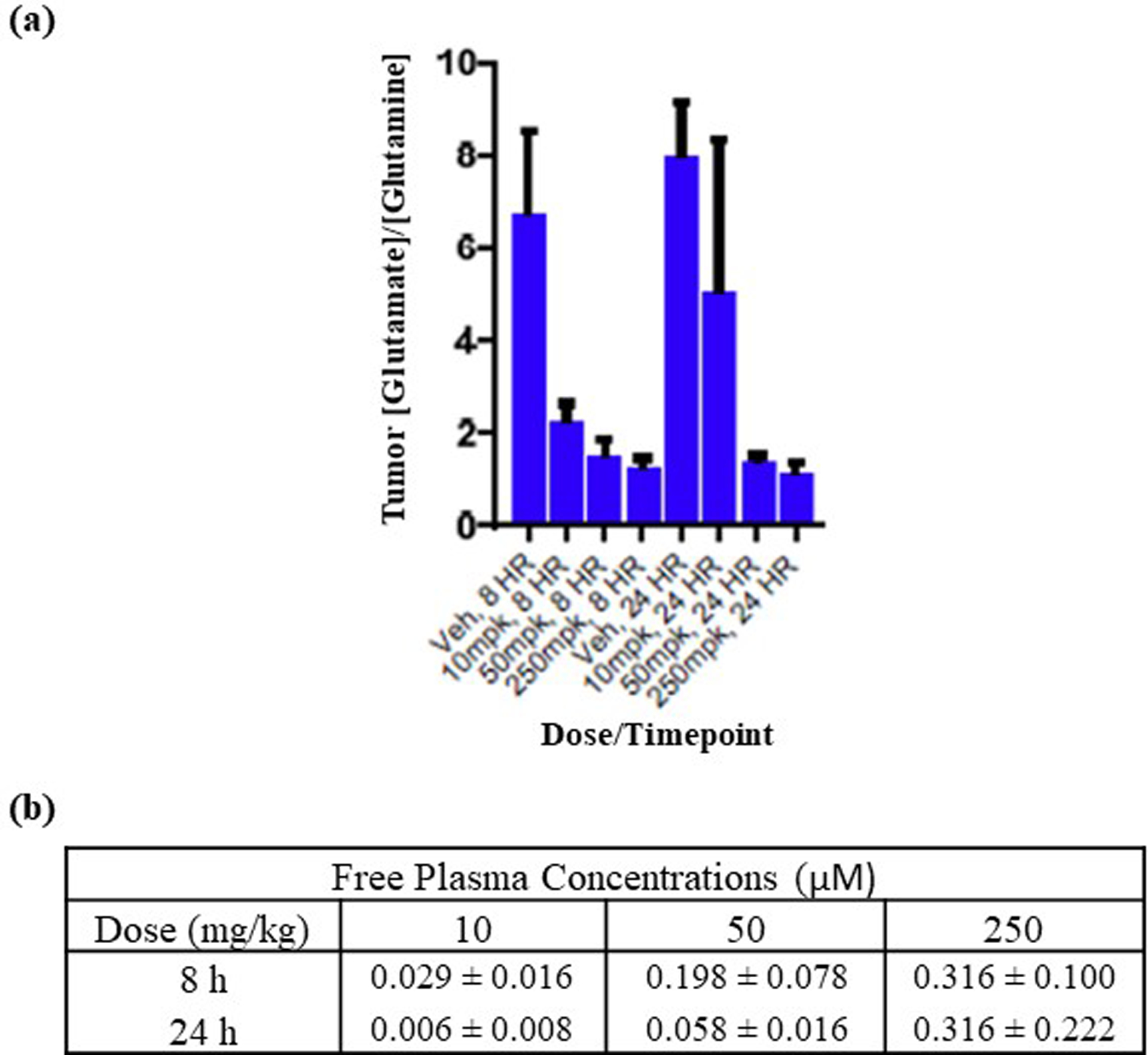
In vivo target engagement and concentrations of compound 27 in an H460 NSG mouse xenograft model. Compound 27 (bis-hydrochloride) was administered orally at the indicated doses in an 0.5% aqueous methylcellulose vehicle. There were six animals per group. (a) Glutamate:glutamine ratios at 8 hours and 24 hours after each dose, with standard deviation bars; (b) Free plasma concentrations, with standard deviations, of compound 27 at 8 hours and 24 hours for each dose.
In Vivo Efficacy of Compound 27.
In a benchmarking study detailed in the supporting information (Figures S1 and S2), we compared compound 27 to clinical stage compound 4 (Calithera’s CB-839) in an H2122 non-small cell lunger cancer cell line-derived xenograft mouse model. The results from that study indicated that compound 27 dosed orally at 100 mg/kg twice daily shows similar efficacy and target engagement to compound 4 dosed orally at 250 mg/kg twice daily.46
To demonstrate anti-tumor efficacy in an additional model, which was also used to confirm a combination therapy hypothesis, 27 was tested in an Ru337 non-small cell lung cancer patient-derived xenograft (PDX) mouse model (Figure 8). Compound 27 demonstrates robust in vivo target engagement when orally dosed to this model at doses of 25–100 mg/kg twice daily, at both 4 days and 28 days (see supporting information, Figure S3). We tested the compound at oral doses of 100 mg/kg twice daily, as both a monotherapy and in combination with the dual TORC1/2 inhibitor TAK-228 (also known as MLN0128, INK128).47 This combination was chosen based on results from translational biology studies to define rational combination strategies (manuscript under revision).48
Figure 8.
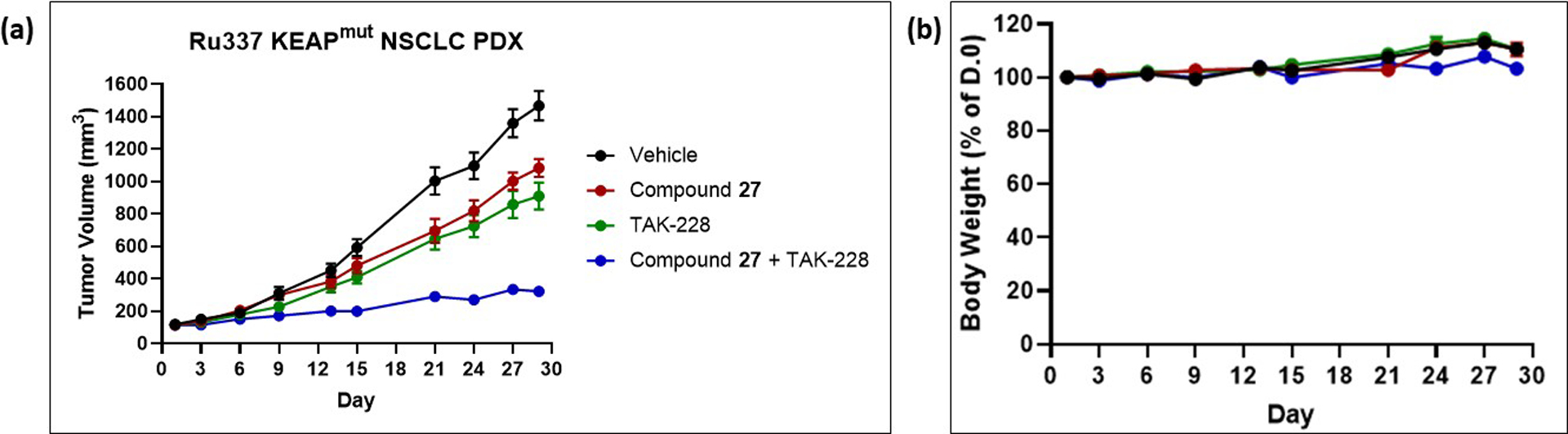
In vivo efficacies in an Ru337 PDX subcutaneous mouse tumor model of compound 27 as monotherapy and in combination with dual TORC inhibitor TAK-228. Compound 27 (neutral compound) was administered orally twice daily at 100 mg/kg in an 0.5% aqueous methylcellulose vehicle; TAK-228 was dosed orally once daily at 1 mg/kg in an 0.5% aqueous methylcellulose, 5% sucrose vehicle. Both compounds were dosed on a schedule of 5 days on / 2 days off,49 and there were 8 animals per group. (a) Tumor volumes plotted as means ± standard deviation; (b) Average % bodyweights relative to day 0 plotted as means ± standard deviation.
As a single agent, 27 caused a 28% tumor growth inhibition. This result is consistent with other reported preclinical studies on GLS inhibitors as monotherapies.9,10,13,31 The effects of TAK-228 administration were likewise modest at the dose tested (41% tumor growth inhibition). However, the combination regimen caused an 85% tumor growth inhibition, an improvement over either single agent. The results strongly support testing this combination clinically.
Chemistry.
The general synthetic routes for preparation of compounds are shown in Schemes 1–5. Scheme 1 shows an early synthesis, which was particularly helpful for exploring variations in the triazole wing. 4-Azido-1-propyne was reacted with tert-butyl propiolate in a selective azide-alkyne Huisgen cyclization to afford triazole 28, and the remaining alkyne was then coupled with 1-iodo-4-aminopyridazine to afford alkynyl aminopyridazine 29. Alkyne 29 was reduced to alkyl aminopyridazine 30, which was converted to varied amide intermediates 31. Finally, the tert-butyl esters of intermediates 31 were deprotected to the corresponding acids, which were coupled with amines to afford final products 5–13.
Scheme 1. General Synthesis of Compounds 5–13a.
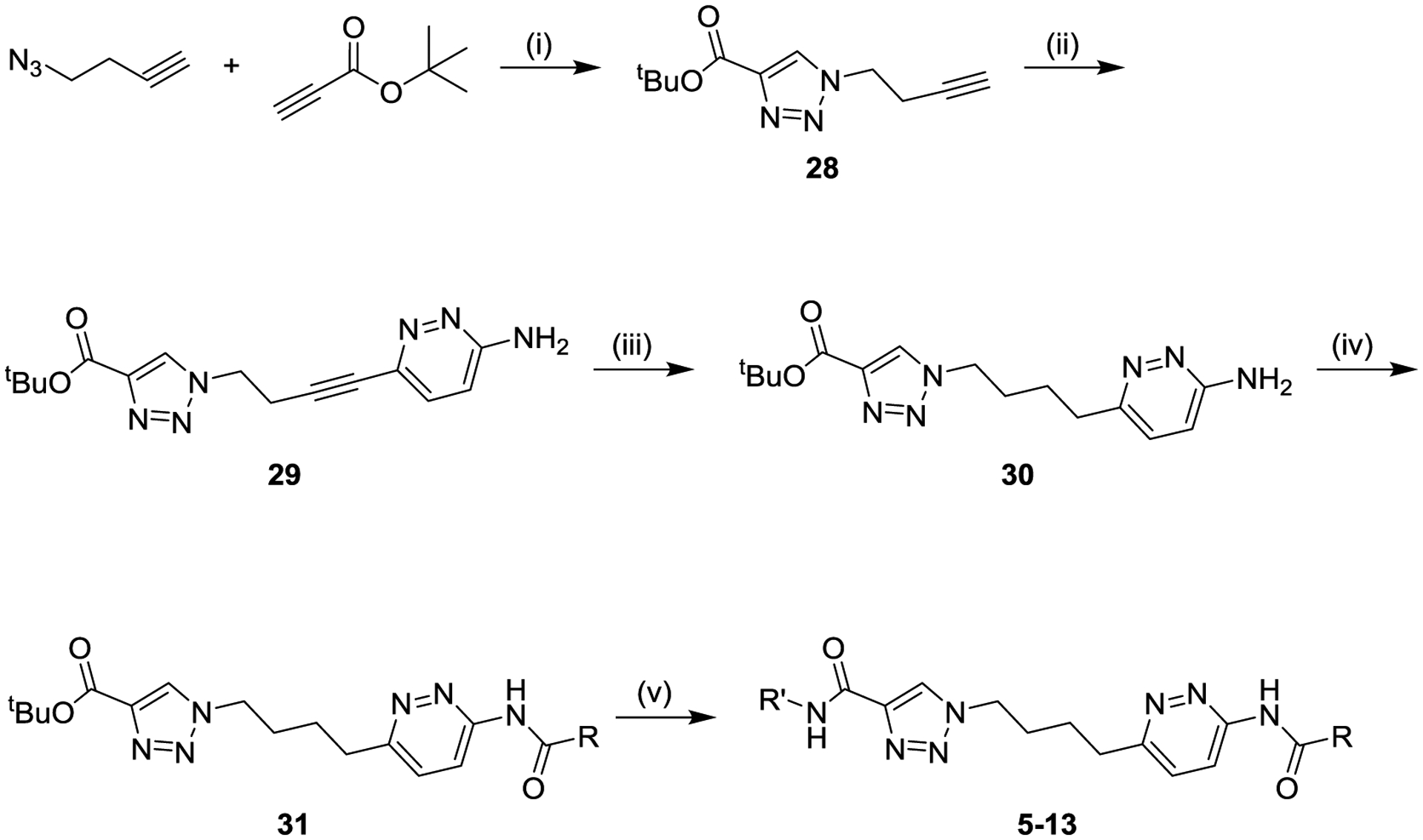
aReagents and conditions: (i) copper sulfate, ascorbic acid, tert-butanol/water (54%); (ii) bis(triphenylphosphine) palladium (II) dichloride, copper iodide, triethylamine, tetrahydrofuran, 60 °C (84%); (iii) Raney nickel, hydrogen, methanol (89%); (iv) R-CO2H, propanephosphonic acid anhydride, pyridine (25–78%); (v) (a) trifluoroacetic acid, dichloromethane, (b) R-NH2, amide coupling reagent, solvent (15–98%).
Scheme 5. General Synthesis of Compounds 24, 27a.
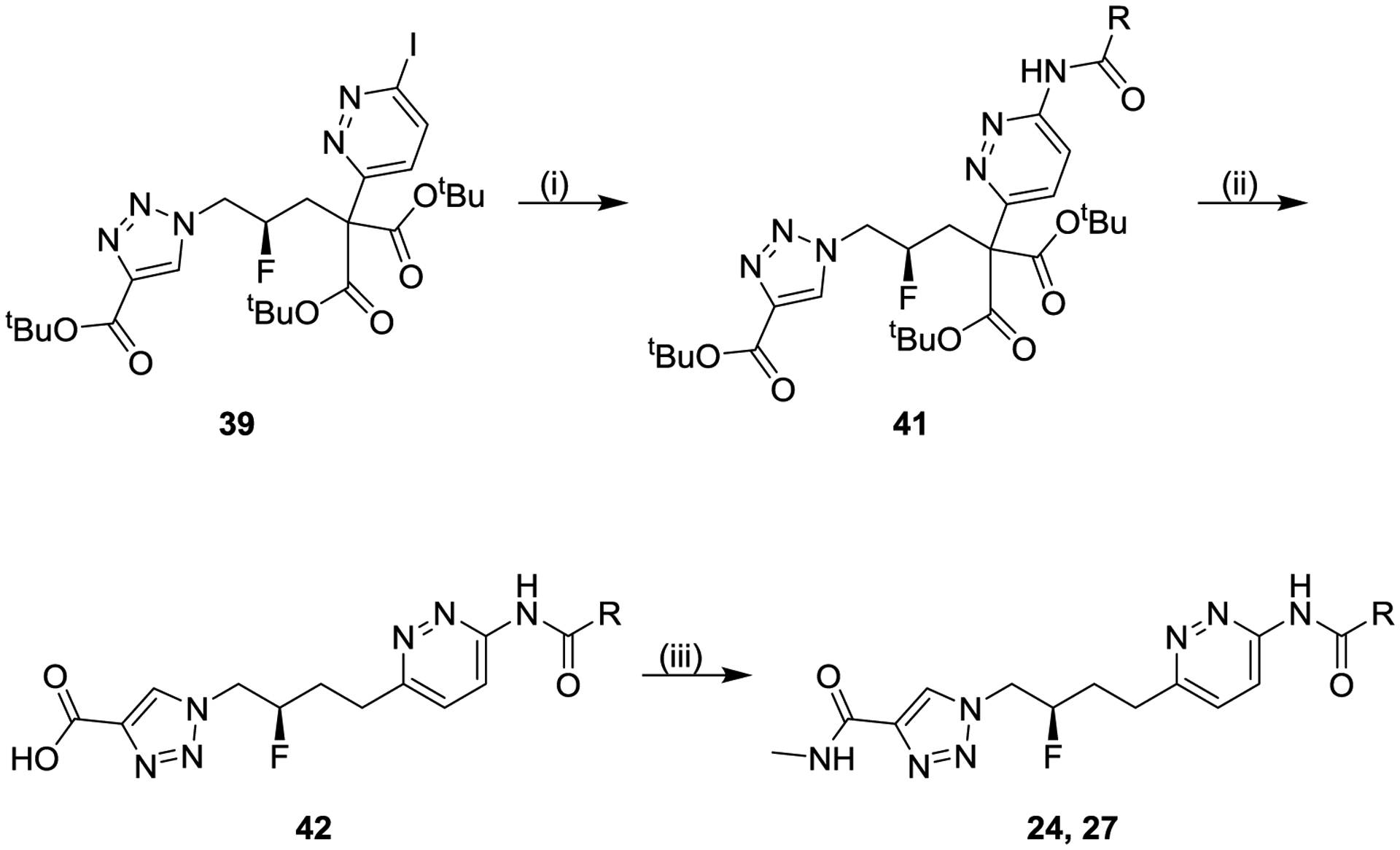
aReagents and conditions: (i) R-CONH2, palladium-catalyzed coupling, solvent (34–86%); (ii) hydrogen chloride, dioxane; (iii) methylamine, N,N-diisopropylethylamine, (1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate, tetrahydrofuran/N,N-dimethylformamide (two-step 56–68%).
Scheme 2 shows the synthesis used to explore variations in the pyridazine wing, in which the triazole wing was kept constant as a methyl amide. The tert-butyl ester of alkyl aminopyridazine 30 was deprotected to the corresponding acid, which was coupled with methylamine to afford methylamide 31, which was subsequently converted to varied final products 14–20.
Scheme 2. General Synthesis of Compounds 14–20a.
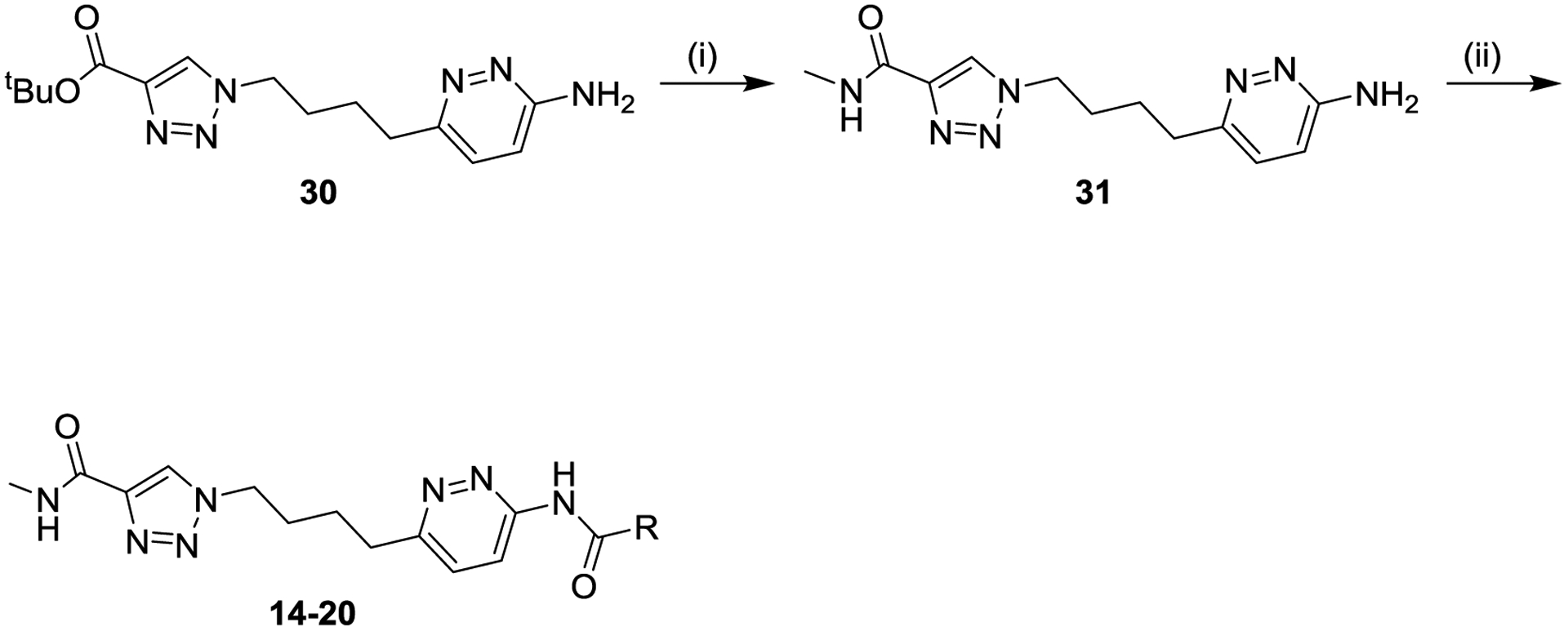
aReagents and conditions: (i) (a) trifluoroacetic acid, dichloromethane, (b) methylamine, N,N-diisopropylethylamine, (1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate, tetrahydrofuran/N,N-dimethylformamide (29%); (ii) R-CO2H, amide coupling reagent, solvent (5–82%).
Synthesis of fluorinated analogs relied on intermediate iodide 32, the synthesis of which is shown in Scheme 3. 2-((benzyloxy)methyl)oxirane was reacted with sodium azide to afford azido alcohol 33, which was reacted with tert-butyl propiolate in an azide-alkyne Huisgen cyclization to afford triazole alcohol 34. Alcohol 34 was converted to fluoride 35, and the benzyl ether of 35 was removed to afford new alcohol 36. The alcohol of intermediate 36 was converted via intermediate tosylate 37 to the corresponding iodide 32. Most of the compounds in this manuscript were synthesized as R-enantiomers, starting from (R)-2-((benzyloxy)methyl)oxirane as shown in the scheme, with the exception of 22, which followed the same synthesis except starting from (S)-2-((benzyloxy)methyl)oxirane.
Scheme 3. Synthesis of Intermediate Iodide 32a.
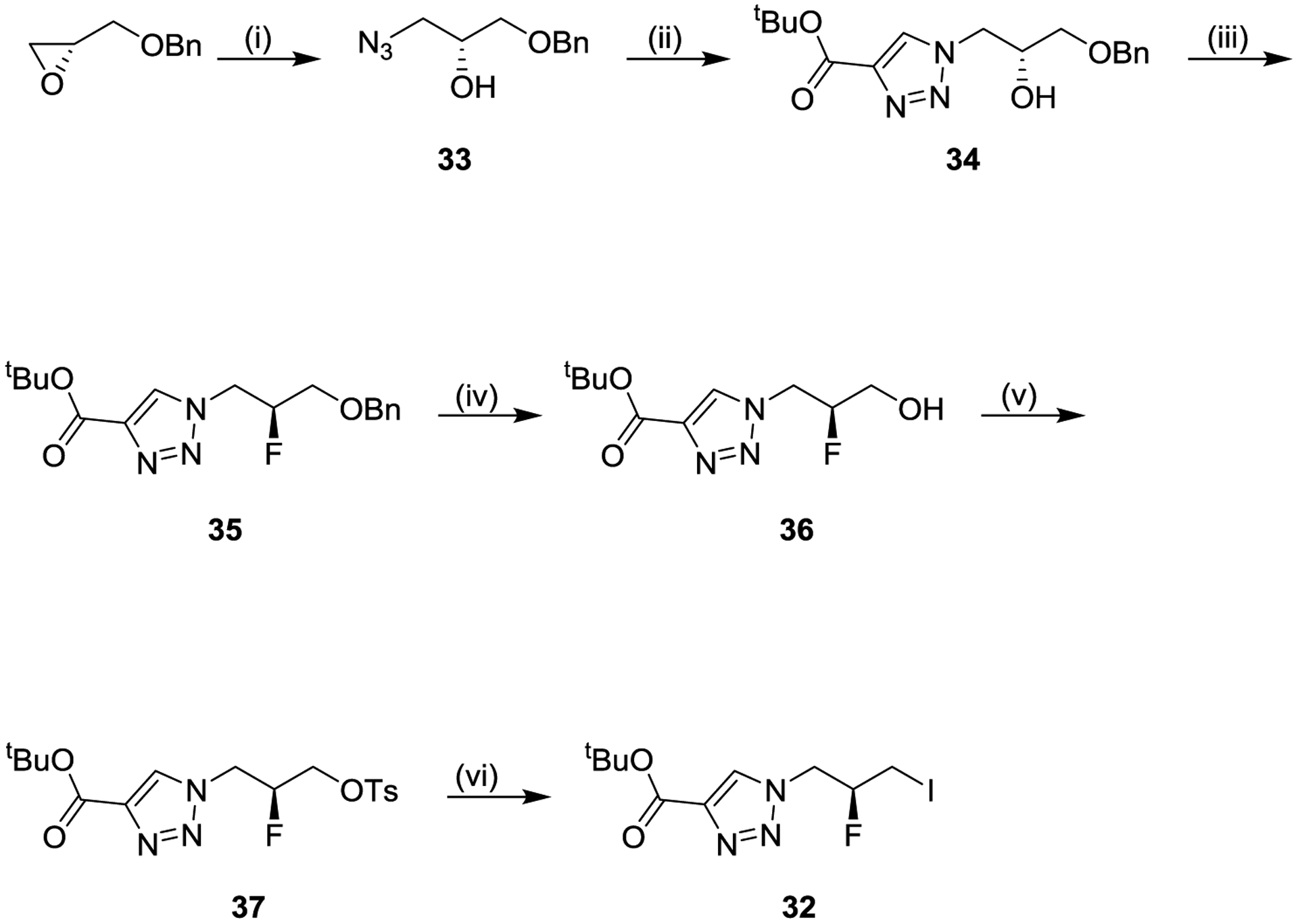
aReagents and conditions: (i) sodium azide, ammonium chloride, methanol/water (91%); (ii) tert-butyl propiolate, N,N-diisopropylethylamine, acetic acid, dichloromethane (82% crude); (iii) diethylaminosulfur trifluoride, pyridine, dichloromethane (45%); (iv) 10% palladium hydroxide on carbon, hydrogen, ethyl acetate (101% crude); (v) 4-toluenesulfonyl chloride, 4-dimethylaminopyridine, dichloromethane (84% crude); sodium iodide, acetone (91%).
Scheme 4 shows one synthetic route to our fluorinated analogs. 3,6-Dichloropyridazine was first treated with hydroiodic acid, and the resulting 3,6-diiodopyridazine was reacted with the anion of tert-butyl malonate to selectively afford mono-substituted malonate 38. Malonate 38 was then coupled with intermediate iodide 32 to afford di-substituted malonate 39. Di-substituted malonate 39 was then treated with trifluoroacetic acid to remove all three tert-butyl esters, with in situ bis-decarboxylation of the alkyl linker, and the remaining carboxylic acid was coupled with methylamine to afford pyridazine iodide 40. The pyridazine iodide was then coupled with varied primary amides, under palladium catalysis conditions, to afford final products 21-23, 25 or 26.
Scheme 4. General Synthesis of Compounds 21–23, 25, 26a,b.
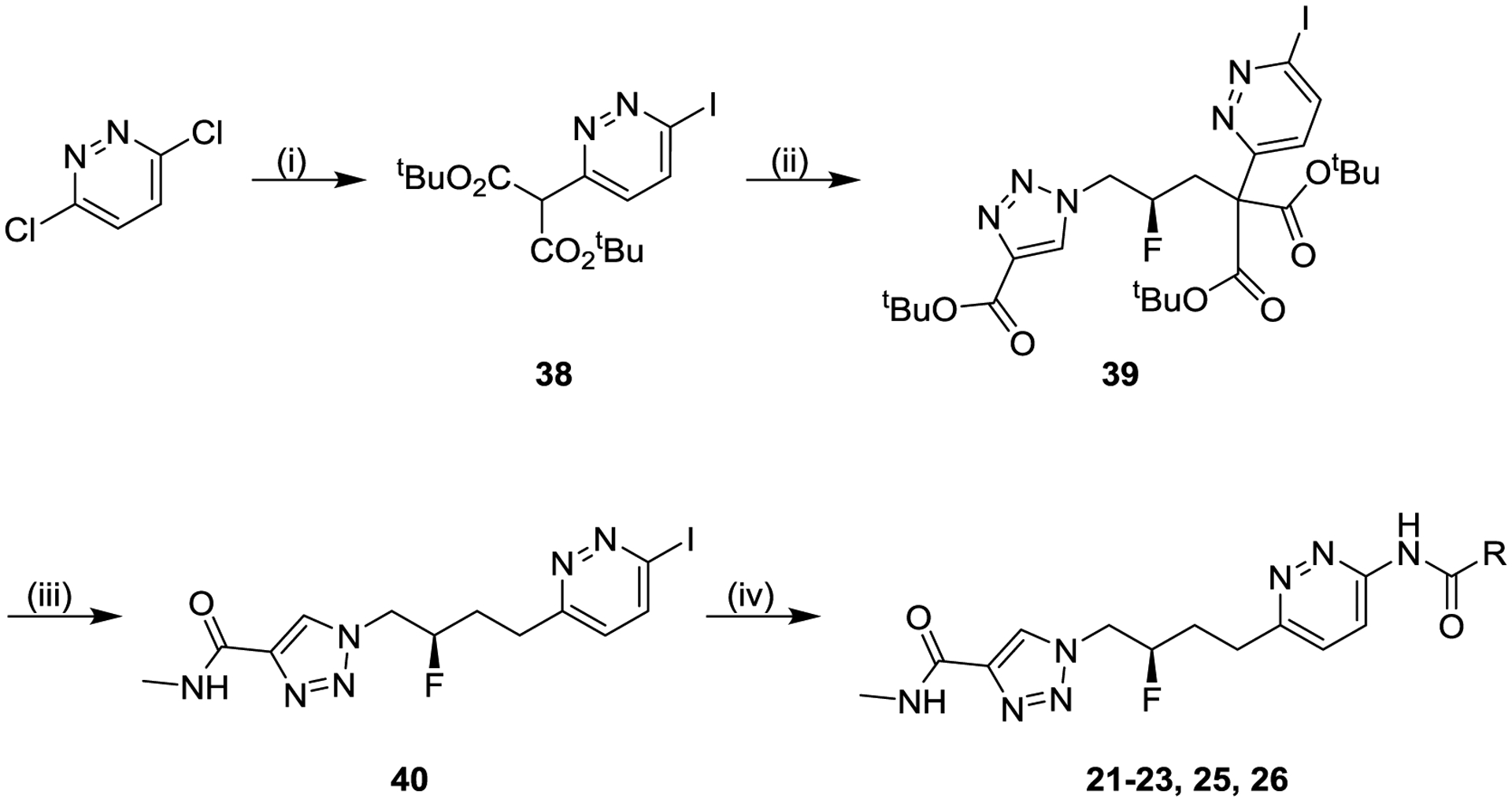
aReagents and conditions: (i) (a) 55% aqueous hydrogen iodide, 90 °C (90%), (b) di-tert-butyl propanedioate, sodium hydride, tetrahydrofuran, 65 °C (78%); (ii) intermediate 32, potassium carbonate, N,N-dimethylformamide (75%); (iii) (a) trifluoroacetic acid, dichloromethane, 40 °C, (b) methylamine, N,N-diisopropylethylamine, (1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate, tetrahydrofuran/N,N-dimethylformamide (56%); (iv) R-CONH2, palladium-catalyzed coupling, solvent (15–57%). bCompound 22, the enantiomer of compound 21, was made using the same synthetic route including preparation of starting material shown in Scheme 3, but starting from (S)-2-((benzyloxy)methyl)oxirane in place of (R)-2-((benzyloxy)methyl)oxirane.
The final coupling step shown in Scheme 4 was sometimes problematic due to the generally poor solubility of intermediate 40. The alternative route shown in Scheme 5 circumvented this issue by instead performing the iodopyridazine-amide coupling on tri-ester 39 (to afford intermediate 41), which was treated with hydrogen chloride to remove all three tert-butyl esters with in situ bis-decarboxylation of the alkyl linker, and the product acid 42 was then coupled with methylamine to afford final compounds 24 or 27.
CONCLUSION
Based on its overall profile, compound 27 (IPN60090) was selected for development and taken into IND-enabling studies. Additional translational research was conducted to inform the future clinical program and is being reported elsewhere.43 Importantly, while this candidate was not the most potent analog created in the series, its excellent physicochemical and pharmacokinetic properties outweighed potency considerations. Compound 27 is currently in the clinic, and robust target engagement is being achieved in human subjects.
EXPERIMENTAL SECTION
Chemistry.
Reagents and solvents were obtained from commercial sources and were used without further purification. Chromatographic purification of compounds were performed by column chromatography utilizing a Biotage system applying Biotage SNAP columns with Biotage KP-Sil silica or Biotage Zip Si columns with Biotage KP-Sil silica, or a Teledyne ISCO system with RediSep Rf normal phase silica cartridges. Other compounds were purified by preparative HPLC in reverse-phase mode using a Waters Autopurify system with a Waters Xbridge Prep C18 5 μm OBD, 19 mm × 150 mm or 50 mm × 100 mm column and SQ detector mass spectrometer with ESI ionization. The identity and purity of inhibitors were confirmed by NMR spectroscopy and Ultra Performance Liquid Chromatography (UPLC) coupled to Low Resolution Mass Spectrometry (LRMS), and were ≥ 95%. High Resolution Mass Spectrometry (HRMS) data were collected for leading compounds 24-27. NMR spectra were recorded on Bruker instruments operating at 500 or 600 MHz. NMR spectra were obtained as CDCl3, CD3OD, D2O, (CD3)2SO, (CD3)2CO, C6D6, or CD3CN solutions (reported in ppm), using tetramethylsilane (0.00 ppm) or residual solvent (CDCl3, 7.26 ppm; CD3OD, 3.31 ppm; D2O, 4.79 ppm; (CD3)2SO, 2.50 ppm; (CD3)2CO, 2.05 ppm; C6D6, 7.16 ppm; CD3CN, 1.94 ppm) as the reference standard. Low-resolution mass spectra were obtained on either a Waters H-class UPLC with a Waters Acquity UPLC BEH C18 1.7 μm, 2.1 mm × 50 mm column, UV detection between 200 and 400 nm, evaporating light scattering detection, and a SQ detector mass spectrometer with ESI ionization; or a Water I class UPLC with a Waters Acquity UPLC CSH C18 1.7 μm, 2.1 mm × 50 mm column, UV detection at 254 and 290 nm, evaporating light scattering detection, and a SQ detector 2 mass spectrometer with ESI ionization. High-resolution mass spectra on leading compounds 24-27 were obtained on a Waters Acquity I-Class UPLC – Thermo LTQ Orbitrap Elite MS instrument, with a Waters Acquity UPLC BEH C18 1.7 μm, 2.1 mm × 100 mm column, ESI ionization.
Di-tert-butyl 2-(6-iodopyridazin-3-yl)malonate (38).
Step 1: 3,6-Diiodopyridazine.
A mixture of 3,6-dichloropyridazine (60.00 g, 402.7 mmol) and 55% aqueous hydrogen iodide solution (30.30 mL, 402.7 mmol) was stirred at 90 °C for 12 h. Solid was isolated by filtration and then suspended in a sat. aq. NaHCO3 solution (300 mL). Solid was isolated by filtration, washing with petroleum ether (2 × 200 mL), to give crude 3,6-diiodopyridazine (120.0 g, 90%) as a yellow solid, which was used without further purification. MS (ES+) C4H2I2N2 requires: 332, found: 333 [M+H]+.
Step 2: Di-tert-butyl 2-(6-iodopyridazin-3-yl)malonate (38).
To a suspension of NaH (60% in mineral oil, 27.12 g, 678.0 mmol) in THF (750 mL) was added di-tert-butyl propanedioate (100.8 mL, 452.0 mmol) and the mixture was stirred at 28 °C for 15 min. To the mixture was added the above-prepared 3,6-diiodopyridazine (75.00 g, 226.0 mmol), and the reaction mixture was stirred at reflux for 8 h. The reaction mixture was quenched with a sat. aq. NH4Cl solution (500 mL) and extracted with 1:1 EtOAc/petroleum ether (3 × 500 mL). The combined organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by SiO2 gel chromatography (10:1 petroleum ether/EtOAc) to give di-tert-butyl 2-(6-iodopyridazin-3-yl)malonate (83.00 g, 87%) as a white solid. MS (ES+) C15H21IN2O4 requires: 420, found: 421 [M+H]+. 1H NMR (DMSO-d6) δ 8.20 (d, J = 8.8 Hz, 1H), 7.47 (d, J = 8.8 H, 1H), 5.03 (s, 1H), 1.43 (s, 18H). 13C NMR (DMSO-d6) δ 165.86, 156.63, 137.92, 129.60, 127.16, 83.03, 59.95, 27.90.
(S)-tert-Butyl 1-(2-fluoro-3-iodopropyl)-1H-1,2,3-triazole-4-carboxylate (32).
Step 1: (R)-1-Azido-3-benzyloxy(propan-2-ol) (33).
To a solution of (R)-2-((benzyloxy)methyl)oxirane (2.423 mL, 15.89 mmol) and NH4Cl (1.70 g, 31.8 mmol) in MeOH (39.5 mL) and water (5.92 mL) was added sodium azide (5.17 g, 79.0 mmol) and the resulting mixture was stirred at RT overnight. The mixture was concentrated under reduced pressure and the residue was partitioned between EtOAc (50 mL) and water (60 mL). The two layers were separated, and the aqueous layer was extracted with EtOAc (3 × 50 mL). The organic layers were combined, dried over MgSO4, filtered, and concentrated under reduced pressure to give (R)-1-azido-3-(benzyloxy)propan-2-ol (3.01 g, 91%) as a colorless oil. MS (ES+) C10H13N3O2 requires: 207, found: 208 [M+H]+.
Step 2: (R)-tert-Butyl 1-(3-benzyloxy)-2-hydroxypropyl)-1H-1,2,3-triazole-4-carboxylate (34).
To a solution of the above-prepared (R)-1-azido-3-(benzyloxy)propan-2-ol (3.01 g, 14.5 mmol), tert-butyl propiolate (2.393 mL, 17.43 mmol), DIEA (0.253 mL, 1.45 mmol), and AcOH (0.083 mL, 1.45 mmol) in DCM (58.1 mL) was added CuI (0.138 g, 0.726 mmol) and the resulting mixture was stirred at RT overnight. SiO2 gel (10 g) was added to the stirring mixture and the resulting suspension was filtered and washed with DCM (20 mL) and EtOAc (20 mL). The filtrate was concentrated under reduced pressure to give crude (R)-tert-butyl 1-(3-(benzyloxy)-2-hydroxypropyl)-1H-1,2,3-triazole-4-carboxylate (3.95 g, 82%) as an orange oil, which was used without further purification. MS (ES+) C17H23N3O4 requires: 333, found: 334 [M+H]+.
Step 3: (S)-tert-Butyl 1-(3-benzyloxy)-2-fluoropropyl)-1H-1,2,3-triazole-4-carboxylate (35).
To a 0 °C solution of the above-prepared (R)-tert-butyl 1-(3-(benzyloxy)-2-hydroxypropyl)-1H-1,2,3-triazole-4-carboxylate (3.95 g, 11.8 mmol) and pyridine (1.909 mL, 23.70 mmol) in DCM (23.70 mL) was added DAST (3.13 mL, 23.7 mmol). The resulting mixture was stirred at RT for 2.5 h, then filtered through a plug of SiO2 gel, rinsing with DCM (50 mL). The filtrate was concentrated under reduced pressure and the residue was adsorbed onto Celite® and purified by SiO2 chromatography (0% to 50% EtOAc in hexanes) to give (S)-tert-butyl 1-(3-(benzyloxy)-2-fluoropropyl)-1H-1,2,3-triazole-4-carboxylate (1.781 g, 45%) as a tan crystalline solid. MS (ES+) C17H22FN3O3 requires: 335, found: 336 [M+H]+.
Step 4: (S)-tert-Butyl 1-(2-fluoro-3-hydroxypropyl)-1H-1,2,3-triazole-4-carboxylate (36).
A reaction vessel was charged with the above-prepared (S)-tert-butyl 1-(3-(benzyloxy)-2-fluoropropyl)-1H-1,2,3-triazole-4-carboxylate (1.78 g, 5.31 mmol) and EtOAc (53.1 mL) under an atmosphere of N2. The solution was purged with N2 for 10 min and then with N2 still flowing, Pd(OH)2 on carbon (0.746 g, 1.06 mmol) was added. The resulting suspension was stirred as it was purged with H2 for 2 min. The reaction mixture was then stirred under an atmosphere of H2 at 1 atm for 12 h, then purged with N2, filtered through Celite® and concentrated under reduced pressure to give crude (S)-tert-butyl 1-(2-fluoro-3-hydroxypropyl)-1H-1,2,3-triazole-4-carboxylate (1.32 g, 101%) as a pale yellow solid, which was used without further purification. (1.32 g, 101%). MS (ES+) C10H16FN3O3 requires: 245, found: 246 [M+H]+.
Step 5: (S)-tert-Butyl 1-(2-fluoro-3-(tosyloxy)propyl)-1H-1,2,3-triazole-4-carboxylate (37).
To a solution of the above-prepared (S)-tert-butyl 1-(2-fluoro-3-hydroxypropyl)-1H-1,2,3-triazole-4-carboxylate (1.32 g, 5.38 mmol) and DMAP (0.986 g, 8.07 mmol) in DCM (26.9 mL) was added 4-methylbenzene-1-sulfonyl chloride (1.23 g, 6.46 mmol) while the solution was maintained at RT by a water bath. The resulting mixture was stirred at RT for 1.5 h, then diluted with EtOAc (100 mL) and washed with sat. aq. NH4Cl (2 × 40 mL). The organic layer was dried over MgSO4, filtered and concentrated under reduced pressure to give crude (S)-tert-butyl 1-(2-fluoro-3-(tosyloxy)propyl)-1H-1,2,3-triazole-4-carboxylate (1.803 g, 84%), which was used without further purification. MS (ES+) C17H22FN3O5S requires: 399, found: 400 [M+H]+.
Step 6: (S)-tert-Butyl 1-(2-fluoro-3-iodopropyl)-1H-1,2,3-triazole-4-carboxylate (32).
To a solution of the above-prepared (S)-tert-butyl 1-(2-fluoro-3-(tosyloxy)propyl)-1H-1,2,3-triazole-4-carboxylate (2.12 g, 5.31 mmol) in acetone (26.5 mL) was added sodium iodide (0.796 g, 5.31 mmol) and the resulting mixture was stirred at 80 °C for 3 h. Additional sodium iodide (1.6 g) was added and the mixture as stirred at 90 °C for 2 h. The mixture was allowed to cool to RT, then diluted with 1:1 EtOAc/hexanes (150 mL) and sequentially washed with water (2 × 50 mL) and a sat. aq. NaCl solution (50 mL). The organic layer was dried over MgSO4, filtered and concentrated under reduced pressure. The residue was adsorbed onto Celite® and purified by SiO2 gel chromatography (0% to 50% EtOAc/hexanes) to give (S)-tert-butyl 1-(2-fluoro-3-iodopropyl)-1H-1,2,3-triazole-4-carboxylate (1.71 g, 91%) as a white crystalline solid. MS (ES+) C10H15FIN3O2 requires: 355, found: 356 [M+H]+. 1H NMR (DMSO-d6) δ 8.69 (s, 1H), 5.09–4.95 (m, 1H), 4.87–4.72 (m, 2H), 3.62–3.54 (m, 1H), 3.48–3.40 (m, 1H), 1.54 (s, 9H). 13C NMR (DMSO-d6) δ 159.87. 140.57, 130.12, 90.42 (d, J = 178.3 Hz), 81.80, 53.52 (d, J = 21.4 Hz), 28.32, 4.26 (d, J = 21.5 Hz).
2-(4-(3,3-Difluorocyclobutoxy)-6-methylpyridin-2-yl)acetamide.
Step 1: 2-Chloro-4-(3,3-difluorocyclobutoxy)-6-methylpyridine.
To a suspension of 3,3-difluorocyclobutanol (590 mg, 5.46 mmol) and 2-chloro-6-methyl-4-nitropyridine (0.660 mL, 5.38 mmol) in THF (5.5 mL) was added cesium carbonate (3562.4 mg, 10.93 mmol) and the resulting orange-yellow mixture was stirred at 65 °C for 24 h. The resulting brown mixture was allowed to cool, then diluted with 11 mL of EtOAc and filtered through Celite 545®, rinsing well with ethyl acetate. The dark yellow filtrate was concentrated to give crude 2-chloro-4-(3,3-difluorocyclobutoxy)-6-methylpyridine (1231.5 mg, 98%) as a dark yellow oil, which was used without further purification. MS (ES+) C10H10ClF2NO requires: 233, found: 234 [M+H]+.
Step 2: tert-Butyl 2-cyano-2-(4-(3,3-difluorocyclobutoxy)-6-methylpyridin-2-yl)acetate.
A mixture of the above-prepared 2-chloro-4-(3,3-difluorocyclobutoxy)-6-methylpyridine (487 mg, 2.08 mmol), tert-butyl 2-cyanoacetate (0.456 mL, 3.13 mmol), cesium carbonate (2048.6 mg, 6.29 mmol), and chloro(2-di-t-butylphosphino-2’.4’,6’-triisopropyl-1,1’-biphenyl)[2-(2-aminoethyl)phenyl]palladium(II) (74.1 mg, 0.108 mmol) in toluene (20 mL) was degassed by blowing N2 through via a needle for 5 min, then put under N2 with an air condensor, heated to 110 °C over 5 min and stirred for 1 h. The orange mixture was allowed to cool, then concentrated to a yellow-orange solid. The residue was purified via SiO2 gel chromatography (0% to 100% EtOAc in hexanes) to give tert-butyl 2-cyano-2-(4-(3,3-difluorocyclobutoxy)-6-methylpyridin-2-yl)acetate (555.6 mg, 79%) as a yellow solid.
Step 3: 2-(4-(3,3-Difluorocyclobutoxy)-6-methylpyridin-2-yl)acetamide.
A flask with stirbar was sequentially charged with water (0.296 mL), conc. aq. HCl (5.616 mL) and tert-butyl 2-cyano-2-(4-(3,3-difluorocyclobutoxy)-6-methylpyridin-2-yl)acetate (203.0 mg, 0.600 mmol). The yellow mixture was stirred at RT for 4 h, in which time it became a yellow solution. The solution was chilled in an ice/water bath then treated with 30 mL of a 2.0 M aqueous NaOH solution, then further adjusted to pH 7 using 2.0 M aqueous NaOH (3–4 mL) then 1 M aqueous HCl (0.5–1 mL). The solution was extracted with EtOAc (3 × 15 mL), and the combined organic layers were dried over Na2SO4, filtered and concentrated to give 2-(4-(3,3-difluorocyclobutoxy)-6-methylpyridin-2-yl)acetamide (133.0 mg, 87%) as an off-white solid, which was used without further purification. MS (ES+) C12H14F2N2O2 requires: 256, found: 257 [M+H]+. 1H NMR (DMSO-d6) δ 7.46 (br. s, 1H), 6.94 (br. s, 1H), 6.69–6.68 (m, 2H), 4.84–4.78 (m, 1H), 3.46 (s, 2H), 3.27–3.19 (m, 2H), 2.74–2.65 (m, 2H), 2.38 (s, 3H). 13C NMR (DMSO-d6) δ 171.57, 163.79, 159.43, 157.74, 119.43 (dd, J = 281.5, 270.9 Hz), 108.55, 107.88, 61.69 (dd, J = 17.0, 10.3 Hz), 45.13, 42.89 (appar. t, J = 22.8 Hz), 24.52.
(R)-1-(4-(6-(2-(4-(3,3-difluorocyclobutoxy)-6-methylpyridin-2-yl)acetamido) pyridazin-3-yl)-2-fluorobutyl)-N-methyl-1H-1,2,3-triazole-4-carboxamide (27).
Step 1: (R)-di-tert-Butyl 2-(3-(4-(tert-butoxycarbonyl)-1H-1,2,3-triazol-1-yl)-2-fluoropropyl)-2-(6-iodopyridazin-3-yl)malonate (39).
A mixture of potassium carbonate (0.412 g, 2.98 mmol), di-tert-butyl 2-(6-iodopyridazin-3-yl)malonate (38) (1.25 g, 2.98 mmol), and (S)-tert-butyl 1-(2-fluoro-3-iodopropyl)-1H-1,2,3-triazole-4-carboxylate (32) (1.00 g, 2.82 mmol) in a vial was degassed and then treated with DMF (9.39 mL). The mixture was degassed and backfilled with N2 for three cycles and then stirred at 25 °C for 80 h. The mixture was diluted with EtOAc/hexanes (1:1, 200 mL) and washed with water (2 × 100 mL). The combined aqueous layers were extracted with EtOAc/hexanes (1:1, 100 mL). The combined organic layers were concentrated, washed with a sat. aq. NaCl solution, and concentrated under reduced pressure. The residue was purified by SiO2 gel chromatography (5% to 60% EtOAc in hexanes) to give (R)-di-tert-butyl 2-(3-(4-(tert-butoxycarbonyl)-1H-1,2,3-triazol-1-yl)-2-fluoropropyl)-2-(6-iodopyridazin-3-yl)malonate (1.36 g, 75%) as a yellow liquid. MS (ES+) C25H35FIN5O6 requires: 647, found: 648 [M+H]+.
Step 2: Di-tert-butyl (R)-2-(3-(4-(tert-butoxycarbonyl)-1H-1,2,3-triazol-1-yl)-2-fluoropropyl)-2-(6-(2-(4-(3,3-difluorocyclobutoxy)-6-methylpyridin-2-yl)acetamido)pyridazin-3-yl)malonate (41).
A degassed solution of (R)-di-tert-butyl 2-(3-(4-(tert-butoxycarbonyl)-1H-1,2,3-triazol-1-yl)-2-fluoropropyl)-2-(6-iodopyridazin-3-yl)malonate (39) (42.4 g, 65.6 mmol), 2-(4-(3,3-difluorocyclobutoxy)-6-methylpyridin-2-yl)acetamide (14.0 g, 54.6 mmol), cesium carbonate (35.6 g, 109 mmol), Xantphos (6.32 g, 10.9 mmol) and allylpalladium chloride dimer (1.00 g, 2.73 mmol) in 1,4-dioxane (300 mL) was stirred at 70 °C for 16 h. The reaction mixture was allowed to cool to RT, then filtered and the filtrate was concentrated under reduced pressure. The residue was purified by SiO2 gel chromatography (0% to 3% MeOH in DCM) to give di-tert-butyl (R)-2-(3-(4-(tert-butoxycarbonyl)-1H-1,2,3-triazol-1-yl)-2-fluoropropyl)-2-(6-(2-(4-(3,3-difluorocyclobutoxy)-6-methylpyridin-2-yl)acetamido)pyridazin-3-yl)malonate (36.5 g, 86%) as a foamy yellow solid. MS (ES+) C37H48F3N7O8 requires: 775, found: 776 [M+H]+.
Step 3: (R)-1-(4-(6-(2-(4-(3,3-difluorocyclobutoxy)-6-methylpyridin-2-yl)acetamido) pyridazin-3-yl)-2-fluorobutyl)-1H-1,2,3-triazole-4-carboxylic acid (42).
A solution of (R)-di-tert-butyl 2-(3-(4-(tert-butoxycarbonyl)-1H-1,2,3-triazol-1-yl)-2-fluoropropyl)-2-(6-(2-(4-(3,3-difluorocyclobutoxy)-6-methylpyridin-2-yl)acetamido)pyridazin-3-yl)malonate (36.0 g, 46.4 mmol) in 4.0 M HCl in 1,4-dioxane (696.0 mL, 2784 mmol) was stirred at 70 °C for 16 h. White precipitate formed. The reaction mixture was allowed to cool to RT, and precipitate was isolated by filtration, washed with EtOAc, and dried in vacuo to give crude (R)-1-(4-(6-(2-(4-(3,3-difluorocyclobutoxy)-6-methylpyridin-2-yl)acetamido) pyridazin-3-yl)-2-fluorobutyl)-1H-1,2,3-triazole-4-carboxylic acid as an off-white solid, which was used without further purification in the next step. MS (ES+) C23H24F3N7O4 requires: 519, found: 520 [M+H]+.
Step 4: (R)-1-(4-(6-(2-(4-(3,3-difluorocyclobutoxy)-6-methylpyridin-2-yl)acetamido) pyridazin-3-yl)-2-fluorobutyl)-N-methyl-1H-1,2,3-triazole-4-carboxamide (27).
To a solution of crude (R)-1-(4-(6-(2-(4-(3,3-difluorocyclobutoxy)-6-methylpyridin-2-yl)acetamido)pyridazin-3-yl)-2-fluorobutyl)-1H-1,2,3-triazole-4-carboxylic acid hydrochloride prepared in the previous step (assumed 46.4 mmol) in DMF (200 mL) at 0 °C was added HATU (17.64 g, 46.4 mmol), DIEA (40.5 mL, 232 mmol) and 2.0 M methylamine in THF (27.8 mL, 55.7 mmol). The resulting mixture was stirred at 20 °C for 1 h, then concentrated under reduced pressure. Water (1000 mL) and DCM (500 mL) were added, and the layers were separated. The aqueous layer was extracted with DCM (3 × 300 mL), and the combined organic layers were washed with sat. aq. NaCl, dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by SiO2 gel chromatography (0% to 8% MeOH in DCM) to give (R)-1-(4-(6-(2-(4-(3,3-difluorocyclobutoxy)-6-methylpyridin-2-yl)acetamido) pyridazin-3-yl)-2-fluorobutyl)-N-methyl-1H-1,2,3-triazole-4-carboxamide (16.8 g, 68%) as a white solid. HRMS (ES+) C24H28F3N8O3 requires: 533.2231 [M+H]+, found: 533.2229 [M+H]+. 1H NMR (DMSO-d6) δ 11.30 (s, 1 H), 8.51 (s, 1 H), 8.47 (q, J = 4.4, 1 H), 8.22 (d, J = 9.1 Hz, 1 H), 7.60 (d, J = 9.3 Hz, 1 H), 6.79 (d, J = 2.5 Hz, 1 H), 6.72 (d, J = 2.5 Hz, 1 H), 5.09–4.96 (m, 1 H), 4.90–4.70 (m, 3 H), 3.87 (s, 2 H), 3.28–3.18 (m, 2 H), 3.08 – 2.98 (m, 2 H), 2.76 (d, J = 4.9 Hz, 3 H), 2.75–2.63 (m, 2 H), 2.39 (s, 3H), 2.20–1.95 (m, 2 H). 13C NMR (DMSO-d6) δ 169.58, 163.45, 160.01, 159.21, 158.70, 156.32, 154.13, 142.99, 128.42, 126.88, 118.94 (dd, J = 281.7, 270.4 Hz), 108.52, 107.73, 91.25 (d, J = 172.3 Hz), 62.31 (dd, J = 16.98, 10.69 Hz), 52.99 (d, J = 21.4 Hz), 45.14, 42.41 (appar. t, J = 22.6 Hz), 31.04 (d, J = 20.1 Hz), 30.07 (d, J = 3.8 Hz), 25.53, 24.07. An assumed bis-hydrochloride salt was used in in vivo experiments described in the text, as well as to confirm enantiopurity using a chiral HPLC method validated with mixtures of the title compound and its (S) enantiomer (synthesized in the same way but starting with (S)-2-((benzyloxy)methyl)oxirane in place of (R)-2-((benzyloxy)methyl)oxirane). The bis-hydrochloride salt was made as follows: To a solution of (R)-1-(4-(6-(2-(4-(3,3-difluorocyclobutoxy)-6-methylpyridin-2-yl)acetamido)pyridazin-3-yl)-2-fluorobutyl)-N-methyl-1H-1,2,3-triazole-4-carboxamide (12.71 g, 23.87 mmol) in MeOH (20 mL) and DCM (60 mL) at 0 °C was added HCl in dioxane (4.0 M, 11.93 mL, 47.70 mmol) and the resulting mixture was stirred for 5 min then concentrated under reduced pressure. The residue was redissolved in ACN and water, lyophilized, and the resulting solid was triturated with EtOAc and dried in vacuo to give (R)-1-(4-(6-(2-(4-(3,3-difluorocyclobutoxy)-6-methylpyridin-2-yl)acetamido) pyridazin-3-yl)-2-fluorobutyl)-N-methyl-1H-1,2,3-triazole-4-carboxamide bis-hydrochloride (14.03 g, 97%) as a white solid. MS (ES+) C24H27F3N8O3 requires: 532, found: 533 [M+H]+. 1H NMR (DMSO-d6) δ 11.66 (s, 1 H), 8.53 (s, 1 H), 8.47 (q, J = 5.3, 1 H), 8.23 (d, J = 9.1 Hz, 1 H), 7.73 (d, J = 9.5 Hz, 1 H), 7.44 (d, J = 2.0 Hz, 1 H), 7.39 (d, J = 2.0 Hz, 1 H), 4.96–5.11 (m, 2 H), 4.67–4.86 (m, 2 H), 4.36 (s, 2 H), 3.34 (m, 2 H), 3.07 (m, 2 H), 2.87 (m, 2 H), 2.76 (d, J = 4.9 Hz, 3 H), 2.68 (s, 3 H), 2.24–1.95 (m, 2 H). The compound (2 mg/mL, 10 μL per injection) was analyzed on a Shimadzu Prominence HPLC system with a Lux Cellulose 4 column (4.6 mm × 150 mm, 5μM, 1 mL/min) using a mobile phase of water:acetonitrile (50:50), and showed an enantiomeric excess of >98%. Retention time: 11.3 min.
In vitro pharmacology.
GLS-1 Enzyme Assay.
The glutaminase dual coupled fluorescence assay was performed in a 384 well, black, Greiner, non-binding, plate (Greiner, Catalog #784900) with assay buffer consisting of 50 mM Hepes (pH 7.4) (Gibco, Catalog #15630–080), 250 μM EDTA (pH 8) (Calbiochem, Catalog #4055), and 0.12 mM Triton-X 100 (Sigma, Catalog #T9284). All final concentrations refer to a 20 μL volume. Stock solutions of the test compounds were prepared in 100% DMSO (Sigma, Catalog #D2650) and serially diluted 1:3 using 100% DMSO. Compounds were additionally diluted 1:50 in assay buffer, and 5 μL/well were transferred to the assay plate. A 4x stock of glutaminase and potassium phosphate dibasic trihydrate (Sigma, Catalog #P5504) diluted in assay buffer was preincubated at RT for 10 min. 5 μL/well of the glutaminase with potassium phosphate dibasic trihydrate were added to the microplate (final concentrations 2 nM and 50 mM, respectively) followed by a 10 min incubation at RT. The coupled reaction consisted of glutamate oxidase (Sigma, Catalog #G5921), Amplex UltraRED (Molecular Probes, Catalog #A36006), glutamine (Sigma, Catalog #G3126), and horseradish peroxidase (Sigma, Catalog #P8375). 5 μL/well of glutamate oxidase and Amplex UltraRED diluted in assay buffer (final concentrations 100 mU/mL and 75 μM, respectively) and 5 μL/well of glutamine and horseradish peroxidase diluted in assay buffer (final concentrations 1 mM and 100 mU/mL, respectively) were added to the microplate under subdued light followed by a 20 min incubation at RT. The resorufin signal was measured using a PerkinElmer Envision plate reader: excitation-535 nm, emission-590 nm. IC50 values were calculated using a four-parameter logistic curve fit using Genedata Screener software.
GLS-2 Enzyme Assay.
The GLS2 dual coupled fluorescence assay was performed in a 384 well, black, Greiner, non-binding, plate (Greiner, Catalog #784900) with assay buffer consisting of 50 mM Hepes (pH 7.4) (Gibco, Catalog #15630–080), 250 μM EDTA (pH 8) (Calbiochem, Catalog #4055), and 0.12 mM Triton-X 100 (Sigma, Catalog #T9284). All final concentrations refer to a 20 μL volume. Stock solutions of the test compounds were prepared in 100% DMSO (Sigma, Catalog #D2650) and serially diluted 1:3 using 100% DMSO. Compounds were additionally diluted 1:50 in assay buffer, and 5 μL/well were transferred to the assay plate. A 4x stock of GLS-2 and potassium phosphate dibasic trihydrate (Sigma, Catalog #P5504) diluted in assay buffer was preincubated at RT for 10 min. 5 μL/well of the GLS-2 with potassium phosphate dibasic trihydrate were added to the microplate (final concentrations 33 nM and 50 mM, respectively) followed by a 10 min incubation at RT. The coupled reaction consisted of glutamate oxidase (Sigma, Catalog #G5921), Amplex UltraRED (Molecular Probes, Catalog #A36006), glutamine (Sigma, Catalog #G3126), and horseradish peroxidase (Sigma, Catalog #P8375). 5 μL/well of glutamate oxidase and Amplex UltraRED diluted in assay buffer (final concentrations 100 mU/mL and 75 μM, respectively) and 5 μL/well of glutamine and horseradish peroxidase diluted in assay buffer (final concentrations 3 mM and 100 mU/mL, respectively) were added to the microplate under subdued light. The resorufin signal was measured continuously for 20 min using a PerkinElmer Envision plate reader: excitation-535 nm, emission-590 nm. IC50 values were calculated using a four-parameter logistic curve fit using Genedata Screener software.
A549 Cell Viability Assay.
A549 cells were routinely maintained in filtered RPMI media (Gibco, Catalog #11875) supplemented with 10% dialyzed FBS (Corning, Catalog #35–071-CV) using a humidified incubator (37 °C, 5% CO2, and ambient O2). In preparation for the viability assay, cells were harvested and resuspended in filtered RPMI media supplemented with 10% dialyzed FBS (Corning, Catalog #35–071-CV). Cells were seeded onto a 384-well black PerkinElmer tissue culture plate (PerkinElmer, Catalog #6007660) at a density of 1,000 cells/well in a volume of 40 μL. The tissue culture plate was incubated for 24 h at 37 °C with 5% CO2 and ambient O2. Stock solutions of the test compounds were prepared in 100% DMSO (Sigma, Catalog #D2650) and serially diluted 1:3 using 100% DMSO. Compounds were additionally diluted 1:40 in culture medium, and 10 μL/well were transferred to the tissue culture plate. Following compound addition, the microplate was incubated at 37 °C for 72 h. 10 μL of Promega’s CellTiter-Fluor reagent (Promega, Catalog #G6082), GF-AFC substrate diluted in assay buffer, was added to the plate for a 1x final concentration. 0.5% DMSO and 20 μM etoposide were used as controls to define 100% and 0% viability, respectively. The plate was then shaken on an orbital shaker at 300 RPM for 15 min at RT followed by a 30 min incubation at 37 °C. The CellTiter-Fluor signal was measured using a PerkinElmer Envision plate reader: excitation-400 nm, emission-505 nm. IC50 values were calculated using a four-parameter logistic curve fit using Genedata Screener software.
A549 Target Engagement Assay.
A549 cells were routinely maintained in filtered RPMI media (Gibco, Catalog 11875) supplemented with 10% dialyzed FBS (Corning, Catalog #35–071-CV) using a humidified incubator (37°C, 5% CO2, and ambient O2). In preparation for the target engagement assay, cells were harvested and resuspended in filtered RPMI media supplemented with 10% dialyzed FBS (Corning, Catalog #35–071-CV). Cells were seeded onto a 96-well tissue culture plate (Falcon, Catalog #353072) at a density of 15,000 cells/well in a volume of 100 μL. The tissue culture plate was incubated for 24 h at 37 °C with 5% CO2 and ambient O2. Stock solutions of the test compounds were prepared in 100% DMSO (Sigma, Catalog #D2650) and serially diluted 1:3 using 100% DMSO. Compounds were additionally diluted 1:200 in culture medium, and 200 μL/well were transferred to the tissue culture plate. Following compound addition, the microplate was incubated at 37 °C for 24 h. The L-glutamine and L-glutamate levels in the media were then measured using the YSI 2900 Biochemistry Analyzer (Xylem). IC50 values were calculated using a four-parameter logistic curve fit using Genedata Screener software.
In vitro Metabolic Stability, Solubility, Caco-2 Permeability, Plasma Protein Binding, hERG and CYP inhibition experiments.
Microsomal stability.
Microsomal stability assays were conducted on a Beckmann Biomek FXp laboratory automation system. The liver microsomal incubation mixture consisted of liver microsomes (0.5 mg microsomal protein/mL), the test compound (1 uM), MgCl2 (3 mM), and EDTA (1 mM) in potassium phosphate buffer (100 mM, pH 7.4). Midazolam and Ketanserin were used as the assay control substrates. The reaction was initiated with the addition of an NADPH regeneration solution (1.3 mM NADPH) and maintained at 37 °C with shaking. At five time points ranging from 0 to 45 min, aliquots (50 uL) were removed and quenched with acetonitrile (100 μL) containing an internal standard (imipramine). After vortex and centrifugation, samples were analyzed by LC-MS/MS. Calculation of the in vitro half-lives and clearance followed literature guidelines.50
Hepatocytes stability.
Hepatocytes were purchased from Bioreclamation IVT, Xenotech, or RILD. Stock solutions were prepared at 10 mM in DMSO for the test compound. Aliquots of the stock solutions were diluted to 200 μM with DMSO and then further diluted to 2 μM with KHB buffer. The procedure was as follows: Count hepatocytes and then dilute the cell suspensions to the appropriate density (viable cell density = 2 × 106 cells/mL). Add 50 μL of pre-warmed (37 °C) 2 μM test compound to the wells designated for different time points. For 0 min, add 100 μL of ACN containing internal standard (IS) to the wells followed by 50 μL of hepatocytes solution and then seal the wells. Add 50 μL of pre-warmed hepatocytes solution to the wells designated for 15 min, 30 min, 60 min and 120 min, and start timing. Place the assay plate in an incubator at 37 °C. At 15 min, 30 min, 60 min and 120 min, add 100 μL of ACN to the wells, respectively, then seal the corresponding wells. After quenching, sonicate the plate for 5 min and then centrifuge at 5594 × g for 15 min (Thermo Multifuge × 3R). Transfer 50 μL of the supernatant from each well into a 96-well sample plate containing 120 μL of ultra-pure water for LC/MS analysis. The peak area response ratio (PARR) to IS of the compounds at 15 min, 30 min, 60 min, and 120 min was compared to the PARR at 0 min to determine the percent of the test compound remaining at each time point. Half-lives were calculated using Excel software, fitting to a single-phase exponential decay equation.
Kinetic solubility.
Studies were conducted in 100 mM phosphate buffer (pH 7.4). Stock solution was prepared at 10 mM in DMSO for each test compound. 10 μL of stock solutions were added into 990 μL of 100 mM phosphate buffer (pH 7.4), for final concentrations of test compounds of 100 μM. The assay plate was shaken on a vibrator (IKA, MTS 2/4) for 1 h at 1000 rpm at RT and then centrifuged at 12,000 rpm (ThermoFisher) for 10 min to precipitate undissolved particles. An aliquot of the supernatant was taken from each well and diluted 10x with buffer, then analysed by LC-MS/MS compared to a standard curve.
Thermodynamic solubility.
Studies were conducted in buffers as listed in Tables 4 and 5. Approximately 2 mg of each test compound was weighed into a 2 mL vial and to the vial was then added 1 mL of buffer. Each sample was vortexed for 30 sec to achieve a homogenous dispersion, and then shaken on a Labquaker for 24 h at RT. If all solid dissolved, then additional test compound was added. After 24 h, the visual appearance of each sample was noted, and the sample was manually filtered through a 0.45 μm nylon filter. The subsequent filtrate, diluted as necessary with suitable solvent, was analyzed by LC-MS/MS compared to a standard curve.
Caco-2 permeability.
Caco-2 cells were obtained from American Tissue Culture Collection (Rockville, MD). The cells were maintained in Modified Eagle’s medium (MEM) containing 10% heat-inactivated FBS and 1% non-essential amino acids, in CO2 at 37 °C. Cells were seeded on polycarbonate filter inserts (Millipore, CAT#PSHT 010 R5). The cells were cultured for 21–28 days prior to the transport experiments. The transepithelial electric resistance (TEER) and Lucifer Yellow permeability were checked before and after the assay. Compounds were dissolved at 10 mM in DMSO and diluted for studies in Hank’s Balanced Salt Solution (HBSS, Invitrogen, Cat# 14025–092) with 25 mM HEPES, pH 7.4. Compounds were tested at 10 μM, in both the apical-to-basolateral (A-B) and basolateral-to-apical (B-A) directions, and were conducted at 37 °C for 90 min. At the end of incubation, donor samples were diluted 10-fold by assay buffer, then 60 μL of receiver and diluted-donor samples were mixed with 60 μL of acetonitrile and concentrations were analyzed by LC-MS/MS compared to a standard curve.
Plasma protein binding.
Plasma protein binding (PPB) assays were conducted using the Rapid Equilibrium Dialysis (RED) device (ThermoFisher Scientific). Warfarin and metoprolol were used as the control substrates. To the receiver side was added 350 μL of phosphate buffered saline (pH 7.4, 1x, Gibco). To the donor side was added 200 μL of plasma (Bioreclamation IVT) spiked with the test compound (5 μM). The same plasma/test compound solution (50 μL) was also used for the recovery sample. The plate was covered with Immunoware sealing tape and was incubated at 37 °C with shaking at 100 rpm for 5 h. After the incubation, both the receiver and donor sides were sampled (50 μL) and matched with the same volume of matrix from the other side. The recovery, donor, and receiver samples were extracted with 300 μL of cold ACN containing imipramine as the internal standard. After vortex and centrifugation, the supernatant (150 μL) was subjected to LC-MS quantitation. PPB (% bound) was calculated as %Bound = 100 × ([Donor]-[Receiver])/[Donor].
hERG Qpatch (Automated Electrophysiology) Assay.
A CHO cell line stably transfected with hERG cDNA and expressing hERG channels was used for studies. Cells were cultured in medium (from Invitrogen) containing Ham’s F12, 10% (v/v) heat inactivated FBS, 100 μg/mL Hygromycin B and 100 μg/mL Geneticin. The cells used in the QPatch study met the following criteria: under microscopy examination, the majority of cells in suspension were single and isolated; their viability was greater than 95%, with only a few debris and cell clumps (which may clog the holes in QPlate during whole-cell clamp recording); cell density ranged within 3–8 × 106 cells/mL in the final suspension before applying to the QPatch stir chamber. After leaving the CO2 incubator, cells were maintained in serum-free medium buffered with HEPES. Cells were used for recording within four hours after harvesting. Whole-cell recordings were performed using automated QPatch (Sophion, Denmark). The cells were voltage clamped at a holding potential of −80 mV. The hERG current was activated by depolarizing at +20 mV for 5 sec, after which the current was taken back to −50 mV for 5 sec to remove the inactivation and observe the outward tail current. The maximum amount of tail current size was used to determine hERG current amplitude. After achieving break-in (whole cell) configuration, the cells were recorded for 120 sec to assess current stability. The voltage protocol described above was then applied to the cells every 15 sec throughout the whole procedure. Only stable cells with recording parameters above threshold were allowed to enter the drug application procedure. All experiments were conducted at RT (about 25 °C). External solution containing 0.1% DMSO (vehicle) was applied to the cells to establish the baseline. After allowing the current to stabilize for 3 min, compound was applied. Compound solution was added, and the cells were kept in the test solution until the compound’s effect reached a steady state or for a maximum of 4 min. For the dose response assay, compound was applied to the cells accumulatively from low to high concentrations. Washout with external solution was performed after compound testing. Positive control cisapride was used to test the same batch of hERG cells for compound testing to ensure the normal response and quality of cells. Data were analyzed using Assay Software provided by Sophion, XLFit and Graphpad Prism.
CYP Inhibition Assays.
Studies were carried out in human liver microsomes (0.1 mg/mL for CYP 1A2/2C9/2D6/3A4; 0.5 mg/mL for CYP 2C19). Liver microsomes were purchased from BD Gentest. Aliquots of the compound stock solutions were diluted to 4 mM with acetonitrile, and then further diluted upon the addition of liver microsomes (0.2 mg/mL). An aliquot of 30 μL of diluted test compound solution was mixed with 15 μL of substrate solution. The plate was pre-warmed to 37 °C, then 15 μL of 8 mM NADPH (also pre-warmed to 37 °C) was added. The plate was incubated at 37 °C for the following incubation times: 5 min for 3A4, 10 min for 1A2/2C9/2D6, and 45 min for 2C19. The reaction was stopped by adding acetonitrile at the designated time point. The assay plates were shaken on a vibrator (IKA, MTS 2/4) for 10 min (600 rpm) and centrifuged at 5,594 g for 15 min (Thermo Multifuge × 3R). Aliquots of the supernatant were taken, diluted 1:3 into distilled water, and metabolite concentrations were analysed by LC-MS/MS compared to internal standards. Substrates used were Phenacetin (30 μM) for 1A2, Diclofenac (10 μM) for 2C9, S-Mephenytoin (35 μM) for 2C19, Midazolam (10 μM) or Testosterone (80 μM) for 3A4, and Bufuralol (10 μM) for 2D6. Metabolites measured were Acetaminophen for 1A2, 4’-Hydroxy-Diclofenac for 2C9, Hydroxy-Mephenytoin for 2C19, 1-Hydroxy-Midazolam or Osalmid for 3A4, and 1-Hydroxy-Bufuralol for 2D6. Positive control inhibitors used were α-Naphthoflavone for CYP1A2, Sulfaphenazole for CYP2C9, Omeprazole for CYP2C19, Quinidine for CYP2D6 and Ketoconazole for CYP3A4. The peak area response ratio (PARR) of metabolite to internal standard of the metabolites in test compound samples at desired time points was compared to the PARR in control samples to determine the percent of control sample (% control) at each time point. % inhibition was calculated as 100-% control.
In vivo pharmacokinetics.
Mouse:
Female mice (CD1 strain, purchased from Shanghai JH Laboratory Animal Co. LTD) weighing 20–30 g were used for studies. Food and water were available to all animals ad libitum. The test article was dosed via tail vein (IV doses) or oral gavage (PO doses), respectively. Blood samples were collected from all animals at predose and at 0.083, 0.25, 0.5, 1, 2, 4, 8, and 24 h postdose into tubes containing the anticoagulant K2EDTA (3 animals per time point with 3 time points collected per animal). Plasma was separated from the blood by centrifugation at 4 °C and stored at −70 °C until analysis. Test article concentrations in plasma were quantified using a liquid chromatography with tandem mass spectrometry (LC-MS/MS) method.
Rat:
Male rats (SD strain, purchased from Shanghai JH Laboratory Animal Co. LTD) weighing 200–300 g were used for studies. Animals were fasted overnight and fed 4 h postdose. Water was available ad libitum for all animals. Test article was dosed via dorsal foot vein (IV doses) or oral gavage (PO doses). Blood samples were collected via tail vein from all animals at predose and at 0.083, 0.25, 0.5, 1, 2, 4, 8, and 24 h postdose into tubes containing the anticoagulant K2EDTA. Plasma was separated from the blood by centrifugation at 4 °C and stored at −70 °C until analysis. Test article concentrations in plasma were quantified using a liquid chromatography with tandem mass spectrometry (LC-MS/MS) method.
Dog:
Male Beagle dogs (purchased from Beijing Marshall Biotechnology Co., Ltd) weighing 7–10 kg were used for studies. Animals were fasted overnight and fed 4 h postdose. Test article was administered to dogs via the cephalic vein (IV doses) or oral gavage (PO doses). Blood samples were collected via the saphenous vein or cephalic vein from all animals at predose and 0.083, 0.25, 0.5, 1, 2, 4, 8, and 24 h postdose into tubes containing the anticoagulant K2EDTA. Plasma was separated from the blood by centrifugation at 4 °C and stored at −70 °C until analysis. Test article concentrations in plasma were quantified using a liquid chromatography with tandem mass spectrometry (LC-MS/MS) method.
Monkey:
Male Cynomolgus monkeys (purchased from Hainan Jingang Biotech. Co., Ltd) weighing 3–5 kg were used for studies. Animals were fasted overnight and fed 4 h postdose. Test article was administered to monkeys via the cephalic vein (IV doses) or nasal gavage (PO doses). Blood samples were collected via the saphenous vein or cephalic vein from all animals at predose and 0.083, 0.25, 0.5, 1, 2, 4, 8, and 24 h postdose into tubes containing the anticoagulant K2EDTA. Plasma was separated from the blood by centrifugation at 4 °C and stored at −70 °C until analysis. Test article concentrations in plasma were quantified using a liquid chromatography with tandem mass spectrometry (LC-MS/MS) method.
In Vivo Models.
All experiments were conducted in compliance with institutional guidelines.
Glutamine and Glutamate Levels in H460 Xenograft Model.
NSG female mice (Jackson Labs) were implanted with H460 cells (5×105 cells/mouse diluted with matrigel 1:1). Mice were between 6–10 weeks old. All animals received LabDiet 5053 chow ad libitum. Tumors were allowed to grow to 300–400 mm3, and animals were treated with compound 27 (bis-hydrochloride) formulated in 0.5% methylcellulose in sterile water. Animals were euthanized via CO2 at 8 or 24 h after a single dose of compound 27 by oral gavage, and tumors were harvested. Each group contained 6 animals. Tumors were weighed and snap frozen. Tumor sections were homogenized using an OmniBEAD Ruptor 24 at 100 mg tissue/mL in MeOH/Water (80:20) containing 13C5-L-glutamine and 13C5-L-glutamate as internal standards. Homogenates were centrifuged at 15,000 rpm at 4 °C for 10 min An aliquot of 10 μL of the supernatant was diluted with 190 μL of 0.1% formic acid in ACN/water (50:50), vortexed for 15 sec, and centrifuged at 15,000 rpm at 4 °C for 5 min Samples were analyzed on an Agilent 1290 infinity LC system coupled with an Agilent 6460 triple quadrupole mass spectrometer operated at positive mode (ESI+). A Waters XBridge Amide column (3.5 μm; 4.6 × 100 mm) was used for analyte separation. The HPLC buffer A was 95% (v/v) water/ACN containing 20 mM ammonium hydroxide and 20 mM ammonium acetate. The HPLC buffer B was 100% ACN. The gradient was 80% B (0–1 min), 80–10% B (1–3 min), 10% B (3–5 min), 10–80% B (5–5.3 min), 80% B (5.3–10 min). The column temperature was 40 °C and the flow rate 0.5 mL/min. The sample injection volume was 2 μL. The detection conditions of the mass spectrometer were as follows: capillary voltage 4000 V, nebulizer pressure 35 psi, cell accelerate voltage 4 V, sheath gas temperature 400 °C, sheath gas flow 11 L/min, source gas temperature 300 °C, source gas flow 11 L/min, fragmenter voltage 80 V, collision energy 26 V (glutamate) and 14 V (glutamine). Metabolites were detected by compound specific multiple reaction monitoring transition (MRM) and retention time (RT): Glutamine (m/z 147>84, RT 4.93 min), 13C5-Glutamine (m/z 152>88, RT 4.93 min), Glutamate (m/z 148>84, RT 4.78 min), 13C5-Glutamate (m/z 153>88, RT 4.78 min). The method was validated with an analytical range of 10 – 5000 ng/mL for both glutamine and glutamate in ACN/Water (1:1). GraphPad Prism was used for generation of graphs, and data is expressed as the mean ± standard deviation.
Efficacy in Ru337 PDX model.
8 week old NSG female mice (Jackson Labs) were implanted with Ru337 (Memorial Sloan Kettering Cancer Center) patient derived xenografts (PDX) subcutaneously on the right flank. Tumors were allowed to grow to an average volume of 100 mm3 as monitored by caliper measurements. Animals were then randomized into groups of 8. All animals received chow ad libitum. Mice were treated with compounds on a 5 day-on/2 day-off schedule. Compound 27 was formulated in 0.5% methylcellulose in sterile water and dosed at 100 mg/kg, PO, BID (doses administered approximately 8 hours apart (8:00 and 16:00) each dosing day followed by a 16 hour gap before the next day’s dose). TAK-228 was formulated in 5% sucrose and 0.5% methylcellulose in sterile water and dosed at 1 mg/kg, PO, QD. Body weights were monitored twice per week. Tumor volume was calculated using the formula: V=l2*L/2 (l=length; L=width). GraphPad Prism was used for generation of graphs, and data is expressed as the mean ± standard deviation. For the combination arms, standard deviations were very low (bars smaller than the size of plotted datapoints).
Molecular Modeling
The molecular model in Figure 3 of compound 5 bound to GLS1 was generated using Molecular Operating Environment (MOE, 2019.01; Chemical Computing Group ULC, 1010 Sherbrooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2019). The X-ray crystal structure of GLS1 with 2 (BPTES) bound (PDB 3UO9) was prepared for modeling using the QuickPrep routine in MOE. 2 was then removed from the structure and 5 was docked using the general docking module within MOE. The top-scoring ligand pose for 5 that replicated the binding mode and interactions between BPTES and GLS1 was subjected to additional energy minimization in MOE while holding the protein coordinates fixed to arrive at the final model.
Supplementary Material
ACKNOWLEDGMENTS
We thank the following chemists at Shanghai Chempartner Co., Ltd for help in synthesizing compounds: Yanbing Ding, Li Wang, Fagui Liu, Baoquan Tian, Xuejie Xuan, Hui Yang, and Chun Zhou. We acknowledge Richard Lewis, Alessia Petrocchi and Naphtali Reyna in our chemistry department for helpful discussions throughout the project. We thank Jason Cross in our structural chemistry department for preparing the modeling snapshot.
ABBREVIATIONS USED
- ACN
acetonitrile
- AcOH
acetic acid
- AUC
area under the curve
- BID
twice daily
- BPTES
bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide
- Cl/CL
clearance
- Cmax
maximum concentration
- CYP
cytochrome P450
- DAST
(diethylamino)sulfur trifluoride
- DCM
dichloromethane
- DIPEA
N,N-diisopropylethylamine
- DMAP
4-dimethylaminopyridine
- DMF
N,N-dimethylformamide
- DMSO
dimethyl sulfoxide
- DON
6-diazo-5-oxy-L-norleucine
- EDC
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- EDTA
ethylenediaminetetraacetic acid
- EtOAc
ethyl acetate
- F%
bioavailability
- FBS
fetal bovine serum
- FESSIF
fed state simulated intestinal fluid
- α-KG
alpha-ketoglutarate
- LC
liquid chromatography
- GLS-1
glutaminase-1
- GLS-2
glutaminase-2
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]- pyridinium 3-oxide hexafluorophosphate
- HBSS
Hank’s balanced salt solution
- HEPES
(4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- hERG
human ether-a-go-go related gene
- HOBt
1H-benzo[d][1,2,3]triazol-1-ol
- IND
investigational new drug
- IS
internal standard
- IV
intravenous
- KEAP
Kelch-like ECH-associated protein
- MeOH
methanol
- MS
mass spectrometry
- NADPH
nicotinamide adenine dinucleotide phosphate
- ND
not determined
- NMR
nuclear magnetic resonance
- NRF2
nuclear factor (erythroid-derived 2)-like 2
- NSCLC
non-small cell lung cancer
- PARR
peak area response ratio
- PDX
patient-derived xenograft
- PK
pharmacokinetic(s)
- PO
per os
- QD
once daily
- ROS
reactive oxygen species
- RPM
revolutions per minute
- RPMI
Roswell Park Memorial Institute
- RT
RT
- SAR
structure activity relationship
- SD
Sprague-Dawley
- SGF
fasted state simulated gastrointestinal fluid
- t1/2
half-life
- TCA
tricarboxylic acid cycle
- T3P
propylphosphonic anhydride
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- Vdss
volume of distribution at steady state
- Xantphos
4,5-bis(diphenylphosphino)-9,9-dimethylxanthene
- X-Phos
2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI:
Experimental details for the preparation of compounds 5-26; UPLC and high resolution mass spectrometry data for componds 24-27; logD, logP and pKa determinations for compound 27; experimental procedues for oral dosing formulations and additional in vivo studies; standard deviations for GLS-1 and A549 assay results for compounds 2, 4 and 5-27; representative IC50 curve for compound 27 in the A549 assay; Kinomescan and CEREP off-target screening data for 27; Figures S1–S3.
Molecular formula strings (CSV)
REFERENCES
- 1.Xu X; Meng Y; Li L; Xu P; Wang J; Li Z; Bian J Overview of the development of glutaminase inhibitors: Achievements and future directions. J. Med. Chem 2019, 62, 1096–1115. [DOI] [PubMed] [Google Scholar]
- 2.Katt WP; Cerione RA Glutaminase regulation in cancer cells: a druggable chain of events. Drug Discovery Today 2014, 19(4), 450–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hensley CT; Wasti AT; DeBerardinis RJ Glutamine and cancer: cell biology, physiology, and clinical opportunities. J. Clin. Invest 2013, 123(9), 3678–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wise DR; Thompson CB Glutamine addiction: a new therapeutic target in cancer. Trends Biochem. Sci 2010, 35, 427–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gross M; Chen J; Englert J; Janes J; Leone R; Rodiriquez M; Mackinnon A; Parlati F; Shwonek P; Powell J Abstract 2329: glutaminase inhibition with CB-839 enhances anti-tumor activity of PD-1 and PD-L1 antibodies by overcoming a metabolic checkpoint blocking T cell activation. Cancer Res 76: 2329, 2016. [Google Scholar]
- 6.Leone RD; Zhao L; Englert JM; Sun I-M;Oh M-H; Sun I-H; Arwood ML; Bettencourt IA; Patel CH; Wen J; Tam A; Blosser RL; Prchalova P; Alt J; Rais R; Slusher BS; Powell JD Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 2019, 366(6468), 1013–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heffernan T; Toniatti C; Kovacs J; Giuliani V; Spencer N; Di Francesco ME; Bristow CA Glutaminase Inhibitor Therapy for Cancer International Patent Application Number WO2016/004418 A1, 2016. [Google Scholar]
- 8.Romero R; Sayin VI; Davidson SM; Bauer MR; Singh SX; LeBoeuf SE; Karakousi TR; Ellis DC; Bhutkar A; Sánchez-Rivera FJ; Subbaraj L; Martinez B; Bronson RT; Prigge JR; Schmidt EE; Thomas CJ; Goparaju C; Davies A; Dolgalev I; Heguy A; Allaj V; Poirier JT; Moreira AL; Rudin CM; Pass HI; Vander Heiden MG; Jacks T; Papgiannakopoulos T Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med 2017, 23(11), 1362–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gross MI; Demo SD; Dennison JB; Chen L; Chernov-Rogan T; Goyal B; Janes JR; Laidig GJ; Lewis ER; Li J; MacKinnon AL; Parlati F; Rodriguez MLM; Shwonek PJ; Sjogren EB; Stanton TF; Wang T; Yang J; Zhao F; Bennett MK Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Molecular Cancer Therapeutics 2014, 13(4), 890–901. [DOI] [PubMed] [Google Scholar]
- 10.Li J; Csibi A; Yang S; Hoffman GR; Li C; Zhang E; Yu JJ; Blenis J Synthetic lethality of combined glutaminase and Hsp90 inhibition in mTORC1-driven tumor cells. Proc. Nat. Acad. Sci 2015, 112(1), E21–E29. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11.Elgogary A; Xu Q; Poore B; Alt J; Zimmermann SC; Zhao L; Fu J; Chen B; Xia S; Liu Y; Neisser M; Nguyen C; Lee R; Park JK; Reyes J; Hartung T; Rojas C; Rais R; Tsukamoto T; Semenza GL; Hanes J; Slusher B; Le A Combination therapy with BPTES nanoparticles and metformin targets the metabolic heterogeneity of pancreatic cancer. Proc. Nat. Acad. Sci 2016, 113(36), E5328–E5336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jacque N; Ronchetti AM; Larrue C; Meunier G; Birsen R; Willems L; Saland E; Decroocq J; Maciel TT; Lambert M; Poulain L; Hospital MA; Sujobert P; Joseph L; Chapuis N; Lacombe C; Moura IC; Demo S; Sarry JE; Recher C; Mayeux P; Tamburini J; Bouscary D Targeting glutaminase has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood 2015, 126(11), 1346–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Momcilovic M; Bailey ST; Lee JT; Fishbein MC; Magyar C; Braas D; Graebor T; Jackson NJ; Czernin J; Emberley E; Gross M; Janes J; Mackinnon A; Pan A; Rodriguez M; Works M; Zhang W; Parlati F; Demo S; Garon E; Krysan K; Walser TC; Dubinett SM; Sadeghi S; Christofk HR; Shackelford DB Targeted inhibition of EGFR and glutaminase induces metabolic crisis in EGFR mutant lung cancer. Cell Reports 2017, 18, 601–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lampa M; Arlt H; He T; Ospina B; Reeves J; Zhang B; Murtie J; Deng G; Barberis C; Hoffmann D; Cheng H; Pollard J; Winter C; Richon V; Garcia-Escheverria C; Adrian F; Wiederschain D; Srinivasan L Glutaminase is essential for the growth of triple-negative breast cancer cells with a deregulated glutamine metabolism pathway and its suppression synergizes with mTOR inhibition. PLOS One, 2017, 12(9), e0185092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sappington DR; Siegel ER; Hiatt G; Desai A; Penney RB; Jamshidi-Parsian A; Griffin RJ; Boysen G Glutamine drives glutathione synthesis and contributes to radiation sensitivity of A549 and H460 lung cancer cell lines. Biochim. Biophys. Acta 2016, 1860(4), 836–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boysen G; Jamshidi-Parsian A; Davis MA; Siegel ER; Simecka CM; Kore RA; Dings RPM; Griffin RJ Glutaminase inhibitor CB-839 increases radiation sensitivity of lung tumor cells and human tumor xenografts in mice. Int. J. Rad. Biol 2019, 95(4), 436–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Willis RC; Seegmiller JE The inhibition by 6-diazo-5-oxo-L-norleucine of glutamine catabolism of the cultured human lymphoblast. J. Cell. Physiol 1977, 93, 375–382. [DOI] [PubMed] [Google Scholar]
- 18.Newcomb RW Preparation of Bis[(carboxamidothiadiazolyl)ethyl] Sulfides and Ethers as Glutaminase Inhibitors US6451828, 2002.
- 19.Wang JB; Erickson JW; Fuji R; Ramachandran S; Gao P; Dinavahi R; Wilson KF; Ambrosio ALB; Dias SMG; Dang CV; Cerione RA Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 2010, 18, 207–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lemberg KM; Vornov JJ; Rais R; Slusher BS We’re not “DON” yet: Optimal dosing and prodrug delivery of 6-diazo-5-oxo-L-norleucine. Molecular Cancer Therapeutics 2018, 17(9), 1824–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Katt WP; Ramachandran S; Erickson JW; Cerione RA Dibenzophenanthridinesas inhibitors of glutaminase C and cancer cell proliferation. Molecular Cancer Therapeutics 2012, 11(6), 1269–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stalnecker CA; Ulrich SM; Li Y; Ramachandran S; McBrayer MK; DeBerardinis RJ; Cerione RA; Erickson JW Mechanism by which a recently discovered allosteric inhibitor blocks glutamine metabolism in transformed cells. Proc. Nat. Acad. Sci 2015, 112(2), 394–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lukey MJ; Cluntun AA; Katt WP; Lin MJ; Druso JE; Ramachandran S; Erickson JW; Le HH; Wang Z-E; Blank B; Greene KS; Cerione RA Liver-type glutaminase GLS2 is a druggable metabolic node in luminal-subtype breast cancer. Cell Rep 2019, 29(1), 76–88.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robinson MM; McBryant SJ; Tsukamoto T; Rojas C; Ferraris DV; Hamilton SK; Hansen JC; Curthoys NP Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES). Biochem. J 2007, 406, 407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeLaBarre B; Gross S; Fang C; Gao Y, Jha A; Jiang F; Song J,J; Wei W; Hurov JB Full-length human glutaminase in complex with an allosteric inhibitor. Biochemistry 2011, 50, 10764–10770. [DOI] [PubMed] [Google Scholar]
- 26.Zimmerman SC; Duvall B; Tsukamoto T Recent progress in the discovery of allosteric inhibitors of kidney-type glutaminase. J. Med. Chem 2019, 62, 46–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shukla K; Ferraris DV; Thomas AG; Stathis M; Duvall B; Delahanty G; Alt J; Rais R; Rojas C; Gao P; Xiang Y; Dang CV; Slusher BS; Tsukamato T Design, synthesis and pharmacological evaluation of bis-2-(5-phenylacetamido-1,2,4-thiadiazolyl-2-yl)ethyl sulfide 3 (BPTES) analogs as glutaminase inhibitors. J. Med. Chem 2012, 55, 10551–10563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McDermott LA; Iyer P; Vernetti L; Rimer S; Sun J; Boby M; Yang T; Fioravanti M; O’Neill J; Wang L; Drakes D; Katt W; Huang Q; Cerione R Design and evaluation of novel glutaminase inhibitors. Biorg. Med. Chem 2016, 24, 1819–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McDermott L; Koes D; Mohammed S; Iyer P; Boby M; Balasubramanian V; Geedy M; Katt W; Cerione R GAC inhibitors with a 4-hydroxypiperidine spacer: Requirements for potency. Biorg. Med. Chem. Lett 2019, 29(19), 126632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Z; Li D; Xu N; Fang J; Yu Y; Hou W; Ruan H; Zhu P; Ma R; Lu S; Cao D; Wu R; Ni M; Zhang W; Su W; Ruan BH Novel 1,3,4-selenadiazole-containing kidney-type glutaminase inhibitors showed improved cellular uptake and antitumor activity. J. Med. Chem 2019, 62, 589–603. [DOI] [PubMed] [Google Scholar]
- 31.Finlay MR; Anderton M; Bailey A; Boyd S; Brookfield J; Cairnduff C; Charles M; Cheasty A; Critchlow SE; Culshaw J; Ekwuru T; Hollingsworth I; Jones N; Leroux F; Littleson M; McCarron H; McKelvie J; Mooney L; Nissink JWM; Perkins D; Powell S; Quesada MJ; Raubo P; Sabin V; Smith J; Smith PD; Stark A; Ting A; Wang P; Wilson Z; Winter-Holt JJ; Wood JM; Wrigley GL; Yu G; Zhang P Discovery of a thiadiazole-pyridazine-based allosteric glutaminase 1 inhibitor series that demonstrates oral bioavailability and activity in tumor xenograft models. J. Med. Chem 2019, 62, 6540–6560. [DOI] [PubMed] [Google Scholar]
- 32.Burns AC; Collins MR; Greasley SE; Hoffman RL; Huang Q; Kania RS; Kung P; Linton MA; Narasimhan LS; Richardson PF; Richter DT; Smith G Cycloalkyl-linked Diheterocycle Derivatives International Patent Application Number WO2015/166373 A1, 2015. [Google Scholar]
- 33.Cianchetta G; Lemieux RM; Cao S; Ding Y; Ye Z Compounds and Their Methods of Use International Patent Application Number WO 2015/143340 A1, 2015. [Google Scholar]
- 34.Bhavar PK; Vakkalanka SK; Viswanadha S; Swaroop MG; Babu G Novel Glutaminase Inhibitors International Patent Application Number WO2015/101958 A2, 2015. [Google Scholar]
- 35.Li J; Chen L; Goyal B; Laidig G; Stanton TF; Sjogren EB Heterocyclic Inhibitors of Glutaminase International Patent Application Number WO2013/078123 A1, 2013. [Google Scholar]
- 36.Vogl DT; Younes A; Stewart K; Orford KW; Bennett M; Siegel D; Berdeja JG Phase 1 study of CB-839, a first-in-class, glutaminase inhibitor in patients with multiple myeloma and lymphoma. Blood 2015, 126(23), 3059. [Google Scholar]
- 37.Kalinsky K; Harding JJ; DeMichele A; Infante JR; Gogineni K; Owonikoko TK; Isakoff S; Iliopuolos O; Patel NR; Munster P; Telli ML; Jenkins Y; Fiji GP; Whiting SH; Meric-Bernstam F Phase 1 study of CB-839, a First-in-Class Inhibitor of Glutaminase, in Combination with Paclitaxel in Patients with Advanced Triple Negative Breast Cancer [abstract]. In: Proceedings of the 2017 San Antonio Breast Cancer Symposium; 2017 Dec 5–9; San Antonio, TX. Philadelphia (PA): AACR; Cancer Res. 2018, 78(4 Suppl), Abstract nr PD3–13. [Google Scholar]
- 38.Meric-Bernstam F; Lee RJ; Carthon BC; Iliopoulos O; Mier JW; Patel MR; Tannir NM; Owonikoko TK; Haas NB; Voss MH; Harding JJ; Srinivasan R; Shapiro G; Telli ML; Munster PN; Carvajal RD; Jenkins Y; Whiting SH; Bendell JC; Bauer TM CB-839, a glutaminase inhibitor, in combination with cabozantanib in patients with clear cell and papillary metastatic renal cell cancer (mRCC): Results of a phase 1 study. J. Clin. Onc 2019, 37 (7 suppl), 549. [Google Scholar]
- 39.DeLaBarre B; Gross S; Fang C; Gao Y, Jha A; Jiang F; Song J,J; Wei W; Hurov JB Full-length human glutaminase in complex with an allosteric inhibitor. Biochemistry 2011, 50, 10764–10770. [DOI] [PubMed] [Google Scholar]
- 40.Soth MJ; Le K; Liu G; Kovacs JJ; Burke JP; Bardenhagen JP; Bristow CA; Czako B; Carroll CL; Di Francesco ME; Hamilton MM; Geck Do M; Harris A; Giuliani V; Huang S; Jiang Y; Johnson T; Kang Z; Liu Z; McAfoos T; Miller M; Morlacchi P; Palmer WS; Pang J; Rogers N; Shepard HE; Spencer ND; Theroff J; Xu Q; Yau A; Draetta G; Toniatti C; Nazarenko N; Heffernan TP; Jones P First-time Disclosure of IPN60090, a Potent and Selective GLS1 Inhibitor with Excellent Physicochemical Properties, Targeting Cancers with Specific Metabolic Vulnerabilities. Abstracts of Papers, 259th ACS National Meeting and Exposition, Philadelphia, PA, United States, March 22–26, 2020; American Chemical Society: Washington, D. C., 2020; MEDI 0288. [Google Scholar]
- 41.Kilbourn MR Thiophenes as phenyl bio-isosteres: Application in radiopharmaceutical design-I. Dopamine uptake antagonists. Nucl. Med. Biol 1989, 16(7), 681–686. [DOI] [PubMed] [Google Scholar]
- 42.Thangavelu K; Pan CQ; Karlberg T; Balaji G; Uttamchandani M; Suresh V; Schüler H; Low BC; Sivaraman J Structural basis for the allosteric inhibitory mechanism of human kidney-type glutaminase (KGA) and its regulation by Raf-Mek-Erk signaling in cancer cell metabolism. Proc. Nat. Acad. Sci 2012, 109(20), 7705–7710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hopkins AL; Groom CR; Alex A Ligand efficiency: a useful metric for lead selection. Drug Discovery Today 2004, 9(10), 430–431. [DOI] [PubMed] [Google Scholar]
- 44.Gillis EP; Eastman KJ; Hill MD; Donnelly DJ; Meanwell NA Applications of fluorine in medicinal chemistry. J. Med. Chem 2015, 58, 8315–8359. [DOI] [PubMed] [Google Scholar]
- 45.This fluorine substitution was inspired by unpublished work in our group on an earlier, structurally related chemical series containing the same four carbon linker. Metabolite identification studies on an analog identified an internal carbon as a site of metabolism, and microsomal stability was improved by adding a fluorine to this carbon.
- 46.The 100 mg/kg BID dose of compound 27 was determined to be the maximally tolerated dose in an earlier model study (unpublished), and we used it as our default dose for subsequent studies, including the ones in this manuscript. The 250 mg/kg BID dose of compound 4 (CB-839) was taken from in-house studies showing consistent maximal in vivo target engagement at this dose, and is slightly higher than the 200 mg/kg BID dose used by Calithera in reference 9.
- 47.Dientsmann R; Rodon J; Serra V; Tabernero J Picking the point of inhibition: A comparative review of PI3K/AKT/mTOR pathway inhibitors. Molecular Cancer Therapeutics 13(5), 2014, 1021–1031. [DOI] [PubMed] [Google Scholar]
- 48.Kovacs JJ; Spencer ND; Bristow CA; Giuliani V; Miller MA; Soth MJ; Harris AL; Le K; Burke JP; Carugo A; Jiang Y; Xu QA; Bardenhagen JP; Carrillo CC; Carroll CL; Chang Q; Cross JB; Czako B; Feng N; Gao G; Garvey JR; Gay JP; Geck Do MK; Gera S; Gogi K; Greer J; Hamilton M; Han M; Herrera ZJ; Huang S; Johnson SB, Johnson T; Kang Z; Kost-Alimova M; Lewis RT; Linares J; Liu C-Y; Liu G; Liu J; Liu Z; Yan Ma X; McAfoos T; Miao L; Minelli R; Molina J; Morlacchi P; Mseeh F; Mullinax RA; Palmer W; Pang J; Parker CA; Peoples MD; Petrocchi A; Quill TM; Ramamoorthy V; Rogers N; Shepard HE; Suarez C; Sun Y; Suzuki E; Tan L; Theroff JP; Wu Q; Yau A; Zhao S; Zuniga AM; Weinstein JN; Lorenzi PL; de Stanchina E; Rudin CM; Toniatti C; Di Francesco ME; Marszalek JR; Draetta G; Yap TA; Jones P; Heffernan TP Unpublished results
- 49.The 5 day / 2 off schedule was standard for the research group running the study, chosen for convenience of dosing. In other studies, including the H2122 study contained in the supporting information, continuous every day schedules were well-tolerated.
- 50.Obach SR Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: An examination on in vitro half-life approach and nonspecific binding to microsomes. Drug Metabolism and Disposition 1999, 27(11), 1350–1359. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.