

Abstract
Conventional angiogenic factors, such as vascular endothelial growth factor (VEGF), regulate both pathological and physiological angiogenesis indiscriminately, and their inhibitors may elicit adverse side effects. Secretogranin III (Scg3) was recently reported to be a diabetes-restricted VEGF-independent angiogenic factor, but the disease selectivity of Scg3 in retinopathy of prematurity (ROP), a retinal disease in preterm infants with concurrent pathological and physiological angiogenesis, was not defined. Here, using oxygen-induced retinopathy (OIR) mice, a surrogate model of ROP, we quantified an exclusive binding of Scg3 to diseased versus healthy developing neovessels that contrasted sharply with the ubiquitous binding of VEGF. Functional immunohistochemistry visualized Scg3 binding exclusively to disease-related disorganized retinal neovessels and neovascular tufts, whereas VEGF bound to both disorganized and well-organized neovessels. Homozygous deletion of the Scg3 gene showed undetectable effects on physiological retinal neovascularization but markedly reduced the severity of OIR-induced pathological angiogenesis. Furthermore, anti-Scg3 humanized antibody Fab (hFab) inhibited pathological angiogenesis with similar efficacy to anti-VEGF aflibercept. Aflibercept dose-dependently blocked physiological angiogenesis in neonatal retinas, whereas anti-Scg3 hFab was without adverse effects at any dose and supported a therapeutic window at least 10X wider than that of aflibercept. Therefore, Scg3 stringently regulates pathological but not physiological angiogenesis, and anti-Scg3 hFab satisfies essential criteria for development as a safe and effective disease-targeted anti-angiogenic therapy for ROP.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00018-021-04111-2.
Keywords: Retinopathy of prematurity, Secretogranin III, Scg3, Pathological angiogenesis, Physiological angiogenesis, Targeted anti-angiogenic therapy
Introduction
Angiogenesis can be classified into two major categories: physiological and pathological angiogenesis [1]. The former involves neovessel formation in embryos, neonates, tissue repair and the female reproductive system, such as the ovary, uterus, and placenta [2, 3]. The latter includes neovascularization in cancer, inflammatory and autoimmune diseases [4]. Pathological angiogenesis is also a common manifestation in multiple ocular diseases, such as proliferative diabetic retinopathy (DR) and retinopathy of prematurity (ROP) with pathological retinal neovascularization (RNV), neovascular age-related macular degeneration (AMD) with choroidal neovascularization (CNV), corneal neovascularization and neovascular glaucoma [5, 6].
Conventional angiogenic factors, such as vascular endothelial growth factor (VEGF), drive both physiological and pathological angiogenesis and play an important role in vasculogenesis and neovessel formation in nearly all types of neovascularization. Despite their broad clinical indications for cancer and ocular neovascular diseases, anti-VEGF drugs inhibit both pathological and physiological angiogenesis indiscriminately and, therefore, exert not only therapeutic benefits on diseased vessels but also adverse side effects on healthy vasculatures [7–9]. Developing disease-targeted VEGF-independent anti-angiogenic therapies with wide safety margins remains a challenge but is of mechanistic and translational importance to unravel disease etiology, improve treatment efficacy and circumvent adverse side effects.
ROP, a retinal vasoproliferative disease primarily occurring in premature infants, is a leading cause of blindness in children [10]. Of all neovascular diseases, ROP is unique because of its concurrent physiological and pathological angiogenesis [11]. Compared with mature and quiescent vessels in adults and the elderly that can tolerate non-specific angiogenic inhibition, neovessels in the developing retina of preterm infants are highly susceptible to such non-specific blockade. In this regard, ROP represents a formidable challenge to develop targeted therapies for preferential inhibition of pathological vs. physiological angiogenesis.
By applying comparative ligandomics profiling we recently reported that secretogranin III (Scg3) is a diabetes-restricted VEGF-independent angiogenic factor that binds selectively to diabetic but not healthy retinal vessels, whereas VEGF binds equally to both diabetic and healthy vasculatures [12]. These distinct binding patterns were independently confirmed by in vivo functional studies, in which Scg3 preferentially stimulated angiogenesis and vascular leakage in diabetic but not healthy vessels, while VEGF-induced angiogenesis and vascular leakage in both diabetic and healthy mice [12, 13]. Furthermore, Scg3-neutralizing monoclonal antibodies (mAbs) alleviated retinal vascular leakage in diabetic mice, pathological RNV in ROP mouse models and CNV in mouse models of neovascular AMD with high efficacy [12, 14, 15]. However, no studies have characterized the disease selectivity of Scg3 in ROP or its surrogate animal models of oxygen-induced retinopathy (OIR). A critical question is whether anti-Scg3 therapy can suppress pathological RNV during OIR without affecting physiological RNV.
Here, we quantitatively compared the binding activity of Scg3 and VEGF to OIR and healthy neovessels in live mice and visualized their distinct binding patterns to different vascular structures by “functional immunohistochemistry (IHC)”. We further investigated the role of Scg3 in physiological and pathological angiogenesis in Scg3-knockout (KO) mice with or without OIR. Despite their similar efficacy to alleviate OIR, dose-escalation studies revealed severe side effects of the anti-VEGF drug aflibercept, including complete blockade of retinal vessel development, but not anti-Scg3 humanized hAb (hAb) Fab fragment (hFab) in neonatal mice, with striking differences in therapeutic windows. Anti-Scg3 hFab is presented with safety and efficacy profiles that satisfy essential criteria for development as a ligand-guided disease-targeted anti-angiogenic therapy for ROP.
Materials and methods
Materials
Anti-Scg3 hFab (Clone EBP2) was generated from anti-Scg3 ML49.3 mAb by Everglades Biopharma, LLC [16]. Briefly, EBP2 hFab recognizes both human (hScg3) and mouse Scg3 (mScg3), which share 88.3% identity in amino acid sequences, and has the binding affinity (KD) of 8.7 nM to hScg3 as compared to the binding affinity of 35.0 nM for the parental ML49.3 mAb. EBP2 hAb and ML49.3 mAb competitively bind to the same site of hScg3 or mScg3. Human retinal microvascular endothelial cells (HRMVECs) were from Cell Systems [12]. The human SK-N-AS neuroblastoma cell line was kindly provided by Dr. Leonid Metelitsa.
Animals and OIR mice
C57BL/6 J mice were purchased from the Jackson Laboratory. Scg3-KO mice from Taconic Bioscience (Model #TF3191) were backcrossed to C57BL/6 J for at least five generations and verified by genotyping. Deletion of the Scg3 gene in Scg3−/− mice was independently confirmed by Western blot using anti-Scg3 polyclonal antibody (unpublished data). OIR mice (males and females) were prepared as described [12, 15]. Briefly, mice (male and female) at P7 were exposed to 75% oxygen for 5 days with their nursing mothers, returned to room air at P12 and analyzed for OIR at indicated time points. OIR mice at P17 with body weights between 6 and 7.5 g were included in the studies [17, 18].
In vitro phage binding assay
Clonal T7 phage displaying VEGF110 (VEGF-Phage) was described in a previous study [12]. Clonal phage displaying full-length hScg3 (Scg3-Phage) was constructed in a similar manner. Scg3-Phage and VEGF-Phage were analyzed for their binding to anti-Scg3 hFab and aflibercept, respectively, in ELISA plates. Briefly, Scg3-Phage, VEGF-Phage and control phage displaying no foreign protein were amplified in BLT5615 bacteria, purified by CsCl gradient centrifugation, dialyzed against PBS and titrated for plaque forming unit (pfu) by phage plaque assay [12]. Purified phages were coated onto ELISA plates (1 × 108 pfu/ml, 100 µl/well), washed, blocked, and incubated with anti-Scg3 hFab or aflibercept (0.1 μg/ml) in the presence or absence of hScg3 or VEGF (1 µg/ml), respectively. After washing, bound anti-Scg3 hFab or aflibercept was detected using horseradish peroxidase-conjugated goat anti-human IgG Ab (Sigma, #A8667), followed by colorimetric assay [19].
In vivo ligand binding assay
Purified Scg3-Phage, VEGF-Phage or control phage was preincubated with anti-Scg3 hFab, aflibercept (4 μg/ml) or PBS for 30 min and injected i.v. to OIR and healthy mice at P17 (1 × 1012 pfu/mouse). After circulating for 20 min, mice were euthanized by CO2 inhalation, immediately followed by intracardial perfusion with 25 ml of PBS for 5 min to remove unbound phages from the blood. The eye, lung, kidney and cortex of the brain were isolated. The retina and lens, including the vitreous, were collected after the removal of the cornea, iris, retinal pigment epithelium, choroid and sclera. All the isolated tissues, including the whole eye and retina/vitreous/lens, were weighed and homogenized in PBS with 1% Triton X-100 using a polytron homogenizer until no solid tissues were visible. Vessel-bound phages were quantified by phage plaque assay and normalized against tissue weights.
In vitro ligand-cell binding assay
HRMVECs and SK-N-AS neuroblastoma cells were seeded in 48-well plates precoated with 1% gelatin at 70–80% confluency and cultured overnight. Purified Scg3-Phage or VEGF-Phage was preincubated with anti-Scg3 hFab, aflibercept (4 µg/ml) or PBS for 30 min and added to cells (1 × 1012 pfu/well). After incubation at 37 ℃ for 30 min in PBS supplemented with 1% BSA, cells were washed three times with PBS/1% BSA and lysed with PBS/0.5% SDS. Cell-bound phages were quantified by plaque assay [19].
Functional IHC
To visualize in vivo phage binding and distribution, Scg3-Phage or VEGF-Phage was amplified in BLT7FLAG bacteria to label each phage particle with ~ 400 FLAG tags [19]. Amplified phages were purified for in vivo binding as above, followed by intracardial perfusion with PBS and subsequent 4% paraformaldehyde (PFA). Retinas were isolated, permeabilized, blocked in Blocking Solution A (5% BSA, 20% goat serum and 1% Triton X-100 in PBS) for 1 h at room temperature (RT) and incubated with anti-FLAG M2 mAb (Sigma, #F1804; 1:200) in Blocking Solution A for 2 days at 4℃. After washing, retinas were incubated with Alexa Fluor 594-conjugated anti-mouse IgG H&L (Cell Signaling, #8890S; 1:1,000) and Alexa Fluor 488-isolectin B4 (AF488-IB4, 10 µg/ml, Thermo Fisher Scientific, #I21411) in Blocking Solution A overnight at 4 °C. After washing, retinas were flat-mounted in 50% glycerol in PBS and analyzed using a Keyence structured illumination fluorescence microscope (SIM, Model BZ-X800). Signal intensity was quantified using ImageJ.
Anti-angiogenic therapy
OIR mice at P14 received intravitreally (ivt) injection of anti-Scg3 hFab, aflibercept, control hFab or PBS at indicated doses (0.5 µl/eye). Eyes were enucleated from euthanized mice at P17 and fixed in 4% PFA (pH 7.4) for 45 min. Retinas were isolated, stained for vessels with AF488-IB4 overnight at RT, washed, flat-mounted and analyzed using the SIM. Images were blind-coded and quantified for neovascularization, neovascular tufts, number of branch points and the central avascular area, as described [12, 15]. Branch points were quantified in a defined area (0.21 mm2), which was 800 μm away from the retinal optic nerve head. Vessel density was quantified as the ratio of a vessel-covered area vs. the same total area. All data were normalized to non-injection control.
Inhibition of physiological angiogenesis
C57BL/6 J mice at P1 were anesthetized by chilling on ice, and an incision was made in the eyelid with a corneal scissor to fully expose the eye. Sharp end glass micropipettes were backfilled with the solution of anti-Scg3 hFab, aflibercept (4–40 μg/μl) or PBS using microloading pipette tips. We injected ivt 0.5 μl/eye solution using a microinjector (Eppendorf FemtoJet 4i Model #5252000021) at 500 hPa for 2 pulses at 0.2 s. Retinas were isolated at P4 from euthanized mice, stained with AF488-IB4, flat-mounted and quantified for the percentage of the vascularized retinal area, radius of vascularized area and vessel density as above. Alternatively, therapeutic agents (5 µg/0.5 µl/eye) were injected ivt at P5, and eyes were isolated at P7, fixed and subjected to the similar analysis.
Supplementary methods
Additional methods for wound healing migration assay, fluorescein angiography (FA), optical coherence tomography (OCT), electroretinography (ERG), IHC, histology and electron microscopy are available online as Supplementary Materials.
Statistics
Results are expressed as means + SEM. Statistical analysis was performed using GraphPad Prism (version 8.0, GraphPad Software). Results were analyzed using Student’s t test (for differences between two groups), one-way ANOVA (for multiple groups). Differences between groups with P < 0.05 were considered statistically significant.
Results
Scg3 expression in OIR retina
We previously reported that Scg3 expression does not increase in the retina and vitreous of OIR mice as measured by Western blot [12]. To confirm this finding, we compared Scg3 expression in OIR and healthy retinas by IHC. The results revealed that Scg3 is predominantly expressed in the retinal ganglion cell layer (GCL), inner plexiform layer (IPL), outer plexiform layer (OPL), and immature photoreceptor segment layer (PRL) (Fig. 1). Scg3 expression is reduced in the inner nuclear layer (INL) and barely detected in the outer nuclear layer (ONL). Scg3 is predominantly expressed in endocrine/neuroendocrine cells and neurons, but not endothelial cells (ECs), where it is directed via a classical signal peptide to secretory vesicles, including neurotransmitter vesicles, to regulate the biogenesis of secretory granules [20]. The results in Fig. 1 are consistent with this expression pattern as well as the pattern in mouse retinas with or without DR [12, 21]. Scg3, primarily expressed in neurotransmitter vesicles, is not expected to be present in the nucleus. Therefore, Scg3 signals are largely absent in the outer nuclear layer and are detected only at reduced levels in the INL, because of the presence of neurites in that layer. Neither the level of Scg3 expression nor its distribution was changed by OIR, and no differences in Scg3 expression were detected between the avascular and vascular retinas of OIR mice (Fig. 1). The specificity of IHC was confirmed by the presence of Scg3 signals only in wild type but not Scg3−/− retinas (Supplementary Fig. 1).
Fig. 1.
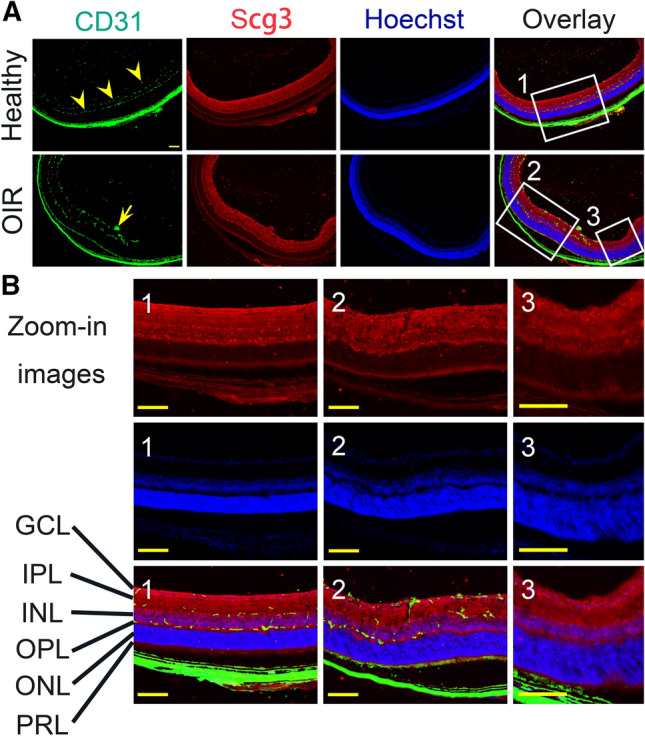
Scg3 expression in healthy and OIR retina. a The expression of Scg3 (red) and endothelial marker CD31 (green) in healthy and OIR retina was detected by IHC. Arrowheads indicate the superficial, intermediate and deep retinal vascular plexuses. Arrow indicates pathological neovascular tufts. GCL, retinal ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer; PRL, immature photoreceptor layer. b Zoom-in images: Image 1 for healthy retina, Image 2 for OIR retina with pathological RNV, and Image 3 for OIR retina in the avascular area. Scale bar = 100 μm
Quantitative comparison of Scg3 and VEGF binding selectivity in OIR and healthy retinal neovessels
Pathological angiogenesis can be driven by the upregulation of angiogenic factors, their cognate receptors or both [22]. Whereas Scg3 expression remains unchanged by OIR (Fig. 1) [12], our data and other reports show that VEGF expression is upregulated in OIR (Supplementary Fig. 2) [23]. As a result, we speculated that OIR increases the expression or availability of an as yet unidentified Scg3 receptor (Scg3R), a condition that would account for the therapeutic activity of Scg3-neutralizing mAbs to alleviate OIR-induced pathological angiogenesis [12, 15]. To address this possibility and measure the binding of Scg3 to healthy versus OIR retinas, we developed a new assay to quantify and compare in vivo ligand binding of Scg3 and VEGF by modifying a previous phage-based ligandomics methodology [12]. Clonal Scg3-Phage and VEGF-Phage displaying full-length Scg3 and VEGF110 were confirmed by ELISA for binding to Scg3-neutralizing hFab and aflibercept, respectively (Supplementary Fig. 3). The ELISA binding profiles were competitively blocked by excess Scg3 or VEGF recombinant protein (Supplementary Fig. 3), consistent with the retention of native conformations of the phage-displayed proteins.
In vivo binding assay was performed by i.v. injection of Scg3-Phage or VEGF-Phage into OIR and healthy mice, followed by 20-min circulation to allow vessel binding and subsequent intracardial perfusion to remove unbound phages (Fig. 2a). Vessel-bound phages in isolated tissues were released by homogenization and quantified by plaque assays. OIR mice develop not only pathological RNV but also intravitreal neovascularization, which is technically difficult to dissect. Therefore, we quantified in vivo ligand binding to vessels of the whole eye as well as a mixture of the retina, vitreous and lens (retina/vitreous/lens). We found that Scg3-Phage binding to the vasculature of the whole eye markedly increased by 7.0-fold in OIR vs. healthy mice, and this increase was fully blocked by Scg3-neutralizing hFab (Fig. 2b). No such differential binding to the OIR eye was detected for VEGF-Phage (Fig. 2d). We further confirmed that Scg3-Phage binding to the OIR retina/vitreous/lens increased by 1.9-fold (Fig. 2c) and was also blocked by anti-Scg3 hFab. Once again, VEGF-Phage binding to the OIR tissues was only marginally increased (Fig. 2e). Minimal Scg3-Phage binding to the healthy eye and retina/vitreous/lens was blocked by anti-Scg3 hFab (Fig. 2b, c). Despite the absence of increased binding to OIR retinas, the basal VEGF-Phage binding to healthy and OIR vasculatures was blocked by aflibercept (Fig. 2d, e). Both Scg3-Phage and VEGF-Phage had similar background binding after blockade by anti-Scg3 hFab and aflibercept, respectively (Fig. 2b vs. 2d and 2c vs. 2e). These findings indicate that Scg3 binds minimally to healthy developing vessels, but the binding to OIR vasculatures is markedly increased. In contrast, VEGF binds equally to both healthy and OIR vessels in the developing eye.
Fig. 2.

In vivo binding selectivity of Scg3 and VEGF to OIR and healthy retinal vessels. a Schematic of the in vivo phage-binding assay. b, c Scg3-Phage binding to the whole eye (b) or retina/vitreous/lens (c) of healthy and OIR mice (pfu/mg tissue) with or without the blockade by anti-Scg3 hFab. d, e VEGF-Phage binding to the whole eye (d) or retina/vitreous/lens (e) of healthy and OIR mice in the presence or absence of aflibercept. + SEM; Sample sizes (n = # mice/group) are indicated below graph bars; **P < 0.01, ***P < 0.001, ****P < 0.0001, ns for not significant; t test. f, g Functional IHC to visualize the binding of FLAG-tagged Scg3-Phage (f) or VEGF-Phage (g) to vessels of flat-mounted OIR retinas with or without the blockade of their cognate inhibitors. Arrows and arrowheads indicate OIR-related disorganized neovessels and neovascular tufts, respectively. Bar = 50 µm
Because pathological neovessels become more permeable, we checked for selective leakage of phage from pathological versus normal vessels that could affect the validity of the binding assay. Although such selective leakage seems unlikely because of the large size of phage particles (see Discussion), we quantified the binding of an empty control phage vector to the eye, retina/vitreous/lens fractions and other organs of both healthy and OIR mice and confirmed similar background binding profiles and minimal increase in phage leakage from OIR eyes (Supplementary Fig. 4a–e).
Exposure of premature infants to hyperoxia may also cause vasculopathy in other organs, including the brain and lung [24]. Interestingly, Scg3-Phage binding to the vasculatures of the brain, lung and kidney in OIR mice minimally increased over healthy controls, and Scg3 binding was not significantly blocked by anti-Scg3-hFab (Supplementary Fig. 5a–c). Similarly, no significant increase in VEGF-Phage binding to the vasculature of the brain, lung and kidney of OIR mice over controls was detected, and excess aflibercept failed to significantly block VEGF-Phage binding to other organs in healthy or OIR mice (Supplementary Fig. 5d–f). These findings suggest that Scg3 is an OIR-inducible, ocular-restricted angiogenic factor.
Visualizing in vivo Scg3 binding to OIR but not healthy neovessels
To independently verify the binding selectivity of Scg3, we visualized the binding of Scg3-Phage to OIR neovessels by “Functional IHC”, which combines in vivo ligand-receptor binding with IHC. To improve detection sensitivity, we amplified clonal phages in BLT7FLAG bacteria, so that each phage particle concurrently displayed 5–15 copies of Scg3 and ~ 400 copies of FLAG tag on different capsid proteins on the phage surface [19]. After in vivo binding and in situ fixation, IHC of flat-mount OIR retinas revealed that clustered signals of FLAG-tagged Scg3-Phage predominantly colocalized with disorganized neovessels and neovascular tufts, rather than well-organized retinal neovessels (Fig. 2f,h). As a control, VEGF-Phage signals clustered and colocalized with all types of the retinal vessels, including large vessels, microcapillaries and tips of neovessels (Fig. 2g,i). VEGF signals were scattered across the OIR vasculature and sporadically associated with both well-organized and disorganized vessels, including neovascular tufts. The binding of both Scg3- and VEGF-Phage was specifically blocked by their cognate antagonists (Fig. 2f–i). Minimal binding of control phage was detected in OIR retinas by functional IHC (Supplementary Fig. 4f). Furthermore, functional IHC detected the binding of VEGF-Phage, but not Scg3-Phage and control phage, to the healthy retina (Supplementary Fig. 6). Differential binding patterns of Scg3-Phage and VEGF-Phage independently confirm that Scg3 selectively binds OIR but not healthy retinal neovessels, whereas VEGF binds to both. Few signals of Scg3-Phage, VEGF-Phage or control phage were detected in the extravascular space, including VEGF-binding retinal neurons [25], consistent with minimal phage leakage during the in vivo ligand binding assay.
Physiological and pathological angiogenesis in Scg3-deficient mice
Scg3 shares no amino acid sequence homology with other proteins, thereby implying an absence of genetic redundancy. Although Scg3-null mice were previously reported to present with normal gross phenotypes, including viability, body weight and fertility [26], vascular and ocular phenotypes of the mice have not been characterized but would be indicative of Scg3 involvement in physiological RNV. Careful comparison of Scg3−/− and Scg3+/+ mice revealed no difference at P6 or P21 by H&E staining, vascular immunostaining, or electron microscopy (Fig. 3a–i and Supplementary Fig. 7). These findings suggest that Scg3 plays no role in physiological angiogenesis and retinal development.
Fig. 3.

Phenotypes of Scg3 KO in healthy and OIR mice. a–e No abnormality in the retinal vascular development in flat-mount retinas of Scg3−/− mice. a Representative images of flat-mount retinas from Scg3+/+ and Scg3−/− mice at P6 and P21 with retinal vessels stained with AF488-IB4. Bar = 500 (top) and 100 μm (bottom). b Percentage of the vascularized area in (a). c Radius of the vascularized area. d Vessel density. e Number of branch points. n = 6 eyes/group in (b–e). f–i Normal phenotype of retinal vascular plexuses in Scg3−/− mice. f Representative images of retinal superficial, intermediate and deep plexuses in retinal sections from Scg3+/+ and Scg3−/− mice at P21. Bar = 100 (top) and 50 μm (bottom). g Number of vessels in the superficial layer (bright spots per 1200 μm). h Number of vessels per 1200 μm in the intermediate layer. i Number of vessels per 1200 μm in the deep layer. n = 6 eyes/group in (g–i). j–n Reduced severity of OIR in Scg3−/− mice. j Representative images of flat-mount retinas stained with AF488-IB4 from Scg3+/+ and Scg3−/− OIR mice at P17. Bar = 500 (top) and 100 μm (bottom). Arrowheads indicate RNV and neovascular tufts. k Quantification of RNV in (j). l Quantification of NV tufts. m Quantification of branch points. n Percentage of avascular area. n = 8 (Scg3+/+) and 12 eyes (Scg3−/−) in (k-n). ± SEM; **P < 0.01, ****P < 0.0001, ns for not significant; t test
To investigate Scg3 contribution to pathological angiogenesis in OIR, we analyzed and compared OIR severity in Scg3-KO vs. healthy mice. The results revealed that Scg3-deficient mice had reduced severity of OIR, including pathological RNV, neovascular tufts and branch points (Fig. 3j–n). These findings confirm that Scg3 plays an important role in OIR-induced pathological RNV.
Anti-Scg3 hAb efficiently alleviates pathological angiogenesis
To facilitate therapeutic translation, we engineered an anti-Scg3 hFab from a previously reported Scg3-neutralizing ML49.3 mAbs [12] (see Methods) and confirmed inhibition of Scg3-induced migration of HRMVECs by the hFab (Supplementary Fig. 8). As a positive control, VEGF-induced HRMVEC migration was blocked by aflibercept.
To analyze therapeutic activities, we induced OIR in mice and ivt injected anti-Scg3 hFab, aflibercept, control hFab or PBS into OIR mice at P14 and analyzed disease status at P17. Scg3-neutralizing hFab (2 µg/eye) significantly inhibited OIR-induced pathological RNV and neovascular tufts and reduced branch points and central avascular region (Fig. 4a–e). As a positive control, aflibercept at the same dose alleviated these symptoms with similar therapeutic efficacy, whereas the control human Fab was without effect (Fig. 4a–e). Interestingly, treatment with anti-Scg3 hFab at P14 did not markedly affect VEGF expression at P17 (Supplementary Fig. 2).
Fig. 4.

Anti-Scg3 hFab and aflibercept to dose-dependently inhibit OIR-induced pathological angiogenesis. a Representative images of flat-mount retinas. Anti-Scg3 hFab, aflibercept, control hFab (2 μg/eye) or PBS was injected ivt into OIR mice at P14. At P17, retinas were isolated, stained with AF488-IB4 to label vessels. Bar = 500 (top row) and 100 μm (bottom row). Arrowheads indicate RNV and neovascular tufts. b Percentage of neovascularization in (a). c Number of neovascular tufts in (a). d Number of branch points. e Percentage of avascular area. n = 12 (non-injection for data normalization), 10 (PBS), 10 (control hFab), 12 (aflibercept) and 15 eyes (anti-Scg3 hFab) in (b–e). f–i Quantifications as in (b–e) at different doses. Representative images of flat-mount OIR retinas, quantifications and sample sizes for 0.5 and 0.1 µg/eye are in Supplementary Fig. 9 and 10, respectively. ± SEM; *P < 0.05, **P < 0.01, *** P < 0.001, ****P < 0.0001, ns for not significant; one-way ANOVA test
To compare dose–response curves, we further titrated therapeutic activity at different doses. At 0.5 µg/eye, both anti-Scg3 hFab and aflibercept significantly inhibited pathological RNV, neovascular tufts and branch points (Fig. 4f–i and Supplementary Fig. 9). At this lower dose, the central avascular area was still significantly reduced by anti-Scg3 hFab (P < 0.05, vs. PBS, Fig. 4i and Supplementary Fig. 9e), but not by aflibercept. At 0.1 µg/eye, anti-Scg3 hFab significantly inhibited branch points (P < 0.01, vs. control hFab, Fig. 4h and Supplementary Fig. 10d), but no other therapeutic activities were detected for either anti-Scg3 hFab or aflibercept (Fig. 4f–i and Supplementary Fig. 10). Control hFab conferred no therapeutic benefits at any doses. These data indicate that anti-Scg3 hFab alleviated OIR with similar efficacy to aflibercept and a minimal effective dose at 0.5 µg/eye for both.
Anti-Scg3 hAb ameliorates retinal arterial tortuosity and vein dilation in OIR mice
Tortuous retinal arteries and dilated veins are the hallmarks of severe ROP called plus disease [10]. After treatments of OIR mice as described above, FA was used to non-invasively visualize retinal arteries and veins in anesthetized mice at P17. FA images confirmed straight retinal arteries in healthy mice but tortuous arteries with dilated veins in OIR mice (Fig. 5a). We quantified arterial tortuosity by measuring the relative length, triangular index and angle metrics of the retinal arteries. Both anti-Scg3 hFab and aflibercept at 2 µg/eye significantly ameliorated tortuosity indices (Fig. 5b–d). We further found that OIR conferred markedly dilated retinal veins that was significantly ameliorated by either anti-Scg3 hAb or aflibercept (Fig. 5a, e).
Fig. 5.

Anti-Scg3 hFab alleviates OIR-induced arterial tortuosity and vein dilation. Anti-Scg3 hFab, aflibercept, control hFab (2 μg/eye) or PBS was injected ivt into OIR mice at P14. At P17, retinas were examined using FA. a Representative FA images. Arrows indicate tortuous arteries, and arrowheads indicate dilated veins. Bar = 200 μm. b Quantification of relative length as a tortuosity index in (a), which is equal to the ratio of curved line/straight line. c Tortuosity index by triangular index. d Tortuosity index by sum of angle metrics. e Relative vein width. n = 14 (non-injection for data normalization), 12 (PBS), 10 (control hFab), 11 (aflibercept) and 16 eyes (anti-Scg3 hFab). ± SEM; **P < 0.01, ***P < 0.001, ****P < 0.0001; one-way ANOVA test
Adverse effects on physiological angiogenesis
Based on their binding selectivity (Fig. 2), we predicted that aflibercept, but not anti-Scg3 hFab, would inhibit physiological angiogenesis in the developing retina. To investigate this, we microinjected ivt anti-Scg3 hFab or aflibercept into healthy mice at P1, a stage where retinal vessels begin to sprout out of the optical nerve head of the avascular retina and expand outward to reach the peripheral retina at approximately P8 [27]. Flat-mount retinas treated with PBS showed a typical pattern of the developing mouse retina with a partially vascularized central region (labeled with green fluorescence, Fig. 6a) and peripheral avascular region at P4 (shadowed area, Fig. 6a). Aflibercept at 2, 5 and 20 µg/eye dose dependently inhibited retinal vascularization in the central area. The total area size of the retina (vascularized + avascular) was not affected by aflibercept treatment. At its highest dose, aflibercept completely abolished physiological angiogenesis of the developing retina, whereas anti-Scg3 hFab at the same dose was without detectable adverse effects. These striking differences were confirmed in multiple parameters of quantifying retinal vascularization, including the percentage of the vascularized area, radius of vascularized area and vessel density (Fig. 6b–d).
Fig. 6.

Aflibercept, but not anti-Scg3 hFab, adversely inhibits physiological retinal angiogenesis. a–d Adverse inhibition at P1–P4. Aflibercept, anti-Scg3 hFab or PBS was microinjected ivt into normal mice at P1. At P4, retinas were isolated and stained with AF488-IB4. a Representative images of flat-mount retinas. Bar = 500 μm. b Percentage of vascularized retinal area in (a). c Radius of vascular area. d Vessel density. n = 15 (PBS), 9 (aflibercept 2 μg/eye), 8 eyes (aflibercept 5 μg/eye), 9 (aflibercept 20 μg/eye) and 10 (anti-Scg3 hFab) in (b–d). e–i Adverse inhibition at P5-P7. Aflibercept, anti-Scg3 hFab (5 μg/0.5 µl/eye) or PBS was injected ivt at P5, and retinas were analyzed at P7 as in (a–d). e Representative images of retinas. Bar = 500 (top) and 100 μm (bottom). f Percentage of vascularized retinal area in (e). g Radius of vascular area. h Vessel density. i Branch points. n = 13 (PBS), 12 (aflibercept) or 12 eyes (anti-Scg3 hFab) in (f–i). ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns for not significant; one-way ANOVA test
To characterize adverse effects at a later developmental stage, we injected ivt anti-Scg3 hFab or aflibercept into healthy mice at P5 and quantified retinal vascularization at P7. Retinal vasculature at P7 covered the entire retina except for a narrow region at the periphery (Fig. 6e). Aflibercept (5 µg/eye) significantly reduced the percentage of the vascularized area, radius of vascularized area, vessel density and branch points, whereas anti-Scg3 hFab at the same dose had no detectable effects (Fig. 6e–i).
Taken together, these two independent studies suggest that blockade of Scg3 has no adverse side effects on physiological angiogenesis in the developing retina even at an excessively high dose. In contrast, aflibercept markedly inhibits physiological angiogenesis at a therapeutic dose.
Adverse effects on retinal structure and function
Inhibition of physiological angiogenesis in the developing retina may adversely impact the development of retinal structures in neonatal mice. Indeed, we found that aflibercept (2 µg/eye) injected ivt at P14 significantly reduced the total retinal thickness and the thickness of the INL and photoreceptor inner/outer segments (PIS/POS), but not the ONL, at P42 (Fig. 7a–e). No such adverse reduction was found for anti-Scg3 hFab at the same dose.
Fig. 7.

Aflibercept, but not anti-Scg3 hFab, decreases the retinal thickness in OIR mice. a–e Histological examination of the retinal thickness. Aflibercept, anti-Scg3 hFab (2 μg/eye) or PBS was injected ivt into OIR mice at P14. Eyes were enucleated at P42, and tissue sections were stained with H&E. Retinal images were analyzed. a Representative images of retina images. Bar = 500 (top) and 100 μm (bottom). b Total retinal thickness in (a). c Thickness of the inner nuclear layer (INL). d Thickness of the outer nuclear layer (ONL). e Thickness of the photoreceptor inner segments and outer segments (PIS/POS). n = 12 (PBS), 13 (aflibercept) and 11 eyes (anti-Scg3 hFab) in (b–e). f–i OCT analysis of the retinal thickness. OIR mice were treated as in (a–e). At P42, retinas were analyzed by OCT. f Representative OCT images. Bar = 200 (top) and 100 μm (bottom). g Total retina thickness (Line 1–3) in (f). h Thickness of the inner retina (Line 1–2). i Thickness of the outer retina (Line 2–3). n = 18 (PBS), 10 (control hFab), 22 (aflibercept) and 18 eyes (anti-Scg3 hFab) in (g–i). ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns for not significant; one-way ANOVA test
The adverse effects of aflibercept on retinal structures were also independently confirmed by OCT imaging (Fig. 7f). The results showed that the same treatment with aflibercept reduced the total retinal thickness (Fig. 7g) as well as the inner retinal thickness (Fig. 7h), but not the outer retinal thickness at P42 (Fig. 7i). Scg3-neutralizing hFab at the same dose had no detectable side effects (Fig. 7f–i).
We predicted that anti-Scg3 hFab with minimal side effects on retinal vessels should not compromise retinal functions. To characterize retinal function, we treated OIR mice ivt with anti-Scg3 hFab, aflibercept (2 µg/eye) or PBS at P14 and performed electroretinography (ERG) at P21, P42 and P60. Aflibercept significantly inhibited a-wave amplitude (i.e., magnitude of photoreceptor response to a flash) at P60 but not at other timepoints (Fig. 8a-d). Aflibercept significantly suppressed b-wave amplitude at P21, P42 and P60 (Fig. 8a–c, f). Aflibercept increased b-wave latency (i.e., time needed to respond a flash) at P42 but not at other time points (Fig. 8g), consistent with previous findings [15, 28]. In contrast, anti-Scg3 hFab had no such adverse effects on the amplitude or latency of a- and b-waves at any timepoint (Fig. 8a–g).
Fig. 8.

Anti-Scg3 hFab has no detectable adverse effects on retinal function. Aflibercept, anti-Scg3 hFab (2 μg/eye) or PBS was injected ivt into OIR mice at P14. ERG was performed at P21, P42 and P60. a–c Average scotopic ERG curves of all mice in each group at P21, P42 and P60, respectively. d a-wave amplitude at P21, P42 and P60. e Latency of a-wave at P21, P42 and P60. f b-wave amplitude at P21, P42 and P60. g Latency of b-wave at P21, P42 and P60. Sample sizes (n = # eyes per group) are indicated at the bottom of the graphs. h Representative images of the OIR retina stained with anti-PKC-α mAb. Arrowheads indicate ectopic dendrites. Bar = 50 μm. i Number of bipolar cells per 500 μm in (h). n = 6 eyes/group. ± SEM; * P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns for not significant; one-way ANOVA test
Suppression of b-wave by aflibercept implies possible disruption of retinal development that affects neuronal impulse transmission from photoreceptors to bipolar cells. Indeed, aflibercept injected ivt into OIR mice at P14 markedly reduced the number of bipolar cells at P42, as detected by IHC using anti-PKC-α mAb (Fig. 8h, i). Furthermore, frequent ectopic dendrites associated with bipolar cells were observed in mice treated with aflibercept (arrowheads, Fig. 8h) [29]. No abnormality in the number of bipolar cells and their ectopic dendrites were detected in mice treated with anti-Scg3 hFab.
Scg3-Phage binding to neuronal cells
The neurotrophic actions of VEGF are well established [25], and the side effects of aflibercept on the retina (Figs. 7 and 8) likely involve direct inhibition of such actions on retinal neurons as well as indirect actions of anti-angiogenesis that restrict blood supply to developing neurons in immature retinas. Whereas anti-Scg3 does not inhibit physiological angiogenesis, it is possible that anti-Scg3 therapy will adversely impact neurons. Therefore, as a possibly prognostic indicator, we investigated putative interactions of Scg3 with retinal neurons by quantitatively comparing the binding of Scg3-Phage and VEGF-Phage to cultured HRMVECs and SK-N-AS neuronal cells in the presence and absence of their cognate blockers. As shown in Fig. 9a–d, the specific binding of Scg3-Phage to HRMVECs was only 10.1% of background (P = 0.023), compared with 54.3% (P = 0.0000004) for VEGF-Phage. Specific binding of Scg3-Phage to SK-N-AS cells was not significant, whereas the equivalent binding of VEGF-Phage was 30.0% (P = 0.0003). The results suggest that Scg3 does not interact directly with neurons, consistent with the apparently normal neuronal development seen in Scg3-knockout mice (Supplementary Fig. 7) [26].
Fig. 9.
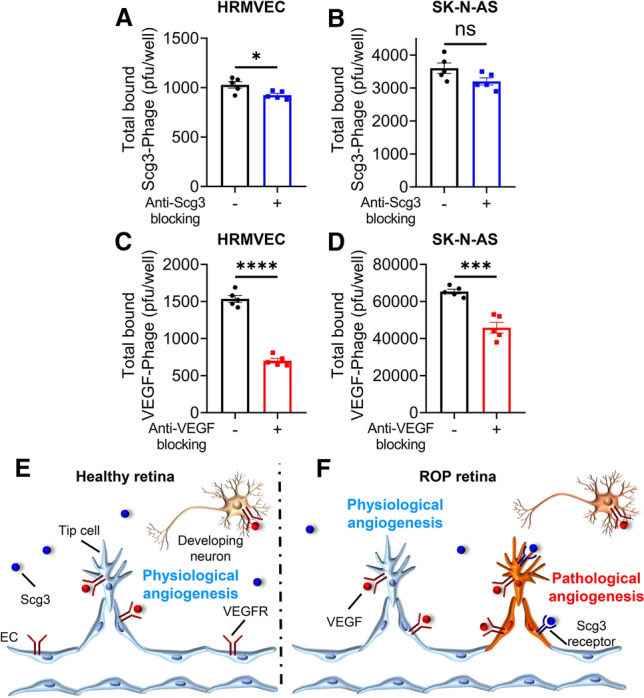
Anti-Scg3 therapy selectively targets pathological, but not physiological, angiogenesis in the OIR retina. a The healthy retina has only physiological angiogenesis. VEGFRs are expressed on the surface of endothelial cells and neurons while Scg3R is not detectable. b The OIR retina has concurrent physiological and pathological angiogenesis. Scg3R selectively expresses on pathological ECs. In contrast, VEGFRs express on both pathological and physiological ECs as well as retinal neurons. Consequently, Scg3 stringently regulates OIR neovessels, whereas VEGF modulates both healthy and OIR neovessels. While anti-Scg3 therapy selectively targets pathological but not physiological angiogenesis with optimal safety, anti-VEGF suppresses both pathological and physiological angiogenesis with adverse side effects
Discussion
Physiological and pathological angiogenesis share many similarities, including the growth of neovessels and regulation by various angiogenic factors, such as VEGF. Despite these common features, some distinctions between these two events are overt, implying subtle differences in underlying mechanisms. For example, physiological angiogenesis forms well-organized vascular networks, whereas pathological neovascularization drives aberrant growth of disorganized vasculatures with abnormal neovascular tufts and increased susceptibility to rupture and hemorrhage, as occurs in ROP, proliferative DR, CNV and cancer (Fig. 4a) [5, 30, 31]. Despite such clear phenotypical distinctions, molecular mechanisms and stimuli that distinguish pathological from physiological angiogenesis are much less clear. Over the past three decades, multiple potential selective regulators of pathological angiogenesis have been proposed, including αvβ3 integrin, rapamycin to inhibit Akt signaling, TEM8, CCR3, CC-chemokine inhibitor “35 K”, prion-like protein doppel, RUNX1, miR-30a-5p, Epac1 and adenosine A2A receptor [32–41]. However, most of these regulators have limited disease selectivity and/or involve VEGF-dependent pathways [40]. In addition, few therapies based on these alternative pathways have been precisely compared with anti-VEGF drugs for disease selectivity and therapeutic windows. Consequently, disease-targeted anti-angiogenic therapies are not currently available, and such feasibility remains uncertain.
All currently approved anti-angiogenic therapies for neovascular AMD and cancer target VEGF and do not discriminate between physiological and pathological angiogenesis [7, 9]. The critical role of VEGF in physiological angiogenesis is demonstrated by the phenotype of mice with deletion of a single VEGF allele or homozygous deletion of either VEGF receptor (VEGFR) 1 or 2, all of which die in utero with severe defects in vasculogenesis [42–44]. Therefore, blockade of VEGF pathways is predicted to elicit side effects for regeneration and repair in adults and especially vascular development in immature subjects. Indeed, aflibercept was reported to inhibit development of the retinal vasculature in neonatal mice and dogs [15, 28, 45]. The developmental disruption of retinal structure and brain function was described in ROP infants treated with anti-VEGF [46, 47]. Consequently, although the anti-VEGF drug ranibizumab is approved for ROP therapy in the European Union, there are currently no approved drug therapies for the disease in the U.S.
Scg3 was recently reported to be a diabetes-restricted angiogenic and vascular leakage factor that binds to DR but not healthy vessels, whereas VEGF binds equally to both diabetic and healthy vasculatures [12]. In contrast to VEGF that is upregulated by neovascular conditions [48, 49], Scg3 is minimally induced during DR or OIR (Fig. 1) [12]. Such differential binding and expression patterns have profound implications for disease therapy. Because secreted VEGF diffuses extracellularly to regulate both diseased and healthy vessels, VEGF inhibitors are predicted to adversely affect healthy vessels in addition to their therapeutic benefits on diseased vasculatures [15]. On the other hand, Scg3 promotes pathological angiogenesis by binding selectively to diseased vasculatures that express elevated Scg3-binding sites stationary on diseased endothelium without cross-reaction with healthy vessels. Therefore, Scg3 inhibition has minimal adverse side effects on healthy vasculatures. However, such correlations between binding selectivity and safety advantage have not been established for any angiogenic diseases and related therapies.
We reported here that Scg3 binding to OIR neovessels increased by 7.0-fold over control neovessels, as opposed to the 1731-fold binding increase to DR vessels (1731:0 for diabetic:healthy) quantified by previous ligandomics profiling [12]. Therefore, it is appropriate to ask why the fold binding increase is much smaller in OIR than that in the DR model, and is the lower Scg3 binding increase sufficient to confer the requisite safety advantages for OIR/ROP therapy?
Reasons for an artificially elevated binding fold in the ligandomics study include an excessively low background binding due to the presence of trillions of cDNA library phage particles that efficiently blocked non-specific binding of Scg3-phage to retinal vessels through phage own surface proteins [12]. Additionally, the high copy number of Scg3-phage detected by ligandomics was arbitrarily exaggerated by PCR amplification of Scg3 cDNA from enriched vessel-bound phages for next-generation sequencing (NGS). An absence to detect Scg3-phage in the healthy vessels by ligandomics was partially due to the limited coverage and depth of the NGS [12]. In contrast, the binding assay in the current study showed a relatively high background binding (Fig. 2b) due to the absence of cDNA library phages that would interfere with quantification of vessel-bound Scg3-Phage by plaque assay. As the high background binding in this study was primarily attributed to the differences in the binding assay platforms, Scg3 binding increase in OIR was arbitrarily reduced to only 7.0-fold. Indeed, the similar binding assay in DR models also detected only 17.8-fold increase in clonal Scg3-Phage binding to retinal vessels in diabetic versus healthy mice (unpublished data). Because the clonal Scg3-Phage binding assay more closely resembles conventional ligand binding than ligandomics, the results of 7.0-fold increase in Scg3 binding to OIR vessels is a more accurate measurement.
In contrast to the relatively low target specificity and high frequent off-target adverse effects of many small molecule drugs, anti-VEGF biologicals have high target specificity, and adverse effects are caused primarily by on-target toxicity, including suppression of developing retinal vessels and neurons (Figs. 6–8) [22]. It is challenging to accurately predict side effects of anti-Scg3 therapy that may be directed at cells and tissues beyond the vasculature. However, quantification of Scg3 binding to different cell types in the presence and absence of neutralizing antibodies may provide indications of such on-target toxicity. Because Scg3 displayed low specific binding to neuronal cells, quantitatively similar to the binding to healthy ECs (Fig. 9a, b), we predict minimal adverse effects of anti-Scg3 therapy on neuronal function. Similarly, minimal binding of Scg3 to healthy and OIR brain, lung or kidney vessels as determined by our functional IHC assay, predicts low to negligible side effects of anti-Scg3 hAb on the vasculatures of these organs. Although these predictions are supported by the normal development of Scg3−/− mice (Fig. 3), it is clear that only clinical trials can provide full validation of the safety in the human context.
Compared to conventional IHC to detect protein expression, functional IHC in this study visualized the location of ligand–receptor interactions by combining IHC with ligand binding. Co-localization of Scg3-Phage signals with disorganized vessels and neovascular tufts in OIR (Fig. 2f) implies that Scg3 likely promotes neovessel sprouting from these vascular structures, which are the hallmarks of ROP vasculature [50]. In contrast, the association of VEGF-Phage signals with large vessels and well-organized microcapillaries implicates that VEGF may drive vascular remodeling and neovessel sprouting from these VEGFR-expressing vasculatures.
The two new binding methods developed in this study represent valuable tools to quantify and visualize endothelial ligand binding. It was technically difficult for conventional approaches to quantify VEGF-cell binding activity, as VEGF had to be labeled with a radioisotope for sensitive detection of in vitro binding [51]. It is even more daunting to quantify VEGF binding in vivo [52], owing to vascular leakage of 45 kDa VEGF homodimer [53]. Healthy vessels are impermeable to serum albumin with 66 kDa, and pathological vasculatures may be permeable to proteins up to 1000–2000 kDa [53]. T7 phage with 55 nm in diameter is equivalent to a protein of approximately 90,000 kDa. Thus, we predicted and confirmed that control phage, Scg3-Phage and VEGF-Phage were not leaky in OIR neovessels (Fig. 2f, g and Supplementary Fig. 4 and 6). In unpublished data (not shown), we confirmed that Scg3-Phage is impermeable even to fenestrated choriocapillaris with or without laser-induced CNV, which is impermeable to proteins larger than 5.2 nm [54]. The new methods will provide much needed convenience to assess the involvement and disease selectivity of Scg3 and VEGF in other vascular diseases, such as CNV, DR and cancer before the expansion of their therapeutic indications. Additionally, we are using this new assay to validate Scg3R candidates.
The normal phenotype of vascular and retinal development in Scg3−/− mice suggest that Scg3 does not regulate physiological angiogenesis (Fig. 3a–i) [26], as opposed to embryonic lethality of VEGF−/+ mice [42]. These findings support the safety advantage of anti-Scg3 therapy, because the homozygous deletion of the Scg3 gene is equivalent to 100% Scg3 blockade. Furthermore, pathological role of Scg3 was revealed by the reduced severity of OIR-induced pathological RNV. Together, these data of Scg3−/− mice support the stringent regulation of Scg3 in pathological but not physiological angiogenesis for targeted anti-Scg3 therapy.
To establish a binding selectivity–safety correlation, this study quantitatively compared therapeutic windows of anti-Scg3 hFab and aflibercept in OIR mice by characterizing their dose–response curves for efficacy and safety (Figs. 4 and 6). We estimate that anti-Scg3 hFab has a therapeutic window of > 40X (20 µg/eye without side effects vs. minimal effective dose 0.5 µg/eye or less), whereas aflibercept has a window of < 4X (2 µg/eye with detectable side effects vs. 0.5 µg/eye) (Fig. 6a–d vs. Figure 4f–i). The safety difference is highlighted by the striking differential effects of aflibercept versus anti-Scg3 hFab on physiological angiogenesis (Fig. 6a). The preferential binding of Scg3 to the OIR retina vs. other non-ocular organs (Fig. 2b, c and Supplementary Fig. 5a–f) and optimal systemic safety profile of anti-Scg3 mAb [15] suggest that this new therapy could be developed for systemic delivery to circumvent ivt injection-related adverse effects in ROP eyes.
In summary, this study provides the following new information: (a) Scg3 is a highly OIR-restricted angiogenic factor that binds only to OIR neovessels, but not to healthy retinal neovessels or other organs subjected to conditions of OIR. (b) Anti-Scg3 hFab stringently targets pathological but not physiological angiogenesis in OIR mice with minimal adverse side effects and at least 10X wider therapeutic window than aflibercept. (c) Two new ligand binding methods described in this study provide valuable tools to quantify and visualize endothelial ligand binding in vivo. Our work confirms the feasibility of developing ligand-guided disease-targeted anti-angiogenic therapies and establishes a framework to develop such therapies by delineating the correlation between binding selectivity and safety, applicable to other signaling pathways and disease conditions. Anti-Scg3 hFab has the potential to be developed not only as a new anti-angiogenic drug, but also a founding member of a new class of selective angiogenesis blockers for targeted therapy of ROP with high efficacy and safety.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
The authors thank Drs. Yingbin Fu and Philp Rosenfeld for scientific discussion and Dr. Yuqin Wang for technical support. Ralph Nichols for TEM at the Imaging Core Facility, Cullen Eye Institute, Baylor College of Medicine.
Abbreviations
- AF488-IB4
Alexa Fluor 488-isolectin B4
- CNV
Choroidal neovascularization
- DR
Diabetic retinopathy
- EC
Endothelial cell
- ERG
Electroretinography
- FA
Fluorescein angiography
- hAb
Humanized antibody
- hFab
Humanized or human antibody Fab fragment
- HRMVEC
Human retinal microvascular endothelial cell
- IHC
Immunohistochemistry
- OCT
Optical coherence tomography
- OCTC
Optimal cutting temperature compound
- OIR
Oxygen-induced retinopathy
- RNV
Retinal neovascularization
- ROP
Retinopathy of prematurity
- Scg3
Secretogranin III
- Scg3R
Secretogranin III receptor
- VEGF
Vascular endothelial growth factor
Author contributions
CD, PW, LJ and CH designed and performed experiments and data analyses. YH and EZ provided technical support. HT provided reagents and technical support. AB, IP, GS and KW provided scientific support. WL conceived and supervised the project and experiments and analyzed data. CD and WL wrote and revised manuscript. KW revised manuscript.
Funding
This work was supported by NIH R01EY027749 (WL), R24EY028764 (WL and KAW), R24EY028764-01A1S1 (WL and KAW), R43EY031238 (HT, KAW and WL), R43EY031643 (HT), R41EY027665 (WL and HT), American Diabetes Association 1-18-IBS-172 (WL), NIH P30EY002520, Knights Templar Eye Foundation Endowment in Ophthalmology (WL) and an unrestricted institutional grant from Research to Prevent Blindness (RPB) to Department of Ophthalmology, Baylor College of Medicine.
Data availability
All data are included in this article and its supplementary files.
Declarations
Conflict of interest
HT and WL are shareholders of Everglades Biopharma, LLC and LigandomicsRx, LLC. The remaining authors declare no competing financial interests.
Ethical approval
All animal studies were approved by the Institutional Animal Care and Use Committee at University of Miami and Baylor College of Medicine.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Chung AS, Ferrara N. Developmental and pathological angiogenesis. Annu Rev Cell Dev Biol. 2011;27:563–584. doi: 10.1146/annurev-cellbio-092910-154002. [DOI] [PubMed] [Google Scholar]
- 2.Shimizu T, Hoshino Y, Miyazaki H, Sato E. Angiogenesis and microvasculature in the female reproductive organs: physiological and pathological implications. Curr Pharm Des. 2012;18:303–309. doi: 10.2174/138161212799040367. [DOI] [PubMed] [Google Scholar]
- 3.Patel-Hett S, D’Amore PA. Signal transduction in vasculogenesis and developmental angiogenesis. Int J Dev Biol. 2011;55:353–363. doi: 10.1387/ijdb.103213sp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aguilar-Cazares D, Chavez-Dominguez R, Carlos-Reyes A, et al. Contribution of Angiogenesis to Inflammation and Cancer. Front Oncol. 2019;9:1399. doi: 10.3389/fonc.2019.01399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campochiaro PA. Molecular pathogenesis of retinal and choroidal vascular diseases. Prog Retin Eye Res. 2015;49:67–81. doi: 10.1016/j.preteyeres.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee P, Wang CC, Adamis AP. Ocular neovascularization: an epidemiologic review. Surv Ophthalmol. 1998;43:245–269. doi: 10.1016/s0039-6257(98)00035-6. [DOI] [PubMed] [Google Scholar]
- 7.Yang Y, Zhang Y, Cao Z, et al. Anti-VEGF- and anti-VEGF receptor-induced vascular alteration in mouse healthy tissues. Proc Natl Acad Sci USA. 2013;110:12018–12023. doi: 10.1073/pnas.1301331110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taugourdeau-Raymond S, Rouby F, Default A, et al. Bevacizumab-induced serious side-effects: a review of the French pharmacovigilance database. Eur J Clin Pharmacol. 2012;68:1103–1107. doi: 10.1007/s00228-012-1232-7. [DOI] [PubMed] [Google Scholar]
- 9.Tanabe K, Wada J, Sato Y. Targeting angiogenesis and lymphangiogenesis in kidney disease. Nat Rev Nephrol. 2020;16:289–303. doi: 10.1038/s41581-020-0260-2. [DOI] [PubMed] [Google Scholar]
- 10.Hellström A, Smith LEH, Dammann O. Retinopathy of prematurity. Lancet. 2013;382:1445–1457. doi: 10.1016/S0140-6736(13)60178-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dai C, Webster KA, Bhatt A, et al. Concurrent physiological and pathological angiogenesis in retinopathy of prematurity and emerging therapies. Int J Mol Sci. 2021;22:4809. doi: 10.3390/ijms22094809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.LeBlanc ME, Wang W, Chen X, et al. Secretogranin III as a disease-associated ligand for antiangiogenic therapy of diabetic retinopathy. J Exp Med. 2017;214:1029–1047. doi: 10.1084/jem.20161802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rong X, Tian H, Yang L, Li W. Function-first ligandomics for ocular vascular research and drug target discovery. Exp Eye Res. 2019;182:57–64. doi: 10.1016/j.exer.2019.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.LeBlanc ME, Wang W, Ji Y, et al. Secretogranin III as a novel target for the therapy of choroidal neovascularization. Exp Eye Res. 2019;181:120–126. doi: 10.1016/j.exer.2019.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang F, LeBlanc ME, Wang W, et al. Anti-secretogranin III therapy of oxygen-induced retinopathy with optimal safety. Angiogenesis. 2019;22:369–382. doi: 10.1007/s10456-019-09662-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ji L, Waduge P, Hao L, et al. Selectively targeting disease-restricted secretogranin III to alleviate choroidal neovascularization. FASEB J. 2022;36:e22106. doi: 10.1096/fj.202101085RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stahl A, Chen J, Sapieha P, et al. Postnatal weight gain modifies severity and functional outcome of oxygen-induced proliferative retinopathy. Am J Pathol. 2010;177:2715–2723. doi: 10.2353/ajpath.2010.100526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Connor KM, Krah NM, Dennison RJ, et al. Quantification of oxygen-induced retinopathy in the mouse: a model of vessel loss, vessel regrowth and pathological angiogenesis. Nat Protoc. 2009;4:1565–1573. doi: 10.1038/nprot.2009.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caberoy NB, Zhou Y, Jiang X, et al. Efficient identification of tubby-binding proteins by an improved system of T7 phage display. J Mol Recognit. 2010;23:74–83. doi: 10.1002/jmr.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li W, Webster KA, LeBlanc ME, Tian H. Secretogranin III: a diabetic retinopathy-selective angiogenic factor. Cell Mol Life Sci. 2018;75:635–647. doi: 10.1007/s00018-017-2635-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hosaka M, Watanabe T. Secretogranin III: a bridge between core hormone aggregates and the secretory granule membrane. Endocr J. 2010;57:275–286. doi: 10.1507/endocrj.k10e-038. [DOI] [PubMed] [Google Scholar]
- 22.Li W, Pang I-H, Pacheco MTF, Tian H. Ligandomics: a paradigm shift in biological drug discovery. Drug Discov Today. 2018;23:636–643. doi: 10.1016/j.drudis.2018.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pierce EA, Avery RL, Foley ED, et al. Vascular endothelial growth factor/vascular permeability factor expression in a mouse model of retinal neovascularization. Proc Natl Acad Sci USA. 1995;92:905–909. doi: 10.1073/pnas.92.3.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kline JE, Illapani VSP, He L, et al. Retinopathy of prematurity and bronchopulmonary dysplasia are independent antecedents of cortical maturational abnormalities in very preterm infants. Sci Rep. 2019;9:19679. doi: 10.1038/s41598-019-56298-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Froger N, Matonti F, Roubeix C, et al. VEGF is an autocrine/paracrine neuroprotective factor for injured retinal ganglion neurons. Sci Rep. 2020;10:12409. doi: 10.1038/s41598-020-68488-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kingsley DM, Rinchik EM, Russell LB, et al. Genetic ablation of a mouse gene expressed specifically in brain. EMBO J. 1990;9:395–399. doi: 10.1002/j.1460-2075.1990.tb08123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sapieha P. Eyeing central neurons in vascular growth and reparative angiogenesis. Blood. 2012;120:2182–2194. doi: 10.1182/blood-2012-04-396846. [DOI] [PubMed] [Google Scholar]
- 28.Tokunaga CC, Mitton KP, Dailey W, et al. Effects of anti-VEGF treatment on the recovery of the developing retina following oxygen-induced retinopathy. Invest Ophthalmol Vis Sci. 2014;55:1884–1892. doi: 10.1167/iovs.13-13397. [DOI] [PubMed] [Google Scholar]
- 29.Liets LC, Eliasieh K, van der List DA, Chalupa LM. Dendrites of rod bipolar cells sprout in normal aging retina. Proc Natl Acad Sci USA. 2006;103:12156–12160. doi: 10.1073/pnas.0605211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnstone C, Rich SE. Bleeding in cancer patients and its treatment: a review. Ann Palliat Med. 2018;7:265–273. doi: 10.21037/apm.2017.11.01. [DOI] [PubMed] [Google Scholar]
- 31.Laude A, Cackett PD, Vithana EN, et al. Polypoidal choroidal vasculopathy and neovascular age-related macular degeneration: same or different disease? Prog Retin Eye Res. 2010;29:19–29. doi: 10.1016/j.preteyeres.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 32.Zhou R, Zhang S, Gu X, et al. Adenosine A2A receptor antagonists act at the hyperoxic phase to confer protection against retinopathy. Mol Med. 2018;24:41. doi: 10.1186/s10020-018-0038-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu H, Mei FC, Yang W, et al. Epac1 inhibition ameliorates pathological angiogenesis through coordinated activation of Notch and suppression of VEGF signaling. Sci Adv. 2020 doi: 10.1126/sciadv.aay3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murinello S, Usui Y, Sakimoto S, et al. miR-30a-5p inhibition promotes interaction of Fas+ endothelial cells and FasL+ microglia to decrease pathological neovascularization and promote physiological angiogenesis. Glia. 2019;67:332–344. doi: 10.1002/glia.23543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Phung TL, Ziv K, Dabydeen D, et al. Pathological angiogenesis is induced by sustained Akt signaling and inhibited by rapamycin. Cancer Cell. 2006;10:159–170. doi: 10.1016/j.ccr.2006.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chaudhary A, Hilton MB, Seaman S, et al. TEM8/ANTXR1 blockade inhibits pathological angiogenesis and potentiates tumoricidal responses against multiple cancer types. Cancer Cell. 2012;21:212–226. doi: 10.1016/j.ccr.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miyoshi T, Hirohata S, Ogawa H, et al. Tumor-specific expression of the RGD-alpha3(IV)NC1 domain suppresses endothelial tube formation and tumor growth in mice. FASEB J. 2006;20:1904–1906. doi: 10.1096/fj.05-5565fje. [DOI] [PubMed] [Google Scholar]
- 38.Takeda A, Baffi JZ, Kleinman ME, et al. CCR3 is a target for age-related macular degeneration diagnosis and therapy. Nature. 2009;460:225–230. doi: 10.1038/nature08151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lam JD, Oh DJ, Wong LL, et al. Identification of RUNX1 as a mediator of aberrant retinal angiogenesis. Diabetes. 2017;66:1950–1956. doi: 10.2337/db16-1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Al-Hilal TA, Chung SW, Choi JU, et al. Targeting prion-like protein doppel selectively suppresses tumor angiogenesis. J Clin Invest. 2016;126:1251–1266. doi: 10.1172/JCI83427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ridiandries A, Tan JTM, Ravindran D, et al. CC-chemokine class inhibition attenuates pathological angiogenesis while preserving physiological angiogenesis. FASEB J. 2017;31:1179–1192. doi: 10.1096/fj.201600540R. [DOI] [PubMed] [Google Scholar]
- 42.Ferrara N, Carver-Moore K, Chen H, et al. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439–442. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- 43.Fong GH, Rossant J, Gertsenstein M, Breitman ML. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature. 1995;376:66–70. doi: 10.1038/376066a0. [DOI] [PubMed] [Google Scholar]
- 44.Shalaby F, Rossant J, Yamaguchi TP, et al. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376:62–66. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 45.Lutty GA, McLeod DS, Bhutto I, Wiegand SJ. Effect of VEGF trap on normal retinal vascular development and oxygen-induced retinopathy in the dog. Invest Ophthalmol Vis Sci. 2011;52:4039–4047. doi: 10.1167/iovs.10-6798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vogel RN, Strampe M, Fagbemi OE, et al. Foveal development in infants treated with bevacizumab or laser photocoagulation for retinopathy of prematurity. Ophthalmology. 2018;125:444–452. doi: 10.1016/j.ophtha.2017.09.020. [DOI] [PubMed] [Google Scholar]
- 47.Morin J, Luu TM, Superstein R, et al. Neurodevelopmental outcomes following bevacizumab injections for retinopathy of prematurity. Pediatrics. 2016 doi: 10.1542/peds.2015-3218. [DOI] [PubMed] [Google Scholar]
- 48.Sato T, Kusaka S, Shimojo H, Fujikado T. Vitreous levels of erythropoietin and vascular endothelial growth factor in eyes with retinopathy of prematurity. Ophthalmology. 2009;116:1599–1603. doi: 10.1016/j.ophtha.2008.12.023. [DOI] [PubMed] [Google Scholar]
- 49.Aiello LP, Avery RL, Arrigg PG, et al. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Engl J Med. 1994;331:1480–1487. doi: 10.1056/NEJM199412013312203. [DOI] [PubMed] [Google Scholar]
- 50.Wallace DK, Kylstra JA, Greenman DB, Freedman SF. Significance of isolated neovascular tufts (“popcorn”) in retinopathy of prematurity. J AAPOS. 1998;2:52–56. doi: 10.1016/s1091-8531(98)90111-2. [DOI] [PubMed] [Google Scholar]
- 51.Gitay-Goren H, Cohen T, Tessler S, et al. Selective binding of VEGF121 to one of the three vascular endothelial growth factor receptors of vascular endothelial cells. J Biol Chem. 1996;271:5519–5523. doi: 10.1074/jbc.271.10.5519. [DOI] [PubMed] [Google Scholar]
- 52.Cooper ME, Vranes D, Youssef S, et al. Increased renal expression of vascular endothelial growth factor (VEGF) and its receptor VEGFR-2 in experimental diabetes. Diabetes. 1999;48:2229–2239. doi: 10.2337/diabetes.48.11.2229. [DOI] [PubMed] [Google Scholar]
- 53.Egawa G, Nakamizo S, Natsuaki Y, et al. Intravital analysis of vascular permeability in mice using two-photon microscopy. Sci Rep. 2013;3:1932. doi: 10.1038/srep01932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pino RM, Essner E. Permeability of rat choriocapillaris to hemeproteins. Restriction of tracers by a fenestrated endothelium. J Histochem Cytochem. 1981;29:281–290. doi: 10.1177/29.2.7252121. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are included in this article and its supplementary files.