

Abstract
Fabry disease (FD) is a rare X‐linked disorder of lipid metabolism, characterized by the accumulation of globotriaosylceramide (Gb3) due to defective the lysosomal enzyme, α‐galactosidase. Gb3 deposits activate immune‐mediated systemic inflammation, ultimately leading to life‐threatening consequences in multiple organs such as the heart and kidneys. Enzyme replacement therapy (ERT), the standard of care, is less effective with advanced tissue injury and inflammation in patients with FD. Here, we showed that MCP‐1 and TNF‐α cytokine levels were almost doubled in plasma from ERT‐treated FD patients. Chemokine receptor CCR2 surface expression was increased by twofold on monocytes from patients with low eGFR. We also observed an increase in IL12B transcripts in unstimulated peripheral blood mononuclear cells (PBMCs) over a 2‐year period of continuous ERT. Apabetalone is a clinical‐stage oral bromodomain and extra terminal protein inhibitor (BETi), which has beneficial effects on cardiovascular and kidney disease related pathways including inflammation. Here, we demonstrate that apabetalone, a BD2‐selective BETi, dose dependently reduced the production of MCP‐1 and IL‐12 in stimulated PBMCs through transcriptional regulation of their encoding genes. Reactive oxygen species production was diminished by up to 80% in stimulated neutrophils following apabetalone treatment, corresponding with inhibition of NOX2 transcription. This study elucidates that inhibition of BET proteins by BD2‐selective apabetalone alleviates inflammatory processes and oxidative stress in innate immune cells in general and in FD. These results suggest potential benefit of BD2‐selective apabetalone in controlling inflammation and oxidative stress in FD, which will be further investigated in clinical trials.
Keywords: apabetalone, BETi, epigenetics, Fabry disease, inflammation, oxidative stress
Inhibition of BET proteins by BD2‐selective apabetalone alleviates inflammatory processes and oxidative stress in activated innate immune cells from FD patients.

Abbreviations
- BD
bromodomain
- BET
bromodomain and extraterminal domain
- BETi
bromodomain and extraterminal domain inhibitor
- BRD
bromodomain and extraterminal domain protein
- CKD
chronic kidney disease
- CVD
cardiovascular disease
- eGFR
estimated glomerular filtration rate
- ERT
enzyme replacement therapy
- FD
Fabry disease
- Gb3
globotriaosylceramide
- PBMC
peripheral blood mononuclear cell
- ROS
reactive oxygen species
1. INTRODUCTION
Fabry disease (FD) is a rare X‐linked lysosomal storage disorder caused by mutations in the GLA gene encoding the lysosomal enzyme α‐galactosidase A (α‐GAL). 1 These mutations result in deficient α‐GLA activity and accumulation of globotriaosyceramide (also known as Gb3) and globotriaosylsphingosine (lyso‐Gb3) in various tissues and cell types. Gb3 deposits activate pathogenic mechanisms such as harmful pro‐inflammatory responses and oxidative stress, 2 , 3 subsequently leading to tissue injury in organs throughout the body including the heart and kidneys. Cardiovascular and renal dysfunction, which commonly manifest as cardiac hypertrophy and a decline in eGFR respectively, remain the leading causes of death in FD. 4 , 5 Enzyme replacement therapy (ERT), the standard of care for nearly 20 years, 6 , 7 reduces Gb3 levels and improves clinical outcomes in the short term. 8 , 9 However, ERT is less effective when tissue injury has developed and abnormal immune responses persist. 2
Activation of the innate component of the immune system by Gb3 through Toll‐like receptor 4 (TLR4) triggers pro‐inflammatory responses in FD. 10 Subsequently, activated innate immune cells (e.g., monocytes and neutrophils) produce deleterious inflammatory cytokines and chemokines, driving FD progression. Elevated levels of interleukin‐6 (IL‐6) and tumor necrosis factor‐α (TNF‐α) have been detected in FD patients’ peripheral blood mononuclear cells (PBMCs) compared with healthy controls. 10 Increased plasma IL‐6 and TNF‐α levels also positively correlate with incidence of left ventricular hypertrophy in patients with chronic kidney disease (CKD) and cardiovascular disease (CVD). 11 , 12 Furthermore, the chemokine monocyte chemoattractant protein‐1 (MCP‐1), involved in monocyte movement, is associated with CVD progression over 1 year of ERT, 12 while interleukin‐12 (IL‐12), a monocyte‐derived T helper 1 (Th1)‐type cytokine that promotes T cell responses, has been proposed to mediate abnormal T cell activity in patients with FD. 13 T cell appearance in damaged myocardium during long‐term ERT 14 implies aberrant T cell infiltration which can result in further tissue damage.
Oxidative stress, associated with excessive production of reactive oxygen species (ROS) is another driver of FD progression. 15 Although ERT has been reported to alleviate oxidative stress in Gb3 stimulated human vascular endothelial cells, 16 recent in vivo studies reveal limitations of ERT in modulating oxidative stress in treated FD patients. 17 , 18 , 19 Pro‐oxidant conditions and oxidative damage correlate with elevated plasma levels of pro‐inflammatory cytokines following continuous ERT (~2 years) 19 demonstrating a potential link between abnormal immune responses and ROS production. These findings suggest that novel therapeutic approaches targeting pathological inflammation and/or oxidative stress are necessary to complement the standard of care in FD to further optimize patient outcomes.
Bromodomain and extra‐terminal (BET) proteins, termed epigenetic “readers”, 20 have been identified as therapeutic targets for disease prevention due to their pivotal role in regulating the transcription of inflammatory genes. 21 , 22 , 23 BET proteins bind acetylated lysine residues on histone tails and other nuclear proteins 24 through their conserved N‐terminal bromodomains (BD1 and BD2) 25 , 26 to recruit and/or facilitate assembly of factors needed for regulation of gene expression. Notably, BET proteins have been shown to cooperate with nuclear factor κ‐light‐chain‐enhancer of activated B (NF‐κB) transcription factor downstream of the TLR4 receptor in cell models including monocytes and macrophages. 27 , 28 , 29 Thus, inhibition of BET proteins could alleviate gene expression activated by Gb3‐TLR4 interactions in FD patients.
Apabetalone (RVX‐208) is a clinical‐stage BET inhibitor (BETi) currently in development for CVD, CKD, 30 , 31 pulmonary arterial hypertension (NCT03655704) and COVID‐19 (NCT04894266). Apabetalone binds preferentially to the second bromodomain (BD2) of BET family members BRD2, BRD3 and BRD4 with >20‐fold selectivity over BD1. 32 , 33 This BD2 dominant binding blocks BET interactions with acetylated lysines on chromatin and transcription factors at latent enhancers and promoters, minimizing maladaptive transcription of disease‐driving genes. 26 Preclinical studies have shown that apabetalone reduces the expression of a variety of markers of CVD, CKD, and inflammation in various cell types and disease models. 27 , 28 , 33 , 34 , 35 , 36 In clinical trials, apabetalone improved cardiac (major adverse cardiovascular events [MACE]) and renal (serum alkaline phosphatase and eGFR) parameters in CVD patients. 30 , 33 , 37 , 38 , 39 These data suggest that treatment with apabetalone may alleviate cardiac, renal, and inflammatory complications in FD patients.
In this study, we first examined the inflammatory status of plasma and PBMCs collected from FD patients. We also tracked immune activation in unstimulated PBMCs over 2 years of continuous ERT. The effects of ex vivo apabetalone treatment on inflammation burden were examined by assessing pro‐inflammatory marker levels in stimulated PBMCs isolated from ERT treated FD patients. Lastly, we investigated apabetalone effects on oxidative stress in stimulated neutrophils, in which the role of BET proteins is currently unknown.
2. MATERIALS AND METHODS
2.1. Patients and samples
Blood samples were collected from eight FD patients receiving ERT therapy at M.A.G.I.C clinic, Calgary, Alberta, Canada. Patients met the following inclusion criteria: existing diagnosis of FD, according to The Canadian Fabry Association Standard and age ≥18 years. Inclusion criteria were limited due to the small population from which to draw participants based on the rarity of the disease and the single‐site study design. The study protocol was reviewed and approved by the HREBA (Health Research Ethics Board of Alberta)—Community Health Committee. Patients declaring interest in study participation provided written informed consent prior to enrollment. Plasma samples from normal donors (age matched) were purchased from StemCell technology. LysoGb3 and Gb3 were detected via untargeted mass spectrometry metabolomic approaches. 40 , 41 , 42 , 43 , 44 , 45 , 46
2.2. Isolation of peripheral blood mononuclear cells and neutrophils from whole blood
Blood samples were collected between 7 and 14 days following ERT treatment, which was administered at 14‐day intervals for each patient. Whole blood samples were collected in BD Vacutainer 1.8 mg/ml ethylenediaminetetre‐acetic acid (EDTA)‐K2 tubes (Dufort et Lavigne) (30 ml maximum per patient). Fresh blood was centrifuged at 2500g for 20 min at room temperature to obtain plasma samples, which were then snap‐frozen in liquid nitrogen, and aliquots stored at −80°C until further use. Pelleted cells were diluted with phosphate buffered saline (PBS, Sigma Aldrich) as 1:1 ratio and then subject to density gradient centrifugation with Histopaque 1077 (Sigma Aldrich) to isolate PBMCs and neutrophils. The layer containing neutrophils was applied to hypotonic lysis of erythrocytes by Red Blood Cell Lysis Buffer. Purified PBMCs and neutrophils were washed and then resuspended in RPMI 1640 medium supplemented with 10% fetal bovine serum. The cells were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum.
2.3. Flow cytometry
Isolated PBMCs or neutrophils were stained with antibodies to assess purity or immune activation status. The antibodies used in this study were V450 anti‐human CD14, APC anti‐human CCR2, PE‐CF594 anti‐human TLR4 (BD Biosciences). Data were acquired with BD FACSCelesta (BD Biosciences) and quantified with FlowJo V10 software (BD Biosciences).
2.4. Ex vivo apabetalone treatment
Isolated PBMCs or neutrophils were stimulated with 1 µg/ml lipopolysaccharide (LPS, Sigma) or 10 ng/ml IFN‐γ (StemCell) and co‐treated with apabetalone, pan BET inhibitor JQ1 or the vehicle control DMSO for 4 h or overnight at 37°C. THP1 cells (ATCC) were stimulated with 20 μM Gb3 (Matreya) with 200 μM 1‐deoxygalactonojirimycin (DGJ, Sigma), 1 μg/ml LPS or 10 ng/ml IFN‐γ with BETi co‐treatments for 4 h at 37°C.
2.5. Real‐time PCR
Relative gene expression was determined by real‐time PCR as previously described. 27 , 34 Briefly, mRNA was isolated using Catcher PLUS kits per manufacturer instructions (ThermoFisher Scientific). Real‐time PCR was performed with Taqman primer probes (ThermoFisher Scientific) to determine the abundance of transcripts relative to the endogenous control cyclophilin. The data were analyzed as 2^ (C T cyclophilin – C T tested marker) and then normalized to DMSO treated samples.
2.6. Extracellular cytokines/chemokines
Cytokine profiles in plasma or undiluted supernatants from stimulated PBMCs post overnight treatment were analyzed with the human cytokine array 42‐plex by Eve Technologies Corp. (Alberta, Canada). Chemokine production was assessed in diluted supernatants with BDTM cytometric bead array human chemokine kit (BD Biosciences). Data were acquired using BD FACSCelesta and quantified with FCAP software (BD Biosciences).
2.7. Western blot
PBMCs were pre‐treated with apabetalone, MZ1 or 0.025% DMSO for 4 h and then stimulated with LPS for an additional 2 h. Cells were lysed and sonicated as previously described. 27 20 μg of total protein was added for protein separation. BET proteins were detected with anti‐BRD2, anti‐BRD3 and anti‐BRD4 mAbs (Bethyl) followed by goat anti‐rabbit IgG H&L chain specific peroxidase (Calbiochem). Actin was stained by anti‐β‐actin conjugated to peroxidase (Sigma).
2.8. Reactive oxygen species
Intracellular ROS levels in LPS‐stimulated neutrophils were detected using CellROX® Green flow cytometry kits (ThermoFisher Scientific) as per manufacturer instruction. Samples were immediately analyzed using BD FACSCelesta Cell Analyzer. The amount of intracellular ROS was quantified as the percentage of cells having positive green fluorescent signal above background in proportion to total live cells using the Flowjo® software (BD biosciences).
2.9. Statistical analyses
Statistical significance was calculated using GraphPad Prism 8.0 with Mann Whitney test, one‐way ANOVA followed by Dunnett's Multiple Comparison Test where appropriate. p < .05 was considered statistically significant.
2.10. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY, 47 and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20. 48
3. RESULTS
3.1. Patient characteristics
In this study, we recruited eight FD patients receiving ERT therapy (44–74 years old) from a single clinical site. Baseline demographic and biochemical data are shown in Table 1. Clinical presentations at the time of enrollment are in Table S1. All patients showed clinical indicators of CVD. Three patients had eGFR below the range of 60 ml/min/1.73 m2 (p = .04), which is indicative of reduced kidney function and vulnerability to rapid deterioration. 1 , 49 Patients also received standard of care medications for management of other complications (such as CVD or CKD) (Table S1). Due to limited blood volume and variation in volume of cells obtained from FD patients, sample number varied in the following analyses.
TABLE 1.
Demographic data and biochemical analysis for patients with fabry disease
| Characteristics |
eGFR > 60 (n = 5, median [IQR]) |
eGFR < 60 (n = 3, median [IQR]) |
p‐value a (Mann Whitney) |
|---|---|---|---|
| Demographic analysis | |||
| Age, years | 64 [23] | 63 [4] | .75 |
| Gender (female/male) | 4F/1 M | 2F/1 M | n/a |
| BMI, kg/cm2 | 23.2 [9.1] | 27.9 [14.7] | .99 |
| Blood pressure, mmHg (systolic/diastolic) | 112 [16]/78 [22] | 124 [32]/80 [14] | .32/.53 |
| Biochemical analysis | |||
| Creatinine, µmol/L | 70 [27] | 103 [55] | .07 |
| eGFR b , ml/min/1.73 m2 | 80 [27] | 50 [13] | .04 |
| ALT, U/L | 21 [19] | 22 [14] | .99 |
| LD, U/L | 206 [56] | 288 [37] | .04 |
| Total bilirubin, µmol/L | 11 [7] | 7 [3] | .39 |
| Gb3, µg/ml | 5 [4] | 3 | .38 |
| Lyso‐Gb3, nmol/L | 11 [15] | 19 [25] | .25 |
p values are calculated by Mann–Whitney U test. Parameters with statistical significance (p < .05) were in bold.
eGFR is calculated by the CKD‐EPI standard.
3.2. Fabry disease patients’ baseline immune status
Pro‐inflammatory cytokines are positively associated with FD progression despite ERT treatment. 10 , 11 , 12 We examined cytokine levels in plasma from ERT‐treated FD patients and commercially available plasma from control donors using multianalyte cytokine profiling. In agreement with previously reported results, 10 , 12 plasma MCP‐1 and TNF‐α concentrations were greater in FD samples compared with normal controls (p = 0.0062 and p = .0246, respectively) (Figure 1A, Table S2). However, plasma cytokine abundance did not differ by eGFR levels (Table S3). We also tracked cytokine expression in PBMCs from four FD patients with eGFR >60 (normal eGFR) on continuous ERT therapy before and after 2 years of treatment. PBMCs showed an ~20‐fold increase in IL12B transcript levels (p = .03) in the most recent visit compared with 2 years prior (Figure 1B). Furthermore, activation markers on monocyte populations (Figure 1C) were assessed. As shown in Figure 1D, the proportion of monocytes expressing CCR2 was greater in patients with low eGFR (eGFR <60, p = .03), implying that innate immune dysregulation may be associated with renal dysfunction. However, the percentage of TLR4 expressing cells did not differ by eGFR levels. Overall, these results suggest the persistence of immune dysregulation during continuous ERT in unstimulated PBMCs.
FIGURE 1.
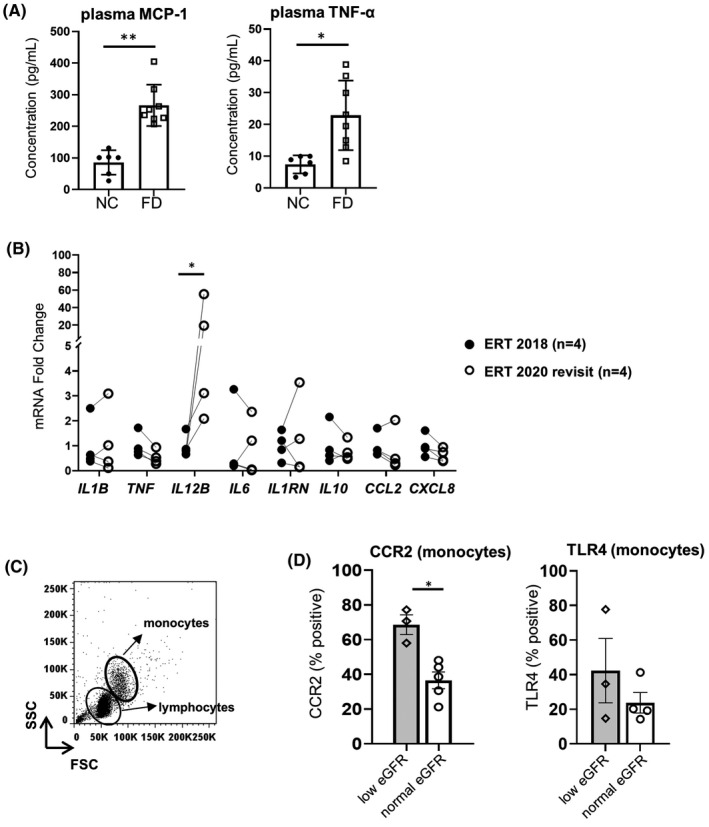
Baseline immune profiles in plasma and peripheral blood mononuclear cells (PBMCs) from enzyme replacement therapy (ERT)‐treated Fabry disease (FD) patients. (A) Plasma MCP‐1 and TNF‐α levels in ERT‐treated FD patients (n = 8) versus normal controls (NCs) (n = 6). (B) Comparison of indicated gene transcript levels in PBMCs from ERT patients with normal eGFR after 2 years of ERT (n = 4). (C) Representative dot plot of gating lymphocyte and monocyte subpopulations in PBMCs isolated from the whole blood of one patient. (D) Monocyte fraction expressing surface CCR2 and TLR4 in ERT‐treated FD patients with indicated eGFR using the same gating strategy as in A. eGFR >60 denotes eGFR levels >60 ml/min/1.73 m2, while eGFR <60 indicates eGFR <60 ml/min/1.73 m2. In (A) and (D), bar graphs show the mean ± SD. Statistical significance was determined by Mann–Whitney or Wilcoxon test. *p < .05, **p < .01
3.3. Ex vivo apabetalone treatment reduces the expression of inflammatory genes in lipopolysaccharide‐stimulated peripheral blood mononuclear cells isolated from Fabry disease patients on enzyme replacement therapy
Immune cell activation observed in FD patients on ERT could potentiate disease progression. BET proteins have been reported to control inflammatory pathways through transcriptional regulation. Here, we treated PBMCs from FD patients with BETi including apabetalone, a BETi in advanced clinical development stages with a favorable safety profile, as well as JQ1, a non‐clinical pan‐BETi comparator with a different chemical scaffold. In unstimulated PBMCs, mRNA levels of all tested genes were not altered by either BETi (Figure S1). We then examined the effects of BETi on the expression of pro‐inflammatory genes induced by TLR4 activation with the bona fide TLR4 ligand, LPS. After 4 h of LPS stimulation, pro‐inflammatory gene levels were robustly induced in PBMCs from all tested patients (Figure S2). Ex vivo apabetalone treatment dose dependently suppressed this induction for all examined genes (Figure 2). Apabetalone at low doses (1 μM and 5 μM) possesses BD2‐selective properties, binding to BET proteins with higher affinity at the BD2 domain over the BD1 domain, while apabetalone at 20 μM (apa 20 μM) or greater concentrations is more similar to a pan‐BETi, binding equally to BD1 and BD2 domains. 27 Notably, BD2‐selective apabetalone (apa 1 μM and 5 μM) downregulated the induced expression of genes tested. For instance, the induced transcription of IL12B and CCL2 was downregulated by >90% with apa 1 μM and apa 5 μM. The percentage of inhibition by pan‐BETi treatment (apa 20 μM) was comparable with JQ1 (Figure 2), a pan‐BETi control, further supporting the engagement of BET proteins in controlling inducible transcription of these genes.
FIGURE 2.
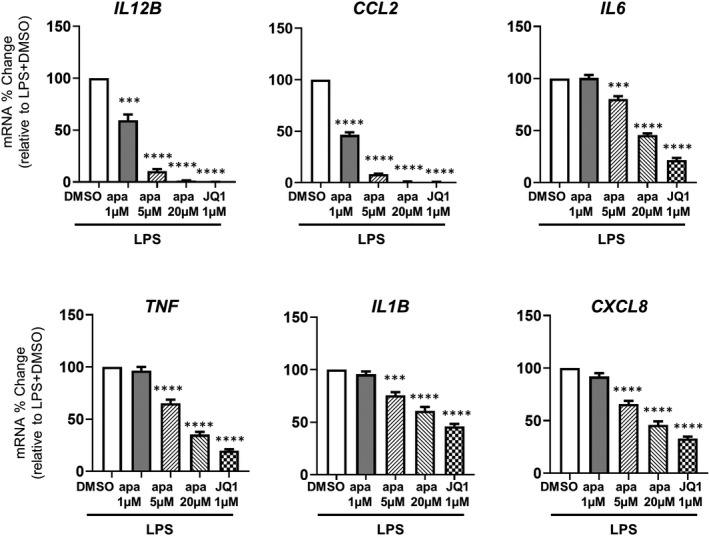
BETi treatment effects on the inducible expression of inflammatory genes in lipopolysaccharide (LPS)‐stimulated peripheral blood mononuclear cells (PBMCs) from Fabry disease patients. Quantification of the indicated gene expression changes driven by apabetalone in PBMCs (enzyme replacement therapy, n = 7) using quantitative RT‐PCR. Bar graphs show the mean ± SEM. On the x‐axis, apa = apabetalone. Statistical significance was determined by one‐way repeated measures ANOVA followed by Dunnett's multiple comparison test compared with LPS+DMSO. *p < .05, **p < .01, ***p < .001, ****p < .0001
3.4. Ex vivo apabetalone treatment attenuates pro‐inflammatory cytokine secretion in lipopolysaccharide‐stimulated peripheral blood mononuclear cells from enzyme replacement theraphy–treated Fabry disease patients
To evaluate apabetalone's effects on protein production, we examined cytokine profiles in supernatants from PBMCs of FD patients on ERT (n = 6) after overnight LPS stimulation and apabetalone co‐treatment. As expected, MCP‐1 (encoded by CCL2 gene), TNF‐α (encoded by TNF gene), IL‐8 (encoded by CXCL8 gene) and IL‐6 (encoded by IL6 gene) were drastically induced (MCP‐1 induced by ~65‐fold, TNF‐α induced by 800‐fold, IL‐8 induced by 3900‐fold, and IL‐6 induced by 72‐fold) (Figure 3A). Apabetalone nearly abrogated MCP‐1 production (>95% reduction, p = .07 in all examined doses), but had no effect on TNF‐α, IL‐6 and IL‐8 production (Figure 3A). Furthermore, using multianalyte immunoprofiling, we identified three important pro‐inflammatory cytokines, IL‐12p40, GM‐CSF and MCP‐3, that were significantly reduced by apabetalone (Figure 3B, Table S4). Secretion of these proteins was robustly induced by LPS stimulation (up to 400‐fold) except for MCP‐3 whose induction appeared less extensive, likely due to large variance in basal levels (2 pg/ml–605 pg/ml) (Figure 3B). Nevertheless, 1µM apabetalone (BD2‐selective) was sufficient to downregulate the induced secretion of IL‐12p40 (encoded by IL12B), GM‐CSF, and MCP‐3 by 52% (p = .02), 25% (p = .04) and 68% (p = .01), respectively. Pan‐BETi treatments (apa 20 μM or JQ1) suppressed the induction of IL‐12p40 and GM‐CSF to basal levels (p = .03 and p = .007, respectively), and reduced MCP‐3 to levels comparable with 5 μM apabetalone (BD2‐selective) (Figure 3B). However, BETi treatments had no effect on LPS‐induced IL‐1β (Figure 3B) despite the significant reduction occurred at gene level (Figure 2).
FIGURE 3.
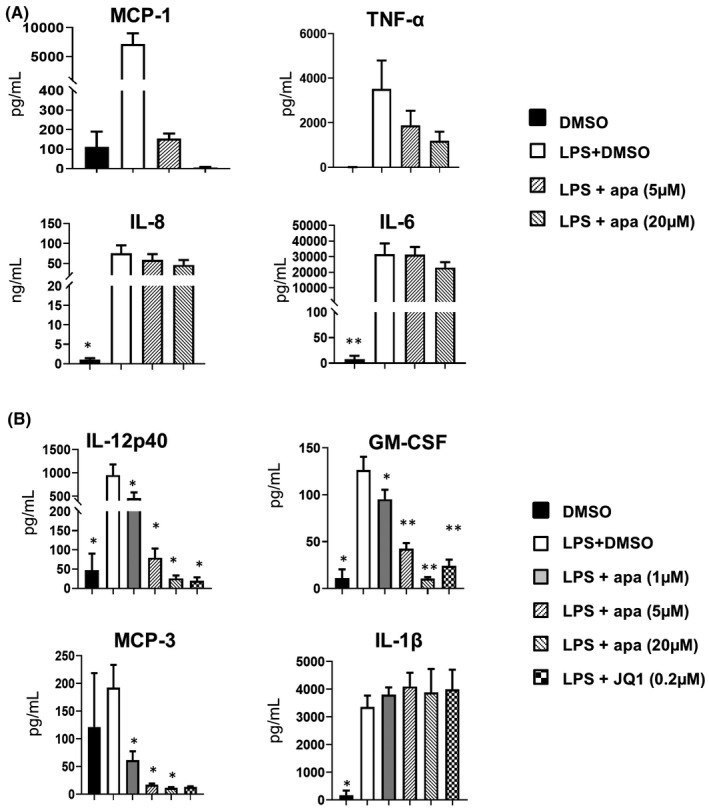
Cytokine profile in supernatants from cultured peripheral blood mononuclear cells from enzyme replacement therapy patients (n = 7) in response to lipopolysaccharide (LPS) stimulation with 18 h apabetalone or JQ1 co‐treatment. Cytokine levels were determined by (A) BDTM Cytometric Bead Array Human Chemokine Kit (BD Biosciences). Bar graphs show the mean ± SEM or (B) Human Cytokine/Chemokine Array 42 plex (Eve Technologies Corp.). In A‐B, apa = apabetalone. Statistical significance was determined by one‐way repeated measures ANOVA followed by Dunnett's multiple comparison test relative to LPS + DMSO. *p < .05, **p < .01
3.5. Apabetalone treatment lowered the inducible expression of inflammatory genes in IFNγ‐stimulated peripheral blood mononuclear cells from enzyme replacement therapy–treated Fabry disease patients
IFN‐γ plays a well‐known role in priming monocytes/macrophages to a pro‐inflammatory phenotype during inflammation. 50 IFN‐γ contributes to pro‐inflammatory signaling in FD patients 13 and its production is also a known downstream effect of TLR4 activation. 51 Here, exposure of FD patients’ PBMCs (normal eGFR) to IFN‐γ resulted in the induction of CCL2, TNF, and IL6 by fivefold, ninefold, and sixfold, respectively, but to a lesser extent than with LPS stimulation (Figure 4). Apabetalone lowered the induced expression of CCL2 and TNF with statistical significance but had little effect on IL6 gene induction. Markedly, the downregulation of CCL2 expression was observed when treating with BD2‐selective (apa 5 μM) and pan‐BETi (apa 20 μM) in LPS or IFN‐γ stimulation conditions, while TNF reduction occurred only with pan‐BETi (apa 20 μM) under IFN‐γ stimulation conditions. Additionally, IL1B was not induced by IFN‐γ stimulation and BET inhibition did not alter this basal IL1B levels, while LPS‐induced IL1B transcription was diminished by ~29% (p = .001) with pan‐BETi (apa 20 μM) (Figure 4).
FIGURE 4.
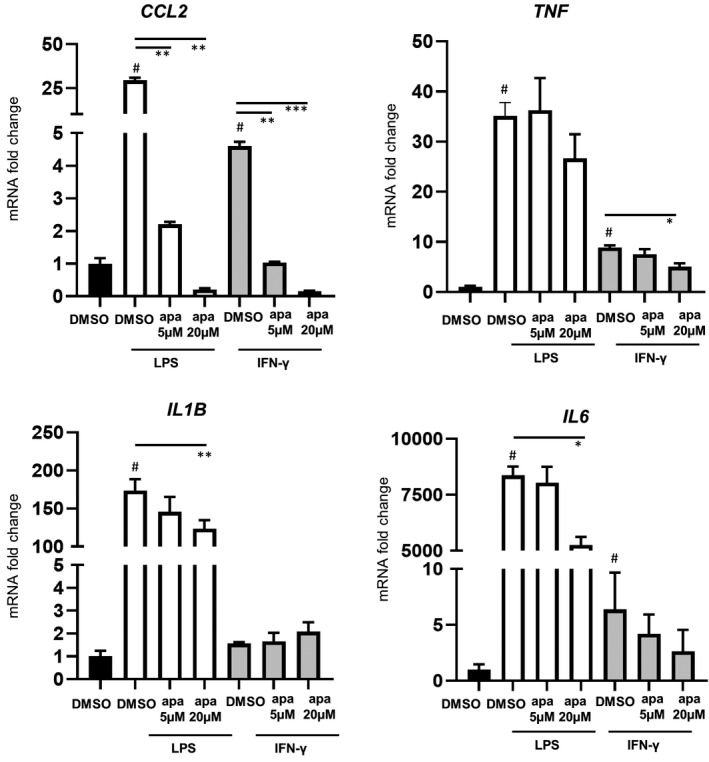
Effects of BETi on the inducible expression of inflammatory genes in stimulated peripheral blood mononuclear cells from Fabry disease patients with lipopolysaccharide (LPS) or IFN‐γ in vitro stimulation (n = 3). Bar graphs show the mean ± SEM. On the x‐axis, apa = apabetalone. Statistical significance was determined by one‐way ANOVA followed by Dunnett's multiple comparison test compared with LPS + DMSO or IFN‐γ+DMSO. *p < .05, **p < .01, ***p < .001. # denotes p < .05 when comparing with DMSO
3.6. Transcription of pro‐inflammatory genes is BET dependent in Fabry disease
To confirm BET protein involvement in the transcription of inflammatory genes in the context of FD, we pretreated PBMCs with MZ‐1, a proteolysis targeting chimera (PROTAC) compound that degrades BET proteins, for 4 h, then measured gene expression following an additional 2‐h stimulation with LPS or IFN‐γ. As expected, MZ‐1 degraded BRD2 and BRD4 proteins (Figure 5A). Upon 2 h of ex vivo LPS stimulation, PBMCs were activated as indicated by elevated mRNA levels of IL12B and IL6 (Figure 5B). MZ‐1 co‐treatment eliminated the induced IL12B expression and partially downregulated IL6 induction (p = .003, p = .003) (Figure 5B). IFN‐γ stimulation also induced the upregulation of CCL2 and IL12B, which was abrogated by MZ‐1 (p = .02, p = .007). These results demonstrate that the transcription of these genes relies on the presence of BRD2 and BRD4 proteins.
FIGURE 5.
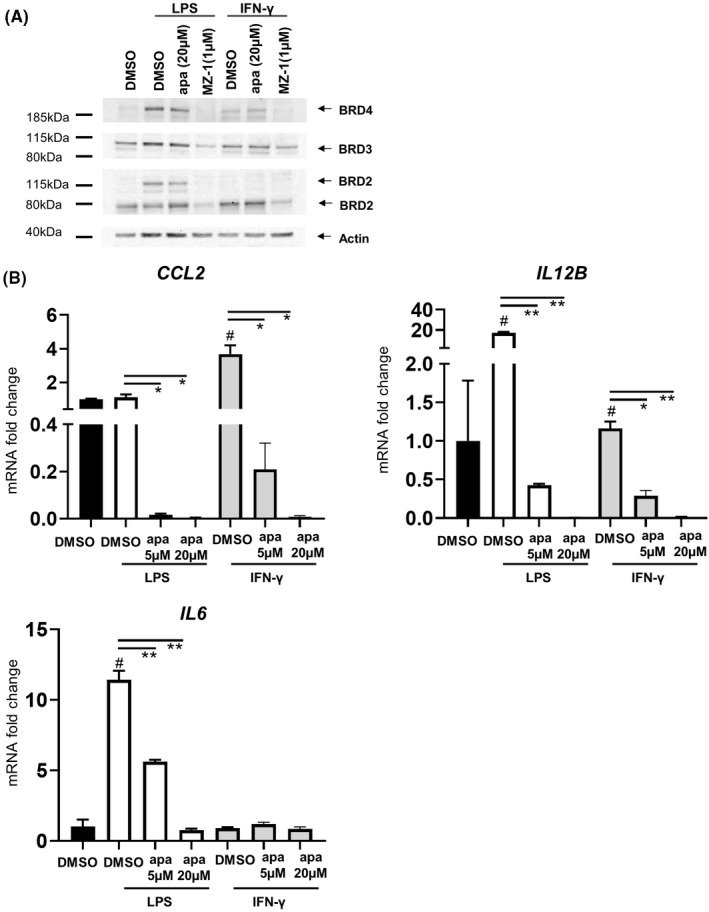
The influence of BET protein knock‐down on the expression of inflammatory genes induced by LPS or IFN‐γ. Representative Western blot analysis of BRD2, BRD3, and BRD4 protein abundance in cell lysates of peripheral blood mononuclear cells (PBMCs) in response to LPS or IFN‐γ stimulation with apabetalone (BET inhibitor) or MZ‐1 (BET degrading PROTAC) co‐treatment. (B) Quantification of pro‐inflammatory gene expression changes in PBMC samples treated with apabetalone or MZ‐1 as indicated in (A). Bar graphs show the mean ± SEM. Apa = apabetalone. Statistical significance was determined by one‐way ANOVA followed by Dunnett's multiple comparison test relative to LPS + DMSO or IFN‐γ + DMSO. *p < .05, **p < .01. # denotes p < .05 when comparing with DMSO
3.7. The effects of apabetalone on the transcription of inflammatory genes in the THP1 monocytic cell line stimulated by Gb3 and DGJ
To further understand the role of BET proteins in monocyte activities in response to Gb3 in FD, we examined the transcription of inducible inflammatory genes in monocyte‐like THP1 cells with Gb3 stimulation ± BETi co‐treatment. Gb3 activates monocyte‐derived cytokine production through the TLR4 pathway, albeit to a lesser extent than LPS. 10 This Gb3‐mediated pro‐inflammatory activation can be detected in cultured cells only in the presence of DGJ, 10 an α‐gal inhibitor, that blocks its enzyme activity and thereby prevents the clearance of its substrates, such as Gb3, and thereby mimicking conditions in FD patient cells. Here, we observed differing transcriptional responses to various stimuli including Gb3+DGJ, LPS or IFN‐γ in THP1 cells. As shown in Figure 6, CCL2 expression was induced in all three stimulation conditions; however, the observed induction was lower with Gb3 + DGJ than with LPS or IFN‐γ. Similarly, a subtle but significant induction was observed for TNF expression by Gb3+DGJ stimulation, but to a lesser extent than with LPS. Importantly, induction of both genes by Gb3+DGJ was downregulated by BD2‐selective apabetalone treatment (apa 1 μM and 5 μM) and by pan‐BETi (apa 20 μM and JQ1 1 μM). These data suggest that apabetalone could downregulate the inducible transcription of inflammatory genes activated in FD.
FIGURE 6.
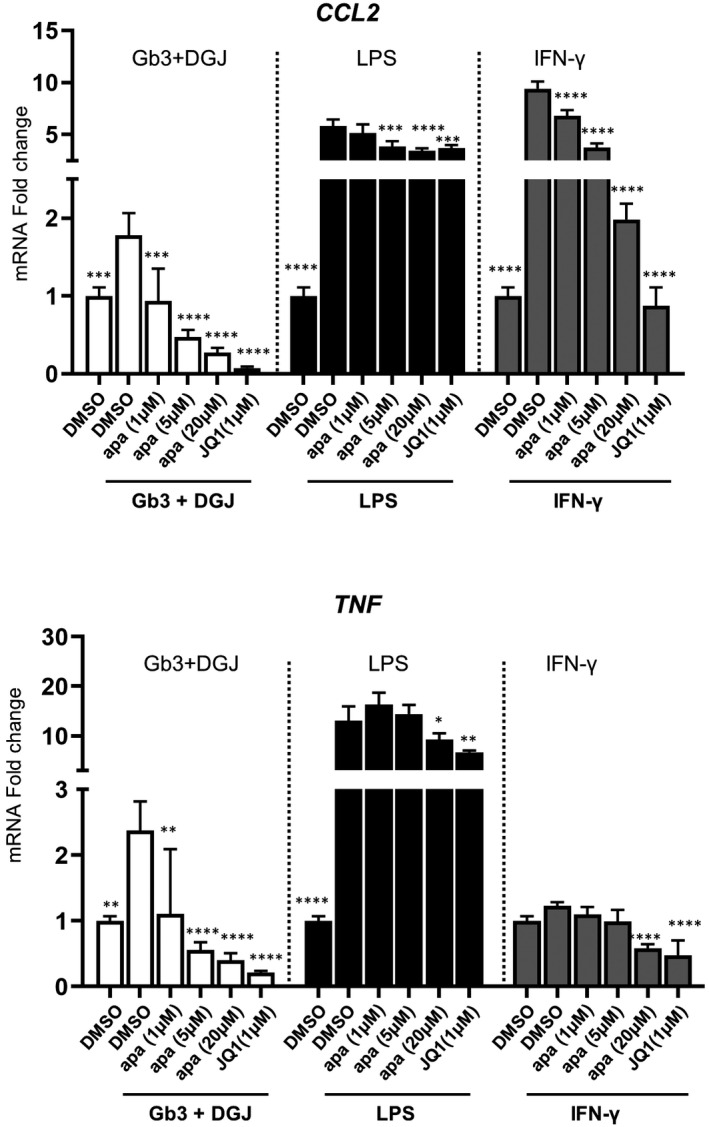
Assessment of inflammatory gene level changes in THP1 cells stimulated with Gb3+DGj, LPS or IFN‐γ with BETi co‐treatment. THP1 cells were stimulated as indicated with BETi co‐treatments for 4 h. Data were normalized to the baseline condition (naive) (n = 4), apa = apabetalone. Bar graphs show the mean ± SD. Statistical significance was calculated by one‐way ANOVA followed by Dunnett's multiple comparison test relative to their corresponding stimulation conditions (GB3 + DGJ + DMSO, LPS + DMSO or IFN‐γ + DMSO). *p < .05, **p < .01, ***p < .001, ****p < .0001
3.8. Apabetalone treatment reduced reactive oxygen species production in neutrophils from Fabry disease patients on enzyme replacement therapy
In FD, Gb3 accumulation stimulates excessive ROS production causing intracellular oxidative damage, and exacerbating inflammation. 17 , 18 , 19 Here we investigated ROS levels in stimulated FD neutrophils with BETi co‐treatment. Apabetalone attenuated LPS‐induced ROS production (Figure S4A and B, an average of 33‐fold) in a dose‐dependent manner (Figure 7A). Pan‐BETi apabetalone (apa 20 μM) countered the induced ROS production to a level comparable with JQ1 (Figure 7A), confirming a BET‐dependent mechanism. IFN‐γ did not alter ROS production from baseline, and apabetalone had no effect on basal ROS levels (Figure 7B). ROS production is generated by cellular enzymes such as nicotinamide adenine dinucleotidephosphate‐oxidase (NADPH) 52 ; NOX2 and NOX4 genes encode two major subunits of the NADPH complex. NOX2 expression was reduced by BD2‐selective BETi treatments (by 29% with apa 1 μM, by 61% with apa 5 μM) and pan‐BETi (by 84% with apa 20 μM, by 93% with JQ1) in all examined patient cells (Figure 7C), though NOX4 was undetectable. This is the first demonstration that neutrophil‐mediated ROS production occurs through a BET‐dependent process at the transcription level.
FIGURE 7.
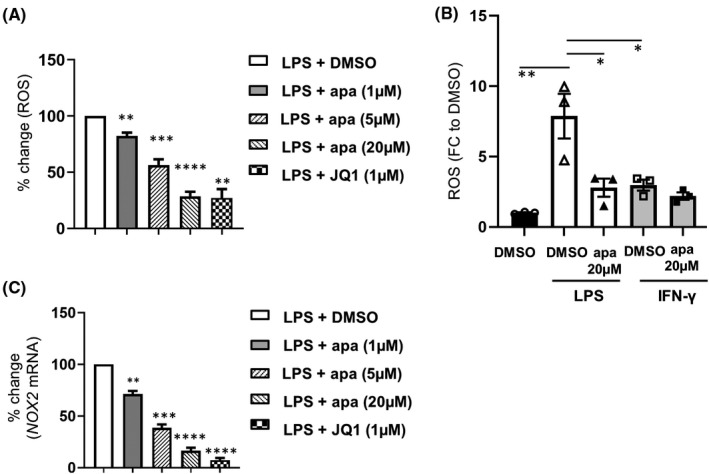
BETi effects on reactive oxygen species (ROS) production in stimulated neutrophils from enzyme replacement therapy–treated Fabry disease patients. (A) percent change in ROS driven by apabetalone. Data were normalized to the stimulation condition (LPS + DMSO) in individual patients (n = 7). (B) ROS level fold change to baseline (DMSO) in LPS or IFN‐γ stimulated neutrophils with apabetalone co‐treatment (n = 3). (C) Apabetalone treatment (4 h) downregulates LPS induced NOX2 gene expression in a dose‐dependent manner. Data were normalized to the LPS + DMSO condition in individual patients. In (A) and (C), Data were normalized to the LPS + DMSO condition in individual patients. Bar graphs show the mean ± SEM. Statistical significance was determined by Mann–Whitney test or one‐way ANOVA followed by Dunnett's multiple comparison test relative to stimulated conditions (LPS + DMSO or IFN‐γ + DMSO). *p < .05, **p < .01, ***p < .001, ****p < .0001
4. DISCUSSION
Immune cell–mediated inflammation is a major factor driving FD progression, despite continuous ERT. Here, we provide baseline inflammatory profiles of plasma and PBMCs from FD patients on ERT in protein and gene expression levels, revealing increased plasma MCP‐1 and TNF‐α, CCR2 overexpression on monocytes from patients with low eGFR and elevated IL12B expression over 2 years of ERT. These baseline results suggest that modulation of ongoing inflammation is necessary to complement long‐term ERT.
Apabetalone inhibits transcription of inducible inflammatory genes in vascular endothelial cells and monocytes from diabetic patients, 27 , 28 suggesting therapeutic potential in controlling chronic inflammation. We further demonstrate that apabetalone at BD2‐selective concentrations counters induction of proinflammatory transcription in Gb3 + DGJ stimulated monocyte‐like THP1 cells, conditions mimicking α‐gal deficiency in FD. Sensitivity of inflammatory genes to apabetalone varies with the magnitude of in vitro immune activation. 27 Therefore, we also used LPS, a potent TLR4‐activator, or IFN‐γ, a cytokine that primes monocytes to pro‐inflammatory responses downstream of TLR4, to stimulate inflammation in PBMCs from FD patients. We show that inhibition of BET proteins, particularly by BD2‐selective apabetalone, attenuates pro‐inflammatory responses in stimulated PBMCs from ERT‐treated patients. Notably, we provide the first evidence linking BET protein function to oxidative stress in stimulated neutrophils from FD patients on long‐term ERT.
4.1. Effects of apabetalone on cytokine secretion in Gb3‐activated THP1 cells and stimulated peripheral blood mononuclear cells from Fabry disease patients
In FD, monocytes use TLR4 to sense Gb3 accumulation, triggering pro‐inflammatory responses including the production of cytokines and chemokines. 10 The chemokine MCP‐1 interacts with its receptor CCR2, promoting monocyte infiltration into injured tissues. 53 , 54 In FD, elevated plasma MCP‐1 correlates with CVD following 1 year of ERT, 12 indicating ongoing immune dysfunction during treatment. We also detect increased plasma MCP‐1 in FD patients undergoing long‐term ERT relative to controls (Figure 1A and Table S2). Increased CCR2 surface expression observed on monocytes from patients with low eGFR (<60 ml/min/1.73 m2) at baseline (Figure 1D) hints at a possible relationship between CCR2 over‐expression and renal dysfunction during FD progression. Increased circulating MCP‐1 and augmented CCR2 on monocytes may enhance FD monocyte chemotaxis. These patients’ immune profiles, together with clinical manifestations of CVD and/or CKD (Table S1), suggest persistent inflammation and immune dysfunction despite continuous ERT.
TLR4‐activated inflammation in FD has been studied in cultured cell models with LPS or concurrent stimulation with Gb3+ DGJ. 10 In THP1 cells, we demonstrate induced transcription of CCL2 by Gb3+DGJ, albeit to a lesser extent than with LPS (Figure 6). BET inhibition using BD2‐selective BETi (apa 1 μM) counters induced CCL2 expression back to basal levels, indicating sensitivity of CCL2 to BETi. Previously, we have documented that pan‐BETi (apa 20 μM) abrogates transcription of CCL2 in line with reduced production of the encoded protein, MCP‐1, in cultured cytokine‐stimulated monocytes from diabetic CVD patients. 28 We substantiate this finding, showing that BD2‐selective BETi (apa 5 μM) had comparable inhibitory effect on CCL2 transcription relative to pan‐BETi (apa 20 μM) (97.8% vs. 99.9%) in LPS or IFN‐γ stimulated PBMCs from ERT‐treated FD patients with clinical indicators of CVD (Table S1; Figures 2 and 4). Results identify a role of BET protein‐BD2 domains in CCL2 regulation, suggesting potential for BD2‐selective apabetalone in reducing MCP‐1 levels in FD. Other chemokines are also involved in monocyte migration/activation. MCP‐3 is important for CCR2‐mediated monocyte recruitment, 55 and GM‐CSF promotes inflammatory activation of monocytes/macrophages. 56 We report increased GM‐CSF in plasma from ERT‐treated FD patients compared with controls (Table S2) and reduced production of both GM‐CSF and MCP‐3 with BD2‐selective apabetalone (1 μM or 5 μM; Figure 3B). Thus, apabetalone may control monocyte migration by lowering cytokine production in FD patients receiving ERT.
TNF‐α and IL‐6 levels are elevated in ERT‐treated FD patient plasma relative to healthy controls. 11 , 12 We also observe increased plasma TNF‐α (twofold) in ERT‐treated FD patients versus controls (Figure 1A, Table S2). BET inhibition opposes induced transcription of these two genes in stimulated cell models. 27 , 28 We further demonstrate that BD2‐selective BETi counter induced transcription of these genes when activated through TLR4 in FD (by LPS or Gb3+DGJ, Figures 2 and 6), as well as via downstream IFN‐γ activation. 51 Furthermore, we broaden our understanding of BET protein‐BD2 domain function in inflammatory signals during acute versus chronic inflammation. We show that BD2‐selective BETi opposed induction of TNF expression at lower grade/chronic‐like inflammatory levels seen with Gb3+DGJ stimulation in THP1 cells (53%–77% reduction by BD2‐selective apabetalone [apa 1 μM, apa 5 μM] on twofold induced TNF, Figure 6) while pan‐BETi suppressed induced IL6 in LPS stimulated conditions, analogous to acute inflammation (~24% reduction by apa 20 μM on >7500‐fold induction by LPS) (Figure 4). Interestingly, apabetalone‐driven inhibition of inflammatory signals at the protein level occurred only in FD patients with low eGFR (Figure S3), who showed higher production of TNF‐α, IL‐6 and IL‐8 in response to LPS (Figure S3). Therefore, higher cytokine levels appear more sensitive to BETi. Clinical studies have demonstrated beneficial effects of in vivo apabetalone treatment in reducing elevated plasma cytokine levels (e.g., TNF‐α and IL‐6) in patients with renal dysfunction. 37 Thus, apabetalone may be particularly effective at reducing hyper‐inflammation in FD patients with renal disease.
IL‐12, a monocyte‐derived cytokine, bridges innate and adaptive immunity to promote Th1 cell‐mediated pro‐inflammatory responses, 57 which aggravate tissue damage. 58 , 59 In FD, IL‐12 has been speculated to link abnormal innate and adaptive immune responses, 13 but its expression pattern remains elusive. After 2 years of continuous ERT, IL12B transcripts (encoding IL‐12p40, a subunit of IL‐12) were elevated in unstimulated PBMCs in four FD patients with normal eGFR (Figure 1B), indicating altered immune status of PBMCs in these patients, even with normal renal function. Depletion of BET proteins by MZ‐1 abrogated induced IL12B expression (Figure 4), confirming a BET‐dependent mechanism. Not surprisingly, both BD2‐selective BETi (apa 1 μM and 5 μM) and pan‐BETi (apa 20 μM or JQ1) attenuated induced expression of IL12B with LPS and IFN‐γ stimulation (Figure 2), as well as IL‐12p40 production (Figure 3). While further investigation of IL‐12 dysregulation in FD is warranted, apabetalone may mitigate increased IL‐12 production via direct inhibition of BET‐dependent IL12B, possibly benefiting long‐term ERT‐treated patients.
4.2. Effect of apabetalone on reactive oxygen species production in stimulated neutrophils
Excessive ROS production drives oxidative stress and exacerbates inflammation 3 , 15 in FD cell models. 17 , 18 , 19 Using cells from ERT‐treated FD patients, we demonstrate neutrophil‐mediated ROS activity and associated aberrant gene expression with LPS or IFN‐γ stimulation (Figure 7, S4A). BETi, particularly BD2‐selective apabetalone (1 μM and 5 μM), reduced ROS production in stimulated FD neutrophils (Figure 7A and B), and regulated transcription of ROS‐related genes such as NOX2 (Figure 7C). Furthermore, our data suggest that neutrophil ROS activity and NOX2 expression may be greater in patients with low eGFR than those with normal eGFR (Figure S4B and D), implying a connection between renal dysfunction and abnormal neutrophil responses during FD progression. These observations suggest that apabetalone can modulate detrimental BET protein activity in FD neutrophils, at least in vitro. To date, work on BET protein involvement in ROS production has focused on fibrosis and cancer. 60 , 61 , 62 Our findings indicate that BD2‐selective BETi prevent neutrophil‐mediated ROS production in the context of FD.
4.3. Study limitations
This study has several limitations. (1). Small sample size due to the rarity of FD, patient availability at a single clinic and logistical challenges in obtaining fresh blood, which may have resulted in underpowered statistical tests; (2). Voluntary patient enrollment resulted in limited representation of “classic” FD in our population, which we attempted to combat by using various stimulants in hopes of more accurately replicating inflammatory status in late‐stage classic FD; (3). A lack of matched controls due to a community‐based clinical site and related ethical restrictions, as well as inaccessibility of commercial fresh blood supply in Canada. Therefore, commercial frozen plasma controls were used for baseline comparison, and Gb3‐stimulated THP1 monocyte‐like cells were tested to confirm the BETi‐driven effect in inflammatory processes within the context of FD; (4). A limited number of ERT‐treated patients were assessed for inflammation progress over time, due to both ethical considerations and the already limited study group; (5). An inability to obtain age and sex‐matched untreated or pre‐ERT FD controls, due to limited numbers of patients with varying degrees of FD at a single clinic and that increased age is associated with disease progression requiring ERT. Therefore, generalized conclusions cannot be drawn without a larger study. Despite these limitations, we showed elevated inflammatory signaling in immune cells from FD patients over time, without clinical presentation or diagnostic signs of renal complications, such as abnormal eGFR. This persistent inflammation in FD patients regardless of ERT indeed warrants further validation in a greater number of patients. Furthermore, we were able to demonstrate the potential for apabetalone in countering inflammatory processes in activated immune cells in FD patients. Despite limitations, this study demonstrates that apabetalone could be a valuable therapeutic for improving FD patient care.
4.4. Conclusions
In summary, our results show that BD2‐selective apabetalone counters inflammation and oxidative stress in stimulated innate immune cells from FD patients undergoing continuous ERT. Potential therapeutic effects of BD2‐selective apabetalone on FD will be further evaluated in the clinical setting.
DISCLOUSRE
The authors declare that LF, SW, LT, BDR, SS, NCWW, JOJ, MS, and EK are employees of Resverlogix Corp. and hold shares. Dr. Aneal Khan is a member of the scientific board of the CFDI (Canadian Fabry Disease Initiative).
AUTHORS’ CONTRIBUTIONS
LF is the principal researcher of the project. LF, SW, and LMT contributed to the experiments. BDR and LF contributed to drafting of the manuscript. BDR, SW, and SCS contributed to project initiation and managed collaboration with AK and CMM. AK and CMM corresponded with the ethics committee, recruited patients, organized sample collection and provided clinical data. EK, NCWW, JOJ, and MS conceived the project. All authors read and approved the final manuscript.
ETHICAL APPROVAL
The study protocol was reviewed and approved by the HREBA (Health Research Ethics Board of Alberta)—Community Health Committee.
CONSENT TO PARTICIPATE
Patients declaring interest in study participation provided written informed consent prior to enrollment.
PERMISSION TO REUSE AND COPYRIGHT
The data sets generated and/or analyzed during the current study are not publicly available. Reasonable requests for data will be considered. All results discussed are provided.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank all the participants who made the study possible, as well as the nurses and staff at M.A.G.I.C. clinic for their valuable contributions. We also acknowledge and thank Dr. Christiane Auray‐Blais (Service of Genetics, Department of Pediatrics, Faculty of Medicine and Health Sciences, Universite ´ de Sherbrooke) and the Canadian Fabry Disease Initiative (CFDI) for the analysis of the globiotriaosylceramides in patient samples.
Fu L, Wasiak S, Tsujikawa LM, et al. Inhibition of epigenetic reader proteins by apabetalone counters inflammation in activated innate immune cells from Fabry disease patients receiving enzyme replacement therapy. Pharmacol Res Perspect. 2022;10:e00949. doi: 10.1002/prp2.949
Funding information
This work was privately funded by Resverlogix Corp.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5:30. doi: 10.1186/1750-1172-5-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rozenfeld P, Feriozzi S. Contribution of inflammatory pathways to Fabry disease pathogenesis. Mol Genet Metab. 2017;122(3):19‐27. doi: 10.1016/j.ymgme.2017.09.004 [DOI] [PubMed] [Google Scholar]
- 3. Simoncini C, Torri S, Montano V, et al. Oxidative stress biomarkers in Fabry disease: is there a room for them? J Neurol. 2020;267(12):3741‐3752. doi: 10.1007/s00415-020-10044-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Desnick RJ, Ioannou YA, Eng CM. α‐Galactosidase A deficiency: Fabry disease. In: Valle DL, Antonarakis S, Ballabio A, Beaudet AL, Mitchell GA, eds. The Online Metabolic and Molecular Bases of Inherited Disease. McGraw‐Hill Education; 2019. [Google Scholar]
- 5. Kok K, Zwiers KC, Boot RG, Overkleeft HS, Aerts JMFG, Artola M. Fabry disease: molecular basis, pathophysiology, diagnostics and potential therapeutic directions. Biomolecules. 2021;11(2):271. doi: 10.3390/biom11020271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Eng CM, Germain DP, Banikazemi M, et al. Fabry disease: guidelines for the evaluation and management of multi‐organ system involvement. Genet Med. 2006;8(9):539‐548. doi: 10.1097/01.gim.0000237866.70357.c6 [DOI] [PubMed] [Google Scholar]
- 7. Desnick RJ, Brady R, Barranger J, et al. Fabry disease, an under‐recognized multisystemic disorder: expert recommendations for diagnosis, management, and enzyme replacement therapy. Ann Intern Med. 2003;138(4):338‐346. doi: 10.7326/0003-4819-138-4-200302180-00014 [DOI] [PubMed] [Google Scholar]
- 8. Eng CM, Guffon N, Wilcox WR, et al. Safety and efficacy of recombinant human alpha‐galactosidase A replacement therapy in Fabry’s disease. New Engl J Medicine. 2001;345(1):9‐16. doi: 10.1056/NEJM200107053450102 [DOI] [PubMed] [Google Scholar]
- 9. Schiffmann R, Kopp JB, Austin HA III, et al. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA. 2001;285(21):2743‐2749. doi: 10.1001/jama.285.21.2743 [DOI] [PubMed] [Google Scholar]
- 10. De Francesco PN, Mucci JM, Ceci R, Fossati CA, Rozenfeld PA. Fabry disease peripheral blood immune cells release inflammatory cytokines: role of globotriaosylceramide. Mol Genet Metab. 2013;109(1):93‐99. doi: 10.1016/j.ymgme.2013.02.003 [DOI] [PubMed] [Google Scholar]
- 11. Yogasundaram H, Nikhanj A, Putko BN, et al. Elevated inflammatory plasma biomarkers in patients with Fabry disease: a critical link to heart failure with preserved ejection fraction. J Am Heart Assoc. 2018;7(21):e009098. doi: 10.1161/JAHA.118.009098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen K‐H, Chien Y, Wang K‐L, et al. Evaluation of proinflammatory prognostic biomarkers for fabry cardiomyopathy with enzyme replacement therapy. Can J Cardiol. 2016;32(10):1221.e1‐1221.e9. doi: 10.1016/j.cjca.2015.10.033 [DOI] [PubMed] [Google Scholar]
- 13. Mauhin W, Lidove O, Masat E, et al. Innate and adaptive immune response in Fabry disease. JIMD Reports. 2015;22:1‐10. doi: 10.1007/8904_2014_371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Frustaci A, Verardo R, Grande C, et al. Immune‐Mediated myocarditis in Fabry disease cardiomyopathy. J Am Heart Assoc. 2018;7(17):e009052. doi: 10.1161/JAHA.118.009052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ravarotto V, Carraro G, Pagnin E, et al. Oxidative stress and the altered reaction to it in Fabry disease: a possible target for cardiovascular‐renal remodeling? PLoS One. 2018;13(9):e0204618. doi: 10.1371/journal.pone.0204618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tseng W‐L, Chou S‐J, Chiang H‐C, et al. Imbalanced production of reactive oxygen species and mitochondrial antioxidant SOD2 in Fabry disease‐specific human induced pluripotent stem cell‐differentiated vascular endothelial cells. Cell Transplant. 2017;26(3):513‐527. doi: 10.3727/096368916X694265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shen J‐S, Meng X‐L, Moore DF, et al. Globotriaosylceramide induces oxidative stress and up‐regulates cell adhesion molecule expression in Fabry disease endothelial cells. Mol Genet Metab. 2008;95(3):163‐168. doi: 10.1016/j.ymgme.2008.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Biancini GB, Moura DJ, Manini PR, et al. DNA damage in Fabry patients: an investigation of oxidative damage and repair. Mutat Res‐Genet Toxicol Environ Mutagen. 2015;784–785:31‐36. doi: 10.1016/j.mrgentox.2015.04.012 [DOI] [PubMed] [Google Scholar]
- 19. Biancini GB, Vanzin CS, Rodrigues DB, et al. Globotriaosylceramide is correlated with oxidative stress and inflammation in Fabry patients treated with enzyme replacement therapy. Biochem Biophys Acta. 2012;1822(2):226‐232. doi: 10.1016/j.bbadis.2011.11.001 [DOI] [PubMed] [Google Scholar]
- 20. Filippakopoulos P, Knapp S. The bromodomain interaction module. FEBS Lett. 2012;586(17):2692‐2704. doi: 10.1016/j.febslet.2012.04.045 [DOI] [PubMed] [Google Scholar]
- 21. Klein K. Bromodomain protein inhibition: a novel therapeutic strategy in rheumatic diseases. RMD Open. 2018;4(2):e000744. eCollection 2018. doi: 10.1136/rmdopen-2018-000744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Morgado‐Pascual J, Rayego‐Mateos S, Tejedor L, Suarez‐Alvarez B, Ruiz‐Ortega M. Bromodomain and extraterminal proteins as novel epigenetic targets for renal diseases. Front Pharmacol. 2019;10:1315. doi: 10.3389/fphar.2019.01315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cochran AG, Conery AR, Sims RJ. Bromodomains: a new target class for drug development. Nat Rev Drug Discovery. 2019;18(8):609‐628. doi: 10.1038/s41573-019-0030-7 [DOI] [PubMed] [Google Scholar]
- 24. Yang XJ. Lysine acetylation and the bromodomain: a new partnership for signaling. BioEssays. 2004;26(10):1076‐1087. doi: 10.1002/bies.20104 [DOI] [PubMed] [Google Scholar]
- 25. Borck PC, Guo LW, Plutzky J. BET epigenetic reader proteins in cardiovascular transcriptional programs. Circ Res. 2020;126(9):1190‐1208. doi: 10.1161/CIRCRESAHA.120.315929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kulikowski E, Rakai BD, Wong NCW. Inhibitors of bromodomain and extra‐terminal proteins for treating multiple human diseases. Med Res Rev. 2021;41(1):223‐245. doi: 10.1002/med.21730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tsujikawa LM, Fu LI, Das S, et al. Apabetalone (RVX‐208) reduces vascular inflammation in vitro and in CVD patients by a BET‐dependent epigenetic mechanism. Clin Epigenetics. 2019;11(1):102‐z. doi: 10.1186/s13148-019-0696-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wasiak S, Dzobo KE, Rakai BD, et al. BET protein inhibitor apabetalone (RVX‐208) suppresses pro‐inflammatory hyper‐activation of monocytes from patients with cardiovascular disease and type 2 diabetes. Clin Epigenetics. 2020;12(1):160‐166. doi: 10.1186/s13148-020-00943-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sun Y, Huang J, Song K. BET protein inhibition mitigates acute myocardial infarction damage in rats via the TLR4/TRAF6/NF‐κB pathway. Exp Ther Med. 2015;10(6):2319‐2324. doi: 10.3892/etm.2015.2789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ray KK, Nicholls SJ, Buhr KA, et al. Effect of apabetalone added to standard therapy on major adverse cardiovascular events in patients with recent acute coronary syndrome and type 2 diabetes: a randomized clinical trial. JAMA. 2020;323(16):1565. doi: 10.1001/jama.2020.3308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kalantar‐Zadeh K, Schwartz GG, Nicholls SJ, et al. Effect of Apabetalone on Cardiovascular Events in Diabetes, CKD, and Recent Acute Coronary Syndrome. Clin J Am Soc Nephrol. 2021;16(5):705‐716. doi: 10.2215/CJN.16751020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McLure KG, Gesner EM, Tsujikawa L, et al. RVX‐208, an inducer of ApoA‐I in humans, is a BET bromodomain antagonist. PLoS One. 2013;8(12):e83190. doi: 10.1371/journal.pone.0083190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kulikowski E, Halliday C, Johansson J, et al. Apabetalone mediated epigenetic modulation is associated with favorable kidney function and alkaline phosphatase profile in patients with chronic kidney disease. Kidney Blood Press Res. 2018;43(2):449‐457. doi: 10.1159/000488257 [DOI] [PubMed] [Google Scholar]
- 34. Gilham D, Tsujikawa LM, Sarsons CD, et al. Apabetalone downregulates factors and pathways associated with vascular calcification. Atherosclerosis. 2019;280:75‐84. doi: 10.1016/j.atherosclerosis.2018.11.002 [DOI] [PubMed] [Google Scholar]
- 35. Wasiak S, Gilham D, Tsujikawa LM, et al. Downregulation of the complement cascade in vitro, in mice and in patients with cardiovascular disease by the BET protein inhibitor apabetalone (RVX‐208). Cardiovasc Transl Res. 2017;10(4):337‐347. doi: 10.1007/s12265-017-9755-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Haarhaus M, Ray KK, Nicholls SJ, et al. Apabetalone lowers serum alkaline phosphatase and improves cardiovascular risk in patients with cardiovascular disease. Atherosclerosis. 2019;290:59‐65. doi: 10.1016/j.atherosclerosis.2019.09.002 [DOI] [PubMed] [Google Scholar]
- 37. Wasiak S, Tsujikawa LM, Halliday C, et al. Benefit of apabetalone on plasma proteins in renal disease. Kid Int Rep. 2017;3(3):711‐721. doi: 10.1016/j.ekir.2017.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nicholls SJ, Ray KK, Johansson JO, et al. Selective BET protein inhibition with apabetalone and cardiovascular events: a pooled analysis of trials in patients with coronary artery disease. Am J Cardiovasc Drugs. 2018;18(2):109‐115. doi: 10.1007/s40256-017-0250-3 [DOI] [PubMed] [Google Scholar]
- 39. Shishikura D, Kataoka YU, Honda S, et al. The effect of bromodomain and extra‐terminal inhibitor apabetalone on attenuated coronary atherosclerotic plaque: insights from the ASSURE trial. Am J Cardiovasc Drugs. 2019;19(1):49‐57. doi: 10.1007/s40256-018-0298-8 [DOI] [PubMed] [Google Scholar]
- 40. Auray‐Blais C, Cyr D, Ntwari A, et al. Urinary globotriaosylceramide excretion correlates with the genotype in children and adults with Fabry disease. Mol Genet Metab. 2008;93(3):331‐340. doi: 10.1016/j.ymgme.2007.10.001 [DOI] [PubMed] [Google Scholar]
- 41. Auray‐Blais C, Boutin M, Gagnon R, Dupont FO, Lavoie P, Clarke JTR. Urinary globotriaosylsphingosine‐related biomarkers for Fabry disease targeted by metabolomics. Anal Chem. 2012;84(6):2745‐2753. doi: 10.1021/ac203433e [DOI] [PubMed] [Google Scholar]
- 42. Auray‐Blais C, Lavoie P, Boutin M, Abaoui M. High‐risk screening for Fabry disease: analysis by Tandem Mass Spectrometry of Globotriaosylceramide (Gb(3)) in Urine Collected on Filter Paper. Curr Protoc Hum Genet. 2017;93:17.26.1‐17.26.12. doi: 10.1002/cphg.34 [DOI] [PubMed] [Google Scholar]
- 43. Dupont FO, Gagnon R, Boutin M, Auray‐Blais C. A metabolomic study reveals novel plasma lyso‐Gb3 analogs as Fabry disease biomarkers. Curr Med Chem. 2013;20(2):280‐288. doi: 10.2174/092986713804806685 [DOI] [PubMed] [Google Scholar]
- 44. Lavoie P, Boutin M, Abaoui M, Auray‐Blais C. Fabry disease biomarkers: analysis of urinary Lyso‐Gb3 and seven related analogs using tandem mass spectrometry. Curr Protoc Human Genet. 2016;90:17.22.1‐17.22.12. doi: 10.1002/cphg.1 [DOI] [PubMed] [Google Scholar]
- 45. Boutin M, Auray‐Blais C. Multiplex tandem mass spectrometry analysis of novel plasma lyso‐Gb₃‐related analogues in Fabry disease. Anal Chem. 2014;86(7):3476‐3483. doi: 10.1021/ac404000d [DOI] [PubMed] [Google Scholar]
- 46. Barr C, Clarke JT, Ntwari A, Drouin R, Auray‐Blais C. Fabry disease urinary globotriaosylceramide/creatinine biomarker evaluation by liquid chromatography‐tandem mass spectrometry in healthy infants from birth to 6 months. Mol Genet Metab. 2009;97(4):278‐283. doi: 10.1016/j.ymgme.2009.04.009 [DOI] [PubMed] [Google Scholar]
- 47. Harding SD, Sharman JL, Faccenda E, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res. 2018;46(D1):D1091‐D1106. doi: 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Alexander SPH, Fabbro D, Kelly E, et al. The Concise Guide to PHARMACOLOGY 2021/22: enzymes. Br J Pharmacol. 2021;178(S1):S313‐S411. doi: 10.1111/bph.15542 [DOI] [PubMed] [Google Scholar]
- 49. Waldek S, Feriozzi S. Fabry nephropathy: a review ‐ how can we optimize the management of Fabry nephropathy? BMC Nephrol. 2014;15:72. doi: 10.1186/1471-2369-15-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nathan C. Metchnikoff’s Legacy in 2008. Nat Immunol. 2008;9(7):695‐698. doi: 10.1038/ni0708-695 [DOI] [PubMed] [Google Scholar]
- 51. Caielli S, Conforti‐Andreoni C, Di Pietro C, et al. On/Off TLR signaling decides proinflammatory or tolerogenic dendritic cell maturation upon CD1d‐mediated interaction with invariant NKT cells. J Immunol. 2010;185(12):7317‐7329. doi: 10.4049/jimmunol.1000400 [DOI] [PubMed] [Google Scholar]
- 52. El‐Benna J, Dang PM, Gougerot‐Pocidalo MA, Marie JC, Braut‐Boucher F. p47phox, the phagocyte NADPH oxidase/NOX2 organizer: structure, phosphorylation and implication in diseases. Exp Mol Med. 2009;41(4):217‐225. doi: 10.3858/emm.2009.41.4.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. França CN, Izar MCO, Hortêncio MNS, et al. Monocyte subtypes and the CCR2 chemokine receptor in cardiovascular disease. Clin Sci (London, England : 1979). 2017;131(12):1215‐1224. doi: 10.1042/CS20170009 [DOI] [PubMed] [Google Scholar]
- 54. O’Connor T, Borsig L, Heikenwalder M. CCL2‐CCR2 signaling in disease pathogenesis. Endocr Metab Immune Disord Drug Targets. 2015;15(2):105‐118. doi: 10.2174/1871530315666150316120920 [DOI] [PubMed] [Google Scholar]
- 55. Jia T, Serbina NV, Brandl K, et al. Additive roles for MCP‐1 and MCP‐3 in CCR2‐mediated recruitment of inflammatory monocytes during Listeria monocytogenes infection. J Immunol (Baltimore, Md : 1950). 2008;180(10):6846‐6853. doi: 10.4049/jimmunol.180.10.6846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bhattacharya P, Budnick I, Singh M, et al. Dual role of GM‐CSF as a pro‐inflammatory and a regulatory cytokine: implications for immune therapy. J Interferon Cytokine Res. 2015;35(8):585‐599. doi: 10.1089/jir.2014.0149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tait Wojno ED, Hunter CA, Stumhofer JS. The Immunobiology of the interleukin‐12 family: room for discovery. Immunity. 2019;50(4):851‐870. doi: 10.1016/j.immuni.2019.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sun L, He C, Nair L, Yeung J, Egwuagu CE. Interleukin 12 (IL‐12) family cytokines: role in immune pathogenesis and treatment of CNS autoimmune disease. Cytokine. 2015;75(2):249‐255. doi: 10.1016/j.cyto.2015.01.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Powell MD, Read KA, Sreekumar BK, Jones DM, Oestreich KJ. IL‐12 signaling drives the differentiation and function of a TH1‐derived TFH1‐like cell population. Sci Rep. 2019;9(1):13991. doi: 10.1038/s41598-019-50614-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhou B, Mu J, Gong YI, et al. Brd4 inhibition attenuates unilateral ureteral obstruction‐induced fibrosis by blocking TGF‐β‐mediated Nox4 expression. Redox Biol. 2017;11:390‐402. doi: 10.1016/j.redox.2016.12.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Stock CJW, Michaeloudes C, Leoni P, et al. Bromodomain and Extraterminal (BET) protein inhibition restores redox balance and inhibits myofibroblast activation. BioMed Res Int. 2019;2019:1‐11. doi: 10.1155/2019/1484736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hussong M, Börno ST, Kerick M, et al. The bromodomain protein BRD4 regulates the KEAP1/NRF2‐dependent oxidative stress response. Cell Death Dis. 2014;5:e1195. doi: 10.1038/cddis.2014.157 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.