

Abstract
Plasma levels of high-density lipoprotein (HDL) inversely correlate with the incidence of cardiovascular diseases (CVD). The causal relationship between plasma HDL-cholesterol levels and CVD has been called into question by Mendelian randomization studies and the majority of clinical trials not showing any benefit of plasma HDL-cholesterol raising drugs on CVD. Nonetheless, recent Mendelian randomization studies including an increased number of CVD cases compared to earlier studies have confirmed that HDL-cholesterol levels and CVD are causally linked. Moreover, several studies in large population cohorts have shown that the cholesterol efflux capacity of HDL inversely correlates with CVD. Cholesterol efflux pathways exert anti-inflammatory and antiatherogenic effects by suppressing proliferation of hematopoietic stem and progenitor cells, and inflammation and inflammasome activation in macrophages. Cholesterol efflux pathways also suppress the accumulation of cholesteryl esters in macrophages, i.e. macrophage foam cell formation. Recent single-cell RNASeq studies on atherosclerotic plaques have suggested that macrophage foam cells have lower expression of inflammatory genes than non-foam cells, probably reflecting liver X receptor activation, upregulation of ATP Binding Cassette A1 and G1 cholesterol transporters and suppression of inflammation. However, when these pathways are defective lesional foam cells may become pro-inflammatory.
Keywords: Atherosclerosis, cholesterol efflux, inflammation, cardiovascular diseases, high-density lipoprotein
Introduction
Plasma levels of high-density lipoprotein (HDL) inversely correlate with the incidence of cardiovascular diseases (CVD) (Castelli et al. 1977; Gordon et al. 1977; Prospective Studies et al. 2007). The causal relationship between plasma HDL-cholesterol levels and CVD has been called into question by Mendelian randomization (MR) studies and the majority of clinical trials not showing any benefit of plasma HDL-cholesterol raising drugs on CVD (Voight et al. 2012; Tall and Rader, 2018). Nonetheless, a recent MR study including an increased number of CVD cases compared to earlier studies and adjustment for metabolic syndrome-related pleiotropic traits has confirmed that HDL-cholesterol levels and CVD are causally linked (Thomas et al. 2021). However, MR at loci linked to direct effects on HDL-cholesterol suggests locus- and mechanism-specific causal effects of these factors on coronary artery disease (CAD) (Thomas et al. 2021), possibly related to impact on cholesterol efflux capacity (CEC) of HDL. In addition, the randomized evaluation of anacetrapib through lipid-modification (REVEAL) trial showed that the cholesteryl ester transfer protein (CETP) inhibitor anacetrapib reduced CVD events. Although this was attributed to a decrease of plasma low-density-lipoprotein (LDL) cholesterol by ~17%, HDL-cholesterol showed a major increase (~104%) (Group et al. 2017), which most likely also contributed to this positive outcome. Effects of HDL raising drugs on CVD have recently been reviewed (Tall and Rader 2018), as has HDL function in the context of several diseases (Rohatgi et al. 2021). Currently, the ApoA-I Event reducing in Ischemic Syndromes II (AEGIS-II) trial evaluating the effect of reconstituted HDL (rHDL; CSL-112) infusions, discoidal particles consisting of phospholipids and apolipoprotein A-I (apoA-I) following an Acute Coronary Syndrome (ACS) event is ongoing (ClinicalTrials.gov Identifier: NCT03473223).
The ability of HDL to act as an acceptor for cholesterol efflux from macrophage foam cells, i.e. macrophages that have accumulated cholesteryl esters, is likely central to its putative beneficial effects in ACS (Didichenko et al. 2016). In addition, several large clinical population studies have shown a strong inverse relationship between HDL CEC and CVD, after correction for plasma HDL-cholesterol levels (Khera et al. 2011; Rohatgi et al. 2014; Saleheen et al. 2015; Shea et al. 2019), suggesting HDL CEC may be superior to plasma HDL-cholesterol as a biomarker of CVD. HDL CEC quantifies cholesterol efflux by apoB depleted serum, which contains HDL. Cholesterol efflux is the first step in reverse cholesterol transport (RCT), the removal of cholesterol from macrophage foam cells in the arterial wall by HDL, transport in plasma, uptake by the liver and ultimate secretion into the bile (Glomset 1968). The scavenger receptor BI (SR-BI) mediates HDL uptake by the liver. While SR-BI accounts for the majority of hepatic cholesterol uptake in RCT, recently, a role for the hepatic LDL receptor in RCT has also been identified (Cedo et al. 2020). Direct uptake of cholesterol by the intestine independent of the liver contributes to RCT as well (van der Velde et al. 2007; Gillard et al. 2018). Interestingly, a rare variant in the SCARB1 gene (encoding SR-BI) exhibiting a loss-of-function and causing increased HDL was associated with increased CAD (Zanoni et al. 2016), lending support for an inverse association between RCT and CAD in humans (Trigatti and Hegele 2016). However, in another study, three rare Icelandic variants in SCARB1 that were associated with elevated HDL-cholesterol did not confer an increased risk of CAD (Helgadottir et al. 2018).
The cholesterol transporter ATP Binding Cassette (ABC) A1 mediates cholesterol efflux to apoA-I and small HDL particles, while ABCG1 mediates cholesterol efflux to mature HDL (Wang et al. 2000, 2004; Kennedy et al. 2005). Studies in macrophages with deficiency of both transporters have shown that these pathways together account for ~60–70% of cholesterol efflux to HDL (Yvan-Charvet et al. 2007; Out et al. 2008; Westerterp et al. 2013). In vivo studies have shown that deficiency of Abca1 and Abcg1 (Abca1/g1) in hematopoietic stem and progenitor cells (HSPCs) accelerates atherogenesis in mice by promoting leukocytosis (Yvan-Charvet, Pagler, Gautier, et al. 2010), and deficiency of Abca1/g1 in myeloid cells (monocytes, macrophages and neutrophils) promotes macrophage foam cell formation, activation of the NLRP3 inflammasome, and atherogenesis (Westerterp et al. 2013, 2018).
The latter result seems to contrast with recent single-cell RNASeq studies that have revealed that macrophage foam cell formation in atherosclerotic plaques, a process that is enhanced by Abca1/g1 deficiency (Westerterp et al. 2013), is accompanied by a decrease in pro-inflammatory gene expression compared to non-foam cells (Kim et al. 2018; Cochain, Vafadarnejad, Arampatzi et al. 2018). However, Abca1/g1 deficient macrophages also show increased free cholesterol accumulation, which enhances inflammation (Yvan-Charvet et al. 2008; Westerterp et al. 2013, 2018).
This review will focus on the role of HDL and cholesterol efflux pathways in CVD in large population cohorts, hematopoietic stem and progenitor cells (HSPCs) in the context of inflammation, and on inflammatory pathways in macrophages of atherosclerotic plaques with a focus on the recent single-cell RNASeq studies.
Cholesterol efflux capacity of HDL and incidence of CVD
The Rader and Rothblat laboratories introduced the concept that HDL CEC might be a better biomarker of CVD than HDL cholesterol levels (Khera et al. 2011). Using a cholesterol efflux assay in J774 murine macrophages incubated with cyclic AMP that upregulates Abca1 expression, they found an inverse relationship between HDL CEC and carotid intima-media thickness (cIMT) in healthy volunteers, and between HDL CEC and CAD, as assessed by coronary angiography, in a US/European cohort (Khera et al. 2011). In both cohorts, this inverse correlation remained significant after multivariate adjustment for plasma HDL-cholesterol levels, suggesting that HDL CEC is a stronger inverse predictor of CVD than plasma HDL-cholesterol (Khera et al. 2011). Subsequently, the relationship between HDL CEC and CVD has been studied more extensively, as summarized in Table 1. The large majority of these studies have confirmed the inverse relationship with CAD originally reported by the Rader/Rothblat laboratories (Table 1). However, some studies while confirming the CAD relationship have found no relationship of CEC to carotid plaque progression, non-hemorrhagic stroke or peripheral artery disease (Shea et al. 2019; Garg et al. 2020).
Table 1.
HDL cholesterol efflux studies in humans and CVD outcomes.
| Authors | Cohort | Cholesterol efflux assay or HDL- particle assay | Outcome |
|---|---|---|---|
|
| |||
| Khera et al. 2011 | 203 healthy white volunteers | Radioactive assay J774 macrophages (cAMP) | Inverse correlation between CEC and cIMTa |
| Khera et al. 2011 | US/European cohortb: 442 cases, 351 controls | Radioactive assay J774 macrophages (cAMP) | Inverse correlation between CEC and CADc |
| Rohatgi et al. 2014 | Dallas heart study: 2924 participants Follow-up period 9.4 years |
Fluorescent assay with BODIPY- cholesterol J774 macrophages (cAMP) |
Inverse correlation between CEC and incident ASCVDd |
| Saleheen et al. 2015 | EPIC-Norfolk Study: 1745 cases, 1749 controls (NCC) Assessed in 1993–97 and follow-up to 2009 |
Radioactive assay J774 macrophages (cAMP) | Inverse correlation between CEC and incident CHDe |
| Li et al. 2013 | Outpatient clinic: 146 cases, 431 controls (NCC) |
Radioactive assay RAW264.7 macrophages (cAMP) | Inverse correlation between CEC and prevalent CADf |
| Li et al. 2013 | GeneBank: 871 cases, 279 controls (NCC) |
Radioactive assay RAW264.7 macrophages (cAMP) | Inverse correlation between CEC and prevalent CADf |
| Li et al. 2013 | GeneBank: 58 MI cases, 113 MACE cases, 279 controls (NCC) Follow-up period 3 years |
Radioactive assay RAW264.7 macrophages (cAMP) | Positive correlation between CEC and incident nonfatal MI/strokef Positive correlation between CEC and incident MACEf |
| Josefs et al. 2020 | CODAM Study: 533 participants | Radioactive assay THP-1 macrophages | No correlation between CEC and (sub)clinical atherosclerosis, in the whole population or in individuals with (pre)diabetes |
| Shea et al. 2019 | MESA Study (cohort 1): 416 cases, 416 controls (NCC) Follow-up period 10.2 years |
Cholesterol mass efflux THP-1 macrophages (T0901317) | Inverse correlation between CMEC and incident CVDg |
| Shea et al. 2019 | MESA Study (subgroup cohort 1): 242 cases, 242 controls (NCC) Follow-up period 10.2 years | Cholesterol mass efflux THP-1 macrophages (T0901317) | Inverse correlation between CMEC and incident CHDg |
| Shea et al. 2019 | MESA Study (subgroup cohort 1): 174 cases, 174 controls (NCC) Follow-up period 10.2 years | Cholesterol mass efflux THP-1 macrophages (T0901317) | No correlation between CMEC and incident strokeg |
| Shea et al. 2019 | MESA Study (cohort 2): 364 cases, 364 controls (NCC) Follow-up period 10.2 years |
Cholesterol mass efflux THP-1 macrophages (T0901317) | Positive correlation between CMEC and carotid plaque progressiong |
| Garg et al. 2020 | MESA study (PAD): 1458 participants 203 clinical PAD subjects at baseline 1255 participants for prospective studies (mean follow-up 6.5 years): 1042 no PAD, 213 clinical PAD | Cholesterol mass efflux THP-1 macrophages (T0901317) | No correlation between CMEC and clinical PAD at baseline, or in prospective studiesh |
| Khera et al. 2017 | JUPITER trial: 310 cases, 312 controls 77 cases, 79 controls |
Radioactive assay J774 macrophages (cAMP) | No correlation between CEC and incident CVD at baselinei Inverse correlation between CEC and incident CVD on statin therapyi |
| Khera et al. 2017 | JUPITER trial: 299 cases, 286 controls 77 cases, 110 controls |
Nuclear magnetic resonance (NMR) spectroscopy, lipoprofile III, LipoScience Inc (now LabCorp Raleigh NC) | Inverse correlation between HDL-P and incident CVD at baseline and on statin therapyj |
| Singh et al. 2020 | Dallas heart study (2535), ARIC study (1595), MESA study (6632), and PREVEND study (5022): total of 15 784 participants | NMR LipoProfile 3 | Inverse correlation between HDL-P and MI/ischemic stroke No association between HDL-c and MI/ischemic strokek |
Adjusted for age, sex, systolic blood pressure, HbA1C, LDL-c, and HDL-c/ApoA-I.
Italian ATVB study, heart attack risk in puget sound, REGICOR, MGH premature coronary artery disease study, FINRISK, and Malmö diet and cancer study.
Adjusted for age, sex, smoking, diabetes, hypertension, LDL-c, and HDL-c/ApoA-I.
Adjusted for age, sex, diabetes, hypertension, smoking, BMI, total cholesterol level, TG level, history of statin use, HDL-c, and HDL-P.
Adjusted for age, sex, batch number, diabetes, hypertension, smoking, alcohol use, waist:hip ratio, BMI, LDL-c, log-TGs, and HDL-c/ApoA-I.
Adjusted for age, sex, smoking, diabetes, hypertension, LDL-c, and HDL-c.
Adjusted for age, sex, race, BMI, site, diabetes, total and HDL-c, statin use, hypertension medication, systolic blood pressure, smoking, alcohol, exercise, and diet.
Adjusted for age, sex, race, BMI, diabetes, total and HDL-c, statin use, hypertension, cigarette smoking, physical activity, eGFR.
Adjusted for age, treatment group, race, smoking status, systolic blood pressure, BMI, fasting glucose, LDL-c, log-TGs, and family history of premature CAD.
Adjusted for cohort and age, hypertension, diabetes, smoking, lipid medications, LCL-c, TG, BMI, waist, hs-CRP and HDL-c.
Adjusted for cohort and age, hypertension, diabetes, smoking, lipid medications, LCL-c, TG, BMI, waist, hs-CRP and HDL-P.
ApoA-I: apolipoprotein A-I; (AS) CVD: (atherosclerotic) cardiovascular disease; BMI: body mass index; CAD: coronary artery disease; cAMP: cyclic adenosine monophosphate; CEC: HDL-cholesterol efflux capacity; CHD: coronary heart disease; cIMT: carotid intima-media thickness; CMEC: HDL-cholesterol mass efflux capacity; eGFR: estimated glomerular filtration rate; HbA1c: hemoglobin A1c; HDL-c: high-density lipoprotein-cholesterol; HDL-P: HDL-particle concentration; hs-CRP: high sensitivity C-reactive protein; LDL-c: low-density lipoprotein-cholesterol; LXR: liver X receptor; MACE: major adverse cardiovascular event (MI, stroke, or death); MI: myocardial infarction; NCC: nested case-control; PAD: peripheral artery disease; TG: triglycerides.
A prospective study with a follow-up period of 9.4 years reported a negative correlation between CEC and incident cardiovascular events in a multiethnic population from the Dallas Heart Study (Rohatgi et al. 2014). This population was free from clinical atherosclerotic CVD (ASCVD) at baseline, supporting the use of HDL CEC as an inverse predictor for ASCVD; however, the number of CVD events in the Dallas Heart Study was relatively low (Rohatgi et al. 2014). For cholesterol efflux assays, J774 macrophages were labeled with BODIPY-fluorescent cholesterol instead of radioactive cholesterol (Rohatgi et al. 2014). BODIPY-cholesterol is a suitable reagent to examine cholesterol efflux (Martel et al. 2013) and may be preferred for high throughput studies. A nested case-control study from the prospective EPIC-Norfolk Study also showed an inverse relationship between CEC and incident coronary heart disease (CHD) (Saleheen et al. 2015), further supporting previous observations (Khera et al. 2011; Rohatgi et al. 2014). However, while an inverse correlation between CEC and prevalent CAD was reported in a convenience sample (outpatient clinic cohort and in patients enrolled in the GeneBank Study) (Li et al. 2013), in the same study, a positive correlation between CEC and a combined endpoint of nonfatal MI, stroke, or death (MACE) was found. Although RAW264.7 macrophages were used in this study, rigorous validation studies have revealed that CEC correlated highly with J774 macrophages in these cells (Li et al. 2013). Hence the positive correlation between HDL CEC and MACE has been attributed to the relatively few MACE events (n = 113) and other potentially high-risk factors in this specific sample (Khera and Rader 2013). Of note, populations from the Dallas Heart Study and EPIC Norfolk study were at low risk of CVD events (Rohatgi et al. 2014; Saleheen et al. 2015), and as such few risk factors in addition to decreased CEC may have contributed to CVD in these populations.
In another study in a selection of participants from the Cohort on Diabetes and Atherosclerosis Maastricht (CODAM) Study that consists of subjects with increased risk of CVD and (pre)diabetes, no association between CEC and markers for (sub)clinical atherosclerosis (cIMT, endothelial dysfunction, prevalent CVD, and history of CV events) was found (Josefs et al. 2020). This may have been the consequence of these subjects carrying several CVD risk factors, especially since ~25% were type II diabetics with increased C-reactive protein (CRP) levels, reflecting increased inflammation, and another ~25% had pre-diabetes (Annema et al. 2016; Josefs et al. 2020). Notably, unlike in other studies (Khera et al. 2011; Rohatgi et al. 2014; Saleheen et al. 2015), no ligand was used to upregulate the expression of ABC transporters in the cholesterol efflux assay. Therefore, the level of cholesterol efflux may have been relatively low, which may compromise the interpretation of these studies.
Further, an inverse correlation between CEC and incident CVD in individuals on statin therapy, but not at baseline was found in a population from the JUPITER (Justification for the Use of statin in Prevention: an intervention trial evaluating Rosuvastatin) trial (Khera et al. 2017). The JUPITER trial included subjects at risk of CVD because of increased plasma C-reactive protein (CRP) levels, reflecting increased inflammation (Ridker et al. 2008). Statin use decreased CRP levels (Ridker et al. 2008), suggesting that CEC may be predictive of CVD in the absence of other risk factors, perhaps similar to findings from the CODAM and a subset of the GeneBank cohort studies (Li et al. 2013; Josefs et al. 2020).
The exchange of cholesterol between HDL and cells is bidirectional. Enhanced free cholesterol bioavailability on HDL, as in the setting of SR-BI deficiency in mice, may contribute to cholesterol influx into cells (Rosales et al. 2019). The use of cholesterol mass efflux assays is the only method that allows for the direct assessment of net cholesterol efflux from cells to HDL. This method was used to study the relation between HDL CEC and CVD in the multi-ethnic study of atherosclerosis (MESA). Similar to the CODAM study (Josefs et al. 2020), for these studies, THP-1 macrophages were used. The expression of ABCA1 and ABCG1 was induced by the Liver X Receptor (LXR) agonist T0901317. HDL cholesterol mass efflux capacity (CMEC) adjusted for other risk factors showed a strong, independent inverse relationship with incident coronary heart disease (CHD) (Shea et al. 2019). This 10-year prospective study involving 465 cases and 465 controls provides powerful support for the protective role of HDL-mediated cholesterol efflux in CVD. Interestingly, there was no correlation of CMEC with carotid plaque progression or incident non-hemorrhagic stroke events in a separate subgroup of the MESA cohort, while the inverse correlation between CEC and CHD was confirmed in this second group (Shea et al. 2019). The differential association of HDL CEC with stroke and CHD was attributed to the differences in the athero-biology of the carotid and coronary arteries, which may rather contribute to stroke and CHD, respectively (Rohatgi 2019).
HDL particle number and CVD
The analysis of the JUPITER trial also revealed an inverse correlation between HDL particle (HDL-P) concentration as estimated by NMR and CVD, which was stronger than for HDL CEC and CVD (Khera et al. 2017). This inverse correlation remained significant after correction for HDL cholesterol. Together, these data suggest that HDL-P concentration may be a stronger inverse predictor for CVD than HDL CEC. The latter is supported by recent studies showing an inverse correlation between HDL-P concentration (as estimated by NMR) and a composite outcome of myocardial infarction (MI) and ischemic stroke in a pooled cohort of 4 large population studies including the Dallas Heart Study, the ARIC (Atherosclerosis Risk in Communities) study, the MESA study, and the PREVEND (Prevention of Renal and Vascular Endstage Disease) study, after correction for HDL-cholesterol (Singh et al. 2020). Surprisingly, HDL-P concentration was not inversely associated with MI among black participants, while the inverse association was strong among white participants irrespective of gender in all studies (Singh et al. 2020). The reason for the differential inverse association of HDL-P for MI by black ethnicity is unclear and would warrant further investigation in view of the suitability of HDL-P concentration as a biomarker for CVD. In addition, further investigation would be required to assess which antiatherogenic properties of HDL are reflected by HDL-P concentration. HDL CEC and HDL-P concentrations have shown moderate correlations in the JUPITER study (r = 0.39) and in a small cohort of subjects without obstructive CAD that were studied for endothelial function (r = 0.54) (Monette et al. 2016; Khera et al. 2017), but only a weak correlation in the Dallas Heart Study (r = 0.15) (Rohatgi et al. 2014). The discrepancy for these findings is unclear and may be related to the patient populations studied.
In sum, several (Khera et al. 2011; Li et al. 2013; Rohatgi et al. 2014; Saleheen et al. 2015; Khera et al. 2017; Shea et al. 2019; Singh et al. 2020) though not all (Li et al. 2013; Josefs et al. 2020) studies emphasize that HDL-CEC and HDL-P are inversely correlated with the incidence of CVD, as summarized in Table 1. Importantly, these inverse correlations persisted after adjustment for plasma HDL-cholesterol levels. In order to establish HDL CEC or HDL-P concentration as biomarkers of CVD, it will be of critical importance to assess the populations and conditions (i.e. inflammation, diabetes), where these parameters may indeed have predictive value. This may have implications for the potential therapeutic benefit of CSL-112 infusions, an rHDL particle that enhances HDL CEC (Didichenko et al. 2016), and is currently evaluated for ACS in the AEGIS-II trial. HDL CEC decreases 2-5 d after ACS before returning to baseline at 30 d after the event (Gibson et al. 2021). The rationale for the AEGIS-II trial is that four weekly 2 h infusions of CSL-112 after ACS would restore HDL CEC and enhance plaque stability to the extent that CSL-112 would suppress CV death and recurrent CV events (MI or stroke). The follow-up period of the AEGIS-II trial is one year (Gibson et al. 2021). While the AEGIS-I trial has shown that CSL-112 infusions over a period of 4 weeks restore HDL CEC (Gille 2018), it is questionable whether HDL CEC remains stable when patients no longer receive CSL-112 infusions. It also remains to be seen whether an increase in HDL CEC over a period of maximally one year suffices to detect effects on atherosclerotic plaque vulnerability. In the largest prospective study in terms of CV events, involving assessment of the relationship between CMEC and CHD in the MESA cohort, a relationship of CMEC with CHD outcome was observed after a follow up period of 10 years (Shea et al. 2019).
Role of cholesterol efflux pathways in hematopoietic stem cell proliferation, inflammation, and atherosclerosis
We have previously reviewed the role of cholesterol efflux pathways mediated by ABCA1, ABCG1, and ABCG4 in inflammation and atherosclerosis (Westerterp et al. 2014). In brief, Abca1 and Abcg1 are highly expressed in macrophages and HSPCs, and Abca1 deficiency leads to increased expression of Abcg1 in these cells and vice versa, suggesting that these transporters have overlapping roles and show mutual compensation (Yvan-Charvet et al. 2007; Westerterp et al. 2014). Hence, to obtain insights into the role of cholesterol efflux pathways in hematopoietic cells in atherogenesis, it is necessary to use mice with combined deficiency of Abca1 and Abcg1. We have shown that cholesterol efflux pathways in myeloid cells suppress atherogenesis (Yvan-Charvet et al. 2007; Yvan-Charvet, Pagler, Gautier, et al. 2010; Westerterp et al. 2013). Deficiency of Abca1 and Abcg1 in HSPCs leads to their expansion due to increased HSPC proliferation, which is associated with increased surface expression of the β subunit common to the receptors for granulocyte-macrophage stimulating factor (GM-CSF) and interleukin-3 (IL-3), due to stabilization of these receptors by increased membrane cholesterol accumulation (Yvan-Charvet, Pagler, Gautier et al. 2010). This leads to leukocytosis, mainly reflected by an expansion of the monocyte and neutrophil population (Yvan-Charvet, Pagler, Gautier et al. 2010). Leukocytosis, and specifically monocytosis, increases atherogenesis in mice and enhances MI in humans (Coller 2005; Yvan-Charvet, Pagler, Gautier et al. 2010; Murphy et al. 2011). Similar to Abca1−/− Abcg1−/− mice, Apoe−/− mice show HSPC expansion (Murphy et al. 2011). HSPCs secrete ApoE, which interacts with Abca1 and Abcg1 at the plasma membrane of HSPCs, and induces cholesterol efflux and suppresses HSPC proliferation in a cell-intrinsic manner (Murphy et al. 2011). Deficiency of the β subunit common to the GM-CSF and IL-3 receptors suppressed HSPC expansion, monocytosis and atherosclerosis in mice transplanted with Apoe−/− bone marrow (Wang et al. 2014), substantiating the causal relationship between HSPC expansion, monocytosis, and atherogenesis.
More recent observations have shown that in a trained immunity model that was triggered by β-glucan and subsequent LPS injections, or in conditions of chronic inflammation as observed in rheumatoid arthritis, expression of Abca1, Abcg1, and Apoe in HSPCs was suppressed (Dragoljevic et al. 2018; Mitroulis et al. 2018). Although speculative, Toll-like receptor signaling in the trained immunity and RA model could suppress cholesterol efflux in HSPCs, as has been observed in macrophages (reviewed below) (Castrillo et al. 2003). Notably, in the trained immunity model, also cholesterol synthesis genes were increased in HSPCs, as were genes involved in the glycolysis and mevalonate pathway (Mitroulis et al. 2018). Glycolysis enhances the formation of acetyl CoA that gives rise to cholesterol in the mevalonate pathway (Mitroulis et al. 2018). This could account for the increase in cholesterol synthesis. The changes in gene expression that favor HSPC cholesterol accumulation were accompanied by increased inflammation, HSPC expansion, increased surface expression of the common β subunit to the GM-CSF and IL-3 receptors, monocytosis, and increased atherogenesis (Dragoljevic et al. 2018; Mitroulis et al. 2018). Importantly, these studies indicate the broader relevance of the previously identified mechanisms in Apoe−/− and Abca1−/− Abcg1−/− mice since they reveal that under conditions of inflammation, defective cholesterol efflux in HSPCs drives HSPC expansion, monocytosis, and atherogenesis. Therefore, it has been postulated that defective cholesterol efflux pathways in HSPCs may be an integral component in myelopoiesis driven CVD (Yvan-Charvet and Swirski 2018).
Inflammation not only suppresses Abca1 and Abcg1 expression in HSPCs, but also in macrophages (Castrillo et al. 2003; Dragoljevic et al. 2018; Mitroulis et al. 2018). Toll-like receptor 3 (TLR3) and TLR4 signaling suppress Abca1 and Abcg1 expression in macrophages via interferon regulatory factor 3 signaling and downstream suppression of their key regulator the liver X receptor (LXR) (Castrillo et al. 2003). Conversely, deficiency of Abca1 and Abcg1 in macrophages increases TLR2, 3, and 4 signalings (Yvan-Charvet et al. 2008). The latter is likely due to increased surface expression of TLR4/mye-loid differentiation-2 (MD-2) complexes as a result of membrane cholesterol accumulation (Yvan-Charvet et al. 2008). Macrophage Abca1/g1 deficiency increases expression of pro-inflammatory cytokines in atherosclerotic plaques, presumably because of increased TLR2, 3, or 4 signalings (Westerterp et al. 2013). Moreover, humans heterozygous for loss-of-function mutations in the ABCA1 gene that show ~50% decreased plasma HDL-cholesterol levels have increased pro-inflammatory cytokine levels and vascular inflammation compared to aged-and-gender-matched control subjects (Bochem et al. 2015), suggesting clinical relevance.
Pro- and anti-inflammatory effects of HDL
While HDL exerts anti-inflammatory effects by a variety of mechanisms (Suzuki et al. 2010; Yvan-Charvet, Kling, Pagler et al. 2010; De Nardo et al. 2014; Fotakis et al. 2019), pro-inflammatory effects of HDL and apoA-I (Smoak et al. 2010; van der Vorst et al. 2017; Fotakis et al. 2019) have also been reported. These have recently been reviewed (Rohatgi et al. 2021). Antiinflammatory effects of HDL mediated by cholesterol efflux include suppression of the surface expression of TLR4/MD-2 complexes and downstream MyD88 and TRIF (TIR-domain-containing adapter-inducing interferon-β) signaling (Yvan-Charvet et al. 2008; Yvan-Charvet, Kling, Pagler, et al. 2010; Fotakis et al. 2019). Extensive transcriptional profiling studies however revealed that HDL exclusively suppressed gene expression downstream of TRIF/TRIF related adaptor molecule (TRAM), independent of cholesterol efflux (Suzuki et al. 2010), reflecting suppression of the type I interferon (IFN) response (Suzuki et al. 2010). HDL-mediated suppression of TRIF signaling may thus occur via cholesterol efflux-dependent and –independent pathways.
Further studies suggested that HDL inhibited inflammation by enhancing activating transcription factor 3 (ATF3) expression, independent of cholesterol efflux (De Nardo et al. 2014). ATF3 is induced by TLR stimulation and acts as a negative-feedback system to limit excessive production of pro-inflammatory cytokines downstream of NF-κB (Labzin et al. 2015). However, several studies could not confirm the upregulation of Atf3 expression in response to HDL (Thacker et al. 2016; van der Vorst et al. 2017; Fotakis et al. 2019), and one study showed that HDL, as a result of suppressing TLR4 surface expression, decreased Atf3 (Fotakis et al. 2019). Hence, the ATF3-mediated anti-inflammatory effects of HDL are controversial. In addition to anti-inflammatory effects, ApoA-I and HDL may also enhance inflammation. This may be beneficial in sepsis, where enhanced inflammation may promote efficient clearance of bacteria (van der Vorst et al. 2017). Under pro-inflammatory conditions, apoA-I may dissociate from HDL (Jahangiri et al. 2009) and directly activate TLR2 and TLR4 (Smoak et al. 2010). Pro-inflammatory effects of HDL in the presence of ligands for almost all TLRs (TLR1-9), including the TLR4 ligand lipopolysaccharide (LPS) have also been reported (van der Vorst et al. 2017). These effects were mediated by PKC activation resulting in STAT-1/ IRF1 activation (van der Vorst et al. 2017). Since HDL and methyl-β-cyclodextrin showed similar effects, it was inferred that PKC was activated downstream of membrane cholesterol depletion (van der Vorst et al. 2017). The exact mechanisms are however unclear, and intriguingly, a more recent study has shown that PKC activation rather drives the LPS response than the proinflammatory HDL response (Fotakis et al. 2019, 2020; van der Vorst et al. 2020).
In an effort to reconcile the divergent views of the anti- and pro-inflammatory effects of HDL, extensive transcriptome profiling studies in macrophages loaded with and without cholesterol in the presence of HDL and LPS have been performed (Fotakis et al. 2019). In line with previous studies (Yvan-Charvet et al. 2008; Yvan-Charvet, Kling, Pagler, et al. 2010), HDL suppressed inflammatory signaling downstream of TLR4 by inducing cholesterol efflux, leading to an early reduced NF-κB response, and a later reduced type I IFN response (Fotakis et al. 2019). Only under conditions of severe cholesterol depletion, HDL became pro-inflammatory by inducing an ER stress response mediated by IRE1a (inositol-requiring enzyme 1a)/ASK1 (apoptosis signal-regulating kinase 1)/p38 MAPK (p38 mitogen-activated protein kinase) signaling (Fotakis et al. 2019). The HDL used in these studies was either native HDL isolated from APOA1TG mice on a background of genetic hyperlipidemia or reconstituted HDL, consisting of apoA-I and phospholipids (Fotakis et al. 2019). These HDL particles are very good cholesterol efflux acceptors, perhaps explaining why the pro- and anti-inflammatory effects of HDL were cholesterol efflux dependent in these particular studies. Importantly, injections of reconstituted HDL only produced anti-inflammatory effects in lesional macrophages, without any evidence for pro-inflammatory effects (Fotakis et al. 2019). This may be of particular relevance in view of the outcomes of the AEGIS-II trial.
Cholesterol efflux pathways and inflammasome activation
We recently found that deficiency of Abca1 and Abcg1 in myeloid cells activates NLRP3 inflammasomes (Westerterp et al. 2017, 2018). NLRP3 inflammasomes regulate the secretion of interleukin-1β (IL-1β) and IL-18. Plasma levels of IL-18 are associated with increased CVD risk (Blankenberg et al. 2002; Ridker et al. 2020). IL-1β is a key regulator of inflammation, and antagonism of IL-1β decreases the incidence of cardiovascular events in humans, as shown in the canakinumab antiinflammatory thrombosis outcomes study (CANTOS) trial (Ridker et al. 2017). These studies illustrate the importance of NLRP3 inflammasome activation for CVD risk. We found that hematopoietic deficiency of the NLRP3 inflammasome components Nlrp3 or caspase-1 suppressed atherosclerosis in mice with myeloid Abca1/g1 deficiency (Westerterp et al. 2018), indicating that in conditions of defective cholesterol efflux in myeloid cells, activation of NLRP3 inflammasomes accelerates atherogenesis. Interestingly, Tangier Disease patients, who carry homozygous loss-of-function mutations in ABCA1, also show increased plasma IL-1β and IL-18 compared to age-and-gender-matched controls or heterozygous ABCA1 mutation carriers (Westerterp et al. 2018), suggesting human relevance. The most consistent phenotype among Tangier Disease patients is peripheral neuropathy (Schaefer et al. 2010). Interestingly, it has been shown that defective myelin clearance due to macrophage Abca1/g1 deficiency promotes inflammasome activation and limits remyelination in the central nervous system of aged mice (Cantuti-Castelvetri et al. 2018). These findings, together with the increase of the inflammasome products IL-1β and IL-18 in Tangier Disease patients, suggest a role for macrophage inflammasome activation in peripheral neuropathy in Tangier Disease.
Two signals are required for NLRP3 inflammasome activation: a priming signal leading to increased mRNA expression of the components of the NLRP3 inflammasome complex, including Nlrp3, Asc, pro-caspase-1, and pro-IL-1β and a second signal that leads to NLRP3 inflammasome assembly and caspase-1 cleavage. Active caspase-1 then cleaves pro-IL-1β and pro-IL-18, required for their secretion. While increased TLR signaling leads to inflammasome priming in the setting of myeloid Abca1/g1 deficiency, the nature of the second activating signal remains unknown. Several pathways including cholesterol crystal formation in the endosomal system or increased ER cholesterol promoting inflammasome assembly are potentially involved (Tall and Westerterp 2019). However, our studies found only minimal cholesterol crystal formation and no evidence of lysosomal damage in macrophage Abca1/g1 deficiency (Westerterp et al. 2018). NLRP3 inflammasome activation contributes to auto-immune lupus glomerulonephritis in the setting of dendritic cell Abca1/g1 deficiency, by augmenting lymph node size and T helper 1 cell polarization, the latter presumably in response to increased IL-18 (Westerterp et al. 2017).
Together, these studies show that inflammation suppresses cholesterol efflux pathways in HSPCs and macrophages (Castrillo et al. 2003; Dragoljevic et al. 2018; Mitroulis et al. 2018). Deficiency of cholesterol efflux pathways in HSPCs contributes to myelopoiesis and atherogenesis (Yvan-Charvet, Pagler, Gautier et al. 2010; Murphy et al. 2011), while deficiency of cholesterol efflux pathways in macrophages enhances inflammation downstream of TLR2, 3, and 4 signalings, and activates NLRP3 inflammasomes, contributing to atherogenesis (Yvan-Charvet et al. 2008; Westerterp et al. 2013, 2018). These effects occur mostly downstream of membrane cholesterol accumulation in HSPCs or macrophages (Yvan-Charvet et al. 2008; Westerterp et al. 2013, 2018). Membrane cholesterol accumulation also contributes to NLRP3 inflammasome priming, while several pathways related to cholesterol accumulation have been proposed as a second signal for the NLRP3 inflammasome in macrophages deficient for Abca1/g1 (Westerterp et al. 2018; Tall and Westerterp 2019).
Since inflammatory pathways suppress cholesterol efflux and reverse cholesterol transport (RCT), which in turn enhances inflammatory responses, there exists a positive feedback loop between inflammation and decreased cholesterol efflux/RCT (Tall and Yvan-Charvet 2015). While this may amplify the innate immune response and help to clear infectious agents, in the setting of chronic sterile inflammation as occurs in atherosclerosis and obesity, there may be an adverse outcome. In addition, recent studies have shown a U-shaped relationship between HDL-cholesterol and infectious disease (Madsen et al. 2018), and, in a presumably more healthy cohort, an inverse relationship between HDL-cholesterol and infectious disease hospitalization (Trinder et al. 2020). Moreover, higher levels of CETP expression in humans and mice are associated with lower levels of HDL cholesterol and increased mortality from sepsis (Trinder et al. 2021). Masucci-Magoulas et al. originally showed that LPS administration reduced hepatic CETP expression as a result of adrenal corticosteroid release and raised plasma HDL-cholesterol levels (Masucci-Magoulas et al. 1995). Dusuel et al. showed that CETP expression was associated with lower HDL-cholesterol, an increased macrophage inflammatory response and increased death during polymicrobial sepsis (Dusuel et al. 2021). It is tempting to speculate that the protective effect of CETP deficiency in sepsis could account for the high frequency of different CETP loss-of-function variants in East Asian populations (Inazu et al. 1990; Hirano et al. 1997). Overall, these findings illustrate the key role of HDL in modulating the inflammatory response to infection and suggest that an imbalance in the homeostatic response in either direction may have adverse consequences.
Foam cell formation, inflammation, and atherosclerotic plaques
We have shown that macrophage Abca1/g1 deficiency increases foam cell formation and accelerates atherosclerosis in mice (Westerterp et al. 2013). These data provide evidence for macrophage foam cell formation driving atherogenesis (Westerterp et al. 2013). In addition, as described above, macrophage Abca1/g1 deficiency increases atherosclerosis by enhancing proinflammatory cytokine expression in atherosclerotic plaques, and inflammasome activation (Westerterp et al. 2013, 2018). Although we found that membrane cholesterol accumulation induces inflammatory gene expression and inflammasome priming in macrophages with Abca1/g1 deficiency (Yvan-Charvet et al. 2008; Westerterp et al. 2013, 2018), these cells also accumulated cholesteryl esters (Westerterp et al. 2013). Hence, the outcome of these studies may be interpreted as foam cell formation, i.e. the accumulation of cholesteryl esters in macrophages, driving inflammation. However, studies by the Glass Laboratory have shown that foam cell formation suppresses inflammatory cytokine expression (Spann et al. 2012). The Glass Laboratory found that thioglycolate-elicited peritoneal macrophages from LDL receptor-deficient (Ldlr−/−) mice fed a high-fat high cholesterol (HFHC) diet for 12 weeks showed decreased inflammatory gene expression compared to macrophages from wild-type mice (Spann et al. 2012). A lipidomic screen revealed that macrophages from HFHC Ldlr−/− mice accumulated high levels of desmosterol. Desmosterol is a precursor of cholesterol in the cholesterol biosynthetic pathway and activates LXR (Yang et al. 2006). In line with previous studies (Joseph et al. 2003; Ghisletti et al. 2007), LXR activation suppressed inflammatory gene expression (Spann et al. 2012). These data introduced the concept that desmosterol accumulates in macrophages during foam cell formation and suppresses inflammatory gene expression via LXR activation.
Recent studies have extended the findings from the Glass Laboratory to macrophage foam cells of atherosclerotic plaques. Using single-cell RNA sequencing, Kim et al. found that a particular cluster of macrophages expressing high levels of Lgals3 (lectin galactoside-binding soluble 3), Abcg1, and Trem2 (triggering receptor on myeloid cells 2), suggestive of macrophage foam cells, was expanded in atherosclerotic plaques of Western-type diet-fed Ldlr−/− mice (Kim et al. 2018). To separate foamy macrophages from non-foamy macrophages in atherosclerotic plaques, Kim et al. used the dye BODIPY493/503 that stains neutral lipids (similar to Oil Red O), including cholesteryl esters (Kim et al. 2018). Similar to the studies by the Glass Laboratory (Spann et al. 2012), Kim et al. found that foamy macrophages showed high levels of LXRα gene expression, as well as its target genes Abca1 and MertK (Kim et al. 2018), suggesting accumulation of LXR ligands. Moreover, foamy macrophages showed decreased expression of proinflammatory genes compared to non-foamy macrophages (Kim et al. 2018). Whether these plaque foamy macrophages, similar to thioglycollate-elicited macrophages (Spann et al. 2012), accumulate desmosterol, remains to be assessed. Interestingly, an earlier single-cell RNA sequencing study on aortas of HFHC fed Ldlr−/− mice identified macrophages with high levels of Trem2 (Cochain, Vafadarnejad, Arampatzi et al. 2018). These Trem2hi macrophages showed high expression of genes involved in cholesterol transport and cholesterol efflux (Cochain, Vafadarnejad, Arampatzi et al. 2018) and were suggested to share similarities with the foamy macrophages described by Kim et al. (Cochain, Saliba, Zernecke 2018). Indeed, independent evaluation of these macrophage populations confirmed that foamy and Trem2hi macrophages are identical (Kim and Choi, 2018; Cochain, Saliba, Zernecke 2018). Recent studies have shown that Trem2hi macrophages expressing high levels of ABCA1 and ABCG1 are also present in human atherosclerotic plaques in carotid arteries from humans (Depuydt et al. 2020). These Trem2hi macrophages showed low expression of pro-inflammatory genes compared to other macrophage populations in these human plaques, suggesting similarities with the Trem2hi macrophage population identified in atherosclerotic plaques of Ldlr−/− mice (Depuydt et al. 2020). Indeed, desmosterol is also present at high levels in human atherosclerotic lesions (Spann et al. 2012), and may accumulate in Trem2hi macrophages.
While these discoveries (Kim et al. 2018; Kim and Choi 2018; Cochain, Saliba, Zernecke 2018, Cochain, Vafadarnejad, Arampatzi, et al. 2018; Depuydt et al. 2020) are descriptive, they challenge the view that foam cells are pro-inflammatory and strongly suggest that LXR activation, presumably by desmosterol, in foam cells suppresses inflammation (Spann et al. 2012). Several mechanisms for the LXR mediated suppression of inflammatory gene expression in macrophages have been proposed (Figure 1). Cholesterol efflux independent mechanisms include trans-repression downstream of NF-κB by SUMOylation of specific NF-κB target genes such as inducible nitric oxide synthase (iNOS) and monocyte chemoattractant protein-1 (MCP-1) (Ghisletti et al. 2007), and closing chromatin accessibility through cis-repression for inflammatory gene enhancers such as IL-1β and cyclooxygenase-2 (COX-2) (Thomas 2018). It has also been proposed that the anti-inflammatory properties of LXR activation are mediated by Abca1 expression (Ito et al. 2015). Abca1 mediated cholesterol efflux suppresses TLR4 induced MyD88 signaling by reducing plasma membrane free cholesterol in macrophages, and Abcg1 or both transporters together have similar effects (Yvan-Charvet et al. 2008; Zhu et al. 2008) (Figure 1). Data showing that LXR activation suppresses inflammatory gene expression and increases the expression of ABC transporters (Joseph et al. 2003) would suggest this is a plausible mechanism for LXR mediated anti-inflammatory effects. Nonetheless, LXR agonists still suppressed TLR induced pro-inflammatory gene expression in Abca1−/− Abcg1−/− macrophages (Kappus et al. 2014), suggesting that the anti-inflammatory effects of LXR agonists can be observed independent of cholesterol efflux. Together, these observations suggest that macrophage foam cells have a limited inflammatory response as a result of LXR activation that has a variety of anti-inflammatory effects including upregulation of cholesterol efflux pathways. However, it is possible that in the setting of defective LXR activation or inadequate up-regulation of cholesterol efflux (e.g. as a result of low HDL levels), foam cells become more pro-inflammatory. This hypothesis remains to be tested.
Figure 1.
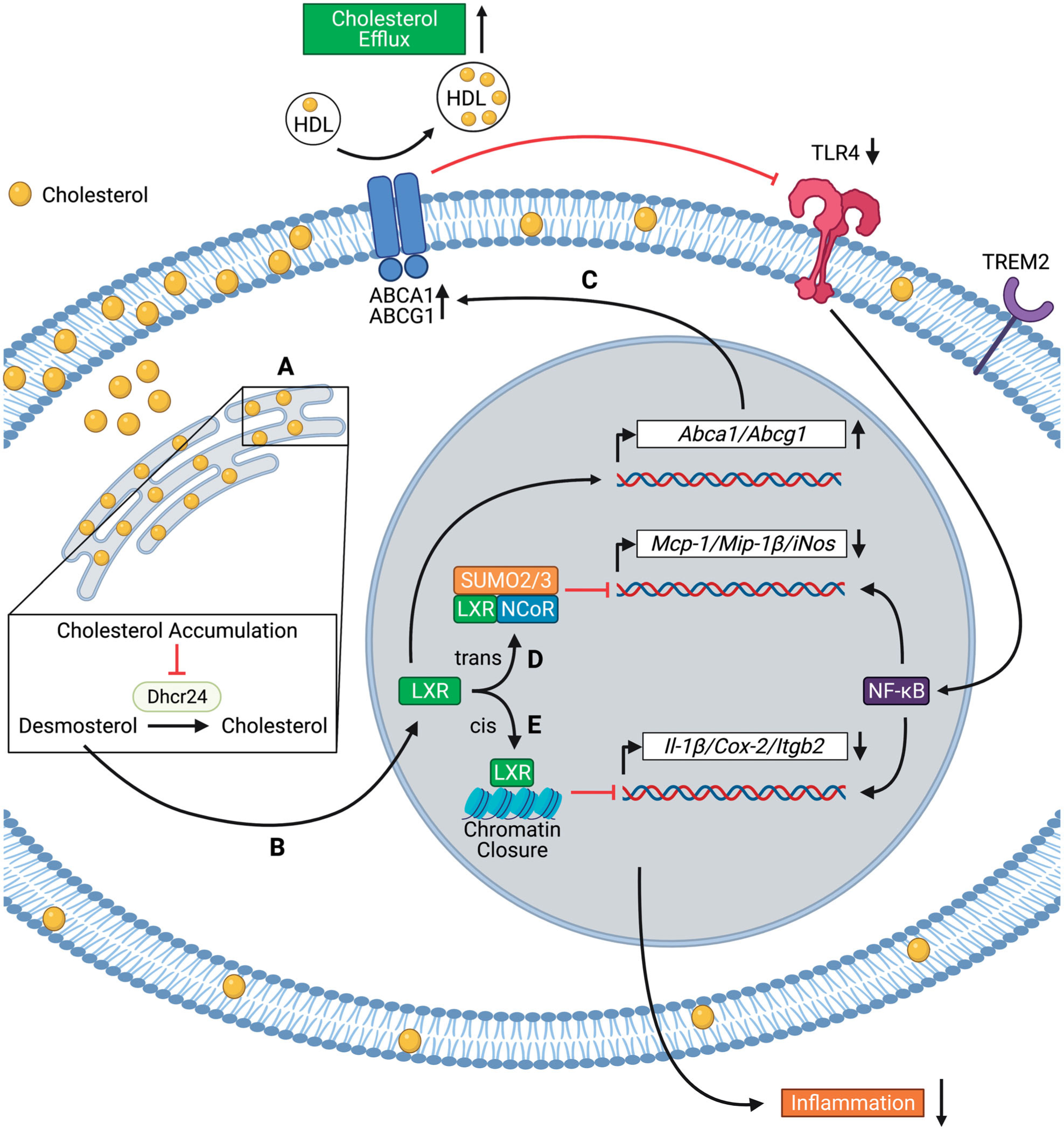
Mechanisms of suppression of inflammatory gene expression by liver X receptor (LXR) activation in foamy macrophages. Foamy macrophages in atherosclerotic plaques express high levels of Trem2. (A) Cholesterol accumulation in the endoplasmic reticulum inhibits the enzymatic activity of 24-dehydrocholesterol reductase (Dhcr24), leading to desmosterol accumulation; (B) Desmosterol activates the transcription factor LXR. LXR activation suppresses inflammation via cholesterol efflux-dependent (C) and independent (D–E) mechanisms; (C) LXR upregulates the expression of ATP-Binding Cassette Transporter A1 and G1 (Abca1 and Abcg1), leading to cholesterol efflux. Cholesterol efflux decreases Toll-like receptor 4 (TLR4) surface expression and NF-κB activation; (D) LXR forms a complex with SUMO-2/3 and NCoR, which trans-represses the transcription of Mcp-1 (monocyte chemoattractant protein-1), Mip-1β (macrophage inflammatory protein-1β), and iNos (inducible nitric oxide synthase); (E) LXR binds to inflammatory gene enhancers, leading to cis-repression of Il-1β (interleukin-1β), Cox-2 (cyclo-oxygenase-2), and Itgb2 (integrin beta 2) through chromatin closure. The figure has been created with Biorender.com.
Conclusions and future directions
Inflammatory pathways suppress cholesterol efflux and reverse cholesterol transport (RCT) (Castrillo et al. 2003; Dragoljevic et al. 2018; Mitroulis et al. 2018), which in turn enhances inflammatory responses, as shown in HSPCs and myeloid cells (Yvan-Charvet et al. 2008; Yvan-Charvet, Pagler, Gautier, et al. 2010; Murphy et al. 2011; Westerterp et al. 2013; Kappus et al. 2014; Bochem et al. 2015; Westerterp et al. 2018), indicating a positive feedback loop between inflammation and cholesterol efflux/RCT (Tall and Yvan-Charvet 2015). While this may amplify the innate immune response and help to clear infectious agents, in the setting of chronic sterile inflammation as occurs in atherosclerosis and obesity, there may be an adverse outcome. Recent studies have also shown that higher levels of CETP expression in humans and mice are associated with lower levels of HDL cholesterol and increased mortality from sepsis (Trinder et al. 2021). While these findings were attributed to low HDL-cholesterol levels, high CETP expression levels also decrease HDL CEC (Matsuura et al. 2006), and hence diminished HDL CEC could have contributed to increased mortality from sepsis.
The majority of studies has shown (Khera et al. 2011; Li et al. 2013; Rohatgi et al. 2014; Saleheen et al. 2015; Khera et al. 2017; Shea et al. 2019; Josefs et al. 2020; Singh et al. 2020) that HDL CEC and HDL-P are stronger inverse predictors of CVD than HDL-cholesterol. It remains to be assessed which anti-atherogenic properties of HDL are reflected by HDL-P concentration. In order to establish HDL CEC or HDL-P concentration as biomarkers of CVD, it will be of critical importance to assess the populations and conditions (i.e. inflammation, diabetes), where these parameters may indeed have predictive value.
Funding
This work was supported by NIH grant HL107653 (A.R.T.), Netherlands Organization of Scientific Research VIDI grant 917.15.350 (M.W.) and a Rosalind Franklin Fellowship from the University Medical Center Groningen (M.W.).
Footnotes
Disclosure statement
A.R.T. is on the scientific advisory board and is a co-founder of Fortico Biotech and Staten Biotech and has consulted from Amgen, CSL, Janssen, the Medicines Company, and AstraZeneca. The other authors report no declarations of interest.
References
- Annema W, Dikkers A, de Boer JF, van Greevenbroek MMJ, van der Kallen CJH, Schalkwijk CG, Stehouwer CDA, Dullaart RPF, Tietge UJF. 2016. Impaired HDL cholesterol efflux in metabolic syndrome is unrelated to glucose tolerance status: the CODAM study. Sci Rep. 6(1):27367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenberg S, Tiret L, Bickel C, Peetz D, Cambien F, Meyer J, Rupprecht HJ, AtheroGene I. 2002. Interleukin-18 is a strong predictor of cardiovascular death in stable and unstable angina. Circulation. 106(1):24–30. [DOI] [PubMed] [Google Scholar]
- Bochem AE, van der Valk FM, Tolani S, Stroes ES, Westerterp M, Tall AR. 2015. Increased systemic and plaque inflammation in ABCA1 mutation carriers with attenuation by statins. Arterioscler Thromb Vasc Biol. 35(7):1663–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantuti-Castelvetri L, Fitzner D, Bosch-Queralt M, Weil MT, Su M, Sen P, Ruhwedel T, Mitkovski M, Trendelenburg G, Lutjohann D, et al. 2018. Defective cholesterol clearance limits remyelination in the aged central nervous system. Science. 359(6376):684–688. [DOI] [PubMed] [Google Scholar]
- Castelli WP, Doyle JT, Gordon T, Hames CG, Hjortland MC, Hulley SB, Kagan A, Zukel WJ. 1977. HDL cholesterol and other lipids in coronary heart disease. The cooperative lipoprotein phenotyping study. Circulation. 55(5):767–772. [DOI] [PubMed] [Google Scholar]
- Castrillo A, Joseph SB, Vaidya SA, Haberland M, Fogelman AM, Cheng G, Tontonoz P. 2003. Crosstalk between LXR and toll-like receptor signaling mediates bacterial and viral antagonism of cholesterol metabolism. Mol Cell. 12(4): 805–816. [DOI] [PubMed] [Google Scholar]
- Cedo L, Metso J, Santos D, Garcia-Leon A, Plana N, Sabate-Soler S, Rotllan N, Rivas-Urbina A, Mendez-Lara KA, Tondo M, et al. 2020. LDL receptor regulates the reverse transport of macrophage-derived unesterified cholesterol via concerted action of the HDL-LDL axis: insight from mouse models. Circ Res. 127(6):778–792. [DOI] [PubMed] [Google Scholar]
- Cochain C, Saliba AE, Zernecke A. 2018. Letter by Cochain et al. regarding article, “transcriptome analysis reveals nonfoamy rather than foamy plaque macrophages are proinflammatory in atherosclerotic murine models.” Circ Res. 123(11):e48–e49. [DOI] [PubMed] [Google Scholar]
- Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, Wolf D, Saliba AE, Zernecke A. 2018. Single-cell RNA-seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ Res. 122(12):1661–1674. [DOI] [PubMed] [Google Scholar]
- Coller BS. 2005. Leukocytosis and ischemic vascular disease morbidity and mortality: is it time to intervene? ATVB. 25(4):658–670. [DOI] [PubMed] [Google Scholar]
- De Nardo D, Labzin LI, Kono H, Seki R, Schmidt SV, Beyer M, Xu D, Zimmer S, Lahrmann C, Schildberg FA, et al. 2014. High-density lipoprotein mediates anti-inflammatory reprogramming of macrophages via the transcriptional regulator ATF3. Nat Immunol. 15(2):152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depuydt MAC, Prange KHM, Slenders L, Ord T, Elbersen D, Boltjes A, de Jager SCA, Asselbergs FW, de Borst GJ, Aavik E, et al. 2020. Microanatomy of the human atherosclerotic plaque by single-cell transcriptomics. Circ Res. 127(11): 1437–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didichenko SA, Navdaev AV, Cukier AM, Gille A, Schuetz P, Spycher MO, Therond P, Chapman MJ, Kontush A, Wright SD. 2016. Enhanced HDL functionality in small HDL species produced upon remodeling of HDL by reconstituted HDL, CSL112: effects on cholesterol efflux, anti-inflammatory and antioxidative activity. Circ Res. 119(6):751–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragoljevic D, Kraakman MJ, Nagareddy PR, Ngo D, Shihata W, Kammoun HL, Whillas A, Lee MKS, Al-Sharea A, Pernes G, et al. 2018. Defective cholesterol metabolism in haematopoietic stem cells promotes monocyte-driven atherosclerosis in rheumatoid arthritis. Eur Heart J. 39(23): 2158–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dusuel A, Deckert V, Pais DEBJP, Van Dongen K, Choubley H, Charron E, Le Guern N, Labbe J, Mandard S, Grober J, et al. 2021. Human CETP lacks lipopolysaccharide transfer activity, but worsens inflammation and sepsis outcomes in mice. J Lipid Res. 62:100011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotakis P, Kothari V, Bornfeldt KE, Tall AR. 2020. Response by Fotakis et al. to letter regarding article, “anti-inflammatory effects of HDL (high-density lipoprotein) in macrophages predominate over proinflammatory effects in atherosclerotic plaques”. Arterioscl Thromb Vasc Biol. 40(2):e33–e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotakis P, Kothari V, Thomas DG, Westerterp M, Molusky MM, Altin E, Abramowicz S, Wang N, He Y, Heinecke JW, et al. 2019. Anti-inflammatory effects of HDL (high-density lipoprotein) in macrophages predominate over proinflammatory effects in atherosclerotic plaques. ATVB. 39(12): e253–e272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg PK, Jorgensen NW, McClelland RL, Allison M, Stein JH, Yvan-Chavret L, Tall AR, Shea S. 2020. Cholesterol mass efflux capacity and risk of peripheral artery disease: the multi-ethnic study of atherosclerosis. Atherosclerosis. 297: 81–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghisletti S, Huang W, Ogawa S, Pascual G, Lin ME, Willson TM, Rosenfeld MG, Glass CK. 2007. Parallel SUMOylation-dependent pathways mediate gene- and signal-specific transrepression by LXRs and PPARgamma. Mol Cell. 25(1): 57–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson CM, Kastelein JJP, Phillips AT, Aylward PE, Yee MK, Tendera M, Nicholls SJ, Pocock S, Goodman SG, Alexander JH, et al. 2021. Rationale and design of ApoA-I event reducing in ischemic syndromes II (AEGIS-II): a phase 3, multicenter, double-blind, randomized, placebo-controlled, parallel-group study to investigate the efficacy and safety of CSL112 in subjects after acute myocardial infarction. Am Heart J. 231:121–127. [DOI] [PubMed] [Google Scholar]
- Gillard BK, Rosales C, Xu B, Gotto AM Jr, Pownall HJ. 2018. Rethinking reverse cholesterol transport and dysfunctional high-density lipoproteins. J Clin Lipidol. 12(4):849–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gille AW, S D, Tortorici M, Duffy D, Liss C, Yee MK, Deckelbaum LI, D’Andrea DM, Gibson CM. 2018. CSL112 restores cholesterol efflux in patients immediately after acute myocardial infarction. Circulation. 136:A16500. [Google Scholar]
- Glomset JA. 1968. The plasma lecithins: cholesterol acyltransferase reaction. J Lipid Res. 9(2):155–167. [PubMed] [Google Scholar]
- Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. 1977. High density lipoprotein as a protective factor against coronary heart disease. The Framingham study. Am J Med. 62(5):707–714. [DOI] [PubMed] [Google Scholar]
- Group HTRC, Bowman L, Hopewell JC, Chen F, Wallendszus K, Stevens W, Collins R, Wiviott SD, Cannon CP, Braunwald E, et al. 2017. Effects of anacetrapib in patients with atherosclerotic vascular disease. New Engl J Med. 377(13): 1217–1227. [DOI] [PubMed] [Google Scholar]
- Helgadottir A, Sulem P, Thorgeirsson G, Gretarsdottir S, Thorleifsson G, Jensson BO, Arnadottir GA, Olafsson I, Eyjolfsson GI, Sigurdardottir O, et al. 2018. Rare SCARB1 mutations associate with high-density lipoprotein cholesterol but not with coronary artery disease. Eur Heart J. 39(23):2172–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano K, Yamashita S, Nakajima N, Arai T, Maruyama T, Yoshida Y, Ishigami M, Sakai N, Kameda-Takemura K, Matsuzawa Y. 1997. Genetic cholesteryl ester transfer protein deficiency is extremely frequent in the Omagari area of Japan. Marked hyperalphalipoproteinemia caused by CETP gene mutation is not associated with longevity. Arterioscler Thromb Vasc Biol. 17(6):1053–1059. [DOI] [PubMed] [Google Scholar]
- Inazu A, Brown ML, Hesler CB, Agellon LB, Koizumi J, Takata K, Maruhama Y, Mabuchi H, Tall AR. 1990. Increased high-density lipoprotein levels caused by a common cholesteryl-ester transfer protein gene mutation. New Engl J Med. 323(18):1234–1238. [DOI] [PubMed] [Google Scholar]
- Ito A, Hong C, Rong X, Zhu X, Tarling EJ, Hedde PN, Gratton E, Parks J, Tontonoz P. 2015. LXRs link metabolism to inflammation through Abca1-dependent regulation of membrane composition and TLR signaling. eLife. 4:e08009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahangiri A, de Beer MC, Noffsinger V, Tannock LR, Ramaiah C, Webb NR, van der Westhuyzen DR, de Beer FC. 2009. HDL remodeling during the acute phase response. ATVB. 29(2):261–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefs T, Wouters K, Tietge UJF, Annema W, Dullaart RPF, Vaisar T, Arts ICW, van der Kallen CJH, Stehouwer CDA, Schalkwijk CG, et al. 2020. High-density lipoprotein cholesterol efflux capacity is not associated with atherosclerosis and prevalence of cardiovascular outcome: the CODAM study. J Clin Lipidol. 14(1):122–132 e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph SB, Castrillo A, Laffitte BA, Mangelsdorf DJ, Tontonoz P. 2003. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat Med. 9(2):213–219. [DOI] [PubMed] [Google Scholar]
- Kappus MS, Murphy AJ, Abramowicz S, Ntonga V, Welch CL, Tall AR, Westerterp M. 2014. Activation of liver X receptor decreases atherosclerosis in Ldlr(−)/(−) mice in the absence of ATP-binding cassette transporters A1 and G1 in myeloid cells. Arterioscler Thromb Vasc Biol. 34(2):279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MA, Barrera GC, Nakamura K, Baldan A, Tarr P, Fishbein MC, Frank J, Francone OL, Edwards PA. 2005. ABCG1 has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulation. Cell Metab. 1(2):121–131. [DOI] [PubMed] [Google Scholar]
- Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, et al. 2011. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. 364(2):127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khera AV, Demler OV, Adelman SJ, Collins HL, Glynn RJ, Ridker PM, Rader DJ, Mora S. 2017. Cholesterol efflux capacity, high-density lipoprotein particle number, and incident cardiovascular events: an analysis from the JUPITER trial (justification for the use of statins in prevention: an intervention trial evaluating rosuvastatin). Circulation. 135(25):2494–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khera AV, Rader DJ. 2013. Cholesterol efflux capacity: full steam ahead or a bump in the road? Arteriosclerosis, Thromb Vasc Biol. 33(7):1449–1451. [DOI] [PubMed] [Google Scholar]
- Kim K, Choi JH. 2018. Response by Kim and Choi to letter regarding article, “transcriptome analysis reveals nonfoamy rather than foamy plaque macrophages are proinflammatory in atherosclerotic murine models.” Circ Res. 123(11): e50. [DOI] [PubMed] [Google Scholar]
- Kim K, Shim D, Lee JS, Zaitsev K, Williams JW, Kim KW, Jang MY, Seok Jang H, Yun TJ, Lee SH, et al. 2018. Transcriptome analysis reveals nonfoamy rather than foamy plaque macrophages are proinflammatory in atherosclerotic murine models. Circ Res. 123(10):1127–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labzin LI, Schmidt SV, Masters SL, Beyer M, Krebs W, Klee K, Stahl R, Lutjohann D, Schultze JL, Latz E, et al. 2015. ATF3 Is a key regulator of macrophage IFN responses. J Immunol. 195(9):4446–4455. [DOI] [PubMed] [Google Scholar]
- Li XM, Tang WH, Mosior MK, Huang Y, Wu Y, Matter W, Gao V, Schmitt D, Didonato JA, Fisher EA, et al. 2013. Paradoxical association of enhanced cholesterol efflux with increased incident cardiovascular risks. Arterioscler Thromb Vasc Biol. 33(7):1696–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen CM, Varbo A, Tybjaerg-Hansen A, Frikke-Schmidt R, Nordestgaard BG. 2018. U-shaped relationship of HDL and risk of infectious disease: two prospective population-based cohort studies. Eur Heart J. 39(14):1181–1190. [DOI] [PubMed] [Google Scholar]
- Martel C, Li W, Fulp B, Platt AM, Gautier EL, Westerterp M, Bittman R, Tall AR, Chen SH, Thomas MJ, et al. 2013. Lymphatic vasculature mediates macrophage reverse cholesterol transport in mice. J Clin Investig. 123(4):1571–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masucci-Magoulas L, Moulin P, Jiang XC, Richardson H, Walsh A, Breslow JL, Tall A. 1995. Decreased cholesteryl ester transfer protein (CETP) mRNA and protein and increased high density lipoprotein following lipopolysaccharide administration in human CETP transgenic mice. J Clin Investig. 95(4):1587–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura F, Wang N, Chen W, Jiang XC, Tall AR. 2006. HDL from CETP-deficient subjects shows enhanced ability to promote cholesterol efflux from macrophages in an apoE-and ABCG1-dependent pathway. J Clin Investig. 116(5): 1435–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitroulis I, Ruppova K, Wang B, Chen LS, Grzybek M, Grinenko T, Eugster A, Troullinaki M, Palladini A, Kourtzelis I, et al. 2018. Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell. 172(1–2): 147–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monette JS, Hutchins PM, Ronsein GE, Wimberger J, Irwin AD, Tang C, Sara JD, Shao B, Vaisar T, Lerman A, et al. 2016. Patients with coronary endothelial dysfunction have impaired cholesterol efflux capacity and reduced hdl particle concentration. Circ Res. 119(1):83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy AJ, Akhtari M, Tolani S, Pagler T, Bijl N, Kuo CL, Wang M, Sanson M, Abramowicz S, Welch C, et al. 2011. ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J Clin Investig. 121(10):4138–4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Out R, Hoekstra M, Habets K, Meurs I, de Waard V, Hildebrand RB, Wang Y, Chimini G, Kuiper J, Van Berkel TJ, et al. 2008. Combined deletion of macrophage ABCA1 and ABCG1 leads to massive lipid accumulation in tissue macrophages and distinct atherosclerosis at relatively low plasma cholesterol levels. ATVB. 28(2):258–264. [DOI] [PubMed] [Google Scholar]
- Lewington S, Whitlock G, Clarke R, Sherliker P, Emberson J, Halsey J, Qizilbash N, Peto R, Collins R. 2007. Blood cholesterol and vascular mortality by age, sex, and blood pressure: a meta-analysis of individual data from 61 prospective studies with 55,000 vascular deaths. Lancet. 370(9602):1829–1839. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM Jr, Kastelein JJ, Koenig W, Libby P, Lorenzatti AJ, MacFadyen JG, et al. 2008. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 359(21):2195–2207. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, et al. 2017. Antiinflammatory therapy with canakinumab for atherosclerotic disease. New Engl J Med. 377(12): 1119–1131. [DOI] [PubMed] [Google Scholar]
- Ridker PM, MacFadyen JG, Thuren T, Libby P. 2020. Residual inflammatory risk associated with interleukin-18 and inter-leukin-6 after successful interleukin-1beta inhibition with canakinumab: further rationale for the development of targeted anti-cytokine therapies for the treatment of atherothrombosis. Eur Heart J. 41(23):2153–2163. [DOI] [PubMed] [Google Scholar]
- Rohatgi A 2019. Reverse cholesterol transport and atherosclerosis. Arterioscler Thromb Vasc Biol. 39(1):2–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi A, Khera A, Berry JD, Givens EG, Ayers CR, Wedin KE, Neeland IJ, Yuhanna IS, Rader DR, de Lemos JA, et al. 2014. HDL cholesterol efflux capacity and incident cardiovascular events. New Engl J Med. 371(25):2383–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi A, Westerterp M, Von Eckardstein A, Remaley AT, Rye KA. 2021. HDL in the 21st century: a multifunctional roadmap for future HDL research. Circulation. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosales C, Gillard BK, Xu B, Gotto AM Jr, Pownall HJ. 2019. Revisiting reverse cholesterol transport in the context of high-density lipoprotein free cholesterol bioavailability. Methodist Debakey Cardiovasc J. 15(1):47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleheen D, Scott R, Javad S, Zhao W, Rodrigues A, Picataggi A, Lukmanova D, Mucksavage ML, Luben R, Billheimer J, et al. 2015. Association of HDL cholesterol efflux capacity with incident coronary heart disease events: a prospective case-control study. Lancet Diabetes Endocrinol. 3(7): 507–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer EJ, Santos RD, Asztalos BF. 2010. Marked HDL deficiency and premature coronary heart disease. Curr Opin Lipidol. 21(4):289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shea S, Stein JH, Jorgensen NW, McClelland RL, Tascau L, Shrager S, Heinecke JW, Yvan-Charvet L, Tall AR. 2019. Cholesterol mass efflux capacity, incident cardiovascular disease, and progression of carotid plaque. ATVB. 39(1): 89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K, Chandra A, Sperry T, Joshi PH, Khera A, Virani SS, Ballantyne CM, Otvos JD, Dullaart RPF, Gruppen EG, et al. 2020. Associations between high-density lipoprotein particles and ischemic events by vascular domain, sex, and ethnicity: a pooled cohort analysis. Circulation. 142(7): 657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoak KA, Aloor JJ, Madenspacher J, Merrick BA, Collins JB, Zhu X, Cavigiolio G, Oda MN, Parks JS, Fessler MB. 2010. Myeloid differentiation primary response protein 88 couples reverse cholesterol transport to inflammation. Cell Metab. 11(6):493–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spann NJ, Garmire LX, McDonald JG, Myers DS, Milne SB, Shibata N, Reichart D, Fox JN, Shaked I, Heudobler D, et al. 2012. Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses. Cell. 151(1):138–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Pritchard DK, Becker L, Hoofnagle AN, Tanimura N, Bammler TK, Beyer RP, Bumgarner R, Vaisar T, de Beer MC, et al. 2010. High-density lipoprotein suppresses the type I interferon response, a family of potent antiviral immunoregulators, in macrophages challenged with lipopolysaccharide. Circulation. 122(19):1919–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tall AR, Yvan-Charvet L. 2015. Cholesterol, inflammation and innate immunity. Nat Rev Immunol. 15(2):104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tall AR, Rader DJ. 2018. Trials and tribulations of CETP inhibitors. Circ Res. 122(1):106–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tall AR, Westerterp M. 2019. Inflammasomes, neutrophil extracellular traps, and cholesterol. J Lipid Res. 60(4): 721–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thacker SG, Zarzour A, Chen Y, Alcicek MS, Freeman LA, Sviridov DO, Demosky SJ Jr, Remaley AT. 2016. High-density lipoprotein reduces inflammation from cholesterol crystals by inhibiting inflammasome activation. Immunology. 149(3):306–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DG, Doran AC, Fotakis P, Westerterp M, Antonson P, Jiang H, Jiang X-C, Gustafsson J-A, Tabas IA, Tall AR. 2018. LXR suppresses inflammatory chromatin and neutrophil migration through a cis-repressive activity. Cell Rep. 25(13):3774–3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DG, Wei Y, Tall AR. 2021. Lipid and metabolic syndrome traits in coronary artery disease: a mendelian randomization study. J Lipid Res. 62(100044):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trigatti BL, Hegele RA. 2016. Rare genetic variants and high-density lipoprotein: marching to a different drum. Arterioscler Thromb Vasc Biol. 36(6):e53–55. [DOI] [PubMed] [Google Scholar]
- Trinder M, Walley KR, Boyd JH, Brunham LR. 2020. Causal inference for genetically determined levels of high-density lipoprotein cholesterol and risk of infectious disease. Arterioscler Thromb Vasc Biol. 40(1):267–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinder M, Wang Y, Madsen CM, Ponomarev T, Bohunek L, Daisely BA, Julia Kong H, Blauw LL, Nordestgaard BG, Tybjaerg-Hansen A, et al. 2021. Inhibition of cholesteryl ester transfer protein preserves high-density lipoprotein cholesterol and improves survival in sepsis. Circulation. 143(9):921–934. [DOI] [PubMed] [Google Scholar]
- van der Velde AE, Vrins CL, van den Oever K, Kunne C, Oude Elferink RP, Kuipers F, Groen AK. 2007. Direct intestinal cholesterol secretion contributes significantly to total fecal neutral sterol excretion in mice. Gastroenterology. 133(3): 967–975. [DOI] [PubMed] [Google Scholar]
- van der Vorst EPC, Biessen EAL, Donners M. 2020. Letter by van der Vorst et al. regarding article, “anti-Inflammatory effects of HDL (high-density lipoprotein) in macrophages predominate over proinflammatory effects in atherosclerotic plaques”. Arterioscler Thromb Vasc Biol. 40(2): e31–e32. [DOI] [PubMed] [Google Scholar]
- van der Vorst EPC, Theodorou K, Wu Y, Hoeksema MA, Goossens P, Bursill CA, Aliyev T, Huitema LFA, Tas SW, Wolfs IMJ, et al. 2017. High-density lipoproteins exert proinflammatory effects on macrophages via passive cholesterol depletion and PKC-NF-kappaB/STAT1-IRF1 signaling. Cell Metab. 25(1):197–207. [DOI] [PubMed] [Google Scholar]
- Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, Jensen MK, Hindy G, Holm H, Ding EL, Johnson T, et al. 2012. Plasma HDL cholesterol and risk of myocardial infarction: a Mendelian randomisation study. Lancet. 380(9841):572–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N, Lan D, Chen W, Matsuura F, Tall AR. 2004. ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. PNAS. 101(26): 9774–9779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N, Silver DL, Costet P, Tall AR. 2000. Specific binding of ApoA-I, enhanced cholesterol efflux, and altered plasma membrane morphology in cells expressing ABC1. J Biol Chem. 275(42):33053–33058. [DOI] [PubMed] [Google Scholar]
- Wang M, Subramanian M, Abramowicz S, Murphy AJ, Gonen A, Witztum J, Welch C, Tabas I, Westerterp M, Tall AR. 2014. Interleukin-3/granulocyte macrophage colony-stimulating factor receptor promotes stem cell expansion, monocytosis, and atheroma macrophage burden in mice with hematopoietic ApoE deficiency. Arterioscler Thromb Vasc Biol. 34(5):976–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerterp M, Bochem AE, Yvan-Charvet L, Murphy AJ, Wang N, Tall AR. 2014. ATP-binding cassette transporters, atherosclerosis, and inflammation. Circ Res. 114(1):157–170. [DOI] [PubMed] [Google Scholar]
- Westerterp M, Fotakis P, Ouimet M, Bochem AE, Zhang H, Molusky MM, Wang W, Abramowicz S, la Bastide-van Gemert S, Wang N, et al. 2018. Cholesterol efflux pathways suppress inflammasome activation, NETosis and atherogenesis. Circulation. 138(9):898–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerterp M, Gautier EL, Ganda A, Molusky MM, Wang W, Fotakis P, Wang N, Randolph GJ, D’Agati VD, Yvan-Charvet L, et al. 2017. Cholesterol accumulation in dendritic cells links the inflammasome to acquired immunity. Cell Metab. 25(6):1294–1304 e1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerterp M, Murphy AJ, Wang M, Pagler TA, Vengrenyuk Y, Kappus MS, Gorman DJ, Nagareddy PR, Zhu X, Abramowicz S, et al. 2013. Deficiency of ATP-binding cassette transporters A1 and G1 in macrophages increases inflammation and accelerates atherosclerosis in mice. Circ Res. 112(11):1456–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, McDonald JG, Patel A, Zhang Y, Umetani M, Xu F, Westover EJ, Covey DF, Mangelsdorf DJ, Cohen JC, et al. 2006. Sterol intermediates from cholesterol biosynthetic pathway as liver X receptor ligands. J Biol Chem. 281(38): 27816–27826. [DOI] [PubMed] [Google Scholar]
- Yvan-Charvet L, Kling J, Pagler T, Li H, Hubbard B, Fisher T, Sparrow CP, Taggart AK, Tall AR. 2010. Cholesterol efflux potential and antiinflammatory properties of high-density lipoprotein after treatment with niacin or anacetrapib. Arterioscler Thromb Vasc Biol. 30(7):1430–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yvan-Charvet L, Pagler T, Gautier EL, Avagyan S, Siry RL, Han S, Welch CL, Wang N, Randolph GJ, Snoeck HW, et al. 2010. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science. 328(5986): 1689–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yvan-Charvet L, Ranalletta M, Wang N, Han S, Terasaka N, Li R, Welch C, Tall AR. 2007. Combined deficiency of ABCA1 and ABCG1 promotes foam cell accumulation and accelerates atherosclerosis in mice. J Clin Investig. 117(12): 3900–3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yvan-Charvet L, Swirski FK. 2018. Is defective cholesterol efflux an integral inflammatory component in myelopoiesis-driven cardiovascular diseases? Eur Heart J. 39(23): 2168–2171. [DOI] [PubMed] [Google Scholar]
- Yvan-Charvet L, Welch C, Pagler TA, Ranalletta M, Lamkanfi M, Han S, Ishibashi M, Li R, Wang N, Tall AR. 2008. Increased inflammatory gene expression in ABC transporter-deficient macrophages: free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation. 118(18):1837–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanoni P, Khetarpal SA, Larach DB, Hancock-Cerutti WF, Millar JS, Cuchel M, DerOhannessian S, Kontush A, Surendran P, Saleheen D, et al. 2016. Rare variant in scavenger receptor BI raises HDL cholesterol and increases risk of coronary heart disease. Science. 351(6278):1166–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Lee JY, Timmins JM, Brown JM, Boudyguina E, Mulya A, Gebre AK, Willingham MC, Hiltbold EM, Mishra N, et al. 2008. Increased cellular free cholesterol in macrophage-specific Abca1 knock-out mice enhances pro-inflammatory response of macrophages. JBiolChem.283(34): 22930–22941. [DOI] [PMC free article] [PubMed] [Google Scholar]