

Abstract
While much effort has been placed on comprehensive quantitative proteome analysis, certain applications demand measurement of only a few target proteins from complex systems. Traditional approaches to targeted proteomics rely on nano-liquid chromatography (nLC) and targeted mass spectrometry (MS) methods, e.g., parallel reaction monitoring (PRM). However, the time requirement for nLC can limit throughput of targeted proteomics. To achieve rapid and high-throughput targeted methods, here we show that nLC separations can be eliminated and replaced with direct infusion shotgun proteome analysis (DISPA) using high-field asymmetric waveform ion mobility spectrometry (FAIMS) with PRM. We demonstrate application of DISPA-PRM for rapid targeted quantification of bacterial enzymes utilized in the production of biofuels by monitoring temporal expression in 72 metabolically engineered bacterial cultures in under 2.5 hours, with a measured dynamic range >1200-fold. We conclude that DISPA-PRM presents a valuable innovative tool with results comparable to nLC-MS/MS, enabling fast and rapid detection of targeted proteins in complex mixtures.
Keywords: nLC-MS/MS, FAIMS, PRM, DISPA, biofuels, targeted proteomics, absolute quantitation, metabolic engineering
Graphical Abstrct

To produce metabolites of interest, metabolic engineering strategies enhance specific metabolic routes by modulating the expression of native enzymes or by introducing entirely new pathways within a host. Broadly described as bioproducts, there is increasing global demand for microbially-derived metabolites from renewable biomass. A general challenge for metabolically engineered microorganisms, is the inability to produce a quantity of bioproduct to be industrially competitive (1,2). To optimize metabolic flux towards the production of bioproducts, high-throughput proteomics technologies can be used to efficiently interrogate protein expression levels in engineered pathways of interest.
Due to humanity’s reliance on fossil fuels and the consequential climate change, there is an urgent need to shift toward renewable biofuels (3–6). The microorganism Zymomonas mobilis is a facultatively anaerobic α-proteobacterium that is increasingly popular in metabolic engineering applications and has shown promise for isoprenoid-based biofuel production (7–9). Isoprenoids, also known as terpenoids, are similar in structure to fossil fuels as they are composed of hydrocarbon-unsaturated alcohols with varying chain lengths. Specifically, the C5 class of isoprenoids provide desirable energy densities and octane numbers comparable to gasoline (10). To enhance the production of isoprenoids in Z. mobilis, metabolic engineering of the methylerythritol phosphate (MEP) pathway is a growing area of research.
The MEP pathway is endogenous to Z. mobilis and theoretically can generate a higher yield of isoprenoids from isopentenyl diphosphate precursors than alternative pathways found in other organisms (11). However, there are significant challenges associated with MEP pathway engineering; in particular, the deleterious effect of oxygen on specific enzymes in the pathway (7,12). Oxygen exposure induces severe but temporary metabolic bottlenecks by oxidatively damaging the final two enzymes (IspG and IspH) in the MEP pathway. Thus, monitoring the protein levels of IspG and IspH is critical for improving isoprenoid production in Z. mobilis.
Metabolic pathway manipulation in Z. mobilis requires quantitation of enzyme abundance. Traditionally, antibodies are used to perform semi-quantitative assessment via Western blotting. Aside from being cumbersome and low throughput, this strategy requires the generation of a target-specific antibody. Instead, protein quantitation via nanoflow-liquid chromatography coupled to tandem mass spectrometry (nLC-MS/MS) circumvents the need for antibodies and can be used to monitor multiple proteins in the same analysis (13). However, the nLC component demands tens of minutes to hours per sample and thus, is not wholly ideal for the large multifactorial arrays of bacterial culture conditions and genetic perturbations that are vital optimization steps for metabolic engineering. Provided sufficient depth can be achieved, direct infusion of tryptic peptide mixtures can substantially increase throughput. (14–18). We recently described a new rapid quantitative direct infusion shotgun proteomic analysis (DISPA) technology that replaces traditional nLC separation with gas-phase separation by ion mobility (19). Here we further develop this concept for targeted proteomics using parallel reaction monitoring (PRM) – DISPA-PRM for rapid monitoring of pathway bioengineering in Z. mobilis. As a case study, we follow overexpression of the final two enzymes in the MEP pathway, IspG and IspH. DISPA-PRM requires less than one minute of analysis per sample, and the quantitative results show excellent agreement with traditional nLC-MS/MS quantification. We further find that spectra generated by DISPA-PRM can be identified using theoretically generated Prosit predictions, abrogating the need for initial nLC-MS/MS analysis to build spectral libraries. We explore how the results are influenced by the inclusion of FAIMS and apply the DISPA-PRM method for high throughput quantification of temporal IspH and IspG overexpression. We expect DISPA-PRM technology to find widespread utility in a diverse array of targeted protein monitoring applications.
Materials and Methods
The generation of the plasmid construct, the strains, and the growth conditions are described in the Supporting Information. For the MS sample preparation, optical density at a wavelength of 600 nm (OD600) was used to estimate the density of ZM4 strain cells for each experiment. We estimated the number of cells in each sample by multiplying the OD600 by 10 (mL) and 8E+08 (cells/mL) where in E. coli cell culture OD600 of 1.0 = 8E+08 cells per milliliter. The protein concentration was calculated by multiply the number of cells by 0.2 (picogram), the estimated amount of protein found in a single bacterial cell. Estimating protein concentration ensures that the appropriate volumes of buffer to create comparative digestions conditions across longitudinal samples. The samples were prepared for tryptic digestion and 45-min nLC-MS/MS, conditions are summarized in the Supporting Information.
Peptide standards.
Two AQUA QuantPro peptides were purchased (Thermo Scientific) and customized with stable isotope-labeled C-terminus amino acids: lysine [13C(6) and 15N(2)] and arginine [13C(6) and 15N(4)]. The standard peptides for ETDIGVTGGGQGK and AIEIVDQALDR are proxies for proteins IspG and IspH, respectively. The heavy standards were spiked into a ZM4 tryptic digest matrix across a 12-fold dilution curve (15.625, 31.25, 62.5, 125, 250, 500 fmol/uL). Three technical injections from each of the concentrations were analyzed by DISPA-PRM.
DISPA-PRM method.
The ZM4 tryptic digest was resuspended in 50% ACN 0.2% FA to a final concentration of 0.500 μg/μL. One μL (500 ng) was loaded onto a mostly empty 30-μm-inside-diameter (i.d.) 15-cm-long capillary shell with an imbedded electrospray emitter. The column tip was lightly packed with a 2 mm plug of 5-μm-particle-size C5 beads. An isocratic flow consisting of 50% ACN in 0.2% FA was maintained during the 2-minute DISPA-PRM, changing only in flow rate as follows: for the first 48.6 seconds a flow rate of 1.50 μL/min was maintained then quickly dropped to 0.30 μL/min for one-minute. The NCP-3200RS unit was used to control the flow rate and isocratic mobile phase composition. The WPS-300-(RS) autosampler loaded the samples onto the infusion column.
The tryptic mixture of peptides was analyzed on an Orbitrap Eclipse (Thermo Fischer Scientific) mass spectrometer. Survey scans were performed at a resolution of 240,000 with an isolation analysis at m/z 300 to 1,350 and 250% normalized automatic gain control (AGC) target. Data-dependent top-speed (1-s) tandem MS (MS/MS) sampling of peptide precursors was enabled with dynamic exclusion set to 10 s for precursors with charge states 2 to 5. Data dependent MS/MS sampling was performed with 0.5 m/z quadrupole isolation, fragmentation by higher-energy collisional dissociation (HCD) with a normal collisional energy (NCE) value of 300%. The mass analysis was performed in the ion trap using the “turbo” scan speed for a mass range of 150–1350 m/z with a maximum inject time of 14 ms, and the normalized AGC target set to 300%.
The FAIMS Pro system was set to Standard Resolution mode and placed between the nano electrospray source and mass spectrometer. The FAIMS-enabled experiment used carrier N2 gas flow at 5 L/min, asymmetric wave form dispersion voltage at −5000 V, and the inner and outer electrode temperature set at 100 °C.
A FAIMS nLC-MS/MS method on the Orbitrap Fusion Lumos MS was designed to determine the optimal compensation voltages (CVs). The acquisition of five scan events were synchronized with an internal stepping of FAIMS compensation voltage (−30, −40, −50, −60, and 70 CV). Each scan event was triggered by m/z from an inclusion list for three IspG peptides and three IspH peptides. Cycling through the CVs, each peptide was isolated using a 2.0 m/z isolation window and subsequently fragmented by HCD at a collision energy of 30%. The mass with a scan range 200–1220 m/z, a maximum injection of 200 ms, and an AGC of 3.0E+04. Supplementary Table 2 summarizes the selected CVs providing the best transmission of signal for the analyte.
Qualitative annotation of target peptides.
The qualitative assessment of targeted peptides from various methods was performed in Skyline-Daily. Raw files were converted to mzML files using the MsConverGUI software prior to processing and searching. The following transition parameters were used: filtering for top 10 transitions (b-ions, y-ions, and precursor), MS/MS mass accuracy of 10 ppm, and product mass analyzer set to centroid. Peptide settings included structural modifications for carbamidomethyl of cysteine, oxidation of methionine, and acetylation and carboxymethylation of the N-terminus. Isotope modifications were manually constructed for stable isotope-labeled lysine [13C(6) and 15N(2)] and arginine [13C(6) and 15N(4)] at the C-terminus. Three reference spectral libraries were constructed for extracting quantitative information of the target peptides. Two of these spectral libraries were generated by the MaxQuant search; one analysis collected with high-resolution MS/MS and the other analysis collected with low-resolution MS/MS. The third library was created from the in-silico fragmentation algorithm Prosit.
Quantitative analysis of target peptides.
The quantification of target peptides was enabled by C# mass spectrometry library, CSMSL. Peptides were constructed in-silico using amino acid sequences unique to IspH and IspG. The intensity of all b- and y-type fragments ions for each target peptide were quantified in each experiment using a ppm mass error of ±10–20 ppm was used. The source code of these scripts has been deposited on GitHub at the following web address: https://github.com/anjitrue/GenerateTheoreticalPeptideFragment_DISPA_PRM.
The spectral purity was determined by first identifying the top ten theoretical y- and b-type ions as determined by Protein Prospector (https://prospector.ucsf.edu/prospector/mshome.htm) for each target peptide. These fragment ions were then searched across the MS/MS spectra and highlighted in red, the remaining ions are considered background. Spectral purity was calculated by determining the ratio of the summed intensity of the top ten ions relative to the background. Next, dot product was generated by previously discussed scoring method, the dot product, to look at spectral similarity as a metric for comparing between spectra. The dot product measures the cosine of the angles between the spectra that are being compared, utilizing the top ten representative ions. Ions in two spectra are aligned and matched within a 10-ppm ion tolerance window. If there are multiple candidate ions within the specified range, the highest value observed is used.
Results and Discussion
Benchmarking the over-expression system with conventional nLC-MS/MS.
To globally assess the ZM4 proteome, we characterized the overexpression systems by shotgun capillary nLC-MS/MS (20). Altogether each single-shot analysis detected 1,523 proteins, covering 85% percent of the ZM4 protein coding genes. With excellent proteome coverage in hand, we then determined which tryptic peptides were most readily identified in these 45-minute shotgun MS analyses and assessed the abundances of IspG and IspH, and their associated peptides. Overall, 38 unique peptides were identified for IspG, representing 89.9% sequence coverage. In contrast, we obtained 58.8% sequence coverage or 22 unique tryptic peptides for IspH (Supplementary Figure 1b). Using the shotgun MS method and MaxQuant for peptide identification by database searching and label free quantitation (LFQ), protein IspG was ~20-fold more abundant in the over-expressed strain than in the WT strain (biological replicate n=3).; comparatively the IspH protein was ~5-fold over-expressed (Supplementary Figure 1a).
Using these data, we next aimed to develop a targeted DISPA method (i.e., DISPA-PRM) that would measure the overexpression of IspG and IspH proteins from representative peptides. Target peptides that could provide robust and reproducible quantification were selected from a pool of peptides mapped to IspG and IspH proteins. Peptide selection was based on the following criteria: 1) reproducible detection across replicates, 2) high quality MS/MS in every replicate, and 3) high signal-to-noise ratio 4) no missed-cleavages. Over half (55%) of the total unique peptides for the IspG protein were reproducibly identified by data-dependent acquisition MS/MS in all replicates. The average normalized LFQ intensities for IspG peptides ranked among the top 8% most abundant among all detected peptides. For the most reproducible IspG peptides, the median coefficient of variation (CV) was 28%, the spread ranged from as low 2% CV to as high as 140 %CV, representing greater variance than IspH peptides (Supplementary Figure 1c). For IspH peptides, 63% triggered an MS/MS in each replicate. The IspH peptides had relatively more consistent intensities with LFQ intensities among the top 12% of all detected peptides. This increased consistency in signal also generated less variability as evinced by the median CV of 10% (Supplementary Figure 1c).
To further evaluate these data, we next ascertained the variance in the quantification between peptides and proteins (21,22). We calculated 95% confidence intervals (CI) around average fold changes (Supplementary Figure 1d) and statistically defined how the fold change of peptides differ from the collated parent protein. The majority of IspG peptides fell outside of the 95% CI fold-change (20- to 25-fold), highlight the high degree of variability in the measured intensity across biological replicates. Note, that this variability did not arise from one specific replicate (Supplemental Figure 1e). Furthermore, when we compare the dynamic range of IspG and IspH respective peptide abundances, we observe that the peptides that originate from IspH have tighter confidence interval bars - the 95% CI of IspH protein fold-change (4.9- to 6-fold).
Development of DISPA-PRM method for quantifying proxy peptides.
To develop the rapid approach, we conducted label-free, DISPA analysis targeting these six peptides from ZM4 tryptic digests. The strategy, DISPA, which we recently described, directly infuses tryptic peptides followed by gas-phase separation and detection by DIA with high resolution MS/MS (19). Here, we explore the utility of DISPA in combination with PRM to analyze targeted peptides at the MS/MS level (23,24). The entirety of our DISPA-PRM experiment takes two minutes, with the first half allocated to the autosampler picking up the sample, filling the loop, and transferring the sample to the infusion emitter. The second half occupies the collection of high resolution PRM MS/MS scans. In PRM, a mass-selected target precursor is isolated, fragmented using beam-type collision-activated dissociation (i.e., HCD) producing b- and y-type product ions, and monitored using high-resolution Orbitrap mass analysis. We hypothesize that the DISPA-PRM method is robust enough to monitor the target peptides from the infusion of the complex tryptic digest of ZM4, providing quantitative measurements on the respective b- and y-type fragments. These fragments can be monitored, summed from all the scans collected over the one-minute, and utilized to calculate fold changes with respect to those measured in the WT strain.
Figure 1a plots the intensities of product ions from the six target peptides following analysis with DISPA-PRM. Similar trends in the relative abundances of the peptides were observed between DISPA-PRM and those collected from nLC-MS/MS analyses. For example, the relative intensities obtained using nLC-MS/MS for the three IspG peptides were greater than the IspH peptides (4:1). This trend is closely mirrored in the DISPA-PRM data: on average the IspG peptides were ~7 times more abundant than the IspH peptides. Subsequently, we identified the transitions for each target peptide that were most abundant and free of interference. For peptide ETDIGVTGGGQGK, a total of 22 b- and y-type fragment ions were detected. Figure 1b plots the extracted ion intensities for the top ten most abundant ETDIGVTGGGQGK transition ions with the most representative, high-mass fragment ions – y7+, y8+, and y9+ - emboldened with color.
Figure 1. Qualitative comparison of DISPA-PRM signals for peptides from IspG and IspH proteins.
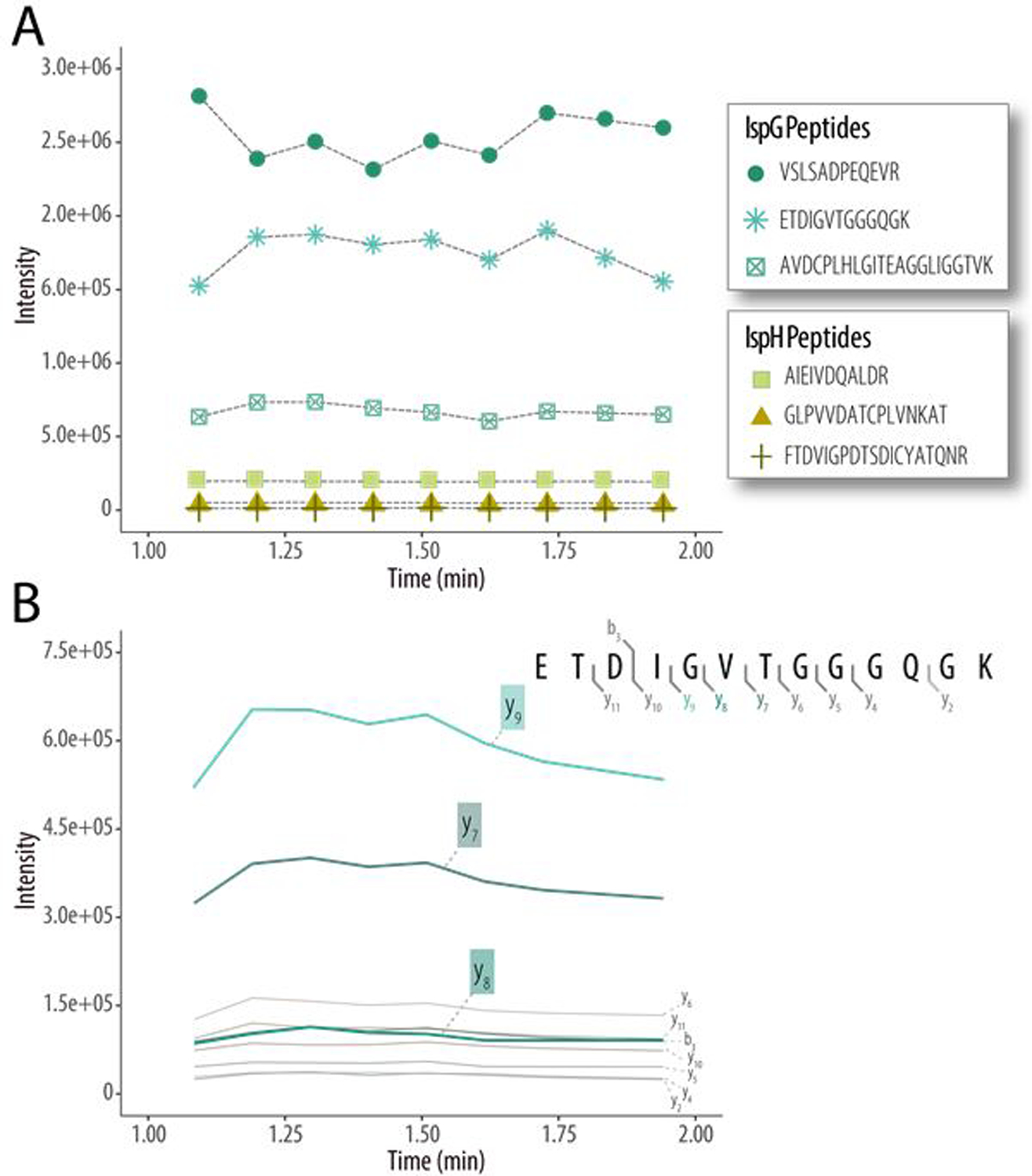
(A) DISPA-PRM quantitative outputs for six target peptides analyzed during a one-minute acquisition. (B) Further evaluation of the proxy ISPG peptide, ETDIGVTGGGQGK, reveals the top ten fragment ions and their intensities across the one-minute analysis. Of the ten most intense fragment ions, the transitions y7+, y8+, and y9+ were used for quantitation of the peptide.
To obtain the best performance of DISPA-PRM, we evaluated several parameters including isolation width, MS/MS max injection time, and mass resolution (Figure 2a). Using a systematic and combinatorial approach, a total of 45 combinations of MS parameters were compared with and without FAIMS, resulting in 270 DISPA-PRM experiments. Note, the FAIMS CVs were set for internal stepping and were determined via nLC-FAIMS-MS/MS (Supplementary Table 1).
Figure 2. Overview of DISPA-PRM method and Systematic Optimization of MS Parameters.

(A) Scheme showing the DISPA-PRM method, and the MS parameters studied: quadrupole isolation width, MS/MS max injection time, and Orbitrap resolving power. Note the red line indicates the optimal parameters that were determined here and utilized for data collection. Heatmaps summarize the coverage and intensity of transition ions for all max injection time and mass resolving powers but with a fixed isolation width of 0.6 m/z for the peptide ETDIGVTGGGQGK (B) and AIEIVDQALDR (D). The intensities are Log2 normalized and represent the average of three technical injection replicates. The raw intensities of each transition ion y7+, y8+, and y9+ for the peptide ETDIGVTGGGQGK (C) and AIEIVDQALDR (E) are plotted. The number of MS/MS scans are appended showcasing how summed intensity decreases with a reduction of scans.
To summarize the data generated, we present examples from the target peptides having the sequence ETDIGVTGGGQGK (Figure 2b) and AIEIVDQALDR (Figure 2d). The data presented in Figure 2 exemplify results for peptides targeted with an isolation width of 0.6 m/z and the heatmaps visualize the summed Log2 intensities for each transition ion using resolutions 120,000, 240,000, and 500,000 @ 200 m/z and max injection times of 502 ms, 1.0 s, and 5.0 s. Note, that the summed Log2 intensities draw the signal for each transition and add them together from each scan acquired during the 60 second DISPA-PRM experiment. Additional heatmaps for isolation widths 0.4, 1.0, and 2.0 m/z for the peptides AIEIVDQALDR and ETDIGVTGGGQGK are found in Supplementary Figure 2. The heatmaps for the remaining four proxy peptides at isolation width 0.6 m/z are summarized in Supplementary Figure 3.
A total of 22 transitions for ETDIGVTGGGQGK and 18 transitions for AIEIVDQALDR were measured with DISPA-PRM across these conditions (Figure 2b–d). From these data we concluded to proceed with an isolation width of 0.6 m/z, as it was neither too narrow which leads to loss of sensitivity nor too wide, resulting in loss of specificity through co-isolation and co-fragmentation which is readily observed especially in the low-mass b- and y-type ions. Next, we examined injection times and mass resolving power and their impact on sensitivity and selectivity. Figure 2b–d demonstrates the effect of increasing selectivity on sensitivity: the summed intensities generally decreased with increased resolution. Notably, summed intensities also decreased when max injection times increased. Taking a closer look, all proxy peptides required greater than 1 second to reach the set AGC target of 106 ions, specifically, for peptides ETDIGVTGGGQGK and AIEIVDQALDR the average ion injection times needed to reach a 106 target were 1.6 s and 2.4 s, respectively. Increasing the max injection to 502 ms or 1.0 s or even the highest possible setting of 5.0 s would mean the isolation, collection, and fragmentation events could slow scan times to well over 2.0 s. For these peptides, longer injection times resulted in fewer MS/MS scans collected. Figure 2c–e plot the raw summed intensities for transition ions y7+, y8+, and y9+ with the number of MS/MS scans appended for peptide ETDIGVTGGGQGK (Figure 2c) and AIEIVDQALDR (Figure 2e). The data confirm that when the max injection time increased and the number of DISPA-PRM scans is reduced, as a result the summed signal drops because fewer scans are recorded. Similarly, with mass resolving powers higher than 240,000 are used, the number of DISPA-PRM scans drops. However, there was minimal change in total number of scans between Orbitrap resolution 120,000 to 240,000. From these data we selected a 502 ms max ion injection time and 240,000 resolving power.
To evaluate the addition of FAIMS to DISPA-PRM, spectral quality was compared for experiments collected at 0.6 m/z and 2.0 m/z isolation widths, respectively (Supplementary Figure 4 and 5). Integrating FAIMS with DISPA-PRM reduced co-isolation of contaminants and improved spectral quality.
Comparing spectral similarity and total ion chromatogram (TIC) explained.
The implementation of DISPA-PRM requires a data analysis component for identifying peptides from raw MS/MS data. To identify the peptides of interest, spectral libraries were generated in Skyline. Although Skyline software is commonly used for targeted MS data, DISPA-PRM challenges traditional Skyline analysis approaches. Skyline peak picking algorithms analyze ions that are selected and extracted from chromatograms, integrating these chromatogram peaks for visualizations. However, DISPA-PRM data does not result from a chromatographic peak, instead MS/MS data is collected from the direct infusion or steady signal of tryptic peptides. Although DISPA-PRM data poses informatic challenges, several Skyline tools and data analysis approaches were employed, for example, the access to Prosit-enabled predicted spectral libraries, the ability to build libraries from data collected by DDA, and the motivation to calculate dot product scores for determining spectral similarities (25–27).
Traditionally, a spectral library is generated from nLC-MS/MS data, however, with the advent of in silico models like Prosit, spectral libraries can be generated to contain predicted fragments and their intensities based on the specific MS/MS settings used. We generated three spectral libraries a high and low-resolution nLC-MS/MS and a theoretical Prosit library, identifying the top 10 most intense transitions highlighted in red (Supplementary Figure 4). For both the peptide AIEIVDQALDR (Supplementary Figure 4a) and ETDIGVTGGGQGK (Supplementary Figure 4b), the Prosit spectra revealed a match to the top 10 transitions and near identical relative product ion intensities as measured by nLC-MS/MS. These data confirm that in silico generated libraries perform well and can eliminate the time and resources required to generate spectral libraries.
To systematically compare the libraries, the dot product was calculated between every possible combination including MS/MS spectra collected with DISPA-PRM (28). Only the top 10 most intense ions were used to calculate the dot product rather than using all available ions collected in the spectrum. This approach known as the projected spectrum concept is extremely relevant for MS/MS spectra where there can be co-isolation of contaminant ions (29). Ranking the fragment ions by intensity and using the top 10 most intense ions alleviates the dot product score from being influenced by contaminants. Supplementary Figure 4a–b shows a heatmap for each dot product, illustrating exceptionally strong agreement between predicted, nLC-MS/MS, and DISPA-PRM collected data confirming the advantage and convenience of using the Skyline Prosit spectral library tool.
Next, the percent total ion chromatogram (i.e., spectral purity) was calculated to describe how much signal is associated with the target peptide transition ion in comparison to the total signal collected in a spectrum. We compared the spectral purity of nLC-MS/MS to DISPA-PRM with and without the FAIMS. As expected, DDA LC-MS/MS spectra had the highest purity scores – i.e., 65.5% and 38.1% of the product ions were explained by the top 10 transition ions for AIEIVDQALDR and ETDIGVTGGGQGK, respectively (Supplementary Figure 4a–b). An advantage of FAIMS coupled to MS is a purer spectrum due to filtering imparted by optimized CV values. The targeted analyzes of AIEIVDQALDR and ETDIGVTGGGQGK by DISPA was more than sufficient to measure and identify the top 10 most intense ions. The FAIMS device improved the purity of the spectrum by greater than 25%. The targeted FAIMS-DISPA method resulted in almost twice as much of the TIC explained by the top 10 transitions, as compared to analysis without the FAIMS unit. We found that the improvement to the spectral purity of DISPA MS/MS data was more pronounced as the isolation width widened and the spectrum was more complex. At the widest isolation width of 2 m/z the spectral purity increased by ~300% for low abundance peptide AIEIVDQALDR and ~200% for overexpressed peptide ETDIGVTGGGQGK (Supplementary Figure 5). Additionally, the dot product score improved for the data collected with the FAIMS device. Altogether, the more complex the spectrum, the greater the benefit of using FAIMS for improving spectral purity and dot product scores.
Investigating the limit of detection and dynamic range of DISPA-PRM.
To investigate the dynamic range of DISPA-PRM we prepared a dilution curve by spiking in heavy labeled peptide standards across a 12-fold dilution into a background matrix of ZM4 tryptic digest. Figure 3 presents the calibration curves generated by comparing the heavy to light ratio for these two peptide standards ETDIGVTGGGQGK and AIEIVDQALDR. The linearity of the curves in Figure 3 showcases the dynamic range accessible when using selection of ions with the ability quantify heavy to light ratios that spans at least three orders of magnitude or ~1200-fold.
Figure 3. DISPA-PRM limit of detection for various transition ions.

DISPA-PRM (w/FAIMS) was implemented on a series of ZM4 digests spiked with a dilution curve of heavy standard peptides for ETDIGVTGGGQGK and AIEIVDQALDR. A response curve assesses the limit of detection across a 12-fold dilution of the spiked standard and helps to estimate dynamic range and method sensitivity. Three technical replicates for each concentration point were generated. The area ratio for heavy (A) ETDIGVTGGGQGK or (B) AIEIVDQALDR to native peptide was determined using either all or extracted transitions, i.e., y9+, y8+, and y7+ transitions respectively for, and plotted versus the spiked-in standard concentration.
To mitigate interference of low-mass transition ions, we performed quantification with y7+, y8+, and y9+ in both the stable isotope-labeled and native peptide for ETDIGVTGGGQGK (Figure 3a) and AIEIVDQALDR (Figure 3b). We observed a significant increase in sensitivity when the heavy to light ratios were calculated with a subset of ions (y7+, y8+, and y9+) versus all. This was particularly emphasized for peptide AIEIVDQALDR; using all the b- and y-type transition ions causes the overall signal of the native peptide transition ions to dampen the heavy to light ratio. The additional interreference, i.e., contamination of low-mass b2+ and b3+, was previously seen in Figure 3b–d, and becomes extremely relevant when using standard peptides. The use of a subset of interference-free transition ions y7+, y8+, and y9+ either individually or as a group, results in a linear relationship between the heavy to light ratios and concentrations. In contrast for the peptide ETDIGVTGGGQGK, there is linearity across all options that include: use of all b- and y-type fragment ions, the subset y7+, y8+, and y9+, and the individual y7+, y8+, and y9+. Regardless of the linearity, there is still evidence of contamination from interfering ions, at the lowest concentrations.
Longitudinal monitoring of IspG and IspH over-expression.
The quantitative performance of DISPA-PRM for real-world applications was tested on a longitudinal study of three ZM4 strains engineered to over-express either IspG or IspH compared to a WT. The cells were induced with 0.5 mM IPTG at time point zero and samples were collected at 7.5, 15, 30, 45, 60, 120, and 180-minutes post induction. A total of eight time points, three ZM4 strains, and three biological replicates resulted in 72 samples (i.e., 8 × 3 × 3) for which IspG and IspH expression was monitored. Each ZM4 tryptic digest was spiked with heavy peptide standards at a concentration of 62.5 fmol/uL prior to analysis. In just over two hours of analysis time, we obtained the abundances of native and synthetic peptides using the y9+, y8+, and y7+ transitions for the target peptides AIEIVDQALDR (Figure 4a) and ETDIGVTGGGQGK (Figure 4b) across all the 72 samples. The ratio between the native (light) and synthetic (heavy) were calculated and plotted across time (Figure 4c–d), suggesting that the IspG protein had higher levels of overexpression as compared to IspH.
Figure 4. Fast DISPA-PRM method enables high throughput monitoring of IspG and IspH over-expression over time.
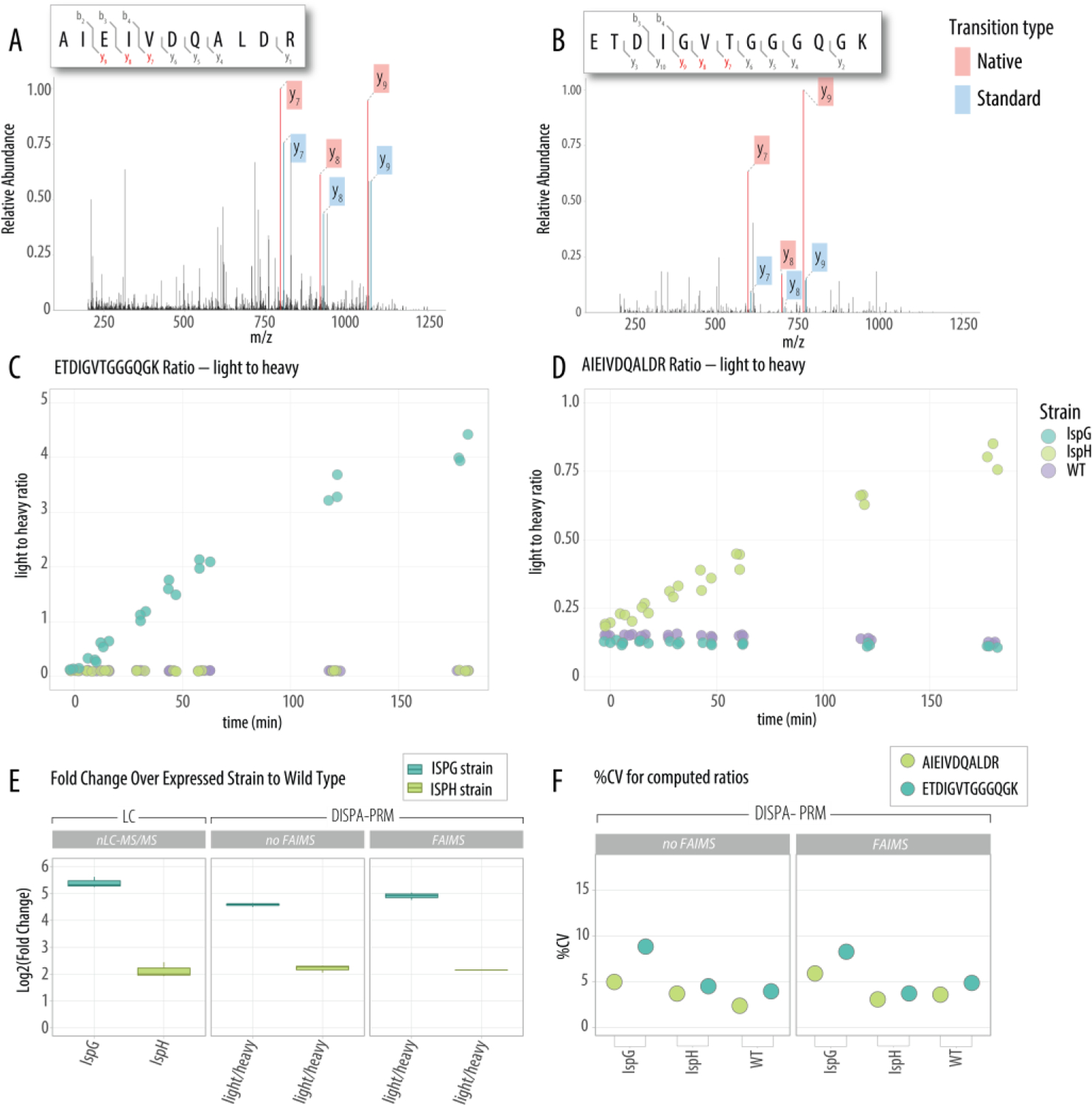
The monitoring and quantitation of over-expressed proteins IspG and IspH is applied to a longitudinal study. A high-resolution MS/MS spectrum snapshot from DISPA-PRM for the proxy peptide AIEIVDQALDR (A) and ETDIGVTGGGQGK (B) highlights the three representative light and heavy ions in red and blue, respectively. Light to heavy ratio for (C) AIEIVDQALDR from IspH and (D) ETDIGVTGGGQGK from IspG are plotted over a 180-minute induction of overexpression after addition of IPTG. (E) Fold change of the overexpression strains at time point 120 minutes compared to wild-type using the nLC-MS/MS and DISPA-PRM with or without FAIMS. (F) Percent coefficient variability for the heavy to light ratio of the proxy peptides measured by DISPA-PRM with and without FAIMS across three biological replicates.
To further evaluate the robustness and reproducibility of DISPA-PRM the fold change of IspG and IspH at time point 120 minutes was compared to measurements obtained by traditional shotgun nLC-MS/MS analysis. DISPA-PRM, with or without the FAIMS, produced remarkably similar quantitative results (IspG fold change of 5.10 and 2.21 for IspH, Figure 4e) as compared to shotgun nLC-MS/MS analysis (IspG fold-change 5.25 and 2.21 for IspH, Figure 4e). We then evaluated the relative variation of the heavy to light ratio by computing the % CV in each of the three strains. We observed an overall median CV of 6% across the DISPA-PRM methods, and improvement over that achieved by nLC-MS/MS (10%). From these data we conclude that DISPA-PRM offers a rapid means to monitor targeted proteins with accuracy and precision comparable to, or more than, traditional nLC-MS/MS approaches.
Conclusion
Here we describe a new method, DISPA-PRM, to quantify the abundance of targeted proteins in as little as two minutes. By leveraging FAIMS for gas-phase fractionation and PRM for sensitive and precise peptide quantification, DISPA-PRM can rapidly reduce the time required to quantify targeted proteins in complex mixtures – i.e., tryptic-digested ZM4 proteome. Specifically, over 20,000 unique peptides, as determined by nLC-MS/MS, were infused during a DISPA-PRM analysis. The effectiveness of monitoring proteins in a more complex mixture – i.e., tryptic-digested human proteomes – was previously shown (19). DISPA paired with PRM holds promise to extend the depth of what proteins can be measured from these earlier works; however, as proteome complexity increases so too does the challenge of detecting low expression level proteins. In this publication, DISPA-PRM enabled the collection of highly accurate and precise quantitative data by leveraging in short order (two minutes) for six ZM4 proxy peptides.
Here we documented the quantitative figures of merit for DISPA-PRM and demonstrated that it generates quantitative results similar in reproducibility to that of a single-shot nLC-MS/MS. Specifically, we examined the ability of DISPA-PRM to monitor overexpression of the IspG and IspH proteins in ZM4 – key proteins in the MEP biosynthetic pathway. We demonstrate a dynamic range of >1200-fold in our experiments, suggesting a robust method for monitoring the temporal expression of proteins, also providing results comparable to nLC-MS/MS. While we could readily detect and quantify these two target proteins without FAIMS, we report that FAIMS provided improved protein quantification by reducing spectral complexity, i.e., reducing co-isolation of contaminants, and improved spectral quality. Note that FAIMS would likely provide even further benefits for protein targets expressed at lower levels. We conclude that DISPA-PRM presents a valuable new tool to enable fast and rapid detection of targeted proteins in complex mixtures.
Supplementary Material
ACKNOWLEDGMENT
A big thank you to Kathrine A. Overmyer, Jean Lodge, and Evgenia Shishkova for the insightful scientific discussions and troubleshooting think-tank sessions. We also thank Daven Khana for preliminary experiment design. This material is based upon work supported by the U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research, Great Lakes Bioenergy Research Center under Award Numbers DE-SC0018409 and DE-FC02-07ER64494. Additional support from Award Number T15LM007359.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI:
Supporting information and methods, Protein and peptide metrics from nLC-MS/MS, Fragment ion intensities measured for isolation widths 0.4, 1.0, and 2.0 m/z for the peptides AIEIVDQALDR and ETDIGVTGGGQGK, Overview of four proxy peptides at isolation width 0.6, Comparison of DISPA-PRM spectral similarity using dot product and percent of total ion chromatogram (TIC) explained, DISPA-PRM spectral similarity and % TIC explained at isolation width 2.0 m/z (PDF).
REFERENCES
- (1).Choi KR, Jang WD, Yang D, Cho JS, Park D, and Lee SY (2019) Systems Metabolic Engineering Strategies: Integrating Systems and Synthetic Biology with Metabolic Engineering. Trends Biotechnol 37, 817–837. [DOI] [PubMed] [Google Scholar]
- (2).Keasling JD (1999) Gene-expression tools for the metabolic engineering of bacteria. Trends Biotechnol 17, 452–460. [DOI] [PubMed] [Google Scholar]
- (3).Krishnan MS, Blanco M, Shattuck CK, Nghiem NP, and Davison BH (2000) Ethanol production from glucose and xylose by immobilized Zymomonas mobilis CP4(pZB5). Appl Biochem Biotech 84–6, 525–541. [DOI] [PubMed] [Google Scholar]
- (4).Kerr RA (2007) Global warming is changing the world. Science 316, 188–190. [DOI] [PubMed] [Google Scholar]
- (5).Atsumi S, and Liao JC (2008) Metabolic engineering for advanced biofuels production from Escherichia coli. Curr Opin Biotechnol 19, 414–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Nielsen J, Larsson C, van Maris A, and Pronk J (2013) Metabolic engineering of yeast for production of fuels and chemicals. Curr Opin Biotechnol 24, 398–404. [DOI] [PubMed] [Google Scholar]
- (7).Martien JI, Hebert AS, Stevenson DM, Regner MR, Khana DB, Coon JJ, and Amador-Noguez D (2019) Systems-Level Analysis of Oxygen Exposure in Zymomonas mobilis: Implications for Isoprenoid Production. mSystems 6, e00987–00921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Panesar PS, Marwaha SS, and Kennedy JF (2006) Zymomonas mobilis: an alternative ethanol producer. J Chem Technol Biot 81, 623–635. [Google Scholar]
- (9).Wang X, He QN, Yang YF, Wang JW, Haning K, Hu Y, Wu B, He MX, Zhang YP, Bao J, Contreras LM, and Yang SH (2018) Advances and prospects in metabolic engineering of Zymomonas mobilis. Metab Eng 50, 57–73. [DOI] [PubMed] [Google Scholar]
- (10).Peralta-Yahya PP, and Keasling JD (2010) Advanced biofuel production in microbes. Biotechnol J 5, 147–162. [DOI] [PubMed] [Google Scholar]
- (11).Chou HH, and Keasling JD (2012) Synthetic pathway for production of five-carbon alcohols from isopentenyl diphosphate. Appl Environ Microbiol 78, 7849–7855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Ong WK, Courtney DK, Pan S, Andrade RB, Kiley PJ, Pfleger BF, and Reed JL (2020) Model-driven analysis of mutant fitness experiments improves genome-scale metabolic models of Zymomonas mobilis ZM4. PLoS Comput Biol 16, e1008137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Aebersold R, Burlingame AL, and Bradshaw RA (2013) Western blots versus selected reaction monitoring assays: time to turn the tables? Mol Cell Proteomics 12, 2381–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Kurulugama RT, Valentine SJ, Sowell RA, and Clemmer DE (2008) Development of a high-throughput IMS-IMS-MS approach for analyzing mixtures of biomolecules. J Proteomics 71, 318–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Chen S (2006) Rapid protein identification using direct infusion nanoelectrospray ionization mass spectrometry. PROTEOMICS 6, 16–25 [DOI] [PubMed] [Google Scholar]
- (16).Xiang Y, and Koomen JM (2012) Evaluation of direct infusion-multiple reaction monitoring mass spectrometry for quantification of heat shock proteins. Anal Chem 84, 1981–1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Chen J, Canales L, Neal RE (2011) Multi-Segment Direct Inject nano-ESI-LTQ-FT-ICR-MS/MS For Protein Identification. Proteome Science 9, 38. 10.1186/1477-5956-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Pereira-Medrano AG, Sterling A, Snijders AP, Reardon KF, and Wright PC (2006) A systematic evaluation of chip-based nanoelectrospray parameters for rapid identification of proteins from a complex mixture. J Am Soc Mass Spectrometry 18, 1714–1725. [DOI] [PubMed] [Google Scholar]
- (19).Meyer JG, Niemi NM, Pagliarini DJ, and Coon JJ (2020) Quantitative shotgun proteome analysis by direct infusion. Nat Methods 17, 1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kolker E, Higdon R, and Hogan JM (2006) Protein identification and expression analysis using mass spectrometry. Trends Microbiol 14, 229–235 [DOI] [PubMed] [Google Scholar]
- (21).Piehowski PD, Petyuk VA, Orton DJ, Xie F, Moore RJ, Ramirez-Restrepo M, Engel A, Lieberman AP, Albin RL, Camp DG, Smith RD, and Myers AJ (2013) Sources of technical variability in quantitative LC-MS proteomics: human brain tissue sample analysis. J Proteome Res 12, 2128–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Tabb DL, et al. (2010) Repeatability and Reproducibility in Proteomic Identifications by Liquid Chromatography-Tandem Mass Spectrometry. Journal of Proteome Research 9, 761–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Peterson AC, Russell JD, Bailey DJ, Westphall MS, and Coon JJ (2012) Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol Cell Proteomics 11, 1475–1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Bourmaud A, Gallien S, and Domon B (2016) Parallel reaction monitoring using quadrupole-Orbitrap mass spectrometer: Principle and applications. Proteomics 16, 2146–2159 [DOI] [PubMed] [Google Scholar]
- (25).Gessulat S, Schmidt T, Zolg DP, Samaras P, Schnatbaum K, Zerweck J, Knaute T, Rechenberger J, Delanghe B, Huhmer A, Reimer U, Ehrlich HC, Aiche S, Kuster B, and Wilhelm M (2019) Prosit: proteome-wide prediction of peptide tandem mass spectra by deep learning. Nat Methods 16, 509–518 [DOI] [PubMed] [Google Scholar]
- (26).Pino LK, Searle BC, Bollinger JG, Nunn B, MacLean B, and MacCoss MJ (2020) The Skyline ecosystem: Informatics for quantitative mass spectrometry proteomics. Mass Spectrom Rev 39, 229–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Egertson JD, MacLean B, Johnson R, Xuan Y, and MacCoss MJ (2015) Multiplexed peptide analysis using data-independent acquisition and Skyline. Nat Protocol 10, 887–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Yen CY, Houel S, Ahn NG, and Old WM (2011) Spectrum-to-spectrum searching using a proteome-wide spectral library. Mol Cell Proteomics 10, M111 007666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Wang J, Perez-Santiago J, Katz JE, Mallick P, and Bandeira N (2010) Peptide identification from mixture tandem mass spectra. Mol Cell Proteomics 9, 1476–1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.