

Abstract

HF transfer reactions between organic substrates are potentially useful transformations. Such reactions require the development of catalytic systems that can promote both defluorination and fluorination steps in a single reaction sequence. Herein, we report a catalytic protocol in which an equivalent of HF is generated from a perfluoroarene | nucleophile pair and transferred directly to an alkyne. The reaction is catalyzed by [Au(IPr)NiPr2] (IPr = N,N′-1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene). HF transfer generates two useful products in the form of functionalized fluoroarenes and fluoroalkenes. Mechanistic studies (rate laws, KIEs, density functional theory (DFT) calculations, competition experiments) are consistent with the Au(I) catalyst facilitating a catalytic network involving both concerted SNAr and hydrofluorination steps. The nature of the nucleophile impacts the turnover-limiting step. The cSNAr step is turnover-limiting for phenol-based nucleophiles, while protodeuaration likely becomes turnover-limiting for aniline-based nucleophiles. The approach removes the need for direct handling of HF reagents in hydrofluorination and offers possibilities to manipulate the fluorine content of organic molecules through catalysis.
Keywords: hydrofluorination, vinyl fluorides, gold catalysis, shuttle catalysis, fluoroarene
Introduction
Hydrofluorination is an essential method in synthesis. The addition of HF to unsaturated functional groups serves as an atom-efficient and expedient way to introduce fluorine atoms into organic molecules.1−10 Such substitutions are highly attractive for drug discovery and agrochemical sciences where the introduction of fluorine is known to block metabolic pathways, improve lipophilicity, modify pKa of adjacent sites, and improve binding through noncovalent interactions.11,12
Hydrofluorination methods are not without their technical challenges. HF is a corrosive gas, and high concentrations can be fatal in contact with skin.13,14 Modified HF reagents, such as pyridinium poly(hydrogen fluoride) (Olah’s reagent) or triethylamine tri(hydrogen fluoride) (TREAT-HF), have been widely adopted, and while not volatile like HF itself, they remain highly toxic and corrosive.8,15,16 Furthermore, these types of fluorinating agents are exclusively derived from HF produced from acidification of fluorite (CaF2). There are concerns over the sustainability of such approaches.17,18 In the long term, the fluorochemicals sector will need to resolve the twin issues of finite raw materials and the damage caused by the release of fluorinated molecules into the environment.
In this paper, we report a new catalytic reaction that results in the transfer of an equivalent of HF between a fluoroarene | nucleophile pair and an alkyne. Transfer functionalizations are an emerging, highly efficient, and powerful class of reactions for synthesis, owing to their high atom economy and potential reversibility.19,20 Recent advances in this field have provided systems for the shuttling of HX (Figure 1a, X = CN,21 Cl, Br,22 I23) between organic fragments.24 Our approach allows the realization of these methods for HF transfer and combines both defluorination and hydrofluorination steps in a single catalytic network. Both products of HF transfer are useful fluorinated synthons, resulting in a highly economic process. The new method removes the need to directly handle HF (or related) reagents in hydrofluorination catalysis, improving safety concerns. It also represents an important step toward the chemical recycling of fluorinated compounds through the reuse of their fluorine content.25
Figure 1.

(a) General reaction scheme for transition metal-catalyzed HX shuttling (X = Cl, Br, CN). (b) HF transfer catalysis. (c) This work.
There is limited precedent for reactions that transfer HF between organic substrates. In 2010, Kalow and Doyle reported the catalytic enantioselective reaction of benzoyl fluoride, 1,1,1,3,3,3-hexafluoro-2-propanol, and cyclohexene oxide (Figure 1b).26−29 The reaction resulted in the net addition of HF to the epoxide. Despite this remarkable result, a general approach to HF transfer for hydrofluorination remains elusive. Indeed, transition metal catalysts developed for HX shuttling (X = CN, Cl, Br, I)21−23 are poor candidates to develop such reactivity due to the reluctance of carbon–fluorine bonds to participate in oxidative addition and reductive elimination steps at transition metal centers.
In 2006, Sadighi and co-workers reported that the gold(I) fluoride complex, [Au(SIPr)F], could catalyze the trans-selective addition of HF to an internal alkyne (SIPr = 1,3-bis(2,6-diisopropylphenyl)-4,5-dihydroimidazol-2-ylidene).1 A gold(I) fluoride complex supported by a bis(phosphine) ligand, [Au(tBuXantPhos)F] has also been shown to be an on-cycle intermediate in the catalytic SNAr of perfluoroarenes (tBuXantPhos = 9,9-dimethyl-4,5-bis(di-tertbutylphosphino)xanthene).30 Both pathways achieve catalytic turnover due to the low fluorophilicity of Au(I). These two key results suggest that Au(I) catalysts may be viable candidates for developing transfer catalysis with HF but only if both types of reactivity can be established within a single catalytical network, ideally by a single catalyst.
Results and Discussion
Catalyst Development
Following a campaign of catalyst screening and reaction optimization, a new catalytic HF transfer reaction was developed (see Supporting Information for further details). The reaction of pentafluoropyridine, 4-methoxyphenol, and diphenylacetylene in toluene at 120 °C was catalyzed by 10 mol % [Au(IPr)NiPr2]31 and led to the formation of corresponding biaryl ether (1a) and fluoroalkene (2a) in >80% spectroscopic yield. This protocol transfers an equivalent of HF from the fluoroarene | nucleophile pair to the alkyne. Hydrofluorination of the alkyne shows complete selectivity for the trans-isomer.1,8 The precatalyst is operationally simple. While a related Au(I) amide has been applied in hydrofluorination catalysis,3 to the best of our knowledge, these types of species are limited to stoichiometric applications in fluoroarene functionalization.32
Optimization of the conditions highlighted the need for the reaction to be performed in a poly(tetrafluoroethylene) lined vessel to exclude the side reactions with borosilicate glassware. The limiting reagent of the reaction is the fluoroarene. This finding exemplifies the difference with traditional hydrofluorination reactions of alkynes, in which an excess of HF-reagent is typical.1,9,33 Here, an excess of alkyne and a slight excess of nucleophile were required for a high yield of fluoroalkene product. Control reactions in the absence of the catalyst show no HF transfer to the alkyne. Furthermore, a background reaction between 4-methoxyphenol and pentafluoropyridine showed no reaction after 16 h at 120 °C. These controls demonstrate that the Au(I) catalyst plays a role in both the hydrofluorination and SNAr steps.
Reaction Scope
A range of substituted phenols and anilines were shown to be effective nucleophiles for the reaction with pentafluoropyridine and diphenylacetylene to form 1a-m (Figure 2). High to modest yields were observed with both electron-rich and electron-deficient nucleophiles; however, a general decrease in the relative yield of fluoroalkene product (2a) was observed with anilines compared to phenols. The scope in fluoroarene was investigated with 4-methoxyphenol as the nucleophile and diphenylacetylene as the HF acceptor to form mixtures of 1n-p and 2a. Lower yields were observed with less electron-deficient fluoroarenes, and the scope is currently limited to systems known to be susceptible to SNAr. Di-substitution products 1o′ and 1p′ were observed with 2,3,4,5,6-pentafluorobenzonitrile and 2,3,4,5,6-pentafluoronitrobenzene allowing >1 equiv of HF to be liberated for each fluoroarene, thereby increasing the yield of the HF transfer product 2a. Multiple SNAr reactions are expected and are consistent with previous reports for these fluoroarenes.34,35 The reaction could also be applied to both symmetric and asymmetric alkyl and aryl internal alkynes, allowing the formation of 2b-i as products of HF transfer. Hydrofluorination tolerates examples of both electron-donating and electron-withdrawing groups, including free alcohol. Attempts to expand the scope to terminal alkynes (e.g., hex-1-yne, ethynylbenzene) or silylated alkynes (e.g., trimethyl(phenylethynyl)silane or 1,2-bis(trimethysilyl)ethyne) did not lead to hydrofluorination products. In every case, the hydrofluorination was 100% trans-selective, and both the scope and selectivity parallels that reported by Sadighi and co-workers using HF-based reagents.1,8 Prior examples of Au(I) catalyzed fluoroarene functionalization are limited to the use of silylated nucleophiles due to the need to create a thermodynamic sink for the liberated fluoride.30,32,36 Hence, HF transfer catalysis allows expansion of the substrate scope to more convenient and synthetically accessible nucleophiles.
Figure 2.
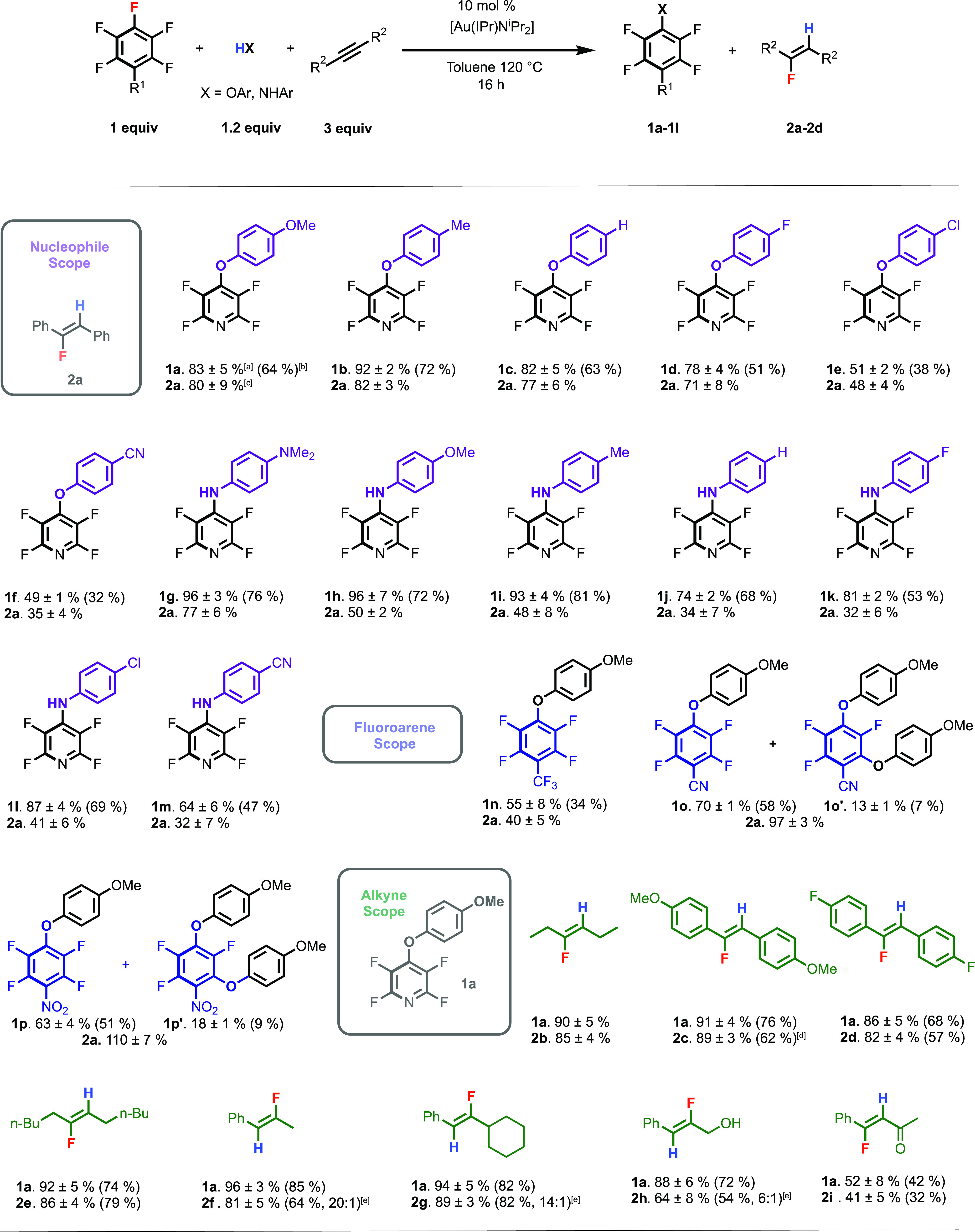
HF transfer reaction scope catalyzed by [Au(IPr)NiPr2]. [a] Reactions were performed with 0.1:1:1.2:3 equivalents of catalyst: fluoroarene (0.04 M): nucleophile: alkyne. Yields of fluoroarene (1a-1p) and fluoroalkene (2a-2d) were calculated from 19F NMR spectroscopy using a fluorobenzene internal standard. Reactions were performed in triplicate, and standard deviations are reported with a 99% confidence level. [b] Isolated yields were obtained from scale-up reactions and are shown in parenthesis. [c] Isolated yields of 2a are not reported due to this compound co-eluting with diphenylacetylene. [d] Due to the challenging isolation, this product was contaminated with ∼20% of unreacted alkyne. [e] Ratio of regioisomers β:α functionalization. Major isomer is shown.
Both fluoroalkene and fluoroarene products have synthetic utility. Substituted fluoroarenes are applied in liquid crystal displays,37,38 light-emitting diodes,39,40 and as precursors for fluorinated synthons.41,421a has also been highlighted as a protected form of phenol and can regenerate the phenol and pentafluoropyridine under mild conditions.43 Vinyl fluoride functional groups, such as those in 2a-2d, are highly desirable due to their role as bioisosteres for amide and enol functional groups.11,44−52
Kinetic Analysis
Kinetic analysis was used to gain insight into the new catalytic protocol. The experimentally determined empirical rate law for the reaction of 4-methoxyphenol (HX), pentafluoropyridine (fluoroarene), and excess diphenylacetylene with 10 mol % [Au(IPr)NiPr2] (cat) is given in eq 1.
| 1 |
The reaction was found to be first order in HX, first order in fluoroarene and first order in the catalyst. Initial rates were used to determine the catalyst order, while pseudo-first-order conditions and timecourse data over 3 half-lives were used to determine the order in HX and fluoroarene.53 Due to the strict requirement to run the reaction in excess of alkyne (>2 equiv, outlined by previous studies1 and confirmed in optimization reactions), orders for this reagent were not determined.
Kinetic experiments were also run using 4-methoxyaniline as a nucleophile in place of 4-methoxyphenol. The interpretation of this data is complicated by the observation that 1h and 2a form at different rates for this nucleophile (vide infra). Despite this limitation, the analysis suggests the formation of 2a is 0th order in fluoroarene and first order in HX and strongly indicates that the mechanism may change, depending on the nature of the nucleophile. This finding is further supported by the measurement of KIEs, which indicate a small isotope effect of 1.1 ± 0.1 for the reaction of 4-methoxyphenol/d1–4-methoxyphenol but a clear primary KIE of 2.8 ± 0.3 for the formation of 2a from 4-methoxyaniline/d2–4-methoxyaniline. These KIE values are consistent across both independent rates measurements and intermolecular competition experiments (Figure 3).
Figure 3.
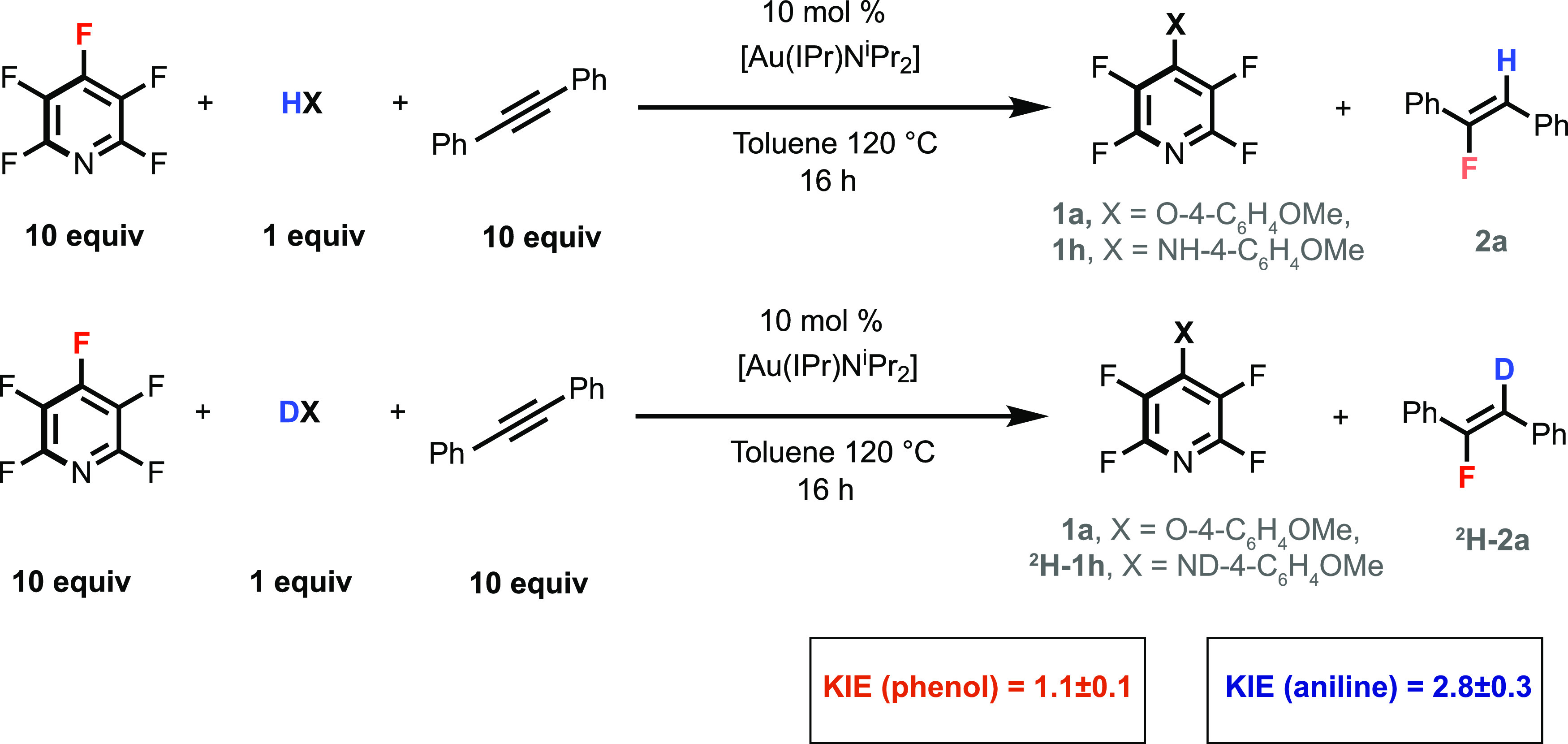
KIEs determined by independent rates.
DFT Calculations
DFT calculations were performed to supplement the kinetic data and used as a foundation with which to build a mechanistic model. The PBE0 functional, which has previously been used to model Au(I) interactions with alkynes,54,55 and 6–311G** basis set were used for all atoms other than Au, for which the SDDAll pseudopotential was applied. A single-point empirical dispersion correction (D3) with Becke–Johnson damping and solvent correction (PCM, ε = 2.38) was applied to the energies of all stationary points. Pathways were calculated for both the reaction of 4-methoxyphenol and 4-methoxyaniline with pentafluoropyridine and diphenylacetylene (Figure 4). Initially, a simple model constructed from a catalytic cycle without considering off-cycle species was considered. Toste, Bergman, and co-workers have demonstrated that [Au(IPr)NiPr2] does not react with diphenylacetylene below 75 °C but that this species is capable of deprotonating weak acids (e.g., fluorene pKa = 23 in THF).31 Precatalyst initiation by protonolysis with HX was considered to be facile, and [Au(IPr)NiPr2] was assumed to react with 4-methoxyphenol (pKa = 19)56 and 4-methoxyaniline (pKa ∼ 30)57 to form [Au(IPr)X] (X = O-4-C6H4OMe, NH-4-C6H4OMe).
Figure 4.
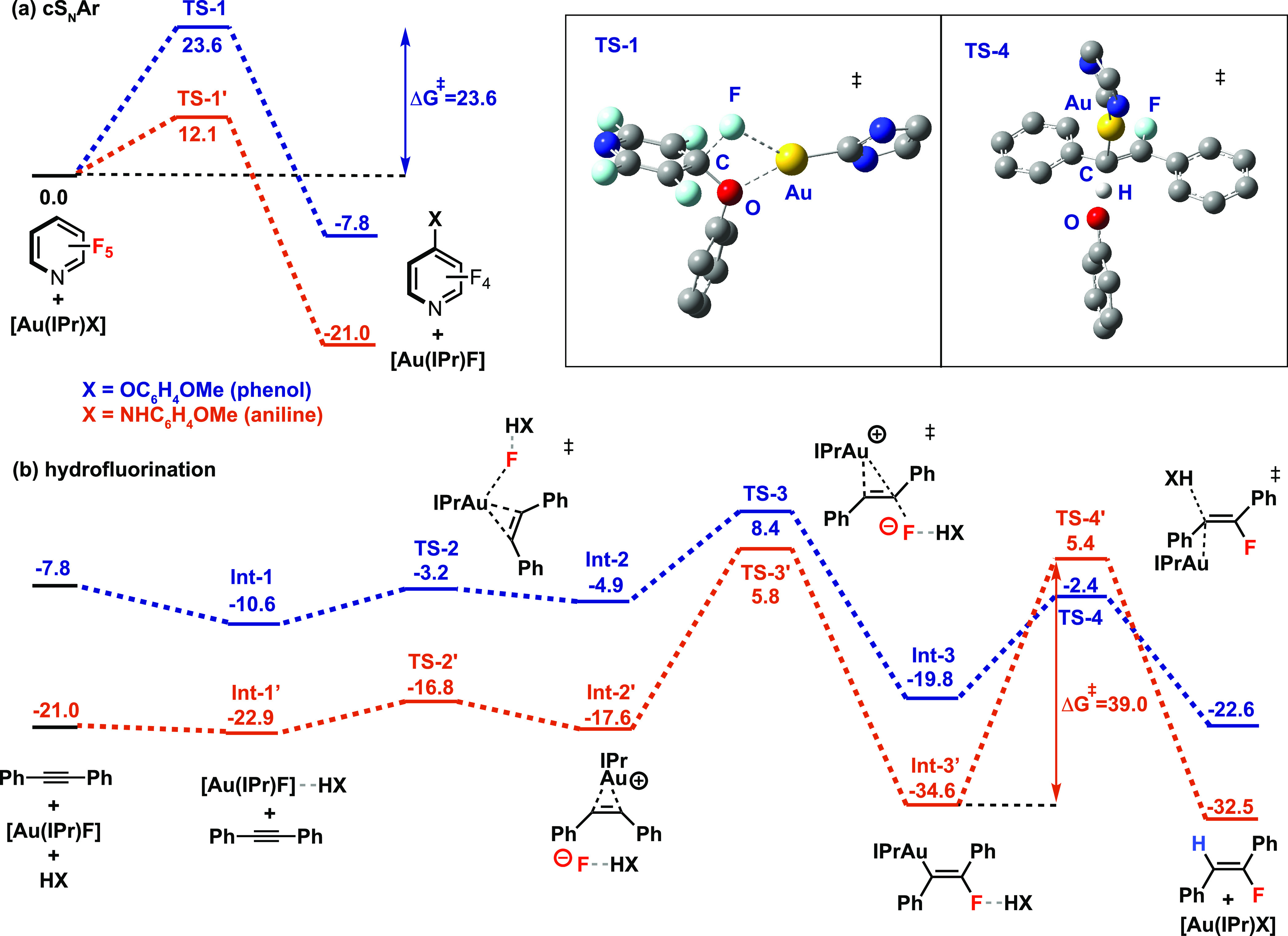
DFT calculated pathways for (a) cSNAr and (b) hydrofluorination reaction pathways. (inset) Models of TS-1 and TS-4 showing geometries of key steps. Free energy values are calculated at 298.15 K.
The reaction of [Au(IPr)X] (X = O-4-C6H4OMe) with pentafluoropyridine is calculated to occur by a concerted SNAr mechanism by TS-1 (ΔG‡ = +23.6 kcal mol–1) to form 1a alongside [Au(IPr)F]. TS-1 bears all features expected for a concerted SNAr (cSNAr) process with charge accumulation and pyramidalization occurring at the ipso-carbon of the electrophile. The {Au(IPr)}+ fragment interacts with both the alkoxide nucleophile and the fluoride leaving group in TS-1, and concerted bond breaking and formation was confirmed by IRC calculations, which show only a single TS connecting starting materials and products. Prior computational studies on Au-catalyzed hydrodefluorination of fluoroarenes have modeled redox pathways but consider trigonal planar three-coordinate Au(I) intermediates supported by bis(phosphine) ligands rather than two-coordinate linear geometries.36
The hydrofluorination sequence evolves from [Au(IPr)F]. Explicit solvation of this species with the nucleophile was considered, leading to a series of structures stabilized by F–H–X hydrogen bonding interactions. Upon addition of diphenylacetylene, fluoride dissociation from Int-1 to form Int-2 is calculated to be endergonic (ΔG°rxn = +5.7 kcal mol–1) and occurs by a facile interchange mechanism by TS-2 (ΔG‡ = +7.4 kcal mol–1). π-Complexation of the alkyne to the cationic Au(I) fragment in Int-2 is supported by NBO calculations; second-order perturbation analysis reveals both σ-donation and π-backdonation components to the bonding (see Supporting Information). This bonding interaction renders the alkyne ligand of Int-2 susceptible to nucleophilic attack by the fluoride ion. TS-3 shows that slippage of the alkyne from an η2 toward an η1 bonding mode occurs during this reaction pathway as evidenced by the asymmetry of the Au–C bond distances (2.13 vs 2.44 Å).58 Fluorination by TS-3 ultimately leads to the vinyl Au(I) species Int-3 with complete stereochemical control. The energy span between Int-1 and TS-3 defines the barrier for the fluorination sequence (ΔG‡ = +19.0 kcal mol–1), which is lower than the barrier for the cSNAr step. Finally, protodeauration of Int-3 by HX occurs via TS-4 (ΔG‡ = +17.4 kcal mol–1) and leads to the hydrofluorination product 2a while regenerating the active catalyst [Au(IPr)X]. NBO analysis reveals partial C–H bond formation and C–Au bond breaking in the transition state, while IRC calculations are consistent with the association of the resulting charged fragments {Au(IPr)}+ and X– occurring afterward.59−64 The overall transformation is calculated to be exergonic (ΔG°rxn = −22.6 kcal mol–1), with the SNAr step being turnover-limiting.
A closely related pathway was calculated for the reaction of [Au(IPr)X] (X = NH-4-C6H4OMe) with pentafluoropyridine and diphenylacetylene, albeit with significantly modified barriers for each of the steps. The improved nucleophilicity of the amide ligand of [Au(IPr)NH-4-C6H4OMe] (NPA charges Au = +0.42, N = −1.00) over the alkoxide of [Au(IPr)O-4-C6H4OMe] (NPA charges Au = +0.45, O = −0.82), alongside the reduced pKa of HO-4-C6H4OMe compared to H2N-4-C6H4OMe impacts key transition state barriers. The SNAr step by TS-1′ is now a low energy process (ΔG‡ = +12.1 kcal mol–1) and, as such, is no longer predicted to be the turnover-limiting step. In contrast, the protodeauration step from the Au(I) vinyl intermediate Int-3′ to TS-4′ involves a large energy barrier (ΔG‡ = +40.0 kcal mol–1) and is not only predicted to be the slowest step of the catalytic sequence, but the activation energy is also large enough to question if it is accessible under the reaction conditions (120 °C, 16 h).
Comparison of the TS-1 and TS-1′ allows identification of the key features that led to the lowering of this barrier for the cSNAr step. The C–X and C–F distances in TS-1 are 1.44 (X = O) and 1.65 Å, respectively, while the X–C–F angle is 89.9°. In contrast, TS-1′ possesses a more open structure with a much longer C–X interaction of 1.92 Å (X = N), shorter C–F distance of 1.36 Å, and obtuse X–C–F angle of 94.1°. These metrics suggest that C–F bond breaking in TS-1′ is less advanced than in TS-1. The accumulation of charge on key moieties in these transition states is consistent with this argument as TS-1 (Δq: Au = +0.07, O = +0.20, Cipso = +0.04, F = −0.23) involves greater charge separation than TS-1′ (Δq: Au = +0.03, N = +0.12, Cipso = +0.03, F = −0.13).65 A competition experiment in which excess 4-methoxyphenol and 4-methoxyaniline were reacted with pentafluoropyridine and 10 mol % [Au(IPr)NiPr2] led exclusively to 1h in preference to 1a. This finding reflects the large energy difference between TS-1′ and TS-1 (ΔΔG‡ = 11.5 kcal mol–1).
Further comparisons can be made between the protodeauration transition states TS-4 and TS-4′. Protodeauration involves direct breaking of the H–X bond (X = O, N) through deprotonation by the Au–C moiety.59−63 In TS-4, the C–H–X bond angle is 174.1° while the H–C–Au angle is 90.7°, and this reflects the orthogonality between the reacting ligand and Au center. The protodeauration transition states TS-4 and TS-4′ both involve isomerization of the vinyl ligand from a σ- to π-coordination mode. This reorganization leads to an elongation of the Au–C bond length (e.g., Int-3, 2.04; TS-4, 2.18) and charge accumulation on the C atom adjacent to Au (e.g., Int-3, −0.44; TS-4, −0.57), both of which facilitate protonation. The acidity of the H–X moiety is a key factor in determining the activation barrier for this step. The transition state geometries would be expected to be consistent with a primary KIE if this step becomes turnover limiting.
In combination, the empirical rate laws, the KIEs, and the DFT calculations are fully consistent with a change of turnover-limiting step depending on the nucleophile. For 4-methoxyphenol, the cSNAr is expected to be turnover-limiting, with a small or no KIE. For 4-methoxyaniline, the protodeuaration step becomes turnover-limiting; the reaction rate no longer depends on [fluoroarene], and a primary KIE is expected. Given the apparent sensitivity of these key steps to the electronics of the nucleophile, further DFT calculations were undertaken in which the nucleophile was modified through variation of the substituent at the 4-position (see Supporting Information for details). These calculations revealed clear free energy relationships between the transition state barriers and Hammett parameters (σp) of the nucleophile. In no case did varying the electronics of phenol or aniline change the predicted turnover-limiting step for each of these systems; the switch-in mechanism requires a complete switch in nucleophile rather than perturbation of its electronic structure.
Catalytic Network
Taken in combination, these data suggest a complex catalytic cycle involving both cSNAr and hydrofluorination mechanisms within a single reaction network. Further insight into the different behavior of the two nucleophile types was obtained by following the concentration of key species (including HF) over the complete timecourse of these reactions (Figure 5). In the case of pentafluoropyridine, 4-methoxyphenol, and diphenylacetylene, the formation of products 1a and 2a was concurrent, and no HF buildup was observed. For the same reaction with 4-methoxyaniline, the formation of product 1h occurred independently of the formation of 2a. HF was identified as a reaction intermediate by a broad and concentration-dependent resonance in the 19F NMR spectrum at δ = 150.5–152.5 ppm. Given the reaction conditions, it remains likely that the HF interacts with 4-methoxyaniline in solution (through proton transfer or hydrogen bonding).66 An induction period was observed for the formation of 2a, and an increase in the rate of formation of this species is observed as the HF concentration increases.
Figure 5.

Plots for the concentration of fluoroarene and fluoroalkene products and HF intermediate over time for the reaction of pentafluoropyridine and diphenylacetylene with (a) 4-methoxyphenol and (b) 4-methoxyaniline.
The catalytic network in Figure 6 is a plausible reaction mechanism. This network explains the combined data and calculations. The reaction potentially operates in two different regimes depending on the nature of the nucleophile. In regime 1, for phenol-based nucleophiles, the cSNAr step is expected to be slow and turnover-limiting. As such, any [Au(IPr)F] generated is expected to be consumed in the onward hydrofluorination sequence. While [Au(IPr)F] can also potentially react with HX to liberate HF and regenerate the active catalyst, this reaction is calculated to be endergonic (X = O-4-C6H4OMe; ΔG° = 9.6 kcal mol–1, ΔG‡ = 12.0 kcal mol–1) and should be reversible under the catalytic conditions. Protodeauration with HX is facile, leading to the synchronous formation of the products 1 and 2 and no buildup of HF during the reaction.
Figure 6.

Plausible catalytic network for HF transfer.
In regime 2, for aniline-based nucleophiles, the cSNAr step is now fast, and the hydrofluorination sequence is slow and likely turnover limiting. The extremely high calculated activation barrier for protodeuaration with HX (ΔG‡ = 40.0 kcal mol–1) suggests that this step may only be a minor contributor under the reaction conditions. Instead, the reaction of [Au(IPr)F] with HX could produce a bypass in the catalytic cycle, resulting in the generation of HF. While again calculated to be endergonic and reversible (X = NH-4-C6H4OMe; ΔG° = 10.5 kcal mol–1, ΔG‡ = 11.3 kcal mol–1), if this bypass step operated in combination with the consumption of HF in the protodeuaration step, it may prove thermodynamically viable. If combined with the fast cSNAr step, this bypass mechanism would lead to the asynchronous production of 1 and 2 along with the potential buildup of HF as a reaction intermediate. Control reactions revealed that under catalytic conditions, 1h forms in near-quantitative yield, alongside HF, even if diphenylacetylene was omitted from the reaction mixture. As HF is susceptible to off-cycle side reactions, this mechanism would explain the lower yield of 2 for aniline-based nucleophiles compared to phenol-based nucleophiles (Figure 2).
This mechanistic hypothesis could be used to improve the yields of the HF transfer product 2 when carrying out reactions with aniline-based nucleophiles. The catalytic reaction to form 1h in 83 ± 5% yield generates 50 ± 2% of 2a as a coproduct (Figure 2). When the standard conditions are repeated but 1.2 equiv of 4-methoxyphenol is added to the reaction mixture, 1h is still formed as the exclusive cSNAr product in 92 ± 5%, but the yield of 2a improves to 72 ± 3%. The finding is consistent with the addition of the phenol-limiting bypass catalysis by accelerating the protodeauration step.
Conclusions
In summary, a Au(I)-catalyzed HF transfer reaction for the tandem hydrofluorination of alkynes and functionalization of fluoroarenes has been developed. HF is generated through the reaction of perfluoroarene with a nucleophile, obviating the need for direct handling of HF-based reagents and providing an operationally simple approach to fluorination catalysis. Through kinetics analysis, competition experiments, and DFT calculations, a detailed understanding of the catalytic network involved in HF transfer has been obtained. These studies showed that the rate of production and distribution of products is dependent on the nature of the nucleophile. This mechanistic understanding was exploited to improve the efficiency of HF transfer catalysis. In the longer term, we believe that these results will provide a foundation for the development of new catalytic approaches to transfer fluorine-containing groups between molecules and recycle fluorinated compounds.
Acknowledgments
The authors are grateful to the European Research Council for generous funding in the form of an ERC StG and ERC CoG.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.1c05474.
Full experimental details: synthetic procedures, computational methods, and analysis; primary data (.mnova,.xyz) are available through the following link: https://data.hpc.imperial.ac.uk/resolve/?doi=9985 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Akana J. A.; Bhattacharyya K. X.; Müller P.; Sadighi J. P. Reversible C–F Bond Formation and the Au-Catalyzed Hydrofluorination of Alkynes. J. Am. Chem. Soc. 2007, 129, 7736–7737. 10.1021/ja0723784. [DOI] [PubMed] [Google Scholar]
- Gorske B. C.; Mbofana C. T.; Miller S. J. Regio- and Stereoselective Synthesis of Fluoroalkenes by Directed Au(I) Catalysis. Org. Lett. 2009, 11, 4318–4321. 10.1021/ol9016782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okoromoba O. E.; Han J.; Hammond G. B.; Xu B. Designer HF-Based Fluorination Reagent: Highly Regioselective Synthesis of Fluoroalkenes and Gem-Difluoromethylene Compounds from Alkynes. J. Am. Chem. Soc. 2014, 136, 14381–14384. 10.1021/ja508369z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He G.; Qiu S.; Huang H.; Zhu G.; Zhang D.; Zhang R.; Zhu H. Cu(I)- or Ag(I)-Catalyzed Regio- and Stereocontrolled Trans-Hydrofluorination of Ynamides. Org. Lett. 2016, 18, 1856–1859. 10.1021/acs.orglett.6b00615. [DOI] [PubMed] [Google Scholar]
- Zhu G.; Qiu S.; Xi Y.; Ding Y.; Zhang D.; Zhang R.; He G.; Zhu H. (IPr)CuF-Catalyzed α-Site Regiocontrolled Trans-Hydrofluorination of Ynamides. Org. Biomol. Chem. 2016, 14, 7746–7753. 10.1039/C6OB01345G. [DOI] [PubMed] [Google Scholar]
- Gómez-Herrera A.; Nahra F.; Brill M.; Nolan S. P.; Cazin C. S. J. Sequential Functionalization of Alkynes and Alkenes Catalyzed by Gold(I) and Palladium(II) N-Heterocyclic Carbene Complexes. ChemCatChem 2016, 8, 3381–3388. 10.1002/cctc.201600868. [DOI] [Google Scholar]
- Zeng X.; Liu S.; Hammond G. B.; Xu B. Divergent Regio- and Stereoselective Gold-Catalyzed Synthesis of α-Fluorosulfones and β-Fluorovinylsulfones from Alkynylsulfones. Chem. - Eur. J. 2017, 23, 11977–11981. 10.1002/chem.201703179. [DOI] [PubMed] [Google Scholar]
- O’Connor T. J.; Toste F. D. Gold-Catalyzed Hydrofluorination of Electron-Deficient Alkynes: Stereoselective Synthesis of β-Fluoro Michael Acceptors. ACS Catal. 2018, 8, 5947–5951. 10.1021/acscatal.8b01341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier R.; Mamone M.; Paquin J.-F. Gold-Catalyzed Hydrofluorination of Internal Alkynes Using Aqueous HF. Org. Lett. 2019, 21, 9024–9027. 10.1021/acs.orglett.9b03425. [DOI] [PubMed] [Google Scholar]
- Guo R.; Qi X.; Xiang H.; Geaneotes P.; Wang R.; Liu P.; Wang Y.-M. Stereodivergent Alkyne Hydrofluorination Using Protic Tetrafluoroborates as Tunable Reagents. Angew. Chem., Int. Ed. 2020, 59, 16651–16660. 10.1002/anie.202006278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meanwell N. A. Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem. 2018, 61, 5822–5880. 10.1021/acs.jmedchem.7b01788. [DOI] [PubMed] [Google Scholar]
- Inoue M.; Sumii Y.; Shibata N. Contribution of Organofluorine Compounds to Pharmaceuticals. ACS Omega 2020, 5, 10633–10640. 10.1021/acsomega.0c00830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E. B. First Aid for a Unique Acid: HF. Chem. Health Saf. 1998, 5, 25–28. 10.1021/acs.chas.8b05511. [DOI] [Google Scholar]
- Segal E. B. First Aid for a Unique Acid, HF: A Sequel. Chem. Health Saf. 2000, 7, 18–23. 10.1016/S1074-9098(99)00077-5. [DOI] [Google Scholar]
- Olah G. A.; Welch J. T.; Vankar Y. D.; Nojima M.; Kerekes I.; Olah J. A. Synthetic Methods and Reactions. 63. Pyridinium Poly(Hydrogen Fluoride) (30% Pyridine-70% Hydrogen Fluoride): A Convenient Reagent for Organic Fluorination Reactions. J. Org. Chem. 1979, 44, 3872–3881. 10.1021/jo01336a027. [DOI] [Google Scholar]
- Kirk K. L. Fluorination in Medicinal Chemistry: Methods, Strategies, and Recent Developments. Org. Process Res. Dev. 2008, 12, 305–321. 10.1021/op700134j. [DOI] [Google Scholar]
- Harsanyi A.; Sandford G. Organofluorine Chemistry: Applications, Sources and Sustainability. Green Chem. 2015, 17, 2081–2086. 10.1039/C4GC02166E. [DOI] [Google Scholar]
- Caron S. Where Does the Fluorine Come From? A Review on the Challenges Associated with the Synthesis of Organofluorine Compounds. Org. Process Res. Dev. 2020, 24, 470–480. 10.1021/acs.oprd.0c00030. [DOI] [Google Scholar]
- Bhawal B. N.; Morandi B. Shuttle Catalysis—New Strategies in Organic Synthesis. Chem. - Eur. J. 2017, 23, 12004–12013. 10.1002/chem.201605325. [DOI] [PubMed] [Google Scholar]
- Bhawal B. N.; Morandi B. Catalytic Transfer Functionalization through Shuttle Catalysis. ACS Catal. 2016, 6, 7528–7535. 10.1021/acscatal.6b02333. [DOI] [Google Scholar]
- Bhawal B. N.; Reisenbauer J. C.; Ehinger C.; Morandi B. Overcoming Selectivity Issues in Reversible Catalysis: A Transfer Hydrocyanation Exhibiting High Kinetic Control. J. Am. Chem. Soc. 2020, 142, 10914–10920. 10.1021/jacs.0c03184. [DOI] [PubMed] [Google Scholar]
- Yu P.; Bismuto A.; Morandi B. Iridium-Catalyzed Hydrochlorination and Hydrobromination of Alkynes by Shuttle Catalysis. Angew. Chem., Int. Ed. 2020, 59, 2904–2910. 10.1002/anie.201912803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrone D. A.; Franzoni I.; Ye J.; Rodríguez J. F.; Poblador-Bahamonde A. I.; Lautens M. Palladium-Catalyzed Hydrohalogenation of 1,6-Enynes: Hydrogen Halide Salts and Alkyl Halides as Convenient HX Surrogates. J. Am. Chem. Soc. 2017, 139, 3546–3557. 10.1021/jacs.7b00482. [DOI] [PubMed] [Google Scholar]
- Wang D.; Astruc D. The Golden Age of Transfer Hydrogenation. Chem. Rev. 2015, 115, 6621–6686. 10.1021/acs.chemrev.5b00203. [DOI] [PubMed] [Google Scholar]
- Cousins I. T.; DeWitt J. C.; Glüge J.; Goldenman G.; Herzke D.; Lohmann R.; Ng C. A.; Scheringer M.; Wang Z. The High Persistence of PFAS Is Sufficient for Their Management as a Chemical Class. Environ. Sci.: Processes Impacts 2020, 22, 2307–2312. 10.1039/D0EM00355G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalow J. A.; Doyle A. G. Enantioselective Ring Opening of Epoxides by Fluoride Anion Promoted by a Cooperative Dual-Catalyst System. J. Am. Chem. Soc. 2010, 132, 3268–3269. 10.1021/ja100161d. [DOI] [PubMed] [Google Scholar]
- Kalow J. A.; Doyle A. G. Mechanistic Investigations of Cooperative Catalysis in the Enantioselective Fluorination of Epoxides. J. Am. Chem. Soc. 2011, 133, 16001–16012. 10.1021/ja207256s. [DOI] [PubMed] [Google Scholar]
- Kalow J. A.; Doyle A. G. Enantioselective Fluoride Ring Opening of Aziridines Enabled by Cooperative Lewis Acid Catalysis. Tetrahedron 2013, 69, 5702–5709. 10.1016/j.tet.2013.01.062. [DOI] [Google Scholar]
- Graham T. J. A.; Lambert R. F.; Ploessl K.; Kung H. F.; Doyle A. G. Enantioselective Radiosynthesis of Positron Emission Tomography (PET) Tracers Containing [18F]Fluorohydrins. J. Am. Chem. Soc. 2014, 136, 5291–5294. 10.1021/ja5025645. [DOI] [PubMed] [Google Scholar]
- Hu J.-Y.; Zhang J.; Wang G.-X.; Sun H.-L.; Zhang J.-L. Constructing a Catalytic Cycle for C–F to C–X (X = O, S, N) Bond Transformation Based on Gold-Mediated Ligand Nucleophilic Attack. Inorg. Chem. 2016, 55, 2274–2283. 10.1021/acs.inorgchem.5b02634. [DOI] [PubMed] [Google Scholar]
- Johnson M. W.; Shevick S. L.; Toste F. D.; Bergman R. G. Preparation and Reactivity of Terminal Gold(I) Amides and Phosphides. Chem. Sci. 2013, 4, 1023–1027. 10.1039/C2SC21519E. [DOI] [Google Scholar]
- Lv H.; Zhan J.-H.; Cai Y.-B.; Yu Y.; Wang B.; Zhang J.-L. π–π Interaction Assisted Hydrodefluorination of Perfluoroarenes by Gold Hydride: A Case of Synergistic Effect on C–F Bond Activation. J. Am. Chem. Soc. 2012, 134, 16216–16227. 10.1021/ja305204y. [DOI] [PubMed] [Google Scholar]
- Nahra F.; Patrick S. R.; Bello D.; Brill M.; Obled A.; Cordes D. B.; Slawin A. M. Z.; O’Hagan D.; Nolan S. P. Hydrofluorination of Alkynes Catalysed by Gold Bifluorides. ChemCatChem 2015, 7, 240–244. 10.1002/cctc.201402891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan R.; Parthiban A. Regioselective Preparation of Functional Aryl Ethers and Esters by Stepwise Nucleophilic Aromatic Substitution Reaction. J. Fluorine Chem. 2014, 162, 17–25. 10.1016/j.jfluchem.2014.03.005. [DOI] [Google Scholar]
- Mulryan D.; White A. J. P.; Crimmin M. R. Organocatalyzed Fluoride Metathesis. Org. Lett. 2020, 22, 9351–9355. 10.1021/acs.orglett.0c03593. [DOI] [PubMed] [Google Scholar]
- Zhan J.-H.; Lv H.; Yu Y.; Zhang J.-L. Catalytic C-F Bond Activation of Perfluoroarenes by Tricoordinated Gold(I) Complexes. Adv. Synth. Catal., 2012, 354, 1529–1541. 10.1002/adsc.201100843. [DOI] [Google Scholar]
- Kirsch P. Fluorine in Liquid Crystal Design for Display Applications. J. Fluorine Chem. 2015, 177, 29–36. 10.1016/j.jfluchem.2015.01.007. [DOI] [Google Scholar]
- Kirsch P.; Bremer M. Nematic Liquid Crystals for Active Matrix Displays: Molecular Design and Synthesis. Angew. Chem., Int. Ed. 2000, 39, 4216–4235. . [DOI] [PubMed] [Google Scholar]
- Kamata T.; Sasabe H.; Watanabe Y.; Yokoyama D.; Katagiri H.; Kido J. A Series of Fluorinated Phenylpyridine-Based Electron-Transporters for Blue Phosphorescent OLEDs. J. Mater. Chem. C 2016, 4, 1104–1110. 10.1039/C5TC03879K. [DOI] [Google Scholar]
- Ragni R.; Punzi A.; Babudri F.; Farinola G. M. Organic and Organometallic Fluorinated Materials for Electronics and Optoelectronics: A Survey on Recent Research. Eur. J. Org. Chem. 2018, 2018, 3500–3519. 10.1002/ejoc.201800657. [DOI] [Google Scholar]
- Dolbier W. R. Fluorine Chemistry at the Millennium. J. Fluorine Chem. 2005, 126, 157–163. 10.1016/j.jfluchem.2004.09.033. [DOI] [Google Scholar]
- Lv H.; Cai Y.-B.; Zhang J.-L. Copper-Catalyzed Hydrodefluorination of Fluoroarenes by Copper Hydride Intermediates. Angew. Chem., Int. Ed. 2013, 52, 3203–3207. 10.1002/anie.201208364. [DOI] [PubMed] [Google Scholar]
- Brittain W. D. G.; Cobb S. L. Tetrafluoropyridyl (TFP): A General Phenol Protecting Group Readily Cleaved under Mild Conditions. Org. Biomol. Chem. 2019, 17, 2110–2115. 10.1039/C8OB02899K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutheuil G.; Couve-Bonnaire S.; Pannecoucke X. Diastereomeric Fluoroolefins as Peptide Bond Mimics Prepared by Asymmetric Reductive Amination of α-Fluoroenones. Angew. Chem., Int. Ed. 2007, 46, 1290–1292. 10.1002/anie.200604246. [DOI] [PubMed] [Google Scholar]
- Burkhart J. P.; Weintraub P. M.; Gates C. A.; Resvick R. J.; Vaz R. J.; Friedrich D.; Angelastro M. R.; Bey P.; Peet N. P. Novel Steroidal Vinyl Fluorides as Inhibitors of Steroid C17(20) Lyase. Bioorg. Med. Chem. 2002, 10, 929–934. 10.1016/S0968-0896(01)00354-6. [DOI] [PubMed] [Google Scholar]
- van Steenis J. H.; der Gen A. van. Synthesis of Terminal Monofluoro Olefins. J. Chem. Soc., Perkin Trans. 1 2002, 2117–2133. 10.1039/b106187a. [DOI] [Google Scholar]
- Zajc B.; Kumar R. Synthesis of Fluoroolefins via Julia-Kocienski Olefination. Synthesis 2010, 11, 1822–1836. 10.1055/s-0029-1218789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landelle G.; Bergeron M.; Turcotte-Savard M.-O.; Paquin J.-F. Synthetic Approaches to Monofluoroalkenes. Chem. Soc. Rev. 2011, 40, 2867–2908. 10.1039/c0cs00201a. [DOI] [PubMed] [Google Scholar]
- Yanai H.; Taguchi T. Synthetic Methods for Fluorinated Olefins. Eur. J. Org. Chem. 2011, 2011, 5939–5954. 10.1002/ejoc.201100495. [DOI] [Google Scholar]
- Pfund Emmanuel.; Lequeux Thierry.; Gueyrard David. Synthesis of Fluorinated and Trifluoromethyl-Substituted Alkenes through the Modified Julia Olefination: An Update. Synthesis 2015, 47, 1534–1546. 10.1055/s-0034-1380548. [DOI] [Google Scholar]
- Drouin Myriam.; Hamel Jean-Denys.; Paquin Jean-Francois. Synthesis of Monofluoroalkenes: A Leap Forward. Synthesis 2018, 50, 881–955. 10.1055/s-0036-1591867. [DOI] [Google Scholar]
- Drouin Myriam.; Paquin Jean-Francois. Recent Progress in the Racemic and Enantioselective Synthesis of Monofluoroalkene-Based Dipeptide Isosteres. Beilstein J. Org. Chem. 2017, 13, 2637–2658. 10.3762/bjoc.13.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- As 1a and 2a form at identical rates in these reactions, orders for the entire process can be confidently assigned by monitoring either starting material consumption or formation of 1a or 2a.
- Kang R.; Chen H.; Shaik S.; Yao J. Assessment of Theoretical Methods for Complexes of Gold(I) and Gold(III) with Unsaturated Aliphatic Hydrocarbon: Which Density Functional Should We Choose?. J. Chem. Theory Comput. 2011, 7, 4002–4011. 10.1021/ct200656p. [DOI] [PubMed] [Google Scholar]
- Li H.; Liu J.; Abosede A. O.; Bao X. Computational Insights into the Mechanisms of Au(i)-Catalysed Intramolecular Addition of the Hydroxylamine Group onto Alkynes. Org. Chem. Front. 2017, 4, 1130–1136. 10.1039/C7QO00072C. [DOI] [Google Scholar]
- Bordwell F. G.; McCallum R. J.; Olmstead W. N. Acidities and Hydrogen Bonding of Phenols in Dimethyl Sulfoxide. J. Org. Chem. 1984, 49, 1424–1427. 10.1021/jo00182a020. [DOI] [Google Scholar]
- Bordwell F. G.; Algrim D. J. Acidities of Anilines in Dimethyl Sulfoxide Solution. J. Am. Chem. Soc. 1988, 110, 2964–2968. 10.1021/ja00217a045. [DOI] [Google Scholar]
- Eisenstein O.; Hoffmann R. Transition-Metal Complexed Olefins: How Their Reactivity toward a Nucleophile Relates to Their Electronic Structure. J. Am. Chem. Soc. 1981, 103, 4308–4320. 10.1021/ja00405a005. [DOI] [Google Scholar]
- Biasiolo L.; Trinchillo M.; Belanzoni P.; Belpassi L.; Busico V.; Ciancaleoni G.; D’Amora A.; Macchioni A.; Tarantelli F.; Zuccaccia D. Unexpected Anion Effect in the Alkoxylation of Alkynes Catalyzed by N-Heterocyclic Carbene (NHC) Cationic Gold Complexes. Chem. - Eur. J. 2014, 20, 14594–14598. 10.1002/chem.201404539. [DOI] [PubMed] [Google Scholar]
- Ciancaleoni G.; Belpassi L.; Zuccaccia D.; Tarantelli F.; Belanzoni P. Counterion Effect in the Reaction Mechanism of NHC Gold(I)-Catalyzed Alkoxylation of Alkynes: Computational Insight into Experiment. ACS Catal. 2015, 5, 803–814. 10.1021/cs501681f. [DOI] [Google Scholar]
- Trinchillo M.; Belanzoni P.; Belpassi L.; Biasiolo L.; Busico V.; D’Amora A.; D’Amore L.; Del Zotto A.; Tarantelli F.; Tuzi A.; Zuccaccia D. Extensive Experimental and Computational Study of Counterion Effect in the Reaction Mechanism of NHC-Gold(I)-Catalyzed Alkoxylation of Alkynes. Organometallics 2016, 35, 641–654. 10.1021/acs.organomet.5b00925. [DOI] [Google Scholar]
- D’Amore L.; Ciancaleoni G.; Belpassi L.; Tarantelli F.; Zuccaccia D.; Belanzoni P. Unraveling the Anion/Ligand Interplay in the Reaction Mechanism of Gold(I)-Catalyzed Alkoxylation of Alkynes. Organometallics 2017, 36, 2364–2376. 10.1021/acs.organomet.7b00377. [DOI] [Google Scholar]
- Lu Z.; Li T.; Mudshinge S. R.; Xu B.; Hammond G. B. Optimization of Catalysts and Conditions in Gold(I) Catalysis—Counterion and Additive Effects. Chem. Rev. 2021, 121, 8452–8477. 10.1021/acs.chemrev.0c00713. [DOI] [PubMed] [Google Scholar]
- Gauthier R.; Tzouras N. V.; Zhang Z.; Bédard S.; Saab M.; Falivene L.; Van Hecke K.; Cavallo L.; Nolan S. P.; Paquin J.-F. Gold N-Heterocyclic Carbene Catalysts for the Hydrofluorination of Alkynes Using Hydrofluoric Acid: Reaction Scope, Mechanistic Studies and the Tracking of Elusive Intermediates. Chem. - Eur. J. 2022, 28, e202103886 10.1002/chem.202103886. [DOI] [PubMed] [Google Scholar]
- Δq Is defined here as the change in NPA charge on an atom, on transitioning from [Au(IPr)X] + pentafluoropyridine to TS-1 or TS-1′.
- Szatyłowicz H.; Krygowski T. M.; Panek J. J.; Jezierska A. H-Bonded Complexes of Aniline with HF/F– and Anilide with HF in Terms of Symmetry-Adapted Perturbation, Atoms in Molecules, and Natural Bond Orbitals Theories. J. Phys. Chem. A 2008, 112, 9895–9905. 10.1021/jp803592v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.