

Abstract
Bacterial chaperones ClpB and DnaK, homologs of the respective eukaryotic heat shock proteins Hsp104 and Hsp70, are essential in the reactivation of toxic protein aggregates that occur during translation or periods of stress. In the pathogen Mycobacterium tuberculosis (Mtb), the protective effect of chaperones extends to survival in the presence of host stresses, such as protein-damaging oxidants. However, we lack a full understanding of the interplay of Hsps and other stress response genes in mycobacteria. Here, we employ genome-wide transposon mutagenesis to identify the genes that support clpB function in Mtb. In addition to validating the role of ClpB in Mtb’s response to oxidants, we show that HtpG, a homolog of Hsp90, plays a distinct role from ClpB in the proteotoxic stress response. While loss of neither clpB nor htpG is lethal to the cell, loss of both through genetic depletion or small molecule inhibition impairs recovery after exposure to host-like stresses, especially reactive nitrogen species. Moreover, defects in cells lacking clpB can be complemented by overexpression of other chaperones, demonstrating that Mtb’s stress response network depends upon finely tuned chaperone expression levels. These results suggest that inhibition of multiple chaperones could work in concert with host immunity to disable Mtb.
Keywords: chaperone, ClpB, heat shock protein, Mycobacterium tuberculosis, Oxidant, proteostasis
1. |. INTRODUCTION
Mycobacterium tuberculosis (Mtb) is the main pathogenic agent of tuberculosis (TB), a leading cause of death due to an infectious disease worldwide (WHO, 2018). About a third of the world’s population is estimated to have latent TB infection, reflecting Mtb’s ability to survive in the human host as bacterial subpopulations in heterogeneous states that range from replicative to non-replicative, with differing sensitivities to antibiotics (Gold and Nathan, 2017). Mtb can reside in acidic host environments, as evidenced by the avirulence of acid-susceptible mutants in mice and the activity of the front-line TB drug Levitte et al., 2016; Yadon et al., 2017; Zhang and Mitchison, 2003). In activated macrophages and in other host microenvironments, Mtb can be exposed to multiple chemical stresses, including reactive oxygen and nitrogen species and antibiotics, as well as the elevated temperature of fever (Dahl et al., 2015; Ehrt and Schnappinger, 2009; Kaufmann et al., 2005; Russell, 2011). Cellular proteins are especially susceptible to unfolding and aggregation under these conditions (Schramm et al., 2020; Vaubourgeix et al., 2015). Chaperones that are conserved from bacteria to higher eukaryotes serve in a dedicated pathway that disaggregates and refolds toxic protein aggregates, maintaining proteostasis, and promoting cell survival (Balchin et al., 2016; Kim et al., 2013). The conserved bacterial pair ClpB/DnaK (homologs of eukaryotic Hsp104/Hsp70) are ATP-powered proteins that form the hub of a protein “bichaperone” network (Calloni et al., 2012; Mogk et al., 1999; Zietkiewicz et al., 2004). In mycobacteria, DnaK is predicted to be essential for growth due to its involvement in folding nascent proteins and reactivating aggregated proteins along with the disaggregase ClpB (Figure 1) (DeJesus et al., 2017; Fay and Glickman, 2014; Lupoli et al., 2016; Lupoli et al., 2018; Sassetti et al., 2003; Vaubourgeix et al., 2015; Yu et al., 2018). Mycobacterial chaperones are also involved in asymmetrically distributing irreversibly damaged, aggregated proteins during cell division as a mechanism of survival during periods of oxidative or antibiotic stress (Fay and Glickman, 2014; Vaubourgeix et al., 2015).
FIGURE 1.
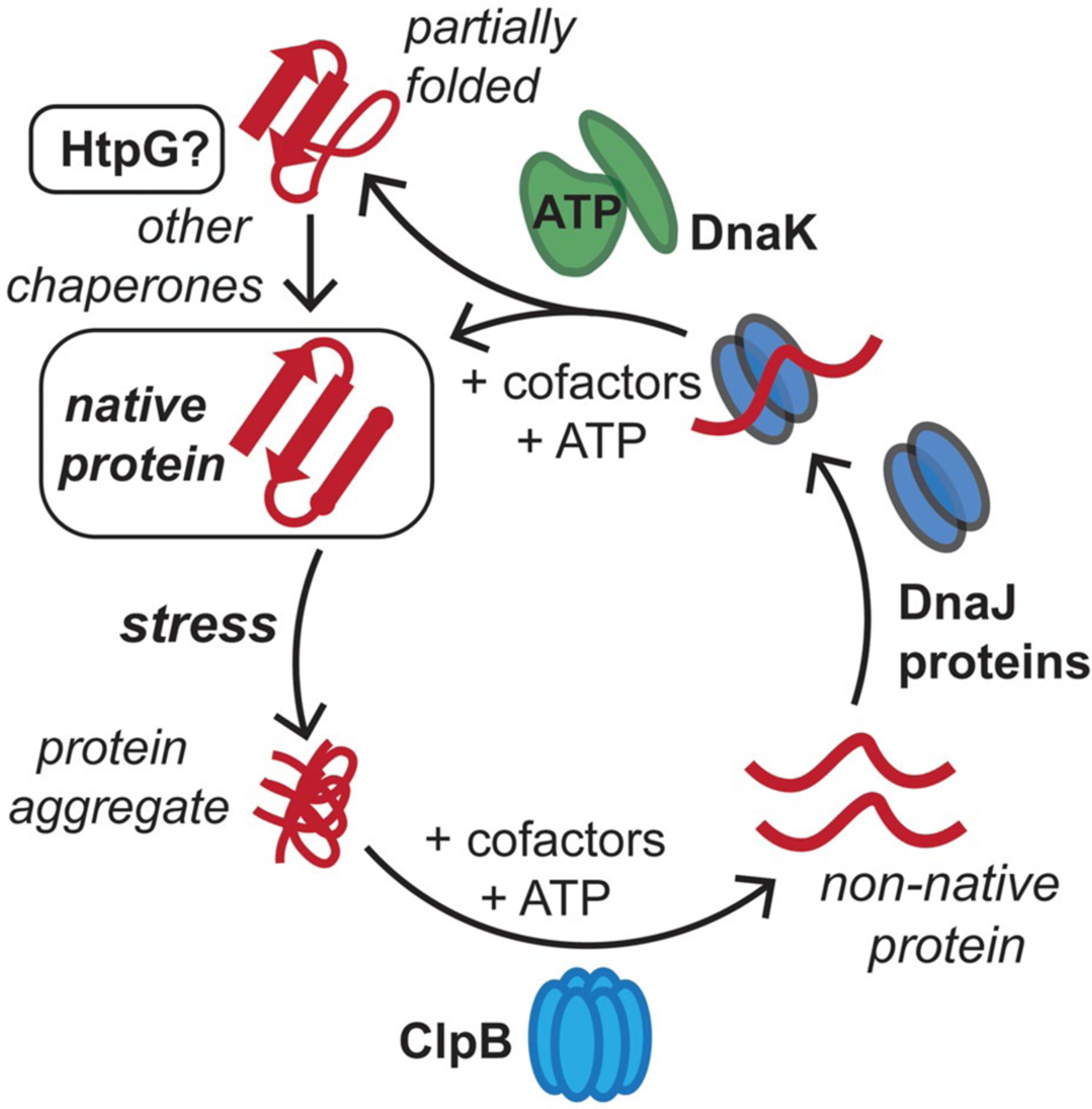
Conserved mycobacterial protein chaperones reactivate aggregated proteins that result from stresses in the host. M. tuberculosis (Mtb) cells are exposed to reactive oxygen and nitrogen species, along with antibiotics, in the host, which can lead to protein aggregation. The conserved bichaperone system consisting of ClpB and DnaK, along with cofactor proteins including DnaJs and GrpE, can help reactivate these proteins to reform native protein folds using ATP as an energy source. Here, we assess whether Mtb HtpG assists the ClpB-DnaK chaperone network in protein folding and homeostasis in response to stress. This figure has been modified from (Lupoli et al., 2018)
Nonreplicating bacterial cells have been shown to contain greater amounts of protein aggregates than those in an actively replicating state (Fay and Glickman, 2014; Kwiatkowska et al., 2008; Maisonneuve et al., 2008; Navarro Llorens et al., 2010; Vaubourgeix et al., 2015). Factors promoting this increase in protein aggregation include suboptimal function of protein quality control machinery, lack of nutrients to support protein synthesis, and an increase in cellular oxidants sufficient to cause irreversible protein modifications (Josefson et al., 2017). Mtb cells lacking clpB exhibit defects in recovery after achieving a stationary growth phase, which is one non-replicative state. In addition, Mtb ΔclpB cells are more sensitive than wild type to isoniazid, a frontline TB drug that induces oxidative damage in mycobacteria, and are attenuated in a mouse infection model (Vaubourgeix et al., 2015).
Given the functional importance of both DnaK and ClpB in mycobacterial survival, both proteins have been suggested as noncanonical targets for antimycobacterial chemotherapy (Lupoli et al., 2018; Mohammadi-Ostad-Kalayeh et al., 2017). Several small molecules inhibit protein degradation machinery in mycobacteria (Compton et al., 2013; Lin et al., 2009) and some natural products may disable chaperones in other bacterial species, such as Escherichia coli (Kragol et al., 2001; Scocchi et al., 2009, 2011). However, we lack a fundamental picture of how other molecular players aid DnaK and ClpB in the bacterial stress response to unfolded proteins. A better understanding of these chaperone networks would grant us insight into mycobacterial protein biochemistry and enhance our ability to target mycobacterial proteostasis. Such an approach could synergize with host-imposed stresses.
Here, we aimed to identify genes that aid or complement clpB function in maintaining protein homeostasis in Mtb. Using Tn-seq (transposon mutagenesis coupled to sequencing) analysis (DeJesus et al., 2017; Long et al., 2015; Sassetti et al., 2001, 2003), we find that many of the genes predicted to be important for survival in cells lacking clpB encode oxidative stress response proteins. We identify a proteostasis-related gene, htpG, that encodes an annotated chaperone (Bardwell and Craig, 1988) whose homologs facilitate protein folding along with DnaK in other organisms (Genest et al., 2011). We show that Mtb ΔclpB cells are sensitive to oxidants, especially diamide, while ΔhtpG cells are not. While htpG, like clpB, is not essential in Mtb (Lopez Quezada et al., 2020; Vaubourgeix et al., 2015), we find that cells lacking both nonessential chaperones are hypersensitive to host-like stresses that induce a nonreplicating state. Finally, a small molecule probe that targets HtpG mimics the effect of htpG disruption in ClpB-deficient Mtb. Our results illuminate the consequences of distorting the levels of Mtb chaperones and suggest targets whose inhibition could increase the vulnerability of Mtb to conditions it encounters in the host.
2 |. RESULTS
2.1 |. Genome-wide transposon mutagenesis analysis suggests a relationship between Mtb clpB function and stress response genes
Tn-seq uses next-generation sequencing methods for whole-genome evaluation of the essentiality of genes by quantifying the frequency of transposon-induced mutations in populations of cells under different conditions (Chao et al., 2016; DeJesus et al., 2017; Sassetti et al., 2003). We used Tn-seq to assess the genetic interactions between clpB and otherwise nonessential genes in Mtb by comparing wild-type and ΔclpB transposon libraries. The mycobacteriophage ϕMycoMarT7 was used to randomly deliver a transposon that carries a kanamycin resistant cassette to one of the greater than 70,000 TA dinucleotide insertion sites in the Mtb genome (Long et al., 2015; Sassetti et al., 2001, 2003) prior to outgrowth of the population on a solid medium under standard conditions for ~22 generations (based on a generation time of ~20 hr for Mtb) (Gill et al., 2009). We prepared two independent libraries in each strain and identified sites of TA insertions (Figure S1, Table S1). Each library covered 50% to 80% of TA sites with 4 to 6 million unique reads (Table S2). Replicate libraries showed good agreement. We combined their data (Figure S2) and analyzed them using published methods that account for stochastic variation between Tn insertion data sets (Figure 2) (Chao et al., 2013; Kieser et al., 2015; Pritchard et al., 2014). As indicated in Figure 2, fewer transposon insertions were isolated in the Mtb ΔclpB background compared to the wild type. One explanation for this outcome is that Mtb ΔclpB cells have a growth defect on agar relative to wild type (Vaubourgeix et al., 2015). Since outgrowth on agar is required for the isolation of transposon mutants, insertions in some genes led to effects that synergized with this ΔclpB deficiency. Based on a p-value cutoff of .001, >100 genes showed interactions with clpB (Table S3), similar to what has been seen for other genes that serve as pathway “hubs” in Mtb (Griffin et al., 2011; Joshi et al., 2006; Nambi et al., 2015).
FIGURE 2.
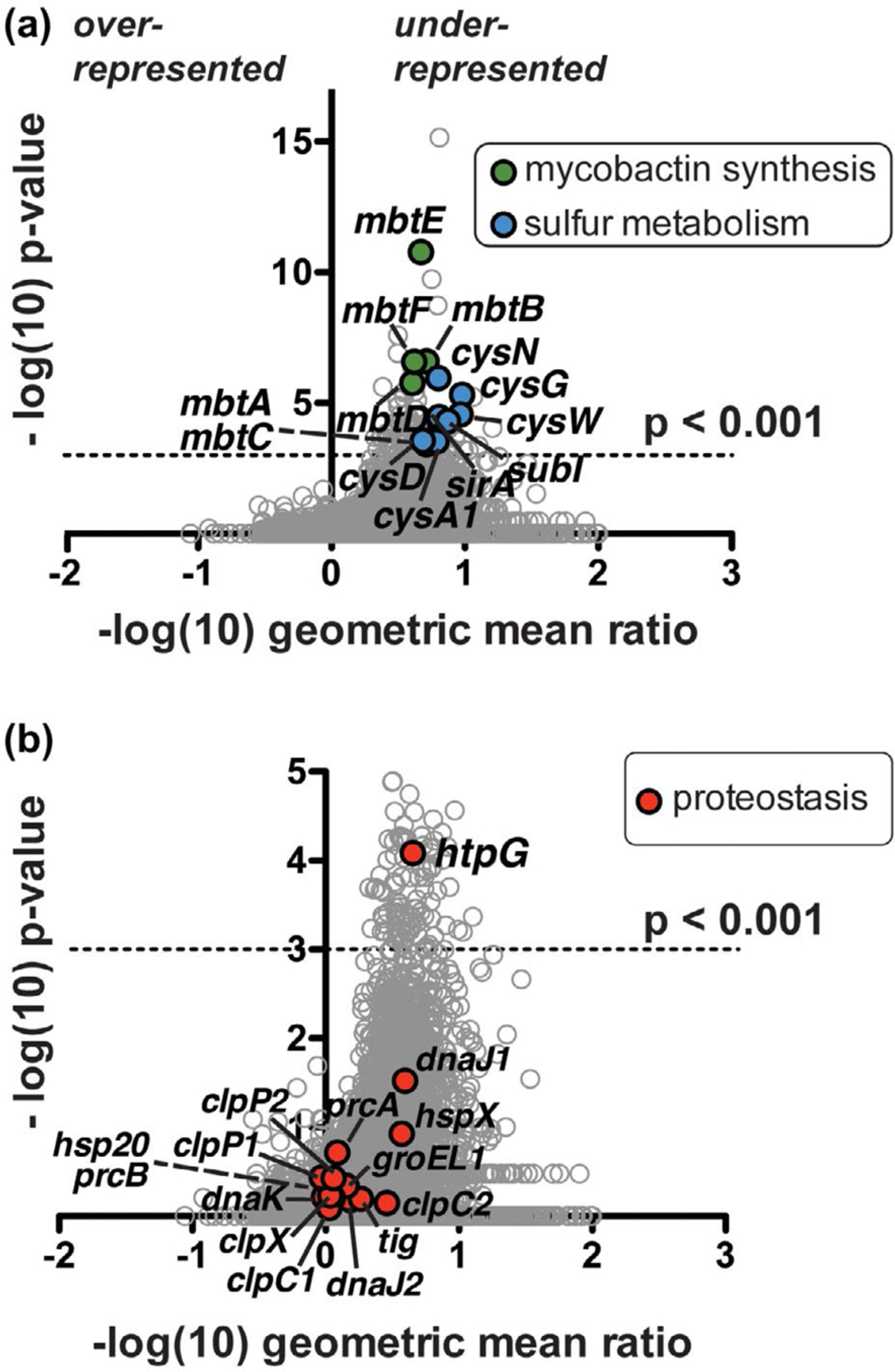
Tn-seq analysis suggests Mtb clpB function is related to oxidative stress response genes and a proposed protein chaperone-encoding gene, htpG. (a) Comparison of Mtb ΔclpB transposon library insertions to that of wild type. Each locus (circle) is plotted based on its p-value, determined by a Mann-Whitney U test, and fold change in sequence reads of the knockout relative to the wild-type background (Pritchard et al., 2014). Over 100 genes are significantly (p < .001) under-represented (right side of y-axis) in the ΔclpB background compared to the wild type; highlighted genes include mycobactin synthetic genes (mbt, green) and sulfur metabolism genes (blue), many of which have been linked to the oxidative stress response. (b) Enlarged representation of data from part A highlighting all annotated proteostasis genes (red), of which only the predicted chaperone gene htpG shows a significant genetic interaction with clpB
Pathway analysis indicated that various mycobacterial metabolic and/or stress response genes had genetic interactions with clpB. Genes implicated in the oxidative stress response (Lin et al., 2016; Voskuil et al., 2011) are highlighted in Figure 2a. A major subset are mycobactin biosynthesis (mbt) genes, in which insertions were under-represented in clpB deficient cells (Figure 2a, green). Mycobactins are small molecule siderophores produced by mycobacteria that recruit ferric ions from the environment during iron starvation, thereby maintaining intracellular Fe2+ (Gold et al., 2001; Quadri et al., 1998; Rodriguez et al., 2002). Lack of insertions in mbt genes suggests that defects in iron acquisition are more compromising to Mtb without clpB than to wild-type cells. The second class of oxidative stress response genes highlighted in Figure 2a (blue) are cysteine biosynthesis and sulfur metabolism genes (cys genes; sirA, subI). Mutations in these genes are likely under-represented in Mtb ΔclpB transposon libraries due to the compensatory effect of free thiols such as cysteine and its intermediates in buffering Mtb against oxidants (Lin et al., 2016; Nambi et al., 2015), which evidently becomes especially important when cells lack clpB.
A search for all annotated mycobacterial proteostasis genes in our data set (Figure 2b, red) revealed that only one gene (Rv2299c) was significantly under-represented in Mtb ΔclpB libraries (Figure S3, Table S4), suggesting that mutation of Rv2299c is detrimental to cells that lack clpB. Rv2299c is annotated as htpG, which encodes a protein chaperone that is the prokaryotic homolog of the well-studied eukaryotic chaperone Hsp90 (Taipale et al., 2010). E. coli HtpG interacts with DnaK (Genest et al., 2011), but mycobacterial HtpG had not yet been characterized. Mycobacterial DnaK cooperates with ClpB in the resolution and reactivation of protein aggregates (Fay and Glickman, 2014; Lupoli et al., 2016; Vaubourgeix et al., 2015). Accordingly, we speculated that HtpG may play a distinct role from ClpB in Mtb proteostasis networks.
2.2 |. Mtb cells lacking clpB, but not htpG, are hypersensitive to heat and oxidative stress
We evaluated the response of Mtb cells lacking either ClpB or HtpG to common proteotoxic stresses. We first verified that HtpG is present in log-phase Mtb cells and a complemented strain carrying a constitutively expressed copy of htpG, but is not present in Mtb ΔhtpG cells (Lopez Quezada et al., 2020) (Figure S4). As seen for Mtb ΔclpB strains (Vaubourgeix et al., 2015), Mtb ΔhtpG cells grew similarly to wild type in nutrient-rich liquid medium (Figure 3a, inset) over nearly a month.
FIGURE 3.
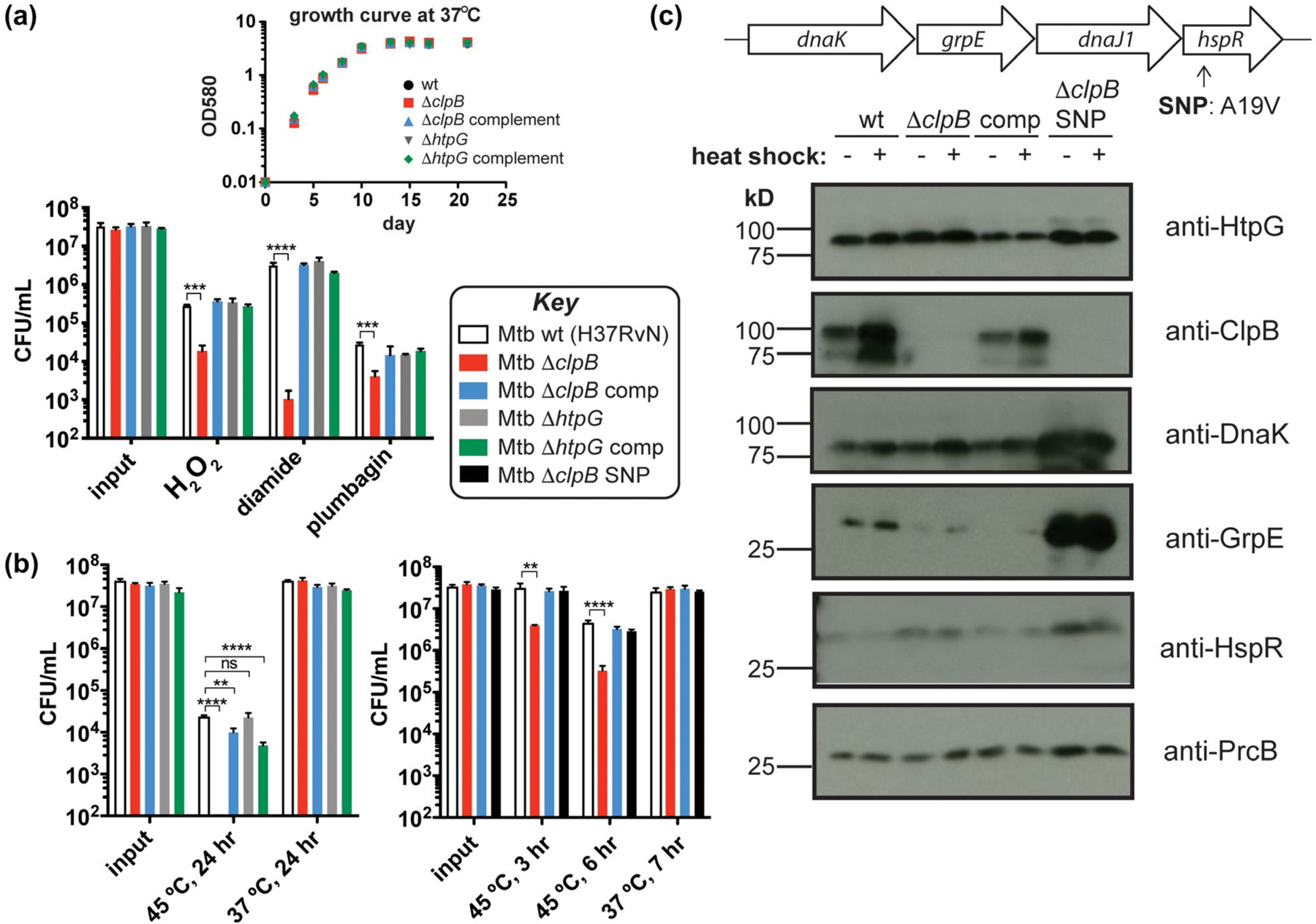
Mtb chaperones play different roles in response to common proteotoxic stresses. (a) Colony-forming unit (CFU) measurements of Mtb wild-type (H37Rv), ΔclpB, ΔhtpG, and corresponding complemented strains treated with H2O2 (5 mM, 4 hr), diamide (50 mM, 8 hr), or plumbagin (0.25 mM, 5 hr) (Lin et al., 2016) show that loss of clpB renders cells more sensitive to various sources of oxidative stress. Inset: growth curves of these strains at 37°C illustrates that there are no defects without added stress. (b) Strains from part A heat shocked at 45°C for 24 hr show that Mtb lacking clpB has a defect in recovery as measured by CFU (left). A ΔclpB strain with a SNP in hspR is protected from this recovery defect following heat stress at shorter time points (right). (c) Western blots of protein chaperone and cofactor levels ± heat shock (45°C, 3 hr) show that point mutation (A19V) of HspR in the ΔclpB SNP strain results in increased amounts of proteins encoded in the dnaK operon compared to strains lacking this SNP (even without heat shock). Wild-type HspR negatively regulates the expression of the dnaK operon (shown on top) and clpB, among other heat shock response genes. **p < .0021, ***p < .0002, ****p < .0001, ns = not significant, one-way ANOVA was used for group comparison (n = 3, and error bars represent standard deviation (SD) for parts A and B)
Several reports have implicated clpB, cys, and mbt genes in the oxidative stress response (Burns-Huang and Mundhra, 2019; Nambi et al., 2015; Tripathi et al., 2020; Vaubourgeix et al., 2015; Voskuil et al., 2011). We exposed Mtb cells with and without clpB or htpG to chemical oxidants using a reported survival assay (Figure 3a) (Lin et al., 2016). Cells lacking htpG behaved like wild type under all conditions tested. In contrast, Mtb ΔclpB cells showed ~1 log10 reduction in survival compared to wild type when treated with H2O2 or the superoxide generator plumbagin, and ~3.5 log10 decrease compared to wild type in the presence of the thiol-specific oxidant diamide. Survival defects in Mtb ΔclpB cells were restored to wild-type levels upon complementation. For comparison, we evaluated the effect of diamide on an Mtb mbt deletion strain, ΔmbtK (Xu et al., 2017), that lacks mycobactins and shows growth defects only in iron-deficient conditions (Madigan et al., 2015). Mtb ΔmbtK behaved similarly to wild type in response to diamide (Figure S5a). These observations reinforce that clpB is important for Mtb survival in the presence of oxidants and suggest that genes interacting with clpB may play different cellular roles depending on the type of proteotoxic stress.
Since protein chaperones are often annotated as “heat shock proteins” (Hsps) for their induction in response to an increase in temperature and their putative ability to counteract unfolding or aggregation resulting from heat shock (Balchin et al., 2016), we evaluated survival of chaperone mutants following heat exposure (Jastrab et al., 2017; Tripathi et al., 2020). After incubating Mtb at 45°C for 24 hr, we observed no colonies of cells deleted of clpB unless the strain was complemented with a wild-type copy of clpB (Figure 3b, left). In contrast, Mtb cells lacking htpG survived as well as wild type. Both of the complemented strains showed some defects in survival, perhaps due to non-native levels of chaperones. Cells lacking an mbt gene showed similar heat sensitivity as wild type (Figure S5b).
Next, we sought to test the effect of loss of htpG on Mtb in the presence of the full range of stresses present in an experimental host. Mtb cells lacking clpB are attenuated in the chronic phase of infection in C57BL/6 mice, a widely used model (Vaubourgeix et al., 2015). In the same model, Mtb ΔhtpG and complemented strains replicated and persisted like wild type for up to 180 days as judged from the numbers of viable Mtb recovered from lung, spleen, and liver. Thus, HtpG does not play a major role in establishing or maintaining infection of Mtb in C57BL/6 mice (Figure S6). It should be noted that the C57BL/6 strain does not form hallmark caseating necrotic granulomas and so this model does not recapitulate critical features of the biology of Mtb infection in Mtb’s natural host, humans (Hoff et al., 2011; Rhoades et al., 1997).
Given that Tn-seq analysis suggested that cells lacking functional copies of both clpB and htpG, and not htpG alone, were defective, we set out to examine the effect of mutating both nonessential chaperones on Mtb cells under conditions that damage the proteome.
2.3 |. Overexpression of protein chaperones rescues cells lacking clpB under proteotoxic stress
As a first step toward assessing the effect of knockdown of htpG in clpB-deficient Mtb, we initially transformed Mtb ΔclpB cells with a plasmid that integrates an additional copy of htpG into the chromosome (Kim et al., 2011). This reverted the slow growth phenotype on solid agar. That observation prompted us to perform whole-genome sequencing (WGS) of the resulting strain, which revealed a single nucleotide polymorphism (SNP) in hspR that resulted in a point mutation (Ala19Val) in the protein HspR (Figure S7). HspR is part of the dnaK operon (Figure 3c, top) and has been shown to act as a repressor of the operon, as well as of other chaperone genes, including clpB (Das Gupta et al., 2008; Grandvalet et al., 1999; Stewart et al., 2002). We confirmed by WGS and targeted sequencing of hspR that none of the other relevant strains (H37RvN wild type, Mtb ΔclpB, or Mtb ΔhtpG) possess this SNP (Figure S7). To assess the phenotypic consequences of the SNP, we removed the extra copy of htpG and isolated the resulting strain Mtb ΔclpB SNP (hspR A19V). Since ΔclpB cells showed significant differences in heat sensitivity compared to wild type (Figure 3b, left), we examined the response of Mtb ΔclpB strains with and without hspR A19V following a briefer heat shock. As seen in Figure 3b (right), Mtb ΔclpB hspR A19V recovered from heat shock (t = 3 and 6 hr) in a similar manner to the wild-type and complemented strains, while Mtb ΔclpB lost ~1 log10 in viability compared to all the other strains. Similarly, ΔclpB hspR A19V behaved similar to the Mtb ΔclpB complemented strain in response to hydrogen peroxide (Figure S8).
A recent report identified suppressor mutations in Mtb hspR in strains lacking the gene pafE, which encodes a proteasome accessory factor involved in cellular protein degradation (Jastrab et al., 2017). These mutations were found to result in overexpression of DnaK, ClpB, and HspR. Since A19V is in the predicted DNA-binding domain of HspR, similar to the observed hspR SNPs (Jastrab et al., 2017), we hypothesized that the Mtb ΔclpB hspR A19V strain may not exhibit properly regulated chaperone expression. This was confirmed by western blot of Mtb lysates after incubation of cells under normal growth conditions (37°C, 3 hr) or following a short heat shock (45°C, 3 hr) (Figure 3c). In wild-type cells, ClpB protein levels were increased after heat shock, as expected. While HtpG and proteasome component PrcB levels were not significantly changed in the tested conditions, there was an excess of DnaK, GrpE, and HspR in the Mtb ΔclpB hspR A19V strain with and without heat compared to the parent strain. These results reinforce the observation that HtpG alone is not involved in the heat shock response (Figure 3b, left, with and without heat shock) and suggest that the HspR A19V mutation leads to loss of repressor function, as seen for other reported HspR point mutants (Jastrab et al., 2017). These phenotypes reinforce the importance of clpB in the heat shock and oxidative stress response and further highlight the differences between clpB and htpG in the cell’s response to proteotoxic stress, as previous work has indicated that htpG expression is not regulated by the same repressor as dnaK and clpB (Stewart et al., 2002).
2.4 |. Mtb cells lacking two nonessential chaperones are hypersusceptible to host stresses
Because allelic exchange strategies led to the isolation of suppressor mutants in Mtb ΔclpB, we resorted to a mycobacterial CRISPRi (clustered regularly interspaced short palindromic repeats interference) approach to deplete htpG upon the addition of anhydrotetracycline (ATc) in cells with and without clpB (Rock et al., 2017) (Figure S9). We first evaluated the effect of heat. While we saw no effect on growth due to lack of both ClpB and HtpG at 37°C (Figure 4a, left, red open squares), under a mild heat stress of 40°C, we saw a prolonged lag phase and decrease in doubling time in cells lacking clpB, which was exacerbated by the depletion of HtpG (Figure 4a, right, compare open and closed red squares). In ΔclpB complemented cells, there was no effect of loss of HtpG under heat stress, underscoring that the growth defect seen was due to combined loss of both chaperones. Western blots confirmed that these strains maintained knockdown of htpG in the presence of ATc over the indicated time period (Figure 4b). In order to determine if this enhanced heat sensitivity was due to increased cellular protein aggregation, we analyzed insoluble and soluble fractions of lysates for changes in reporter chaperone levels following heat shock. We found that the essential chaperone DnaK, and a small heat shock protein (sHsp20) known to be upregulated upon heat stress (Stewart et al., 2002), both accumulate in the insoluble fraction in heat-treated cells lacking ClpB and HtpG compared to those lacking HtpG alone or in wild-type cells (Figure S10), suggesting an increase in total protein aggregation. Hence, upon loss of ClpB, HtpG function becomes important in maintaining proteostasis following heat shock.
FIGURE 4.
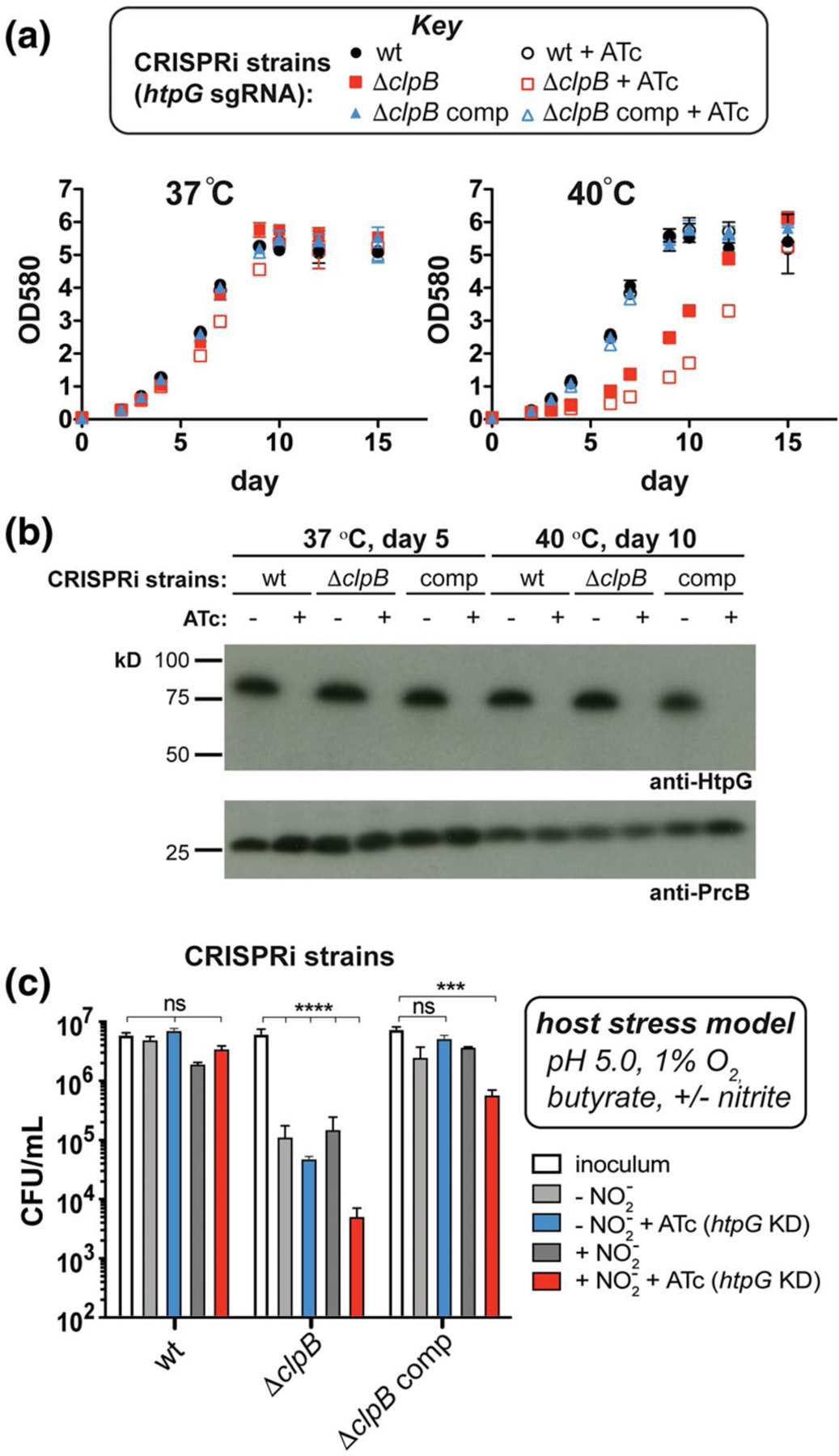
Depletion of HtpG from Mtb cells lacking clpB exacerbates survival defects under model host stresses. (a) Growth curves measured at 37°C and 40°C of Mtb wild-type, ΔclpB, and complemented strains containing sgRNA targeting htpG co-expressed with dCas9 (+ATc) to facilitate CRISPRi-mediated transcriptional repression of htpG. While strains grow similarly at 37°C, there is an increased lag phase in ΔclpB compared to wild type at 40°C that is enhanced when htpG is repressed. (b) Western blots of lysates purified from strains in part (a) at indicated time points and temperatures verify that HtpG is not expressed in cells when ATc is added (PrcB was used as a loading control). (c) Survival of Mtb wild-type, ΔclpB, and complemented CRISPRi strains (described in part a) after t = 7 days under stresses that mimic the host environment (Gold et al., 2015) show that strains lacking both ClpB and HtpG are more sensitive to these stresses than wild type, especially with nitric oxide generated (inoculum was taken at day 0). ***p < .0002, ****p < .0001, one-way ANOVA was used for group comparison (n = 3, error bars represent SD, some error bars may be too small to visualize)
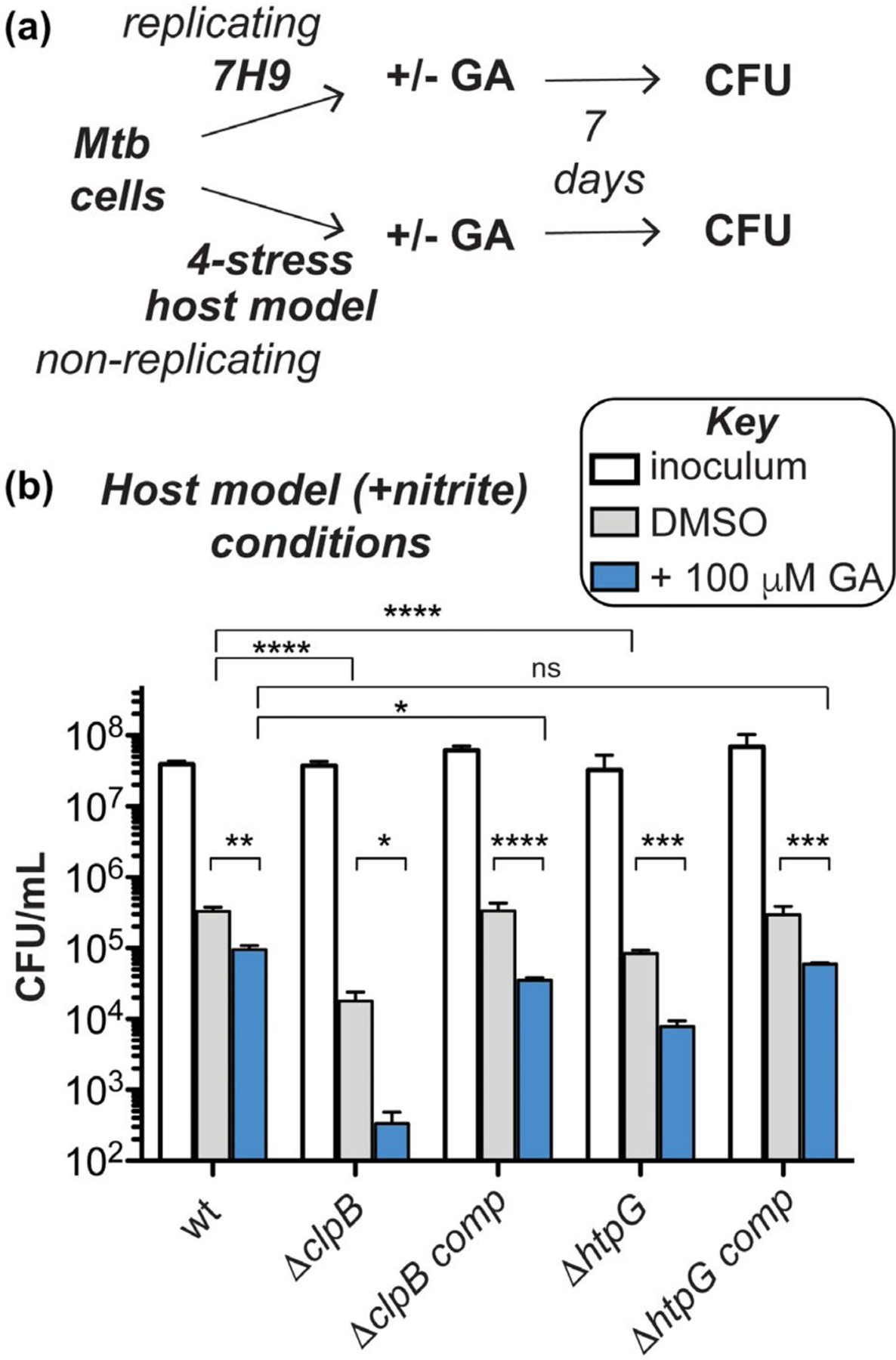
To evaluate the importance of clpB and htpG under more physiologically relevant conditions, we assessed survival of the CRISPRi-mediated depletion strains following exposure to stresses like those experienced in the host. Diamide and hydrogen peroxide affected the ClpB-deficient strain similarly with and without htpG knockdown (Figure S11). We then exposed Mtb htpG depletion strains to a three- or four-stress model of host-relevant stresses (Bryk et al., 2008; Gold et al., 2015) involving hypoxic conditions (1% O2) in an acidic medium (pH 5.0) containing a fatty acid (butyrate) as the sole carbon source, with or without nitrite (Ehrt and Schnappinger, 2009; Gold et al., 2015). As seen in Figure 4c, wild-type cells did not grow during 7 days of observation under these conditions, demonstrating a bacteriostatic response (compare inoculum to all conditions tested in wt). There was no significant change in survival when htpG alone was depleted (compare wt inoculum (white) to plus ATc (blue and red)). In contrast, Mtb ΔclpB cells showed a reduction (>1.5 log10 relative to the inoculum) in survival without nitrite (light gray, ΔclpB) and with nitrite (dark gray, ΔclpB). This phenotype was exacerbated in cells lacking both ClpB and HtpG under conditions containing nitrite (red, ΔclpB) (>3 log10 decrease relative to the inoculum) compared to those in the absence of nitrite (blue, ΔclpB) (>2 log10 decrease relative to the inoculum). The survival defects were largely abrogated in the Mtb ΔclpB htpG knockdown strains upon complementation with clpB (see all conditions for ΔclpB comp). Loss of htpG in the ΔclpB complemented strain in the presence of nitrite (red, ΔclpB comp) led to >1 log10 decrease in cell count relative to the inoculum. Similarly, lack of full complementation has been observed during recovery from stationary phase in ΔclpB comp cells (Vaubourgeix et al., 2015), perhaps due to non-native levels of ClpB in this strain under some stress conditions (as seen with added heat in Figure 3c). These data demonstrate that loss of both chaperones leads to synergistic survival defects in response to host-relevant stresses.
Mycobacterial HtpG interacts with DnaK but not ClpB. Because htpG and clpB appear to play different roles in proteostasis in stressed cells, we next sought to assess biochemical interactions involving HtpG within the mycobacterial chaperone network. We purified His-HtpG and performed pull-downs in Mtb cell lysates. SDS-PAGE showed that a protein band at the approximate molecular weight of DnaK (~70 kD) became enriched when His-HtpG was affinity purified after incubation with cell lysates (Figure S12a). Western blot (Figure S12b) and mass spectrometry (Table S5) verified that this band was Mtb DnaK. We did not detect enrichment of bands migrating with apparent molecular weights similar to that of ClpB (~93 kD), nor did we detect a direct interaction between HtpG and ClpB in in vitro pull-down assays (Figure S13). In many organisms, Hsp70s (DnaK) interact with Hsp90s (HtpG) and Hsp104s (ClpB), and are aided by other cofactor proteins that regulate their activity (Genest et al., 2019; Schumacher et al., 1996; Taipale et al., 2010). Our results suggest that ClpB and HtpG are not involved in a single proteostasis pathway in mycobacteria and are instead part of a broader network that is connected by DnaK.
2.5 |. The Hsp90 probe geldanamycin disrupts Mtb HtpG in vitro
Mtb HtpG shares considerable homology with the well-characterized E. coli HtpG (~45% sequence identity). However, some mycobacterial chaperones function in a distinct manner from their E. coli counterparts (Lupoli et al., 2016; Raju et al., 2012; Rock et al., 2015; Srivastava et al., 2016). Since mycobacterial HtpGs had not yet been studied in vitro, we assessed the biochemical features of Mtb HtpG with experiments informed by studies on Hsp90s.
Like many chaperones, Hsp90 proteins function in an ATP-dependent manner in a multimeric state (Ali et al., 2006; Shiau et al., 2006). By size exclusion chromatography, purified Mtb HtpG eluted at the molecular weight of an oligomer (Figure S14). It is difficult to assess the oligomeric state of Hsp90s by size exclusion analysis using known standards because Hsp90 dimers are known to exhibit extended rather than globular structures (Jakob et al., 1995; Koyasu et al., 1986; Spence and Georgopoulos, 1989). To better assess the oligomeric state of Mtb HtpG, we examined contacts between HtpG monomers using site-specific protein cross-linking. We inserted the UV-activatable unnatural amino acid para-benzoyl-phenylalanine (BpF or B) into HtpG by amber suppression (Chin et al., 2002). Based on a structural model of HtpG (Kelley et al., 2015), we chose 17 sites for insertion distributed around the surface of the protein, with a bias toward aromatic or hydrophobic residues likely to be present at protein–protein interfaces (Figure 5a). As expected, the resulting mutants were only expressed as full-length proteins when BpF was present in the growth medium (Figure S15). We then assessed cross-linking by SDS-PAGE analysis with or without preceding UV irradiation (Figure 5b), using a standard protein ladder to approximate molecular weight (Figure S16). As seen in Figures 5a,b and S17, only residues F10, W197, and F635 along one interface of Mtb HtpG consistently formed covalent dimers (Figure 5b, lanes 4, 6, and 12). This is a homologous interface to that in E. coli HtpG dimers (Shiau et al., 2006).
FIGURE 5.
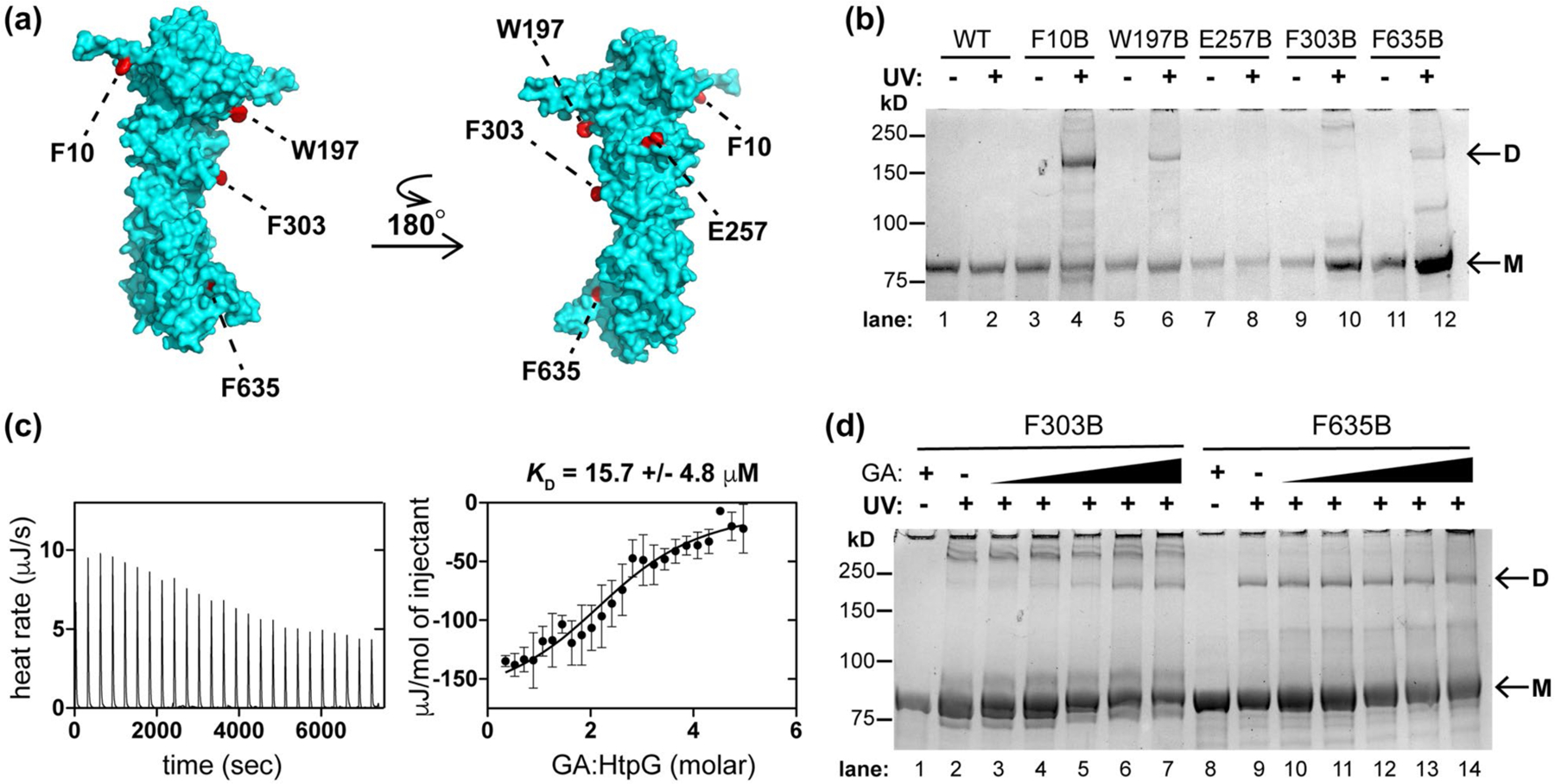
Mtb HtpG dimerizes in vitro and interacts with Hsp90 probe geldanamycin (GA). (a) Model of Mtb HtpG (Phyre2) (Kelley et al., 2015) with relevant residues highlighted that are sites of point mutation to the UV-activatable crosslinking amino acid para-benzoyl phenyalanine (BpF or B). (b) SDS-PAGE analysis of Mtb HtpG BpF mutants at indicated residues with and without prior UV irradiation (monomer bands are indicated as “M” and dimer bands are indicated as “D”). F10, W197, and F635 consistently mediate the formation of dimer. (c) ITC analysis of Mtb HtpG (47 µM) with GA (raw data is shown on left, analysis of average of three replicates with SD shown on the right) illustrates µM affinity. (d) SDS-PAGE analysis of indicated Mtb HtpG BpF mutants without (plus 400 µM GA) and with UV irradiation (in the presence of 0, 10, 50, 100, 200, and 400 µM GA) suggests that F303 turns into the dimer interface upon binding GA, while F635 is oriented in the dimer interface regardless of ligand binding. At least three identical analyses were performed for each experiment shown
The natural product geldanamycin (GA) was isolated and characterized as an antibiotic (DeBoer et al., 1970). It binds the N-terminal nucleotide-binding domain of eukaryotic Hsp90s (Prodromou et al., 1997; Stebbins et al., 1997) but does not bind all bacterial HtpGs (Millson et al., 2011). We next assessed the interactions of this chemical probe with mycobacterial HtpG by isothermal titration calorimetry (ITC) of Mtb HtpG in the presence of excess MgCl2 after extensive dialysis to remove bound nucleotide (Rauch and Gestwicki, 2014). GA was calculated to bind Mtb HtpG with a KD ~ 15.7 µM (Figures 5c and S18), an affinity ~10-fold less than that measured for GA with purified yeast Hsp90 (KD ~ 1.2 µM) (Roe et al., 1999) and ~3- fold less than that found with E. coli HtpG (KD ~ 5.5 µM) (Millson et al., 2011). The differences in affinity are likely due to sequence variation in the binding sites (E. coli and Mtb HtpG N-terminal domains are each <50% identical in sequence to that of yeast Hsp90). We purified yeast Hsp90 (S. cerevisiae Hsp82) to compare its activity to that of mycobacterial HtpG (Obermann et al., 1998). Both proteins exhibited low ATPase activity that was inhibited by GA (Figure S19), as seen for yeast Hsp90 (Lotz et al., 2003; Obermann et al., 1998; Owen et al., 2002).
GA induces changes in the overall structure of Hsp90s (Grenert et al., 1997; Stebbins et al., 1997). Therefore, we examined the effect of GA on BpF-mediated cross-linking of Mtb HtpG. Addition of increasing concentrations of GA enhanced dimerization of HtpG in the F303B mutant (Figure 5d, lanes 2–7), which typically forms a previously noted higher order oligomer (Jakob et al., 1995) following irradiation (Figure 5b), suggesting a conformational change in the N-terminal domain upon binding to GA. In contrast, there was no observed change in HtpG dimerization via a BpF residue in the C-terminus (F635, lanes 9–14) or other residues that typically mediate dimer formation in the absence of GA (Figure S20); since, C-terminal domains of Hsp90s are denoted “oligomerization” domains (Stebbins et al., 1997; Wearsch and Nicchitta, 1996). These data demonstrate that GA binds to dimeric Mtb HtpG and modulates its ATPase activity and structure.
2.6 |. Small molecule inhibition of HtpG in stressed Mtb cells causes survival defects
The foregoing results encouraged us to test the impact of GA on HtpG’s function in Mtb. We assessed cell survival in the presence of GA upon loss of htpG or clpB under replicating conditions or the nonreplicating conditions of the four-stress host model (Figure 6a). Under replicating conditions, GA did not affect mycobacterial cell growth (Table S6) or survival (Figure S21), even at high concentrations, as suggested (Johnson et al., 2019). However, GA reduced survival by >50-fold of Mtb cells lacking clpB under stress conditions (Figure 6b), compared to a ~3-fold loss in cell count for wild-type cells and ~10-fold loss for ΔhtpG cells. The result for ΔhtpG cells establishes that GA has off-target effects in Mtb under nonreplicating conditions. Cell death was slightly enhanced in cells with restored, but non-native concentrations of ClpB (compare wt to ΔclpB comp or ΔhtpG comp with GA added), as seen in Figure 4c. Overall, the survival trends seen in Mtb htpG genetic depletion strains (Figure 4c) were phenocopied by the small molecule inhibitor.
FIGURE 6.
Stressed Mtb cells lacking clpB are hypersensitive to GA-mediated inhibition of HtpG. (a) Schematic of experimental set-up for evaluating chemical inhibition of HtpG by GA with and without nonessential chaperones under replicating (nutrient-rich) and nonreplicating (host stress model with nitrite) conditions as shown in Figure 4c. (b) CFU analysis demonstrates that survival is significantly reduced under nonreplicating conditions for all strains tested, but there is no change under replicating conditions (Figure S21). Compared to Figure 4c, these results suggest that GA addition phenocopies genetic depletion experiments, but might exhibit off-target effects or enhance cell death through different modes of action. *p < .0332, **p < .0021, ***p < .0002, ****p < .0001, two-way ANOVA or student’s t test was used for comparison (n = 3, error bars represent SD)
3 |. DISCUSSION
Information has been lacking on the biological contributions of individual mycobacterial molecular chaperones to survival during host infection. We took a genetic approach to identify the molecular players in Mtb that function in addition to ClpB to maintain protein homeostasis. Comparison of Mtb wild-type and ΔclpB mutant libraries by Tn-seq predicted that loss of many genes involved in the cell’s oxidative stress response would aggravate defects due to loss of clpB. These genes include those responsible for mycobactin biosynthesis (mbt) and cysteine biosynthesis (cys and others). Transcriptomic experiments have shown that clpB along with several mbt and cys genes are upregulated in cells exposed to chemical oxidants (Hatzios and Bertozzi, 2011; Manganelli et al., 2002; Pinto et al., 2004; Voskuil et al., 2011). As we and others have observed (Lin et al., 2016; Nambi et al., 2015), decreases in free cysteine or thiols derived from cysteine lead to greater defects in cells that already lack a component of the oxidative stress response. Indeed, we found that Mtb ΔclpB cells are more sensitive to certain oxidants, especially the thiol-specific oxidant diamide. Diamide is a reactive electrophile that thiolates cysteine residues in bacterial proteins (Pöther et al., 2009). Our results suggest that Mtb cells lacking ClpB are compromised in their ability to protect or repair susceptible proteins and may rely heavily on free thiols to provide a buffer from oxidants.
In addition to a purported buffering capacity of thiols whose production is dependent on cys genes, both cys and mbt genes are upregulated when cells are exposed to intermediate levels of peroxide and thereby contribute to repair of iron-sulfur cluster proteins (Dragset et al., 2019; Voskuil et al., 2011). Fe-S prosthetic groups in these proteins undergo redox reactions that fulfill critical biological roles but also render them more susceptible to oxidative damage than most other protein classes (Beinert and Kiley, 1999). Repair of these damaged redox proteins requires precursors for the prosthetic groups, which include sulfur-containing components dependent on cys gene products and iron recruited by mbt gene products (Pinto et al., 2004; Rodriguez et al., 2002). While increased production of mycobactins can also be detrimental to cells, since enhanced cellular uptake of Fe2+ can enhance oxidative stress via the Fenton reaction (Winterbourn, 1995), we postulate that increased oxidative damage via the mbt pathway does not occur in cells lacking clpB.
The observation that mbt mutations are underrepresented in ΔclpB strains hints that ClpB and mycobactin function might be related. mbt genes are known to be upregulated not only after oxidative stress, but also under non-replicative (Muttucumaru et al., 2004) and iron starvation (Gold et al., 2001) conditions. Accordingly, a recent report has shown that iron acquisition is defective in Mtb ΔclpB cells starved for iron, perhaps because the production of mycobactins is somehow disrupted (Kurthkoti et al., 2017). Microbial iron scavenging appears to be important in the host, as an Mtb ΔmbtK strain deficient in mycobactin production was attenuated in mice (Madigan et al., 2015). Since our Tn-seq experiment was performed in ironrich conditions, our findings highlight the importance of mycobactin function under additional stresses, such as loss of clpB, which affects cellular proteostasis. We are currently investigating the role of ClpB in regulation of mbt gene expression and/or mycobactin biosynthesis, which may provide greater insight into the distinct role that ClpB plays in the oxidative stress response.
Strikingly, only one annotated proteostasis gene, htpG, showed a significant decrease in transposon insertions in cells lacking clpB compared to wild type. Other proteostasis gene products are known to function with mycobacterial ClpB, such as the protein folding-associated chaperone DnaK, and cofactors DnaJ1, DnaJ2, and GrpE (Fay and Glickman, 2014; Lupoli et al., 2016). Since dnaK and grpE are predicted to be essential in pathogenic mycobacteria (DeJesus et al., 2017; Fay and Glickman, 2014), and dnaJ1 and dnaJ2 are known to be synthetic lethal in M. smegmatis (Lupoli et al., 2016), we observed a low frequency of mutations in these genes, as expected. However, it was unexpected that the nonessential chaperone groEL1 did not show a genetic interaction with clpB. Like clpB, groEL genes are upregulated in response to heat (Stewart et al., 2002) as well as oxidative (Dosanjh et al., 2005) and macrophage-imposed stresses (Monahan et al., 2001). However, Tn-seq analysis does not necessarily link genes that function in identical cellular pathways. Instead, this analysis often facilitates the identification of mutations that show synthetic phenotypes. Hence, as we anticipated from our Tn-seq analysis, our data collectively suggests that ClpB and HtpG play different roles in cellular proteostasis (see Figure 1).
HtpG it is part of the highly conserved Hsp90 family of protein chaperones. E. coli HtpG has been shown to play a role in protein folding and stabilization in concert with DnaK and cofactors (Figure 1) (Genest et al., 2011), as well as prevention of protein aggregation (Thomas and Baneyx, 2000). As we observed here, variation in heat shock recovery phenotypes between cells lacking htpG and clpB has been observed in E. coli. Since Mtb cells lacking clpB and htpG responded differently under the variety of stress conditions tested, these chaperones likely have differing roles in proteostasis depending on the type of stress experienced by the bacterium. Namely, our data support the idea that clpB is involved in the oxidative stress response and htpG is not.
A combination of modeling and experimental work has suggested that DnaK is the main bacterial “guardian” chaperone in response to oxidative stress or other damage to the proteome (Santra et al., 2018). While we found that HtpG interacts with DnaK in mycobacterial cell lysates, we did not detect a direct interaction between HtpG and ClpB, as seen in other organisms (Genest et al., 2019; Rosenzweig et al., 2013). We isolated a point mutation in hspR in Mtb ΔclpB cells that led to loss of function of the encoded repressor and overexpression of chaperone genes. This mutation reduced sensitivity of cells to heat, as seen for hspR suppressor mutations in an Mtb proteasome cofactor knockout strain (Jastrab et al., 2017), as well as peroxide. These observations can be rationalized by the ability of DnaK and cofactor proteins to perform “holdase” functions, by which proteins are protected from unfolding or aggregation by binding to chaperones under stress conditions (Perales-Calvo et al., 2018). Our experiments showed that HtpG cannot compensate for loss of ClpB, and that HtpG levels remain constant in wild-type and ΔclpB hspR A19V cells with and without heat treatment. Taken together, these results reinforce that DnaK acts as a hub through which HtpG and ClpB indirectly interact, and that DnaK can compensate for loss of ClpB in stressed cells when overexpressed with cofactors (Thomas and Baneyx, 2000). We circumvented the generation of hspR mutations to generate double chaperone mutants using a CRISPRi approach. We commend this approach to others studying chaperone biology in mycobacteria.
While deletion of hspR improves Mtb’s heat tolerance, it has been shown to decease Mtb’s survival in mice (Stewart et al., 2001). The attenuated growth of Mtb ΔhspR strains during infection was attributed to the overexpression and secretion of Mtb DnaK into macrophages, which is thought to promote secretion of other proteins, leading to a heightened immune response (Barrios et al., 1992). Hence, besides inhibiting the combinations of chaperones, another possible mechanism to promote mycobacterial death in the host could be to disrupt regulatory factors such as HspR to alter the native levels of chaperone expression.
In mycobacteria, htpG is not encoded in avirulent, species such as M. smegmatis, but is conserved in pathogenic species such as Mtb, M. leprae, M. bovis and M. marinum, suggesting that HtpG may be important for virulence. Our in vivo infection experiments were subject to limitations of the standard mouse model that was used. As a complementary approach, we used in vitro experiments that were compatible with mycobacterial CRISPRi strategies, and found that cells lacking clpB that were depleted of htpG or treated with the HtpG inhibitor GA showed significant loss in viability compared to wild type when exposed to multi-stress conditions that mimic those in the host, especially in the presence of reactive nitrogen species that induce a non-replicative state. This result indicates that htpG aids in Mtb’s response to nitrosative damage (Ehrt and Schnappinger, 2009), which was not predicted by transcriptomic analysis (Wattam et al., 2017). While GA treatment demonstrated off-target effects in cells treated under nonreplicating conditions, a wealth of available GA analogs can be tested for cellular HtpG selectivity (Hadden et al., 2006; Ueda et al., 2019). We recently reported that Mtb ΔhtpG shows fourfold more cidality than wild type in the presence of select cephalosporins under nonreplicating conditions (Lopez Quezada et al., 2020). Perhaps this family of beta-lactams will synergize with GA analogs to kill Mtb.
This study evaluated the genetic depletions and an active site inhibitor to probe chaperone biology and biochemistry in mycobacteria. A promising alternative approach to blocking individual chaperones’ activity is to interfere with the protein–protein interactions (PPIs) that are central to the process of protein folding by networks of chaperones, substrates, and cofactor proteins. These methods have been applied to eukaryotic Hsp90s for cancer treatments (Dutta Gupta et al., 2019). Our site-specific UV cross-linking results indicate dimerization sites and exposed surfaces that can be used for studies aimed at targeting PPIs of HtpG. Similar experiments could identify other PPIs in proteostasis pathways.
In sum, we have shown that disruption of two nonessential chaperones increases the death of Mtb under conditions designed to mimic those in the human host, that GA can be used to inhibit one of these nonessential chaperones in Mtb, and that Mtb cells lacking ClpB are highly susceptible to a thiol-based oxidant. It has been proposed that balanced chaperone expression is important for Mtb’s virulence (Stewart et al., 2001). If so, then even a disturbance in chaperone function that results in an upregulation of remaining cellular chaperones could grant the host an advantage over the pathogen. Strategies aimed at disabling several components of the proteostasis machinery by targeting PPIs are under investigation.
4 |. EXPERIMENTAL PROCEDURES
4.1 |. General materials, bacterial strains and culture conditions
M. tuberculosis H37Rv North (H37RvN) and mutant strains were cultured at 37°C with 5% CO2 in Middlebrook 7H9 medium (BD Difco) supplemented with 0.2% glycerol, 0.05% Tween 80, 0.5% bovine serum albumin (Roche), 0.2% dextrose, and 0.085% NaCl (called 7H9 complete/tween unless otherwise noted). E. coli strains were grown in Luria–Bertani (LB) medium (BD Biosciences). Antibiotics were used at the following concentrations in liquid culture unless otherwise noted: nourseothricin (25 µg/ml), hygromycin B (50 µg/ml for mycobacteria, 200 µg/ml for E. coli), zeocin (25 µg/ml), kanamycin (25 µg/ml), and carbenicillin (50 µg/ml). Primers were purchased from Invitrogen, and DNA sequencing was performed by Macrogen and Genewiz. Vectors and expression hosts were obtained from Novagen and Addgene. Relevant plasmids (Table S7), strains (Table S8) and primers (Table S9) are listed in the Supporting Information. PBS buffer (calcium chloride and magnesium chloride free) was from Gibco. Polyvinylidene fluoride (PVDF) membranes (Bio-Rad) and ECL Western blotting substrate (Pierce) or Clarity Western ECL substrate (Bio-Rad) were used for immunoblot experiments. All other chemicals were purchased from Sigma-Aldrich unless otherwise noted. Graphical data were plotted and analyzed using Prism software (GraphPad), unless otherwise noted.
4.2 |. Source or generation of antibodies
Anti-PrcB (Gandotra et al., 2010), anti-ClpB (Vaubourgeix et al., 2015), and anti-HspR (Jastrab et al., 2017) have been described previously. Anti-His-HRP (Penta-His HRP Conjugate, Qiagen, Cat# 34460) was used according to the manufacturer’s protocols. Anti-rabbit-HRP was a gift from the Glickman lab (Sloan-Kettering).
Recombinant tagless HtpG (~1.4 mg) was purified as previously described (Lopez Quezada et al., 2020) and used to immunize rabbits without Freund’s incomplete adjuvant, instead using TiterMax (Pierce). HtpG antiserum was further purified using purified HtpG with the AminoLink Plus Immobilization Kit (Thermo Scientific) following the manufacturer’s protocols.
For DnaK and GrpE antibodies, M. smegmatis dnaK and grpE were cloned into digested pET-His-SUMO vectors. Briefly, primers 44 and 45 (Table S9) and restriction endonucleases SacI/XhoI for grpE, and primers 43 and 46 and restriction endonuclease BamHI/HindIII for dnaK with M. smegmatis mc2155 genomic DNA as template were used to yield pAJF550 and pAJF551 (Table S7) prior to transformation into BL21-CodonPlus (DE3)-RIPL cells (Agilent). For each, a 500 ml LB culture containing kanamycin (40 µg/ml) and chloramphenicol (25 µg/ml) was grown to an OD600 of 0.175 and induced for 1 hr with 50 mM IPTG (isopropyl β-D-1-thiogalactopyranoside) at 37°C. Cell pellets were harvested by centrifugation (3,700g, 15 min, 4°C) and washed with Buffer A (50 mM Tris (tris(hydroxymethyl) aminomethane), 100mM NaCl, 10% glycerol, and 1mM EDTA (ethylenediaminetetraacetic acid), pH 8.0). Pellets were then frozen for storage overnight at −20°C. Frozen pellets were resuspended in 7 ml of Buffer A and lysed by sonication (Branson Digital Sonifier, 30 s, 1 s on/1 s off, 50% amplitude, four times). Debris was cleared by centrifugation at 3,700g for 10 min at 4°C. The supernatant was then spun at 20k × g for 20 min at 4°C. This supernatant was collected and brought to a final volume of 10 ml with Buffer A and then, passed over a pre-washed Ni-NTA (Qiagen) column. The resin was then washed twice with 5 ml Buffer A, twice with 5 ml Buffer B (50 mM Tris, 200 mM NaCl, pH 8.0), and then, protein was eluted with three aliquots of 1 ml Buffer B containing 200 mM imidazole. About 3 mg/ml of Ulp1 (30 µl) was added to pooled elution fractions and dialyzed (7 kD MWCO Slide-A-Lyzer, ThermoScientific) against Buffer B overnight at 4°C. Dialyzed samples were then passed over a Ni-NTA column and the flow through containing cleaved protein of interest was collected. Purified DnaK and GrpE were diluted to 1 mg/ml concentration in Buffer B for antisera production (Pocono Rabbit Farm & Laboratory).
4.3 |. Generation and sequencing of transposon mutant libraries in Mtb wild type and ΔclpB
Duplicate transposon libraries were generated in Mtb H37Rv and ΔclpB backgrounds by himar1 mutagenesis as previously reported (Long et al., 2015). Briefly, 50 ml of each strain was grown in 7H9 complete/tween (with nourseothricin, hygromycin B, and zeocin for the knockout strain) with rolling at 37°C to an optical density at 580 nm (OD580) of 4.0–4.4 prior to washing with MP buffer (50 mM Tris (pH 7.5), 150 mM NaCl, 10 mM MgSO4, and 2 mM CaCl2). Note that we prepared libraries after the background strains had reached stationary phase growth conditions. Samples were centrifuged at 3,100g for 5 min for each wash step. Pellets were resuspended in 15.5 ml of MP buffer and were incubated with equal volumes of 2.95 × 1010 PFU/ml (library 1) or 2.95 × 1010 PFU/ml (library 2) of ΦMycoMarT7 phage (Sassetti et al., 2003) at 37°C for 4 hr (PFU = plaque forming units). The cultures were then washed (with 7H9 containing detergent) and plated on Middlebrook 7H10 supplemented with oleic acid-albumin-dextrose-catalase (BD BBL), 0.5% glycerol, and 0.05% Tween 80 and kanamycin (25 µg/ml), in addition to nourseothricin (25 µg/ml), hygromycin B (50 µg/ml), and zeocin (25 µg/ml) for the knockout background, and incubated for 18 days at 37°C, yielding libraries that contained approximately 76k–85k mutants in the wild-type background and 18k–85k mutants in the ΔclpB background based on crude counts of visible colonies. The library was harvested by scraping and stored as frozen stocks in Middlebrook 7H9 medium with 15% glycerol at −80°C. Genomic DNA was extracted from a mixture of each library using standard mycobacterial protocols and the composition of each library was determined by Illumina sequencing amplicons of transposon-genome junctions as described below. Sequencing data has been deposited in the Sequence Read Archive (SRA).
4.4 |. Mapping, counting, and analysis of transposon libraries
Previously described methods were used to analyze sequences obtained from Illumina sequencing of transposon libraries (Kieser et al., 2015; Pritchard et al., 2014). Briefly, DNA sequence data were filtered for transposon motifs and trimmed of the transposon sequence except for TA sites that contained an insertion using reported Python scripts. Trimmed reads were mapped to the H37Rv genome using Bowtie2, and insertions at each TA site were tallied using reported Python scripts. The ARTIST pipeline was used to evaluate loci that were disrupted differently between wild-type and mutant backgrounds, which entails use of the Mann-Whitney U test and simulation-based normalization of reads. For each locus in each library, the geometric mean of the sequence reads was determined with a described MATLAB script (Kieser et al., 2015).
4.5 |. Isolation of hspR SNP in Mtb ΔclpB strain
In order to introduce additional mutations into clpB-deficient strains of Mtb, we first generated a knockout of clpB using a kanamycin resistant cassette (kanR) because we sought to have a single antibiotic resistant strain in place of the multiple-drug resistant Mtb ΔclpB used in all other experimental protocols. Mtb ΔclpB kanR was generated using a suicide plasmid approach, which enlists Gateway cloning techniques and vectors (Ganapathy et al., 2015; Kim et al., 2011; Pelicic et al., 1997). Briefly, upstream and downstream of clpB (rv0384c) fragments that have been described (Vaubourgeix et al., 2015) were cloned into pDE43-XSTS (a temperature-sensitive plasmid) containing kanR to generate a pKO-XSTS-clpB-tb. Mtb H37RvN was transformed with pKO-XSTS-clpB-tb and plated on 7H10 agar containing kanamycin (25 µg/ml), followed by incubation at the permissive temperature of 37°C. Resulting transformants were then inoculated into 7H9 complete containing kanamycin (25 µg/ml) at 37°C and grown to stationary phase. Cells were periodically plated on 7H10 agar with kanamycin (25 µg/ml) containing 10% sucrose and incubated at the restrictive temperature of 40°C. 0.5 M pyrocatechol was added to plates that contained colonies and white colonies were inoculated into 7H9 complete/tween containing kanamycin (25 µg/ml) and grown at 37°C prior to purification of DNA and confirmation of allelic exchange by PCR. Deletion of clpB and insertion of kanR was confirmed by WGS.
To control the expression of htpG in Mtb, we generated vectors with different promoters. We cloned pMCH_pre500_SD_htpG using primers 1 and 2 (Table S9) and Gateway vector pDO23A using published techniques (Kim et al., 2011; White et al., 2018), which integrates htpG into the L5 site of the Mtb chromosome under the control of its predicted native promoter region. Upon isolation of Mtb ΔclpB kanR clones carrying pMCH_pre500_SD_htpG and subsequent sequencing of the hspR region using primers 3 and 4, we confirmed that analyzed clones carrying an extra copy of htpG contained a SNP corresponding to an Ala19Val mutation in the gene product HspR (Figure S7). As noted above, the parent strain Mtb ΔclpB kanR did not contain this SNP. In order to examine the behavior of the Mtb ΔclpB hspR SNP without an extra copy of htpG, Mtb ΔclpB hspR SNP pMCH_pre500_SD_htpG was transformed with pTCS-mcs1 to replace htpG at the L5 site with a streptomycin resistant cassette. The resulting strain, called Mtb ΔclpB hspR SNP, exhibited both streptomycin and kanamycin resistance and contained the hspR SNP, as confirmed by sequencing using primers 3 and 4 (Table S9, Figure S7).
4.6 |. Construction of CRISPRi strains for targeted htpG knockdown
A previously described method was used to clone sgRNA sequences into a CRISPRi vector backbone (PLJR965, Table S7) for gene knockdown in mycobacteria (Rock et al., 2017). To construct CRISPRi strains that targeted the expression of htpG, we inserted a single guide RNA (sgRNA) targeting different regions (20–30 nucleotides in length) of htpG near permissive protospacer adjacent motifs (PAMs) found in htpG that could be recognized by the Streptococcus thermophilus dCas9 system controlled by the inducer ATc. sgRNAs were designed to contain BsmBI restriction sites on the 5’ end of each sequence (listed in Table S9 as primers 5–8). Complementary targeting sequences were then ligated into BsmBI-treated PLJR965 using T4 ligase (New England BioLabs) and resulting vectors (pTL_965_ htpG_crispr_1 and pTL_965_htpG_crispr_2) were sequenced prior to transformation into competent Mtb wild-type (H37RvN), ΔclpB, and ΔclpB complemented strains using standard mycobacterial protocols. Resulting colonies were grown in 7H9 complete/tween containing kanamycin (25 µg/ml) in addition to hygromycin (50 µg/ml) for strains in the ΔclpB and ΔclpB complemented backgrounds. Strains carrying different sgRNAs were compared based on growth experiments (Figure S9) prior to choosing one sgRNA sequence for further experiments.
4.7 |. Growth curve analysis of Mtb strains and lysate purification for western blot analysis
For growth curve measurements, indicated strains were grown to late log or early stationary phase (OD580 ~ 1.2) prior to dilution to OD580 in 8–10 ml 7H9/complete with appropriate antibiotics in indicated replicates and incubated at 37 or 40°C. Antibiotics used for initial cultures of bacterial strains were as follows: ΔclpB and ΔclpB complemented strains (hygromycin 50 µg/ml was used in place of all three antibiotics since each resistance cassette was integrated into the chromosome), ΔhtpG (zeocin 25 µg/ml), ΔhtpG complemented (zeocin 25 µg/ml, hygromycin 50 µg/ml), and CRISPRi strains (kanamycin 25 µg/ml and appropriate antibiotics for background strains). To compare the growth of CRISPRi strains with and without mediated knockdown, cultures were grown with and without ATc (100 ng/ml), respectively, starting at day 0. OD580 values were measured at indicated days. Lysates were purified at indicated time points using a modification of a previously published protocol for mycobacteria (Jastrab et al., 2017). Briefly, OD580 values of cultures were measured and a volume of culture equivalent to four OD580 units was taken, pelleted (3,100g, 5 min), and resuspended in 500 µl lysis buffer (25 mM tris (pH 7.5), 1 mM EDTA (pH 8), and 1 mM phenylmethylsulfonyl fluoride (PMSF)). Cell suspensions were added to sealed 2.0 ml microtubes (Sarstedt) with approximately 250 µl of zirconia/silica beads (1 mm, BioSpec) prior to lysis by three rounds (high setting for 30 s) of bead beating (Mini-Beadbeater-1, BioSpec) with cooling on ice in between each round. Samples were centrifuged at 20k × g (4 min), and resulting supernatants (~300–350 µl) were transferred to new tubes prior to addition of 50 µl of laemmli buffer (4x) containing beta-mercaptoethanol and boiling at 100°C for >15 min. For western blot analysis, 50 µl of each sample was loaded on 12% (w/v) polyacrylamide gels and run at 150 V for 50 min. Subsequent immunoblotting was performed with the following dilutions of primary antibodies: anti-DnaK (1:20K), anti-GrpE (1:20K), anti-HtpG (1:2.5K), anti-HspR (1:1K), or anti-ClpB (1:5K). For each, the secondary antibody anti-rabbit HRP (1:20K) was used.
4.8 |. Oxidative, heat, model host stress, and GA experiments on Mtb strains
To examine the effect of different oxidants on viability, a published method was slightly modified (Lin et al., 2016). Indicated strains were grown to log phase (OD580 ~ 0.5) in 7H9 complete (5 ml). Cultures were centrifuged (3,100g, 5 min), washed with 7H9 complete and resuspended in fresh 7H9 complete prior to generating a single cell suspension (spin at 120g, 2 min, no break). Cells were diluted to OD580 = 0.1 in a total volume of 300 µl (in 24-well plates, Corning) and 10x stocks of plumbagin, H2O2, or diamide were diluted into cells followed by incubation at 37°C for indicated time points. For colony forming unit (CFU) determination, cells were serially diluted (10-fold) in PBS containing 0.05% Tween-80, 10 µl of each (100–10−5 dilutions) was plated on 7H11 (BD Difco), and plates were incubated for > 14 days at 37°C (plates were taken out when colony sizes were approximately equivalent to input since delayed growth on plates was seen for Mtb ΔclpB and for all strains treated with oxidants). Cells were then counted to determine the CFU/ml value.
To assess the effect of heat shock on indicated strains, strains were grown to mid- to late-log phase (OD580 = 0.4–1.0), centrifuged (3,100g, 5 min), and resuspended in fresh 7H9 complete prior to dilution in the same medium to OD580 = 0.08, as described (Jastrab et al., 2017). Strains (1 ml) were placed in sealed 2.0 ml microtubes (Sarstedt) and incubated at indicated temperatures for the described amount of time in a heating block. Cells were then diluted and plated as described above to determine CFU/ml except 7H10 plates were used and cells were incubated at 37°C for 21 days prior to counting. The 24 hr heat-treated Mtb ΔclpB plated samples were incubated at 37°C for ~1 month and no colonies were observed. Additional M. smegmatis strains were examined for heat shock sensitivity as detailed above, except strains were grown to log phase (OD580 of 0.5–1.2) and diluted to OD580 = 0.08 prior to incubation of 0.25 ml of culture at 53°C or 37°C for indicated time periods. Cell survival was determined by spot assays (see Supporting Information) in which 5 µl of indicated dilutions of each strain were added to 7H10 agar plates and incubated for t = 3 days at 37°C under standard conditions.
CRISPRi strains were evaluated for survival after treatment with a well-described three- or four-stress host model by modifying a published protocol (Gold et al., 2015). Briefly, CRISPRi strains were inoculated in 7H9 complete at a final OD580 = 0.05 and incubated at 37°C with or without ATc (100 ng/µl). On day 5, the four-stress model experiment was performed, in which replicating cells were washed and diluted to an OD580 = 0.1 in nonreplicating medium composed of modified Sauton’s base (0.05% potassium dihydrogen phosphate (KH2PO4), 0.05%MgSO4, and 0.005% ferric ammonium citrate) at a pH 5.0 with 0.02% tyloxapol, 0.5% bovine serum albumin (BSA), 0.0001% ZnSO4, 0.05% (NH4)2SO4, 0.085% NaCl, and 0.05% butyrate as the carbon source (Gold et al., 2016; Warrier et al., 2015). About 100 ng/µl of ATc was added where specified and the nitric oxide stress was generated by adding 0.5 mM of NaNO2. Mtb cells were then incubated in 5% CO2 and 1.0% O2 at 37°C for 7 days. After the incubation period, cell viability was determined as described above. Peroxide exposure was also performed on CRISPRi strains on day 5: cells were diluted to OD580 = 0.1 in 7H9 complete and treated with hydrogen peroxide as described above for other strains and shown in Figure S11b.
To assess the effect of GA on replicating and nonreplicating cells, indicated strains were diluted to an OD580 = 0.1 in either the four-stress model medium described above (non-replicating) or 7H9 complete medium (replicating) and 100 µM GA or DMSO was added. For nonreplicating conditions, Mtb cells were incubated at 37°C for 7 days under 5% CO2 and 1.0% O2 (host model) or under standard conditions (replicating) prior to quantification of cell viability by CFU.
4.9 |. Cloning, expression, and purification of Mtb HtpG-His6 containing a photoactivatable amino acid
Mutations of codons encoding indicated residues to the amber stop codon (TAG) were introduced into a plasmid encoding pET27b-HtpG-His (Table S7). Briefly, pET27-HtpG-His was cloned using overlap extension PCR cloning techniques (Bryksin and Matsumura, 2010) with pET27b plasmid (Novagen) and primer pairs 51 and 52 (Table S9). Following PCR steps and DpnI treatment, DNA samples were purified using a PCR purification kit (Qiagen) and transformed into Mach1 competent cells (Invitrogen). After confirmation of gene insertion by DNA sequencing, indicated mutations were introduced using QuickChange Site-Directed Mutagenesis Kit (Agilent) with appropriate forward and reverse primer pairs 9–42 (Table S9). Wild-type HtpG-His6 encoding plasmid was transformed into BL21 competent cells (Novagen) for overexpression and purification following the same protocol used for wild-type His6-HtpG protein in the Supporting Information. For HtpG-His6 proteins containing para-benzoyl phenylalanine (BpF), plasmids encoding HtpG with a single TAG mutation (Table S7) were each co-transformed with pE-VOLV-pBpF plasmid into BL21 competent cells (Chin and Schultz, 2002). For overexpression, overnight cultures of each were used to inoculate 500 ml LB (1:100 dilution) containing 25 µg/ml kanamycin and 30 µg/ml chloramphenicol with 0.8 mM BpF added. Cells were grown and HtpG-His6 mutants were over-expressed and purified as described for wild type except both 1 mM IPTG and 0.02% arabinose were used for induction. Protein concentrations were determined using BSA as a standard with the DC Assay (Bio-Rad).
4.10 |. Analysis of HtpG oligomeric state by photocrosslinking
Photocrosslinking reactions were carried out with 2 µM wild-type or BpF-containing Mtb HtpG in 100 µl buffer (1x PBS, 2 mM MgCl2 and 1% dimethyl sulfoxide (DMSO)) with excess nucleotide (5 mM ATP). Each protein solution was prepared at 4°C prior to incubation with or without UV irradiation at 365 nm for 30 min at 4°C, following a similar method to other reports (Chin and Schultz, 2002; Freinkman et al., 2012). Reactions were diluted with 2x laemmli sample buffer (+0.05% beta-mercaptoethanol), boiled for 30 min, and analyzed by SDS-PAGE (9% (w/v) polyacrylamide gel) prior to Coomassie staining. Photocrosslinking reactions in the presence of GA with Mtb wild-type or selected BpF mutants were performed as described above except in the presence of GA (at final concentrations of 0, 10, 50, 100, 200, and 400 µM) with no added nucleotides.
4.11 |. Isothermal Titration Calorimetry (ITC) analysis of Mtb HtpG with GA
Tagless recombinant HtpG was purified from 4 L of culture (strain 4, Table S8) using a previously described method (Lopez Quezada et al., 2020). Nucleotide free Mtb HtpG (1.6 ml) was obtained by following a previously described 4-day dialysis protocol (Rauch and Gestwicki, 2014) using 1 L each of the following buffers: (1) 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 150 mM NaCl, 5 mM EDTA (pH 8), (2) 25 mM HEPES, 150 mM NaCl, 1 mM EDTA (pH 8), (3) 10 mM KCl, 5 mM MgCl2 (pH 8), and (4) 25 mM HEPES, 150 mM NaCl, 10% glycerol, 1% DMSO, 5 mM MgCl2 (pH 8). ITC was carried out at 25°C in Buffer 4 using a low volume Nano ITC calorimeter (TA Instruments). About 750 µM GA solution in Buffer 4 was injected into 200 µl of 47 µM Mtb HtpG. Twenty-five injections (1.78 µl/injection) were performed to achieve a fully saturated binding curve. The heat of dilution was recorded by each injection of the ligand into buffer under the same titration conditions. The value was subtracted from the heat of reaction to determine the final thermodynamic values. KD and the N-value were evaluated using Nano ITC software and fit with a one-site binding model.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Kan Lin, Michael Chao, Kristin Burns-Huang, Ben Gold, Dirk Schnappinger, Michael Glickman, Gloria Marcela Rodriguez, and Sunil Kumar for assistance with experimental interpretation and set-up, Heran Darwin for anti-HspR, Gang Lin for anti-PrcB, and Jeremy Rock for CRISPRi plasmids and protocols. Rockefeller University Proteomics Resource Center (Joseph Fernandez) performed the proteomics analysis. Next-generation sequencing was performed at the Weill Cornell Genomics Resources Core Facility (Jenny Xiang). Jamie Bean performed genomic variant analysis to identify SNPs and verify deletion strains. pEVOL-pBpF was a gift from Peter Schultz (Addgene plasmid # 31190; http://n2t.net/addgene:31190; RRID:Addgene_31190). pET His6 Sumo TEV LIC cloning vector (2S-T) was a gift from Scott Gradia (Addgene plasmid # 29711 http://n2t.net/addgene:29711; RRID:Addgene_29711). pHYRS52 was a gift from Hideo Iwai (Addgene plasmid # 31122; http://n2t.net/addgene:31122; RRID:Addgene_31122). pACYC-T7 was a gift from Dan Bolon (Addgene plasmid # 41187; http://n2t.net/addgene:41187; RRID:Addgene_41187). This work was supported by NIH grant U19 AI111143 (CN, PI); Milstein Program in Chemical Biology and Translational Medicine (CN, PI); Bill & Melinda Gates Foundation (EJR, PI). K.J.K. was partially supported by a National Science Foundation Graduate Research Fellowship (Grants DGE1144152 and DGE0946799). The Department of Microbiology and Immunology at WCM is supported by the William Randolph Hearst Trust. TL acknowledges the Helen Hay Whitney and Simons Foundation, as well as NYU FAS for additional support.
Funding information
Helen Hay Whitney Foundation; Bill and Melinda Gates Foundation; National Science Foundation, Grant/Award Number: DGE1144152 and DGE0946799; William Randolph Hearst Trust; Simons Foundation; Center for Scientific Review, Grant/Award Number: U19 AI111143; Milstein Program in Chemical Biology and Translational Medicine
Footnotes
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest with the contents of this article.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section.
DATA AVAILABILITY STATEMENT
The Tn-Seq data that support the findings of this study are available for review in the SRA databank at accession number PRJNA597221 and will be made publicly available upon publication. Public link: https://dataview.ncbi.nlm.nih.gov/object/PRJNA597221?reviewer=c8b14lrjnj9cu94qh9b8te6han
REFERENCES
- Ali MM, Roe SM, Vaughan CK, Meyer P, Panaretou B, Piper PW, et al. (2006) Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature, 440(7087), 1013–1017. 10.1038/nature04716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker JJ, Dechow SJ and Abramovitch RB (2019) Acid fasting: modulation of Mycobacterium tuberculosis metabolism at acidic pH. Trends in Microbiology, 27(11), 942–953. 10.1016/j.tim.2019.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balchin D, Hayer-Hartl M and Hartl FU (2016) In vivo aspects of protein folding and quality control. Science, 353(6294), aac4354. 10.1126/science.aac4354 [DOI] [PubMed] [Google Scholar]
- Bardwell JC and Craig EA (1988) Ancient heat shock gene is dispensable. Journal of Bacteriology, 170(7), 2977–2983. 10.1128/jb.170.7.2977-2983.1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrios C, Lussow AR, Van Embden J, Van der Zee R, Rappuoli R, Costantino P, et al. (1992) Mycobacterial heat-shock proteins as carrier molecules. II: the use of the 70-kDa mycobacterial heat-shock protein as carrier for conjugated vaccines can circumvent the need for adjuvants and Bacillus Calmette Guérin priming. European Journal of Immunology, 22(6), 1365–1372. 10.1002/eji.1830220606 [DOI] [PubMed] [Google Scholar]
- Beinert H and Kiley PJ (1999) Fe-S proteins in sensing and regulatory functions. Current Opinion in Chemical Biology, 3(2), 152–157. 10.1016/S1367-5931(99)80027-1 [DOI] [PubMed] [Google Scholar]
- Bryk R, Gold B, Venugopal A, Singh J, Samy R, Pupek K, et al. (2008) Selective killing of nonreplicating mycobacteria. Cell Host & Microbe, 3(3), 137–145. 10.1016/j.chom.2008.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryksin AV and Matsumura I (2010) Overlap extension PCR cloning: a simple and reliable way to create recombinant plasmids. BioTechniques, 48(6), 463–465. 10.2144/000113418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns-Huang K and Mundhra S (2019) Mycobacterium tuberculosis cysteine biosynthesis genes mec+-cysO-cysM confer resistance to clofazimine. Tuberculosis (Edinb), 115, 63–66. 10.1016/j.tube.2019.02.002 [DOI] [PubMed] [Google Scholar]
- Calloni G, Chen T, Schermann SM, Chang HC, Genevaux P, Agostini F, et al. (2012) DnaK functions as a central hub in the E. coli chaperone network. Cell Reports, 1(3), 251–264. 10.1016/j.celrep.2011.12.007 [DOI] [PubMed] [Google Scholar]
- Chao MC, Abel S, Davis BM and Waldor MK (2016) The design and analysis of transposon insertion sequencing experiments. Nature Reviews Microbiology, 14(2), 119–128. 10.1038/nrmicro.2015.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao MC, Pritchard JR, Zhang YJ, Rubin EJ, Livny J, Davis BM, et al. (2013) High-resolution definition of the Vibrio cholerae essential gene set with hidden Markov model-based analyses of transposon-insertion sequencing data. Nucleic Acids Research, 41(19), 9033–9048. 10.1093/nar/gkt654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin JW, Martin AB, King DS, Wang L and Schultz PG (2002) Addition of a photocrosslinking amino acid to the genetic code of Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America, 99(17), 11020–11024. 10.1073/pnas.172226299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin JW and Schultz PG (2002) In vivo photocrosslinking with unnatural amino acid mutagenesis. ChemBioChem, 3(11), 1135–1137. [DOI] [PubMed] [Google Scholar]
- Compton CL, Schmitz KR, Sauer RT and Sello JK (2013) Antibacterial activity of and resistance to small molecule inhibitors of the ClpP peptidase. ACS Chemical Biology, 8(12), 2669–2677. 10.1021/cb400577b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl JU, Gray MJ and Jakob U (2015) Protein quality control under oxidative stress conditions. Journal of Molecular Biology, 427(7), 1549–1563. 10.1016/j.jmb.2015.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das Gupta T, Bandyopadhyay B and Das Gupta SK (2008) Modulation of DNA-binding activity of Mycobacterium tuberculosis HspR by chaperones. Microbiology, 154(Pt 2), 484–490. 10.1099/mic.0.2007/012294-0 [DOI] [PubMed] [Google Scholar]
- DeBoer C, Meulman PA, Wnuk RJ and Peterson DH (1970) Geldanamycin, a new antibiotic. The Journal of Antibiotics (Tokyo), 23(9), 442–447. 10.7164/antibiotics.23.442 [DOI] [PubMed] [Google Scholar]
- DeJesus MA, Gerrick ER, Xu W, Park SW, Long JE, Boutte CC, et al. (2017) Comprehensive essentiality analysis of the Mycobacterium tuberculosis genome via saturating transposon mutagenesis. mBio, 8(1), e02133–16. 10.1128/mBio.02133-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosanjh NS, Rawat M, Chung JH and Av-Gay Y (2005) Thiol specific oxidative stress response in Mycobacteria. FEMS Microbiology Letters, 249(1), 87–94. 10.1016/j.femsle.2005.06.004 [DOI] [PubMed] [Google Scholar]
- Dragset MS, Ioerger TR, Zhang YJ, Mærk M, Ginbot Z, Sacchettini JC, et al. (2019) Genome-wide phenotypic profiling identifies and categorizes genes required for mycobacterial low iron fitness. Scientific Reports, 9(1), 11394. 10.1038/s41598-019-47905-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta Gupta S, Bommaka MK and Banerjee A (2019) Inhibiting protein-protein interactions of Hsp90 as a novel approach for targeting cancer. European Journal of Medicinal Chemistry, 178, 48–63. 10.1016/j.ejmech.2019.05.073 [DOI] [PubMed] [Google Scholar]
- Ehrt S and Schnappinger D(2009) Mycobacterialsurvivalstrategiesinthe phagosome: defence against host stresses. Cellular Microbiology, 11(8), 1170–1178. 10.1111/j.1462-5822.2009.01335.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay A and Glickman MS (2014) An essential nonredundant role for mycobacterial DnaK in native protein folding. PLoS Genetics, 10(7), e1004516. 10.1371/journal.pgen.1004516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freinkman E, Okuda S, Ruiz N and Kahne D (2012) Regulated assembly of the transenvelope protein complex required for lipopolysaccharide export. Biochemistry, 51(24), 4800–4806. 10.1021/bi300592c [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganapathy U, Marrero J, Calhoun S, Eoh H, de Carvalho LPS, Rhee K, et al. (2015) Two enzymes with redundant fructose bisphosphatase activity sustain gluconeogenesis and virulence in Mycobacterium tuberculosis. Nature Communications, 6, 7912. 10.1038/ncomms8912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandotra S, Lebron MB and Ehrt S (2010) The Mycobacterium tuberculosis proteasome active site threonine is essential for persistence yet dispensable for replication and resistance to nitric oxide. PLoS Pathogens, 6(8), e1001040. 10.1371/journal.ppat.1001040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genest O, Hoskins JR, Camberg JL, Doyle SM and Wickner S (2011) Heat shock protein 90 from Escherichia coli collaborates with the DnaK chaperone system in client protein remodeling. Proceedings of the National Academy of Sciences of the United States of America, 108(20), 8206–8211. 10.1073/pnas.1104703108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genest O, Wickner S and Doyle SM (2019) Hsp90 and Hsp70 chaperones: collaborators in protein remodeling. Journal of Biological Chemistry, 294(6), 2109–2120. 10.1074/jbc.REV118.002806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill WP, Harik NS, Whiddon MR, Liao RP, Mittler JE and Sherman DR (2009) A replication clock for Mycobacterium tuberculosis. Nature Medicine, 15(2), 211–214. 10.1038/nm.1915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold B and Nathan C (2017) Targeting phenotypically tolerant Mycobacterium tuberculosis. Microbiology Spectrum, 5(1), TBTB2–0031-2016. 10.1128/microbiolspec.TBTB2-0031-2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold B, Rodriguez GM, Marras SA, Pentecost M and Smith I (2001) The Mycobacterium tuberculosis IdeR is a dual functional regulator that controls transcription of genes involved in iron acquisition, iron storage and survival in macrophages. Molecular Microbiology, 42(3), 851–865. 10.1046/j.1365-2958.2001.02684.x [DOI] [PubMed] [Google Scholar]
- Gold B, Smith R, Nguyen Q, Roberts J, Ling Y, Lopez Quezada L, et al. (2016) Novel cephalosporins selectively active on nonreplicating Mycobacterium tuberculosis. Journal of Medicinal Chemistry, 59(13), 6027–6044. 10.1021/acs.jmedchem.5b01833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold B, Warrier T and Nathan C (2015) A multi-stress model for high throughput screening against non-replicating Mycobacterium tuberculosis. Methods in Molecular Biology, 1285, 293–315. 10.1007/978-1-4939-2450-9_18 [DOI] [PubMed] [Google Scholar]
- Grandvalet C, de Crécy-Lagard V and Mazodier P (1999) The ClpB ATPase of Streptomyces albus G belongs to the HspR heat shock regulon. Molecular Microbiology, 31(2), 521–532. 10.1046/j.1365-2958.1999.01193.x [DOI] [PubMed] [Google Scholar]
- Grenert JP, Sullivan WP, Fadden P, Haystead TA, Clark J, Mimnaugh E, et al. (1997) The amino-terminal domain of heat shock protein 90 (hsp90) that binds geldanamycin is an ATP/ADP switch domain that regulates hsp90 conformation. Journal of Biological Chemistry, 272(38), 23843–23850. 10.1074/jbc.272.38.23843 [DOI] [PubMed] [Google Scholar]
- Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ and Sassetti CM (2011) High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathogens, 7(9), e1002251. 10.1371/journal.ppat.1002251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadden MK, Lubbers DJ and Blagg BS (2006) Geldanamycin, radicicol, and chimeric inhibitors of the Hsp90 N-terminal ATP binding site. Current Topics in Medicinal Chemistry, 6(11), 1173–1182. 10.2174/156802606777812031 [DOI] [PubMed] [Google Scholar]
- Hatzios SK and Bertozzi CR (2011) The regulation of sulfur metabolism in Mycobacterium tuberculosis. PLoS Pathogens, 7(7), e1002036. 10.1371/journal.ppat.1002036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoff DR, Ryan GJ, Driver ER, Ssemakulu CC, De Groote MA, Basaraba RJ, et al. (2011) Location of intra- and extracellular M. tuberculosis populations in lungs of mice and guinea pigs during disease progression and after drug treatment. PLoS One, 6(3), e17550. 10.1371/journal.pone.0017550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakob U, Meyer I, Bügl H, André S, Bardwell JC and Buchner J (1995) Structural organization of procaryotic and eucaryotic Hsp90. Influence of divalent cations on structure and function. Journal of Biological Chemistry, 270(24), 14412–14419. 10.1074/jbc.270.24.14412 [DOI] [PubMed] [Google Scholar]
- Jastrab JB, Samanovic MI, Copin R, Shopsin B and Darwin KH (2017) Loss-of-function mutations in HspR rescue the growth defect of a Mycobacterium tuberculosis proteasome accessory factor E. Journal of Bacteriology, 199(7), e00850–16. 10.1128/JB.00850-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EO, LaVerriere E, Office E, Stanley M, Meyer E, Kawate T, et al. (2019) Large-scale chemical-genetics yields new M. tuberculosis inhibitor classes. Nature, 571(7763), 72–78. 10.1038/s41586-019-1315-z [DOI] [PubMed] [Google Scholar]
- Josefson R, Andersson R and Nyström T (2017) How and why do toxic conformers of aberrant proteins accumulate during ageing? Essays in Biochemistry, 61(3), 317–324. 10.1042/EBC20160085 [DOI] [PubMed] [Google Scholar]
- Joshi SM, Pandey AK, Capite N, Fortune SM, Rubin EJ and Sassetti CM (2006) Characterization of mycobacterial virulence genes through genetic interaction mapping. Proceedings of the National Academy of Sciences of the United States of America, 103(31), 11760–11765. 10.1073/pnas.0603179103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann SH, Cole ST, Mizrahi V, Rubin E and Nathan C (2005) Mycobacterium tuberculosis and the host response. Journal of Experimental Medicine, 201(11), 1693–1697. 10.1084/jem.20050842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN and Sternberg MJ (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nature Protocols, 10(6), 845–858. 10.1038/nprot.2015.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieser KJ, Baranowski C, Chao MC, Long JE, Sassetti CM, Waldor MK, et al. (2015) Peptidoglycan synthesis in Mycobacterium tuberculosis is organized into networks with varying drug susceptibility. Proceedings of the National Academy of Sciences of the United States of America, 112(42), 13087–13092. 10.1073/pnas.1514135112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Wei JR, Wallach JB, Robbins RS, Rubin EJ and Schnappinger D (2011) Protein inactivation in mycobacteria by controlled proteolysis and its application to deplete the beta subunit of RNA polymerase. Nucleic Acids Research, 39(6), 2210–2220. 10.1093/nar/gkq1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YE, Hipp MS, Bracher A, Hayer-Hartl M and Hartl FU (2013) Molecular chaperone functions in protein folding and proteostasis. Annual Review of Biochemistry, 82, 323–355. 10.1146/annurev-biochem-060208-092442 [DOI] [PubMed] [Google Scholar]
- Koyasu S, Nishida E, Kadowaki T, Matsuzaki F, Iida K, Harada F, et al. (1986) Two mammalian heat shock proteins, HSP90 and HSP100, are actin-binding proteins. Proceedings of the National Academy of Sciences of the United States of America, 83(21), 8054–8058. 10.1073/pnas.83.21.8054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kragol G, Lovas S, Varadi G, Condie BA, Hoffmann R and Otvos L (2001) The antibacterial peptide pyrrhocoricin inhibits the ATPase actions of DnaK and prevents chaperone-assisted protein folding. Biochemistry, 40(10), 3016–3026. 10.1021/bi002656a [DOI] [PubMed] [Google Scholar]
- Kurthkoti K, Amin H, Marakalala MJ, Ghanny S, Subbian S, Sakatos A, et al. (2017) The capacity of. mBio, 8(4). 10.1128/mBio.01092-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowska J, Matuszewska E, Kuczyńska-Wiśnik D and Laskowska E (2008) Aggregation of Escherichia coli proteins during stationary phase depends on glucose and oxygen availability. Research in Microbiology, 159(9–10), 651–657. 10.1016/j.resmic.2008.09.008 [DOI] [PubMed] [Google Scholar]
- Levitte S, Adams KN, Berg RD, Cosma CL, Urdahl KB and Ramakrishnan L (2016) Mycobacterial acid tolerance enables phagolysosomal survival and establishment of tuberculous infection in vivo. Cell Host & Microbe, 20(2), 250–258. 10.1016/j.chom.2016.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin G, Li D, de Carvalho LP, Deng H, Tao H, Vogt G, et al. (2009) Inhibitors selective for mycobacterial versus human proteasomes. Nature, 461(7264), 621–626. 10.1038/nature08357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K, O’Brien KM, Trujillo C, Wang R, Wallach JB, Schnappinger D, et al. (2016) Mycobacterium tuberculosis thioredoxin reductase is essential for thiol redox homeostasis but plays a minor role in antioxidant defense. PLoS Pathogens, 12(6), e1005675. 10.1371/journal.ppat.1005675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JE, DeJesus M, Ward D, Baker RE, Ioerger T and Sassetti CM (2015) Identifying essential genes in Mycobacterium tuberculosis by global phenotypic profiling. Methods in Molecular Biology, 1279, 79–95. 10.1007/978-1-4939-2398-4_6 [DOI] [PubMed] [Google Scholar]
- Lopez Quezada L, Li K, Lupoli T, Edoo Z, Li X, Gold B, et al. (2020) Activity-based protein profiling reveals that cephalosporins selectively active on non-replicating Mycobacterium tuberculosis bind multiple protein families and spare peptidoglycan transpeptidases. Frontiers in Microbiology, 11, 1248. 10.3389/fmicb.2020.01248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotz GP, Lin H, Harst A and Obermann WM (2003) Aha1 binds to the middle domain of Hsp90, contributes to client protein activation, and stimulates the ATPase activity of the molecular chaperone. Journal of Biological Chemistry, 278(19), 17228–17235. 10.1074/jbc.M212761200 [DOI] [PubMed] [Google Scholar]
- Lupoli TJ, Fay A, Adura C, Glickman MS and Nathan CF (2016) Reconstitution of a Mycobacterium tuberculosis proteostasis network highlights essential cofactor interactions with chaperone DnaK. Proceedings of the National Academy of Sciences of the United States of America, 113(49), E7947–E7956. 10.1073/pnas.1617644113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupoli TJ, Vaubourgeix J, Burns-Huang K and Gold B (2018) Targeting the proteostasis network for mycobacterial drug discovery. ACS Infectious Diseases, 4(4), 478–498. 10.1021/acsinfecdis.7b00231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madigan CA, Martinot AJ, Wei JR, Madduri A, Cheng TY, Young DC, et al. (2015) Lipidomic analysis links mycobactin synthase K to iron uptake and virulence in M. tuberculosis. PLoS Pathogens, 11(3), e1004792. 10.1371/journal.ppat.1004792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisonneuve E, Ezraty B and Dukan S (2008) Protein aggregates: An aging factor involved in cell death. Journal of Bacteriology, 190(18), 6070–6075. 10.1128/JB.00736-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manganelli R, Voskuil MI, Schoolnik GK, Dubnau E, Gomez M and Smith I (2002) Role of the extracytoplasmic-function sigma factor sigma(H) in Mycobacterium tuberculosis global gene expression. Molecular Microbiology, 45(2), 365–374. 10.1046/j.1365-2958.2002.03005.x [DOI] [PubMed] [Google Scholar]
- Millson SH, Chua CS, Roe SM, Polier S, Solovieva S, Pearl LH, et al. (2011) Features of the Streptomyces hygroscopicus HtpG reveal how partial geldanamycin resistance can arise with mutation to the ATP binding pocket of a eukaryotic Hsp90. The FASEB Journal, 25(11), 3828–3837. 10.1096/fj.11-188821 [DOI] [PubMed] [Google Scholar]
- Mogk A, Tomoyasu T, Goloubinoff P, Rüdiger S, Röder D, Langen H, et al. (1999) Identification of thermolabile Escherichia coli proteins: prevention and reversion of aggregation by DnaK and ClpB. EMBO Journal, 18(24), 6934–6949. 10.1093/emboj/18.24.6934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammadi-Ostad-Kalayeh S, Hrupins V, Helmsen S, Ahlbrecht C, Stahl F, Scheper T, et al. (2017) Development of a microarray-based assay for efficient testing of new HSP70/DnaK inhibitors. Bioorganic & Medicinal Chemistry, 25(24), 6345–6352. 10.1016/j.bmc.2017.10.003 [DOI] [PubMed] [Google Scholar]
- Monahan IM, Betts J, Banerjee DK and Butcher PD (2001) Differential expression of mycobacterial proteins following phagocytosis by macrophages. Microbiology, 147(Pt 2), 459–471. 10.1099/00221287-147-2-459 [DOI] [PubMed] [Google Scholar]
- Muttucumaru DG, Roberts G, Hinds J, Stabler RA and Parish T (2004) Gene expression profile of Mycobacterium tuberculosis in a non-replicating state. Tuberculosis (Edinb), 84(3–4), 239–246. 10.1016/j.tube.2003.12.006 [DOI] [PubMed] [Google Scholar]
- Nambi S, Long JE, Mishra BB, Baker R, Murphy KC, Olive AJ, et al. (2015) The oxidative stress network of Mycobacterium tuberculosis reveals coordination between radical detoxification systems. Cell Host & Microbe, 17(6), 829–837. 10.1016/j.chom.2015.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro Llorens JM, Tormo A and Martínez-García E (2010) Stationary phase in gram-negative bacteria. FEMS Microbiology Reviews, 34(4), 476–495. 10.1111/j.1574-6976.2010.00213.x [DOI] [PubMed] [Google Scholar]
- Obermann WM, Sondermann H, Russo AA, Pavletich NP and Hartl FU (1998) In vivo function of Hsp90 is dependent on ATP binding and ATP hydrolysis. Journal of Cell Biology, 143(4), 901–910. 10.1083/jcb.143.4.901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen BA, Sullivan WP, Felts SJ and Toft DO (2002) Regulation of heat shock protein 90 ATPase activity by sequences in the carboxyl terminus. Journal of Biological Chemistry, 277(9), 7086–7091. 10.1074/jbc.M111450200 [DOI] [PubMed] [Google Scholar]
- Pelicic V, Jackson M, Reyrat JM, Jacobs WR, Gicquel B and Guilhot C (1997) Efficient allelic exchange and transposon mutagenesis in Mycobacterium tuberculosis. Proceedings of the National Academy of Sciences of the United States of America, 94(20), 10955–10960. 10.1073/pnas.94.20.10955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perales-Calvo J, Giganti D, Stirnemann G and Garcia-Manyes S (2018) The force-dependent mechanism of DnaK-mediated mechanical folding. Proceedings of the National Academy of Sciences of the United States of America, 4(2), eaaq0243. 10.1126/sciadv.aaq0243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto R, Tang QX, Britton WJ, Leyh TS and Triccas JA (2004) The Mycobacterium tuberculosis cysD and cysNC genes form a stress-induced operon that encodes a tri-functional sulfate-activating complex. Microbiology, 150(Pt 6), 1681–1686. 10.1099/mic.0.26894-0 [DOI] [PubMed] [Google Scholar]
- Pöther DC, Liebeke M, Hochgräfe F, Antelmann H, Becher D, Lalk M, et al. (2009) Diamide triggers mainly S thiolations in the cytoplasmic proteomes of Bacillus subtilis and Staphylococcus aureus. Journal of Bacteriology, 191(24), 7520–7530. 10.1128/JB.00937-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JR, Chao MC, Abel S, Davis BM, Baranowski C, Zhang YJ, et al. (2014) ARTIST: high-resolution genome-wide assessment of fitness using transposon-insertion sequencing. PLoS Genetics, 10(11), e1004782. 10.1371/journal.pgen.1004782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prodromou C, Roe SM, O’Brien R, Ladbury JE, Piper PW and Pearl LH (1997) Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell, 90(1), 65–75. 10.1016/s0092-8674(00)80314-1 [DOI] [PubMed] [Google Scholar]
- Quadri LE, Sello J, Keating TA, Weinreb PH and Walsh CT (1998) Identification of a Mycobacterium tuberculosis gene cluster encoding the biosynthetic enzymes for assembly of the virulence-conferring siderophore mycobactin. Chemistry & Biology, 5(11), 631–645. 10.1016/s1074-5521(98)90291-5 [DOI] [PubMed] [Google Scholar]
- Raju RM, Unnikrishnan M, Rubin DH, Krishnamoorthy V, Kandror O, Akopian TN, et al. (2012) Mycobacterium tuberculosis ClpP1 and ClpP2 function together in protein degradation and are required for viability in vitro and during infection. PLoS Pathogens, 8(2), e1002511. 10.1371/journal.ppat.1002511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch JN and Gestwicki JE (2014) Binding of human nucleotide exchange factors to heat shock protein 70 (Hsp70) generates functionally distinct complexes in vitro. Journal of Biological Chemistry, 289(3), 1402–1414. 10.1074/jbc.M113.521997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoades ER, Frank AA and Orme IM (1997) Progression of chronic pulmonary tuberculosis in mice aerogenically infected with virulent Mycobacterium tuberculosis. Tubercle and Lung Disease, 78(1), 57–66. 10.1016/s0962-8479(97)90016-2 [DOI] [PubMed] [Google Scholar]
- Rock JM, Hopkins FF, Chavez A, Diallo M, Chase MR, Gerrick ER, et al. (2017) Programmable transcriptional repression in mycobacteria using an orthogonal CRISPR interference platform. Nature Microbiology, 2, 16274. 10.1038/nmicrobiol.2016.274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock JM, Lang UF, Chase MR, Ford CB, Gerrick ER, Gawande R, et al. (2015) DNA replication fidelity in Mycobacterium tuberculosis is mediated by an ancestral prokaryotic proofreader. Nature Genetics, 47(6), 677–681. 10.1038/ng.3269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez GM, Voskuil MI, Gold B, Schoolnik GK and Smith I (2002) ideR, An essential gene in mycobacterium tuberculosis: role of IdeR in iron-dependent gene expression, iron metabolism, and oxidative stress response. Infection and Immunity, 70(7), 3371–3381. 10.1128/iai.70.7.3371-3381.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe SM, Prodromou C, O’Brien R, Ladbury JE, Piper PW and Pearl LH (1999) Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. Journal of Medicinal Chemistry, 42(2), 260–266. 10.1021/jm980403y [DOI] [PubMed] [Google Scholar]
- Rosenzweig R, Moradi S, Zarrine-Afsar A, Glover JR and Kay LE (2013) Unraveling the mechanism of protein disaggregation through a ClpB-DnaK interaction. Science, 339(6123), 1080–1083. 10.1126/science.1233066 [DOI] [PubMed] [Google Scholar]
- Russell DG (2011) Mycobacterium tuberculosis and the intimate discourse of a chronic infection. Immunological Reviews, 240(1), 252–268. 10.1111/j.1600-065X.2010.00984.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santra M, Dill KA and de Graff AMR (2018) How do chaperones protect a cell’s proteins from oxidative damage? Cell Systems, 6(6), 743–751.e743. 10.1016/j.cels.2018.05.001 [DOI] [PubMed] [Google Scholar]
- Sassetti CM, Boyd DH and Rubin EJ (2001) Comprehensive identification of conditionally essential genes in mycobacteria. Proceedings of the National Academy of Sciences of the United States of America, 98(22), 12712–12717. 10.1073/pnas.231275498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassetti CM, Boyd DH and Rubin EJ (2003) Genes required for mycobacterial growth defined by high density mutagenesis. Molecular Microbiology, 48(1), 77–84. [DOI] [PubMed] [Google Scholar]
- Schramm FD, Schroeder K and Jonas K (2020) Protein aggregation in bacteria. FEMS Microbiology Reviews, 44(1), 54–72. 10.1093/femsre/fuz026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher RJ, Hansen WJ, Freeman BC, Alnemri E, Litwack G and Toft DO (1996) Cooperative action of Hsp70, Hsp90, and DnaJ proteins in protein renaturation. Biochemistry, 35(47), 14889–14898. 10.1021/bi961825h [DOI] [PubMed] [Google Scholar]
- Scocchi M, Lüthy C, Decarli P, Christen P and Gennaro R (2009) The proline-rich antibacterial peptide Bac7 binds to and inhibits in vitro the molecular chaperone DnaK. International Journal of Peptide Research and Therapeutics, 15, 147–155. [Google Scholar]
- Scocchi M, Tossi A and Gennaro R (2011) Proline-rich antimicrobial peptides: converging to a non-lytic mechanism of action. Cellular and Molecular Life Sciences, 68(13), 2317–2330. 10.1007/s00018-011-0721-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiau AK, Harris SF, Southworth DR and Agard DA (2006) Structural analysis of E. coli hsp90 reveals dramatic nucleotide-dependent conformational rearrangements. Cell, 127(2), 329–340. 10.1016/j.cell.2006.09.027 [DOI] [PubMed] [Google Scholar]
- Spence J & Georgopoulos C (1989) Purification and properties of the Escherichia coli heat shock protein, HtpG. Journal of Biological Chemistry, 264(8), 4398–4403. [PubMed] [Google Scholar]
- Srivastava A, Asahara H, Zhang M, Zhang W, Liu H, Cui S, et al. (2016) Reconstitution of protein translation of mycobacterium reveals functional conservation and divergence with the gram-negative bacterium Escherichia coli. PLoS One, 11(8), e0162020. 10.1371/journal.pone.0162020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stebbins CE, Russo AA, Schneider C, Rosen N, Hartl FU and Pavletich NP (1997) Crystal structure of an Hsp90-geldanamycin complex: targeting of a protein chaperone by an antitumor agent. Cell, 89(2), 239–250. [DOI] [PubMed] [Google Scholar]
- Stewart GR, Snewin VA, Walzl G, Hussell T, Tormay P, O’Gaora P, et al. (2001) Overexpression of heat-shock proteins reduces survival of Mycobacterium tuberculosis in the chronic phase of infection. Nature Medicine, 7(6), 732–737. 10.1038/89113 [DOI] [PubMed] [Google Scholar]
- Stewart GR, Wernisch L, Stabler R, Mangan JA, Hinds J, Laing KG, et al. (2002) Dissection of the heat-shock response in Mycobacterium tuberculosis using mutants and microarrays. Microbiology, 148(Pt 10), 3129–3138. 10.1099/00221287-148-10-3129 [DOI] [PubMed] [Google Scholar]
- Taipale M, Jarosz DF and Lindquist S (2010) HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nature Reviews Molecular Cell Biology, 11(7), 515–528. 10.1038/nrm2918 [DOI] [PubMed] [Google Scholar]
- Thomas JG and Baneyx F (2000) ClpBand HtpGfacilitate de novoprotein folding in stressed Escherichia coli cells. Molecular Microbiology, 36(6), 1360–1370. 10.1046/j.1365-2958.2000.01951.x [DOI] [PubMed] [Google Scholar]
- Tripathi P, Singh LK, Kumari S, Hakiem OR and Batra JK (2020) ClpB is an essential stress regulator of Mycobacterium tuberculosis and endows survival advantage to dormant bacilli. International Journal of Medical Microbiology, 310(3), 151402. 10.1016/j.ijmm.2020.151402 [DOI] [PubMed] [Google Scholar]
- Ueda T, Tamura T and Hamachi I (2019) Development of a cell-based ligand-screening system for identifying Hsp90 inhibitors. Biochemistry, 59(2), 179–182. 10.1021/acs.biochem.9b00781 [DOI] [PubMed] [Google Scholar]
- Vaubourgeix J, Lin G, Dhar N, Chenouard N, Jiang X, Botella H, et al. (2015) Stressed mycobacteria use the chaperone ClpB to sequester irreversibly oxidized proteins asymmetrically within and between cells. Cell Host & Microbe, 17(2), 178–190. 10.1016/j.chom.2014.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voskuil MI, Bartek IL, Visconti K and Schoolnik GK (2011) The response of mycobacterium tuberculosis to reactive oxygen and nitrogen species. Frontiers in Microbiology, 2, 105. 10.3389/fmicb.2011.00105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrier T, Martinez-Hoyos M, Marin-Amieva M, Colmenarejo G, Porras-De Francisco E, Alvarez-Pedraglio AI, et al. (2015) Identification of novel anti-mycobacterial compounds by screening a pharmaceutical small-molecule library against nonreplicating Mycobacterium tuberculosis. ACS Infectious Diseases, 1(12), 580–585. 10.1021/acsinfecdis.5b00025 [DOI] [PubMed] [Google Scholar]
- Wattam AR, Davis JJ, Assaf R, Boisvert S, Brettin T, Bun C, et al. (2017) Improvements to PATRIC, the all-bacterial Bioinformatics Database and Analysis Resource Center. Nucleic Acids Research, 45(D1), D535–D542. 10.1093/nar/gkw1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wearsch PA and Nicchitta CV (1996) Endoplasmic reticulum chaperone GRP94 subunit assembly is regulated through a defined oligomerization domain. Biochemistry, 35(51), 16760–16769. 10.1021/bi962068q [DOI] [PubMed] [Google Scholar]
- White DW, Elliott SR, Odean E, Bemis LT and Tischler AD (2018) Mycobacterium tuberculosis Pst/SenX3-RegX3 regulates membrane vesicle production independently of ESX-5 activity. mBio, 9(3), e00778–18. 10.1128/mBio.00778-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO. (2018) Global Tuberculosis Report 2018 Geneva, Switzerland: WHO Press. [Google Scholar]
- Winterbourn CC (1995) Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicology Letters, 82–83, 969–974. 10.1016/0378-4274(95)03532-x [DOI] [PubMed] [Google Scholar]
- Xu W, DeJesus MA, Rücker N, Engelhart CA, Wright MG, Healy C, et al. (2017) Chemical genetic interaction profiling reveals determinants of intrinsic antibiotic resistance in Mycobacterium tuberculosis. Antimicrobial Agents and Chemotherapy, 61(12), 10.1128/AAC.01334-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadon AN, Maharaj K, Adamson JH, Lai YP, Sacchettini JC, Ioerger TR, et al. (2017) A comprehensive characterization of PncA polymorphisms that confer resistance to pyrazinamide. Nature Communications, 8(1), 588. 10.1038/s41467-017-00721-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Lupoli TJ, Kovach A, Meng X, Zhao G, Nathan CF, et al. (2018) ATP hydrolysis-coupled peptide translocation mechanism of. Proceedings of the National Academy of Sciences of the United States of America, 115(41), E9560–E9569. 10.1073/pnas.1810648115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y and Mitchison D (2003) The curious characteristics of pyrazinamide: a review. The International Journal of Tuberculosis and Lung Disease, 7(1), 6–21. [PubMed] [Google Scholar]
- Zietkiewicz S, Krzewska J and Liberek K (2004) Successive and synergistic action of the Hsp70 and Hsp100 chaperones in protein disaggregation. Journal of Biological Chemistry, 279(43), 44376–44383. 10.1074/jbc.M402405200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Tn-Seq data that support the findings of this study are available for review in the SRA databank at accession number PRJNA597221 and will be made publicly available upon publication. Public link: https://dataview.ncbi.nlm.nih.gov/object/PRJNA597221?reviewer=c8b14lrjnj9cu94qh9b8te6han