

Abstract
This study provides a whole‐body physiologically‐based pharmacokinetic (PBPK) model of dextromethorphan and its metabolites dextrorphan and dextrorphan O‐glucuronide for predicting the effects of cytochrome P450 2D6 (CYP2D6) drug‐gene interactions (DGIs) on dextromethorphan pharmacokinetics (PK). Moreover, the effect of interindividual variability (IIV) within CYP2D6 activity score groups on the PK of dextromethorphan and its metabolites was investigated. A parent‐metabolite‐metabolite PBPK model of dextromethorphan, dextrorphan, and dextrorphan O‐glucuronide was developed in PK‐Sim and MoBi. Drug‐dependent parameters were obtained from the literature or optimized. Plasma concentration‐time profiles of all three analytes were gathered from published studies and used for model development and model evaluation. The model was evaluated comparing simulated plasma concentration‐time profiles, area under the concentration‐time curve from the time of the first measurement to the time of the last measurement (AUClast) and maximum concentration (Cmax) values to observed study data. The final PBPK model accurately describes 28 population plasma concentration‐time profiles and plasma concentration‐time profiles of 72 individuals from four cocktail studies. Moreover, the model predicts CYP2D6 DGI scenarios with six of seven DGI AUClast and seven of seven DGI Cmax ratios within the acceptance criteria. The high IIV in plasma concentrations was analyzed by characterizing the distribution of individually optimized CYP2D6 kcat values stratified by activity score group. Population simulations with sampling from the resulting distributions with calculated log‐normal dispersion and mean parameters could explain a large extent of the observed IIV. The model is publicly available alongside comprehensive documentation of model building and model evaluation.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Dextromethorphan is a substrate of cytochrome P450 2D6 (CYP2D6) and is consequently subject to considerable drug‐gene interaction (DGI) effects. High interindividual variability (IIV) in dextromethorphan plasma concentrations is apparent, even within activity score groups.
WHAT QUESTION DID THIS STUDY ADDRESS?
The objective of this study was to develop a physiologically‐based pharmacokinetic (PBPK) model that can describe and predict the effect of CYP2D6 DGIs on the pharmacokinetics (PK) of dextromethorphan and its metabolites dextrorphan and dextrorphan O‐glucuronide.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
This study presents a PBPK model of dextromethorphan and its major metabolites that integrates current knowledge on relevant PK processes and DGIs. The model can accurately describe and predict the impact of CYP2D6 DGIs on the PK of the modeled analytes and was applied to explain a large extent of observed IIV in dextromethorphan plasma concentrations.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
The developed PBPK model serves as a prototype for the development of PBPK models for other CYP2D6 substrates. Modeling provides valuable insights regarding the extent of observed overall IIV in plasma concentrations of CYP2D6 substrates as well as the observed IIV within activity score groups.
INTRODUCTION
Dextromethorphan is a widely used over‐the‐counter cough suppressant and a common ingredient of cold medicines marketed toward children and adults. 1 The mechanisms of action of dextromethorphan and its major metabolite dextrorphan are multifarious and include antagonism of σ1‐ and N‐methyl‐D‐aspartate (NMDA) receptors as well as inhibition of serotonin reuptake transporters (SERTs) and norepinephrine reuptake transporters (NERTs). 2 Dextrorphan has a higher affinity to NMDA receptors than dextromethorphan and is considered to be mainly responsible for the psychoactive and euphoric effects when dextromethorphan is ingested in supratherapeutic doses as a recreational drug. 3
Dextromethorphan is typically administered as its hydrobromide salt, which is considered a Biopharmaceutics Drug Disposition Classification System (BDDCS) class I drug with high solubility and permeability. 4 After oral administration, dextromethorphan is rapidly absorbed. Next, dextromethorphan undergoes an extensive first‐pass metabolism, predominately mediated by CYP2D6, reducing the bioavailability to 1%–2% in CYP2D6 extensive metabolizers (EMs) and 80% in CYP2D6 poor metabolizers (PMs). 5 Unbound dextromethorphan accounts for 35% of the total drug plasma concentration. 2 Dextromethorphan‐O‐demethylation via CYP2D6 leads to the formation of the major active metabolite dextrorphan. Dextrorphan subsequently undergoes rapid glucuronidation via uridine diphosphate‐glucuronosyltransferases 2B (UGT2Bs), namely UGT2B15, or N‐demethylation via CYP3A4. 6 Alternatively, dextromethorphan is N‐demethylated by CYP3A4, which was found to be the main pathway of dextromethorphan metabolism in CYP2D6 PMs. 2 Depending on the CYP2D6 phenotype, up to 50% of orally administered dextromethorphan is excreted unchanged in urine. 5 , 7 Because the CYP2D6 gene is prone to genetic alterations, dextromethorphan pharmacokinetics (PK) is subject to considerable drug‐gene interaction (DGI) effects. For instance, the dextromethorphan area under the plasma concentration‐time curve (AUC) in CYP2D6 PMs was reported to be 26‐fold higher than that of CYP2D6 EMs. 8 Hence, the US Food and Drug Administration (FDA) lists dextromethorphan as a sensitive substrate of CYP2D6 and recommends its usage in clinical drug‐drug interaction studies and dextromethorphan O‐demethylation as an in vitro marker reaction for CYP2D6 metabolism. 9 Furthermore, the dextromethorphan/dextrorphan metabolic ratio is frequently used to determine the CYP2D6 phenotype in vivo. 10 , 11 Hence, dextromethorphan is frequently included in different phenotyping cocktails. 12 , 13
To date, more than 140 alleles of the CYP2D6 gene are known, some of which have only been discovered in recent years. 14 With well over 10,000 potential CYP2D6 diplotypes, investigating the effect of every genotype on a drug’s PK is an unfeasible task for clinical researchers. 15 Consequently, an activity score system is in place to facilitate the process of translating the CYP2D6 diplotype into a patient’s phenotype. 15 , 16 This process has since been harmonized between pharmacogenomics laboratories and between clinical guidelines of the Dutch Pharmacogenomics Working Group (DPWG) and the Clinical Pharmacogenetics Implementation Consortium (CPIC). 17 Here, a patients’ activity score is defined as the sum of activity values assigned to the patients’ alleles with values encoding for no (0), decreased (0.25–0.5), or normal function (1), or a copy number variation of a normal function allele (>2). 15 The activity score system is an eminently useful concept for grouping study subjects based on their genotypes. However, a large interindividual variability (IIV) in the PK of CYP2D6 substrates in subjects with an identical activity score remains largely unexplained and requires further research. 16
The objectives of this study were (1) to develop and evaluate a physiologically‐based pharmacokinetic (PBPK) parent‐metabolite DGI model of dextromethorphan, dextrorphan, and dextrorphan O‐glucuronide, (2) to describe the effects of different CYP2D6 activity scores on the PK of dextromethorphan by implementing specific CYP2D6 activity score‐dependent metabolic processes, and (3) to apply the developed model to explain the observed IIV in individual subjects sharing the same CYP2D6 activity score. The final PBPK model will be publicly available in the Open Systems Pharmacology (OSP) repository (www.open‐systems‐pharmacology.org) 18 as a clinical research tool. Moreover, the Supplementary document (Supplementary S1) to this article provides an in‐depth evaluation of the model performance and can be used as a model reference manual.
METHODS
Software
The dextromethorphan PBPK model was developed using PK‐Sim and MoBi (Open Systems Pharmacology Suite 9.1, www.open‐systems‐pharmacology.org). Model parameter optimizations via Monte Carlo algorithm and local sensitivity analyses were conducted in PK‐Sim. Published clinical study data were digitized according to the recommended practice 19 using GetData Graph Digitizer 2.26.0.20 (© S. Fedorov). PK parameters, model performance metrics, and plots were calculated and generated using Python (version 3.9.1; Python Software Foundation, Wilmington, DE). Regression analyses were performed using ordinary least squares utilizing the statsmodels package (version 0.12.2) in Python. 20
Clinical study data
Published clinical studies were obtained from the literature, including aggregated plasma concentration‐time profiles after intravenous and oral administrations in single and multiple dose regimens of dextromethorphan alone or various phenotyping cocktails. It was assumed that there were no relevant mutual interactions between the cocktail compounds affecting dextromethorphan PK. 12 , 21 The composition of phenotyping cocktails used in the respective studies is provided in Section S1.1 of Supplementary S1. All collected dextromethorphan plasma concentration‐time profiles were split into a training dataset, for model building and a test dataset, for model evaluation. Studies for model training were selected to include different routes of administration (intravenous and oral), a wide range of administered doses as well as data covering all investigated CYP2D6 genotypes or activity scores. The training dataset was used for estimation of model input parameters which could not be obtained from the literature. Studies were complemented by individual dextromethorphan, dextrorphan, and total dextrorphan (dextrorphan and dextrorphan O‐glucuronide) plasma profiles from 72 study participants. The respective data was reported in a PhD thesis by Frank in 2009 as a compilation of four clinical cocktail studies (studies A–E). 22 Study B was excluded from the dataset due to inconsistencies between the reported individual genotypes and the corresponding plasma concentrations of dextromethorphan, which may be explained by the limited set of genetic CYP2D6 variants assessed (see Section S6.1 of Supplementary S1 for a detailed analysis). Sections S2.2, S4.2, and S6.3 of Supplementary S1 provide comprehensive information on population and individual demographics (sex, age, weight, and height), analyzed compounds, CYP2D6 activity (CYP2D6 phenotype, genotype, and activity score, if available), drug dosing regimens and the assignment to the respective test and training datasets for all modeled studies and individual profiles.
PBPK base model building
The dextromethorphan PBPK model building process started with an extensive literature search to obtain physicochemical data on dextromethorphan, dextrorphan, and dextrorphan O‐glucuronide as well as information on absorption, distribution, metabolism, and excretion. The dextromethorphan PBPK model was developed using individual simulations based on typical mean individuals for the respective study populations (see Section S1.3 of Supplementary S1). First, a combination of quantitative structure‐activity relationship methods implemented in PK‐Sim was selected for the estimation of cellular permeabilities and organ/plasma partition coefficients. Here, the selection of the optimal combination was based on the minimum residual error for parameter estimations fitting intravenous dextromethorphan administration simulations to their respective observed data. Subsequently, studies of orally administered dextromethorphan in PMs were used to optimize model parameters independent of CYP2D6 metabolism, as the CYP2D6 activity of poor metabolizers was assumed to be 0% due to the lack of expression of functional CYP2D6 protein in carriers of two CYP2D6 loss‐of‐function alleles (e.g., CYP2D6*3, *4, and *6). 10 Finally, CYP2D6 catalytic rate constant (kcat) values were optimized for EMs by fitting to EM plasma concentration‐time profiles of the training dataset. Here, the historical term “extensive metabolizer” was used to describe populations which were either not phenotyped or phenotyped via classical phenotyping methods, such as measurements of metabolic ratios or screening for CYP2D6 null alleles. Genotyped populations possessing activity scores ranging from 1.25–2.25 were considered “normal metabolizers.” 17
Overall, the minimal number of processes necessary to mechanistically describe the PK of dextromethorphan, dextrorphan, and dextrorphan O‐glucuronide were implemented to limit the number of unknown parameter values to be optimized. Total dextrorphan was calculated as the sum of simulated dextrorphan and dextrorphan O‐glucuronide. System‐dependent parameters and details on the implementation of CYP2D6, CYP3A4, and UGT2B15 are presented in Section S7 of Supplementary S1.
PBPK model evaluation
Performance of the PBPK model regarding the prediction of dextromethorphan and its metabolites dextrorphan and dextrorphan O‐glucuronide was evaluated using graphical and statistical methods.
First, simulated population plasma concentrations (arithmetic mean ± SD) were compared graphically to observed data of the respective clinical studies. For this, virtual populations of 1000 individuals were created using the mode of reported sex and ethnicity as well as mean values for age, weight, and height from each study protocol. Sections S1.3 and S1.4 of Supplementary S1 provide a comprehensive description of virtual individuals and virtual populations.
Second, the arithmetic mean of population simulations or individual predictions for all plasma concentration‐time profiles were plotted against their corresponding observed values in goodness‐of‐fit plots.
Third, predicted and observed AUC values and maximum plasma concentration (Cmax) values were graphically compared. Here, all AUC values (predicted as well as observed) were calculated from the time of the first measurement to the time of the last measurement (AUClast).
Finally, as quantitative measures of the model performance, the mean relative deviation (MRD) of all predicted plasma concentrations (Equation 1) and the geometric mean fold error (GMFE) of all predicted AUClast and Cmax values (Equation 2) were calculated.
| (1) |
where = predicted plasma concentration that corresponds to the i‐th observed concentration, = i‐th observed plasma concentration, k = number of observed values.
| (2) |
where = predicted AUClast or Cmax value of study = corresponding observed AUClast or Cmax value of study i, m = total number of studies.
Local sensitivity of the AUC0–24 h of dextromethorphan, dextrorphan, and dextrorphan O‐glucuronide to single parameter changes was analyzed for a simulation of 30 mg orally administered dextromethorphan hydrobromide as a single dose (standard dose). Parameters were included if they have been optimized (kcat values and dextromethorphan intestinal permeability), if they are associated with optimized parameters (KM values) or if they might have a strong impact due to calculation methods used (lipophilicity, fraction unbound, and pKa values). A detailed description is provided in Section S1.6 of Supplementary S1 and a list of all parameters included in the sensitivity analysis is given in Section S3.6 of Supplementary S1.
DGI model building
The principal pathway of dextromethorphan metabolism is the CYP2D6‐mediated O‐demethylation, leading to the formation of dextrorphan. This pathway was implemented using Michaelis‐Menten kinetics according to Equation 323 :
| (3) |
where v = reaction velocity at substrate concentration S, Vmax = maximum reaction velocity, KM = Michaelis‐Menten constant, kcat = catalytic rate constant, and E = enzyme concentration.
For DGI modeling, the CYP2D6 Michaelis‐Menten constant (KM) values for the dextromethorphan O‐demethylation were kept constant over the whole range of modeled activity scores. 24 CYP2D6 kcat values were optimized separately for each activity score. CYP2D6 PMs (activity score = 0) were assumed to show no CYP2D6 activity (0%), whereas populations with two wildtype alleles (activity score = 2) were assumed to possess normal CYP2D6 activity (100%). Activity scores were assigned according to Caudle et al. 17
DGI model evaluation
Modeled DGIs were evaluated by comparison of predicted versus observed plasma concentration‐time profiles of dextromethorphan and its metabolites. Plasma concentration‐time profiles for populations displaying variant phenotypes were compared to those of the EM phenotype, whereas plasma concentration‐time profiles for populations with a variant activity score were compared to profiles of a population with normal activity (activity score = 2) in studies reporting activity scores or genotypes. Similarly, predicted DGI AUClast ratios (Equation 4) and DGI Cmax ratios (Equation 5) were evaluated for study populations with different CYP2D6 activity scores or phenotypes.
| (4) |
Here, AUClast, DGI = AUClast of variant activity score or phenotype, AUClast, reference = AUClast of activity score = 2 or EM phenotype.
| (5) |
with Cmax, DGI = Cmax of variant activity score or phenotype, Cmax, reference = Cmax of activity score = 2 or EM phenotype.
Additionally, GMFE values of the predicted DGI AUClast ratios and DGI Cmax ratios were calculated according to Equation 2 as a quantitative measure of prediction accuracy.
Assessment of interindividual variability within activity score groups
To assess the impact of IIV on the PK of dextromethorphan, CYP2D6 kcat values were optimized separately, using their respective observed data, for all individual plasma concentration‐time profiles of the four cocktail studies. Activity scores for all genotyped subjects were calculated according to Caudle et al. 17 Subjects with the same activity scores were grouped and geometric means and standard deviations were calculated from the optimized individual CYP2D6 kcat values. Subsequently, these values were graphically compared to the population kcat values, obtained in the model building process. Finally, an ordinary least squares regression analysis was applied between individual optimized kcat and their population kcat counterpart for the respective activity score.
RESULTS
PBPK base model building
The dextromethorphan PBPK model was developed using a total of 28 clinical studies where dextromethorphan was administered as an intravenous infusion (one study), orally in single (26 studies), or multiple doses (one study), alone (17 studies) or as part of a phenotyping cocktail (11 studies). Doses ranged between 5 and 80 mg of administered dextromethorphan. Table 1 provides an overview of demographics and CYP2D6 activity for all modeled studies.
TABLE 1.
Summary of demographic parameters and CYP2D6 activity for all modeled studies
| Dataset I: Base model building and evaluation | Dataset II: DGI model building and evaluation | Dataset III: Studies reporting individual data | |||||
|---|---|---|---|---|---|---|---|
| A | B | C | D | E | |||
| Number of studies | 15 | 13 | |||||
| Number of individuals | 309 | 120 | 16 | 12 | 16 | 12 | 28 |
| Demographics | |||||||
| Females, % | 27 | 37 | 0 | 0 | 0 | 0 | 25 |
| Age, years | 31 (19–74) | 26 (18–55) | 29 (23–42) | 36 (24–49) | 29 (21–43) | 32 (18–48) | 40 (25–60) |
| Weight, kg | 74 (49–110) | 69 (60–79) | 78 (65–101) | 80 (60–103) | 80 (66–99) | 72 (60–82) | 72 (49–106) |
| Height, cm | * | * | 182 (171–195) | 181 (165–198) | 182 (173–194) | 180 (171–190) | 174 (156–190) |
| CYP2D6 activity | |||||||
| Phenotypea | |||||||
| PM | 1 | 2 | 1 | 1 | – | – | 1 |
| IM | – | 3 | 7 | 5 | 2 | 4 | 13 |
| EM | 14 | 2 | – | – | 4 | 1 | – |
| NM | – | 6 | 4 | 6 | 9 | 7 | 13 |
| UM | – | – | 2 | – | 1 | – | 1 |
| Activity scorea | |||||||
| 0 | – | – | 1 | 1 | – | – | 1 |
| 0.25 | – | 2 | 1 | – | – | – | – |
| 0.5 | – | 2 | – | 1 | 1 | – | – |
| 1 | – | 1 | 7 | 3 | 1 | 4 | 13 |
| 1.25 | – | 2 | – | – | – | – | – |
| 1.5 | – | – | 1 | 1 | 4 | 2 | – |
| 2 | – | 4 | 4 | 5 | 4 | 5 | 13 |
| 3 | – | – | 2 | – | 1 | – | 1 |
| Dextromethorphan observed AUClast, ng⋅h/ml | |||||||
| Phenotype | |||||||
| PM | 981.2 | 756.7 (547.7) | |||||
| EM | 39.2 (30.1) | 40.4 (25.8) | |||||
| Activity score | |||||||
| 0 | 162.4 | 3.1 | 97.6 | ||||
| 0.25 | 146.3 | ||||||
| 0.5 | 73.5 (5.8) | 3.9 (2.5) | 63.7 | ||||
| 1 | 63.54 | 5.7 (1.5) | 8.2 (5.2) | 5.1 | 14.6 (15.3) | 36.4 (57.4) | |
| 1.25 | 22.0 (1.1) | ||||||
| 1.5 | 17.3 | 5.9 | 3.5 (2.3) | 16.4 (16.2) | |||
| 2 | 10.2 (3.7) | 0.6 (0.5) | 49.3 (53.6) | 0.5 (0.2) | 4.3 (5.2) | 8.7 (7.0) | |
| 3 | 0.4 (0.2) | 2.3 | |||||
AUClast values are given as arithmetic mean (SD), demographic parameters are given as mean (range). *Insufficient data available to calculate mean values; –, not available, agiven as mode of every study for datasets I and II, or number of individuals with the respective activity score for dataset III.
Abbreviations: AUClast, area under the plasma concentration‐time curve from the time of the first concentration measurement to the time of the last concentration measurement; DGI, drug‐gene interaction.
For dextromethorphan, the PBPK model implements metabolism via CYP2D6 (leading to the formation of dextrorphan) and CYP3A4 as well as excretion via passive glomerular filtration. To emulate the effect of lysosomal trapping in the gastrointestinal mucosa, 25 , 26 a binding process was included in the model that is comprehensively described in Section S1.5 of Supplementary S1.
The primary metabolite dextrorphan is metabolized via CYP3A4 and UGT2B15. The latter serves as a surrogate pathway in the model for the glucuronidation via multiple UGT2B enzymes, as UGT2B15 was reported to have the largest contribution of all involved UGTs. 6 Dextrorphan O‐glucuronide is renally eliminated via passive glomerular filtration and active secretion to the urine. Other dextromethorphan metabolites, such as 3‐methoxymorphinan or 3‐hydroxymorphinan, were not included as model compounds due to the limited number of published plasma concentration‐time profiles for these analytes.
An overview of the implemented model compounds and pathways is provided in Figure 1. For dextromethorphan, dextrorphan, and dextrorphan O‐glucuronide, the drug‐dependent model input parameters are provided in Section S2.1 of Supplementary S1.
FIGURE 1.
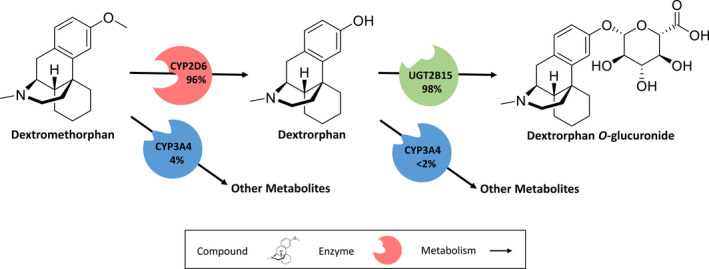
Implemented dextromethorphan metabolic pathways. Dextromethorphan is O‐demethylated by CYP2D6 and N‐demethylated by CYP3A4. The metabolite dextrorphan is further metabolized via CYP3A4 (N‐demethylation) and UGT2B15 (O‐glucuronidation). Dextrorphan O‐glucuronide is excreted in the urine. Percentages shown refer to the fraction metabolized by the respective enzyme, calculated for extensive metabolizers of CYP2D6. CYP2D6: cytochrome P450 2D6, CYP3A4: cytochrome P450 3A4, UGT2B15: Uridine 5'‐diphospho‐glucuronosyltransferase 2B15
PBPK base model evaluation
Overall, the PBPK model accurately predicted dextromethorphan, dextrorphan, and dextrorphan O‐glucuronide plasma concentrations after intravenous and oral administration with a selection of predicted compared to observed plasma concentration time‐profiles presented in Figure 2. The simulations of all 28 modeled population studies are shown in sections S3.1 and S5.1 of Supplementary S1.
FIGURE 2.
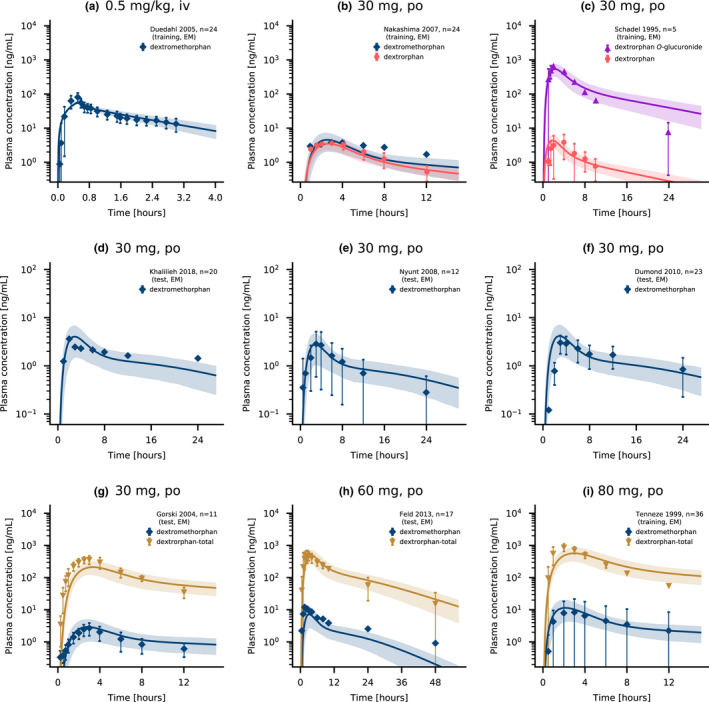
Dextromethorphan and dextrorphan plasma concentrations. Model predictions of dextromethorphan and its metabolites dextrorphan and dextrorphan O‐glucuronide as well as total dextrorphan (dextrorphan + dextrorphan O‐glucuronide) plasma concentration‐time profiles of selected intravenous (a) and oral studies (b–i) from the training and test datasets, compared to observed data. 7 , 8 , 45 , 46 , 47 , 48 , 49 , 50 , 51 Population predictions (n = 1000) are shown as lines with ribbons (arithmetic mean ± SD), symbols present the corresponding observed data ±SD. Detailed information on all clinical studies is listed in sections S2.2 and S4.2 of Supplementary S1. iv, intravenous; po, oral
Goodness‐of‐fit plots comparing predicted and observed plasma concentrations, AUClast and Cmax values are presented in Figure 3. Overall, 70.6% of predicted plasma concentrations were within the two‐fold range of the corresponding observed concentrations. Furthermore, 35 of 42 of the predicted AUClast values (several studies included measurements of multiple analytes) and 35 of 41 of the predicted Cmax values were within two‐fold range with model GMFE values of 1.53 (range 1.01–3.45) for predicted AUClast and 1.46 (range 1.01–2.97) for predicted Cmax values. MRD values of predicted plasma concentrations as well as AUClast and Cmax ratios for all 28 clinical studies and all measured analytes are provided in sections S3.3, S3.5, S5.3, and S5.5 of Supplementary S1.
FIGURE 3.
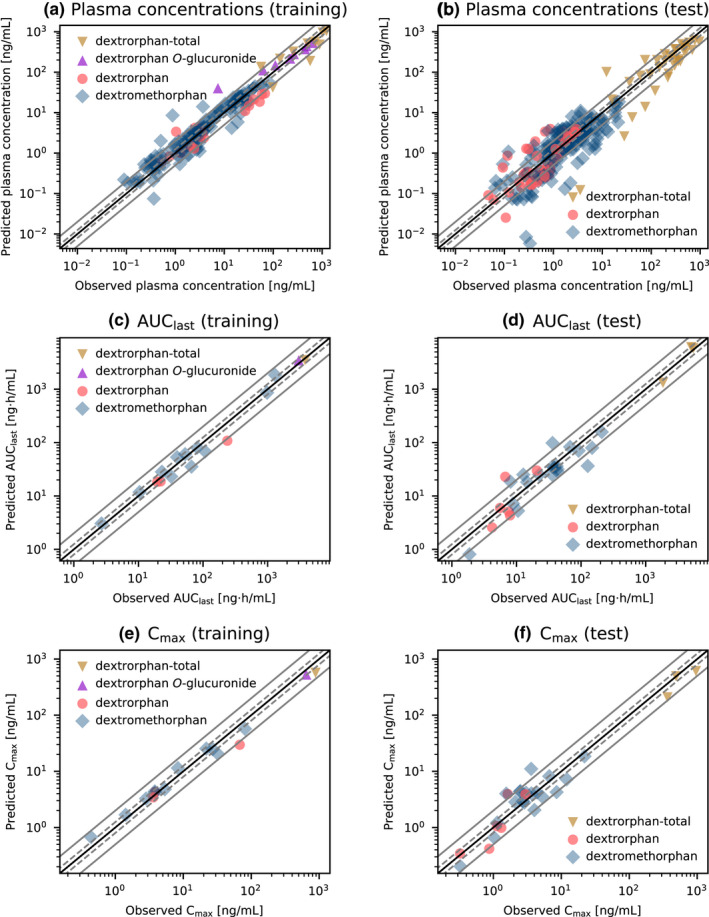
Goodness‐of fit plots for the final dextromethorphan model. Predicted versus observed plasma concentrations (a, b), AUClast values (c, d) and Cmax values (e, f) for the training (left column) and test (right column) datasets. The solid black line indicates the line of identity, solid gray lines show two‐fold deviation, dashed gray lines indicate 1.25‐fold deviation. Detailed information on all clinical studies is listed in sections S2.2 and S4.2 of Supplementary S1. AUClast, area under the plasma concentration‐time curve from the time of the first concentration measurement to the time of the last concentration measurement; Cmax, maximum plasma concentration, dextrorphan‐total: sum of dextrorphan and dextrorphan O‐glucuronide concentrations
A simulation of 30 mg dextromethorphan hydrobromide administered orally (standard dose) was used for local sensitivity analysis. Parameters with associated sensitivity values greater than 0.5 (100% parameter value perturbation resulting in a greater than 50% change of predicted AUC) were considered sensitive. Sensitive parameters were, in order of highest to lowest impact, fu (literature value), CYP2D6 kcat (optimized value), lipophilicity (literature value), CYP2D6 KM (literature value), and intestinal permeability (optimized value). A quantitative and visual representation of the local sensitivity analysis is provided in Section S3.6 of Supplementary S1.
DGI model building
The DGI model training dataset consisted of four studies that reported CYP2D6 activity scores or genotypes of their respective study populations. To complement these studies, 24 individual plasma concentration‐time profiles were included. The assignment of studies and individual profiles to the respective datasets is listed in sections S4.2 and S6.3 of Supplementary S1.
Overall, activity scores in the DGI model training dataset ranged from 0 (PM) to 3 (ultrarapid metabolizer) and covered a total of eight activity scores. This dataset was used to optimize population kcat values for the activity scores of the respective studies or individual profiles (see Section S4.1 of Supplementary S1).
DGI model evaluation
The DGI model was evaluated using a total of 13 clinical population studies, which stratified their subjects by CYP2D6 activity score or phenotype. These studies either provided the CYP2D6 phenotype (4 studies) or comprehensive information on the CYP2D6 genotype of individuals (9 studies). Simulations were performed using the corresponding kcat values with respect to activity score (Section S4.1 of Supplementary S1) or phenotype (Section S2.1 of Supplementary S1).
The good performance of the final dextromethorphan DGI model is demonstrated in Figure 4a–e depicting predicted dextromethorphan plasma concentration‐time profiles of populations with different activity scores compared to their respective observed data. Plots documenting the model performance of all 15 DGI studies are provided in Section S5.1 of Supplementary S1.
FIGURE 4.
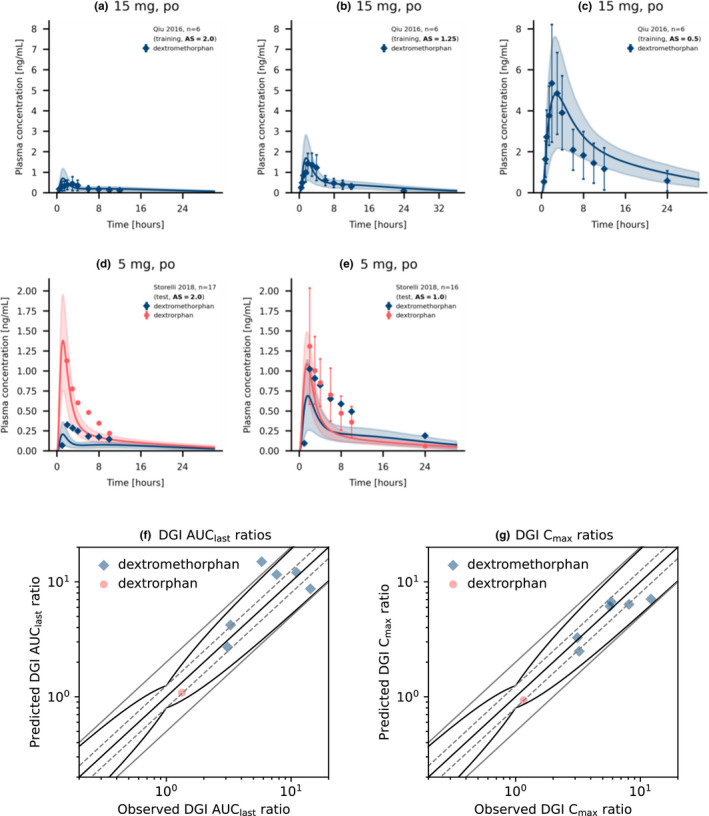
Simulated dextromethorphan and dextrorphan plasma concentrations and DGI ratios for different CYP2D6 activity scores. Upper panel: Dextromethorphan (a–c) as well as Dextromethorphan and dextrorphan (d, e) plasma concentration‐time profiles of selected dextromethorphan CYP2D6 DGI studies, compared to observed data. 52 , 53 Population predictions (n = 1000) are shown as lines with ribbons (arithmetic mean ± SD), symbols present the corresponding observed data ±SD. Lower panel: comparison of predicted versus observed DGI AUClast ratios (f) and DGI Cmax ratios (g) for all analyzed dextromethorphan CYP2D6 DGI studies. The straight black line indicates the line of identity, curved black lines show prediction success limits proposed by Guest et al. including 1.25‐fold variability. 27 Solid gray lines indicate two‐fold deviation, dashed gray lines show 1.25‐fold deviation. Detailed information on all DGI studies as well as the plotted values are given in section S4.1 and S5.4 of Supplementary S1, respectively. AS, activity score; AUC, area under the plasma concentration‐time curve; AUClast, AUC from the time of the first concentration measurement to the time of the last concentration measurement; Cmax, maximum plasma concentration; DGI, drug‐gene interaction; po, oral
Predicted DGI AUClast and Cmax ratios were in good agreement with observed DGI ratios, demonstrating that the effect of different CYP2D6 activity scores on the PK of dextromethorphan and dextrorphan was well‐described by the model. Specifically, six of seven AUClast and six of six Cmax ratios were within the prediction success limits suggested by Guest et al. adopted for DGI evaluations, 27 as visualized in Figure 4f,g. The predicted DGI AUClast ratios showed an overall GMFE of 1.45 (range 1.04–2.84) and the overall GMFE of predicted DGI Cmax ratios was calculated as 1.21 (range 1.02–1.40). Predicted to observed DGI AUClast and Cmax ratios for all studies are provided in Section S5.5 of Supplementary S1. Predictions of dextromethorphan, dextrorphan, and dextrorphan O‐glucuronide exposure in individuals with different activity scores after a single oral dose of 30 mg dextromethorphan hydrobromide and a comparison of the corresponding AUC values are given in Figure 5.
FIGURE 5.
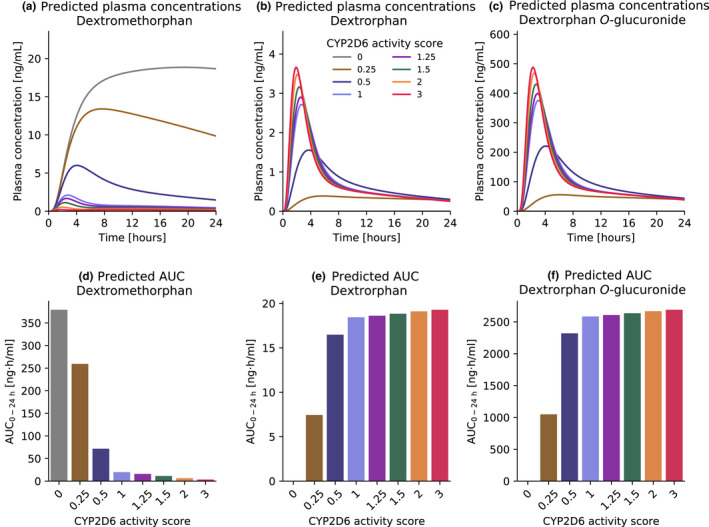
Predicted dextromethorphan, dextrorphan and dextrorphan O‐glucuronide exposure in individuals with different activity scores. Simulations were performed for a single oral dose of 30 mg dextromethorphan hydrobromide in healthy male individuals. Top row: dextromethorphan (a), dextrorphan (b) and dextrorphan O‐glucuronide (c) plasma concentrations. Bottom row: Dextromethorphan (d), dextrorphan (e) and dextrorphan O‐glucuronide (f) AUC0–24 h values for different activity scores. AUC, area under the plasma concentration‐time curve
Interindividual variability within activity score groups
The individual profiles from four cocktail studies were used to assess the extent of IIV within activity score groups. For 66 of the 72 study subjects, the CYP2D6 genotype was provided. Six subjects were not genotyped and consequently excluded from this analysis.
The distribution of activity scores from the dataset is listed in Section S6.2 of Supplementary S1. Plasma concentration‐time profiles of dextromethorphan, dextrorphan, and total dextrorphan were simulated using the population kcat values given in Section S4.1 of Supplementary S1. Additionally, the profiles were simulated using individually optimized kcat values and the geometric mean with geometric standard deviation of the individual kcat values were calculated for all activity score groups with n greater than 2 (see Section S6.2 of Supplementary S1).
A representative selection of predictions using individual and model CYP2D6 kcat values is visualized in Figure 6. Furthermore, Section S6.4 of Supplementary S1 includes plots with model and individual predictions for all 66 genotyped individuals alongside model predictions for the six non‐genotyped individuals. The latter were simulated using the population kcat value for EMs (see Section S2.1 of Supplementary S1).
FIGURE 6.
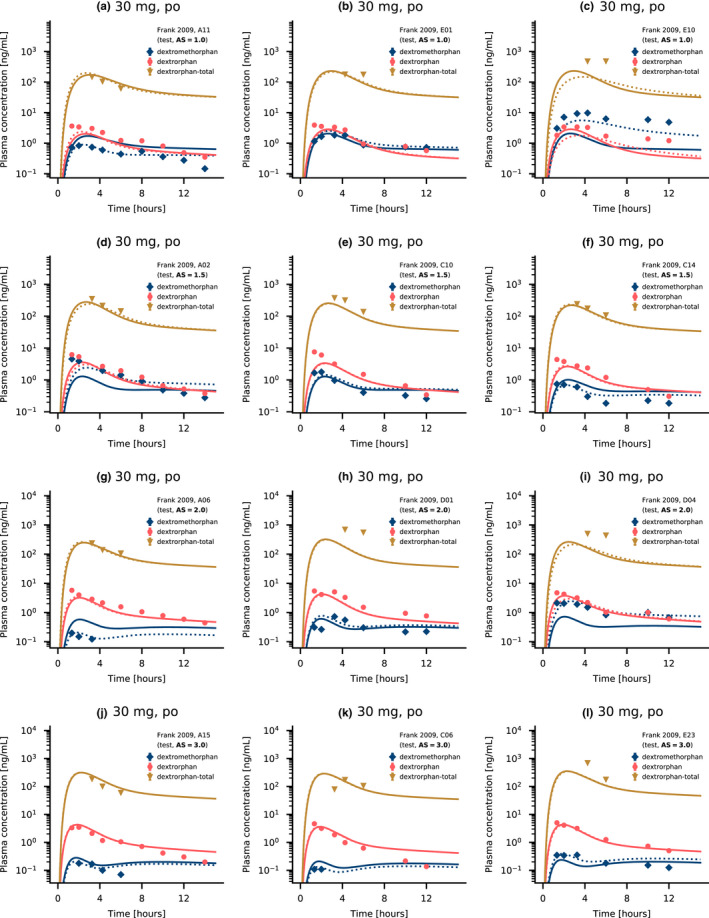
Dextromethorphan and dextrorphan plasma concentrations for individuals of several activity score groups. Selected dextromethorphan, dextrorphan, and total dextrorphan (dextrorphan + dextrorphan O‐glucuronide) plasma concentration‐time profiles compared to observed data reported by Frank 2009. 22 Predictions are shown as lines. Solid lines represent model predictions, dotted lines represent individual predictions. Symbols present the corresponding observed data. Detailed information on all individual profiles is listed in Sections S6.1, S6.2, and S6.3 of Supplementary S1. AS, activity score; po, oral
The predictive performance using model kcat was compared to using the individual optimized kcat values by calculating the GMFE for all individual plasma concentration‐time profiles (see Sections S6.7, S6.8, and S8 of Supplementary S1). Generally, model performance improved for simulations of dextromethorphan and dextrorphan plasma concentration‐time profiles using the individually optimized kcat when compared to simulations, where population kcat values were used across all activity scores and analyzed studies. However, total dextrorphan AUClast and Cmax values were markedly underpredicted for studies D and E (GMFEs of 3.93 and 3.28 for study D and 2.81 and 2.69 for study E) compared to studies A and C (GMFEs of 1.30 and 1.44 for study A and 1.20 and 1.24 for study C). Predicted to observed AUClast and Cmax ratios for all individual simulations using the model kcat and the individual optimized kcat are listed in Section S6.7 of Supplementary S1. Section S6.8 of Supplementary S1 gives a detailed breakdown of AUClast and Cmax ratios grouped by study and activity score.
Moreover, the optimized individual kcat values for the different activity score groups were plotted against their activity score to visualize the distribution of individual kcat values in the respective activity score groups (see Figure 7a). A regression analysis of model kcat values compared to the geometric mean of optimized individual kcat values revealed a high correlation (R 2 = 0.9988). Consequently, the individual profiles were sufficiently well‐described with the model kcat values. The results of the regression analysis are illustrated in Figure 7b.
FIGURE 7.
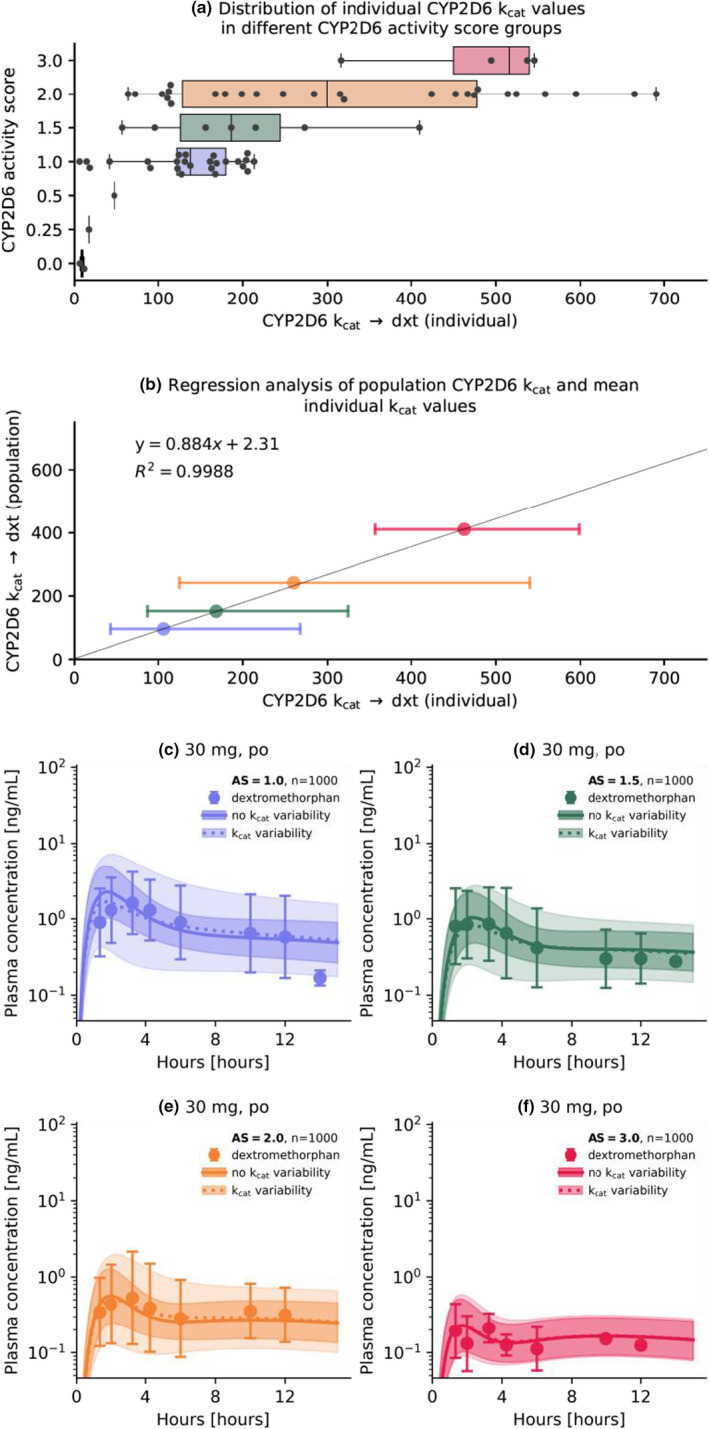
Analysis of optimized individual CYP2D6 kcat values for the different activity scores and population simulations for different activity score groups. (a) Box‐ and scatterplots for optimized individual kcat values in the respective activity score groups. Boxes represent interquartile ranges, lines within boxes represent median values. (b) Comparison of model kcat and optimized geometric mean kcat values and regression analysis. Colored circles represent the geometric mean kcat value for an activity score group compared to the population kcat value. Error bars represent the geometric standard deviation. Simulations were performed with the population kcat values using a standard administration protocol (a single dose of 30 mg dextromethorphan hydrobromide) for populations with an CYP2D6 activity score of 1 (c), 1.5 (d), 2 (e), and 3 (f) with no variability and variability (calculated geometric standard deviation) on the CYP2D6 population kcat. Population predictions (n = 1000) are represented as lines with ribbons (geometric mean with geometric standard deviation), symbols represent the corresponding observed data (geometric mean with geometric standard deviation) for the population reported by Frank 2009. 22 AS, activity score; CYP2D6, cytochrome p450 2D6; dxt, dextrorphan; kcat, catalytic rate constant; R 2, coefficient of determination
Finally, population simulations were performed with sampling from a log‐normal distribution with mean and dispersion parameters calculated from the samples of optimized individual kcat values (see Section S6.2 of Supplementary S1) to analyze the simulated coverage of IIV observed in dextromethorphan plasma concentrations from the study populations.
Subsequently, predictions were compared graphically in population simulations with no variability of the CYP2D6 population kcat. As expected, model predictions including the kcat variability improved describing the large extent of IIV within an activity score group compared to predictions with no variability on the CYP2D6 kcat (see Figure 7c–f).
DISCUSSION
In this study, a whole‐body PBPK model of dextromethorphan and its metabolites dextrorphan and dextrorphan O‐glucuronide was developed and evaluated to predict drug plasma concentrations over a wide dosing range (5–80 mg). A CYP2D6 activity score‐dependent metabolism of dextromethorphan was implemented to describe the effect of CYP2D6 DGIs on the PK of the modeled compounds. Moreover, the model was applied to investigate the IIV of dextromethorphan PK within different activity score groups.
Three previously published PBPK models of dextromethorphan were found in the literature that focused on different aspects of PBPK modeling, specifically cross‐species modeling, 28 investigation of pregnancy effects, 29 and the impact of formulations (and, by extension, lysosomal trapping) 26 on dextromethorphan pharmacokinetics. Two studies included either dextrorphan 29 or dextrorphan and dextrorphan O‐glucuronide 26 as model compounds. These studies also included “traditional” phenotypes (EMs and PMs) in the model and did not further differentiate between CYP2D6 activity scores. Consequently, our model is the first whole‐body parent‐metabolite‐metabolite PBPK model of dextromethorphan, aiming to investigate the effect of CYP2D6 activity scores on dextromethorphan PK, with a total of eight different activity scores implemented.
In our model, the dextromethorphan CYP2D6 DGIs were described without explicitly modeling distinct CYP2D6 genotypes. Although a wide variety of relevant genotype‐specific in vitro parameters, such as KM and Vmax are available in the literature, 30 , 31 , 32 implementing all possible genotypes using a genotype‐specific approach would be infeasible due to the large (and still growing) amount of known CYP2D6 alleles. 33 Thus, a CYP2D6 activity score‐specific approach was developed. As a result, this PBPK model cannot further differentiate between different genotypes within the same activity score group, for instance, CYP2D6*1/*1 and CYP2D6*2/*2. However, the model could be readily extended to include a genotype‐specific CYP2D6 metabolism in the future.
Moreover, CYP2D6 metabolism for different activity scores was implemented with a fixed KM literature value 6 for all covered activity scores. However, in vitro data shows that KM may vary between different genotypes and activity scores. 31 , 32 , 34 Nonetheless, a study investigating the effect of activity scores on the CYP2D6‐dependent metabolism of dextromethorphan in vitro found no significant correlation between activity score and CYP2D6 KM. 30 Most studies reported a reduction of CYP2D6‐dependent clearance (CLint and Vmax/KM) when comparing reduced function alleles (*10 and *17) to the wildtype *1 allele. 30 , 31 , 32 Additionally, analyses of CYP2D6 content in HLMs showed a high positive correlation between CYP2D6 abundances and activity score, albeit substantial IIV in CYP2D6 content within activity score groups and even in groups sharing the same CYP2D6 diplotype has been observed. 16 These trends in CYP2D6 content in HLMs and CYP2D6 CLint are reflected in the final dextromethorphan PBPK model with higher CYP2D6 activity scores inferring higher population kcat values (see Section S4.1 of Supplementary S1). A similar modeling approach was also utilized for previously developed PBPK models of CYP2D6 substrates. 24 The CYP2D6 kcat value for populations grouped as EMs was observed to be lower than for genotyped normal metabolizers with activity scores ranging from 1.25–2.25 (compare sections S2.1 and S4.1 of Supplementary S1). Typically, study subjects in the literature were either phenotyped via measurements of urinary metabolic ratio, often using arbitrary cutoff points for poor metabolizers, 15 or via screening for null alleles. 35 Thus, there is only a limited intersection between the broad EM phenotype category and the genetically determined NMs. 36 Overall, the presented model was able to accurately describe DGI AUClast and Cmax ratios as well as the plasma concentration‐time profiles of all analyzed clinical studies.
The final dextromethorphan PBPK model was applied to investigate the effect of IIV on the PK of dextromethorphan with a total of 72 individual plasma concentration‐time profiles of dextromethorphan, dextrorphan, and total dextrorphan. A substantial variability was observed within activity scores 1–3 (geometric standard deviation range of 1.29–2.52). For activity scores less than 1, the number of individual profiles per score (less than 5) was insufficient to make meaningful assessments of the IIV. The large extent of IIV in the PK of CYP2D6 substrates within activity score groups or even within subjects possessing the same CYP2D6 genotype, is a well‐documented phenomenon. 16 A twin study on the heritability of metoprolol PK, concluded that genetic components independent of the CYP2D6 gene may be responsible for the IIV in CYP2D6 activity. 37 Indeed, the rs5758550 single‐nucleotide polymorphism (SNP) was identified as an enhancer SNP and may, in the future, even lead to a reclassification of activity scores based on CYP2D6 and rs5758550 genotype. 38 Currently published literature lacks clinical in vivo studies describing the effect of the rs5758550 genotype on the PK of dextromethorphan. Other genetic factors, such as regulation of CYP2D6 expression via transcription factors or miRNA, are also likely to contribute to IIV and intraindividual variability. 16 Additionally, genetic and non‐genetic variability in enzymes other than CYP2D6 are expected to contribute to the IIV in dextromethorphan PK, specifically for CYP2D6 PMs, as the fraction metabolized by CYP2D6 decreases for dextromethorphan from greater than 95% for EMs 5 to 0% for PMs of CYP2D6, 10 consequently increasing the fraction of dextromethorphan metabolized by CYP3A4. Additionally, IIV can be observed in plasma concentrations of dextrorphan and dextrorphan O‐glucuronide, possibly caused by variability in CYP3A4 and UGT enzymes. As genotypic data for CYP3A and UGT was unavailable for study subjects, the analysis of IIV was performed for dextromethorphan plasma concentrations purely in the context of CYP2D6 activity score groups. However, as new data emerges, the presented PBPK model can mechanistically be adapted to describe these genotypic effects of CYP2D6 and other pharmacogenes affecting the PK of dextromethorphan and its metabolites. A large extent of IIV in plasma concentrations and CYP2D6 activity was observed and quantified in this study. To reflect this in the model, the distributions of CYP2D6 kcat values for activity scores 1, 1.5, 2, and 3 were characterized from kcat optimizations in 72 individuals to improve population predictions, as demonstrated in Figure 7c–f, and may be used in future PBPK models of CYP2D6 substrates.
To supplement the limited number of studies in which dextromethorphan was administered alone (14 studies), studies in which dextromethorphan was administered as part of a phenotyping cocktail (11 studies and the studies compiled by Frank et al. 22 ) were included in the model dataset. All modeled cocktail studies administered either the “Cologne” cocktail, 21 , 39 the “Cooperstown 5+1” 12 cocktail, or minor variations thereof (see Section 1.1 of Supplementary S1). No relevant mutual interactions have been observed for these cocktails, although sample sizes for these assessments were often small. 21 Additionally, assessments of these interactions are generally concerned with the effect of the cocktail on primary pathways of the cocktail compounds (i.e., dextromethorphan O‐demethylation). 40 Here, additional in vitro experiments are needed to evaluate possible effects of phenotyping cocktails on other model pathways, such as dextrorphan O‐glucuronidation. Overall, plasma concentration‐time profiles were well‐predicted for all population studies regardless of whether dextromethorphan was administered alone or as part of a phenotyping cocktail (see Sections S3.1–S3.5 and Sections S5.1–S5.5).
Overall, model predictions were considered adequate for all population studies regardless of whether the study was a cocktail study or not (see Sections S3.2–S3.5 and S5.2–5.7 of Supplementary S1). For studies reporting individual plasma concentration‐time profiles, the model performed comparably well across all activity scores. However, a large interstudy variability was observed for dextromethorphan and total dextrorphan AUClast and Cmax values (see Section S6.8 of Supplementary S1). For instance, studies D and E reported up to four‐fold higher AUClast and Cmax values for total dextrorphan compared with studies A and C. As these studies were comparable in study design, cocktail composition, and sample analysis, as well as dextromethorphan and dextrorphan plasma concentrations, this apparent discrepancy was attributed to relatively small study cohorts and the large extent of IIV in CYP2D6 activity (see Figure 7a,b) described in the published literature. 16
Finally, the developed and evaluated PBPK model of dextromethorphan is a useful tool for clinicians to investigate the effect of CYP2D6 DGIs and the associated IIV on the PK of dextromethorphan and its metabolites. The mechanistical model can be extended to be used in other PBPK modeling scenarios, such as the prediction of drug‐drug interaction and DGI effects 41 and scaling to special populations, such as pediatrics, 42 geriatrics, 43 or patients with renal or hepatic impairment. 44 Moreover, the modeling approach presented in this study can serve as a blueprint to develop PBPK models of other CYP2D6 substrates.
CONFLICT OF INTEREST
The authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
S.R., D.S., U.F., M.S., and T.L. wrote the manuscript. T.L., D.S., and S.R. designed the research. S.R. performed the research. S.R., D.S., and T.L. analyzed the data.
Supporting information
Supplementary Material1
Supplementary Material2
Supplementary Material3
ACKNOWLEDGEMENTS
Open Access funding enabled and organized by Projekt DEAL.
Rüdesheim S, Selzer D, Fuhr U, Schwab M, Lehr T. Physiologically‐based pharmacokinetic modeling of dextromethorphan to investigate interindividual variability within CYP2D6 activity score groups. CPT Pharmacometrics Syst Pharmacol. 2022;11:494‐511. doi: 10.1002/psp4.12776
Funding information
M.S. was supported by the Robert Bosch Stiftung (Stuttgart, Germany), the European Commission Horizon 2020 UPGx grant 668353, a grant from the German Federal Ministry of Education and Research (BMBF 031L0188D), and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy—EXC 2180—390900677. T.L. was supported by the German Federal Ministry of Education and Research (BMBF, Horizon 2020 INSPIRATION grant 643271), under the frame of ERACoSysMed
REFERENCES
- 1. Guenin E, Armogida M, Riff D. Pharmacokinetic profile of dextromethorphan hydrobromide in a syrup formulation in children and adolescents. Clin Drug Investig. 2014;34:609‐616. [DOI] [PubMed] [Google Scholar]
- 2. Taylor CP, Traynelis SF, Siffert J, Pope LE, Matsumoto RR. Pharmacology of dextromethorphan: Relevance to dextromethorphan/quinidine (Nuedexta®) clinical use. Pharmacol Ther. 2016;164:170‐182. [DOI] [PubMed] [Google Scholar]
- 3. Zawertailo LA, Kaplan HL, Busto UE, Tyndale RF, Sellers EM. Psychotropic effects of dextromethorphan are altered by the CYP2D6 polymorphism. J Clin Psychopharmacol. 1998;18:332‐337. [DOI] [PubMed] [Google Scholar]
- 4. Benet LZ, Broccatelli F, Oprea TI. BDDCS applied to over 900 drugs. AAPS J. 2011;13:519‐547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Capon DA, Bochner F, Kerry N, Mikus G, Danz C, Somogyi AA. The influence of CYP2D6 polymorphism and quinidine on the disposition and antitussive effect of dextromethorphan in humans. Clin Pharmacol Ther. 1996;60:295‐307. [DOI] [PubMed] [Google Scholar]
- 6. Lutz JD, Isoherranen N. Prediction of relative in vivo metabolite exposure from in vitro data using two model drugs: dextromethorphan and omeprazole. Drug Metab Dispos. 2012;40:159‐168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schadel M, Wu D, Otton SV, Kalow W, Sellers EM. Pharmacokinetics of dextromethorphan and metabolites in humans. J Clin Psychopharmacol. 1995;15:263‐269. [DOI] [PubMed] [Google Scholar]
- 8. Gorski JC, Huang S‐M, Pinto A, et al. The effect of echinacea (Echinacea purpurea root) on cytochrome P450 activity in vivo. Clin Pharmacol Ther. 2004;75:89‐100. [DOI] [PubMed] [Google Scholar]
- 9. FDA . FDA Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers. https://www.fda.gov/drugs/drug‐interactions‐labeling/drug‐development‐and‐drug‐interactions‐table‐substrates‐inhibitors‐and‐inducers#table2‐1.
- 10. Zanger UM, Raimundo S, Eichelbaum M. Cytochrome P450 2D6: overview and update on pharmacology, genetics, biochemistry. Naunyn Schmiedebergs Arch Pharmacol. 2004;369:23‐37. [DOI] [PubMed] [Google Scholar]
- 11. Cicali EJ, Smith DM, Duong BQ, Kovar LG, Cavallari LH, Johnson JA. A scoping review of the evidence behind cytochrome P450 2D6 isoenzyme inhibitor classifications. Clin Pharmacol Ther. 2020;108:116‐125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chainuvati S, Nafziger AN, Steven Leeder J, et al. Combined phenotypic assessment of cytochrome P450 1A2, 2C9, 2C19, 2D6, and 3A, N‐acetyltransferase‐2, and xanthine oxidase activities with the ‘Cooperstown 5+1 cocktail’. Clin Pharmacol Ther. 2003;74:437‐447. [DOI] [PubMed] [Google Scholar]
- 13. Ryu JY, Song IS, Sunwoo YE, et al. Development of the ‘Inje cocktail’ for high‐throughput evaluation of five human cytochrome P450 isoforms in vivo. Clin Pharmacol Ther. 2007;82:531‐540. [DOI] [PubMed] [Google Scholar]
- 14. Pharmacogene Variation Consortium (PharmVar) CYP2D6 gene . Gaedigk al. 2018, CPT 103399; Gaedigk al. 2019, CPT 10529 at https://www.pharmvar.org/gene/CYP2D6, (Gaedigk et al 2018, CPT 103:399; Gaedigk et al 2019, CPT 105:29).
- 15. Gaedigk A, Simon SD, Pearce RE, Bradford LD, Kennedy MJ, Leeder JS. The CYP2D6 activity score: translating genotype information into a qualitative measure of phenotype. Clin Pharmacol Ther. 2008;83:234‐242. [DOI] [PubMed] [Google Scholar]
- 16. Gaedigk A, Dinh JC, Jeong H, Prasad B, Ten Leeder JS. years’ experience with the CYP2D6 activity score: a perspective on future investigations to improve clinical predictions for precision therapeutics. J Pers Med. 2018;8:1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Caudle KE, Sangkuhl K, Whirl‐Carrillo M, et al. Standardizing CYP 2D6 genotype to phenotype translation: consensus recommendations from the clinical pharmacogenetics implementation consortium and Dutch pharmacogenetics working group. Clin Transl Sci. 2020;13:116‐124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lippert J, Burghaus R, Edginton A, et al. Open systems pharmacology community—an open access, open source, open science approach to modeling and simulation in pharmaceutical sciences. CPT Pharmacometrics Syst Pharmacol. 2019;8:878‐882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wojtyniak JG, Britz H, Selzer D, Schwab M, Lehr T. Data digitizing: accurate and precise data extraction for quantitative systems pharmacology and physiologically‐based pharmacokinetic modeling. CPT Pharmacometrics Syst Pharmacol. 2020;9:322‐331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Seabold S, Perktold J. Statsmodels: econometric and statistical modeling with Python. Proc 9th Python Sci Conf. 2010:92‐96. https://conference.scipy.org/proceedings/scipy2010/seabold.html [Google Scholar]
- 21. Fuhr U, Jetter A, Kirchheiner J. Appropriate phenotyping procedures for drug metabolizing enzymes and transporters in humans and their simultaneous use in the ‘cocktail’ approach. Clin Pharmacol Ther. 2007;81:270‐283. [DOI] [PubMed] [Google Scholar]
- 22. Frank D. Bewertung von pharmakokinetischen Parametern zur Phänotypisierung des menschlichen Cytochrom P450 Enzyms CYP2D6 mittels Dextromethorphan. (2009). https://bonndoc.ulb.uni‐bonn.de/xmlui/bitstream/handle/20.500.11811/4056/1707.pdf?sequence=1&isAllowed=y
- 23. Open Systems Pharmacology Suite Community Open Systems Pharmacology Suite Manual, Version 7.4. (2018). https://github.com/Open‐Systems‐Pharmacology/OSPSuite.Documentation/blob/master/OpenSystemsPharmacologySuite.pdf
- 24. Rüdesheim S, Wojtyniak J‐G, Selzer D, et al. Physiologically based pharmacokinetic modeling of metoprolol enantiomers and α‐hydroxymetoprolol to describe CYP2D6 drug‐gene interactions. Pharmaceutics. 2020;12:1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kazmi F, Hensley T, Pope C, et al. Lysosomal sequestration (trapping) of lipophilic amine (cationic amphiphilic) drugs in immortalized human hepatocytes (Fa2N‐4 cells). Drug Metab Dispos. 2013;41:897‐905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bolger MB, Macwan JS, Sarfraz M, Almukainzi M, Löbenberg R. The irrelevance of in vitro dissolution in setting product specifications for drugs like dextromethorphan that are subject to lysosomal trapping. J Pharm Sci. 2019;108:268‐278. [DOI] [PubMed] [Google Scholar]
- 27. Guest EJ, Aarons L, Houston JB, Rostami‐Hodjegan A, Galetin A. Critique of the two‐fold measure of prediction success for ratios: application for the assessment of drug‐drug interactions. Drug Metab Dispos. 2011;39:170‐173. [DOI] [PubMed] [Google Scholar]
- 28. Thiel C, Schneckener S, Krauss M, et al. A systematic evaluation of the use of physiologically based pharmacokinetic modeling for cross‐species extrapolation. J Pharm Sci. 2015;104:191‐206. [DOI] [PubMed] [Google Scholar]
- 29. Ke AB, Nallani SC, Zhao P, Rostami‐Hodjegan A, Isoherranen N, Unadkat JD. A physiologically based pharmacokinetic model to predict disposition of CYP2D6 and CYP1A2 metabolized drugs in pregnant women. Drug Metab Dispos. 2013;41:801‐813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Storelli F, Desmeules J, Daali Y. Genotype‐sensitive reversible and time‐dependent CYP2D6 inhibition in human liver microsomes. Basic Clin Pharmacol Toxicol. 2019;124:170‐180. [DOI] [PubMed] [Google Scholar]
- 31. Marcucci KA, Pearce RE, Crespi C, Steimel DT, Steven Leeder J, Gaedigk A. Characterization of cytochrome P450 2D6.1 (CYP2D6.1), CYP2D6.2, and CYP2D6.17 activities toward model CYP2D6 substrates dextromethorphan, bufuralol, and debrisoquine. Drug Metab Dispos. 2002;30:595‐601. [DOI] [PubMed] [Google Scholar]
- 32. Shen H, He MM, Liu H, et al. Comparative metabolic capabilities and inhibitory profiles of CYP2D6.1, CYP2D6.10, and CYP2D6.17. Drug Metab Dispos. 2007;35:1292‐1300. [DOI] [PubMed] [Google Scholar]
- 33. Nofziger C, Turner AJ, Sangkuhlet K, et al. PharmVar GeneFocus: CYP2D6. Clin Pharmacol Ther. 2020;107:154‐170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bapiro TE, Hasler JA, Ridderström M, Masimirembwa CM. The molecular and enzyme kinetic basis for the diminished activity of the cytochrome P450 2D6.17 (CYP2D6.17) variant: Potential implications for CYP2D6 phenotyping studies and the clinical use of CYP2D6 substrate drugs in some African populations. Biochem Pharmacol. 2002;64:1387‐1398. [DOI] [PubMed] [Google Scholar]
- 35. Heim M, Meyer U. Genotyping of poor metabolisers of debrisoquine by allele‐specific PCR amplification. Lancet. 1990;336:529‐532. [DOI] [PubMed] [Google Scholar]
- 36. Lu S, Nand RA, Yang JS, Chen G, Gross AS. Pharmacokinetics of CYP2C9, CYP2C19, and CYP2D6 substrates in healthy Chinese and European subjects. Eur J Clin Pharmacol. 2018;74:285‐296. [DOI] [PubMed] [Google Scholar]
- 37. Matthaei J, Brockmöller J, Tzvetkov MV, et al. Heritability of metoprolol and torsemide pharmacokinetics. Clin Pharmacol Ther. 2015;98:611‐621. [DOI] [PubMed] [Google Scholar]
- 38. Thomas CD, Mosley SA, Kim S, et al. Examination of metoprolol pharmacokinetics and pharmacodynamics across CYP2D6 genotype‐derived activity scores. CPT Pharmacometrics Syst Pharmacol. 2020;9:678‐685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gazzaz M, Kinzig M, Schaeffeler E, et al. Drinking ethanol has few acute effects on CYP2C9, CYP2C19, NAT2, and P‐glycoprotein activities but somewhat inhibits CYP1A2, CYP2D6, and intestinal CYP3A: so what? Clin Pharmacol Ther. 2018;104:1249‐1259. [DOI] [PubMed] [Google Scholar]
- 40. Frank D, Jaehde U, Fuhr U. Evaluation of probe drugs and pharmacokinetic metrics for CYP2D6 phenotyping. Eur J Clin Pharmacol. 2007;63:321‐333. [DOI] [PubMed] [Google Scholar]
- 41. Türk D, Hanke N, Lehr T. A physiologically‐based pharmacokinetic model of trimethoprim for MATE1, OCT1, OCT2, and CYP2C8 drug–drug–gene interaction predictions. Pharmaceutics. 2020;12:1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Edginton AN, Schmitt W, Willmann S. Development and evaluation of a generic physiologically based pharmacokinetic model for children. Clin Pharmacokinet. 2006;45:1013‐1034. [DOI] [PubMed] [Google Scholar]
- 43. Schlender J‐F, Meyer M, Thelen K, et al. Development of a whole‐body physiologically based pharmacokinetic approach to assess the pharmacokinetics of drugs in elderly individuals. Clin Pharmacokinet. 2016;55:1573‐1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hanke N, Türk D, Selzer D, et al. A comprehensive whole‐body physiologically based pharmacokinetic drug–drug–gene interaction model of metformin and cimetidine in healthy adults and renally impaired individuals. Clin Pharmacokinet. 2020;59:1419‐1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Duedahl TH, Dirks J, Petersen KB, et al. Intravenous dextromethorphan to human volunteers: relationship between pharmacokinetics and anti‐hyperalgesic effect. Pain. 2005;113:360‐368. [DOI] [PubMed] [Google Scholar]
- 46. Nakashima D, Takama H, Ogasawara Y, et al. Effect of cinacalcet hydrochloride, a new calcimimetic agent, on the pharmacokinetics of dextromethorphan. in vitro and clinical studies. J Clin Pharmacol. 2007;47:1311‐1319. [DOI] [PubMed] [Google Scholar]
- 47. Khalilieh S, Hussain A, Montgomery D, et al. Effect of tildrakizumab (MK‐3222), a high affinity, selective anti‐IL23p19 monoclonal antibody, on cytochrome P450 metabolism in subjects with moderate to severe psoriasis. Br J Clin Pharmacol. 2018;84:2292‐2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nyunt MM, Becker S, MacFarland RT, et al. Pharmacokinetic effect of AMD070, an Oral CXCR4 antagonist, on CYP3A4 and CYP2D6 substrates midazolam and dextromethorphan in healthy volunteers. JAIDS J Acquir Immune Defic Syndr. 2008;47:559‐565. [DOI] [PubMed] [Google Scholar]
- 49. Dumond JB, Vourvahis M, Rezk NL, et al. A phenotype‐genotype approach to predicting CYP450 and P‐glycoprotein drug interactions with the mixed inhibitor/inducer tipranavir/ritonavir. Clin Pharmacol Ther. 2010;87:735‐742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Feld R, Woo MM, Leighl N, et al. A clinical investigation of inhibitory effect of panobinostat on CYP2D6 substrate in patients with advanced cancer. Cancer Chemother Pharmacol. 2013;72:747‐755. [DOI] [PubMed] [Google Scholar]
- 51. Tennezé L, Verstuyft C, Becquemont L, Poirier JM, Wilkinson GR, Funck‐Brentano C. Assessment of CYP2D6 and CYP2C19 activity in vivo in humans: a cocktail study with dextromethorphan and chloroguanide alone and in combination. Clin Pharmacol Ther. 1999;66:582‐588. [DOI] [PubMed] [Google Scholar]
- 52. Qiu F, Liu S, Miao P, et al. Effects of the Chinese herbal formula “Zuojin Pill” on the pharmacokinetics of dextromethorphan in healthy Chinese volunteers with CYP2D6*10 genotype. Eur J Clin Pharmacol. 2016;72:689‐695. [DOI] [PubMed] [Google Scholar]
- 53. Storelli F, Matthey A, Lenglet S, et al. Impact of CYP2D6 functional allelic variations on phenoconversion and drug‐drug interactions. Clin Pharmacol Ther. 2018;104:148‐157. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material1
Supplementary Material2
Supplementary Material3