

Abstract
Recent data suggest that most genotoxic agents in cancer therapy can lead to shock of genome and increase in cell size, which leads whole genome duplication or multiplication, formation of polyploid giant cancer cells, activation of an early embryonic program, and dedifferentiation of somatic cells. This process is achieved via the giant cell life cycle, a recently proposed mechanism for malignant transformation of somatic cells. Increase in both cell size and ploidy allows cells to completely or partially restructures the genome and develop into a blastocyst-like structure, similar to that observed in blastomere-stage embryogenesis. Although blastocyst-like structures with reprogrammed genome can generate resistant or metastatic daughter cells or benign cells of different lineages, they also acquired ability to undergo embryonic diapause, a reversible state of suspended embryonic development in which cells enter dormancy for survival in response to environmental stress. Therapeutic agents can activate this evolutionarily conserved developmental program, and when cells awaken from embryonic diapause, this leads to recurrence or metastasis. Understanding of the key mechanisms that regulate the different stages of the giant cell life cycle offers new opportunities for therapeutic intervention.
Keywords: polyploid giant cancer cells; giant cell life cycle (giant cell cycle); embryonic diapause; dormancy, therapeutic resistance
1. Introduction
Despite limited successes in certain subtypes of cancer, the death rates associated with most late-stage cancers remain high. Many cancers quickly acquire resistance to drug therapy. Others show a remarkable initial clinical response, such that the patient is considered “cancer-free,” but many of these cancers have actually gone dormant and can then recur or metastasize after months and even years [1–7]. Despite intensive studies of these dormant cells over the past few decades, the mechanisms that allow these cells to become dormant and re-awaken for recurrence or metastasis are largely unknown. Dormancy has been listed as one of nine cancer “grand challenge problems” for which the National Cancer Institute and Cancer Research UK have called for an international effort to address [8].
Although long ignored, polyploid giant cancer cells (PGCCs) have recently become recognized as a culprit for cancer initiation, therapeutic resistance, and metastasis [9–16]. The origin of cancer is remarkably similar to early embryogenesis and can be explained by the newly proposed “life code” theory [11, 12, 17]. Multiple excellent reviews from various authors have been devoted to this topic recently and highlight the excitement of this new field of cancer biology [9–29]. In this review, we focus on developmental aspects of PGCCs and their relationship with normal embryogenesis and embryonic diapause, an evolutionarily conserved suspension mechanism for embryonic development at the blastocyst stage. We discuss how the development of PGCCs leads to the generation of blastocyst-like structures that can then undergo dormancy, leading to therapy resistance, recurrence, and metastasis. We also discuss challenges and opportunities for cancer therapy along different stages of the giant cell life cycle.
2. Blastocyst formation and embryonic diapause
The egg cell is the largest cell in humans. Following fertilization by sperm, the zygote within the zona pellucida undergoes progressive cleavage to form 2-cell and 4-cell blastomeres with progressively decreased cell size and increased nuclear:cytoplasmic ratio to activate the embryonic genome. The blastomeres at the 8- to 16-cell stage then compact to form the morula. The morula then develops into the blastocyst, which hatches out from the zona pellucida and implants into the uterus for normal embryonic development. The detailed embryogenesis mechanism and embryo-specific heredity explained by Barbara McClintock is described in an accompanying paper [17]. The giant egg cell and the associated compaction and morula stages are not commonly classified by embryologists as examples of classic polyploidy. However, compacted embryonic cells or morulae with high nuclear content within the defined space formed by the zona pellucida activate the embryonic program and develop into a blastocyst composed of the trophectoderm and inner cell mass. The life cycle of PGCCs has been shown to recapitulate this embryonic process for malignant transformation of somatic cells [11, 12].
Reproduction is a costly process in terms of energy requirements, and it is beneficial to have ideal conditions such as adequate food, regulated temperature, and all previous offspring weaned to ensure survival of the developing offspring before giving birth. Embryonic diapause is a period of suspension in blastocyst development following fertilization in response to a variety of environmental stresses [30, 31]. Diapause can occur in mammals or insects in response to unfavorable biological and/or environmental conditions that could represent a risk to the mother’s life. In many mammalian species, development of the early embryo ceases or implantation is delayed, but although the embryo stops proliferating, it remains viable for a short period [30]. Although this was initially considered a rare phenomenon, increasing evidence has shown that this may be a general mechanism in much of the animal kingdom in response to an unfavorable environment [30, 31]. Depending on the species, this delay can range from a few days to 11 months.
Diapause is extremely common in insects and includes several distinct phases [32, 33]. Before the process of diapause begins, the insect undergoes pre-diapause, which includes induction and preparation. The pre-diapause induction phase occurs at a genetically predetermined stage of life; insects are responsive to external cues that trigger the switch from direct development pathways to diapause pathways. During the pre-diapause preparation phase, insects accumulate and store molecules such as lipids, proteins, and carbohydrates to provide fuel for developmental diapause.
The process of diapause includes initiation, maintenance, and termination phases [33]. The initiation phase of diapause often starts when the insects begin to aggregate, migrate, or search for suitable overwintering sites. During the maintenance phase of diapause, insects have low metabolic activity and arrested development, and the insects are unresponsive to changes in the environment. The termination phase begins following certain stimuli such as chilling, freezing, or contact with water, and then development can resume over time. The termination phase of diapause is often followed by a state of quiescence [33], which provides a transition period until full development resumes, should conditions change to become more favorable.
3. PGCCs as somatic “blastomeres”
PGCCs have been long observed pathology and described in pathology textbook for medical students [34]. PGCCs can contain a single giant nucleus or multiple nuclei, referred to as mononucleated PGCCs or multinucleated PGCCs, respectively. The nuclei in PGCCs are usually irregularly shaped. PGCCs are commonly found in high-grade cancers (Fig. 1). At the histopathologic level, PGCCs can be observed at different stages of their life cycle. PGCCs may be mononucleated (Fig. 1A). Nuclear replication within the giant cell can be asynchronous. As shown in Fig. 1B, part of the nucleus can undergo condensation (Fig. 1B, red arrow) while the remaining part of the giant nucleus can be cleaved into small nuclei of various sizes (Fig. 1B, black arrows). Or, the giant part of the nucleus can undergo multipolar open mitosis (Fig. 1C, red arrow) while other parts of the nucleus at different stages of chromosomal condensation to undergo karyokinesis and form nuclei (Fig. 1C, black arrows). The giant nucleus can undergo multipolar mitosis with complete breakdown of nuclear membrane (Fig. 1D), followed by complete karyokinesis with individually separated nuclei within the giant cancer cell without cytokinesis (Fig. 1E). In addition, the nuclei within giant cells display different levels of proliferative activity. As shown in a representative example in Fig. 1F, immunohistochemical staining for proliferative Ki-67 antigen shows positive staining in small daughter nuclei (black arrows) but negative staining in giant nuclei (red arrow).
Fig. 1. Polyploid giant cancer cells (PGCCs) observed in high-grade serous carcinoma.
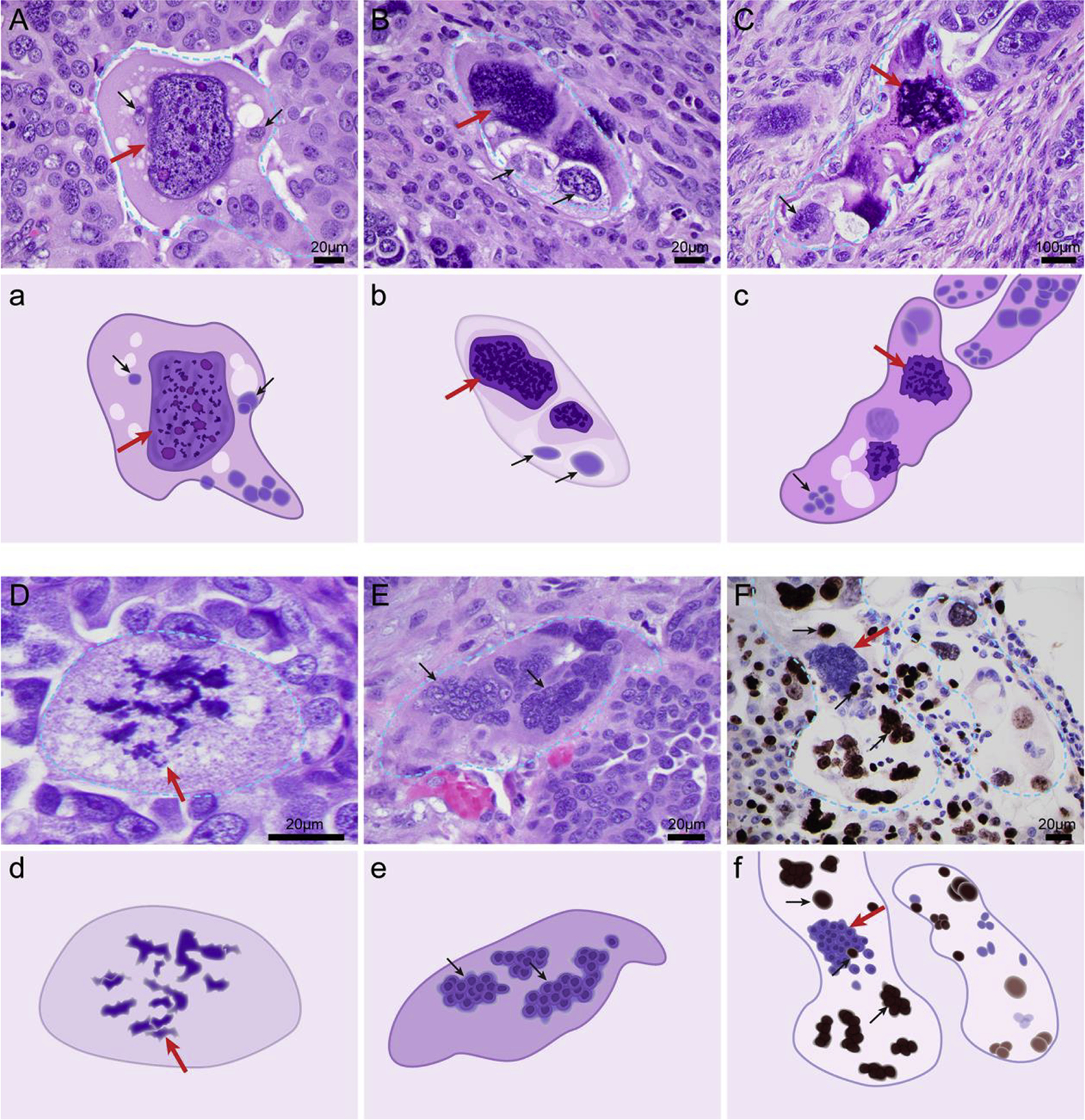
A. Mononucleated giant nucleus at interphase (red arrow). B. Giant nucleus with part of the nucleus undergoing chromosomal condensation (red arrow) and other parts of the nucleus showing maturation and budding of daughter cells (lower part of the giant nucleus, black arrows). C. A nucleus in which the top part of the nucleus is undergoing multipolar mitosis (red arrow) while the lower parts of the nucleus undergo asynchronous karyokinesis and generate large nuclei composed of multiple daughter nuclei (black arrows). D. Giant nucleus undergoing complete multipolar mitosis (red arrow). E. PGCCs with multinucleated giant cells. The giant nucleus has undergone karyokinesis (red arrow) and developed into multiple nuclear membrane–wrapped nuclei (black arrows). F. The giant nucleus is negative for Ki-67 (red arrow) and the budded nuclei show high levels of Ki-67 expression (black arrows). The slides were stained with hematoxylin and eosin (A-E) or Ki-67 proliferative nuclear antigen. F. Hematoxylin and eosin stain and Ki-67 activity by antibody are described as previously reported [162]. a-f. The schematic that highlights key morphologic feature of individual PGCC corresponding to A through F.
The generation of nuclear atypia in PGCCs represents the most important hallmark of cancer in diagnostic pathology [34]. PGCCs are generated via whole genome duplication or multiplication, in which the whole genome is reshaped in response to sudden and unexpected catastrophic genomic shock, a hereditary principle described by Nobel Laureate Barbara McClintock [35]. This process mimics early embryonic development [17, 35]. A high nuclear grade largely correlates with more copies of genomic DNA, which allows cells to acquire nuclear pleomorphism. These traits often form the basis of a pathologic cancer diagnosis and are used to predict clinical behavior [36]. The presence of PGCCs in cancer tissues usually signals a high grade and poor prognosis [36–38]. PGCCs have been observed in approximately 37% of human tumors [39]. The number of PGCCs may be significantly increased following chemotherapy, radiotherapy, and hypoxia mimetic CoCl2 [40]. PGCCs contribute to cancer-acquired therapeutic resistance in multiple cancer types, including ovarian [41, 42], prostate [43, 44], breast [37, 38, 45, 46], and colorectal cancers [47] and melanoma [48].
Traditionally, PGCCs were considered nonviable owing to their inability to execute mitosis and therefore have been largely ignored. However, we and others have observed that PGCCs can rejuvenate themselves via multiple modes of amitotic division, including budding, splitting, and burst-like division, similar to processes observed in primitive organisms such as fungi or protozoa [42, 49–53]. These PGCCs can further develop into blastocyst-like structures and generate a variety of high-grade cancers without proper differentiation [11, 12, 17]. PGCCs can generate daughter cells via this amitotic budding to generate blastomere-like cancer stem cells, which leads to the formation of blastocyst-like structures that are capable of tumor initiation and aberrant differentiation of different lineages [41, 42, 54–57]. Therefore, cancer can be considered as a disease in which “development” goes awry and arrests at a specific developmental stage, resulting in cells that do not differentiate into the appropriate cells for a specific organ site. This can occur at any timepoint of development following implantation of a blastocyst in the uterus, or it may occur as part of the somatic cell derived embryogenesis without proper differentiation [11, 12, 17]. These findings have fundamentally changed our understanding of PGCCs and open a new door for investigation of the cancer biology and therapeutic implications of PGCCs.
4. The giant cell life cycle and dormancy
To further delineate the detailed cellular mechanism underlying PGCC growth and division, we used fluorescent protein–labeled single-cell time-lapse images to track the dynamic processes of mitotic spindle and chromosome formation in PGCCs [53]. The fluorescence-labeled chromosomes and tubulin provided a detailed live image view of the production of daughter cells at the subcellular structure level and defined the life cycle of PGCCs, which is called the giant cell life cycle (or giant cell cycle). The giant cell life cycle is asexual and represents the very beginning of development, generating neoplastic life using mechanisms similar to those of early embryogenesis [53, 54].
One of major questions that have not been addressed in cancer biology involve the concept of dormancy. Why would some patients have disease that responds to therapy initially but then becomes dormant and recurs many months or years later? What is the relationship between dormancy and PGCCs? What mechanisms awaken the dormant cancer cells for recurrence or metastasis? We propose that dormancy in cancer can be explained by the giant cell life cycle, which can generate blastocyst-like structures capable of embryonic diapause similar to that observed insects and mammals [32, 33, 58, 59]. Here we describe each phase of the giant cell life cycle and compare it to phases of normal early embryogenesis and embryonic diapause and discuss how our cancer cells use such evolutionarily conserved mechanism for their survival under the emergency conditions posed the genotoxic therapeutic agents [32, 33].
4.1. The initiation phase: “somatic blastomeres” for dedifferentiation
The initiation phase of the giant cell life cycle starts with massive cell death induced by severe genotoxic damage, followed by activation of a senescence program, which plays a critical role in this phase. The initiation phase of the giant cell life cycle is very similar to both the induction and initiation phases of embryonic diapause in insects [32, 33]. The senescent cells increase its size and lose their ability to divide, leading to a state of growth arrest. However, this state of growth arrest is not permanent, as was previously believed. Whether senescence is reversible or irreversible depends on the availability of p53 and p16 [24, 60–62]. Senescence itself promotes tissue remodeling and mediates the regenerative potential and function of tissues, serving as a positive regulator for preventing tumorigenesis [63–66]. The polyploid cells stop proliferating but remain metabolically active and viable in a “dormant” state [67].
Following therapy-induced genomic shock, a subset of cancer cells that have survived the genotoxic damage and entered a reversible senescent state transform into polyploid cells, and these cells exhibit several characteristics of senescence, including β-galactosidase expression and flattened morphology. The increase in cancer cell size shuts down the traditional mitotic cycle and prepares the cell for endoreplication to initiate the giant cell life cycle [53].
4.2. The self-renewal phase: erasing parental memory
The self-renewal phase occurs when surviving cells have become PGCCs via senescence escape, a basic mechanism for survival and resistance [24]. In senescence escape, a small minority of cancer cells successfully evades apoptosis and death, enters the S phase, and accumulates in G2. This results in a mixed cell population consisting of cycling, cell cycle–arrested, and senescent cells [68–70]. Other mechanisms to increase ploidy include cell fusion and entosis [71–74]. The self-renewal phase of the giant cell life cycle is associated with morphologic transition of diploid large senescent cells to polyploid cell via endoreplication, either mononucleated or multinucleated, and these cells can further develop and form a cystic structure within the giant cells to acquire the ability to compact, form morulae, and exhibit blastocyst-like morphology [54] (Fig. 2). As shown in Fig. 2, the morphologic characteristics of PGCCs as visualized by scanning electron microscopy are remarkably similar to those of egg cells and 4-cell and 8-cell embryonic morulae (Fig. 2A) and blastocyst-like structures (Fig. 2B and 2C).
Fig. 2. Growth and division of polyploid giant cancer cells (PGCCs) mimics that observed in blastomere-stage embryos.
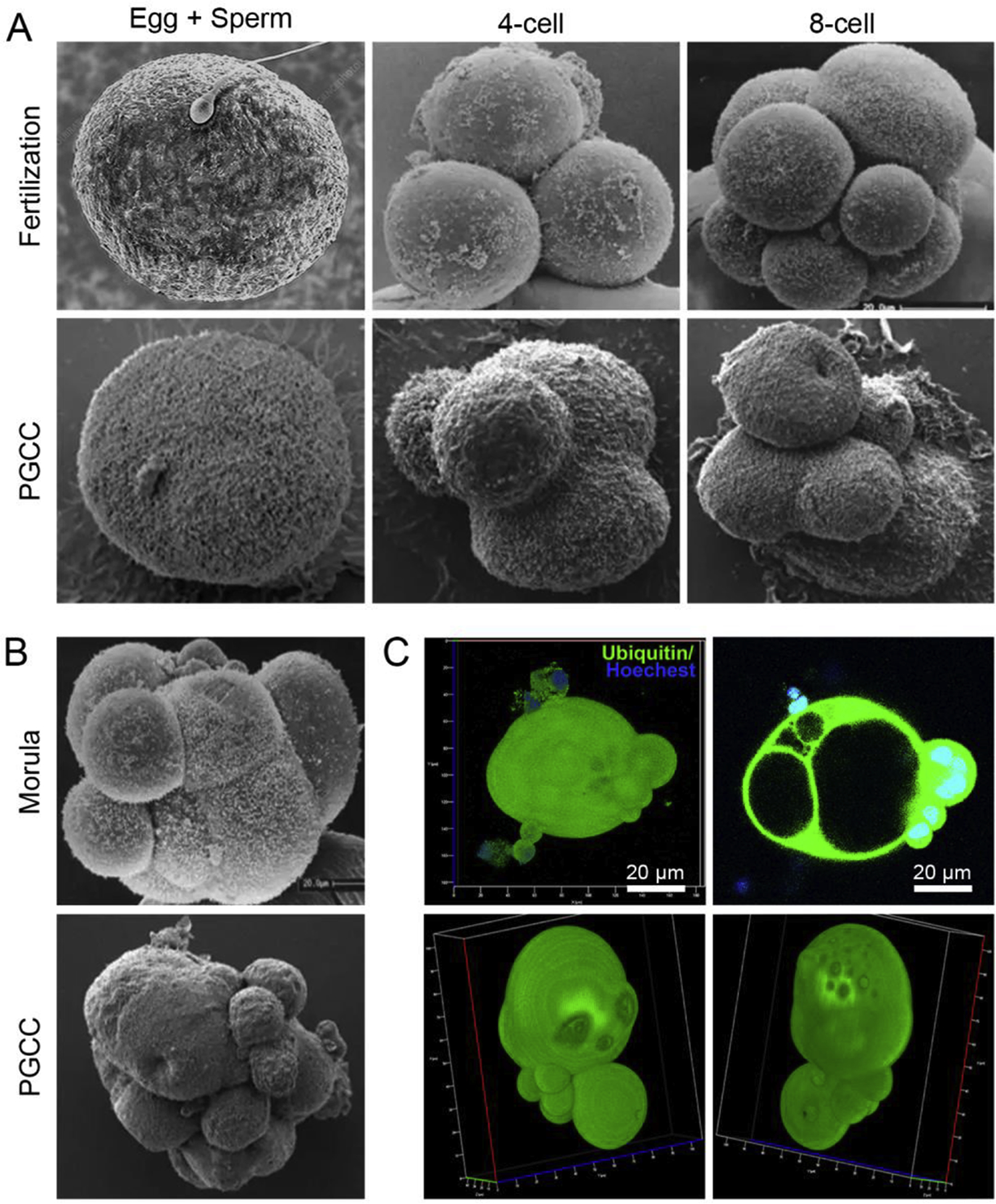
A. Stages of cleaved blastomeres following fertilization. The lower panels of A and B showing the morphology of PGCCs mimicking blastomeres were visualized by scanning electron microscopy and are adapted from our previous publication [54]. B. Scanning confocal microscopy images showing PGCCs with a blastocyst-like structure as compared with human morulae. C. 3D confocal scanning images of Hey-derived PGCC (upper panel) and a spheroid derived from Hey-derived PGCC (low panel). The zygote image is adapted from https://www.sciencephoto.com/media/873684/view/human-egg-and-sperm-sem (credit, Dennis Kunkel, microscope/science photo library with permission). The 4-cell, 8-cell, and morula stage embryo images are adapted from a figure previously published in Biology of Reproduction [163], reproduced with permission from Dr. George Nikas and the journal. The images of PGCCs were adapted from our previous publication [54].
In the self-renewal phase, a subpopulation of cancer cells enters a state of endoreduplication to generate mononucleated or multinucleated PGCCs [53]. These cells first replicate via the endoreplication cycle to generate cells with a single polyploid nucleus and no features of mitosis. Next, the cells enter endomitosis, in which cells execute abortive mitosis and subsequently re-enter the S phase to develop multinucleated cells. This blastocyst stage corresponds to the maintenance phase of embryonic diapause in insects [32, 33].
YAP is a vital Hippo pathway protein involved in the regulation of stem cell differentiation and cancer development, and the multilineage differentiation potential of PGCCs depends on nuclear localization of YAP [54, 75]. YAP has been shown to be located mainly in the cytoplasm of regular ovarian cancer cells and daughter cells but predominantly in the nuclei of PGCCs, and abrogation of nuclear expression of YAP was shown to decrease the capability of multilineage differentiation in ovarian cancer [54]. In addition, PGCCs activate the expression of cancer stem cell markers CD44 and CD133 [42, 76, 77]. Time- and spatial-dependent activation of several embryonic cell markers, including SSEA1, OCT4, Nanog, and SOX2, is observed during the formation of PGCCs [45, 54, 78]. Furthermore, expression of Xist, a biomarker for inactivation of the X chromosome, was lost in PGCCs after chemotherapy [54].
During this stage, PGCCs undergo amitotic cell division to generate different forms of slow-proliferating polyploid cells [11, 12, 54]. The formation of polyploidy allows the initiation of genomic chaos and macroevolution of the cancer genome to achieve dedifferentiation via activation of embryonic reprogramming factors [53, 54]. At the genomic level, following therapeutic stress, cancer cells undergo massive genomic reorganization via a variety of mechanisms, including the breakage-fusion-break cycle, whole genome duplication [39, 79], chromoplexy [80], chromothripsis [81], methylation and demethylation, and activation of transposable elements [82, 83]. In particular, chromothripsis has been shown to be a major mechanism for DNA amplification and drug resistance [84].
PGCCs grow autonomously with traditional mitotic checkpoints in diploid cells and develop into compaction-, morula-, and blastocyst-like somatic embryos. From a developmental perspective, the life cycle of PGCCs recapitulates a blastomere-like embryonic cycle, and the resulting cells can develop into various morphological heterogeneous forms of cancer histology for proliferative growth via mitosis to generate resistant or metastatic cancer cells, while a subset of blastocyst-like structures potentially undergo embryonic diapause in response to unfavorable environmental conditions [30, 31, 85, 86]. These changes work together to erase the structure of previous cancer genomes and create a new cancer genome for better survival [35].
4.3. The termination phase: parturition for a new “life”
The termination phase starts when PGCCs have formed a blastocyst-like structure, which undergoes depolyploidization to generate daughter cells with largely diploid, tetraploid, aneuploid, polyploid, or other chaotic karyotypes, which is largely like an inner cell mass in normal embryogenesis. Although a blastocyst is programmed for embryogenesis and fetal development following implantation in the uterus, somatically derived PGCCs vary significantly in terms of genomic copy number and can generate daughter cells by massive protozoan-like budding, fungus-like bursting, or viral-like budding in the termination phase [42]. PGCCs thus form reproductive blastocyst-like structures but without proper differentiation [54].
The renascent embryonic-like blastocysts acquire genetic or epigenetic mutations and are arrested at different stages of development, gradually acquiring competence in mitosis and becoming proliferative for malignant growth [11, 12, 54]. Owing to multiple copies of centrosomes and multipolar spindle structures in PGCCs, the fragments of genomes reassemble in a random order, resulting in the formation of daughter cells with a massively reorganized genome. The chromosomes are not evenly distributed into the daughter cells, leading to high levels of chromosomal instability and a different chromosome combination from the original parental cells [87]. These daughter cells show massive gain or loss of partial or whole chromosomes from translocations, duplications, or deletions of chromosomal regions [53]. The newly formed tumor cells tolerate aneuploidy and genomic instability without death or senescence. We have shown that new chromosome recombination occurs more frequently in daughter cells than in parental ovarian cancer cells, indicating that daughter cells obtain new cancer genomes and are distinct from parental PGCCs [53].
This newborn “life” comprises not only newly programmed neoplastic cells but also supporting stroma. The stroma co-evolves with neoplastic cells via multiple endocrine and paracrine pathways to create a range of new “organisms,” which may be malignant or benign, resistant or sensitive to drugs, and metastatic or not metastatic. The new organisms develop and evolve together with the microenvironment and eventually develop into a clinically visible tumor. This phase corresponds to the termination phase of diapause in insects [32, 33].
4.4. The stability phase: maturation into an “organism”
The stability phase is characterized by many diploid, tetraploid, or aneuploid progeny cells with novel or altered genotypes via genomic remodeling, point mutations, or altered karyotype or epigenetic modification after reprogramming through the giant cell life cycle. These processes lead to stable growth of new, resistant neoplastic life. This phase corresponds to awakening of diapause in insects [32, 33]. The reconstructed diploid cells gradually become more stable; restart the mitotic cell cycle with bipolar, tripolar, or multipolar mitosis [53]; and differentiate into a variety of cell types with variable levels of developmental potency [54]. These daughter cells gradually achieve stability following genomic reorganization, point mutations, or altered karyotype and then resume mitosis and start proliferating. This stable growth may ensure tumor cell survival with a newly acquired genome under “new normal” conditions.
Functionally, PGCCs behave like a blastomere-stage embryo for dedifferentiation of the parental genome [54]. Their offspring are capable of long-term proliferation and can further develop into resistant, metastatic, benign, and even germ cell tumors [42, 54, 56]. The progeny cells can grow into tumors with different histologic characteristics or grades, and one of these subsets of progeny cells may become clonally dominant or develop a mixed histologic type over time, which can be observed under a light microscope.
Further supporting the concept of activation of a blastomere-stage embryo via the giant cell life cycle, Nehme et al recently described human cytomegalovirus-induced transformation of human mammary epithelial cells, leading to a malignant formation via formation of blastomere-like PGCCs [88]. These findings provide additional evidence that PGCCs use an activated blastomere-stage program to achieve malignant transformation.
4.5. Differentiation of multilineage benign stromal cells in PGCCs
Developmentally, if PGCCs recapitulate the blastomere-stage embryo, they should acquire the ability to differentiate into any lineage of tissue. Indeed, we found that PGCCs are capable of generating not only cancer cells with new karyotypes but also multilineage benign differentiation, including stromal fibroblasts [55], endothelial cells [41, 57], chondrocytes, adipose tissue, embryonic erythroid cells [56], neutrophils [57], and even germ cell lineages [54]. PGCC-derived erythrocytes bind oxygen with high affinity by expressing fetal and embryonic hemoglobins [56]. Different components of stromal cells provide blood flow, nutrition, and oxygen supply to cancer cells as a mechanism of vascular mimicry, contributing to the ineffectiveness of anti-angiogenesis therapies in some cancer patients.
The appearance of different lineages via transdifferentiation can be observed within PGCCs. In high-grade serous carcinoma specimens exposed to chemotherapy (Fig. 3), neutrophil-like cells, fibroblasts, erythrocytes, histiocytes, chondrocytes, and neuronal cells can be directly observed within PGCCs. The daughter nuclei within PGCCs can go in a specific direction while the mother nuclei retain the properties of undifferentiated cancer. These observations indicate that different parts of nuclei have the potential for differentiation into various cell lineages, supporting the concept that PGCCs are blastomere-like cancer stem cells with pluripotent stemness. The specific lineage potential of a nucleus is likely to depend on the genetic makeup of the specific tumor, its microenvironment, and type of therapeutic agents. In addition, the lineage probably also depends on the nature and timing of the inducing factors, although the underlying mechanisms of benign differentiation are entirely unknown.
Fig. 3. Polyploid giant cancer cells (PGCCs) are capable of differentiation into morphologically different benign lineages.
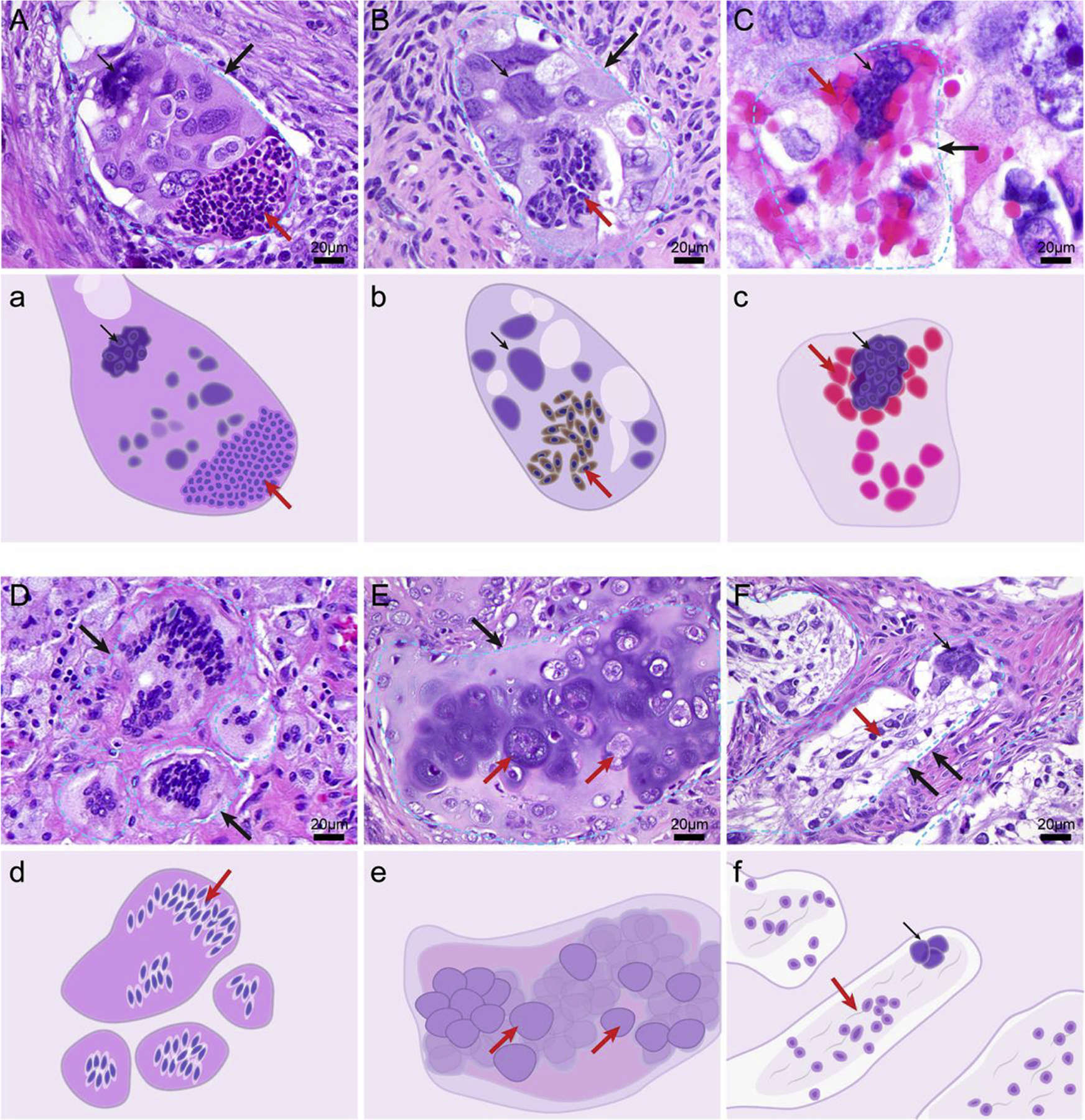
A. A PGCC with half nuclei differentiating into neutrophil-like cells. B. A PGCC differentiating into fibroblasts. C. A PGCC generating hemoglobin-forming erythroid cells. D. A PGCC differentiating into multinucleated foamy histiocytes. E. A PGCC forming chondrocytes. F. A PGCC differentiating into neuronal cells. Large black arrows indicate the outline of a PGCC; small black arrows, the giant nucleus of a PGCC; red arrows, the differentiated benign lineage. The slides were stained with hematoxylin and eosin. a-f. The schematic that highlights key morphologic feature of individual PGCC corresponding to A through F.
4.6. PGCCs and metastasis
Although early studies showed that PGCCs were capable of budding and generating viable progeny cells in vitro tissue-cultured cells [49–51, 89, 90], the role of PGCCs in tumorigenesis and metastasis in vivo was largely unknown. The metastatic ability of PGCCs was first identified by Weihua et al, who showed that a single PGCC was capable of generating distant metastases in nude mice [42, 52]. Subsequently, we confirmed this finding by showing that individual PGCCs derived from ovarian cancer cell lines were capable of tumor initiation in nude mice. Surprisingly, in addition to the tumor initiating ability, we also found that PGCCs were capable of multilineage differentiation including bone, cartilage, adipose, and fibroblasts in the peritoneal cavity of nude mice [41, 42], suggesting the embryonic-like properties are associated with PGCCs during tumor initiation and metastasis. Subsequently, an increased number of PGCCs was also reported in association with high metastatic potential and resistance in prostate cancer cell lines [91].
Interestingly, several groups have reported detecting giant cells, referring to them as cancer-associated macrophage-like cells (CAMLs), in blood samples from patients with breast and pancreatic cancer but not in blood samples from healthy individuals [92]. CAMLs have been found to be associated with poor prognosis in breast, lung, and esophageal cancers [93–95]. CAMLs express epithelial, monocytic, and endothelial protein markers and can be potentially used as a biomarker of solid tumors. However, CAMLs were characterized using only fluorescence markers rather than histologic characteristics, and the relationship between CAMLs and PGCCs remains unclear [92, 96]. It will be important to characterize these cells at a histopathologic level to determine whether CAMLs are really macrophages or PGCCs with macrophage-like differentiation, and whether CAMLs are associated with histologic characteristics of primary tumors. Functionally, it will be important to determine whether CAMLs are tumorigenic or capable of metastasis in nude mice, like PGCCs. If these atypical CAMLs are truly PGCCs, these data would provide additional evidence that PGCCs can shed into the bloodstream for metastasis to distant organs in patients, which may provide a new opportunity for early detection and therapeutic intervention before the primary tumor mass can be detected.
5. Embryonic diapause: the root of dormancy
As discussed in section 2, embryonic diapause is an evolutionarily conserved developmental mechanism that arises in response to unfavorable environmental conditions [31, 33]. Although phases of embryonic diapause in humans have not been studied, it has been proposed that such a mechanism is likely to exist in humans [97], given that the early phase of embryogenesis in humans is evolutionarily conserved from insects and other mammals, although no definite cases have been reported owing to ethical issues [97].
Unexpectedly, evidence supporting the existence of embryonic diapause-like mechanism in humans did not come from human reproductive biologists or in vitro fertilization physicians, but rather from cancer biologists. Recently, two independent groups reported molecular evidence that drug-tolerant persister (DTP) cells recapitulate the transcriptional signature of embryonic diapause [85, 86]. Rehman et al showed that all colorectal cancer cells possess an equipotent capacity to enter a DTP state and maintain clonal complexity. These DTP-state tumors show characteristics similar to embryonic diapause and are dependent on autophagy and suppression of mTOR for survival [86]. Dhimolea et al showed that chemo-persister cells have a suppressed Myc program with a diapause-like molecular adaptation, which allows the cells to survive via reduced redox stress and an apoptotic program that can be revised by CDK9 inhibitors [85]. This phenotype links to the well-described embryonic diapause phenotype induced by Myc depletion, which can induce a reversible, pluripotent diapause-like stage in blastocysts [98].
These findings strongly establish a relationship between DTP or dormant cancer cells and embryonic diapause and support the possible existence of embryonic diapause in human normal embryogenesis. However, these two reports did not discuss the developmental mechanisms and potential origins of embryonic diapause. Work on PGCCs from our group [54] and subsequently that of Was et al [99] provided this missing link. As described in Fig. 2, the early embryonic structures of PGCCs described in our early study provided a developmental and structural basis to embryonic diapause. PGCCs acquire stemness through a reprogramming mechanism mimicking the blastomere-stage embryo to generate a blastocyst, which can diapause for dormancy in response to therapeutic stress. Thus, the giant cell life cycle may explain dormancy via its link to blastocyst-like structure and embryonic diapause. The activation of a blastomere-stage embryo explains the mechanism needed to generate a malignant case of arrested development [100].
6. Warburg effect in blastomeres and cancer
The link between PGCCs and blastomere-stage embryos provides a satisfactory explanation for a puzzling observation first reported by Warburg in 1956, known as the Warburg effect [101]: cancer cells use glycolysis and the production of lactate rather than oxidative phosphorylation for ATP, even though oxygen is plentiful. It has long been observed that giant cells in pre-implantation embryos use the Warburg effect for metabolism, similar to cancer cells [101–103]. The commonality between pre-implantation embryos and cancer cells in terms of glucose, amino acids, and fatty acids is evident [102–104]. A blastomere-stage embryo uses nuclear transformation for new life, and cancer cells simply recapitulate the developmental program of a pre-implantation embryo, which represents an evolutionarily conserved mechanism for reproduction. PGCCs most likely use a similar Warburg effect for their metabolism.
7. An organismal model for dormancy, resistance, and metastasis
Fig. 4 shows a schematic model of the germ cell life cycle that hypothesizes diapause, along with a schematic model of the giant cell life cycle that recapitulates embryonic diapause in response to the therapeutic stress of cancer. In the germ cell life cycle, cell size changes and diapause could occur following blastocyst formation, as shown in Fig. 4A. The female primordial germ cell is set aside for gametogenesis during the second or third week of life. During puberty, the oocyte grows in size and matures into an ovum for fertilization. The zygote starts the blastomere cleavage and generates compaction and the morular-stage embryo before developing into the blastocyst for development of different germ layers in the uterus, and this blastocyst can potentially undergo embryonic diapause in response to environmental stress.
Fig. 4. Schematic of embryonic diapause and therapy-induced diapause.
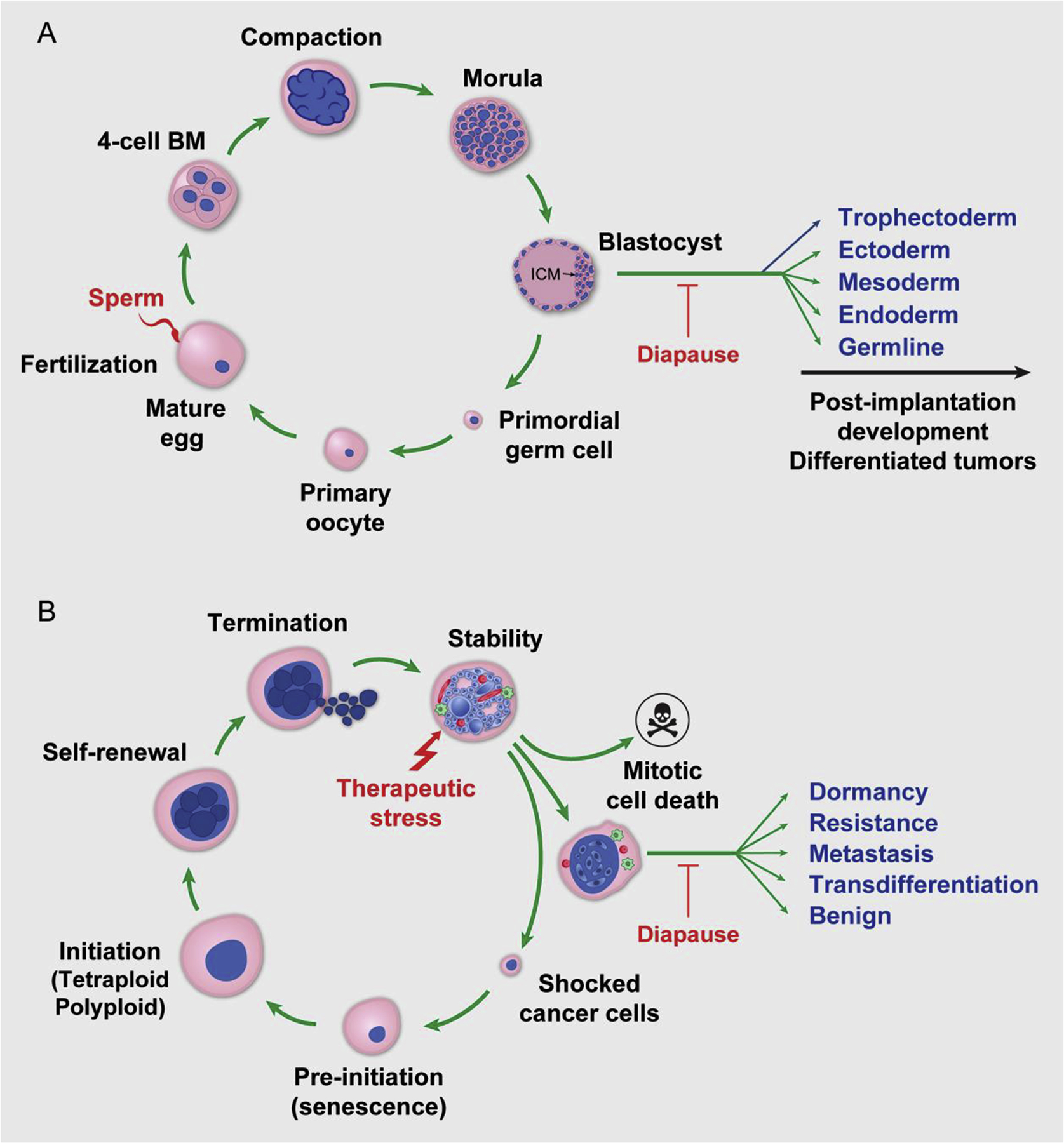
A. Schematic of human early embryogenesis and hypothesized diapause. Following fertilization, the egg cleaves to generate the blastomere (BM), compaction, morula, and blastocyst and then for post-implantation development or arrested at specific developmental stage for development of differentiated tumors. B. Schematic of the giant cell life cycle and diapause in response to treatment. The different phases of giant cell life cycle following the therapy induced mitotic death and genomic shock is shown in diagram. A subset of blastocyst-like structure undergoes diapause for dormancy. Awakening of diapause leads to birth of resistant or metastatic cells or transdifferentiation into various benign lineages.
Similarly, as illustrated in Fig. 4B, the giant cell life cycle recapitulates the size and ploidy augmentation in the blastomere-stage embryo and develops into a blastocyst-like structure that can undergo embryonic diapause in response to therapeutic stress. In the initiation phase, tenacious cancer cells that survived genomic shock begin to increase in size and undergo endoreplication and/or cell fusion to escape severe genotoxic damage. In the self-renewal phase, these cells sequentially become PGCCs and enter a blastocyst-like state for embryonic diapause. In the termination phase, PGCCs produce depolyploidized offspring via genome reductional division. In the stability phase, the diploid/aneuploid progeny cells achieve stable tumor expansion with newly acquired genotypes. The tumor cells then return to the mitotic cell cycle for growth and development.
PGCCs also contribute to stromal generation via a variety of mechanisms involving changes in cell identity, most notably epithelial-to-mesenchymal transition, epithelial-to-endothelial transition, and epithelial-to-hematopoietic transition, to generate the stromal cells needed for tumor development. Epithelial-to-hematopoietic transition generates various tissue structures such as lumen and glands, which provide a novel mechanism for tissue differentiation; this mechanism plays an important role in the development of benign tumors [12].
The level of dedifferentiation can vary depending on the type of stress, duration of stress, and type of tumor cells. The longer the period of the giant cell life cycle, the closer dedifferentiation is to the early stage of embryonic development and the greater the potential for tumors to be malignant and high grade. Genetically, there is significant genomic instability in both blastomeres and PGCCs. Biologically, there are frequent mitotic or cytokinetic failures and endoreplication during the formation of both PGCCs and blastomeres.
Thus, the giant cell life cycle largely represents a default emergency that genome of cells to respond to life-threatening stress that leads to a shock to the genome. Such a massive genomic response triggered by a single catastrophic event was observed by Barbara McClintock nearly 70 years ago [35, 105–107]. Years later, Henry Heng provided a detailed account of how the genome responds to therapeutic stress and how this leads to macroevolution of the cancer genome via genomic chaos in cancer [27, 108–111]. The cyclical change of cell size in giant cells, together with ploidy changes, provides a link between embryogenesis and the macroevolution of cancer, including dormancy via embryonic diapause. This may represent a universally conserved mechanism through millions of years of evolution on Earth [17].
8. Targeting the giant cell life cycle
PGCCs represent a tremendously exciting target for therapy. We and others have proposed various strategies to target these cells [10–12, 112, 113]. Our understanding of giant cells as progressing through their life cycle at an organismal level provides both challenges and opportunities for therapy; each stage of the giant cell life cycle provides a potential opportunity for therapeutic intervention (Fig. 5). The stress that can initiate the giant cell life cycle varies by tumor histologic type, intrinsic genetic makeup, and microenvironment. Therefore, appropriately targeted agents may be selected according to tumor type, timing of administration, and duration of administration.
Fig. 5. Strategies that target the giant cell life cycle for prevention and therapy.
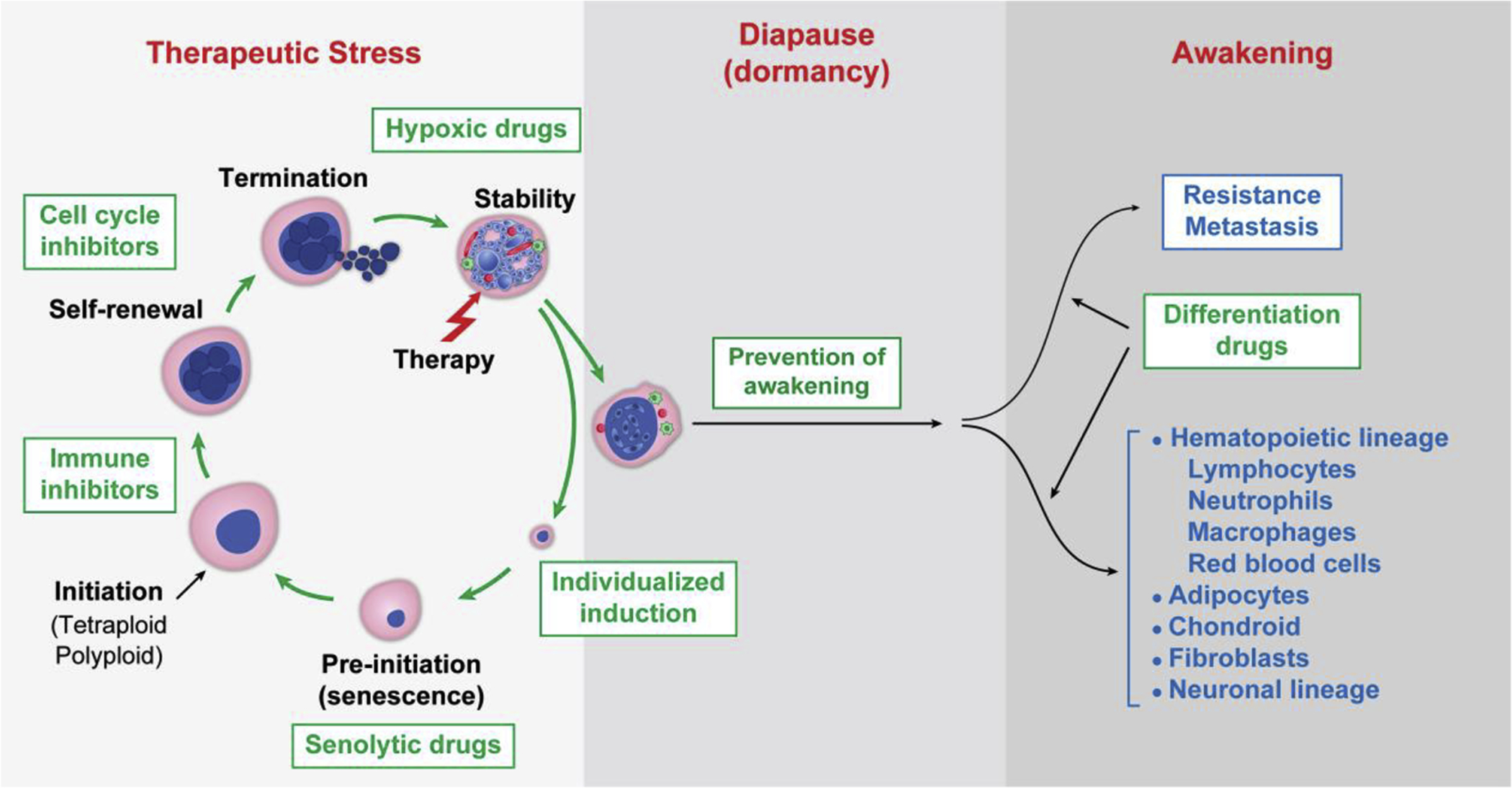
Phases of the giant cell life cycle are illustrated in response to therapeutic stress, followed formation of blastocyst-like structure for embryonic diapause for dormancy, and then awakening for resistance and metastasis. Specific strategies to block each phase of the giant cell life cycle including the differentiation therapy are shown in the diagram.
8.1. Prevention of giant cell life cycle activation: individualized induction
Clearly, therapeutic agents are a double-edged sword: they can reduce the tumor volume by killing mitotically active cells, but they may also induce the onset of the giant cell life cycle, which can generate resistance, metastasis, or dormancy. However, most US Food and Drug Administration–approved drugs are designed to decrease tumor volume without consideration of the giant cell life cycle. Changing this approach will require changing the assessment criteria used during the early stages of drug development. Oncologists and investigators should consider not only tumor size but also the formation of polyploidy when assessing the efficacy of drugs. In addition, because activation of the giant cell life cycle is specific to the reagent, dose, and timing of treatment for each tumor, therapy should be individualized to achieve maximum tumor cell killing with minimal or no induction of the giant cell life cycle.
Moreover, polyploidy formation should be included in the assessment of new treatments in clinical trials. Scribano et al demonstrated that the pre-existing chromosomal instability as indicated by multipolar division can predict the sensitivity to paclitaxel treatment in breast cancer [114]. Although remains to be validated, as multipolar division occurs in PGCCs, these results raise the question whether pre-existing PGCCs can serve as a predictive marker for paclitaxel response in breast and other cancer patients. Increasing importance of polyploidy in cancer also call for reliable methods to quantitate the number and type of PGCCs in cancer. Toward this end, Saini et al recently proposed an artificial intelligence–based approach to predict therapy response in breast cancer [115]. Certainly, this type of approach and other approaches that may predict an individual patient’s response will be highly valuable to determine the optimal type of drug and timing of administration.
8.2. Abortion of pre-initiation: pro-senescence and senolytic therapies
Transition of senescence activation to escape is the initial phase of PGCC formation. Thus, pro-senescence treatment that inhibits cancer cell proliferation and escape could be a promising therapeutic strategy to suppress PGCC formation and chemoresistance [116, 117]. Selective CDK4/6 inhibitors are currently the most promising pro-senescence drugs associated with a high response rate in patients with breast cancer [118] and ovarian cancer [119]. Various types of Myc inactivation also promote a cellular senescence response in many cancers. In addition, inactivation of Myc appears to be required to induce embryonic diapause [85, 86, 98] and keep blastocysts in a dormant state. Therefore, Myc inhibitors may induce both senescence and diapause, and targeting Myc may serve as a beneficial approach to drive senescence and keep PGCCs in a dormant state [120]. Following induction of senescence in aging-prone senescent somatic cells with administration of senolytic drugs may be effective in eliminating senescent cells [121, 122]. The combination of induction of senescence with administration of senolytic drugs may abort the pre-initiation phase of the giant cell life cycle, which may be effective in cancer therapy and cancer prevention.
Thura et al showed that phosphatase of generating live 3 (PRL-3), a member of a unique family of C-terminal prenylated phosphatases within the protein tyrosine phosphatase superfamily, can induce the formation of PGCCs [123]. PRL3+ cells express embryonic stem cell markers OCT4 and SOX2 and have decreased levels of apoptosis in response to genotoxic chemotherapy. Most excitingly, the antibody against PRL-3 can reduce tumor relapse, suggesting that blocking the formation of PGCCs may prevent disease relapse.
Our most recent work suggested that the inflammatory cytokines in senescence associated secretory phenotype play a critical role in the initiation of PGCC formation [124]. The interleukin-6, together with multiple cytokines involved in senescence and associated with a secretory phenotype, including IL-8, IL-1ꞵ, and Gro-1, is markedly activated in PGCCs in response to paclitaxel-induced genomic shock. This leads to not only activation of embryogenic progress of cancer cells but also reprogramming of the tumor microenvironment by converting normal fibroblasts to cancer-associated fibroblasts and GPR77+/CD10 fibroblasts, increasing collagen synthesis and vascular endothelial cell growth factor to promote tumor growth. Blocking IL-6 function using an antibody against its receptor (tocilizumab) decreased tumor growth in patient-derived xenograft models. These results suggest that blocking the formation of PGCCs by targeting the key protein involved in their formation could block tumor growth in mouse models, although such antitumor effects remain to be tested in future human trials.
8.3. Eliminating immune privilege in newborn cancer cells
The giant cell life cycle is enriched in pro-inflammatory cytokines that are immunosuppressive. Therefore, it is critical to block such immunosuppressive molecules in order to achieve the desired therapeutic effect of immune checkpoint blockers. There are numerous immune cells in the tumor microenvironment. Recently, it has been shown that IL-33 can induce the formation of polyploidy and lead to immune suppression [125]. Blocking IL-33 was shown to elicit antitumor immunity in mice with IL-33+ tumors; tumor-specific CD8+ T cells were increased within the tumors, and splenic CTL activity was significantly elevated [125]. Other studies have shown that hypoxia is involved in the immunosuppressive tumor microenvironment; HIF-1 can directly induce PD-L1 expression or upregulate the CTLA-4 receptor [126, 127]. Therefore, the combination of anti-hypoxia therapy with immunotherapy could be an ideal method to facilitate the survival of T cells in the tumor microenvironment created by PGCCs and effectively prevent inactivation of T cell functions [128].
8.4. Suppression of renewal and termination of PGCCs
Self-renewal is one of the critical steps employed by PGCCs to maintain the polyploid state. During this phase, the genome is restructured, which is associated with activation of the embryonic program. On the one hand, this reprogramming allows cancer cells to generate new progeny; on the other hand, the tumor cells are highly chaotic and plastic during this phase, which can provide a therapeutic window for intervention. During this phase, it may be possible to re-direct the differentiation toward benign or normal development rather than to generate new and more aggressive giant cells that are capable of dormancy or travel to different sites.
Sustained aberrant activation of self-renewal is associated with anomalous regulation of homeostasis, endoreplication, and cell fusion. Blocking endoreplication could be an effective strategy to inhibit self-renewal in PGCCs. The switch from mitosis to endoreplication in polyploidization depends on downregulation of mitotic cyclin-dependent kinase (M-CDK), whose activity is provided by CDK1 bound to cyclin B or A. CDK1 can be forcibly activated by wee1 inhibitors, serving as a potential therapeutic approach to abrogate endoreplication of PGCCs [129]. The Hippo pathway could be another potential target for anti-PGCC therapy, because activation of this pathway leads to the formation of polyploidy, especially in the presence of p53 mutation or deficiency [130].
It appears that the polyploid genome, in contrast to the diploid genome, uses a distinct set of genes to maintain the polyploidy. Recent analysis of roughly 10,000 primary human samples spanning 32 distinct tumor types from The Cancer Genomic Atlas revealed that at least 36% of tumors had at least one whole genome duplication during their evolution [131]. In addition, gene expression analysis revealed that multiple specific genes are expressed in the polyploid genome but not in the diploid genome. Functional analysis revealed that KIF18A, which encodes a mitotic kinesin protein, is required for viability of polyploid cells, but not for diploid cells [131], demonstrating that the polyploid genome has unique cell replication and division mechanisms, which can be potentially explored as a vulnerability for therapeutic targets. In addition, actin and vimentin have been shown to play an important role in the volume and migratory phenotype of PGCCs [132–134], suggesting that PGCCs are regulated through a unique mechanistic regulatory network to promote tumor initiation and chemoresistance.
Voelkel-Johnson’s group reported a series of elegant studies on the role of lipids in the life cycle of PGCCs. The sphingolipid enzyme acid ceramidase (ASAH1), an enzyme involved in oocyte maturation and blastomere cleavage [135], is required for amitotic division of PGCCs [136]. Inactivation of this enzyme prevented the production of budded progeny cells from PGCCs [85, 136]. Ceramide synthase 6 can synergize with p53 protein and prevent the formation of progeny from PGCCs [137]. Tamoxifen, a commonly used anti-estrogen agent, inhibits the generation of PGCC offspring in prostate cancer, glioblastoma, and melanoma cells via an estrogen-independent mechanism, potentially through inhibition of acid ceramidase [138]. Supporting the idea that an acid ceramidase inhibitor can act in conjunction with radiation in a preclinical model of prostate cancer, Cheng et al showed that acid ceramidase inhibition had no significant impact on tumor growth or radiation therapy response but completely prevented relapse, resulting in durable cures in 100% of the mice [139]. Although these results are preliminary, they certainly point to the potential for repurposing various existing drugs to inhibit certain phases of the giant cell life cycle.
8.5. Targeting metabolic vulnerability
Rapid tumor growth is accompanied by insufficient blood flow and a hypoxic tumor microenvironment [140]. Hypoxia is one of the main causes of therapy resistance, and a hypoxic tumor microenvironment was shown to promote the formation of PGCCs [42, 47, 141, 142], indicating that targeting the hypoxic tumor microenvironment could inhibit the formation of PGCCs. HIF-1 is the core regulator in hypoxia, and many of its inhibitors have exhibited promising anticancer effectiveness [143]. PGCCs with budding progeny can differentiate into erythroid and vascular endothelial cells, restoring the blood supply and promoting chemoresistance. Therefore, anti-angiogenesis therapy combined with chemotherapy may be an effective combination treatment strategy.
The metabolism of tumor cells is distinct from that of normal cells, which makes metabolic vulnerability a promising target to overcome chemoresistance and metastasis in cancer therapy [144–146]. Sirois et al evaluated metabolic reprogramming in PGCCs in breast cancer and noted several metabolic vulnerabilities, including dependence on PLIN4, a perilipin coating the observed lipid droplets in chemoresistant tumors, and these findings offer new therapeutic targets associated with PGCCs [46]. PGCCs exhibit a higher metabolic rate than normal cells owing to the Warburg effect [101, 147]. Several studies have revealed that PGCCs have increased dependence on glycolysis and are sensitive to glycolysis inhibition [147].
Starvation has been shown to induce diapause via upregulation of the glutamine transporter. In addition, diapause is associated with increased lipolysis via downregulation of mTORC2. Inhibition of glutamine transporters leads to exit from the diapause epigenetic state [148]. Intriguingly, mTOR inhibitors can also induce cell death or reduce the formation of PGCCs via regulation of metabolism [149, 150].
8.6. Prevention of awakening
Diapause has been shown to be reversible, and diapause could be induced in mouse blastocysts by inhibition of the rapamycin (mTOR) pathway and activation of the autophagic pathway [151]. Exiting of diapause is associated with increased mTOR pathway activity [152]. Persistence of diapause is associated with increased glycolytic and lipolytic pathways [148]. In mouse embryonic stem cells, deletion of Myc allows the cell to enter a pluripotent dormant state mimicking embryonic diapause [98]. Therefore, various drugs to block Myc or autophagy activity may prevent awakening of blastocysts, which may represent a novel therapeutic approach. PGCCs generate progeny through budding or bursting, like bacteria, fungi, and protozoa; therefore, antibacterial, antifungal, or anti-protozoan therapy may be a good choice for decreasing the formation of progeny [153]. Similarly, filovirus VP40 matrix protein was found to drive the budding process, and this may also represent a novel and rational target to prevent the formation of progeny [154].
8.7. Differentiation toward benign lineages
Another way to limit PGCC budding progeny is differentiation therapy, which induces differentiation of arrested malignant tumors into benign tissues [155]. The embryonic environment has been shown to reprogram malignant cells into a benign phenotype [156]. Although multiple obstacles remain, accumulated evidence suggests that inducing reversion can be used as a strategy to control cancer [157]. Several studies revealed that either retinoic acid or arsenic promoted differentiation in acute promyelocytic leukemia, and histone deacetylase or isocitrate dehydrogenase inhibitors may play an important role in leukemia or solid tumors as differentiation therapy [158, 159]. MEK inhibitors and the anti-diabetic drug rosiglitazone can block metastasis by promoting transdifferentiation into adipose cells [160]. Overexpressing single neural transcription factor Neurogenic differentiation 1 (NeuroD1), neurogenin-2 (Neurog2), or Achaete-scute homolog 1 (Ascl1) can induce neuronal conversion from human GBM cells [161]. The giant cell life cycle suggests that differentiation therapy may be most effective if it is combined with dedifferentiation agents to activate embryonic stemness.
9. Conclusion
PGCCs have emerged in recent years as an important new field of cancer biology with tremendous potential, and we are just at the very beginning stages of understanding the life cycle of PGCCs. This newly revealed information allows us to move from understanding cancer cell dormancy as part of a dysregulated mitotic cell cycle to embryonic diapause at an organismal level. The giant cell life cycle provides a unified mechanism for cancer initiation, rapid drug resistance, dormancy, recurrence, and metastasis. Attacking different stages of the giant cell life cycle offers new therapeutic strategies for cancer to improve patient survival.
Acknowledgments:
The authors thank Ms. Kim Vu for graphic rendering. This work was supported, in part, by the Moonshot program in Ovarian Cancer at The University of Texas MD Anderson Cancer Center, SPORE in Ovarian Cancer (CA217685), and the American Cancer Society. We thank Erica Goodoff, Senior Scientific Editor in the Research Medical Library at The University of Texas MD Anderson Cancer Center, for editing this article, and two reviewers for their constructive comments to improve this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
This paper is the fourth in a series of papers on the enigma of the origin of human tumors.
Conflict of interest statement: The authors declare that there are no conflicts of interest.
References
- [1].Jahanban-Esfahlan R, Seidi K, Manjili MH, Jahanban-Esfahlan A, Javaheri T, Zare P, Tumor Cell Dormancy: Threat or Opportunity in the Fight against Cancer, Cancers (Basel) 11(8) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Park SY, Nam JS, The force awakens: metastatic dormant cancer cells, Exp Mol Med 52(4) (2020) 569–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Phan TG, Croucher PI, The dormant cancer cell life cycle, Nature reviews. Cancer 20(7) (2020) 398–411. [DOI] [PubMed] [Google Scholar]
- [4].Recasens A, Munoz L, Targeting Cancer Cell Dormancy, Trends Pharmacol Sci 40(2) (2019) 128–141. [DOI] [PubMed] [Google Scholar]
- [5].Shen S, Vagner S, Robert C, Persistent Cancer Cells: The Deadly Survivors, Cell 183(4) (2020) 860–874. [DOI] [PubMed] [Google Scholar]
- [6].Yadav AS, Pandey PR, Butti R, Radharani NNV, Roy S, Bhalara SR, Gorain M, Kundu GC, Kumar D, The Biology and Therapeutic Implications of Tumor Dormancy and Reactivation, Front Oncol 8 (2018) 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Boire A, Coffelt SB, Quezada SA, Vander Heiden MG, Weeraratna AT, Tumour Dormancy and Reawakening: Opportunities and Challenges, Trends Cancer 5(12) (2019) 762–765. [DOI] [PubMed] [Google Scholar]
- [8].Foulkes I, Sharpless NE, Cancer Grand Challenges: Embarking on a New Era of Discovery, Cancer discovery 11(1) (2021) 23–27. [DOI] [PubMed] [Google Scholar]
- [9].Amend SR, Torga G, Lin KC, Kostecka LG, de Marzo A, Austin RH, Pienta KJ, Polyploid giant cancer cells: Unrecognized actuators of tumorigenesis, metastasis, and resistance, Prostate (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chen J, Niu N, Zhang J, Qi L, Shen W, Donkena KV, Feng Z, Liu J, Polyploid Giant Cancer Cells (PGCCs): The Evil Roots of Cancer, Curr Cancer Drug Targets 19(5) (2019) 360–367. [DOI] [PubMed] [Google Scholar]
- [11].Liu J, The dualistic origin of human tumors, Seminars in cancer biology 53 (2018) 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Liu J, The “life code”: A theory that unifies the human life cycle and the origin of human tumors, Seminars in cancer biology 60 (2020) 380–397. [DOI] [PubMed] [Google Scholar]
- [13].Mirzayans R, Andrais B, Murray D, Roles of Polyploid/Multinucleated Giant Cancer Cells in Metastasis and Disease Relapse Following Anticancer Treatment, Cancers (Basel) 10(4) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Moein S, Adibi R, da Silva Meirelles L, Nardi NB, Gheisari Y, Cancer regeneration: Polyploid cells are the key drivers of tumor progression, Biochim Biophys Acta Rev Cancer (2020) 188408. [DOI] [PubMed] [Google Scholar]
- [15].White-Gilbertson S, Voelkel-Johnson C, Giants and monsters: Unexpected characters in the story of cancer recurrence, Advances in cancer research 148 (2020) 201–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Richards JS, Candelaria NR, Lanz RB, Polyploid Giant Cancer cells and ovarian Cancer: New insights into mitotic regulators and polyploidy, Biol Reprod (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Liu J, Giant cells: Linking McClintock’s heredity to early embryogenesis and tumor origin throughout millennia of evolution on Earth, Seminars in cancer biology (2021). [DOI] [PubMed] [Google Scholar]
- [18].Erenpreisa J, Salmina K, Anatskaya O, Cragg MS, Paradoxes of cancer: Survival at the brink, Seminars in cancer biology (2020). [DOI] [PubMed] [Google Scholar]
- [19].Erenpreisa J, Salmina K, Huna A, Jackson TR, Vazquez-Martin A, Cragg MS, The “virgin birth”, polyploidy, and the origin of cancer, Oncoscience 2(1) (2015) 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Heng E, Moy A, Liu G, Heng HH, Zhang K, ER Stress and Micronuclei Cluster: Stress Response Contributes to Genome Chaos in Cancer, Front Cell Dev Biol 9 (2021) 673188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Herbein G, Nehme Z, Polyploid Giant Cancer Cells, a Hallmark of Oncoviruses and a New Therapeutic Challenge, Front Oncol 10 (2020) 567116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Pienta KJ, Hammarlund EU, Austin RH, Axelrod R, Brown JS, Amend SR, Cancer cells employ an evolutionarily conserved polyploidization program to resist therapy, Seminars in cancer biology (2020). [DOI] [PubMed] [Google Scholar]
- [23].Pienta KJ, Hammarlund EU, Axelrod R, Brown JS, Amend SR, Poly-aneuploid cancer cells promote evolvability, generating lethal cancer, Evol Appl 13(7) (2020) 1626–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sikora E, Czarnecka-Herok J, Bojko A, Sunderland P, Therapy-induced polyploidization and senescence: Coincidence or interconnection?, Seminars in cancer biology (2020). [DOI] [PubMed] [Google Scholar]
- [25].Song Y, Zhao Y, Deng Z, Zhao R, Huang Q, Stress-Induced Polyploid Giant Cancer Cells: Unique Way of Formation and Non-Negligible Characteristics, Front Oncol 11 (2021) 724781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ye CJ, Sharpe Z, Alemara S, Mackenzie S, Liu G, Abdallah B, Horne S, Regan S, Heng HH, Micronuclei and Genome Chaos: Changing the System Inheritance, Genes (Basel) 10(5) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ye JC, Horne S, Zhang JZ, Jackson L, Heng HH, Therapy Induced Genome Chaos: A Novel Mechanism of Rapid Cancer Drug Resistance, Front Cell Dev Biol 9 (2021) 676344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Was H, Borkowska A, Olszewska A, Klemba A, Marciniak M, Synowiec A, Kieda C, Polyploidy formation in cancer cells: How a Trojan horse is born, Seminars in cancer biology (2021). [DOI] [PubMed] [Google Scholar]
- [29].Zhang J, Qiao Q, Xu H, Zhou R, Liu X, Human cell polyploidization: The good and the evil, Seminars in cancer biology (2021). [DOI] [PubMed] [Google Scholar]
- [30].Fenelon JC, Renfree MB, The history of the discovery of embryonic diapause in mammals, Biol Reprod 99(1) (2018) 242–251. [DOI] [PubMed] [Google Scholar]
- [31].Renfree MB, Fenelon JC, The enigma of embryonic diapause, Development 144(18) (2017) 3199–3210. [DOI] [PubMed] [Google Scholar]
- [32].Kostal V, Stetina T, Poupardin R, Korbelova J, Bruce AW, Conceptual framework of the eco-physiological phases of insect diapause development justified by transcriptomic profiling, Proc Natl Acad Sci U S A 114(32) (2017) 8532–8537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kostal V, Eco-physiological phases of insect diapause, J Insect Physiol 52(2) (2006) 113–27. [DOI] [PubMed] [Google Scholar]
- [34].Kumar VA, AK; Fausto N; Aster JC, Robbins and Contran Pathologic Basis of Disease, Sauders Elsevier Chapter 7 (2010) 262–270. [Google Scholar]
- [35].McClintock B, The significance of responses of the genome to challenge, Science 226(4676) (1984) 792–801. [DOI] [PubMed] [Google Scholar]
- [36].Malpica A, Deavers MT, Lu K, Bodurka DC, Atkinson EN, Gershenson DM, Silva EG, Grading ovarian serous carcinoma using a two-tier system, The American journal of surgical pathology 28(4) (2004) 496–504. [DOI] [PubMed] [Google Scholar]
- [37].Fei F, Zhang D, Yang Z, Wang S, Wang X, Wu Z, Wu Q, Zhang S, The number of polyploid giant cancer cells and epithelial-mesenchymal transition-related proteins are associated with invasion and metastasis in human breast cancer, Journal of experimental & clinical cancer research : CR 34(1) (2015) 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lv H, Shi Y, Zhang L, Zhang D, Liu G, Yang Z, Li Y, Fei F, Zhang S, Polyploid giant cancer cells with budding and the expression of cyclin E, S-phase kinase-associated protein 2, stathmin associated with the grading and metastasis in serous ovarian tumor, BMC Cancer 14 (2014) 576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhang CZ, Wala J, Mermel CH, Sougnez C, Gabriel SB, Hernandez B, Shen H, Laird PW, Getz G, Meyerson M, Beroukhim R, Pan-cancer patterns of somatic copy number alteration, Nature genetics 45(10) (2013) 1134–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zhang Z, Feng X, Deng Z, Cheng J, Wang Y, Zhao M, Zhao Y, He S, Huang Q, Irradiation-induced polyploid giant cancer cells are involved in tumor cell repopulation via neosis, Mol Oncol (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zhang S, Mercado-Uribe I, Liu J, Tumor stroma and differentiated cancer cells can be originated directly from polyploid giant cancer cells induced by paclitaxel, Int J Cancer 134(3) (2013) 508–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zhang S, Mercado-Uribe I, Xing Z, Sun B, Kuang J, Liu J, Generation of cancer stem-like cells through the formation of polyploid giant cancer cells, Oncogene 33(1) (2014) 116–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Mittal K, Donthamsetty S, Kaur R, Yang C, Gupta MV, Reid MD, Choi DH, Rida PCG, Aneja R, Multinucleated polyploidy drives resistance to Docetaxel chemotherapy in prostate cancer, British journal of cancer 116 (2017) 1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lin KC, Torga G, Sun Y, Axelrod R, Pienta KJ, Sturm JC, Austin RH, The role of heterogeneous environment and docetaxel gradient in the emergence of polyploid, mesenchymal and resistant prostate cancer cells, Clin Exp Metastasis 36(2) (2019) 97–108. [DOI] [PubMed] [Google Scholar]
- [45].Lagadec C, Vlashi E, Della Donna L, Dekmezian C, Pajonk F, Radiation-induced reprogramming of breast cancer cells, Stem cells 30(5) (2012) 833–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sirois I, Aguilar-Mahecha A, Lafleur J, Fowler E, Vu V, Scriver M, Buchanan M, Chabot C, Ramanathan A, Balachandran B, Legare S, Przybytkowski E, Lan C, Krzemien U, Cavallone L, Aleynikova O, Ferrario C, Guilbert MC, Benlimame N, Saad A, Alaoui-Jamali M, Saragovi HU, Josephy S, O’Flanagan C, Hursting SD, Richard VR, Zahedi RP, Borchers CH, Bareke E, Nabavi S, Tonellato P, Roy JA, Robidoux A, Marcus EA, Mihalcioiu C, Majewski J, Basik M, A Unique Morphological Phenotype in Chemoresistant Triple-Negative Breast Cancer Reveals Metabolic Reprogramming and PLIN4 Expression as a Molecular Vulnerability, Mol Cancer Res 17(12) (2019) 2492–2507. [DOI] [PubMed] [Google Scholar]
- [47].Lopez-Sanchez LM, Jimenez C, Valverde A, Hernandez V, Penarando J, Martinez A, Lopez-Pedrera C, Munoz-Castaneda JR, De la Haba-Rodriguez JR, Aranda E, Rodriguez-Ariza A, CoCl2, a mimic of hypoxia, induces formation of polyploid giant cells with stem characteristics in colon cancer, PLoS One 9(6) (2014) e99143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Meierjohann S, Effect of stress-induced polyploidy on melanoma reprogramming and therapy resistance, Seminars in cancer biology (2021). [DOI] [PubMed] [Google Scholar]
- [49].Erenpreisa JA, Cragg MS, Fringes B, Sharakhov I, Illidge TM, Release of mitotic descendants by giant cells from irradiated Burkitt’s lymphoma cell line, Cell Biol Int 24(9) (2000) 635–48. [DOI] [PubMed] [Google Scholar]
- [50].Sundaram M, Guernsey DL, Rajaraman MM, Rajaraman R, Neosis: a novel type of cell division in cancer, Cancer Biol Ther 3(2) (2004) 207–18. [DOI] [PubMed] [Google Scholar]
- [51].Walen KH, The origin of transformed cells: studies of spontaneous and induced cell transformation in cell cultures from marsupials, a snail, and human amniocytes, Cancer genetics and cytogenetics 133(1) (2002) 45–54. [DOI] [PubMed] [Google Scholar]
- [52].Weihua Z, Lin Q, Ramoth AJ, Fan D, Fidler IJ, Formation of solid tumors by a single multinucleated cancer cell, Cancer 117(17) (2011) 4092–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Niu N, Zhang J, Zhang N, Mercado-Uribe I, Tao F, Han Z, Pathak S, Multani AS, Kuang J, Yao J, Bast RC, Sood AK, Hung MC, Liu J, Linking genomic reorganization to tumor initiation via the giant cell cycle, Oncogenesis 5(12) (2016) e281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Niu N, Mercado-Uribe I, Liu J, Dedifferentiation into blastomere-like cancer stem cells via formation of polyploid giant cancer cells, Oncogene 36 (2017) 4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Jia L, Zhang S, Ye Y, Li X, Mercado-Uribe I, Bast RC Jr., Liu J, Paclitaxel inhibits ovarian tumor growth by inducing epithelial cancer cells to benign fibroblast-like cells, Cancer Lett 326(2) (2012) 176–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Zhang S, Mercado-Uribe I, Liu J, Generation of erythroid cells from fibroblasts and cancer cells in vitro and in vivo, Cancer Lett 333(2) (2013) 205–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Zhang S, Mercado-Uribe I, Sood A, Bast RC, Liu J, Coevolution of neoplastic epithelial cells and multilineage stroma via polyploid giant cells during immortalization and transformation of mullerian epithelial cells, Genes & cancer 7(3–4) (2016) 60–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Murphy BD, Embryonic diapause: advances in understanding the enigma of seasonal delayed implantation, Reprod Domest Anim 47 Suppl 6 (2012) 121–4. [DOI] [PubMed] [Google Scholar]
- [59].Murphy BD, Under Arrest: The Embryo in Diapause, Developmental cell 52(2) (2020) 139–140. [DOI] [PubMed] [Google Scholar]
- [60].Chakradeo S, Elmore LW, Gewirtz DA, Is senescence reversible?, Current drug targets (2015). [DOI] [PubMed] [Google Scholar]
- [61].Ewald JA, Desotelle JA, Wilding G, Jarrard DF, Therapy-induced senescence in cancer, Journal of the National Cancer Institute 102(20) (2010) 1536–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Wang Q, Wu PC, Dong DZ, Ivanova I, Chu E, Zeliadt S, Vesselle H, Wu DY, Polyploidy road to therapy-induced cellular senescence and escape, Int J Cancer 132(7) (2013) 1505–15. [DOI] [PubMed] [Google Scholar]
- [63].Campisi J, Aging, cellular senescence, and cancer, Annu Rev Physiol 75 (2013) 685–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Mosteiro L, Pantoja C, Alcazar N, Marion RM, Chondronasiou D, Rovira M, Fernandez-Marcos PJ, Munoz-Martin M, Blanco-Aparicio C, Pastor J, Gomez-Lopez G, De Martino A, Blasco MA, Abad M, Serrano M, Tissue damage and senescence provide critical signals for cellular reprogramming in vivo, Science 354(6315) (2016). [DOI] [PubMed] [Google Scholar]
- [65].Munoz-Espin D, Serrano M, Cellular senescence: from physiology to pathology, Nat Rev Mol Cell Biol 15(7) (2014) 482–96. [DOI] [PubMed] [Google Scholar]
- [66].Hernandez-Segura A, Nehme J, Demaria M, Hallmarks of Cellular Senescence, Trends in cell biology 28(6) (2018) 436–453. [DOI] [PubMed] [Google Scholar]
- [67].Bharadwaj D, Mandal M, Senescence in polyploid giant cancer cells: A road that leads to chemoresistance, Cytokine Growth Factor Rev 52 (2020) 68–75. [DOI] [PubMed] [Google Scholar]
- [68].Mosieniak G, Sikora E, Polyploidy: the link between senescence and cancer, Curr Pharm Des 16(6) (2010) 734–40. [DOI] [PubMed] [Google Scholar]
- [69].Mosieniak G, Sliwinska MA, Alster O, Strzeszewska A, Sunderland P, Piechota M, Was H, Sikora E, Polyploidy Formation in Doxorubicin-Treated Cancer Cells Can Favor Escape from Senescence, Neoplasia (New York, N.Y.) 17(12) (2015) 882–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Rohnalter V, Roth K, Finkernagel F, Adhikary T, Obert J, Dorzweiler K, Bensberg M, Muller-Brusselbach S, Muller R, A multi-stage process including transient polyploidization and EMT precedes the emergence of chemoresistent ovarian carcinoma cells with a dedifferentiated and pro-inflammatory secretory phenotype, Oncotarget (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Overholtzer M, Brugge JS, The cell biology of cell-in-cell structures, Nat Rev Mol Cell Biol 9(10) (2008) 796–809. [DOI] [PubMed] [Google Scholar]
- [72].Krajcovic M, Overholtzer M, Mechanisms of ploidy increase in human cancers: a new role for cell cannibalism, Cancer Res 72(7) (2012) 1596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Fais S, Overholtzer M, Cell-in-cell phenomena in cancer, Nature reviews. Cancer 18(12) (2018) 758–766. [DOI] [PubMed] [Google Scholar]
- [74].Demin S, Berdieva M, Goodkov A, Cell-cell fusions and cell-in-cell phenomena in healthy cells and cancer: Lessons from protists and invertebrates, Seminars in cancer biology (2021). [DOI] [PubMed] [Google Scholar]
- [75].Arun RP, Sivanesan D, Patra B, Varadaraj S, Verma RS, Simulated microgravity increases polyploid giant cancer cells and nuclear localization of YAP, Scientific reports 9(1) (2019) 10684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Díaz-Carballo D, Saka S, Klein J, Rennkamp T, Acikelli AH, Malak S, Jastrow H, Wennemuth G, Tempfer C, Schmitz I, Tannapfel A, Strumberg D, A Distinct Oncogenerative Multinucleated Cancer Cell Serves as a Source of Stemness and Tumor Heterogeneity, Cancer Research 78(9) (2018) 2318–2331. [DOI] [PubMed] [Google Scholar]
- [77].Jiang Q, Zhang Q, Wang S, Xie S, Fang W, Liu Z, Liu J, Yao K, A Fraction of CD133+ CNE2 Cells Is Made of Giant Cancer Cells with Morphological Evidence of Asymmetric Mitosis, Journal of Cancer 6(12) (2015) 1236–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Salmina K, Jankevics E, Huna A, Perminov D, Radovica I, Klymenko T, Ivanov A, Jascenko E, Scherthan H, Cragg M, Erenpreisa J, Up-regulation of the embryonic self-renewal network through reversible polyploidy in irradiated p53-mutant tumour cells, Experimental cell research 316(13) (2010) 2099–112. [DOI] [PubMed] [Google Scholar]
- [79].Santaguida S, Amon A, Short- and long-term effects of chromosome mis-segregation and aneuploidy, Nat Rev Mol Cell Biol 16(8) (2015) 473–85. [DOI] [PubMed] [Google Scholar]
- [80].Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, Park K, Kitabayashi N, MacDonald TY, Ghandi M, Van Allen E, Kryukov GV, Sboner A, Theurillat JP, Soong TD, Nickerson E, Auclair D, Tewari A, Beltran H, Onofrio RC, Boysen G, Guiducci C, Barbieri CE, Cibulskis K, Sivachenko A, Carter SL, Saksena G, Voet D, Ramos AH, Winckler W, Cipicchio M, Ardlie K, Kantoff PW, Berger MF, Gabriel SB, Golub TR, Meyerson M, Lander ES, Elemento O, Getz G, Demichelis F, Rubin MA, Garraway LA, Punctuated evolution of prostate cancer genomes, Cell 153(3) (2013) 666–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Cortes-Ciriano I, Lee JJ, Xi R, Jain D, Jung YL, Yang L, Gordenin D, Klimczak LJ, Zhang CZ, Pellman DS, Group PSVW, Park PJ, Consortium P, Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing, Nature genetics 52(3) (2020) 331–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Jang HS, Shah NM, Du AY, Dailey ZZ, Pehrsson EC, Godoy PM, Zhang D, Li D, Xing X, Kim S, O’Donnell D, Gordon JI, Wang T, Transposable elements drive widespread expression of oncogenes in human cancers, Nature genetics 51(4) (2019) 611–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Lynch-Sutherland CF, Chatterjee A, Stockwell PA, Eccles MR, Macaulay EC, Reawakening the Developmental Origins of Cancer Through Transposable Elements, Front Oncol 10 (2020) 468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Shoshani O, Brunner SF, Yaeger R, Ly P, Nechemia-Arbely Y, Kim DH, Fang R, Castillon GA, Yu M, Li JSZ, Sun Y, Ellisman MH, Ren B, Campbell PJ, Cleveland DW, Chromothripsis drives the evolution of gene amplification in cancer, Nature (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Dhimolea E, de Matos Simoes R, Kansara D, Al’Khafaji A, Bouyssou J, Weng X, Sharma S, Raja J, Awate P, Shirasaki R, Tang H, Glassner BJ, Liu Z, Gao D, Bryan J, Bender S, Roth J, Scheffer M, Jeselsohn R, Gray NS, Georgakoudi I, Vazquez F, Tsherniak A, Chen Y, Welm A, Duy C, Melnick A, Bartholdy B, Brown M, Culhane AC, Mitsiades CS, An Embryonic Diapause-like Adaptation with Suppressed Myc Activity Enables Tumor Treatment Persistence, Cancer Cell 39(2) (2021) 240–256 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Rehman SK, Haynes J, Collignon E, Brown KR, Wang Y, Nixon AML, Bruce JP, Wintersinger JA, Singh Mer A, Lo EBL, Leung C, Lima-Fernandes E, Pedley NM, Soares F, McGibbon S, He HH, Pollet A, Pugh TJ, Haibe-Kains B, Morris Q, Ramalho-Santos M, Goyal S, Moffat J, O’Brien CA, Colorectal Cancer Cells Enter a Diapause-like DTP State to Survive Chemotherapy, Cell 184(1) (2021) 226–242 e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Chen S, Stout JR, Dharmaiah S, Yde S, Calvi BR, Walczak CE, Transient endoreplication down-regulates the kinesin-14 HSET and contributes to genomic instability, Molecular biology of the cell 27(19) (2016) 2911–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Nehme Z, Pasquereau S, Haidar Ahmad S, Coaquette A, Molimard C, Monnien F, Algros MP, Adotevi O, Diab Assaf M, Feugeas JP, Herbein G, Polyploid giant cancer cells, stemness and epithelial-mesenchymal plasticity elicited by human cytomegalovirus, Oncogene 40(17) (2021) 3030–3046. [DOI] [PubMed] [Google Scholar]
- [89].Illidge TM, Cragg MS, Fringes B, Olive P, Erenpreisa JA, Polyploid giant cells provide a survival mechanism for p53 mutant cells after DNA damage, Cell Biol Int 24(9) (2000) 621–33. [DOI] [PubMed] [Google Scholar]
- [90].Walen KH, Spontaneous cell transformation: Karyoplasts derived from multinucleated cells produce new cell growth in senescent human epithelial cell culutres, In Vitro Cellular & Developmental Biology - Animal 40(5 & 6) (2004) 150–158. [DOI] [PubMed] [Google Scholar]
- [91].Zhang L, Wu C, Hoffman RM, Prostate Cancer Heterogeneous High-Metastatic Multi-Organ-Colonizing Chemo-Resistant Variants Selected by Serial Metastatic Passage in Nude Mice Are Highly Enriched for Multinucleate Giant Cells, PLoS One 10(11) (2015) e0140721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Adams DL, Martin SS, Alpaugh RK, Charpentier M, Tsai S, Bergan RC, Ogden IM, Catalona W, Chumsri S, Tang CM, Cristofanilli M, Circulating giant macrophages as a potential biomarker of solid tumors, Proc Natl Acad Sci U S A 111(9) (2014) 3514–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Gironda DJ, Adams DL, He J, Xu T, Gao H, Qiao Y, Komaki R, Reuben JM, Liao Z, Blum-Murphy M, Hofstetter WL, Tang CM, Lin SH, Cancer associated macrophage-like cells and prognosis of esophageal cancer after chemoradiation therapy, J Transl Med 18(1) (2020) 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Augustyn A, Adams DL, He J, Qiao Y, Verma V, Liao Z, Tang CM, Heymach JV, Tsao AS, Lin SH, Giant Circulating Cancer-Associated Macrophage-Like Cells Are Associated With Disease Recurrence and Survival in Non-Small-Cell Lung Cancer Treated With Chemoradiation and Atezolizumab, Clin Lung Cancer 22(3) (2021) e451–e465. [DOI] [PubMed] [Google Scholar]
- [95].Mu Z, Wang C, Ye Z, Rossi G, Sun C, Li L, Zhu Z, Yang H, Cristofanilli M, Prognostic values of cancer associated macrophage-like cells (CAML) enumeration in metastatic breast cancer, Breast Cancer Res Treat 165(3) (2017) 733–741. [DOI] [PubMed] [Google Scholar]
- [96].Adams DL, Adams DK, Alpaugh RK, Cristofanilli M, Martin SS, Chumsri S, Tang CM, Marks JR, Circulating Cancer-Associated Macrophage-Like Cells Differentiate Malignant Breast Cancer and Benign Breast Conditions, Cancer Epidemiol Biomarkers Prev 25(7) (2016) 1037–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Ptak GE, Modlinski JA, Loi P, Embryonic diapause in humans: time to consider?, Reprod Biol Endocrinol 11 (2013) 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Scognamiglio R, Cabezas-Wallscheid N, Thier MC, Altamura S, Reyes A, Prendergast AM, Baumgartner D, Carnevalli LS, Atzberger A, Haas S, von Paleske L, Boroviak T, Worsdorfer P, Essers MA, Kloz U, Eisenman RN, Edenhofer F, Bertone P, Huber W, van der Hoeven F, Smith A, Trumpp A, Myc Depletion Induces a Pluripotent Dormant State Mimicking Diapause, Cell 164(4) (2016) 668–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Was H, Czarnecka J, Kominek A, Barszcz K, Bernas T, Piwocka K, Kaminska B, Some chemotherapeutics-treated colon cancer cells display a specific phenotype being a combination of stem-like and senescent cell features, Cancer Biol Ther 19(1) (2018) 63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Lin YH, Zhu H, A Malignant Case of Arrested Development: Cancer Cell Dormancy Mimics Embryonic Diapause, Cancer Cell 39(2) (2021) 142–144. [DOI] [PubMed] [Google Scholar]
- [101].Warburg O, On the origin of cancer cells, Science 123(3191) (1956) 309–14. [DOI] [PubMed] [Google Scholar]
- [102].Smith DG, Sturmey RG, Parallels between embryo and cancer cell metabolism, Biochem Soc Trans 41(2) (2013) 664–9. [DOI] [PubMed] [Google Scholar]
- [103].Krisher RL, Prather RS, A role for the Warburg effect in preimplantation embryo development: metabolic modification to support rapid cell proliferation, Mol Reprod Dev 79(5) (2012) 311–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Miyazawa H, Aulehla A, Revisiting the role of metabolism during development, Development 145(19) (2018). [DOI] [PubMed] [Google Scholar]
- [105].McClintock B, The Stability of Broken Ends of Chromosomes in Zea Mays, Genetics 26(2) (1941) 234–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].McClintock B, The Fusion of Broken Ends of Chromosomes Following Nuclear Fusion, Proc Natl Acad Sci U S A 28(11) (1942) 458–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].McClintock B, Induction of Instability at Selected Loci in Maize, Genetics 38(6) (1953) 579–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Heng HH, Genomic chaos: rethinking genetics, evolution, and molecular medicine, Elsevier inc; academic press; (2019). [Google Scholar]
- [109].Heng HH, The genome-centric concept: resynthesis of evolutionary theory, BioEssays : news and reviews in molecular, cellular and developmental biology 31(5) (2009) 512–25. [DOI] [PubMed] [Google Scholar]
- [110].Heng J, Heng HH, Genome chaos: Creating new genomic information essential for cancer macroevolution, Seminars in cancer biology (2020). [DOI] [PubMed] [Google Scholar]
- [111].Heng J, Heng HH, Two-phased evolution: Genome chaos-mediated information creation and maintenance, Prog Biophys Mol Biol (2021). [DOI] [PubMed] [Google Scholar]
- [112].Mirzayans R, Andrais B, Scott A, Wang YW, Kumar P, Murray D, Multinucleated Giant Cancer Cells Produced in Response to Ionizing Radiation Retain Viability and Replicate Their Genome, International journal of molecular sciences 18(2) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Zhang JZ, S; Shi Qiong; Yang Dun, Allen TD, Emerging Strategies to Attack Polyploid Cancer Cells, Journal of Cancer Immunology 2(4): 199–206. (2020). [Google Scholar]
- [114].Scribano CM, Wan J, Esbona K, Tucker JB, Lasek A, Zhou AS, Zasadil LM, Molini R, Fitzgerald J, Lager AM, Laffin JJ, Correia-Staudt K, Wisinski KB, Tevaarwerk AJ, O’Regan R, McGregor SM, Fowler AM, Chappell RJ, Bugni TS, Burkard ME, Weaver BA, Chromosomal instability sensitizes patient breast tumors to multipolar divisions induced by paclitaxel, Science translational medicine 13(610) (2021) eabd4811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Saini G, Joshi S, Garlapati C, Li H, Kong J, Krishnamurthy J, Reid MD, Aneja R, Polyploid giant cancer cell characterization: New frontiers in predicting response to chemotherapy in breast cancer, Seminars in cancer biology (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Nardella C, Clohessy JG, Alimonti A, Pandolfi PP, Pro-senescence therapy for cancer treatment, Nature reviews. Cancer 11(7) (2011) 503–11. [DOI] [PubMed] [Google Scholar]
- [117].Ovadya Y, Krizhanovsky V, Strategies targeting cellular senescence, The Journal of clinical investigation 128(4) (2018) 1247–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].O’Leary B, Finn RS, Turner NC, Treating cancer with selective CDK4/6 inhibitors, Nat Rev Clin Oncol 13(7) (2016) 417–30. [DOI] [PubMed] [Google Scholar]
- [119].Iyengar M, O’Hayer P, Cole A, Sebastian T, Yang K, Coffman L, Buckanovich RJ, CDK4/6 inhibiti maintenance and combination therapy for high grade serous ovarian cancer, Oncotarget 9(21) (2018) 15658–15672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Wu CH, van Riggelen J, Yetil A, Fan AC, Bachireddy P, Felsher DW, Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation, Proc Natl Acad Sci U S A 104(32) (2007) 13028–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Kirkland JL, Tchkonia T, Senolytic drugs: from discovery to translation, Journal of internal medicine 288(5) (2020) 518–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Saleh T, Carpenter VJ, Bloukh S, Gewirtz DA, Targeting tumor cell senescence and polyploidy as potential therapeutic strategies, Seminars in cancer biology (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Thura M, Ye Z, Al-Aidaroos AQ, Xiong Q, Ong JY, Gupta A, Li J, Guo K, Ang KH, Zeng Q, PRL3 induces polypoid giant cancer cells eliminated by PRL3-zumab to reduce tumor relapse, Commun Biol 4(1) (2021) 923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Niu NY, Jun; Bast Robert, C; Sood Anil K; and Liu Jinsong, IL-6 promotes drug resistance through formation of polyploid giant cancer cells and stromal fibroblast reprogramming, Oncogenesis in press (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Kudo-Saito C, Miyamoto T, Imazeki H, Shoji H, Aoki K, Boku N, IL33 Is a Key Driver of Treatment Resistance of Cancer, Cancer Res 80(10) (2020) 1981–1990. [DOI] [PubMed] [Google Scholar]
- [126].Barsoum IB, Smallwood CA, Siemens DR, Graham CH, A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells, Cancer Res 74(3) (2014) 665–74. [DOI] [PubMed] [Google Scholar]
- [127].Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E, Johnson RS, Goldrath AW, Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen, Nat Immunol 14(11) (2013) 1173–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Sanmamed MF, Chen L, A Paradigm Shift in Cancer Immunotherapy: From Enhancement to Normalization, Cell 175(2) (2018) 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Krajewska M, Heijink AM, Bisselink YJ, Seinstra RI, Sillje HH, de Vries EG, van Vugt MA, Forced activation of Cdk1 via wee1 inhibition impairs homologous recombination, Oncogene 32(24) (2013) 3001–8. [DOI] [PubMed] [Google Scholar]
- [130].Ganem NJ, Cornils H, Chiu SY, O’Rourke KP, Arnaud J, Yimlamai D, Thery M, Camargo FD, Pellman D, Cytokinesis failure triggers hippo tumor suppressor pathway activation, Cell 158(4) (2014) 833–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Quinton RJ, DiDomizio A, Vittoria MA, Kotynkova K, Ticas CJ, Patel S, Koga Y, Vakhshoorzadeh J, Hermance N, Kuroda TS, Parulekar N, Taylor AM, Manning AL, Campbell JD, Ganem NJ, Whole-genome doubling confers unique genetic vulnerabilities on tumour cells, Nature 590(7846) (2021) 492–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Xuan B, Ghosh D, Dawson MR, Contributions of the distinct biophysical phenotype of polyploidal giant cancer cells to cancer progression, Seminars in cancer biology (2021). [DOI] [PubMed] [Google Scholar]
- [133].Xuan B, Ghosh D, Jiang J, Shao R, Dawson MR, Vimentin filaments drive migratory persistence in polyploidal cancer cells, Proc Natl Acad Sci U S A 117(43) (2020) 26756–26765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Xuan B, Ghosh D, Cheney EM, Clifton EM, Dawson MR, Dysregulation in Actin Cytoskeletal Organization Drives Increased Stiffness and Migratory Persistence in Polyploidal Giant Cancer Cells, Scientific reports 8(1) (2018) 11935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Voelkel-Johnson C, Sphingolipids in embryonic development, cell cycle regulation, and stemness - Implications for polyploidy in tumors, Seminars in cancer biology (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].White-Gilbertson S, Lu P, Norris JS, Voelkel-Johnson C, Genetic and pharmacological inhibition of acid ceramidase prevents asymmetric cell division by neosis, Journal of lipid research (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Lu P, White-Gilbertson S, Beeson G, Beeson C, Ogretmen B, Norris J, Voelkel-Johnson C, Ceramide Synthase 6 Maximizes p53 Function to Prevent Progeny Formation from Polyploid Giant Cancer Cells, Cancers (Basel) 13(9) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].White-Gilbertson S, Lu P, Jones CM, Chiodini S, Hurley D, Das A, Delaney JR, Norris JS, Voelkel-Johnson C, Tamoxifen is a candidate first-in-class inhibitor of acid ceramidase that reduces amitotic division in polyploid giant cancer cells-Unrecognized players in tumorigenesis, Cancer medicine 9(9) (2020) 3142–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Cheng JC, Bai A, Beckham TH, Marrison ST, Yount CL, Young K, Lu P, Bartlett AM, Wu BX, Keane BJ, Armeson KE, Marshall DT, Keane TE, Smith MT, Jones EE, Drake RR Jr., Bielawska A, Norris JS, Liu X, Radiation-induced acid ceramidase confers prostate cancer resistance and tumor relapse, The Journal of clinical investigation 123(10) (2013) 4344–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Petrova V, Annicchiarico-Petruzzelli M, Melino G, Amelio I, The hypoxic tumour microenvironment, Oncogenesis 7(1) (2018) 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Sullivan R, Pare GC, Frederiksen LJ, Semenza GL, Graham CH, Hypoxia-induced resistance to anticancer drugs is associated with decreased senescence and requires hypoxia-inducible factor-1 activity, Molecular cancer therapeutics 7(7) (2008) 1961–73. [DOI] [PubMed] [Google Scholar]
- [142].Barker HE, Paget JT, Khan AA, Harrington KJ, The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence, Nature reviews. Cancer 15(7) (2015) 409–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Yu T, Tang B, Sun X, Development of Inhibitors Targeting Hypoxia-Inducible Factor 1 and 2 for Cancer Therapy, Yonsei Med J 58(3) (2017) 489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Wolpaw AJ, Dang CV, Exploiting Metabolic Vulnerabilities of Cancer with Precision and Accuracy, Trends in cell biology 28(3) (2018) 201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Fendt SM, Metabolic vulnerabilities of metastasizing cancer cells, BMC Biol 17(1) (2019) 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Obrist F, Michels J, Durand S, Chery A, Pol J, Levesque S, Joseph A, Astesana V, Pietrocola F, Wu GS, Castedo M, Kroemer G, Metabolic vulnerability of cisplatin-resistant cancers, The EMBO journal 37(14) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Donovan P, Cato K, Legaie R, Jayalath R, Olsson G, Hall B, Olson S, Boros S, Reynolds BA, Harding A, Hyperdiploid tumor cells increase phenotypic heterogeneity within Glioblastoma tumors, Molecular bioSystems 10(4) (2014) 741–58. [DOI] [PubMed] [Google Scholar]
- [148].Hussein AM, Wang Y, Mathieu J, Margaretha L, Song C, Jones DC, Cavanaugh C, Miklas JW, Mahen E, Showalter MR, Ruzzo WL, Fiehn O, Ware CB, Blau CA, Ruohola-Baker H, Metabolic Control over mTOR-Dependent Diapause-like State, Developmental cell 52(2) (2020) 236–250 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Sharma S, Zeng JY, Zhuang CM, Zhou YQ, Yao HP, Hu X, Zhang R, Wang MH, Small-molecule inhibitor BMS-777607 induces breast cancer cell polyploidy with increased resistance to cytotoxic chemotherapy agents, Molecular cancer therapeutics 12(5) (2013) 725–36. [DOI] [PubMed] [Google Scholar]
- [150].Zeng JY, Sharma S, Zhou YQ, Yao HP, Hu X, Zhang R, Wang MH, Synergistic activities of MET/RON inhibitor BMS-777607 and mTOR inhibitor AZD8055 to polyploid cells derived from pancreatic cancer and cancer stem cells, Molecular cancer therapeutics 13(1) (2014) 37–48. [DOI] [PubMed] [Google Scholar]
- [151].Bulut-Karslioglu A, Biechele S, Jin H, Macrae TA, Hejna M, Gertsenstein M, Song JS, Ramalho-Santos M, Inhibition of mTOR induces a paused pluripotent state, Nature 540(7631) (2016) 119–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].He B, Zhang H, Wang J, Liu M, Sun Y, Guo C, Lu J, Wang H, Kong S, Blastocyst activation engenders transcriptome reprogram affecting X-chromosome reactivation and inflammatory trigger of implantation, Proc Natl Acad Sci U S A 116(33) (2019) 16621–16630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Ogden A, Rida PC, Knudsen BS, Kucuk O, Aneja R, Docetaxel-induced polyploidization may underlie chemoresistance and disease relapse, Cancer Lett 367(2) (2015) 89–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Harty RN, No exit: targeting the budding process to inhibit filovirus replication, Antiviral Res 81(3) (2009) 189–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].de The H, Differentiation therapy revisited, Nature reviews. Cancer 18(2) (2018) 117–127. [DOI] [PubMed] [Google Scholar]
- [156].Zhou S, Abdouh M, Arena V, Arena M, Arena GO, Reprogramming Malignant Cancer Cells toward a Benign Phenotype following Exposure to Human Embryonic Stem Cell Microenvironment, PLoS One 12(1) (2017) e0169899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Tripathi A, Kashyap A, Tripathi G, Yadav J, Bibban R, Aggarwal N, Thakur K, Chhokar A, Jadli M, Sah AK, Verma Y, Zayed H, Husain A, Bharti AC, Kashyap MK, Tumor reversion: a dream or a reality, Biomark Res 9(1) (2021) 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Yen K, Travins J, Wang F, David MD, Artin E, Straley K, Padyana A, Gross S, DeLaBarre B, Tobin E, Chen Y, Nagaraja R, Choe S, Jin L, Konteatis Z, Cianchetta G, Saunders JO, Salituro FG, Quivoron C, Opolon P, Bawa O, Saada V, Paci A, Broutin S, Bernard OA, de Botton S, Marteyn BS, Pilichowska M, Xu Y, Fang C, Jiang F, Wei W, Jin S, Silverman L, Liu W, Yang H, Dang L, Dorsch M, Penard-Lacronique V, Biller SA, Su SM, AG-221, a First-in-Class Therapy Targeting Acute Myeloid Leukemia Harboring Oncogenic IDH2 Mutations, Cancer discovery 7(5) (2017) 478–493. [DOI] [PubMed] [Google Scholar]
- [159].Bots M, Verbrugge I, Martin BP, Salmon JM, Ghisi M, Baker A, Stanley K, Shortt J, Ossenkoppele GJ, Zuber J, Rappaport AR, Atadja P, Lowe SW, Johnstone RW, Differentiation therapy for the treatment of t(8;21) acute myeloid leukemia using histone deacetylase inhibitors, Blood 123(9) (2014) 1341–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Ishay-Ronen D, Diepenbruck M, Kalathur RKR, Sugiyama N, Tiede S, Ivanek R, Bantug G, Morini MF, Wang J, Hess C, Christofori G, Gain Fat-Lose Metastasis: Converting Invasive Breast Cancer Cells into Adipocytes Inhibits Cancer Metastasis, Cancer Cell 35(1) (2019) 17–32 e6. [DOI] [PubMed] [Google Scholar]
- [161].Wang X, Pei Z, Hossain A, Bai Y, Chen G, Transcription factor-based gene therapy to treat glioblastoma through direct neuronal conversion, Cancer Biol Med (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Salem A, Pinto K, Koch M, Liu J, Silva EG, Are polyploid giant cancer cells in high grade serous carcinoma of the ovary blastomere-like cancer stem cells?, Ann Diagn Pathol 46 (2020) 151505. [DOI] [PubMed] [Google Scholar]
- [163].Nikas G, Ao A, Winston RM, Handyside AH, Compaction and surface polarity in the human embryo in vitro, Biol Reprod 55(1) (1996) 32–7. [DOI] [PubMed] [Google Scholar]