

Abstract
Objective
Infants with focal-onset epilepsy are an understudied population, requiring additional evaluation for clinical assessment and prognostication. Our goal was to characterize the etiology and natural history of infantile-onset focal epilepsy.
Methods
We retrospectively identified all infants (0-24 months) with onset of focal epilepsy while resident in Olmsted Co, MN between 1980-2018, using the Rochester Epidemiology Project Database. We assessed the impact of etiology on both seizure and neurodevelopmental outcome, and mortality.
Results
Of 686 children with epilepsy onset <18 years, 125 (18.2%) presented with focal-onset seizures in infancy. Median follow-up for this group was 10.9 years (IQR 6.2, 19.3). Etiology was identified in 65.6% (structural N = 62, genetic N = 13, both structural and genetic N = 3, metabolic N = 4). Of 107 patients followed >2 years, 38 (35.5%) developed drug-resistant epilepsy (DRE). DRE was more likely with younger age at onset, known etiology, and presence of epileptic spasms. Sixty-eight (63.0% of those with follow-up) were developmentally delayed at last follow-up, and known etiology, DRE, and presence of epileptic spasms were significantly associated with delay (P < 0.001 for all). Fifteen (12.0%) patients expired at a median age of 7.1 years (IQR 1.7, 21.7) but only 1 death was seizure-related (suspected SUDEP). Of 20 infants with normal development at onset and no known etiology with >2 years follow-up, none developed DRE, all were seizure free at last follow-up (95% off ASMs), and all remained developmentally normal.
Significance
Infantile-onset focal epilepsy accounts for 18% of all epilepsy in childhood, is frequently due to known etiologies and has a high rate of DRE. However, developmentally normal infants without known cause appear to have a very favorable course.
INTRODUCTION
Though estimates of the incidence of pediatric epilepsy have shown some variation across populations and methodologies, it is commonly accepted that the incidence is highest within the first two years of life (1–5). Epilepsy is a particularly heterogeneous disorder, with marked range in underlying etiology, seizure type, frequency, severity, and impact on neurological development and quality of life, however, children with focal seizures remain an understudied population. The relative dearth of clinical data in the literature has limited the ability of clinicians to identify children at risk of adverse outcomes and provide accurate prognostic counseling to families.
In the present study, we address this unmet need by analysis of a retrospective cohort of infants who had onset of focal epilepsy up to and including 24 months of age, while resident in Olmsted County, Minnesota (USA). Specifically, our goal was to address several outstanding questions, including:
What are the important causes of infantile-onset focal epilepsy?
Does epilepsy which is focal at onset remain focal?
How commonly do infants presenting with focal-onset seizures evolve to epileptic spasms and what are the risk factors for that evolution
What is the long-term seizure and developmental outcome in infants with focal epilepsy, and how well can this be predicted?
Study participants were followed up through April 15, 2020, or last available follow-up, with periodic assessment of seizure type, frequency, and medication responsiveness as well as functional outcomes.
METHODS
Study design and population
We analyzed data available in the Olmsted County Rochester Epidemiology Project (REP) Database from 1980 to 2018 in a retrospective analysis of infants (age <24 months) with onset of focal epilepsy. The REP is a well-documented, record-linkage system for retrospective studies which tracks demographic characteristics, diagnostic and procedure codes as well as all paper and electronic medical records for a dynamic cohort of 502,820 unique individuals who resided in Olmsted County at some point after 1966 (6–8). A total of 686 individuals with childhood-onset epilepsy were identified within the study period. These records were individually reviewed by pediatric epileptologists to identify subjects who met study criteria: 1) diagnosis of epilepsy, defined as 2 or more unprovoked, afebrile seizures, 2) onset at age <24 months, and 3) seizures which were focal at onset.
Seizure classification was performed by a qualified pediatric epileptologist, based on recorded clinical descriptions of semiology by licensed child neurologists, EEG reports and imaging studies. For inclusion in the study, seizure type at time of diagnosis had to be categorized as focal-onset. Seizures were deemed to be focal in onset if the semiology was consistent with a focal seizure – focal tonic or clonic activity, staring/altered awareness with a postictal state, or focal to bilateral tonic-clonic and the EEG showed either focal or multifocal abnormalities or did not show clear epileptiform discharges. Bilateral convulsive seizures with focal epileptiform discharge on EEG were also classified as focal-onset. Children with epileptic spasms were also classified as having focal epilepsy if they had clear focal features to the spasms, if clusters began or terminated with focal seizures, if they had a focal lesion on imaging that was deemed to be causal for the epilepsy, or if the EEG showed clearly focal or hemispheric (not multifocal or generalized) epileptiform discharges. Children who presented with both focal and generalized onset seizures were excluded.
The study was approved by the Institutional Review Boards at both Mayo Clinic and Olmsted Medical Center.
Demographics and Etiology
We recorded basic demographic data, epilepsy characteristics and results of clinical evaluations (Table 1). For subjects who expired during the study, date and cause of death were also recorded.
Table 1. Characteristics of the 125 Study Cases.
Table lists general study characteristics, including demographics, birth history, and neurological exam at presentation. Sum of individual brain injuries sum to >34 due to N = 5 cases with multiple subtypes (E.G. head injury with subsequent meningitis). Miscellaneous brain injuries include infarction (N = 4), periventricular leukomalacia (N = 3), and N =1 each of ruptured aneurysm, traumatic brain injury from motor vehicle accident, prenatal methamphetamine exposure, and hypoglycemic brain injury. In 1 case of HIE and 1 case of encephalitis, a variant of unknown significance was also identified on genetic testing. The case of prenatal drug exposure ultimately had no abnormalities noted on MRI. Non-focal motor difficulties included changes in motor tone or coordination. Follow-up duration, age at onset, and gestational age are reported as median (IQR); all other variables shown as N (%).
| General Study Population Characteristics Total N = 125 | |
|---|---|
| Male / Female | 61 (48.8%) / 64 (51.2%) |
| Duration Follow-up | 10.9 years (6.2, 19.3) |
| Age Onset | 7 months (2.0, 15.5) |
| Gestational Age | 40 weeks (37.0, 40.0) |
| Early Pre-term Births (<34 wks) | 15 (12.0%) |
| Late Pre-term Births (34 to 36 6/7) | 9 (7.2%) |
| Prenatal Complications Requiring Hospitalization | 15 (12.0%) |
| Postnatal Complications Requiring ≥ 7 Days in NICU | 48 (38.4%) |
| Presence of Neonatal Seizures | 28 (22.4%) |
| Presence of Febrile Seizures | 16 (12.8%) |
| Family History of Seizures in 1st Degree Relative | 18 (14.4%) |
| Brain Injury | 34 (27.2%) |
| Hypoxic-Ischemic Encephalopathy | 14 (11.2%) |
| Intraventricular Hemorrhage | 8 (6.3%) |
| Head Injury | 1 (0.8%) |
| Meningitis | 3 (2.4%) |
| Encephalitis | 2 (1.6%) |
| Miscellaneous | 11 (8.8%) |
| Abnormal Findings on Neurologic Exam | 64 (51.2%) |
| Quadriparesis | 27 (21.6%) |
| Hemi/paraparesis | 20 (16.0%) |
| Extrapyramidal Signs | 2 (1.6%) |
| Non-focal Motor Difficulties | 15 (12.0%) |
Seizure type(s) were recorded at the time of initial presentation and at time of final follow-up, noting all seizure types that had emerged with time. Seizures were divided into focal aware, focal with impaired awareness, focal undifferentiated (if awareness could not be determined), focal to bilateral convulsive seizure, epileptic spasms, as well as co-existing generalized seizure types including myoclonic, tonic, atonic, absence or generalized tonic clonic.
Etiology class was divided into genetic, structural, metabolic, infectious, immune or unknown. Where more than one category applied (e.g. DCX mutation with cortical malformation), both etiologies were assigned. The specific etiology was also noted.
Clinical work-up including results of brain imaging (MRI and CT), electroencephalograms (EEGs) (presence and location of background slowing and epileptiform discharges)—as well as any genetic, metabolic or other investigations were noted.
Developmental Outcomes
We selected cases followed for longer than 1 year to assess the correlation of infantile-onset focal epilepsy on cognitive development. Study participants were categorized as either normal function (measured or estimated development quotient (DQ) ≥ 80), mild/borderline (DQ 50-79), or moderate to severe disability (DQ < 50) from clinical abstraction, based on developmental milestones, clinical evaluation and when performed, formal neuropsychological evaluation. Scores were applied both at time of epilepsy onset, and at most recent follow-up. Given the young age of this study population, few underwent formalized psychoeducational testing prior to seizure onset.
Epilepsy Outcome
Seizure frequency at final follow-up was recorded categorically for each of the first three years after diagnosis, and then at year 5, 10, 15, and 20 years (as available) as well as most recent follow-up. At each of those time points, the number of currently used antiseizure medications (ASMs) was also recorded, as well as the number of ASMs discontinued due to either intolerable side-effects or lack of efficacy. These data were then used to identify individuals with drug-resistant epilepsy (DRE), defined as those with ongoing seizures at final follow-up despite trials of two or more ASMs (either current or failed due to lack of efficacy)(9) and those who underwent epilepsy surgery for DRE, regardless of seizure frequency at last follow-up. In determining the proportion with DRE, we only included cases with >24 months follow-up (N=108) to allow adequate time for medication trials and assessment of efficacy.
Statistical analysis
All statistical analysis was performed using the IBM® SPSS® software platform. Statistical comparison between groups was done by two-sided Chi-squared test. The P values for each test are reported in the Results. Paired two-tailed Student’s t test was used to assess changes in frequencies of seizure types at onset compared to time of follow-up; specific tests used are indicated in individual figure legends. For all tests, statistical significance was set at P ≤ 0.05. Figures were generated using Biorender.com.
RESULTS
Study Population
A total of 686 cases of new-onset pediatric epilepsy (onset prior to 18 years of age) were identified. Of these, 167 (24.3%) had onset in infancy (<24 months of age). The majority, 125 of the 167 infantile cases (74.9%), had focal epilepsy and were included in this study.
Demographic data and details of epilepsy and clinical investigations are shown in Table 1. Median age at onset was 7 months, with 48.0% diagnosed at or prior to 6 months of age. Twenty-nine (23.2%) had documented history of neonatal seizures. At time of epilepsy diagnosis, 31 (24.8%) had at least one episode of status epilepticus (SE). Structural brain injury was common, being present in 34 subjects (27.2%). Over half of cases (51.2%) had abnormalities detected on neurological exam at the time of epilepsy diagnosis.
Investigations
Almost all children (122/125, 97.6%) had brain imaging; including MRI (N=110) or CT head alone (N=12). Of those who underwent brain MRI, 60 (54.5%) studies revealed significant causal abnormalities. For those with only CT, 3 (25.0%) had clear structural findings. The use of MRI significantly increased over time: in the first ten years of the study, 24 out of 34 cases (70.6%) underwent MRI imaging compared to 51 of 54 (94.4%) in the last ten years (P < 0.01).
All participants had at least one EEG performed. A total of 31 (24.8%) had normal initial EEG, with no background slowing or epileptiform discharges. Background slowing on initial EEG was noted in 49.6% (generalized in 44.8% and only focal in 4.8%). Epileptiform discharges were present on 68.0% of initial recordings. In most cases these were either multifocal (N = 47, 37.6%) or unifocal (N = 20, 16.0%). The remainder were classified as having 2 foci (N = 7), hemispheric (N = 4), or both focal and generalized (N = 1). Hypsarrhythmia was observed in 6 cases (4.8%). Of these, 4 evolved into multifocal epileptiform discharges over time, 1 had 2 foci, and 1 still demonstrated hypsarrhythmia at most recent EEG. Two cases developed hypsarrhythmia which was not present at initial recording. Of the 20 with a single discharge focus at diagnosis, only 3 progressed to multifocal or generalized discharges. The remaining 5 did not have another recording during the study period.
Only 23 individuals (18.4%) had documented results from genetic testing (karyotype analysis, fluorescent in situ hybridization, chromosomal microarray, whole exome sequencing, or specific gene panel tests). All but 3 of these were completed in cases diagnosed after the year 2005, representing a notable increase in utilization of genetic testing in focal epilepsy.
Seizure Type at Presentation and Evolution with Time
At epilepsy onset, 79 (63.2%) presented with only focal seizures (focal aware, impaired awareness, or focal undifferentiated), 36 (28.8%) presented as focal to bilateral tonic-clonic seizures (with or without other focal aware or focal impaired awareness), and 10 (8.0%) with both focal onset seizures and epileptic spasms (Figure 1). The median age of those who presented with only focal seizures was 4 months (IQR 0, 14), 11.5 months (IQR 6.3, 17.8) in cases with focal to bilateral tonic-clonic (P < 0.01 compared to focal only), and 6 months (IQR 2.3, 9.5) in those with spasms at onset (P = 0.16 compared to focal only).
Figure 1. Seizure type at Presentation and Evolution with Time.
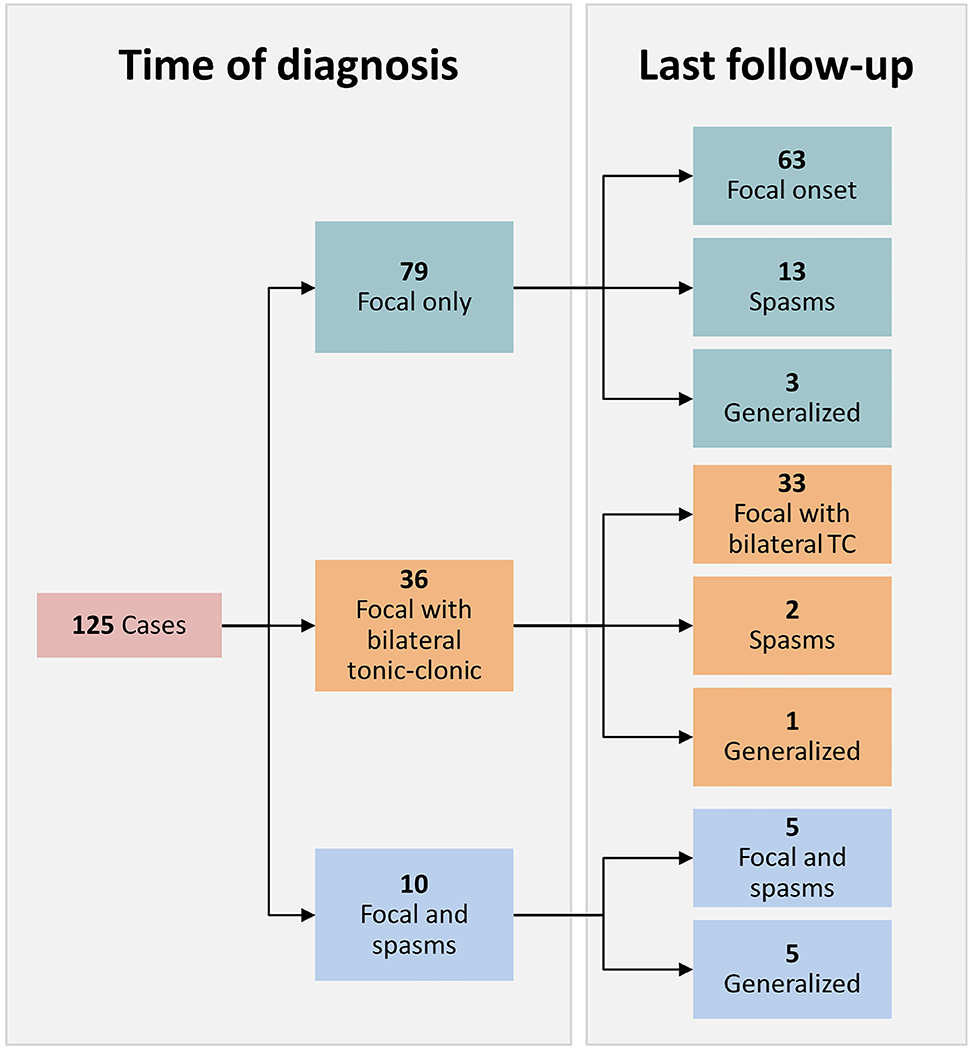
Frequency of seizure semiology is shown both at time of diagnosis and at last follow-up. The 63 cases with only focal seizures at onset that remain focal onset include 47 with only focal seizures and 16 with evolution to bilateral tonic-clonic activity. Of the 15 that developed epileptic spasms at follow-up, 5 of them also demonstrated generalized onset seizures. The generalized semiologies that evolved over time include: generalized tonic-clonic, tonic, atonic, and myoclonic.
Importantly, of those with only focal seizures at presentation, 47/79 (59.5%) continued with this seizure type long term while an additional 16 (20.3%) developed focal to bilateral tonic-clonic seizures (Figure 1). Two developed generalized seizure types including generalized tonic-clonic, tonic, atonic, and myoclonic seizures. Those that developed generalized seizures (including focal to bilateral tonic-clonic) were more likely to have abnormal findings on neurological exam (P < 0.01) and were less likely to be categorized as etiology unknown, developmentally normal (P < 0.05). Thirteen cases (16.5%) eventually developed spasms. The median time to development of spasms was 5.3 months, with the longest interval between onset of focal seizures and development of spasms being 12.1 months. Within this group, younger age of onset was significantly associated with eventual development of epileptic spasms (mean age at onset of 3.9 months in those evolving to spasms vs 8.1 months without spasms, P < 0.01); 10 out of the 49 cases (20.4%) who presented <6 months of age with only focal seizures would develop spasms, compared to only 3 of the 30 (10.0%) diagnosed ≥6 months. Development of epileptic spasms was significantly associated with cortical dysplasia (P < 0.05), where 44.4% of cases developed spasms, as well as abnormal neurological exam (P = 0.03) and development (P < 0.001) at onset (Table 2). Reassuringly, no children with unknown etiology and typical development evolved to spasms.
Table 2. Development of Spasms after Presentation.
Table lists general study characteristics, including age onset, birth history, and neurological exam at presentation for cases presenting with only focal seizures, subdivided into those who remained with only focal seizures, those that developed generalized seizures, and those that developed spasms. Age onset shown as median (IQR), all other variables reported as N (%). P values represent comparison between group that remained focal only vs developed spasms; cases that developed generalized seizures were not included due to small sample size. P values <0.05 shown in bold text, statistical significance determined by Student’s two-tailed T-test for age onset, Chi-Square for all other variables.
| Evolution of Seizure Types from Focal Only Onset Total N = 79 | ||||
|---|---|---|---|---|
| Focal Only | Focal+Gen’d | Focal+Spasms | P Value | |
| Number Cases | 63 | 3 | 13 | -- |
| Age Onset | 6 months (1.0, 16.0) | 2 months (0.0, 2.0) | 2 months (0.0, 8.5) | 0.05 |
| Abnormal Neurological Exam | 31 (49.2%) | 3 (100.0%) | 11 (84.6%) | 0.02 |
| Moderate-Severe Developmental Delay | 9 (14.3%) | 0 (0.0%) | 9 (69.2%) | <0.001 |
| Prenatal Complications Requiring Hospitalization | 7 (11.1%) | 0 (0.0%) | 1 (6.7%) | 0.71 |
| Postnatal Complications Requiring ≥ 7 Days in NICU | 24 (38.1%) | 0 (0.0%) | 7 (53.8%) | 0.29 |
| Presence of Neonatal Seizures | 15 (23.8%) | 0 (0.0%) | 7 (53.8%) | 0.03 |
| Mortality | 6 (9.5%) | 0 (0.0%) | 4 (30.8%) | 0.04 |
| Etiology | ||||
| Structural—Acquired | 23 (36.5%) | -- | 4 (30.8%) | 0.69 |
| Structural—Congenital | 18 (28.6%) | 1 (33.3%) | 7 (53.8%) | 0.08 |
| Metabolic | 1 (1.6%) | -- | 1 (7.7%) | 0.21 |
| Unknown | 21 (33.3%) | 2 (66.6%) | 1 (7.7%) | 0.06 |
Evolution to epileptic spasms was much less likely in cases who initially presented with focal to bilateral tonic-clonic seizures, where only 2/36 (5.5%) of cases evolved to spasms. Of the 10 cases presenting with both focal seizures and spasms, half developed generalized-onset seizures over time and 4 met criteria for Lennox-Gastaut syndrome (Figure 1).
Etiology
An etiology was found in 82 (65.6%) cases (Table 3). Structural etiologies were the most common cause, comprising 65 (79.3%) cases with known etiology. Two of those with cortical dysplasias and 1 with brain injury also had a pathogenic genetic variant. A genetic etiology was found in 16 (19.5%), which is 69.6% of the 23 with genetic testing. Four had metabolic causes.
Table 3. Causes of Focal Infantile-Onset Epilepsy.
A specific cause can be identified in two thirds of cases. Perinatal brain injury includes stroke, hypoxic-ischemic encephalopathy, intracranial hemorrhage, trauma. Monogenic disorders identified include mutations in DCX, CHD7, Gli3, NSD1, SCN5A, STXBP1; microdeletions were seen at 2q31.1, 15q11.2, 15q13.3, 18p11.2, and there was one duplication at 10q23.1. Trisomies: one case was of trisomy 21, the other was a partial trisomy 15q. The final case categorized as genetic presented with multiple congenital anomalies including microcephaly and presented prior to wider availability of detailed genetic tests. Counts sum to >125 due to 3 cases of genetic+structural etiology counted in both categories. Variables reported as N (%).
| Etiology of Focal Infantile-Onset Epilepsy Total N = 125 | |
|---|---|
| Structural – Acquired | 39 (31.2%) |
| Brain Injury | 34 (27.2%) |
| Vascular Malformation | 2 (1.6%) |
| Mesial Temporal Sclerosis | 2 (1.6%) |
| Tumor | 1 (0.8%) |
| Structural – Congenital | 26 (20.8%) |
| Cortical Dysplasia | 18 (14.4%) |
| Tuberous Sclerosis/Neurocutaneous | 8 (6.4%) |
| Genetic | 16 (12.8%) |
| Monogenic Disorder | 8 (6.4%) |
| Microdeletion/duplication | 5 (4.0%) |
| Trisomy | 2 (1.6%) |
| Unable to Determine | 1 (0.8%) |
| Metabolic | 4 (3.2%) |
| Mitochondrial | 1 (0.8%) |
| Peroxisomal | 1 (0.8%) |
| Mucopolysaccharidosis | 1 (0.8%) |
| Amino Acid Disorder | 1 (0.8%) |
| Unknown Cause | 43 (34.4%) |
| Developmentally Normal | 31 (24.8%) |
| Developmentally Delayed | 12 (9.6%) |
Of the 43 (34.4%) with unknown cause, 31 (72.1%) were developmentally normal at diagnosis. The rate of etiology identification only slightly increased through the study period: 15 of 34 (44.1%) of cases diagnosed in the first fifteen years (1980-1995) were classified as unknown, compared to 20 of the 70 (28.6%) cases diagnosed in the last fifteen years (2003-2018) (P = 0.12).
Mortality
Fifteen children (12.0%) died over the duration of the study (5 males and 10 females), at a median age of 7.1 years (IQR 1.7, 21.7). Most deaths (14/15) were due to cardiorespiratory failure due to profound neurological impairment, the remaining case was suspected sudden unexpected death in epilepsy (SUDEP). Mortality was highly associated with abnormalities on neurological exam (P < 0.001), intellectual disability at diagnosis (P < 0.001) and epileptic spasms (P < 0.001). Slowing of background activity on initial EEG (P < 0.001) and abnormal MRI (P = 0.03) were both associated with mortality. Known etiology was similarly a poor prognostic factor (P = 0.03). Malformations of cortical development were significantly associated with death in analysis of both imaging findings and etiology, as was metabolic etiology. Among cases with known etiology, mortality was significantly more likely in those with moderate-severe developmental delay (10/26, 38.5%) compared to those without (2/48, 4.5%, P < 0.001). Two deaths occurred in cases reported only to have mild delay at diagnosis; these cases both had a known etiology, other abnormalities noted on neurological exam, and intractable seizures. No deaths occurred in children with a normal examination and normal development at onset. All 28 developmentally normal children with unknown etiology were living at most recent follow-up.
Developmental Outcome (Figure 2)
Figure 2. Evolution of Developmental Function Over Time.
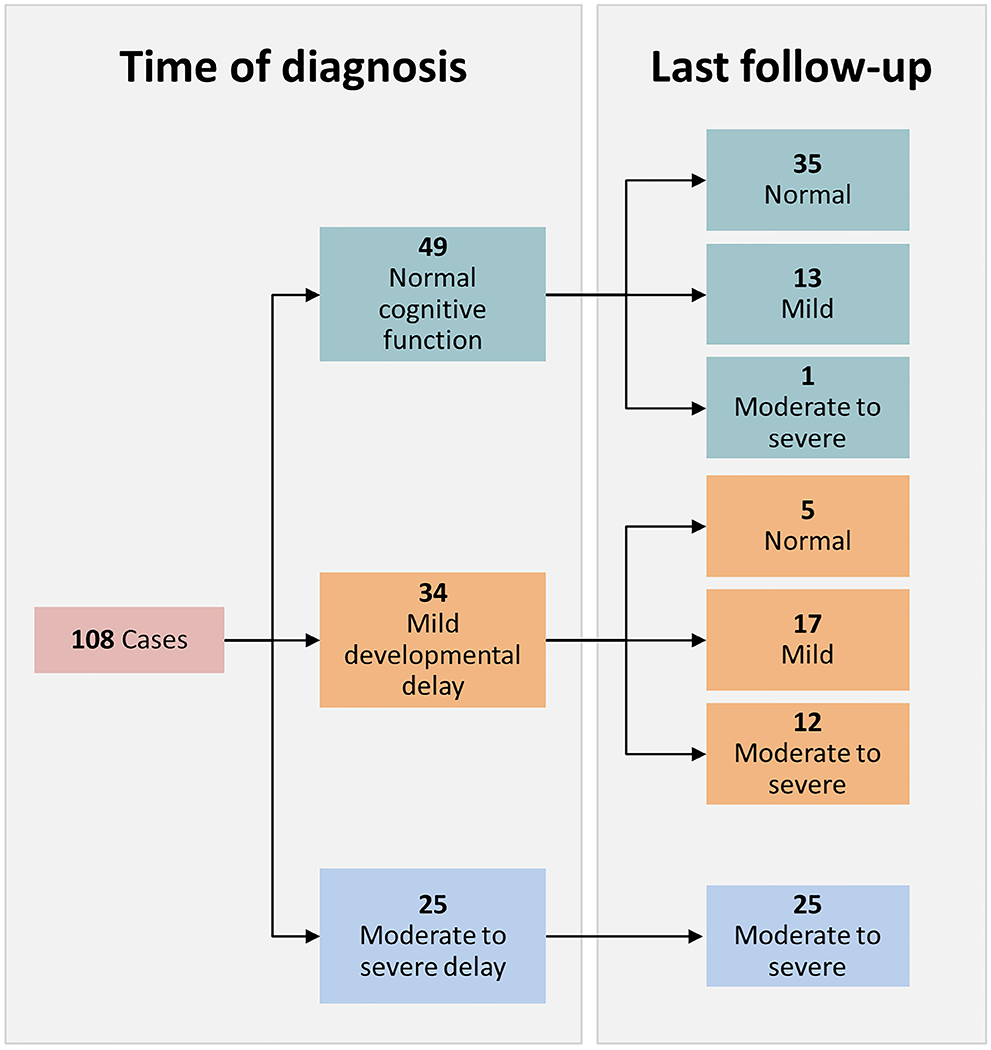
Developmental function was assessed at time of diagnosis and recent follow-up and classified as normal, mildly impaired, or moderate to severe disability. Seventy-one percent of those with normal development at onset progressed without accumulation of disability. In total, 24.1% of cases with >12 months follow-up demonstrated increase in level of disability during the study.
Of the 108 individuals (56 male and 52 female) with more than 12 months of follow-up who had assessment of development, 45.4% had normal cognitive function at time of epilepsy onset. Of these, 71.4% remained normal at follow-up. Five cases with mild disability at onset showed improved function over time. Those with moderate to severe levels of impairment remained in that category throughout the study duration. Only 2 (1.7%) cases had undergone formal psychoeducational testing of any time prior to diagnosis, though 36 (33.3%) did by time of last follow-up. Within this group, the median full-scale intelligence quotient (FSIQ) was 67 (IQR 49.0, 78.0).
Sixty-eight (63.0%) were developmentally delayed at last follow-up. Developmental status at last follow-up was significantly associated with function at epilepsy onset (P < 0.001). Other correlates with moderate-severe intellectual disability at follow-up included abnormal neurological exam at diagnosis (P < 0.001), development of spasms (P < 0.001), abnormalities on MRI (P = 0.002), background slowing on initial EEG (P < 0.001) and known etiology (P < 0.001). Regarding specific etiologies, both genetic disorders and brain injury were significantly increased in those with cognitive dysfunction. The 26 who exhibited decline over time were more likely to have abnormalities on neurological exam at diagnosis (P = 0.001) and generalized slowing on first EEG (P = 0.001). Of these, 6 had subsequently documented encephalopathy, 1 Ohtahara syndrome, 1 Lennox-Gastaut syndrome, and the other 4 with unspecified static encephalopathy. All children with unknown etiology who were developmentally on track at diagnosis remained cognitively normal at follow-up.
Epilepsy Outcome (Figure 3)
Figure 3. Seizure Frequency through Time.
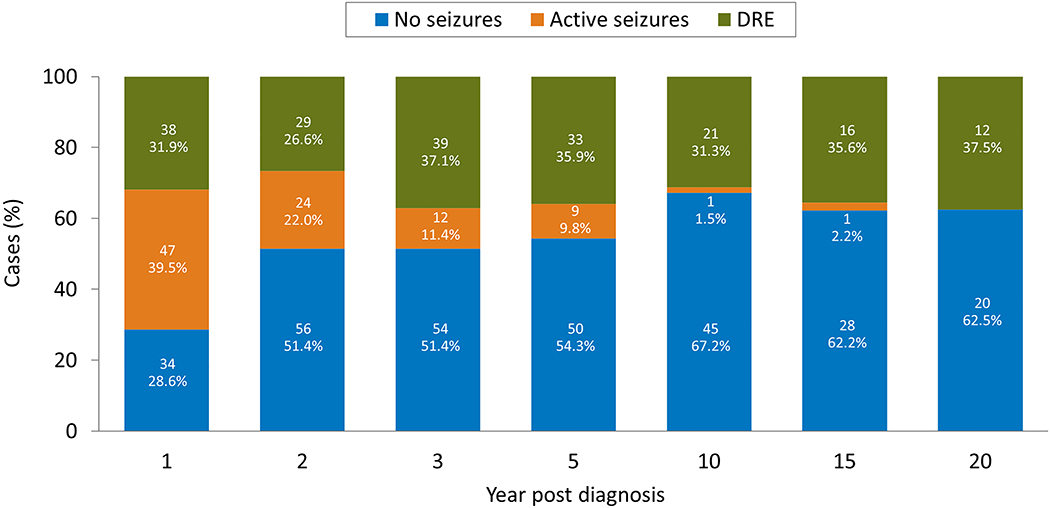
Cumulative percentages of seizure activity (Y-axis) is shown in years following epilepsy diagnosis along the X-axis. The cases that have been seizure free for 12 months, with or without ASMs, are designated “No Sz.” The orange bar shows cases that have on-going seizures (“Active Sz”) but have not reached the criteria for being considered drug-resistant. The remainder of cases are those with drug-resistant epilepsy (“DRE”), meaning ongoing seizures with 2 or more ASMs failed for either side effects of lack of efficacy. By 3 years, almost 90% of cases have either achieved excellent seizure control or become intractable.
Of the 107 patients (55 male, 52 female) with more than 24 months of follow-up, 38 (35.5%) had drug-resistant epilepsy at final follow-up; including 7 who underwent epilepsy surgery. Four of the 7 who underwent epilepsy surgery were seizure-free at last follow-up. A total of 41 (38.3%) were seizure-free and off ASMs for at least the previous 12 months and another 20 (18.7%) were seizure free but remained on ASMs. The remaining 8 had continuing seizures but did not meet criteria for drug-resistant epilepsy (Figure 3).
a. Drug-Resistant Epilepsy
Development of DRE was correlated with abnormal neurological exam at presentation (P < 0.001), any degree of cognitive impairment at onset (P < 0.001) and presence of epileptic spasms (P < 0.001). Background slowing on initial EEG (P = 0.007) and status epilepticus (P = 0.003) were also associated with drug-resistance at follow-up. The association between known etiology and development of drug-resistant epilepsy did not reach statistical significance (P = 0.08).
Seven children (6.5%) underwent epilepsy surgery; 2 of these had a repeat procedure due to seizure recurrence. Within the surgically managed group, 3 still had intractable seizures at follow-up; 2 out of the 4 without ongoing seizures had successfully discontinued ASMs without seizure recurrence for >12 months. These cases were all caused by focal structural alterations.
b. Seizure Freedom and Epilepsy Remission
Normal cognition (P < 0.01), normal neurological exam at onset (P < 0.001), normal MRI (P = 0.03), absence of generalized slowing on initial EEG (P = 0.01) and unknown etiology (P < 0.01) were significant predictors of achieving seizure freedom for at least 12 months at last follow-up. Of 20 infants who were developmentally normal at seizure onset and had no known etiology for their epilepsy, all were seizure-free for at least one year at last follow-up, and 19 (95%) had successfully weaned off all ASM.
Of cases that were seizure-free at last follow-up, normal exam (P = 0.01), normal cognition (P < 0.001), absence of structural findings on MRI (P < 0.001) and unknown etiology while developmentally normal (P = 0.004) were all significantly enriched in those that had weaned off ASMs entirely.
DISCUSSION
Infantile-onset, focal epilepsy is an important though understudied category of childhood epilepsy. In our dataset, it comprises 18% of all childhood epilepsy and is by far the most common cause of infantile epilepsy, making up 75% of epilepsy cases diagnosed before 24 months of age.
The outcome of focal epilepsy in infancy is often worrisome. Compared to the overall pediatric epilepsy population, we found that infants with focal-onset epilepsy had increased mortality, developmental delay, drug resistance, and likelihood of etiology identification. Over one third of our cohort developed drug resistant epilepsy compared to 22% in our study of focal epilepsy (excluding those with self-limited focal epilepsy syndromes) across the pediatric age range (12) and 13.8% in a prospective, community-based cohort of all children with epilepsy (10). Conversely, over half of infants became seizure-free by one year, and by 10 years, two-thirds achieved that outcome. Overall mortality in pediatric epilepsy is 228 per 100,000 person-years (11), significantly lower than in our study, where 12.0% of infants with focal onset epilepsy died over the course of follow-up. We identified that 23.9% of our infantile cohort had notable developmental delay, increased from 17.9% in pediatric focal epilepsy of all ages (12).
We found that initial clinical evaluation provided critically important information to inform long-term outcome. Key poor prognostic features include significant developmental delay at onset, abnormal neurological examination, presence of epileptic spasms and specific etiologies.
A specific etiology was found in two thirds of our cohort. This rate of identification is higher in infants compared to the wider focal pediatric epilepsy population, in whom we previously reported that 46% of children (age 1 month to 17 years) with focal seizures had identified etiology (12). In the current study, we found structural abnormalities were the most common cause, emphasizing the need for high-quality neuroimaging in infantile-onset focal epilepsy (13–15). Our detected rate of known etiologies is similar to a recent population-based study from Sweden (4) which enrolled all infants with epilepsy presenting in the first 2 years of life and found a known etiology in 59% (including structural in 34%, genetic in 15% and metabolic in 8.6%). The rate of structural etiologies in our cohort was higher (52%), likely as we included only those with focal onset seizures. It is probable that the rate of genetic etiologies in focal epilepsy is higher than our reported rate of 12.7% as only a minority of our cases underwent genetic testing. In a recent population-based study which evaluated yield of genetic testing in children with new-onset epilepsy before 36 months of age, Symonds et al. found genetic etiologies in 24% of cases (16). It is well recognized that genetic etiologies are important for both self-limited epilepsies in early life (i.e. PRRT2, SCN2A and KCNQ2 pathogenic variants) as well as for the developmental and epileptic encephalopathies (i.e. KCNT1, CDKL5 or STXBP1 pathogenic variants). Genetic testing should be strongly considered in all cases of early-onset epilepsy, given the high yield and prognostic and therapeutic implications associated with many of these genes. Identification of a reassuring genetic variant in an infant with focal epilepsy lacking any poor prognostic factors will confirm the diagnosis of a self-limited focal epilepsy and avoid the need for further extensive evaluations.
Infants with poor prognostic factors at presentation should be strongly considered for expedient referral to a comprehensive epilepsy center, as they have a high risk of developmental and epileptic encephalopathy, with drug resistant seizures and poor developmental outcome. Careful delineation of epilepsy syndrome and etiology can enhance the selection of most efficacious therapies. Early resective surgery is an important treatment for cases with a surgically-amenable lesion, and can result in both improved epilepsy and developmental outcomes (17–19). Increasingly, new and repurposed precision and other antiseizure therapies are being developed for specific syndromes or etiologies (20).
Early detection of epileptic spasms is of critical clinical importance, as decreased lead time to treatment has been associated with modest improvements in neurodevelopmental outcomes (21). Our data demonstrate that we can predict which children are at higher risk of developing spasms, specifically, those with younger age of onset and known etiology. In particular, 44.4% of cortical dysplasia cases went on to develop spasms, significantly higher than other groups. Median time to evolution of spasms was 5.3 months, and all who developed spasms did so within a year of diagnosis. We suggest that infants that present with focal seizures before 6 months of age, especially those with identified structural abnormalities should have close neurological follow-up and parents and primary care providers should be counseled to monitor closely for subtle, brief abnormal movements. Spasms are known to be associated with increased mortality (22), a finding which was consistent with our data as well.
Children with unknown etiology and typical development had excellent long-term outcomes. None of them evolved to spasms or expired during the study. These cases also had normal cognitive development. All cases with more than two years of follow-up had achieved seizure freedom at last follow-up, with 95% of these also having weaned off ASMs entirely.
As noted above, recent advances in genetic research and technology have accelerated our identification of epilepsy-associated genes (23, 24). The most significant weakness of our study is that genetic testing was done in only a minority of cases. Identifying a specific genetic etiology, when present, is key to select optimal treatment and provide families with the most up-to-date prognostic information.
Figure 4. Outcomes of Focal Infantile-Onset Epilepsy.
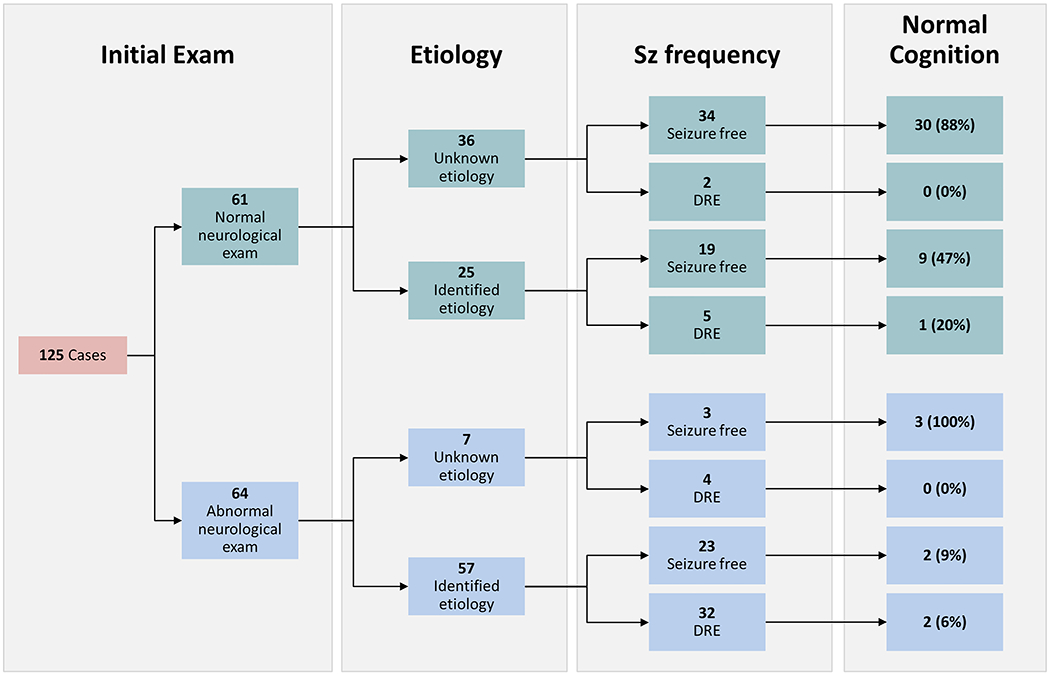
Chart tracks outcomes based on initial exam and etiology. Initial exam refers to overall neurological exam; 73.8% of those with normal exam were also cognitively normal, the remainder had only mild delay. Only 13.8% of those with abnormal initial exam were classified as developmentally normal, 44.6% had severe delay. Seizure frequency shows cases which had been seizure free for >12 months at last follow-up with or without ASMs and those with DRE. Any remaining cases had ongoing seizures with ASM not yet meeting criteria for drug-resistance (not shown). The percent of each group that was developmentally normal at last follow-up is also shown, the remainder of each group had at least some degree of delay; the highest rates of moderate/severe delay were in those cases with identified etiology.
Key Points.
Focal epilepsy accounts for 75% of all cases of epilepsy with onset prior to 24 months of age.
Of infants presenting with focal-onset seizures, 59.5% continue with this seizure type long-term
16.5% of infants presenting with focal onset seizures develop epileptic spasms after a median duration of 5.3 months
71% of infants who were developmentally normal at seizure onset remain that way long term
Drug-resistant epilepsy is seen in 35.5% of all focal epilepsy beginning prior to 24 months.
ACKNOWLEDGEMENTS
This study used the resources of the Rochester Epidemiology Project (REP) medical records-linkage system, which is supported by the National Institute on Aging (NIA; AG 058738), by the Mayo Clinic Research Committee, and by fees paid annually by REP users. The content of this article is solely the responsibility of the authors and does not represent the official views of the National Institutes of Health (NIH) or the Mayo Clinic.
Footnotes
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Disclosures
E Wirrell has served as a paid consultant for Encoded Therapeutics and Biomarin. She is the Editor-in-Chief of Epilepsy.com.
E Triplet, K Nickels, L Wong-Kisiel and A Fine have no conflicts of interest.
Contributor Information
Erin M Triplet, Mayo Clinic, Rochester MN.
Katherine Nickels, Mayo Clinic, Rochester MN.
Lily Wong-Kisiel, Mayo Clinic, Rochester MN.
Anthony Fine, Mayo Clinic, Rochester MN.
Elaine C Wirrell, Mayo Clinic, Rochester MN.
REFERENCES
- 1.Camfield CS, Camfield PR, Gordon K, Wirrell E, Dooley JM. Incidence of epilepsy in childhood and adolescence: a population-based study in Nova Scotia from 1977 to 1985. Epilepsia. 1996;37(1):19–23. [DOI] [PubMed] [Google Scholar]
- 2.Hauser WA, Annegers JF, Kurland LT. Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota: 1935-1984. Epilepsia. 1993;34(3):453–68. [DOI] [PubMed] [Google Scholar]
- 3.Olafsson E, Ludvigsson P, Gudmundsson G, Hesdorffer D, Kjartansson O, Hauser WA. Incidence of unprovoked seizures and epilepsy in Iceland and assessment of the epilepsy syndrome classification: a prospective study. Lancet Neurol. 2005;4(10):627–34. [DOI] [PubMed] [Google Scholar]
- 4.Stodberg T, Tomson T, Barbaro M, Stranneheim H, Anderlid BM, Carlsson S, et al. Epilepsy syndromes, etiologies, and the use of next-generation sequencing in epilepsy presenting in the first 2 years of life: A population-based study. Epilepsia. 2020;61(11):2486–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wirrell EC, Grossardt BR, Wong-Kisiel LC, Nickels KC. Incidence and classification of new-onset epilepsy and epilepsy syndromes in children in Olmsted County, Minnesota from 1980 to 2004: a population-based study. Epilepsy Res. 2011;95(1-2):110–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rocca WA, Yawn BP, St Sauver JL, Grossardt BR, Melton LJ, 3rd. History of the Rochester Epidemiology Project: half a century of medical records linkage in a US population. Mayo Clin Proc. 2012;87(12):1202–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.St Sauver JL, Grossardt BR, Yawn BP, Melton LJ 3rd, Pankratz JJ, Brue SM, et al. Data resource profile: the Rochester Epidemiology Project (REP) medical records-linkage system. Int J Epidemiol. 2012;41(6):1614–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.St Sauver JL, Grossardt BR, Yawn BP, Melton LJ, 3rd, Rocca WA. Use of a medical records linkage system to enumerate a dynamic population over time: the Rochester epidemiology project. Am J Epidemiol. 2011;173(9):1059–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kwan P, Arzimanoglou A, Berg AT, Brodie MJ, Allen Hauser W, Mathern G, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia. 2010;51(6):1069–77. [DOI] [PubMed] [Google Scholar]
- 10.Berg AT, Vickrey BG, Testa FM, Levy SR, Shinnar S, DiMario F, et al. How long does it take for epilepsy to become intractable? A prospective investigation. Ann Neurol. 2006;60(1):73–9. [DOI] [PubMed] [Google Scholar]
- 11.Berg AT, Nickels K, Wirrell EC, Geerts AT, Callenbach PM, Arts WF, et al. Mortality risks in new-onset childhood epilepsy. Pediatrics. 2013;132(1):124–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wirrell EC, Grossardt BR, So EL, Nickels KC. A population-based study of long-term outcomes of cryptogenic focal epilepsy in childhood: cryptogenic epilepsy is probably not symptomatic epilepsy. Epilepsia. 2011;52(4):738–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gaillard WD, Chiron C, Cross JH, Harvey AS, Kuzniecky R, Hertz-Pannier L, et al. Guidelines for imaging infants and children with recent-onset epilepsy. Epilepsia. 2009;50(9):2147–53. [DOI] [PubMed] [Google Scholar]
- 14.Hirtz D, Ashwal S, Berg A, Bettis D, Camfield C, Camfield P, et al. Practice parameter: evaluating a first nonfebrile seizure in children: report of the quality standards subcommittee of the American Academy of Neurology, The Child Neurology Society, and The American Epilepsy Society. Neurology. 2000;55(5):616–23. [DOI] [PubMed] [Google Scholar]
- 15.Hourani R, Nasreddine W, Dirani M, Hmaimess G, Sabbagh S, El Tourjuman O, et al. When Should a Brain MRI Be Performed in Children with New-Onset Seizures? Results of a Large Prospective Trial. AJNR Am J Neuroradiol. 2021;42(9):1695–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Symonds JD, Zuberi SM, Stewart K, McLellan A, O’Regan M, MacLeod S, et al. Incidence and phenotypes of childhood-onset genetic epilepsies: a prospective population-based national cohort. Brain. 2019;142(8):2303–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Braun KPJ. Influence of epilepsy surgery on developmental outcomes in children. Eur J Paediatr Neurol. 2020;24:40–2. [DOI] [PubMed] [Google Scholar]
- 18.Jonas R, Asarnow RF, LoPresti C, Yudovin S, Koh S, Wu JY, et al. Surgery for symptomatic infant-onset epileptic encephalopathy with and without infantile spasms. Neurology. 2005;64(4):746–50. [DOI] [PubMed] [Google Scholar]
- 19.Wilmshurst JM, Gaillard WD, Vinayan KP, Tsuchida TN, Plouin P, Van Bogaert P, et al. Summary of recommendations for the management of infantile seizures: Task Force Report for the ILAE Commission of Pediatrics. Epilepsia. 2015;56(8):1185–97. [DOI] [PubMed] [Google Scholar]
- 20.Johannessen Landmark C, Potschka H, Auvin S, Wilmshurst JM, Johannessen SI, Kasteleijn-Nolst Trenite D, et al. The role of new medical treatments for the management of developmental and epileptic encephalopathies: Novel concepts and results. Epilepsia. 2021;62(4):857–73. [DOI] [PubMed] [Google Scholar]
- 21.Widjaja E, Go C, McCoy B, Snead OC. Neurodevelopmental outcome of infantile spasms: A systematic review and meta-analysis. Epilepsy Res. 2015;109:155–62. [DOI] [PubMed] [Google Scholar]
- 22.Harini C, Nagarajan E, Bergin AM, Pearl P, Loddenkemper T, Takeoka M, et al. Mortality in infantile spasms: A hospital-based study. Epilepsia. 2020;61(4):702–13. [DOI] [PubMed] [Google Scholar]
- 23.Ellis CA, Petrovski S, Berkovic SF. Epilepsy genetics: clinical impacts and biological insights. Lancet Neurol. 2020;19(1):93–100. [DOI] [PubMed] [Google Scholar]
- 24.Scheffer IE. Epilepsy genetics revolutionizes clinical practice. Neuropediatrics. 2014;45(2):70–4. [DOI] [PubMed] [Google Scholar]