

Abstract
Background:
The locus coeruleus (LC) is one of the earliest brain regions to accumulate hyperphosphorylated tau, but a lack of animal models that recapitulate this pathology has hampered our understanding of its contributions to Alzheimer’s disease (AD) pathophysiology.
Objective:
We previously reported that TgF344-AD rats, which overexpress mutant human amyloid precursor protein and presenilin-1, accumulate early endogenous hyperphosphorylated tau in the LC. Here, we used TgF344-AD rats and a wild-type (WT) human tau virus to interrogate the effects of endogenous hyperphosphorylated rat tau and human tau in the LC on AD-related neuropathology and behavior.
Methods:
Two-month old TgF344-AD and WT rats received bilateral LC infusions of full-length WT human tau or mCherry control virus driven by the noradrenergic-specific PRSx8 promoter. Rats were subsequently assessed at 6 and 12 months for arousal (sleep latency), anxiety-like behavior (open field, elevated plus maze, novelty-suppressed feeding), passive coping (forced swim task), and learning and memory (Morris water maze and fear conditioning). Hippocampal microglia, astrocyte, and AD pathology were evaluated using immunohistochemistry.
Results:
In general, the effects of age were more pronounced than genotype or treatment; older rats displayed greater hippocampal pathology, took longer to fall asleep, had reduced locomotor activity, floated more, and had impaired cognition compared to younger animals. TgF344-AD rats showed increased anxiety-like behavior and impaired learning and memory. The tau virus had negligible influence on most measures.
Conclusion:
Effects of hyperphosphorylated tau on AD-like neuropathology and behavioral symptoms were subtle. Further investigation of different forms of tau is warranted.
Keywords: Alzheimer’s disease, locus coeruleus, tau, rat, transgenic, behavior, cognition
INTRODUCTION
Alzheimer’s disease (AD) is the most common neurodegenerative disorder in the world and is characterized by two protein aggregates: amyloid-β (Aβ) plaques and neurofibrillary tangles composed of hyperphosphorylated and misfolded tau. Initiation of AD has been attributed to Aβ [1], which subsequently leads to tau hyperphosphorylation [2–4], oxidative stress and mitochondrial dysfunction [5–9], and synaptic impairments [10–12]. However, antibody treatments targeting late-stage Aβ deposition have been largely unsuccessful and/or controversial, highlighting the pressing need to investigate other therapeutic avenues that focus on earlier stages of disease prior to catastrophic neuronal loss and significant cognitive impairment.
Neurofibrillary tangles (NFT) have garnered increasing attention in recent years because tau load is consistently reported as a better predictor of cognitive decline and neuronal degeneration compared with Aβ [13–15]. Although Aβ plaques appear prior to NFT, recent evidence places the deposition of “pretangle” hyperphosphorylated tau prior to both pathologies. Specifically, Braak and colleagues identified the locus coeruleus (LC) as the first brain region to accumulate hyperphosphorylated tau [16], a finding that has since been independently confirmed by other groups [17, 18]. The LC is the primary source of norepinephrine (NE) in the brain, which has potent anti-inflammatory/neuroprotective properties and regulates attention, arousal, and stress responses [19–22], all of which often go awry in AD [23–29]. Although tau pathology can cause cell death and there is evidence for some early reduction of LC volume (which likely corresponds to loss of proximal axons/dendrites) [18, 30–32], frank LC cell body degeneration is not evident until mid- to late-stages of AD [18, 33, 34]. Thus, detrimental effects of hyperphosphorylated tau pathology on LC function could persist for years or even decades, and may contribute to behavioral abnormalities in prodromal AD. Indeed, aggregation of hyperphosphorylated tau in the LC coincides with the emergence of non-cognitive behavioral impairments commonly observed in prodromal AD that are consistent with LC dysfunction [33, 35, 36]. Lower LC integrity as measured with MRI contrast has been linked with depression, sleep disorders, and impaired cognition at stages when hyperphosphorylated tau is expected in the LC but few other regions [37–41]. LC post-mortem integrity and tangle load [42, 43] and BOLD activation/functional connectivity [44–48] have also been implicated in cognitive reserve and/or behavioral abnormalities, highlighting a central role for the LC in their dysregulation. One study even identified the LC as the origin of functional connectivity deficits in AD patients [49]. Separately, tau abundance in cerebrospinal fluid and positron emission tomography levels have been associated with non-cognitive behavioral impairments in healthy adults and in populations at risk for developing AD [50–54]. Together, these data suggest an association between the development of LC tau neuropathology and behavioral impairments in early AD, but a causal link remains speculative due to limitations in the ability for imaging techniques to localize and quantify pathology in small nuclei such as the LC [55].
Animal models are valuable tools for establishing mechanistic relationships between pathology and behavior in AD. These models have provided crucial information regarding the consequences of LC perturbations in the context of disease that are largely congruent with human studies. For example, neurotoxic LC lesions exacerbate AD neuropathology, neurodegeneration, inflammation, lethality, and/or cognitive impairment in transgenic rodent AD models [56–58]. Even in the absence of experimental LC lesions, noradrenergic denervation, altered NE signaling, and LC cell loss have been reported in these models [59–63], but there are important limitations to consider. Amyloidosis models generally fail to recapitulate tau pathology in the LC. Even the P301S mouse, which does accumulate hyperphosphorylated tau in the LC, employs a ubiquitous promoter and a mutant form of tau that causes frontotemporal dementia that simultaneously produces tau pathology all over the brain and more closely mimics pure tauopathies [56, 64]. To isolate the effects of aberrant tau in the LC, we and others have used viral vectors to express various forms of tau specifically in the LC of mice and rats [32, 65], which triggers LC degeneration, transsynaptic tau spread, and cognitive deficits. These manipulations, however, were not performed in the context of Aβ, primarily utilized mutant forms of tau not known to occur in AD, and did not assess behaviors reminiscent of non-cognitive prodromal AD symptoms.
Our lab has previously demonstrated that the TgF344-AD rat, which overexpresses mutant human amyloid precursor protein and presenilin-1 (APP/PS1), develops endogenous, age-related accumulation of hyperphosphorylated tau in the LC prior to appreciable plaque or tangle pathology in the forebrain [63]. This model provides a unique resource for understanding the consequences of hyperphosphorylated tau in the LC in the earliest phases of AD because the tau pathology (1) is triggered by AD-causing mutant Aβ, (2) is endogenous (i.e. there is no human tau transgene), and (3) follows a spatial pattern of appearance reminiscent of human disease. Dysregulation of the noradrenergic system in TgF344-AD rats is evidenced by forebrain denervation and reduced NE levels in the absence of frank neuronal loss. Behavioral characterizations of these rats have shown impaired learning and memory, hyposmia, and anxiety-like phenotypes [63, 66–69], and we have shown that chemogenetic LC activation rescues reversal learning deficits [63]. However, few studies have comprehensively tested this strain on behaviors influenced by the LC along disease progression, beginning with the appearance of tau in the LC (~6 months) and at more advanced stages, when forebrain pathology is present (~12 months). Moreover, LC tau in this model is limited to a mild hyperphosphorylated state reminiscent of prodromal AD, limiting our understanding of how more mature forms of tau affect LC function.
The goals of this study were two-fold: (1) to expand the characterization of TgF344-AD rats across a battery of AD- and LC-relevant behaviors at time points when hyperphosphorylated tau in the LC is the only detectable AD-like neuropathology (6 months) and when Aβ and tau pathology is evident in the forebrain (12 months), and (2) to assess whether viral-mediated expression of wild-type (WT) human tau (hTau) in the LC that is prone to more advanced states of hyperphosphorylation exacerbates AD-like behavioral phenotypes and pathology.
MATERIALS AND METHODS
Animals:
A total of 81 TgF344-AD rats hemizygous for the APPsw/PS1ΔE9 transgene and WT littermates were housed in the animal facilities at Emory University. Rats were housed in groups of 2–3 on a 12-h light/dark cycle (lights on at 7:00 am) and given ad libitum access to food and water except during behavioral testing or otherwise specified. All procedures were approved by the Institutional Animal Care and Use Committee of Emory University.
Stereotaxic Injections:
At two months of age, rats underwent stereotaxic, sterile-tip surgery. Rats were anesthetized with isoflurane and given meloxicam or ketoprofen (2 mg/kg or 5 mg/kg, s.c., respectively) prior to receiving bilateral injections (1.3 ul/hemisphere) of virus expressing either full-length WT hTau under control of a noradrenergic-specific promoter (AAV9-PRSx8-hTau(WT)-WPRE-SV40) or mCherry control (AAV9-PRSx8-mCherry-WPRE-rBG) targeting the LC (AP: −3.8, ML: +/−1.2mm, DV: −7.0mm from lambda with a 15 degree downward head-tilt). Viral expression was driven by the noradrenergic-specific PRSx8 promoter [70]. Following the infusion, the injection syringe was left in place for 5 min before being moved dorsally 1mm and waiting 2 additional min to ensure diffusion of the virus at the site of injection. Behavioral assays were conducted 4 or 10 months following surgery (i.e. at 6 or 12 months of age).
Behavioral Assays
General:
A variety of assays were chosen to assess changes in behaviors commonly associated with prodromal and later stage AD including arousal (sleep latency), general activity levels (23-h locomotor activity), anxiety-like behaviors (open field, elevated plus maze, and novelty-suppressed feeding), passive coping that may reflect aspects of depression (forced swim test), and learning and memory (Morris water maze, cue- and context-dependent-fear conditioning). Rats were tested on behavioral tasks in the following order, from least to most stressful/invasive: sleep latency, 23-h locomotor activity, open field, elevated plus maze, forced swim task, Morris water maze, novelty-suppressed feeding, and fear conditioning. Rats were singly-housed 3–7 days before sleep latency and subsequently re-paired following completion of the 23-h locomotor test. Behavioral tests were separated by at least 1 day. All behavioral tests were conducted during the light phase under white light, unless otherwise reported.
Sleep latency:
Approximately 2–3 hrs following lights on, rats were gently handled for 2–3 s and placed back into their home cage for 4 h and recorded with video cameras mounted above the home cage. Latency to fall asleep was quantified as the duration of time (min) until the rat had its first sleep bout. Sleep bouts were defined as periods of time that rats exhibited a sleep position for at least 75% of a 10 min time period that began with at least 2 mins of uninterrupted sleep, as previously described [71], which our lab has validated with EEG [72].
23-h locomotor activity:
Rats were placed into a clean cage with free access to food and water for 23 h. Cages were surrounded with 11.5 × 20” Photobeam Activity System-Home Cage infrared beams to measure locomotor activity. Total number of ambulations were binned in 30-min intervals over the 23 h testing period. Testing for all cohorts began between 9:15–10:15 am. Analyses were performed on novelty-induced locomotion, defined as occurring within the first 30 min interval, locomotion during the light and dark phases, and total locomotion over the entire 23-h period.
Open field:
Rats were individually placed into the center of a 39” outer diameter circular arena with plastic floors and 12” gray plexiglass walls and allowed to freely explore the arena for 5 min. Activity was measured and recorded with a ceiling-mounted camera and TopScan software. Analyses focused on the duration of time spent in the inner 50% of the circle area and total distance travelled. Time spent in the inner circle is interpreted as lower anxiety-like behavior.
Elevated plus maze:
Rats were individually placed into the center of an elevated plus maze (Harvard Apparatus) with two arms enclosed by 50 cm high walls and two open arms with no walls (each 50 cm × 10 cm) and allowed to freely explore for 5 min. Activity was measured and recorded with a ceiling-mounted camera and TopScan software. Rats were tested during the light phase under red lighting. Analyses focused on the time spent in the open arms and total distance travelled. Time spent in the open arms is interpreted as lower anxiety-like behavior.
Forced swim test:
Rats were individually placed in a clear plastic cylinder filled two-thirds with water at 25°C for 10 min. Behavior was recorded by video cameras, and immobility, defined as when the rats made only those movements necessary to keep their heads above water, was manually scored using BORIS software [73]. Immobility is indicative of passive coping behavior and was calculated using the average of scores from two blinded experimenters.
Morris water maze:
Acquisition – Rats underwent four training sessions in which they were placed in a different quadrant of a 75” diameter plastic circular arena surrounded by several extra-maze cues and filled with opaque water, with a hidden platform just below the surface of the water. The platform remained in the same location throughout the acquisition sessions. Rats remained in the water until locating the platform or 60 s had elapsed, whichever came first. If 60 s elapsed, the rat was placed on the platform for 10 s. Rats completed four training sessions every day for four consecutive days. Behavior was tracked and recorded by a ceiling-mounted camera and TopScan software. Analyses were performed on distance travelled, latency to find the hidden platform, and total velocity averaged across training sessions for each day.
Acquisition Probe – The day following the final acquisition training day, rats were placed into the circular arena with no platform for 60 s. Analyses were performed on time spent and distance travelled in the quadrant of the maze where the platform was located during acquisition training.
Reversal Training – Rats underwent the same training they received during the acquisition training, but the hidden platform was moved to a different quadrant.
Reversal Probe – Following reversal training, rats underwent a reversal probe under identical conditions to the acquisition probe.
Novelty-suppressed feeding:
Rats were food-deprived for 24 h and then placed in a 24 × 12 × 12” rectangular arena that contained a pre-weighed food pellet at the center of the arena during the light phase under red lighting. The session ended when the rat picked up and bit into the pellet, or when 15 min elapsed, whichever came first. Rats were subsequently placed into their home cages with the same pellet. The primary outcome measure was latency to eat in the novel environment. Shorter latency to eat the pellet in a novel environment was interpreted as lower anxiety-like behavior. To control for the possibility that there were group differences in overall hunger levels or motivation to consume food, latency to eat in the home cage immediately following the experimental session and the amount of pellet consumed after 1 h was recorded.
Fear conditioning:
Testing was performed over three consecutive days. After each session, rats were returned to their home cage. Freezing levels for each session were recorded with FreezeFrame software.
Training – For the training phase, rats were placed in a sound-enclosed metal chamber illuminated with a single light with stainless-steel bar flooring capable of delivering footshocks. Rats were allowed to habituate to the chamber for 3 min which was followed by a 2000 Hz, 80 dB tone for 20 s that co-terminated with a 2 s, 0.5 mA shock. This procedure was repeated three times with inter-tone intervals set at 60 s. Chambers were cleaned with RB10 between sessions.
Contextual fear conditioning – To test memory for the shock-context pairing, rats were placed in the training context 24 h later for 8 min in the absence of tone or footshock.
Cued fear conditioning – To test memory for the association between the cue-shock pairing, 24 h after contextual fear conditioning rats were placed in a novel chamber with a different texture (metal grid flooring) and odor (chambers cleaned with 70% ethanol before and after each session). After 1 min, rats were played the same tone from the training sessions a total of three times, with 60 s intertrial intervals in the absence of footshocks.
Primary outcome measures were freezing levels throughout contextual and cued fear conditioning. Higher freezing levels are indicative of better memory for the association between the original context and/or cue-shock pairing.
Tissue Preparation & Immunohistochemistry:
Following the conclusion of behavioral testing, rats were overdosed with isoflurane and perfused with ice cold potassium phosphate-buffered saline (KPBS) followed by 4% paraformaldehyde. Brains were removed and stored overnight in 4% PFA before being transferred to 30% sucrose until sectioning. Brains were sectioned at 40 μm and stored in cryoprotectant until staining.
All antibody information can be found in Supplemental Table 1. Sections from the LC were stained with anti-norepinephrine transporter (NET) and anti-AT8 or dsRED antibodies to confirm viral expression. Sections from the hippocampus were stained for AT8 and 4G8 immunoreactivity to assess AD-related pathology. Hippocampal sections were stained with anti-ionized calcium binding adaptor molecule 1 (IBA1) and anti-glial fibrillary acidic protein (GFAP) antibodies to mark microglia and astrocytes, respectively. Hippocampal sections were also stained with anti-NET antibody to assess LC innervation. Briefly, sections were washed 3×5 min in 0.1 M PBS and then incubated for 1 h at room temperature in blocking solution (2% normal goat serum, 1% bovine serum albumin in 0.1% Triton PBS). Sections were then incubated for 24 h at 4 degrees Celsius in blocking solution with a 1:1000 dilution of primary antibody. Sections were subsequently washed 3×5 min in PBS and incubated for 1 h at room temperature in blocking solution with an appropriate secondary antibody (1:500 dilution of goat anti-rabbit 568 for IBA1 and GFAP in the hippocampus, and dsRED, goat anti-rabbit 633 for IBA1 and GFAP in the LC, goat anti-mouse 488 for AT8, 4G8, and NET). Sections were washed 3×5 min in PBS, mounted on SuperFrost+ slides, and coverslipped with DAPI+fluoromount.
Image Analysis:
For each rat, 2–3 brain sections were stained, analyzed, averaged for each animal, and subsequently averaged with animals of the same age, genotype, and virus to obtain group means. All image analysis was performed in ImageJ by blinded experimenters. In hippocampal regions a threshold was applied (Otsu), and a region of interest (ROI) of a standard size across sections (95,381 pixels2) was used to count AT8-, IBA1-, and GFAP-positive cells in CA1, CA3, and the dentate gyrus (DG) using the analyze particles function. Plaques were analyzed as a percent area in a hippocampal subregion following thresholding. NET images were processed and analyzed as previously described [63, 74]. Briefly, the FeatureJ Hessian plugin was used to process the image, selecting the largest eigenvalue of hessian tensor, absolute eigenvalue comparison, and a smoothing scale of 0.5. An ROI of standard size (2970 pixels2) was used to assess the mean and standard deviation background fluorescent intensity. Two lines that were perpendicular in orientation and of standard length (208.75 pixels) were used to find peaks using the plot profile and find peaks function, which corresponded to NET fibers. Peaks were defined as fluorescent intensity values greater than the mean plus two standard deviations above background.
Statistics and Analysis:
All data are reported as mean ± SEM and were analyzed using a 3-way ANOVA with age, genotype, and virus as factors. When interactions between the main effects were present, post-hoc analyses with Šidák correction were performed between groups differing by a single factor (age, genotype, or virus) with statistical significance set at α = 0.05. All statistical information regarding main and interaction effects for behavior and immunohistochemistry are available in Supplementary Tables 2 and 3, respectively. Meanwhile, significant effects are noted in-text and bolded in Supplementary Tables. Information for significant post-hoc comparisons, when appropriate, are also noted in-text and appear in Supplementary Table 4. Statistical analysis was performed, and data were graphed using Gaphpad Prism. For 23-h locomotion, Morris water maze, and fear conditioning, initial analyses considered interval/time period/training day as a factor. The area under the curve function in GraphPad prism was used to compare across ages with a 3-way ANOVA for novelty-induced, light, and dark phase in the 23-h locomotion paradigm, Morris water maze acquisition and reversal acquisition, and cued and contextual fear condition training and test days.
RESULTS
Confirmation of viral expression:
We confirmed robust expression of WT hTau and the mCherry control virus in the LC of WT and TgF344-AD rats at 6 and 12 months of age (Fig. 1). Rats that did not show mCherry expression in either hemisphere were categorized as control animals. Rats that demonstrated unilateral expression of WT hTau animals were designated as such, and there were no animals that did not have at least unilateral expression of WT hTau. There was a main effect of genotype of AT8 fluorescence levels, where WT hTau injected TgF344-AD rats had elevated AT8 staining at both ages (Supplemental Fig. 1). As previously reported [63], the LC of mCherry-infected TgF344-AD rats did not stain positive utilizing the AT8 antibody, but robust AT8 immunoreactivity was observed in both WT and TgF344-AD rats that received the WT hTau virus (Fig. 1).
Fig. 1.
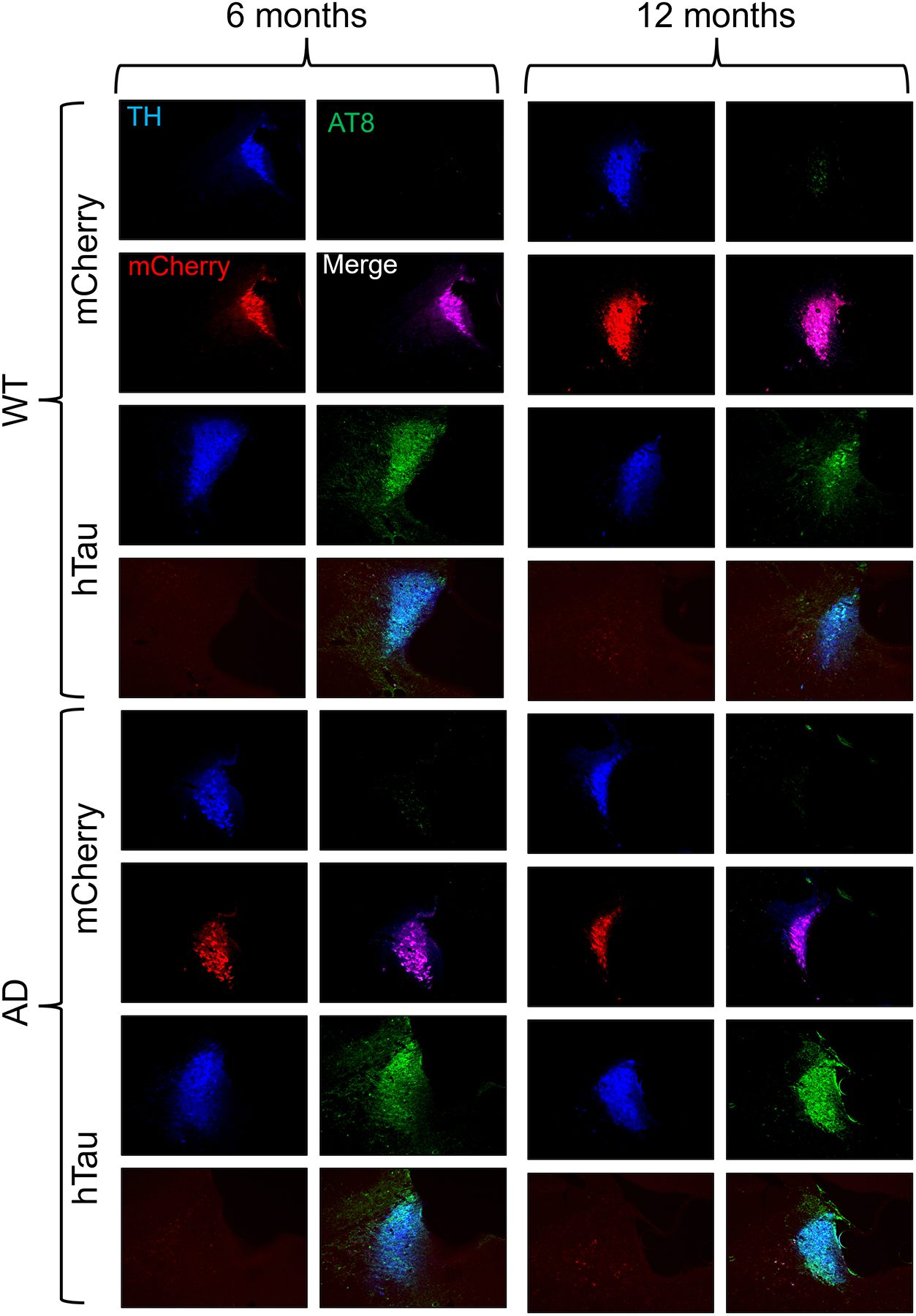
Verification of viral-mediated hTau and mCherry expression in the rat LC. Wild-type (WT) and TgF344-AD (AD) rats were injected bilaterally into the LC with AAV-PRSx8-hTau (hTau) or AAV-PRSx8-mCherry (mCherry) at 2 months of age and assessed for hTau and mCherry expression by immunohistochemistry at 6 months or 12 months. Shown are representative 10x immunofluorescent images for the LC marker tyrosine hydroxylase (TH; blue), the hyperphosphorylated tau marker AT8 (green), mCherry (red), and merged signals.
General Arousal and Locomotion
Sleep Latency:
Sleep disturbances are common in AD, and often emerge coincident with tau pathology in the LC [24–26, 35]. We first sought to determine whether age, genotype, virus, or their interactions influenced wakefulness/arousal by measuring latency to fall asleep after gentle handling, and found a main effect of age, where older animals took longer to fall asleep (Fig. 2A). There was also an age × genotype × virus interaction (Fig. 2A); at 12 months, hTau virus appeared to increase sleep latency in WT rats and decrease sleep latency in TgF344-AD rats compared to their mCherry-expressing counterparts, but no post hoc tests were significant.
Fig. 2.
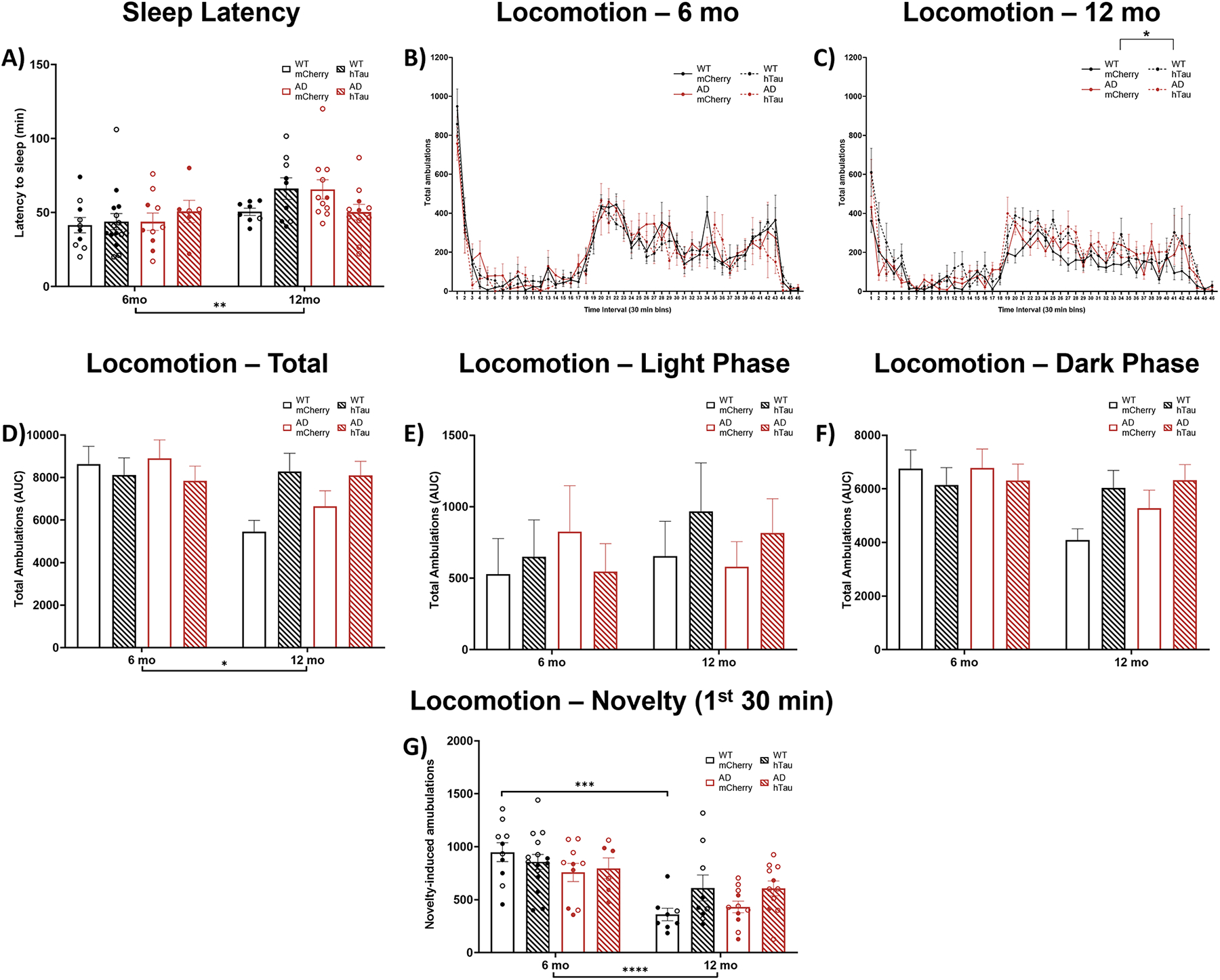
Assessment of arousal and locomotor activity. Wild-type (WT, black) and TgF344-AD (AD, red) rats were injected bilaterally into the LC with AAV-PRSx8-hTau (hTau, hashed) or AAV-PRSx8-mCherry (mCherry, solid) at 2 months of age, and assessed for sleep latency and locomotor activity at 6 months or 12 months. N = 6–15 per group. Shown are latency to fall asleep after gentle handling (A), time course of 23-h locomotor activity in 6 (B) and 12 (C) month rats, total ambulations (D), light phase ambulations (E), dark phase ambulations (F), and novelty-induced ambulations during the first 30 min of the test (G). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Males are represented by closed circles, females by open circles.
23-h locomotion:
We next used chambers equipped with photobeams to assess overall arousal and locomotor activity patterns over a full day. There was a main effect of time on locomotor activity in both 6- and 12-month rats, indicating that rats had normal circadian rhythms across the light/dark cycle. While there were no main effects of genotype, virus, or an interaction on ambulations in 6-month animals (Fig. 2B), we did detect a main effect of virus and a virus × time interaction on locomotor activity in 12-month animals (Fig. 2C). We next performed an area under the curve analysis to include age as a variable. A main effect of age and an age × virus interaction was apparent for locomotion over the full 23-h period (Fig. 2D). Parsing the data by light/dark phase, there were no main effects during the light period (Fig. 2 E), but there was a main effect of age and an age × virus interaction during the dark period (Fig. 2F). While no comparisons survived post-hoc correction, there was a consistent pattern for 12-month mCherry rats of both genotypes to show decreased locomotor activity compared with 6-month animals (Fig. 2D), whereas 12-month hTau rats either maintained or even increased ambulations compared to the younger rats. Finally, we assessed novelty-induced locomotion (Fig. 2G), defined as the number of amublations within the first 30 min following placement in the chambers, and found a main effect of age and an age × virus interaction. Older rats generally moved less than younger animals, but only WT mCherry-injected rats met statistical significance in post-hoc analysis.
Anxiety- and active/passive coping-like behavior
Open field:
We investigated anxiety-like behavior, which can be induced by augmenting LC activity [75–79], using a battery of canonical tests beginning with the open field test. There was a significant main effect of genotype on percent of time spent in the inner 50% of an open field, which was primarily driven by younger TgF344-AD rats (Fig. 3A), indicative of increased anxiety-like behavior [80]. There was also a significant effect of age on total distance traveled in the open field, where older animals moved less compared with their younger counterparts (Fig. 3B), similar to results we obtained in the locomotor activity experiment (Fig. 2).
Fig. 3.
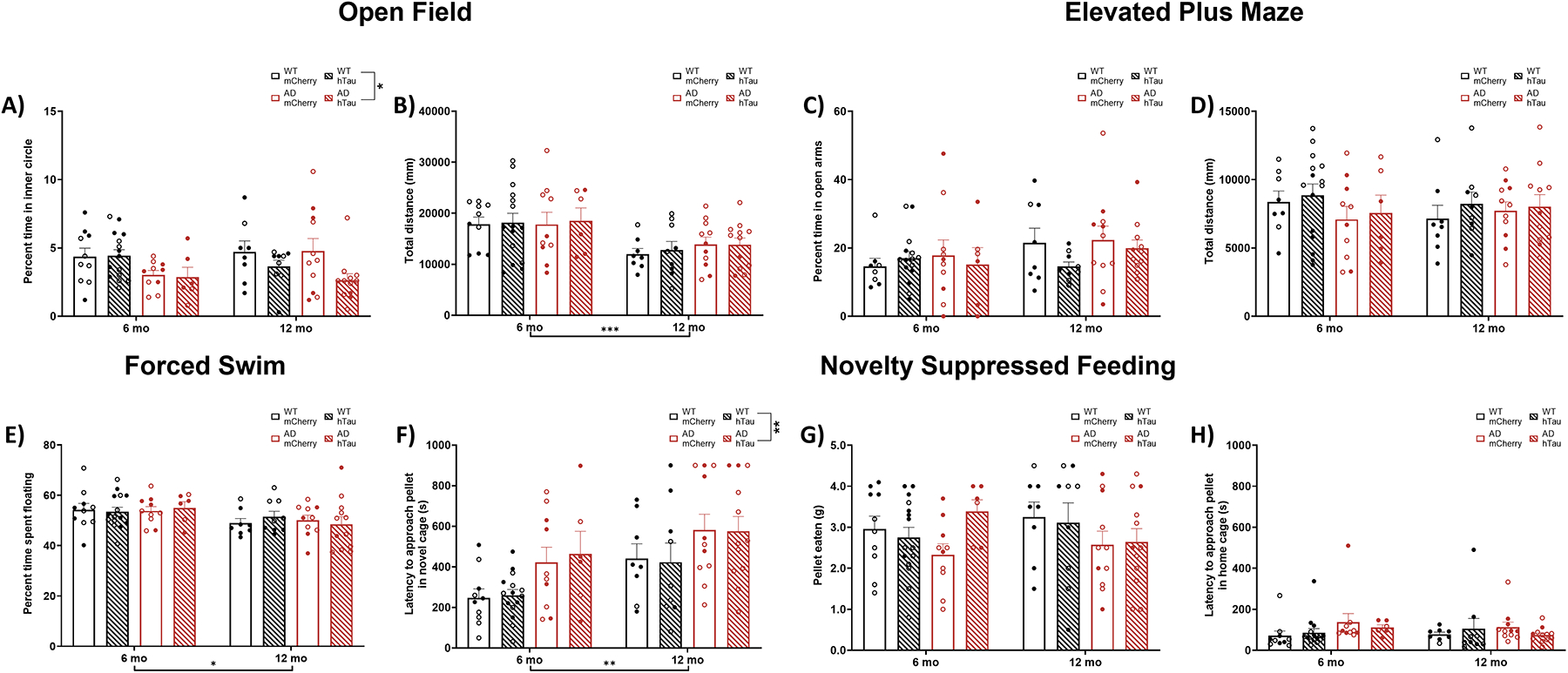
Assessment of anxiety-like behaviors and coping strategies. Wild-type (WT, black) and TgF344-AD (AD, red) rats were injected bilaterally into the LC with AAV-PRSx8-hTau (hTau, hashed bars) or AAV-PRSx8-mCherry (mCherry, solid) at 2 months of age, and assessed for open field, elevated plus maze, novelty-suppressed feeding, and forced swim at 6 months or 12 months. N = 6–15 per group. Shown is percent time in the inner portion (A) and total distance travelled (B) in the open field test, percent time in the open arms (C) and total distance travelled (D) in the elevated plus maze, latency to eat in a novel (E) or home/familiar (F) cage and amount of food consumed (G) in the novelty-suppressed feeding test, and time spent floating (H) in the forced swim test. *p<0.05, **p<0.01, ***p<0.001. Males are represented by closed circles, females by open circles.
Elevated plus maze:
We next tested rats on the elevated plus maze. There were no main effects on percent time spent in the open arms (Fig. 3C) or distance traveled (Fig. 3D).
Novelty-suppressed feeding:
Novelty-suppressed feeding (NSF) pits a hungry animal’s appetitive drive against its fear of exposure in unfamiliar open spaces, and longer latencies to eat are interpreted as increased anxiety-like behavior [81, 82]. Importantly, our lab has recently shown this task to be especially sensitive to dysregulation of LC-NE transmission [82]. There were significant main effects of age and genotype, where it took longer for older and TgF344-AD animals to eat the food pellet in a novel cage (Fig. 3E). To control for possible differences in hunger, we recorded the latency to eat a food pellet in the home cage (Fig. 3F) and amount of pellet eaten within 1 h (Fig. 3G). There were no main effects of age, genotype, or virus on either measure. These data are consistent with increased anxiety-like behavior in TgF344-AD rats and in older animals in general.
Forced swim test:
The forced swim test is thought to reflect passive (floating) vs active (struggling or swimming) behavior under conditions of inescapable stress [83, 84]. There were no main effects on percent of time struggling or swimming in the forced swim test (data not shown). There was a significant effect of age on percent of time spent floating whereby older animals floated less than younger animals (Fig. 3H).
Learning and Memory
Morris water maze:
We have previously reported that 6- and 16-month TgF344-AD rats exhibit modest deficits in acquisition of spatial learning in the Morris water maze but have profound impairments in reversal learning [63]. We again employed this task to assess hippocampal-dependent learning and memory in WT and TgF344-AD rats with and without hTau expression in the LC. During acquisition, a main effect of day on distance travelled to find the hidden platform was apparent in 6- (Fig. 4A) and 12-month animals (Fig. 4B), indicating that animals of both ages were able to learn the location of the hidden platform with training. 6-month animals also displayed a significant effect of genotype, with impaired learning in TgF344-AD rats (Fig. 4A). For 12-month animals, a main effect of virus indicated that hTau expression was associated with reduced performance (Fig. 4B). Collapsing across training days and factoring in age revealed no main effects of age, genotype, virus, or interactions on distance travelled (Fig. 4C). For the probe trial, there was a main effect of age, where older animals spent less time in the target quadrant compared with younger animals (Fig. 4D).
Fig. 4.
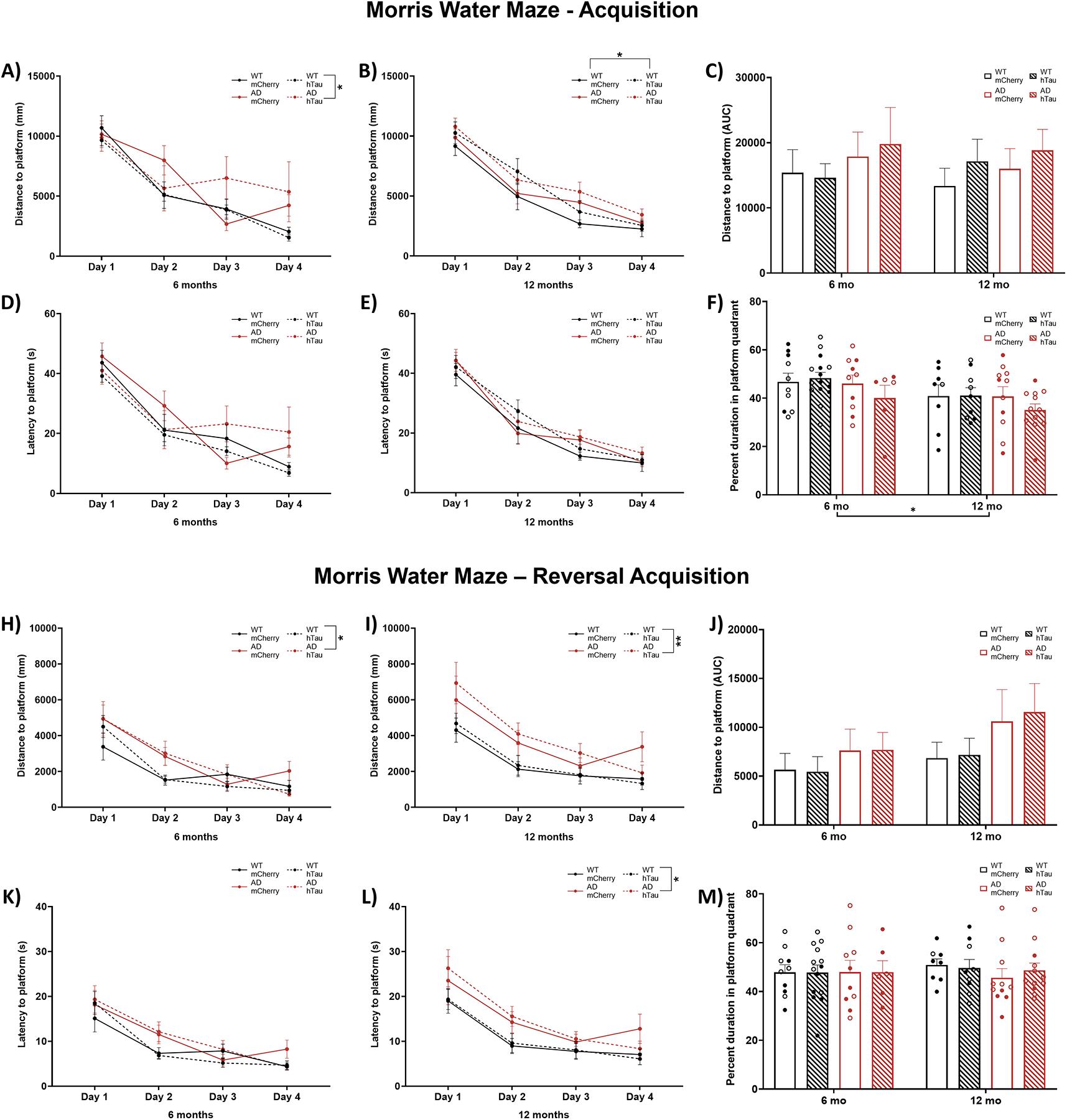
Assessment of hippocampal-dependent spatial learning and memory in the Morris water maze. Wild-type (WT, black) and TgF344-AD (AD, red) rats were injected bilaterally into the LC with AAV-PRSx8-hTau (hTau, hashed) or AAV-PRSx8-mCherry (mCherry, solid) at 2 months of age, and assessed for performance in the Morris water maze at 6 months or 12 months. N = 6–15 per group. Shown are total distance to find the hidden platform by day at 6 months (A) and 12 months (B), total area under the curve distance collapsed across days at both ages (C), percent time spent in target quadrant during the probe trial (D), total distance during reversal learning at 6 months (E) and 12 months (F), total area under the curve distance during reversal learning collapsed across days at both ages (G), and percent time spent in target quadrant during the probe trial after reversal learning (H). *p<0.05, **p<0.01. Males are represented by closed circles, females by open circles.
For reversal acquisition, main effects of day were again present in both 6- and 12-month animals on distance travelled to find the hidden platform (Fig. 4E & F), with rats improving over training. Consistent with our previous study, reversal learning was impaired in TgF344-AD rats at both 6 (Fig. 4E) and 12 months (Fig. 4F). No differences were observed after collapsing days (Fig. 4G) or during the the probe trial (Fig. 4H).
Fear conditioning:
To determine whether the impairment in spatial cognition we observed in the TgF344-AD rats in the Morris water maze extended to associative learning and memory, we performed cued and contextual fear conditioning experiments. During shock-tone pairing, a main effect of interval was apparent for both 6- and 12- month rats (Fig. 5A & B), demonstrating that all animals learned to associate the application of a tone with shock. An effect of genotype and a genotype × interval interaction were also apparent in 12-month animals, with TgF344-AD animals freezing less during the tone/shock pairings compared with WT animals. No post-hoc tests were significant, and no differences were apparent when groups were compared across age (Fig. 5C). During subsequent exposure to the shock-paired context in the absence of the tone, there was a main effect of interval in both 6- and 12-month rats (Fig. 5D & E). Likewise, there was a main effect of interval present in 6- and 12-month rats when presented with the shock-paired tone cue in a novel environment (Fig. 5G & H). These results demonstrate maintenance of the association between context/cue and shock regardless of genotype or virus group, and there were no other main effects on freezing behavior to either the context (Fig. 5D & E) or cue (Fig. 5G & H). Area under the curve analysis indicated a significant effect of age on freezing, where older animals froze less than younger ones under both context (Fig. 5F) and cue (Fig. 5I) conditions.
Fig. 5.
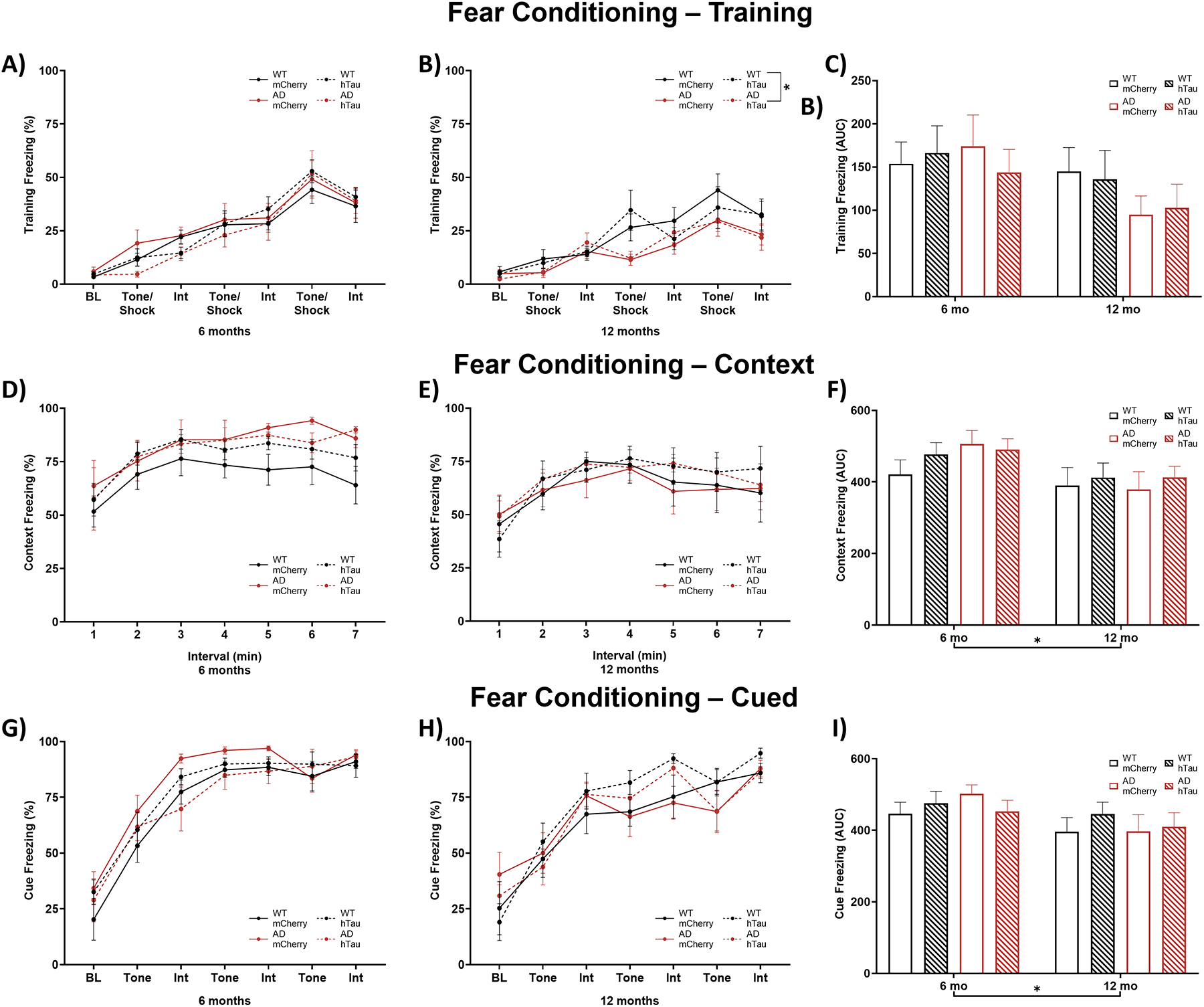
Assessment of contextual and cued fear conditioning. Wild-type (WT, black) and TgF344-AD (AD, red) rats were injected bilaterally into the LC with AAV-PRSx8-hTau (hTau, hashed) or AAV-PRSx8-mCherry (mCherry, solid) at 2 months of age, and assessed for contextual and cued fear conditioning at 6 months or 12 months. N = 6–15 per group. Shown is % freezing during shock-tone pairing in 6-month (A) and 12-month (B) rats, area under the curve collapsed across both ages (C), during subsequent context exposure in 6-month (D) and 12-month (E) rats, area under the curve collapsed across both ages (F), during subsequent cue exposure in 6-month (G) and 12-month (H) rats, and area under the curve collapsed across both ages (I) BL, baseline; Int, interval. *p<0.05.
Hippocampus Pathology:
AD-like neuropathology has previously been reported in the hippocampus of TgF344-AD rats [63, 66]. To expand on these analyses and determine the effect of hTau expression in the LC, we performed immunohistochemistry for Aβ (4G8), hyperphosphorylated tau (AT8), and neuroinflammation (GFAP and IBA1). Representative images of amyloid pathology in the DG, CA3, and CA1 are shown in Fig. 6A and in Supplemental Fig. 2A & B. There were main effects of age, genotype, and an age × genotype interaction on the percentage of plaque-positive area in all three hippocampal subregions (Fig. 6B–D). Post-hoc analyses indicated 12-month TgF344-AD rats had elevated plaque burden when compared with 6-month TgF344-AD rats and age- and virus-matched WT littermates across all hippocampal subregions. There were no effects of hTau expression on plaque pathology across age or genotype.
Fig. 6.
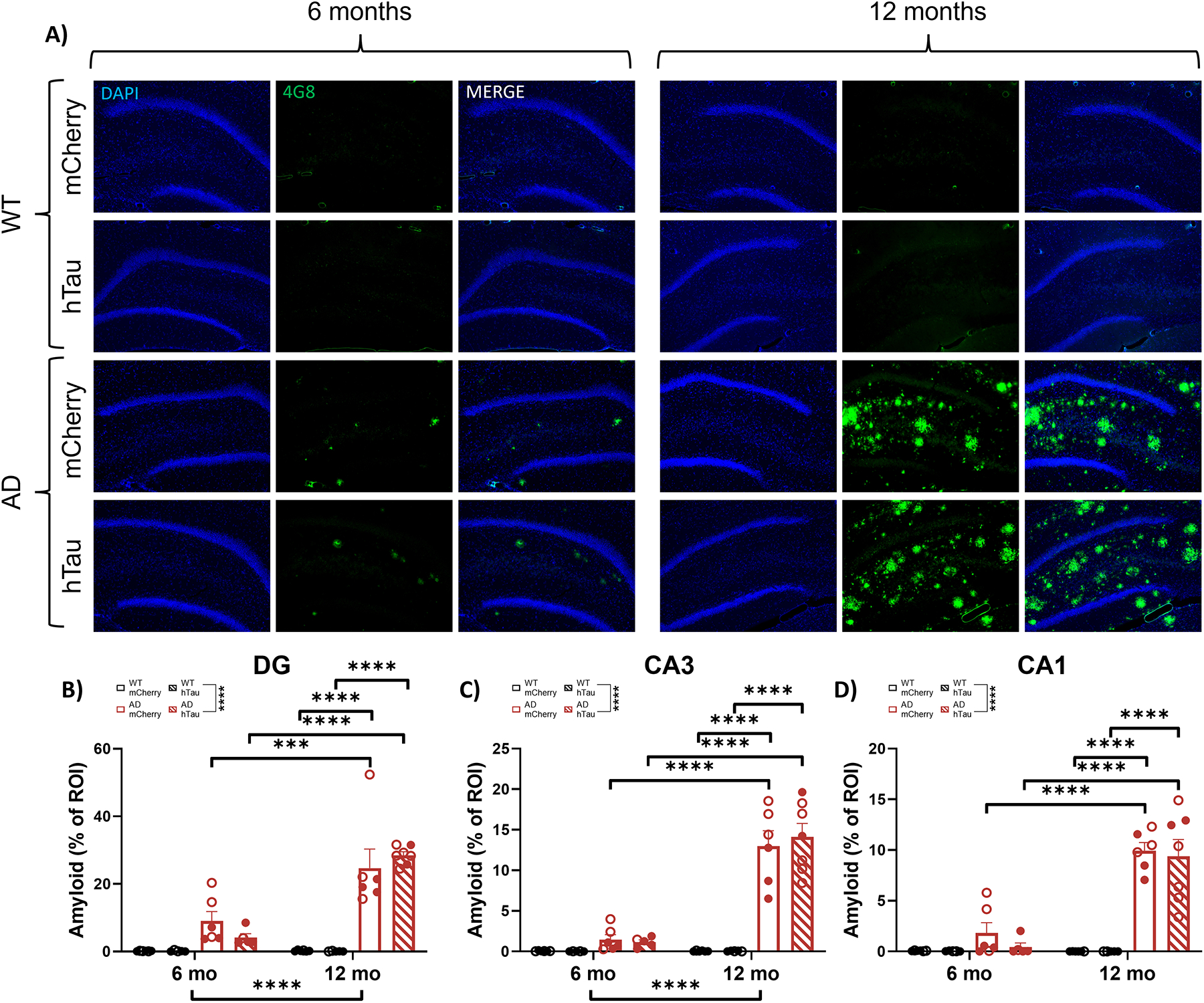
Amyloid pathology in the hippocampus. Wild-type (WT, black) and TgF344-AD (AD, red) rats were injected bilaterally into the LC with AAV-PRSx8-hTau (hTau, hashed) or AAV-PRSx8-mCherry (mCherry, solid) at 2 months of age, and assessed for 4G8 (i.e., Aβ) immunoreactivity at 6 months or 12 months. N = 5–7 per group. Shown are representative images of amyloid pathology in the DG subregion of the hippocampus (A), and quantification of amyloid pathology in the DG (B), CA3 (C), and CA1 (D) expressed as % region of interest (ROI). Main images and insets taken at 10x and 40x magnification, respectively. ***p<0.001, ****p<0.0001. Males are represented by closed circles, females by open circles.
To assess potential contribution of hTau expression in the LC to hippocampal tau pathology, AT8 was used to visualize hyperphosphorylated tau. Representative images of AT8 pathology in the DG CA3, and CA1 are shown in Fig. 7A and in Supplemental Fig. 3A & B. There was a main effect of genotype in the DG, where TgF344-AD rats had higher levels of hyperphosphorylated tau pathology compared with their WT littermates (Fig. 7B), as well as an age × genotype interaction in CA3, where older TgF344-AD rats had elevated AT8 staining compared to their WT littermates (Fig. 7C). However, for CA3, no significant differences were evident during post-hoc comparisons. In CA1, there was a main effect of age and an age × genotype interaction (Fig. 7D). Post-hoc comparisons revealed increased AT8+ cells in 12-month mCherry injected TgF344-AD animals compared with their 6-month TgF344-AD counterparts. Similar to plaque pathology, there were no effects of hTau expression on AT8 immunoreactivity across age or genotype.
Figure 7.
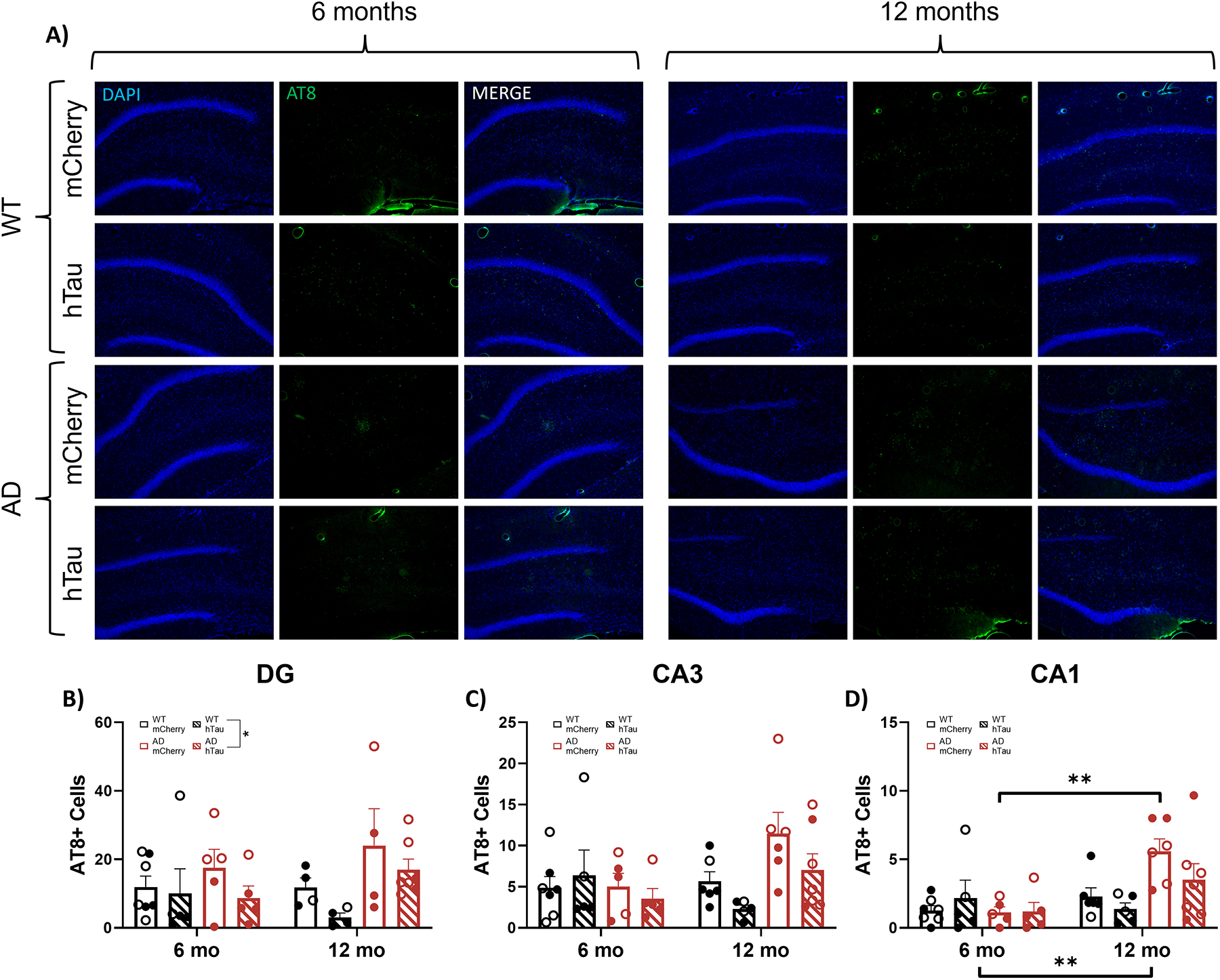
Assessment of hyperphosphorylated tau. Wild-type (WT, black) and TgF344-AD (AD, red) rats were injected bilaterally into the LC with AAV-PRSx8-hTau (hTau, hashed) or AAV-PRSx8-mCherry (mCherry, solid) at 2 months of age, and assessed for AT8 (i.e., hTau) immunoreactivity at 6 months or 12 months. N = 4–7 per group. Shown are representative images of hyperphosphorylated tau pathology in the DG subregion of the hippocampus (A), and quantification of hyperphosphorylated tau pathology in the DG (B), CA3 (C), and CA1 (D) expressed as the number of AT8+ cells. Main images and insets taken at 10x and 40x magnification, respectively. *p<0.05, **p<0.01. Males are represented by closed circles, females by open circles.
We next assessed neuroinflammatory astrocyte (GFAP) and microglia (IBA1) markers that are noted in AD. Representative images of GFAP staining in the DG, CA3, and CA1 are shown in Fig. 8A and in Supplemental Fig. 4A & B. There was a main effect of age, genotype, and an age × genotype interaction on number of GFAP+ cells in the DG (Fig. 8B), CA3 (Fig. 8C), and CA1 (Fig. 8D). Post-hoc analysis of the DG revealed elevated GFAP+ cells in 12-month mCherry- and hTau-injected TgF344-AD rats compared with their 6-month counterparts. In addition, 12-month mCherry- and hTau-injected TgF344-AD rats had elevated GFAP+ cells in the DG compared with their WT age-matched littermates. Similarly, 12-month mCherry- and hTau-injected TgF344-AD rats showed increased GFAP+ cells in CA3 compared with 6-month animals. 12-month mCherry- and hTau-injected TgF344-AD rats also demonstrated elevated GFAP+ cells in the CA3 compared with WT age-matched littermates. Within CA1, 12-month mCherry injected TgF344-AD rats had more GFAP+ cells compared with 6-month TgF344-AD rats and 12-month WT controls.
Fig. 8.
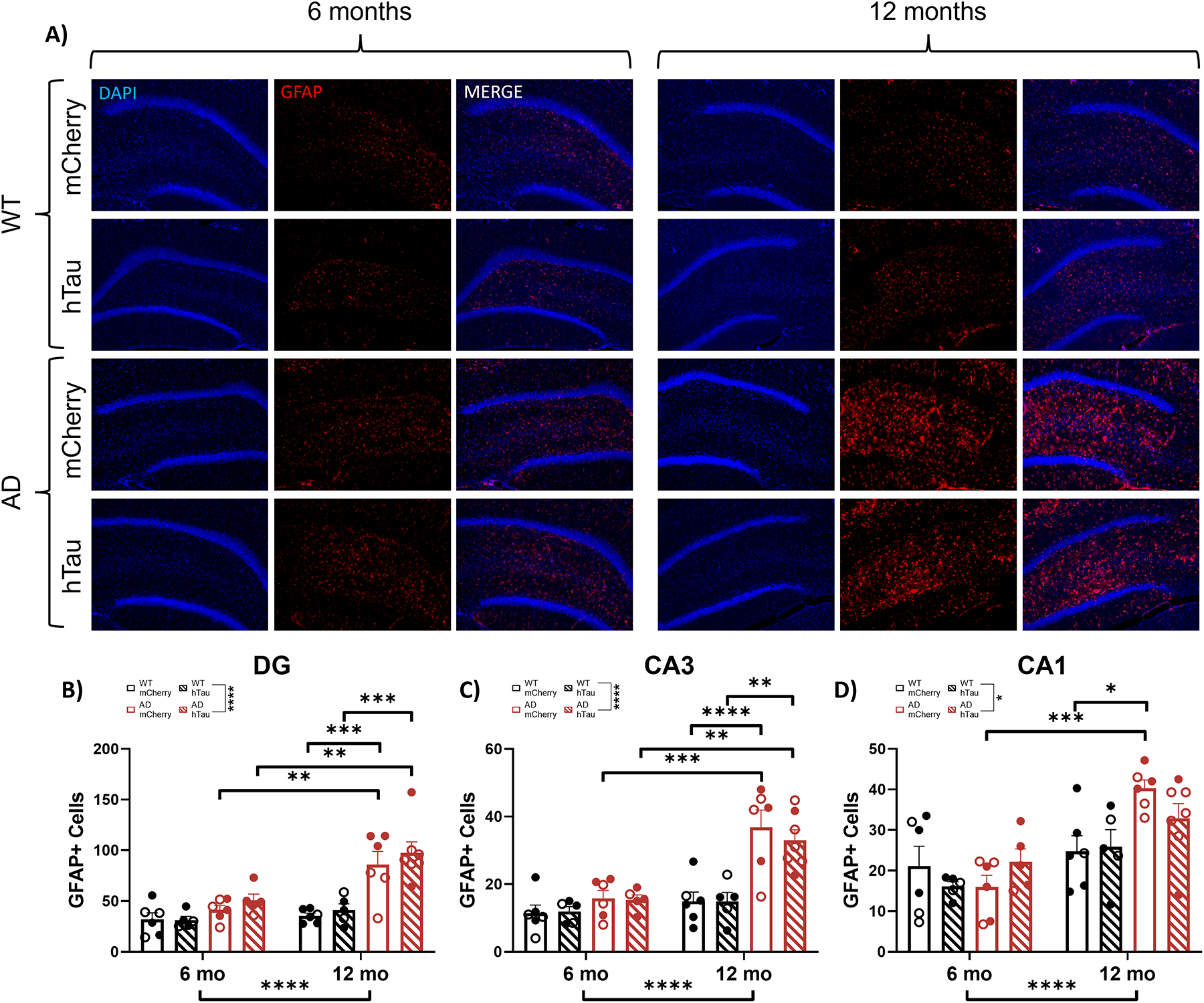
Assessment of astrocyte inflammation. Wild-type (WT, black) and TgF344-AD (AD, red) rats were injected bilaterally into the LC with AAV-PRSx8-hTau (hTau, hashed) or AAV-PRSx8-mCherry (mCherry, solid) at 2 months of age, and assessed for GFAP immunoreactivity at 6 months or 12 months. N = 5–7 per group. Shown are representative images of astrocyte inflammation in the DG subregion of the hippocampus (A), and quantification of astrocyte inflammation in the DG (B), CA3 (C), and CA1 (D) expressed as the number of GFAP+ cells. Main images and insets taken at 10x and 40x magnification, respectively. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Males are represented by closed circles, females by closed circles.
Representative images of IBA1 staining in the DG, CA3, and CA1 are shown in Fig. 9A and in Supplemental Fig. 5A and B. There was a main effect of genotype in the DG, where transgenic animals had more IBA1+ cells than their WT littermates (Fig. 9B). No main effects of age, genotype, or virus were observed on IBA1+ cells in CA3 (Fig. 9C). There was a main effect of age on IBA1+ cells in CA1 (Fig. 9D).
Fig. 9.
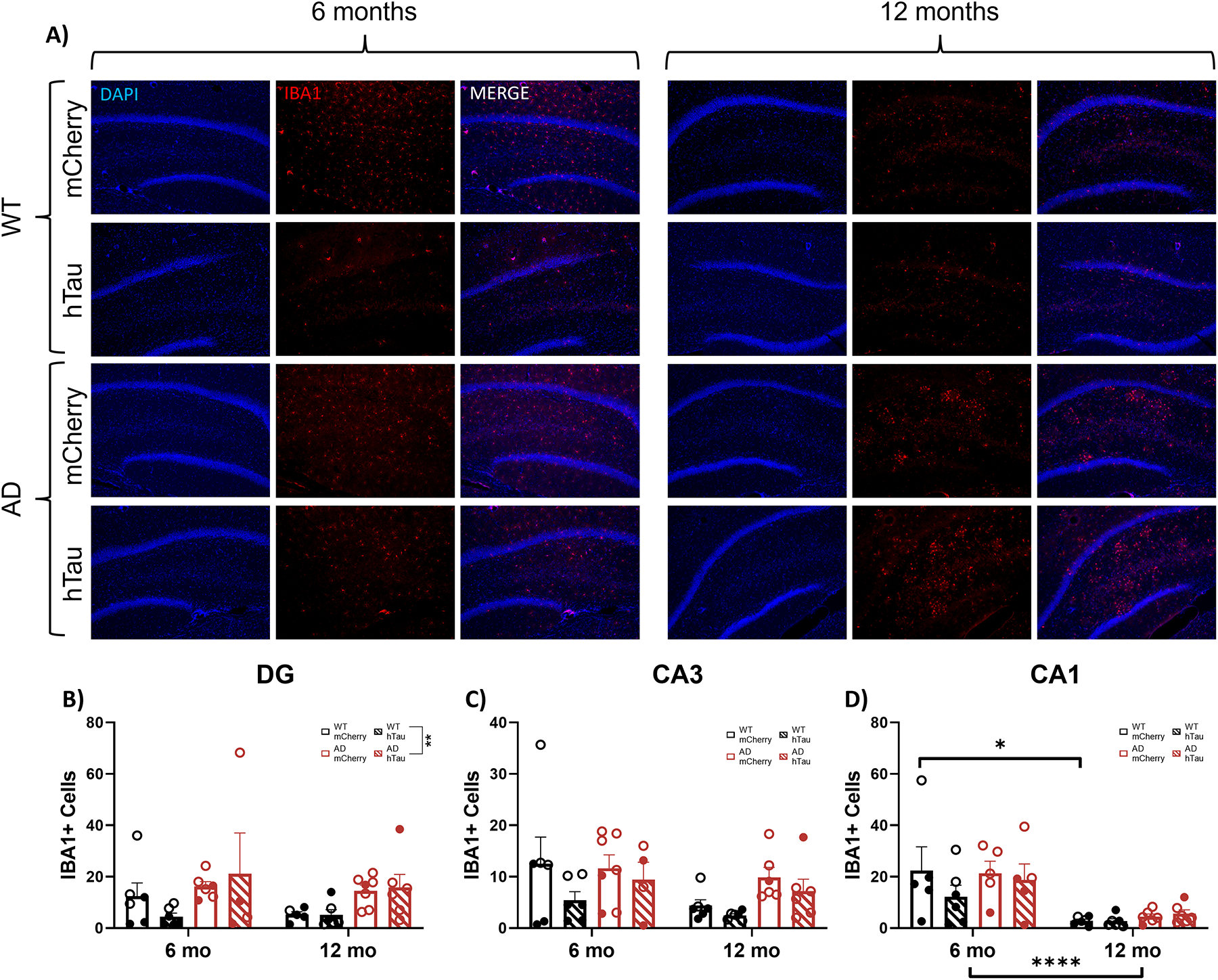
Assessment of microglial inflammation. Wild-type (WT, black) and TgF344-AD (AD, red) rats were injected bilaterally into the LC with AAV-PRSx8-hTau (hTau, hashed) or AAV-PRSx8-mCherry (mCherry, solid) at 2 months of age, and assessed for IBA1 immunoreactivity at 6 months or 12 months. N = 4–7 per group. Shown are representative images of microglial inflammation in the DG subregion of the hippocampus (A), and quantification of astrocyte inflammation in the DG (B), CA3 (C), and CA1 (D) expressed as the number of IBA1+ cells. Main images and insets taken at 10x and 40x magnification, respectively. Males are represented by closed circles, females by open circles.
Hippocampus NE Innervation:
Noradrenergic projections from the LC to the hippocampus, particularly the DG, deteriorate over time in TgF344-AD rats, which contributes to cognitive impairment [63, 85]. Here, we used NET+ fiber density to assess noradrenergic innervation to the distinct hippocampal subfields. Representative images of NET staining in the DG, CA3, and CA1 are shown in Fig. 10A and in Supplemental Fig. 6A & B. There was a significant effect of genotype in the DG (Fig. 10B). In CA3, there was a main effect of age, where older animals generally had lower NET fiber density compared to younger animals (Fig. 10C), while there were no main effects in CA1 (Fig. 10D). These data confirm the loss of LC innervation to the hippocampus in TgF344-AD rats, with the DG exhibiting the highest susceptibility.
Fig. 10.
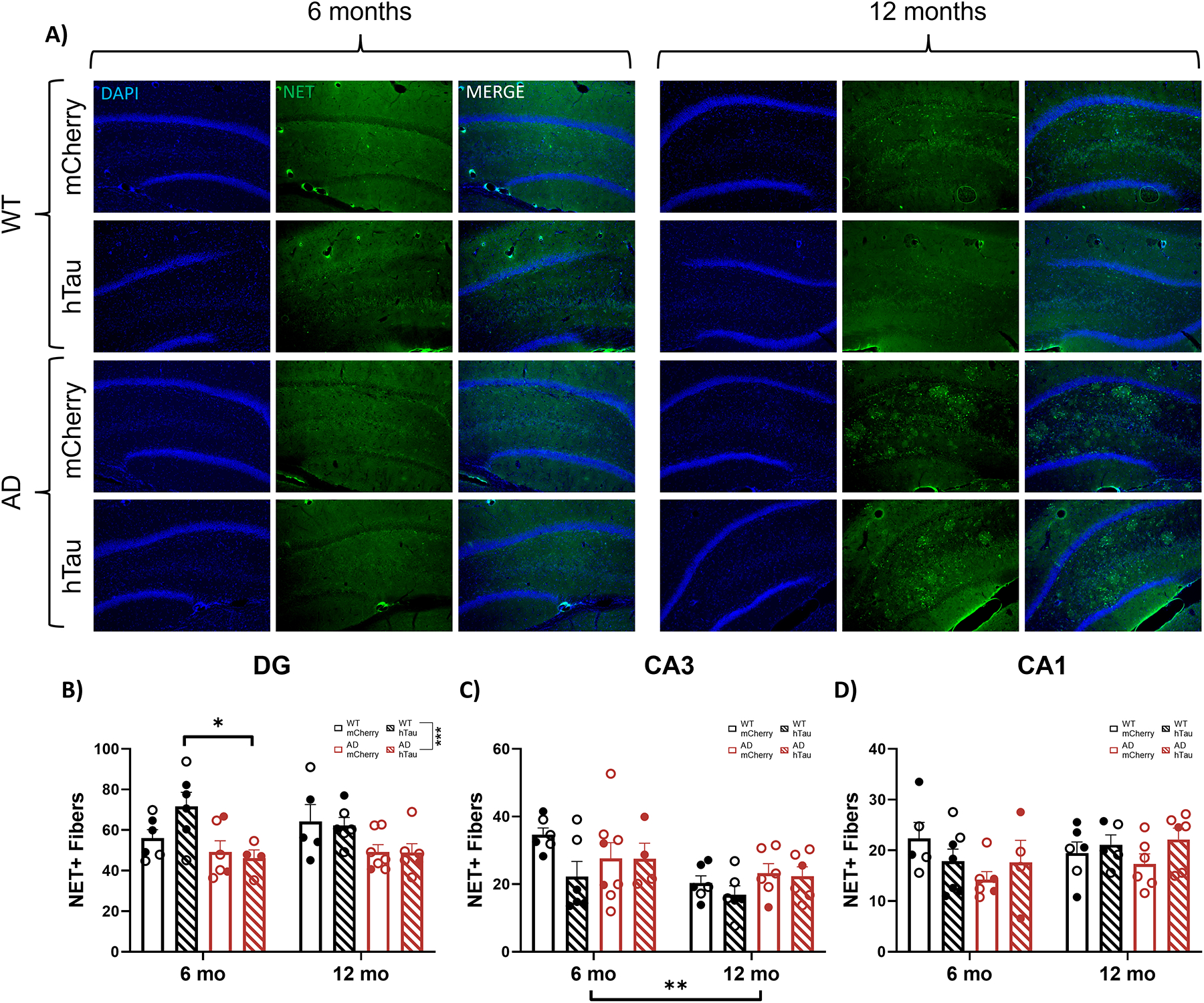
Assessment of LC fiber density in the hippocampus. Wild-type (WT, black) and TgF344-AD (AD, red) rats were injected bilaterally into the LC with AAV-PRSx8-hTau (hTau, hashed) or AAV-PRSx8-mCherry (mCherry, solid) at 2 months of age, and assessed for NET immunoreactivity at 6 months or 12 months. N = 4–8 per group. Shown are representative images in the DG subregion of the hippocampus (A), and quantification in the DG (B), CA3 (C), and CA1 (D) expressed as the number of NET+ fibers. (D). Main images and insets taken at 10x and 40x magnification, respectively. **p<0.01, ***p<0.001. Males are represented by closed circles, females by open circles.
DISCUSSION
The LC is one of the first brain regions that accumulates hyperphosphorylated “pretangle” tau pathology at a time when notable behavioral changes begin to emerge, years or even decades prior to cognitive impairment. In this study, we set out to determine the effects of age, Aβ-triggered endogenous rat tau pathology, and virally-induced WT hTau pathology in the LC on AD-relevant behavioral phenotypes, pathology, and neuroinflammation. We observed significant effects of age and TgF344-AD genotype on behavioral and pathological markers of AD, whereas WT hTau expression in the LC was mostly inert. In general, older rats took longer to fall asleep, had reduced locomotor activity, mixed phenotypes in tests of anxiety-like behavior and passive coping, and impaired learning and memory compared with younger animals. The presence of AD-like neuropathology tended to increase anxiety-like behavior and impair cognitive performance. There are mixed reports on sex differences in behavioral phenotypes in these rats, and we were underpowered to detect differences based on sex. However, beyond differences in general locomotion, where females appeared to move more than males (see Fig. 2G and Fig. 3B, D), we did not observe obvious trends in other behavioral measures.
Changes in arousal and sleep disturbances are nearly ubiquitous in AD [24–26]. A previous study reported that 17-month TgF344-AD rats exhibited atypical EEG sleep/wake features compared to age-matched controls [86], but did not include a younger cohort for comparison. We chose to perform the sleep latency test which is an EEG-validated measure of arousal sensitive to noradrenergic manipulation [71, 72, 87]. We observed a main effect of age (older animals took longer to fall asleep) and a complex interaction between age, genotype, and virus on sleep latency of questionable physiological significance due to its modest influence. We also observed effects of age on 23-h locomotion over the total time of the task, similar to a previous report [67], and within the first 30-min. Together, results indicate that 12-month animals took longer to fall asleep but were generally less mobile. Moreover, in 12-month animals specifically, the presence of WT hTau induced an increase in locomotor activity over the entire light/dark cycle, one of the few viral-expression induced phenotypes seen here. This phenotype mimics those reported in mouse models of tauopathy [88–90], indicating these effects maybe be isoform- and/or time-specific or depend on additional influence of forebrain pathology.
Anxiety and depression are highly prevalent in prodromal populations and increase risk of developing AD [28, 29, 35, 91, 92]. When testing for anxiety-like phenotypes, we observed that 6-month TgF344-AD rats spent less time in the center of an open field compared with littermate controls, while no differences were observed in time spent in the open arms of the elevated plus maze. It has previously been reported that TgF344-AD rats show increased anxiety-like behaviors at 12, but not 2 months of age in these tasks [93]. However, these canonical tests of anxiety-like behaviors assume that rodents are motivated to explore novel environments. A lack of anxiety-like phenotypes in these tasks could be masked by differences in exploratory behavior and/or general locomotion, which we and others have observed over the course of aging and in TgF344-AD rats [67, 93]. We therefore employed the NSF task, where the main conflict is fear of a novel environment versus drive to consume food in a hungry animal. The NSF task has previously been validated as a NE-sensitive anxiety-like task by our lab using dopamine-β hydroxylase knockout mice, which lack phenotypes in canonical exploration-based anxiety paradigms [82]. We observed that both older and transgenic rats took longer to eat the food pellet in a novel environment compared with younger and age-matched WT littermates, respectively. These effects are unlikely to be mediated by differences in hunger levels because rats did not differ in the total amount of pellet eaten or latency to eat the pellet in their home cage. Therefore, accumulating evidence from our lab suggests that NSF may be particularly useful at gauging noradrenergic-specific impacts on anxiety-like behaviors.
In the forced swim task, we observed that 12-month rats spent less time spent floating than younger animals. Given the acute nature of this paradigm and the fact that older rats generally moved less in land-based locomotion assays, these results support the notion of an increase in active coping behaviors in older rats [83, 84]. A previous report demonstrated an increase in immobility in TgF344-AD rats at 12 months of age [93], which is opposite of the effects reported here. Differences may be attributed to multiple exposures to forced swim, influences of sex, or other factors known to influence coping-like phenotypes in the forced swim task [83, 94].
Spatial learning and memory deficits, which are hallmark behavioral phenotypes of AD, presented here are largely congruent with previous studies of the TgF344-AD strain, where the most robust genotype differences arise in reversal learning [63, 66]. These differences were primarily observed in acquisition of reversal learning and were exacerbated by age in TgF344-AD rats. Interestingly, we also detected deficits in initial acquisition in 6-month TgF344-AD rats, and hTau virus-induced acquisition deficits in 12-month WT rats that were reminiscent of those we observed in 6-month transgenic rats. In both of those cases (6-month Tg rats and 12-month WT rats with hTau), the main detectable AD-like neuropathology present in the brains was hyperphosphorylated tau in the LC, suggesting a causal relationship. We sought to further categorize the nature and anatomical specificity of these deficits by testing these rats on cued and contextual fear conditioning, which are associative learning tasks as opposed to spatial. Additionally, cued fear conditioning is non-hippocampal dependent, unlike both the Morris water maze and contextual fear conditioning. During training, 12-month TgF344-AD rats froze less than WT animals, which suggests a lower ability to form initial associations between the tone/shock pairing. However, genotype differences were not observed during cued or contextual fear conditioning, indicating that recall of these associations once formed was intact. Main effects of age were evident where older animals froze less to both the cue and the context than younger counterparts, suggesting a modest age-related impairment of associative memory.
In this study, we expanded on our previous work demonstrating that TgF344-AD rats have progressive noradrenergic fiber loss [63] by also showing lower NET+ fiber density in TgF344-AD rats, specifically in the DG. Recent studies further confirm a reduction in LC innervation to the DG [85], suggesting selective vulnerability of NE fibers within this hippocampal subfield. We also reaffirmed an age-dependent decrease in NE fibers within CA3, 4 months earlier than previously reported [63], in addition to confirming a lack of change in NE innervation to CA1. Given the loss of DBH+ fibers and reduction in NE content in the hippocampus, a loss of NET+ fibers could be indicative of compensatory mechanisms downregulating NE reuptake to increase noradrenergic signaling. Indeed, other noradrenergic compensatory changes, including enhanced β-adrenergic receptor function and axonal sprouting, have been noted in TgF344-AD rats [85] and human AD/dementia cases [95, 96].
We did observe increasing levels of amyloid deposition beginning at 6 months in TgF344-AD rats across hippocampal subregions, which is slightly earlier than previously reported [66, 67]. By contrast, there were low levels of endogenous hyperphosphorylated tau across all hippocampal subregions and lack of support for transsynaptic spread of hTau from the LC. This was somewhat surprising, given the synergistic effects of amyloid and tau deposition [97]. We based our viral expression and testing paradigm on previous literature, where pseudophosphorylated tau demonstrated transsynaptic spread and induced cognitive deficits 7 months post-injection in ~10 month old rats [32]. The viral form of tau used here (WT hTau) is a milder version from a pathological perspective compared to that reported previously (pseudophosphorylated at 14 sites), and the endogenous LC tau pathology in TgF344-AD rats is an even milder form that only reacts with the CP13 antibody (Ser202), which does not mature into more toxic species observed in the typical progression of AD [63]. Although we observed no differences in the level of hTau expression within the LC between 6 and 12 month rats, it is possible that longer expression time (e.g. 14 or more months post-infusion) or initial injection of the virus into aged rats could be necessary to trigger appreciable effects on behavior and pathology. Indeed, we did observe a potential “seeding” effect, whereby TgF344-AD rats developed more AT8 pathology in the LC compared to WT littermates.
Behavioral differences between genotypes are likely attributable to development of AD-like neuropathology and inflammation over the course of aging. The effects of tau pathology on neural activity are mixed, as both hyper- and hypo-activity have been reported [98–101]. These likely depend on specific tau isoform and neuronal population, and changes in LC activity throughout the course of AD have yet to be characterized. Furthermore, the TgF344-AD rats harbor the same mutations as the APP/PS1 mouse counterpart, and APP/PS1 mice develop intraneuronal Aβ oligomers within the LC that induce noradrenergic neuron hyperactivity [102]. Given that the transgenes are expressed under the same promoter in TgF344-AD rats [66], they also likely develop Aβ oligomers. The additional accumulation of hyperphosphorylated tau within the LC could lead to a complex interaction between the two AD-like pathologies [97]. Thus, it will be important for future studies to chronicle the time course of AD-like neuropathology accumulation within the LC of TgF344-AD rats and directly measure LC firing.
Changes in LC activity can also be speculated based on behavioral measures and align with the predicted accumulation and effects of LC pathology. Generally, anxiety-like behaviors are promoted by augmented basal LC firing (3–5 Hz) [75–79]. In addition, the LC responds with phasic discharge to novelty [103, 104], and probable/confirmed AD patients show impaired novelty processing and perform worse on tasks incorporating novelty [105–107]. Similarly, aged TgF344-AD rats also demonstrate reduced ability to distinguish novel objects [66], which may be partially driven by novelty-induced anxiety-like phenotypes, as demonstrated by the NSF task. The opposite effect is observed in young TgF344-AD rats, which present with enhanced novelty detection that is mediated by β-adrenergic receptors [85], suggesting an augmented phasic response. Overall, results from the NSF task lend support to an age- and AD-like neuropathology-dependent alteration in LC activity in TgF344-AD rats [108]. Specifically, in young rats, enhanced signal-to-noise ratio (phasic:tonic firing) could support maintenance of novelty detection, but may lead to anxiety-like phenotypes. This effect may be reversed in 12-month rats, where signal-to-noise ratio of the LC is diminished, resulting in reduced novelty detection, and heightened basal LC activity, which also prompts expression of anxiety-like behaviors. This phenotype might be masked in other canonical tasks, such as the open field and elevated plus maze, due to differences in exploratory behavior and general locomotion, which we and others have observed over the course of aging and in TgF344-AD rats [66, 67]. We have previously reported impairments in acquisition and reversal acquisition of the Morris water maze in TgF344-AD rats [63], and here demonstrate impairments in training during fear conditioning, but only in 12-month TgF344-AD rats. However, we saw no impairments in probe trials of the Morris water maze or freezing in the contextual and cued fear conditioning paradigms, suggesting intact memory retrieval mechanisms. TgF344-AD rats present with decreased noradrenergic forebrain innervation regions early in disease [63, 85]. Spatial and associative hippocampal-dependent and independent learning and memory are supported by LC-NE release and are sensitive to noradrenergic perturbations [85, 109–111]. The loss of forebrain LC fibers could engage compensatory mechanisms, such as increased β-adrenergic receptor function, and potentially increased firing could support the maintenance of cognition in prodromal phases of AD. For example, heightened β-adrenergic receptor function facilitates extinction learning and novel object recognition [85] in younger (6–9 month) TgF344-AD rats, but these studies should be expanded to additional paradigms.
Independent of genotype effects, we observed aging effects on behavior that are suggestive of altered LC activity. 12-month rats spent less time floating in the forced swim test, indicative of an increase in active coping behaviors in response to stress. Wistar Kyoto rats, a common rodent model of depression, display increased immobility and enhanced LC activity when tested on the forced swim [112, 113]. Moreover, coping behaviors mediated by the LC may be partially dependent on galanin co-release [114], which is expressed in a high percentage of LC neurons [115]. Eliminating galanin from the LC promotes active coping behaviors in mice [114], an effect that is mimicked in older rats tested in the forced swim task. Though we cannot completely rule out frank LC cell loss over the course of aging, 16-month TgF344-AD rats do not exhibit LC degeneration compared with WT littermates [63]. Given that galanin co-expressing LC neurons are protected in AD [116], we instead theorize that changes in firing rates could reduce galanin co-release and promote active coping behaviors. Although we did not observe changes in sleep latency that would be indicative of increased LC activity, other arousal systems that have yet to be explored in these rats could compensate for LC-specific abnormalities. Alternatively, there is evidence for decreased LC activity with aging [117], but the effects may be strain-dependent. The presence of AD-like neuropathology could further modulate age-related changes in LC activity, but this has not been investigated.
Our results expand the characterization of behavior and AD-like neuropathology characterization in the TgF344-AD rat that reflect preclinical and prodromal AD. Further studies are necessary to investigate more subtle behavioral phenotypes such as detailed sleep architecture. Impulse dyscontrol and agitation are also commonly observed in prodromal AD [35, 50], and can be improved by anti-adrenergic drugs [118, 119], which warrants further study in TgF344-AD rats. Changes in behavior in 6-month animals are highly suggestive of altered LC activity in the presence of AD-like hyperphosphorylated tau pathology but in the absence of appreciable forebrain pathology. Current AD therapeutics target late-stage pathology, but targeting prodromal stages with drugs that modulate activity or signaling of early affected structures, such as the LC-NE system, may be more effective at slowing the progression of AD. A recent study reported that atomoxetine, a NE reuptake inhibitor, normalized multiple biomarkers of AD in a cohort of patients with mild cognitive impairment [120]. However, successful development and implementation of these therapies will require a precise understanding of the physiological changes of LC activity across disease stages.
Supplementary Material
Acknowledgements
This study was supported by funding from the National Institute of Aging (AG062581 to DW, AG069502 to MAK), the National Institute of Neurological Disorders and Stroke (NS96050 to MAK), and the Eli Lilly Innovation Fellowship Award (CRA and JMR).
The authors of this study would like to thank Ryan McCann and Shivaang Chawla who assisted with rat husbandry.
This study was supported in part by the Rodent Behavioral Core (RBC), which is subsidized by the Emory University School of Medicine and is one of the Emory Integrated Core Facilities. Additional support was provided by the Emory Neuroscience NINDS Core Facilities (P30NS055077). Further support was provided by the Georgia Clinical & Translational Science Alliance of the National Institutes of Health under Award Number UL1TR002378. The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institutes of Health.
This research project was supported in part by the Viral Vector Core of the Emory Center for Neurodegenerative Disease Core Facilities and the Vector Core of the University of Pennsylvania.
Footnotes
Conflict of Interest/Disclosure Statement
The authors have no conflict of interest to report.
References
- [1].Hardy JA, Higgins GA (1992) Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185. [DOI] [PubMed] [Google Scholar]
- [2].Jin M, Shepardson N, Yang T, Chen G, Walsh D, Selkoe DJ (2011) Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci U S A 108, 5819–5824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Busciglio J, Lorenzo A, Yeh J, Yankner BA (1995) beta-amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron 14, 879–888. [DOI] [PubMed] [Google Scholar]
- [4].Zhang F, Gannon M, Chen Y, Yan S, Zhang S, Feng W, Tao J, Sha B, Liu Z, Saito T, Saido T, Keene CD, Jiao K, Roberson ED, Xu H, Wang Q (2020) beta-amyloid redirects norepinephrine signaling to activate the pathogenic GSK3beta/tau cascade. Sci Transl Med 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH (2006) Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet 15, 1437–1449. [DOI] [PubMed] [Google Scholar]
- [6].Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, Xu HW, Stern D, McKhann G, Yan SD (2005) Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J 19, 2040–2041. [DOI] [PubMed] [Google Scholar]
- [7].Mao P, Reddy PH (2011) Aging and amyloid beta-induced oxidative DNA damage and mitochondrial dysfunction in Alzheimer’s disease: implications for early intervention and therapeutics. Biochim Biophys Acta 1812, 1359–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Leuner K, Muller WE, Reichert AS (2012) From mitochondrial dysfunction to amyloid beta formation: novel insights into the pathogenesis of Alzheimer’s disease. Mol Neurobiol 46, 186–193. [DOI] [PubMed] [Google Scholar]
- [9].Chauhan V, Chauhan A (2006) Oxidative stress in Alzheimer’s disease. Pathophysiology 13, 195–208. [DOI] [PubMed] [Google Scholar]
- [10].Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM (2009) Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457, 1128–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ (2002) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539. [DOI] [PubMed] [Google Scholar]
- [12].Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL (2007) Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J Neurosci 27, 796–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT (1992) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 42, 631–639. [DOI] [PubMed] [Google Scholar]
- [14].Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM (2007) Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol 64, 343–349. [DOI] [PubMed] [Google Scholar]
- [15].Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT (1997) Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol 41, 17–24. [DOI] [PubMed] [Google Scholar]
- [16].Braak H, Thal DR, Ghebremedhin E, Del Tredici K (2011) Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol 70, 960–969. [DOI] [PubMed] [Google Scholar]
- [17].Pletnikova O, Kageyama Y, Rudow G, LaClair KD, Albert M, Crain BJ, Tian J, Fowler D, Troncoso JC (2018) The spectrum of preclinical Alzheimer’s disease pathology and its modulation by ApoE genotype. Neurobiol Aging 71, 72–80. [DOI] [PubMed] [Google Scholar]
- [18].Theofilas P, Ehrenberg AJ, Dunlop S, Di Lorenzo Alho AT, Nguy A, Leite REP, Rodriguez RD, Mejia MB, Suemoto CK, Ferretti-Rebustini REL, Polichiso L, Nascimento CF, Seeley WW, Nitrini R, Pasqualucci CA, Jacob Filho W, Rueb U, Neuhaus J, Heinsen H, Grinberg LT (2017) Locus coeruleus volume and cell population changes during Alzheimer’s disease progression: A stereological study in human postmortem brains with potential implication for early-stage biomarker discovery. Alzheimers Dement 13, 236–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Poe GR, Foote S, Eschenko O, Johansen JP, Bouret S, Aston-Jones G, Harley CW, Manahan-Vaughan D, Weinshenker D, Valentino R, Berridge C, Chandler DJ, Waterhouse B, Sara SJ (2020) Locus coeruleus: a new look at the blue spot. Nat Rev Neurosci 21, 644–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chandler DJ, Jensen P, McCall JG, Pickering AE, Schwarz LA, Totah NK (2019) Redefining Noradrenergic Neuromodulation of Behavior: Impacts of a Modular Locus Coeruleus Architecture. J Neurosci 39, 8239–8249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sara SJ (2009) The locus coeruleus and noradrenergic modulation of cognition. Nat Rev Neurosci 10, 211–223. [DOI] [PubMed] [Google Scholar]
- [22].Aston-Jones G, Cohen JD (2005) An integrative theory of locus coeruleus-norepinephrine function: adaptive gain and optimal performance. Annu Rev Neurosci 28, 403–450. [DOI] [PubMed] [Google Scholar]
- [23].Hamann SB, Monarch ES, Goldstein FC (2000) Memory enhancement for emotional stimuli is impaired in early Alzheimer’s disease. Neuropsychology 14, 82–92. [PubMed] [Google Scholar]
- [24].Ju YE, McLeland JS, Toedebusch CD, Xiong C, Fagan AM, Duntley SP, Morris JC, Holtzman DM (2013) Sleep quality and preclinical Alzheimer disease. JAMA Neurol 70, 587–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Winsky-Sommerer R, de Oliveira P, Loomis S, Wafford K, Dijk DJ, Gilmour G (2019) Disturbances of sleep quality, timing and structure and their relationship with other neuropsychiatric symptoms in Alzheimer’s disease and schizophrenia: Insights from studies in patient populations and animal models. Neurosci Biobehav Rev 97, 112–137. [DOI] [PubMed] [Google Scholar]
- [26].Yesavage JA, Friedman L, Kraemer H, Tinklenberg JR, Salehi A, Noda A, Taylor JL, O’Hara R, Murphy G (2004) Sleep/wake disruption in Alzheimer’s disease: APOE status and longitudinal course. J Geriatr Psychiatry Neurol 17, 20–24. [DOI] [PubMed] [Google Scholar]
- [27].Berardi AM, Parasuraman R, Haxby JV (2005) Sustained attention in mild Alzheimer’s disease. Dev Neuropsychol 28, 507–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Pietrzak RH, Lim YY, Neumeister A, Ames D, Ellis KA, Harrington K, Lautenschlager NT, Restrepo C, Martins RN, Masters CL, Villemagne VL, Rowe CC, Maruff P, Australian Imaging B, Lifestyle Research G (2015) Amyloid-beta, anxiety, and cognitive decline in preclinical Alzheimer disease: a multicenter, prospective cohort study. JAMA Psychiatry 72, 284–291. [DOI] [PubMed] [Google Scholar]
- [29].Teri L, Ferretti LE, Gibbons LE, Logsdon RG, McCurry SM, Kukull WA, McCormick WC, Bowen JD, Larson EB (1999) Anxiety of Alzheimer’s disease: prevalence, and comorbidity. J Gerontol A Biol Sci Med Sci 54, M348–352. [DOI] [PubMed] [Google Scholar]
- [30].Gendron TF, Petrucelli L (2009) The role of tau in neurodegeneration. Mol Neurodegener 4, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, Feany MB (2001) Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science 293, 711–714. [DOI] [PubMed] [Google Scholar]
- [32].Ghosh A, Torraville SE, Mukherjee B, Walling SG, Martin GM, Harley CW, Yuan Q (2019) An experimental model of Braak’s pretangle proposal for the origin of Alzheimer’s disease: the role of locus coeruleus in early symptom development. Alzheimers Res Ther 11, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Arendt T, Bruckner MK, Morawski M, Jager C, Gertz HJ (2015) Early neurone loss in Alzheimer’s disease: cortical or subcortical? Acta Neuropathol Commun 3, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Busch C, Bohl J, Ohm TG (1997) Spatial, temporal and numeric analysis of Alzheimer changes in the nucleus coeruleus. Neurobiol Aging 18, 401–406. [DOI] [PubMed] [Google Scholar]
- [35].Ehrenberg AJ, Suemoto CK, Franca Resende EP, Petersen C, Leite REP, Rodriguez RD, Ferretti-Rebustini REL, You M, Oh J, Nitrini R, Pasqualucci CA, Jacob-Filho W, Kramer JH, Gatchel JR, Grinberg LT (2018) Neuropathologic Correlates of Psychiatric Symptoms in Alzheimer’s Disease. J Alzheimers Dis 66, 115–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Matchett BJ, Grinberg LT, Theofilas P, Murray ME (2021) The mechanistic link between selective vulnerability of the locus coeruleus and neurodegeneration in Alzheimer’s disease. Acta Neuropathol 141, 631–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Shibata E, Sasaki M, Tohyama K, Otsuka K, Endoh J, Terayama Y, Sakai A (2008) Use of neuromelanin-sensitive MRI to distinguish schizophrenic and depressive patients and healthy individuals based on signal alterations in the substantia nigra and locus ceruleus. Biol Psychiatry 64, 401–406. [DOI] [PubMed] [Google Scholar]
- [38].Shibata E, Sasaki M, Tohyama K, Otsuka K, Sakai A (2007) Reduced signal of locus ceruleus in depression in quantitative neuromelanin magnetic resonance imaging. Neuroreport 18, 415–418. [DOI] [PubMed] [Google Scholar]
- [39].Knudsen K, Fedorova TD, Hansen AK, Sommerauer M, Otto M, Svendsen KB, Nahimi A, Stokholm MG, Pavese N, Beier CP, Brooks DJ, Borghammer P (2018) In-vivo staging of pathology in REM sleep behaviour disorder: a multimodality imaging case-control study. Lancet Neurol 17, 618–628. [DOI] [PubMed] [Google Scholar]
- [40].Ehrminger M, Latimier A, Pyatigorskaya N, Garcia-Lorenzo D, Leu-Semenescu S, Vidailhet M, Lehericy S, Arnulf I (2016) The coeruleus/subcoeruleus complex in idiopathic rapid eye movement sleep behaviour disorder. Brain 139, 1180–1188. [DOI] [PubMed] [Google Scholar]
- [41].Olivieri P, Lagarde J, Lehericy S, Valabregue R, Michel A, Mace P, Caille F, Gervais P, Bottlaender M, Sarazin M (2019) Early alteration of the locus coeruleus in phenotypic variants of Alzheimer’s disease. Ann Clin Transl Neurol 6, 1345–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wilson RS, Nag S, Boyle PA, Hizel LP, Yu L, Buchman AS, Schneider JA, Bennett DA (2013) Neural reserve, neuronal density in the locus ceruleus, and cognitive decline. Neurology 80, 1202–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kelly SC, He B, Perez SE, Ginsberg SD, Mufson EJ, Counts SE (2017) Locus coeruleus cellular and molecular pathology during the progression of Alzheimer’s disease. Acta Neuropathol Commun 5, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Jacobs HIL, Muller-Ehrenberg L, Priovoulos N, Roebroeck A (2018) Curvilinear locus coeruleus functional connectivity trajectories over the adult lifespan: a 7T MRI study. Neurobiol Aging 69, 167–176. [DOI] [PubMed] [Google Scholar]
- [45].Jacobs HI, Priovoulos N, Poser BA, Pagen LH, Ivanov D, Verhey FR, Uludag K (2020) Dynamic behavior of the locus coeruleus during arousal-related memory processing in a multi-modal 7T fMRI paradigm. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Jacobs HI, Wiese S, van de Ven V, Gronenschild EH, Verhey FR, Matthews PM (2015) Relevance of parahippocampal-locus coeruleus connectivity to memory in early dementia. Neurobiol Aging 36, 618–626. [DOI] [PubMed] [Google Scholar]
- [47].Del Cerro I, Martinez-Zalacain I, Guinea-Izquierdo A, Gascon-Bayarri J, Vinas-Diez V, Urretavizcaya M, Naval-Baudin P, Aguilera C, Rene-Ramirez R, Ferrer I, Menchon JM, Soria V, Soriano-Mas C (2020) Locus coeruleus connectivity alterations in late-life major depressive disorder during a visual oddball task. Neuroimage Clin 28, 102482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Grueschow M, Stenz N, Thorn H, Ehlert U, Breckwoldt J, Brodmann Maeder M, Exadaktylos AK, Bingisser R, Ruff CC, Kleim B (2021) Real-world stress resilience is associated with the responsivity of the locus coeruleus. Nat Commun 12, 2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zhao S, Rangaprakash D, Venkataraman A, Liang P, Deshpande G (2017) Investigating Focal Connectivity Deficits in Alzheimer’s Disease Using Directional Brain Networks Derived from Resting-State fMRI. Front Aging Neurosci 9, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Johansson M, Stomrud E, Insel PS, Leuzy A, Johansson PM, Smith R, Ismail Z, Janelidze S, Palmqvist S, van Westen D, Mattsson-Carlgren N, Hansson O (2021) Mild behavioral impairment and its relation to tau pathology in preclinical Alzheimer’s disease. Transl Psychiatry 11, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Babulal GM, Roe CM, Stout SH, Rajasekar G, Wisch JK, Benzinger TLS, Morris JC, Ances BM (2020) Depression is Associated with Tau and Not Amyloid Positron Emission Tomography in Cognitively Normal Adults. J Alzheimers Dis 74, 1045–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Pichet Binette A, Vachon-Presseau E, Morris J, Bateman R, Benzinger T, Collins DL, Poirier J, Breitner JCS, Villeneuve S, Dominantly Inherited Alzheimer N, Group P-AR (2021) Amyloid and Tau Pathology Associations With Personality Traits, Neuropsychiatric Symptoms, and Cognitive Lifestyle in the Preclinical Phases of Sporadic and Autosomal Dominant Alzheimer’s Disease. Biol Psychiatry 89, 776–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].d’Oleire Uquillas F, Jacobs HIL, Biddle KD, Properzi M, Hanseeuw B, Schultz AP, Rentz DM, Johnson KA, Sperling RA, Donovan NJ (2018) Regional tau pathology and loneliness in cognitively normal older adults. Transl Psychiatry 8, 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Gatchel JR, Donovan NJ, Locascio JJ, Schultz AP, Becker JA, Chhatwal J, Papp KV, Amariglio RE, Rentz DM, Blacker D, Sperling RA, Johnson KA, Marshall GA (2017) Depressive Symptoms and Tau Accumulation in the Inferior Temporal Lobe and Entorhinal Cortex in Cognitively Normal Older Adults: A Pilot Study. J Alzheimers Dis 59, 975–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kelberman M, Keilholz S, Weinshenker D (2020) What’s That (Blue) Spot on my MRI? Multimodal Neuroimaging of the Locus Coeruleus in Neurodegenerative Disease. Front Neurosci 14, 583421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Chalermpalanupap T, Schroeder JP, Rorabaugh JM, Liles LC, Lah JJ, Levey AI, Weinshenker D (2018) Locus Coeruleus Ablation Exacerbates Cognitive Deficits, Neuropathology, and Lethality in P301S Tau Transgenic Mice. J Neurosci 38, 74–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Heneka MT, Ramanathan M, Jacobs AH, Dumitrescu-Ozimek L, Bilkei-Gorzo A, Debeir T, Sastre M, Galldiks N, Zimmer A, Hoehn M, Heiss WD, Klockgether T, Staufenbiel M (2006) Locus ceruleus degeneration promotes Alzheimer pathogenesis in amyloid precursor protein 23 transgenic mice. J Neurosci 26, 1343–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Jardanhazi-Kurutz D, Kummer MP, Terwel D, Vogel K, Dyrks T, Thiele A, Heneka MT (2010) Induced LC degeneration in APP/PS1 transgenic mice accelerates early cerebral amyloidosis and cognitive deficits. Neurochem Int 57, 375–382. [DOI] [PubMed] [Google Scholar]
- [59].Cao S, Fisher DW, Rodriguez G, Yu T, Dong H (2021) Comparisons of neuroinflammation, microglial activation, and degeneration of the locus coeruleus-norepinephrine system in APP/PS1 and aging mice. J Neuroinflammation 18, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Mehla J, Lacoursiere SG, Lapointe V, McNaughton BL, Sutherland RJ, McDonald RJ, Mohajerani MH (2019) Age-dependent behavioral and biochemical characterization of single APP knock-in mouse (APP(NL-G-F/NL-G-F)) model of Alzheimer’s disease. Neurobiol Aging 75, 25–37. [DOI] [PubMed] [Google Scholar]
- [61].O’Neil JN, Mouton PR, Tizabi Y, Ottinger MA, Lei DL, Ingram DK, Manaye KF (2007) Catecholaminergic neuronal loss in locus coeruleus of aged female dtg APP/PS1 mice. J Chem Neuroanat 34, 102–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Liu Y, Yoo MJ, Savonenko A, Stirling W, Price DL, Borchelt DR, Mamounas L, Lyons WE, Blue ME, Lee MK (2008) Amyloid pathology is associated with progressive monoaminergic neurodegeneration in a transgenic mouse model of Alzheimer’s disease. J Neurosci 28, 13805–13814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Rorabaugh JM, Chalermpalanupap T, Botz-Zapp CA, Fu VM, Lembeck NA, Cohen RM, Weinshenker D (2017) Chemogenetic locus coeruleus activation restores reversal learning in a rat model of Alzheimer’s disease. Brain 140, 3023–3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM (2007) Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53, 337–351. [DOI] [PubMed] [Google Scholar]
- [65].Kang SS, Liu X, Ahn EH, Xiang J, Manfredsson FP, Yang X, Luo HR, Liles LC, Weinshenker D, Ye K (2020) Norepinephrine metabolite DOPEGAL activates AEP and pathological Tau aggregation in locus coeruleus. J Clin Invest 130, 422–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Cohen RM, Rezai-Zadeh K, Weitz TM, Rentsendorj A, Gate D, Spivak I, Bholat Y, Vasilevko V, Glabe CG, Breunig JJ, Rakic P, Davtyan H, Agadjanyan MG, Kepe V, Barrio JR, Bannykh S, Szekely CA, Pechnick RN, Town T (2013) A transgenic Alzheimer rat with plaques, tau pathology, behavioral impairment, oligomeric abeta, and frank neuronal loss. J Neurosci 33, 6245–6256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Sare RM, Cooke SK, Krych L, Zerfas PM, Cohen RM, Smith CB (2020) Behavioral Phenotype in the TgF344-AD Rat Model of Alzheimer’s Disease. Front Neurosci 14, 601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Munoz-Moreno E, Tudela R, Lopez-Gil X, Soria G (2018) Early brain connectivity alterations and cognitive impairment in a rat model of Alzheimer’s disease. Alzheimers Res Ther 10, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Pentkowski NS, Berkowitz LE, Thompson SM, Drake EN, Olguin CR, Clark BJ (2018) Anxiety-like behavior as an early endophenotype in the TgF344-AD rat model of Alzheimer’s disease. Neurobiol Aging 61, 169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Hwang DY, Carlezon WA Jr., Isacson O, Kim KS (2001) A high-efficiency synthetic promoter that drives transgene expression selectively in noradrenergic neurons. Hum Gene Ther 12, 1731–1740. [DOI] [PubMed] [Google Scholar]
- [71].Hunsley MS, Palmiter RD (2004) Altered sleep latency and arousal regulation in mice lacking norepinephrine. Pharmacol Biochem Behav 78, 765–773. [DOI] [PubMed] [Google Scholar]
- [72].Porter-Stransky KA, Centanni SW, Karne SL, Odil LM, Fekir S, Wong JC, Jerome C, Mitchell HA, Escayg A, Pedersen NP, Winder DG, Mitrano DA, Weinshenker D (2019) Noradrenergic Transmission at Alpha1-Adrenergic Receptors in the Ventral Periaqueductal Gray Modulates Arousal. Biol Psychiatry 85, 237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Friard O, Gamba M (2016) BORIS: a free, versatile open-source event-logging software for video/audio coding and live observations. British Ecological Society 7, 1325–1330. [Google Scholar]
- [74].Sathyanesan A, Ogura T, Lin W (2012) Automated measurement of nerve fiber density using line intensity scan analysis. J Neurosci Methods 206, 165–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].McCall JG, Al-Hasani R, Siuda ER, Hong DY, Norris AJ, Ford CP, Bruchas MR (2015) CRH Engagement of the Locus Coeruleus Noradrenergic System Mediates Stress-Induced Anxiety. Neuron 87, 605–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Curtis AL, Lechner SM, Pavcovich LA, Valentino RJ (1997) Activation of the locus coeruleus noradrenergic system by intracoerulear microinfusion of corticotropin-releasing factor: effects on discharge rate, cortical norepinephrine levels and cortical electroencephalographic activity. J Pharmacol Exp Ther 281, 163–172. [PubMed] [Google Scholar]
- [77].Curtis AL, Leiser SC, Snyder K, Valentino RJ (2012) Predator stress engages corticotropin-releasing factor and opioid systems to alter the operating mode of locus coeruleus norepinephrine neurons. Neuropharmacology 62, 1737–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Page ME, Abercrombie ED (1999) Discrete local application of corticotropin-releasing factor increases locus coeruleus discharge and extracellular norepinephrine in rat hippocampus. Synapse 33, 304–313. [DOI] [PubMed] [Google Scholar]
- [79].Valentino RJ, Foote SL (1988) Corticotropin-releasing hormone increases tonic but not sensory-evoked activity of noradrenergic locus coeruleus neurons in unanesthetized rats. J Neurosci 8, 1016–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Pentkowski NS, Rogge-Obando KK, Donaldson TN, Bouquin SJ, Clark BJ (2021) Anxiety and Alzheimer’s disease: Behavioral analysis and neural basis in rodent models of Alzheimer’s-related neuropathology. Neurosci Biobehav Rev 127, 647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Dulawa SC, Hen R (2005) Recent advances in animal models of chronic antidepressant effects: the novelty-induced hypophagia test. Neurosci Biobehav Rev 29, 771–783. [DOI] [PubMed] [Google Scholar]
- [82].Lustberg D, Tillage RP, Bai Y, Pruitt M, Liles LC, Weinshenker D (2020) Noradrenergic circuits in the forebrain control affective responses to novelty. Psychopharmacology (Berl) 237, 3337–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Commons KG, Cholanians AB, Babb JA, Ehlinger DG (2017) The Rodent Forced Swim Test Measures Stress-Coping Strategy, Not Depression-like Behavior. ACS Chem Neurosci 8, 955–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].de Kloet ER, Molendijk ML (2016) Coping with the Forced Swim Stressor: Towards Understanding an Adaptive Mechanism. Neural Plast 2016, 6503162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Goodman AM, Langner BM, Jackson N, Alex C, McMahon LL (2021) Heightened Hippocampal beta-Adrenergic Receptor Function Drives Synaptic Potentiation and Supports Learning and Memory in the TgF344-AD Rat Model during Prodromal Alzheimer’s Disease. J Neurosci 41, 5747–5761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Kreuzer M, Keating GL, Fenzl T, Hartner L, Sinon CG, Hajjar I, Ciavatta V, Rye DB, Garcia PS (2020) Sleep/Wake Behavior and EEG Signatures of the TgF344-AD Rat Model at the Prodromal Stage. Int J Mol Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Butkovich LM, Houser MC, Chalermpalanupap T, Porter-Stransky KA, Iannitelli AF, Boles JS, Lloyd GM, Coomes AS, Eidson LN, De Sousa Rodrigues ME, Oliver DL, Kelly SD, Chang J, Bengoa-Vergniory N, Wade-Martins R, Giasson BI, Joers V, Weinshenker D, Tansey MG (2020) Transgenic Mice Expressing Human alpha-Synuclein in Noradrenergic Neurons Develop Locus Ceruleus Pathology and Nonmotor Features of Parkinson’s Disease. J Neurosci 40, 7559–7576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Scattoni ML, Gasparini L, Alleva E, Goedert M, Calamandrei G, Spillantini MG (2010) Early behavioural markers of disease in P301S tau transgenic mice. Behav Brain Res 208, 250–257. [DOI] [PubMed] [Google Scholar]
- [89].Wang X, Smith K, Pearson M, Hughes A, Cosden ML, Marcus J, Hess JF, Savage MJ, Rosahl T, Smith SM, Schachter JB, Uslaner JM (2018) Early intervention of tau pathology prevents behavioral changes in the rTg4510 mouse model of tauopathy. PLoS One 13, e0195486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Jul P, Volbracht C, de Jong IE, Helboe L, Elvang AB, Pedersen JT (2016) Hyperactivity with Agitative-Like Behavior in a Mouse Tauopathy Model. J Alzheimers Dis 49, 783–795. [DOI] [PubMed] [Google Scholar]
- [91].Becker E, Orellana Rios CL, Lahmann C, Rucker G, Bauer J, Boeker M (2018) Anxiety as a risk factor of Alzheimer’s disease and vascular dementia. Br J Psychiatry 213, 654–660. [DOI] [PubMed] [Google Scholar]
- [92].Rasmussen H, Rosness TA, Bosnes O, Salvesen Ø, Knutli M, Stordal E (2018) Anxiety and Depression as Risk Factors in Frontotemporal Dementia and Alzheimer’s Disease: The HUNT Study. Dementia and Geriatric Cognitive Disorders Extra 8, 414–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Wu C, Yang L, Li Y, Dong Y, Yang B, Tucker LD, Zong X, Zhang Q (2020) Effects of Exercise Training on Anxious-Depressive-like Behavior in Alzheimer Rat. Med Sci Sports Exerc 52, 1456–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Bogdanova OV, Kanekar S, D’Anci KE, Renshaw PF (2013) Factors influencing behavior in the forced swim test. Physiol Behav 118, 227–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Szot P, White SS, Greenup JL, Leverenz JB, Peskind ER, Raskind MA (2006) Compensatory changes in the noradrenergic nervous system in the locus ceruleus and hippocampus of postmortem subjects with Alzheimer’s disease and dementia with Lewy bodies. J Neurosci 26, 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Szot P, White SS, Greenup JL, Leverenz JB, Peskind ER, Raskind MA (2007) Changes in adrenoreceptors in the prefrontal cortex of subjects with dementia: evidence of compensatory changes. Neuroscience 146, 471–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Busche MA, Hyman BT (2020) Synergy between amyloid-beta and tau in Alzheimer’s disease. Nat Neurosci 23, 1183–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Crimins JL, Rocher AB, Luebke JI (2012) Electrophysiological changes precede morphological changes to frontal cortical pyramidal neurons in the rTg4510 mouse model of progressive tauopathy. Acta Neuropathol 124, 777–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Holth JK, Bomben VC, Reed JG, Inoue T, Younkin L, Younkin SG, Pautler RG, Botas J, Noebels JL (2013) Tau loss attenuates neuronal network hyperexcitability in mouse and Drosophila genetic models of epilepsy. J Neurosci 33, 1651–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Huijbers W, Schultz AP, Papp KV, LaPoint MR, Hanseeuw B, Chhatwal JP, Hedden T, Johnson KA, Sperling RA (2019) Tau Accumulation in Clinically Normal Older Adults Is Associated with Hippocampal Hyperactivity. J Neurosci 39, 548–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Busche MA, Wegmann S, Dujardin S, Commins C, Schiantarelli J, Klickstein N, Kamath TV, Carlson GA, Nelken I, Hyman BT (2019) Tau impairs neural circuits, dominating amyloid-beta effects, in Alzheimer models in vivo. Nat Neurosci 22, 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Kelly L, Seifi M, Ma R, Mitchell SJ, Rudolph U, Viola KL, Klein WL, Lambert JJ, Swinny JD (2021) Identification of intraneuronal amyloid beta oligomers in locus coeruleus neurons of Alzheimer’s patients and their potential impact on inhibitory neurotransmitter receptors and neuronal excitability. Neuropathol Appl Neurobiol 47, 488–505. [DOI] [PubMed] [Google Scholar]
- [103].Vankov A, Herve-Minvielle A, Sara SJ (1995) Response to novelty and its rapid habituation in locus coeruleus neurons of the freely exploring rat. Eur J Neurosci 7, 1180–1187. [DOI] [PubMed] [Google Scholar]
- [104].Herve-Minvielle A, Sara SJ (1995) Rapid habituation of auditory responses of locus coeruleus cells in anaesthetized and awake rats. Neuroreport 6, 1363–1368. [DOI] [PubMed] [Google Scholar]
- [105].Amanzio M, Geminiani G, Leotta D, Cappa S (2008) Metaphor comprehension in Alzheimer’s disease: novelty matters. Brain Lang 107, 1–10. [DOI] [PubMed] [Google Scholar]
- [106].Daffner KR, Mesulam MM, Cohen LG, Scinto LF (1999) Mechanisms underlying diminished novelty-seeking behavior in patients with probable Alzheimer’s disease. Neuropsychiatry Neuropsychol Behav Neurol 12, 58–66. [PubMed] [Google Scholar]
- [107].Fritsch T, Smyth KA, Debanne SM, Petot GJ, Friedland RP (2005) Participation in novelty-seeking leisure activities and Alzheimer’s disease. J Geriatr Psychiatry Neurol 18, 134–141. [DOI] [PubMed] [Google Scholar]
- [108].Weinshenker D (2018) Long Road to Ruin: Noradrenergic Dysfunction in Neurodegenerative Disease. Trends Neurosci 41, 211–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Hagena H, Hansen N, Manahan-Vaughan D (2016) beta-Adrenergic Control of Hippocampal Function: Subserving the Choreography of Synaptic Information Storage and Memory. Cereb Cortex 26, 1349–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Hansen N, Manahan-Vaughan D (2015) Locus Coeruleus Stimulation Facilitates Long-Term Depression in the Dentate Gyrus That Requires Activation of beta-Adrenergic Receptors. Cereb Cortex 25, 1889–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Giustino TF, Maren S (2018) Noradrenergic Modulation of Fear Conditioning and Extinction. Front Behav Neurosci 12, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Nam H, Kerman IA (2016) A2 noradrenergic neurons regulate forced swim test immobility. Physiol Behav 165, 339–349. [DOI] [PubMed] [Google Scholar]
- [113].Rittenhouse PA, Lopez-Rubalcava C, Stanwood GD, Lucki I (2002) Amplified behavioral and endocrine responses to forced swim stress in the Wistar-Kyoto rat. Psychoneuroendocrinology 27, 303–318. [DOI] [PubMed] [Google Scholar]
- [114].Tillage RP, Sciolino NR, Plummer NW, Lustberg D, Liles LC, Hsiang M, Powell JM, Smith KG, Jensen P, Weinshenker D (2020) Elimination of galanin synthesis in noradrenergic neurons reduces galanin in select brain areas and promotes active coping behaviors. Brain Struct Funct 225, 785–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Holets VR, Hokfelt T, Rokaeus A, Terenius L, Goldstein M (1988) Locus coeruleus neurons in the rat containing neuropeptide Y, tyrosine hydroxylase or galanin and their efferent projections to the spinal cord, cerebral cortex and hypothalamus. Neuroscience 24, 893–906. [DOI] [PubMed] [Google Scholar]
- [116].Miller MA, Kolb PE, Leverenz JB, Peskind ER, Raskind MA (1999) Preservation of noradrenergic neurons in the locus ceruleus that coexpress galanin mRNA in Alzheimer’s disease. J Neurochem 73, 2028–2036. [PubMed] [Google Scholar]
- [117].Olpe HR, Steinmann MW (1982) Age-related decline in the activity of noradrenergic neurons of the rat locus coeruleus. Brain Res 251, 174–176. [DOI] [PubMed] [Google Scholar]
- [118].Peskind ER, Tsuang DW, Bonner LT, Pascualy M, Riekse RG, Snowden MB, Thomas R, Raskind MA (2005) Propranolol for disruptive behaviors in nursing home residents with probable or possible Alzheimer disease: a placebo-controlled study. Alzheimer Dis Assoc Disord 19, 23–28. [DOI] [PubMed] [Google Scholar]
- [119].Wang LY, Shofer JB, Rohde K, Hart KL, Hoff DJ, McFall YH, Raskind MA, Peskind ER (2009) Prazosin for the treatment of behavioral symptoms in patients with Alzheimer disease with agitation and aggression. Am J Geriatr Psychiatry 17, 744–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Levey AI, Qiu D, Zhao L, Hu WT, Duong DM, Higginbotham L, Dammer EB, Seyfried NT, Wingo TS, Hales CM, Gamez Tansey M, Goldstein DS, Abrol A, Calhoun VD, Goldstein FC, Hajjar I, Fagan AM, Galasko D, Edland SD, Hanfelt J, Lah JJ, Weinshenker D (2021) A phase II study repurposing atomoxetine for neuroprotection in mild cognitive impairment. Brain. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.