

Abstract
Background and aims:
Isolation of cellular constituents from the mouse aorta is commonly used for expression or functional analyses in atherosclerosis research. However, current procedures to isolate primary cells are difficult, inefficient, and require separate mice. RNA extraction from aortic intima and media for transcriptomic analysis is also considered difficult with mixed RNA yields. To address these gaps, we provide: 1) a rapid, efficient protocol to isolate and culture diverse cell types concomitantly from the mouse aorta using immunomagnetic cell isolation; and 2) an optimized aortic intimal peeling technique for efficient RNA isolation from the intima and media.
Methods and Results:
Aortic cells were obtained using an enzymatic solution and different cell types were isolated by magnetic beads conjugated to antibodies targeting endothelial cells (CD31+), leukocytes (CD45+), and fibroblast cells (CD90.2+), and smooth muscle cells were isolated by negative selection. Our protocol allows the isolation of relatively large numbers of cells (10,000 cells per aorta) in a predictable manner with high purity (>90%) verified by cell-marker gene expression, immunofluorescence, and flow cytometry. These cells are all functionally active when grown in cell culture. We also provide a rapid method to collect aortic intima-enriched RNA from Ldlr−/− mice utilizing an intima peeling approach and assess transcriptomic profiling associated with accelerated lesion formation.
Conclusions:
This protocol provides an effective means for magnetic bead-based isolation of different cell types from the mouse aortic wall, and the isolated cells can be utilized for functional and mechanistic studies for a range of vascular diseases including atherosclerosis.
Keywords: Atherosclerosis, aortic primary cells, magnetic bead cell isolation, intima peeling
Graphical Abstract
1. INTRODUCTION
Atherosclerosis and its attendant ischemic cardiovascular complications have emerged as the leading causes of death in industrialized countries.1, 2 The increased prevalence of cardiovascular disease has become a global health crisis in recent years owing to the progressive aging of our societies and the consequences of modern unhealthy lifestyles, posing a serious threat to public health. Atherosclerosis is a chronic inflammatory disease, characterized by the formation and growth of a lipid-laden plaque within the vasculature. This multifactorial process is characterized by endothelial cell (EC) dysfunction, monocyte recruitment and adhesion, foam cell formation, smooth muscle cell (SMC) migration and remodeling, and extracellular matrix remodeling and fibrosis.2, 3 Isolation of the cellular constituents of the mouse aorta is commonly used for expression or functional analyses to investigate different aspects of plaque formation in experimental atherosclerosis models. However, research in this area is hampered by the inefficiency of current isolation protocols, often requiring the use of separate mice for each cell type desired.
To date, there are several reported protocols for obtaining different cell types from mouse aorta; however, these procedures are either simply focused on isolation of one specific cell type by selection for specific markers or require long periods (weeks) of time for the isolated primary cells to grow and proliferate in culture.4–6 Herein, our protocol provides a rapid and efficient method to simultaneously isolate mouse aortic cells including ECs, leukocytes, SMCs, and fibroblasts. Furthermore, we were able to obtain increased numbers of viable cells in a predictable manner (approximately 10,000 cells per mouse aorta after enzymatic digestion) compared to conventional methods, and over 90% purity of the magnetic beads-selected cells as verified by immunofluorescence staining, transcript expression, and flow cytometry.
In addition, we also provide a rapid and simple method to collect aortic intima-enriched RNA utilizing an intima peeling approach, which was used to assess transcriptomic profiling from Ldlr−/− mice on a high fat, high sucrose-containing (HFSC) diet in a model of accelerated lesion formation. 7
We anticipate that this magnetic bead-based protocol will provide a more effective means for simultaneous isolation of different cell types from the mouse aortic wall. The isolated aortic cells, together with the RNA collected from aortic intima and media, can be utilized for functional and mechanistic studies in a broad range of applications for vascular diseases including atherosclerosis.
2. MATERIALS AND METHODS
All methods are described in detail. The data that support the findings of this study are available from the corresponding author on reasonable request.
Detailed methods are provided in the online Supplementary Material.
2.1. Enzymatic digestion of aorta
One isolated aorta was placed into a 1.5 ml tube containing 1 ml of enzyme mix (Supplementary Table 1) and was chopped into small pieces with scissors in the enzyme mix. Aorta were further digested at 37°C for 20-45min at 750 rpm, stopping as soon as digestion completed with the addition of 10ml of 0.4% BSA-DPBS. After washing a 70 μm filter with 2 ml 0.4% BSA-DPBS, the digested cell suspensions were then filtered. After centrifugation for 5 min at 1750 RPM at 4 °C, digestions were resuspended in 10 ml 0.4% BSA-DPBS buffer. After centrifugation for 5 min at 1750 RPM, digested cells were resuspended in 100 μl of 0.4% BSA-DPBS. The single cell suspensions were stained with trypan blue and live cells counted with hemocytometer. Approximately 100,000 cells can be isolated from one mouse aorta.
2.2. CD45+ leukocyte isolation
After resuspension of the cell pellet in 90 μl of buffer per 107 total cells, 10 μl of anti-CD45 MACS MicroBeads was added, mixed, and incubated for 15 min at 4°C. After washing cells by adding 2 mL of buffer and centrifuging at 300xg for 10 min, supernatants were completely removed, and cell pellets were resuspended in 500 μl of buffer. After placement of an LS column in the magnetic field of a suitable MACS Separator, the columns were washed by rinsing with 3 ml of buffer, and the cell suspension was applied into the column. After pre-washing the column 3 times with 0.4% BSA-DPBS, the total effluent was collected by passing the cell suspension through the column as CD45- cells. After removal of the column from the separator and placing it on 15 ml tube, 3 ml of buffer was pipetted onto the column, and the magnetically labeled CD45+ cells were immediately flushed out by pushing the plunger into the column.
2.3. CD90.2+ cell isolation
After centrifugation of CD45- cell suspensions at 300xg for 10 min and removal of supernatants, the cell pellets were resuspended in 100 μl of buffer. Cells were stained with anti-CD90.2 by adding 10 μl CD90.2-PE antibody, mixing well, and incubating for 15 min in the dark at 4°C. Cells were washed by adding 2 ml of buffer, centrifuging at 300xg for 10 min, and resuspending cell pellets in 80 μL of buffer. 20 μl of anti-PE MACS MicroBeads were added, mixed, and incubated for another 15 min at 4°C. Cells were washed by adding 2 ml of buffer, centrifuging at 300xg for 10 min and resuspending the cell pellet in 500 μl of buffer. After pre-washing the column 3 times with 0.4% BSA-DPBS, the total effluent was collected by passing the cell suspension through the column as CD45- CD90.2- cell. After removal of the column from the separator and placing it on a 15 ml tube, 3 ml of buffer was added onto the column and the magnetically labeled CD90.2+ cells were flushed out by pushing the plunger into the column.
2.4. CD31+ EC and smooth muscle cell (CD45-CD90.2-CD31- cell) isolation
After centrifugation of the CD45-CD90.2- cell suspension at 300xg for 10 min, the cell pellet was resuspended in 90 μl of buffer, 10 μl of anti-CD31 MACS MicroBeads were added, mixed, and incubated for 15min at 4°C. Cells are washed by adding 2 ml of buffer and centrifugation at 300xg for 10 min and resuspension of cell pellet in 500 μl of buffer. The column was pre-washed 3 times with 0.4% BSA-DPBS prior to the selection of the cells, and total effluent was collected by passing the cell suspension through the column as CD45-CD90.2-CD31- cells, which were considered smooth muscle cells. After removal of the column from the separator, the magnetically labeled CD31+ cells were flushed out with buffer by pushing the plunger into the column.
2.5. Intimal RNA isolation from aorta tissue
After preparing a 5 ml syringe with cold DPBS with winged infusion set needle, the isolated aorta (from ascending to diaphragm) was placed in a 100 mm culture dish. Under the dissecting microscopy, the winged infusion set needle was inserted into the diaphragm end of the aorta and the aorta carefully flushed with cold DPBS for 10 s. The aorta should be blood free (clean and white) after the DPBS perfusion. After preparing a 5 ml syringe filled with TRIzol reagent (Invitrogen) and connecting the syringe with winged infusion set needle, the prepared winged infusion set needle was inserted into the diaphragm end of aorta and the aorta was carefully flushed with TRIzol for 10 s, followed by a 10 s pause, and flushed another 10 s. The TRIzol was collected in an Eppendorf tube (~700 μl in total), and snap-frozen in liquid nitrogen (related to Fig. 3A).
Fig. 3.
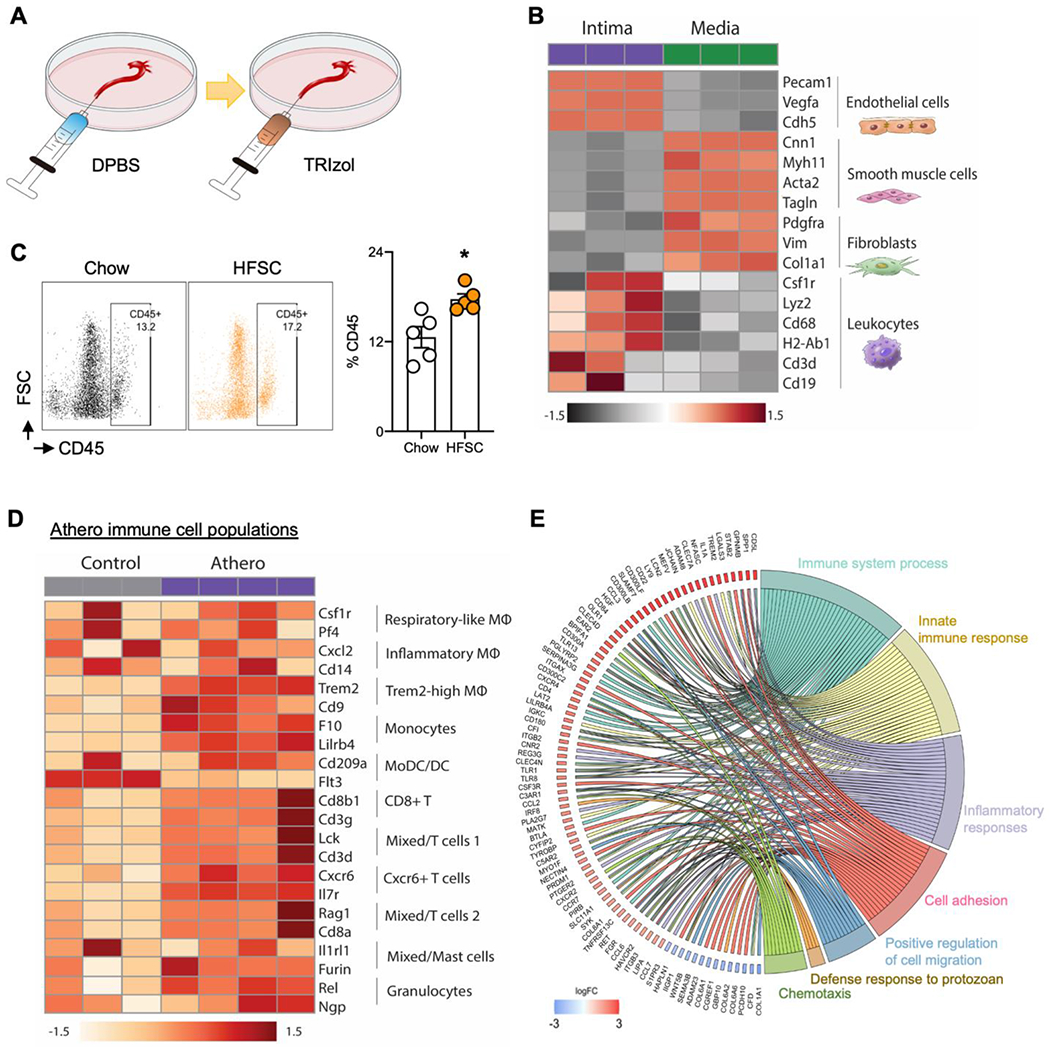
Intima peeling for isolation of mouse aortic intima and media alongside cell heterogeneity and gene enriched pathway analyses from control and HFSC atherosclerotic mouse aortas.
(A)Schematic illustration of intima peeling using TRIzol in aorta of eight-week-old C57BL/6 mice. (B) Heatmap showing differentially expressed aortic major cell marker genes in aortic intima and media by bulk RNA-seq analysis (FDR<0.05). (C) Flow cytometry showing the CD45+ leukocytes in comparison of aortic cell digestions from atherosclerosis (LDLR−/− mice fed with HFSC for 12 weeks) and chow diet controls. (D) Heatmap showing immune cells heterogeneity in atherosclerotic aorta (LDLR−/− mice fed with HFSC for 12 weeks) compared to chow control (FDR, <0.05). (E) GOChord plot showing the significantly regulated genes (log2 fold change, >1.5; FDR, <0.05) involved in the top 7 enriched pathways in the aortic intima from the HFSC atherosclerotic aorta and chow control.
3. Results
3.1. Aortic digestion protocol for FACS and single cell profiling
Our digestion protocol from aorta of eight-week-old C57BL/6 mice yielded relatively large numbers of cells (100,000 per mouse aorta) and high viability (over 90%) was confirmed by trypan blue (Fig. 1A and B). To identify the diverse types of aortic cell populations under different diet or disease models, flow cytometry and single-cell transcriptomic analysis are two commonly used methods in which the quality of the single cell suspension is one of the most important key steps for orchestrating both analyses. Flow cytometry profiling and gating strategy allowed for targeting major cellular constituents of the vessel wall including CD45+ cells (leukocytes), CD90.2+ cells (fibroblasts), CD31+ (endothelial cells), α-SMA (smooth muscle cells), F4/80 (macrophage), CD3+ cells (total T cells), CD8+ and CD4+ T cells (Supplementary Fig. 1A). We next performed a single-cell analysis on digested aorta. A total of 71,446 cells from 8 mouse aortas met quality control metrics. By using the digestion protocol, we obtained high quality of single-cell sequencing data (Supplementary Fig. 2). The clusters were identified based on the identified markers genes (Supplementary Table II) and was visualized using embedding algorithms uniform manifold approximation and projection (UMAP) that classified the cells into 8 major subtypes (Supplementary Fig. 3A–D). The aortic gene markers expression in each cell type was further confirmed in UMAP and violin plot (Supplementary Fig. 4 and 5). Taken together, our optimized digestion protocol provides an opportunity to obtain high-quality living cells for subsequent research including both flow cytometry and single-cell transcriptomic analysis.
Fig. 1.
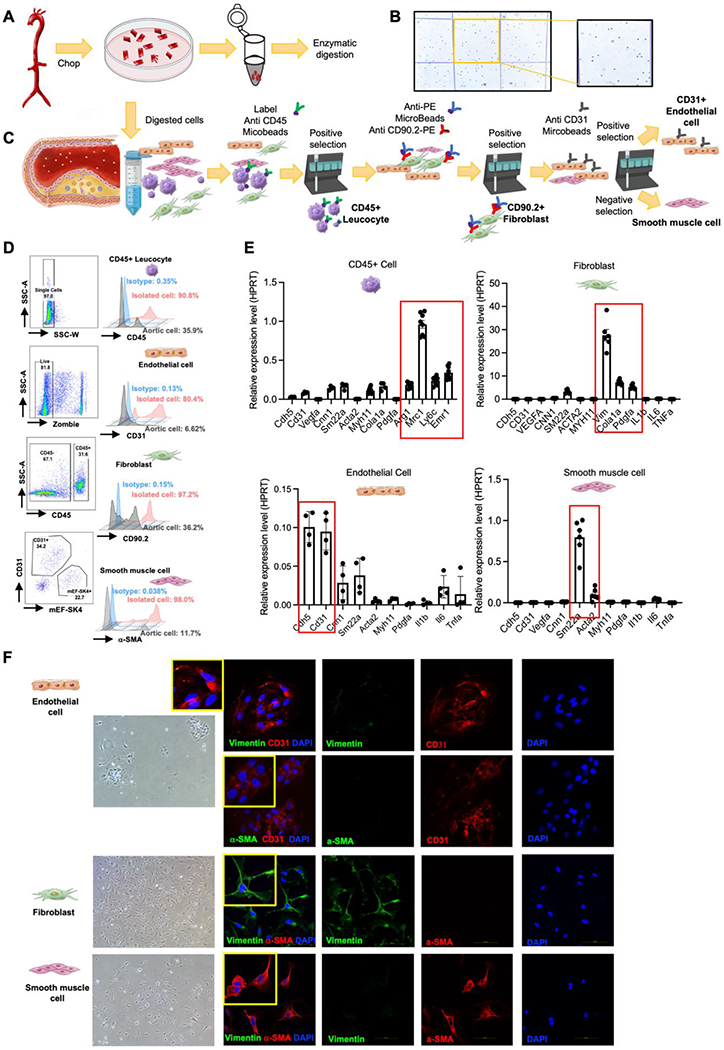
Enzymatic digestion of aortic cells and magnetic beads-based isolation of primary aortic cells.
(A) Schematic illustration of the enzymatic digestion process of aortic cells from eight-week-old C57BL/6 mice. (B) Digested aortic cell suspension stained with trypan blue. (C) Schematic of aortic primary cell isolation. (D) Purity verification by flow cytometry: analysis strategy, representative flow cytometry histogram plot for CD90.2, mEF-SK4, α-SMA, and CD31 staining in isolated CD45+ leucocyte, representative flow cytometry histogram plot for CD45, CD90.2, mEF-SK4, and α-SMA staining in isolated ECs, representative flow cytometry histogram plot for CD45, mEF-SK4, α-SMA, and CD31 staining in isolated fibroblast cells, representative flow cytometry histogram plot for CD45, CD90.2, mEF-SK4, and CD31 staining in isolated smooth muscle cells. (E) Expression of different gene markers in isolated CD45+ leucocytes, fibroblasts, ECs and SMCs from aorta. Normalized by HPRT. (F) Representative Immunofluorescence staining for vimentin and CD31, and α-SMA and CD31 in isolated CD45-CD90.2-CD31+ ECs. Representative immunofluorescence staining for vimentin and α-SMA in isolated CD45-CD90.2+ fibroblasts. Representative Immunofluorescence staining for vimentin and α-SMA in isolated CD45-CD90.2-CD31- SMCs.
3.2. Development of a rapid, high yield, high purity approach to isolate aortic cells using magnetic beads
To develop a more efficient strategy to isolate specific aortic cell types without the need for flow cytometry, we applied a magnetic beads-based isolation approach with the anticipation that such a rapid approach would provide high yields and purity. To confirm the purity of targeted aortic cells isolated by magnetic beads from eight-week-old C57BL/6 mice (Fig. 1C), a comprehensive set of methods including flow cytometry, RT qPCR, and immunofluorescent staining was performed. Flow cytometry analysis showed the purity of isolated leukocytes expressed high levels of CD45 (90.8%), and Isolated endothelial cells were enriched in CD31 (80.4%). Similarly, isolated CD90.2+ fibroblasts (97.2%) showed specific enrichment for fibroblast markers CD90.2 and mEF-SK4, and isolated vascular smooth muscle cells had expression of α-SMA (~98%) (Fig. 1D and Supplementary Fig. 1B). We next measured the expression of cell markers for each type of aortic cells and found isolated leukocytes highly expressed Mrc1, Arg1, Ly6c and Emr1; fibroblasts highly expressed Vim, Cola1a, and Pdgfa; endothelial cells highly expressed Cdh5 and Cd31; and smooth muscle cells highly expressed Sm22a and Acta2 (Fig. 1E). Additionally, isolated cells were cultured in the dish for 1 passage and stained with cell markers including CD31, Vimentin, and α-SMA (Fig. 1F). The immunofluorescent staining results showed that endothelial cells specifically expressed CD31, fibroblast cells expressed Vimentin, and smooth muscle cells expressed α-SMA. Collectively, these isolated aortic cells showed high purity by using the magnetic beads-based isolation method.
3.4. Functional characterization of magnetic beads isolated primary aortic cells
To confirm the phenotypic function of aortic cells after magnetic beads-based isolation, we performed different functional assays for each type of cells. We treated primary ECs with lipopolysaccharide (LPS) (1μg/ml, 24 h) and found that endothelial cell adhesion molecules including Sele, Icam-1 and Vcam-1 were significantly upregulated (Supplementary Fig. 6A).
Vascular endothelial growth factor (VEGF) is a potent angiogenic factor and was first described as an essential growth factor for vascular ECs.8 VEGF signaling increases production of nitric oxide in vascular ECs via activation of eNOS.9 To assess the functionality of ECs after magnetic beads-based isolation, primary ECs were treated with VEGF (100 ng/ml) for 10 minutes and eNOS phosphorylation was examined by Western blot analysis with a phospho-eNOS-specific (Ser1177) antibody. Isolated ECs showed increased eNOS activation after 10 minutes in response to VEGF stimulation (Fig. 2A and Supplementary Fig. 6B). The tube-like network formation assay in Matrigel is a rapid and quantitative method for determining the angiogenic function of ECs10. As shown in Figure 2B and Supplementary Figure 7, isolated ECs demonstrated typical network tube-like structures in Matrigel within 6-8 hours.
Fig. 2.
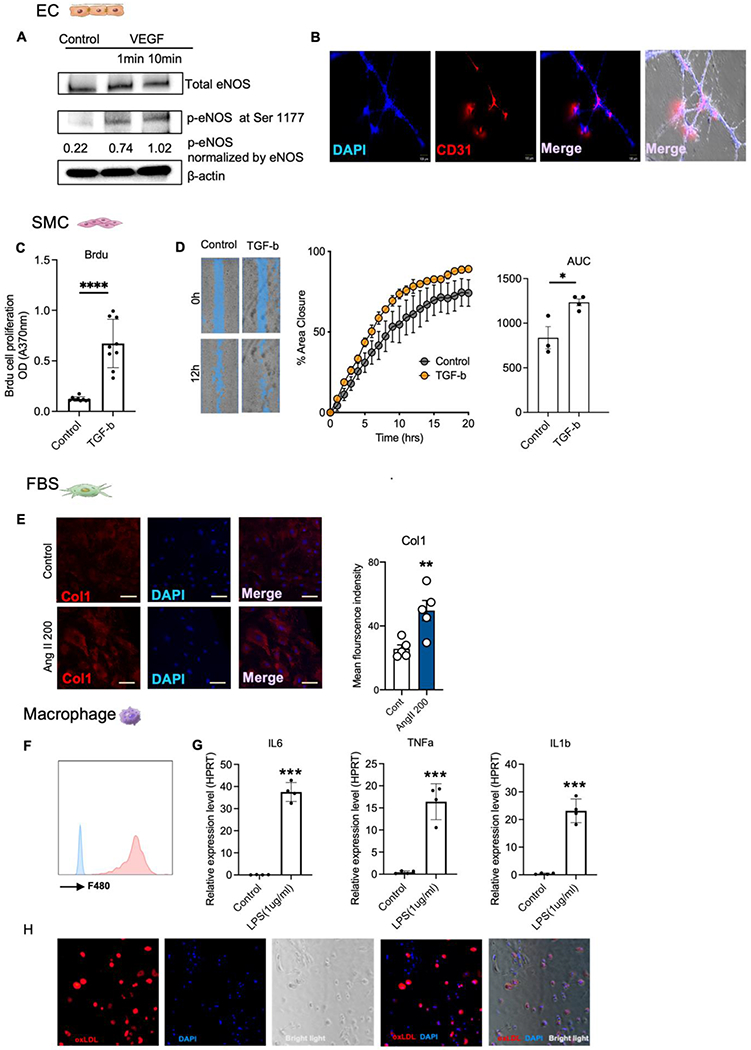
Functional identification of isolated primary aortic cells.
(A) Mouse primary ECs were treated with VEGF (100 ng/ml) for 1 and 10 min at 37 °C. Cell lysates were analyzed by immunoblot using polyclonal anti-phospho-serine 1179 eNOS or monoclonal eNOS antibody. (B) Mouse primary ECs were used for tube-like network formation assay. 30,000 cells were plated into 48 well plate coated with Matrigel (n = 6) and tube-formation were measures from 0 to 16h. (C) BrdU cell proliferation assay and quantitative analysis were performed to detect primary vascular smooth muscle cell proliferation in response to TGF-β1 treatment (4 ng/ml, 24h) (n = 9). (D) Cell migration assay and quantitative analysis were performed to detect primary vascular smooth muscle cell migration under TGF-β1 treatment (4 ng/ml, 24h) (n=3). (E) Immunofluorescent staining of Col1a1 in fibroblast treated with angiotensin II (200 nM, 24h). (F) Flow cytometry identification of primary aortic macrophages by F4/80. (G) A battery of pro-inflammatory genes were measure in primary aortic macrophages treated with LPS (1μg/ml, 24h). (H) Induction of foam-cell formation in primary aortic macrophages treated with Dil-oxLDL (100 μg/ml, 24h) in vitro.
We next tested primary SMCs in two related functional assays – scratch and BrdU assays. Both assays demonstrated that primary SMCs significantly increased proliferation and growth in response to transforming growth factor beta (TGF-β1) treatment (4 ng/mL for 24 h; Fig. 2C and D). Consistently Sm22a and Myh11 expression were significantly increased in SMCs under TGF-β1 treatment as demonstrated by RT-qPCR (Supplementary Fig. 6C). We treated primary fibroblasts with angiotensin II (up to 200nM for 24 h) and observed a robust increase in extracellular matrix associated genes including Col1a1, Col1a3, Col1a5, and matrix metalloproteinase-2 (Mmp2) as shown by RT-qPCR (Supplementary Fig.6D).11 Immunofluorescent staining also showed that angiotensin II (200 nM, 24h) significantly increased the presence of Cola1a1 in fibroblasts (Fig. 2E).
Finally, leukocytes were cultured in macrophage conditioned medium for 3 days prior to further characterization. The attached cells were found to be positive for the macrophage marker F4/80 as examined by flow cytometry (Fig. 2F). We treated the attached aortic macrophages with LPS (1 ug/ml, 24h) and found that the pro-inflammatory genes including Il6, Tnf-α, and Il1β were significantly increased (Fig. 2G). The attached aortic macrophages were also assessed for foam-cell formation.12, 13 After treatment with Dil-oxLDL (100 ug/ml, 24h), macrophages exhibited robust Dil-oxLDL uptake (red) as indicated by fluorescent lipid droplet presence (Fig. 2H). Taken together, these classical functional assays demonstrate that our approach of utilizing magnetic beads isolated a diverse set of primary aortic cells which maintain phenotypic and functional characteristics relevant to ex vivo studies used in atherosclerosis research.
3.5. Identification of aortic intima and media gene markers using intima peeling separation of aortic intima and media
An alternative rapid approach to identify transcripts only within the intima of the blood vessel wall is using an aortic intima peeling strategy where TRIzol is pumped through the aorta and lysates are collected (Fig. 3A). Using this separation method, we collected RNA and performed a transcriptomic analysis of the aortic intima and media from Ldlr−/− mice fed with either a chow diet (age-matched control) or a HFSC diet for 12 weeks. RNA sequencing (RNA-seq) profiling captured 10,033 differentially expressed genes comparing the aortic intima and media (false discovery rate (FDR), <0.05). Among the differentially expressed genes, we found that EC and leukocyte cell markers were significant enriched in the intima, while SMC and fibroblast gene markers were significantly enriched in the media (Fig. 3B, Supplementary Table II). Interestingly, different subsets of immune cells including macrophages, monocytes, dendritic cells, T cells, and natural killer cells were shown highly enriched in the intima, while mast cells were enriched in the media (Supplementary Fig. 9A, Supplementary Table III). These RNA-seq results demonstrate that the aortic intima peeling method is an effective strategy for separating the aortic intima and media.
3.6. Cell heterogeneity changes and dysregulated cell processes classically involved in atherosclerosis in mice discovered by using enzymatic digestion and intima peeling isolation of intima and media
To gain insights on dynamic transcript changes in the aorta in response to an accelerated high fat high sucrose (HFSC) containing diet in Ldlr−/− mice.7 We first used the aortic digestion protocol and compared the cell heterogeneity changes of using flow cytometry. As expected, leukocyte CD45-positive cells were significantly increased in Ldlr−/− mice fed a HFSC for 12 weeks compared to a regular chow diet group (age-matched control, 12 weeks on chow diet) (Fig. 3C), while CD31-positive (EC) and CD90.2-positive cells (fibroblast) showed no difference (Supplementary Fig. 8). Using the intima peeling method from both HFSC- and chow-diet fed Ldlr−/− groups, the aortic intima RNA was collected and RNA-seq profiling captured a total of 2,902 differentially expressed genes comparing the 2 groups (false discovery rate (FDR), <0.05). Heatmaps demonstrate that specific subsets of leukocyte markers were significantly increased in the HFSC atherosclerosis group, indicating a pro-inflammatory state within the intima layer in this mouse atherosclerosis model (Fig. 3D). To identify enriched pathways derived from these intimal transcripts, we performed a gene-annotation enrichment analysis on the set of differentially expressed genes using the DAVID functional annotation tool in combination with R package GOplot14 (adjusted P-value < 0.05). GOcircle plots, dot plot and GOchord plots of enriched pathways showed increased cell adhesion, innate immune response, positive regulation of cell migration, and chemotaxis in the intimal HFSC atherosclerotic group compared to the chow control group (Fig. 3E, Supplementary Fig 9B and C). Collectively, these data highlight the utility of using the intimal peeling technique to capture distinct transcripts and signaling pathways in response to an accelerated model of atherosclerosis.
4. DISCUSSION
Isolation of cellular constituents of the arterial wall is an integral part of vascular disease research, including atherosclerosis. The present study demonstrates two strategies: 1) a simple and rapid method to isolate and culture a diverse range of aortic cell types including endothelial cells, smooth muscle cells, fibroblasts, and leukocytes from the mouse arterial wall; and 2) an aortic intimal peeling technique using TRIzol to separate the intima and media in order to more effectively capture relevant transcripts in these distinct arterial wall compartments. Using our aortic digestion protocol, it is possible to investigate the extent of cellular heterogeneity within different cell populations through applying different assays including flow cytometry, immunofluorescent staining, and single-cell transcriptome analysis. We found that the specific enzymatic preparation is critically important for the viability and quality of aortic cells, and we specified the optimal enzymatic cocktail recipe and key steps in the methods section. Notably, the enzymatic digestion procedure in our protocol preserved the surface antigens of aortic cells well. The quality of single-cell suspension is the most important key steps for orchestrating further single-cell analysis and the digested cell suspension using our protocol aligns with the high-quality standard for droplet-based single-cell sequencing.
A unique aspect of this protocol is the ability to capture non-activated aortic fibroblasts. While CD90.2 can recognize both T cells and fibroblasts, our isolation method excludes the potential mix of T cells and fibroblasts. We first utilized anti-CD45 bead isolation to isolate leukocytes (including T lymphocytes) from aortic cells. The CD45-negative cells were then used for further isolation thereby enriching for fibroblasts. After isolation, we used DMEM with 10% FBS to culture the isolated fibroblasts and washed after 24h. Consequently, T cells will be nonadherent and cannot survive in the growing medium conditions. As shown in Figure. 1 and Supplementary Fig. 1, the purity of isolated fibroblasts in this method is over 97.2% and they did not express any CD45. In theory, other fibroblast markers could be considered such as fibroblast-specific protein 1 (FSP1) or collagen type 1 alpha 1 chain (Col1a1). However FSP-1 has been challenged for the specificity as a fibroblast marker since other cell types infiltrating injured tissues can also express FSP1 including vascular smooth muscle cells 15, macrophages16, and dendritic cells 17. Given that vascular smooth muscle cells are a major subtype of aortic cells and are isolated by negative selection in our method, FSP1 may not be a good choice in aortic fibroblast isolation. Similarly, Col1a1 is considered as an activation signature marker 18. For in vitro experiments, non-activated fibroblasts are typically preferred and can then be activated by diverse stimuli. In addition, during endothelial-mesenchymal transition, the expression of Col1a1 is increased in endothelial cells 19. Hence, it is not feasible to use Col1a1 as a selection marker to isolate fibroblast in this protocol.
Several previously published protocols have been focused on isolating and culture of mouse aortic cells.20 There were two commonly proposed approaches: one approach involved explanting fragmented vascular tissues and then allowing time for aortic cells (i.e. smooth muscle cells) to migrate out from the explanted vascular tissue fragments.21, 22 The other approach involved isolating aortic cells via enzymatic digestion of the aortic walls, followed by immunomagnetic cell sorting.11,1 Regarding the first approach, a higher risk of contamination is incurred since it requires several days for the cells to migrate from the chopped vascular tissue. Previously reported methods using the second common immunomagnetic cell sorting approach to isolate mouse aortic cells was developed to only focus on one specific cell type, i.e. ECs 23, or fibroblast cells.24 To our knowledge, no comparable mouse aortic cell isolation and culture protocol has so far allowed the successful simultaneous separation and yield of the four major cell types (ECs, SMCs, leukocytes, and fibroblasts) from the arterial wall. Our digestion and magnetic bead-based protocol allows isolation and comparison of the proportion of distinct cell types in aortic walls from mice with different genetic backgrounds or in response to diet or drug treatment. Our protocol also provides an option to utilize primary aortic cells including ECs, SMCs, and fibroblasts in culture for functional cell-based assays. From an individual aorta, yield of ECs using the isolation protocol is limited since the percentage of ECs in the aortic wall is relatively low compared to other constituents of the vessel wall. For the purpose of performing EC related functional assays, we would suggest to pool 10 aortas to achieve a reasonable number of ECs (typically yields 2^106 ECs). The isolated ECs could also be cultured in the dish for 3-5 days to achieve better yield for experimental purpose.
Lastly, we provide a quick and simple method to collect aortic intima-enriched RNA utilizing an intima peeling approach with TRIzol. This rapid and efficient RNA isolation technique allows investigation of transcriptome analysis in the aortic intima and media layers in different mouse models, which has been applied in our recent screening of novel non-coding transcripts in experimental atherosclerotic mice.25–30
4.1. Conclusion
We developed an improved, rapid, and effective method for magnetic beads-based isolation of different cell types from the mouse aorta and a quick RNA isolation technique from both aortic intima and media. The isolated and purified primary aortic cells maintain phenotypic characteristics in cell culture. The isolated aortic intima and media can be utilized for a range of transcriptomic analyses. Taken together, the magnetic beads-based isolation of aortic cells and intima and media RNA separation approach provide powerful tools for functional and mechanistic studies in vascular diseases including atherosclerosis.
Supplementary Material
Highlights.
We provide an optimized enzymatic digestion strategy for aortic cells, allowing a relatively large yield of aortic cells with over 90% viability. The cell suspension is ideal for further single cell transcriptomic analysis, flow cytometry analysis, and primary aortic cell isolation.
A new magnetic bead based-aortic cell selection method can provide a rapid and efficient approach to simultaneously isolate primary ECs, SMCs, leukocytes, and fibroblasts from the same mouse aorta.
The isolated primary cells can be cultured and utilized for further functional assays related to vascular biology and atherosclerosis research.
An optimized intima peeling approach enables efficient collection and separation of the aortic intima and media, providing enriched RNA from atherosclerotic lesions of the mouse aorta. The isolated aortic intima and media can be utilized for a range of transcriptomic analyses.
Acknowledgements
The authors would like to thank Ana Lay-Hong and Aniket P. Gad for their assistance with immunofluorescence imaging (Harvard Digestive Disease Center, NIH P30DK034854).
Financial support
This work was supported by the National Institutes of Health (HL115141, HL134849, HL148207, HL148355, and HL153356 to M.W. Feinberg), and the American Heart Association (18SFRN33900144 and 20SFRN35200163 to M.W. Feinberg).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Reference
- 1.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325 [DOI] [PubMed] [Google Scholar]
- 2.Libby P. Inflammation in atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:2045–2051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berliner JA, Navab M, Fogelman AM, Frank JS, Demer LL, Edwards PA, Watson AD, Lusis AJ. Atherosclerosis: Basic mechanisms: Oxidation, inflammation, and genetics. Circulation. 1995;91:2488–2496 [DOI] [PubMed] [Google Scholar]
- 4.Wang J-M, Chen AF, Zhang K. Isolation and primary culture of mouse aortic endothelial cells. JoVE (Journal of Visualized Experiments). 2016:e52965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hubert MO, Rodriguez-Vita J, Wiedmann L, Fischer A. Isolation of murine primary aortic smooth muscle cells. Bio-protocol. 2021;11:e3907–e3907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Metz RP, Patterson JL, Wilson E. Vascular smooth muscle cells: Isolation, culture, and characterization. Cardiovascular development. Springer; 2012:169–176. [DOI] [PubMed] [Google Scholar]
- 7.Neuhofer A, Wernly B, Leitner L, Sarabi A, Sommer NG, Staffler G, Zeyda M, Stulnig TM. An accelerated mouse model for atherosclerosis and adipose tissue inflammation. Cardiovascular diabetology. 2014;13:1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duffy AM, Bouchier-Hayes DJ, Harmey JH. Vascular endothelial growth factor (vegf) and its role in non-endothelial cells: Autocrine signalling by vegf. Madame curie bioscience database [internet]. Landes Bioscience; 2013. [Google Scholar]
- 9.Fukumura D, Gohongi T, Kadambi A, Izumi Y, Ang J, Yun C-O, Buerk DG, Huang PL, Jain RK. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proceedings of the National Academy of Sciences. 2001;98:2604–2609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arnaoutova I, George J, Kleinman HK, Benton G. The endothelial cell tube formation assay on basement membrane turns 20: State of the science and the art. Angiogenesis. 2009;12:267–274 [DOI] [PubMed] [Google Scholar]
- 11.Singh S, Torzewski M. Fibroblasts and their pathological functions in the fibrosis of aortic valve sclerosis and atherosclerosis. Biomolecules. 2019;9:472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu S, Huang Y, Xie Y, Lan T, Le K, Chen J, Chen S, Gao S, Xu X, Shen X. Evaluation of foam cell formation in cultured macrophages: An improved method with oil red o staining and dii-oxldl uptake. Cytotechnology. 2010;62:473–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mori M, Itabe H, Higashi Y, Fujimoto Y, Shiomi M, Yoshizumi M, Ouchi Y, Takano T. Foam cell formation containing lipid droplets enriched with free cholesterol by hyperlipidemic serum. Journal of lipid research. 2001;42:1771–1781 [PubMed] [Google Scholar]
- 14.Walter W, Sánchez-Cabo F, Ricote M. Goplot: An r package for visually combining expression data with functional analysis. Bioinformatics. 2015;31:2912–2914 [DOI] [PubMed] [Google Scholar]
- 15.Brisset AC, Hao H, Camenzind E, Bacchetta M, Geinoz A, Sanchez J-C, Chaponnier C, Gabbiani G, Bochaton-Piallat M-L. Intimal smooth muscle cells of porcine and human coronary artery express s100a4, a marker of the rhomboid phenotype in vitro. Circulation research. 2007;100:1055–1062 [DOI] [PubMed] [Google Scholar]
- 16.Österreicher CH, Penz-Österreicher M, Grivennikov SI, Guma M, Koltsova EK, Datz C, Sasik R, Hardiman G, Karin M, Brenner DA. Fibroblast-specific protein 1 identifies an inflammatory subpopulation of macrophages in the liver. Proceedings of the National Academy of Sciences. 2011;108:308–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boomershine CS, Chamberlain A, Kendall P, Afshar-Sharif A-R, Huang H, Washington MK, Lawson WE, Thomas JW, Blackwell TS, Bhowmick NA. Autoimmune pancreatitis results from loss of tgfβ signalling in s100a4-positive dendritic cells. Gut. 2009;58:1267–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F, Chu SG, Raby BA, Deluliis G, Januszyk M. Single-cell rna-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Science advances. 2020;6:eaba1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pinto MT, Melo FUF, Malta TM, Rodrigues ES, Placa JR, Silva WA Jr, Panepucci RA, Covas DT, de Oliveira Rodrigues C, Kashima S. Endothelial cells from different anatomical origin have distinct responses during snail/tgf-β2-mediated endothelial-mesenchymal transition. American Journal of Translational Research. 2018;10:4065. [PMC free article] [PubMed] [Google Scholar]
- 20.Ray JL, Leach R, Herbert J-M, Benson M. Isolation of vascular smooth muscle cells from a single murine aorta. Methods in Cell Science. 2001;23:185–188 [DOI] [PubMed] [Google Scholar]
- 21.Ross R. The smooth muscle cell ii. Growth of smooth muscle in culture and formation of elastic fibers. Journal of Cell Biology. 1971;50:172–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kobayashi M, Inoue K, Warabi E, Minami T, Kodama T. A simple method of isolating mouse aortic endothelial cells. Journal of atherosclerosis and thrombosis. 2005;12:138–142 [DOI] [PubMed] [Google Scholar]
- 23.Ni C-W, Kumar S, Ankeny G, Jo H. Development of immortalized mouse aortic endothelial cell lines. Vascular cell. 2014;6:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuwabara JT, Tallquist MD. Tracking adventitial fibroblast contribution to disease: A review of current methods to identify resident fibroblasts. Arteriosclerosis, thrombosis, and vascular biology. 2017;37:1598–1607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ni H, Haemmig S, Deng Y, Chen J, Simion V, Yang D, Sukhova G, Shvartz E, Wara AK, Cheng HS. A smooth muscle cell–enriched long noncoding rna regulates cell plasticity and atherosclerosis by interacting with serum response factor. Arteriosclerosis, Thrombosis, and Vascular Biology. 2021;41:2399–2416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Simion V, Zhou H, Haemmig S, Pierce JB, Mendes S, Tesmenitsky Y, Pérez-Cremades D, Lee JF, Chen AF, Ronda N. A macrophage-specific lncrna regulates apoptosis and atherosclerosis by tethering hur in the nucleus. Nature communications. 2020;11:1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou H, Simion V, Pierce JB, Haemmig S, Chen AF, Feinberg MW. Lncrna-map3k4 regulates vascular inflammation through the p38 mapk signaling pathway and cis-modulation of map3k4. The FASEB Journal. 2021;35:e21133. [DOI] [PubMed] [Google Scholar]
- 28.Simion V, Zhou H, Pierce JB, Yang D, Haemmig S, Tesmenitsky Y, Sukhova G, Stone PH, Libby P, Feinberg MW. Lncrna vinas regulates atherosclerosis by modulating nf-κb and mapk signaling. JCI insight. 2020;5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun X, He S, Wara A, Icli B, Shvartz E, Tesmenitsky Y, Belkin N, Li D, Blackwell TS, Sukhova GK. Systemic delivery of microrna-181b inhibits nuclear factor-Kb activation, vascular inflammation, and atherosclerosis in apolipoprotein e–deficient mice. Circulation research. 2014;114:32–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haemmig S, Yang D, Sun X, Das D, Ghaffari S, Molinaro R, Chen L, Deng Y, Freeman D, Moullan N. Long noncoding rna snhg12 integrates a DNA-pk–mediated DNA damage response and vascular senescence. Science translational medicine. 2020;12 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.