

Abstract
Histone deacetylases (HDACs) are a family of 18 epigenetic modifiers that fall into 4 classes. Histone deacetylase inhibitors (HDACi) are valid tools to assess HDAC functions. HDAC6 and HDAC10 belong to the class IIb subgroup of the HDAC family. The targets and biological functions of HDAC10 are ill-defined. This lack of knowledge is due to a lack of specific and potent HDAC10 inhibitors with cellular activity. Here, we have synthesized and characterized piperidine-4-acrylhydroxamates as potent and highly selective inhibitors of HDAC10. This was achieved by targeting the acidic gatekeeper residue Glu274 of HDAC10 with a basic piperidine moiety that mimics the interaction of the polyamine substrate of HDAC10. We have confirmed the binding modes of selected inhibitors using X-ray crystallography. Promising candidates were selected based on their specificity by in vitro profiling using recombinant HDACs. The most promising HDAC10 inhibitors 10c and 13b were tested for specificity in acute myeloid leukemia (AML) cells with the FLT3-ITD oncogene. By immunoblot experiments we assessed the hyperacetylation of histones and tubulin-α, which are class I and HDAC6 substrates, respectively. As validated test for HDAC10 inhibition we used flow cytometry assessing autolysosome formation in neuroblastoma and AML cells. We demonstrate that 10c and 13b inhibit HDAC10 with high specificity over HDAC6 and with no significant impact on class I HDACs. The accumulation of autolysosomes is not a consequence of apoptosis and 10c and 13b are not toxic for normal human kidney cells. These data show that 10c and 13b are nanomolar inhibitors of HDAC10 with high specificity. Thus, our new HDAC10 inhibitors are tools to identify the downstream targets and functions of HDAC10 in cells.
Keywords: Histone deacetylases (HDAC), HDAC10, drug design, ligand docking, chronic lymphoid leukemia, acute myeloid leukemia (AML), autophagy, lysosomes
1. Introduction
Histone deacetylases (HDACs) are epigenetic modifiers that control the acetylation of histones and non-histone proteins. This posttranslational modification regulates a plethora of physiologically relevant processes including cell proliferation, survival, genomic integrity, and protein homeostasis. Accordingly, HDACs are often dysregulated in transformed cells.1, 2 HDACs are grouped into 4 classes, class I (HDAC1, HDAC2, HDAC3, HDAC8), class IIa (HDAC4, HDAC5, HDAC7, HDAC9), class IIb (HDAC6, HDAC10), class III (sirtuins SIRT1–7), and class IV (HDAC11)1, 2 (Fig. 1). Sirtuins use NAD+ for the deacetylation of acetyl-lysine residues and all other HDACs catalyze this reaction via a Zn2+ located within their catalytic pockets.3 Meanwhile, the catalytic domains of HDAC subtypes display a high degree of homology whereas adjacent regions show greater structural differences as shown by solved crystal structures. HDAC6 and HDAC10 belong to the class IIb subgroup. While both catalytic domains are active in HDAC6, HDAC10 has an active domain and an inactive domain.
Figure 1:

Zn2+-dependent HDACs and examples of histone deacetylase inhibitors (HDACi). The catalytic domain(s) in the different Zn2+-dependent HDACs are represented in color. Examples of developed inhibitors are named.
HDAC inhibitors (HDACi) have been developed to address diseases which have been linked to overactive and overexpressed HDACs. The FDA has approved four pan-HDACi as salvage therapy for the treatment of cutaneous T-cell lymphoma and multiple myeloma. However, the lack of selectivity of these clinically approved drugs frequently causes dose limiting side-effects.1, 2, 4, 5 The inhibitory mechanism of HDACi relies on the complexation of their warheads (e.g., hydroxamic acid, thiol) with the Zn2+ ion in the catalytic pockets of HDACs.3 The benzamide derivative entinostat (MS-275), the aliphatic fatty acid valproic acid (VPA), and the two hydroxamic acid derivatives panobinostat (LBH589) and vorinostat (SAHA) are class-specific and pan-HDACi (Fig. 2).
Figure 2.
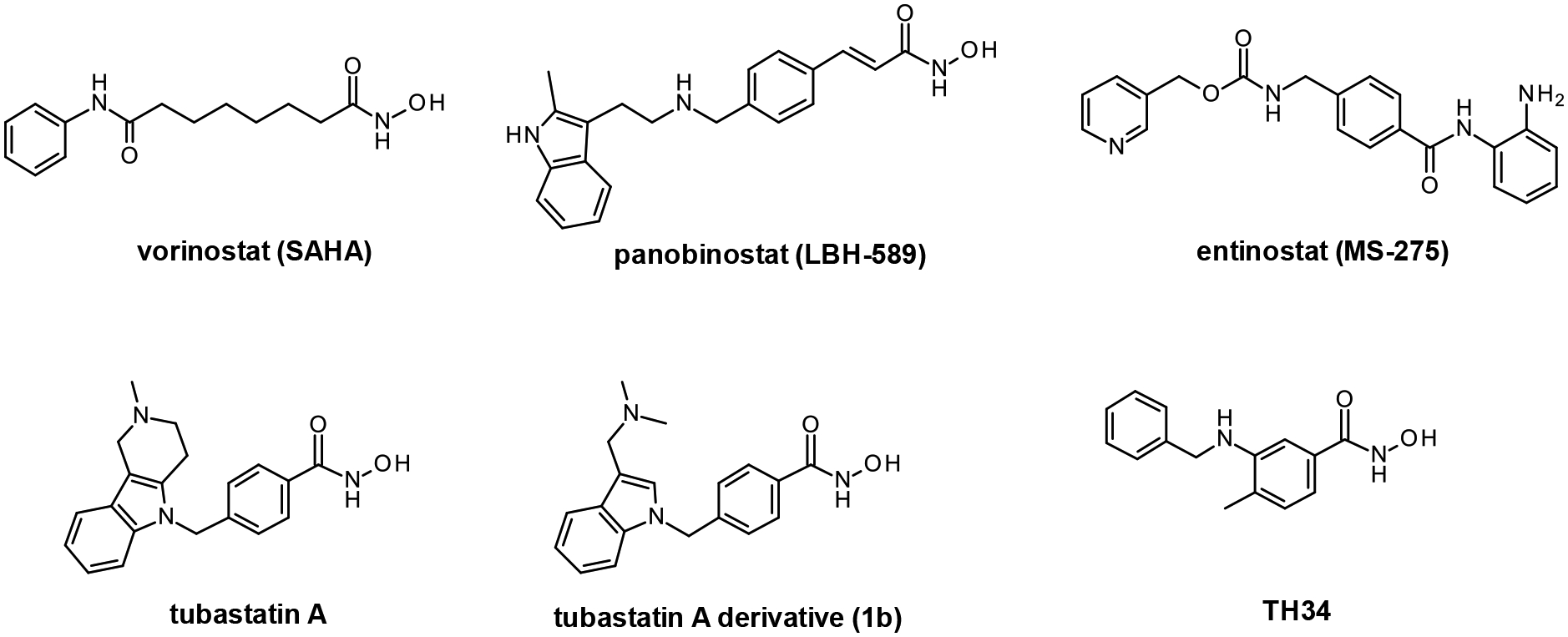
Structures of discussed HDAC inhibitors.
HDAC10 has unique functions and structural features in comparison with other HDAC isozymes, such as an active and an inactive deacetylase domain (Fig. 1). Recently, the substrate specificity of HDAC10 was determined to contrast with that of other isozymes: HDAC10 was discovered to be a polyamine deacetylase.6 Thus, HDAC10 has important non-protein, non-lysine deacetylase activity. Polyamines are regulators of protein homeostasis through macroautophagy (hereafter termed autophagy).6–8 This evolutionarily conserved pathway is activated upon starvation but also upon other types of stress, including DNA replication stress and DNA damage.9 During autophagy, cytoplasmic proteins and structures are embedded in autophagosomes that are subsequently digested with the help of lysosomal components.10 In neuroblastoma cells, the catalytic activity of HDAC10 is necessary for the process of autophagy (also termed autophagic flux) and inhibition of HDAC10 can cause an accumulation of autolysosomes.7 Moreover, inhibition of HDAC10 causes an accumulation of lysosomes in neuroblastoma cells.11 In cervical carcinoma cells, genetic knockout of HDAC10 disrupts chaperone-mediated autophagy (CMA). This process depends on lysosome-associated protein type 2A and promotes the lysosomal degradation of the glycolytic enzyme GAPDH.12
There is particularly little knowledge about the role of HDAC10 in leukemia.8 It was shown that an overexpression of HDAC10 in cultured chronic lymphoid leukemia (CLL) and mantle cell lymphoma cells (transformed B lymphocytes) caused cell cycle arrest and ultimately apoptosis.13 Whether this is due to non-physiological levels of HDAC10 is currently unknown. Analysis of HDAC expression in 32 primary CLL cells and normal lymphoid cells revealed that HDAC1, HDAC3, HADC6, HDAC7, HDAC9, HDAC10, SIRT1, and SIRT6 are overexpressed CLL cells. The high expression of these enzymes was associated with advanced disease and poor prognosis.14, 15 A further study compared 200 CLL patient samples at diagnosis and after relapse with normal B cells. This work revealed an association of poor prognosis with overexpression of HDAC7 and HDAC10 and reduced expression of HDAC6 and SIRT3 in CLL cells.16 While the impact of HDACs on the clinical course of CLL seems to be complex, both studies agree on a linkage between HDAC10 and worse prognosis. Since a full-body genetic elimination of HDAC10 in mice is not toxic for normal tissues,17 HDAC10 inhibition could be a safe strategy to combat tumor cells that depend on it. To date, nothing is published about the relevance of HDAC10 in other types of leukemia.
The use of HDAC10 inhibitors can help elucidate the biological relevance of this enzyme and whether it is a valid pharmacological target in disease. However, only a few inhibitors that target HDAC10 have been identified to date (Fig. 2).18 For example, tubastatin A and recently reported hybrid inhibitors based on the tubastatin A scaffold have been reported (1b).19 Additionally, TH34 (3-(N-benzylamino)-4-methylbenzhydroxamic acid) is a pan-HDAC6/8/10 inhibitor that was recently reported by us.20 Compounds containing a 2-(oxazol-2-yl)phenol moiety were also reported to inhibit HDAC1, HDAC6, and HDAC10.21 Thus, HDAC10 inhibitors with selectivity over the class I isozymes are available but the main challenge lies in achieving selectivity over the other class IIB member HDAC6.
2. Results and Discussion
2.1. Synthesis and in vitro testing of novel inhibitors
Based on the observation that TH34 is an inhibitor of HDAC10, but equally active on HDAC6,20 we started with the synthesis of a first series of benzhydroxamic acid derivatives that bear a basic amine in the capping group. The selection of capping groups as well as the position of the basic amine was guided by docking the compounds into the available X-ray structures of danio rerio HDAC10 (drHDAC10).18, 22, 23 The drHDAC10 isozyme exhibits essentially identical catalytic activity and substrate specificity in comparison with human HDAC10; moreover, drHDAC10 is more stable and amenable to crystallographic analysis for the determination of structure-function relationships. Additionally, a “humanized” version of drHDAC10 containing the A24E and D94A substitutions yields an active site contour that better mimics that of human HDAC10.18
Introducing a methylene-group between the benzhydroxamic core and the basic amine resulted in hits that showed a salt-bridge to the gatekeeper residue Glu274 of HDAC10 (Fig. S1, Supplementary Information) and were therefore prepared as described in the Methods section.
To evaluate the HDAC selectivity profile, we tested all synthesized compounds against drHDAC10 as well as human HDACs 1, 6, and 8 in vitro. In case of drHDAC10 we recently developed an enzymatic in vitro assay that was used in the current study.24 Compound 6a, bearing an N-piperidinomethylene capping group, showed only weak HDAC10 inhibition and also very low activity against other HDACs. Meanwhile, replacement of the piperidine ring of 6a with an N-methylpiperazine moiety yielded compound 6b which was found to a potent inhibitor against drHDAC10 (IC50 43 ± 7 nM). Substitution of the secondary amino group with different arylmethyl moieties similarly yielded compounds with strong inhibitory activity against drHDCA10 (4a-4c). Among these benzhydroxamic acid derivatives, 4c bearing a 3-thienylmethyl capping group showed the strongest HDAC10 inhibition (Table 1) with an IC50 of 24 ± 5 nM. The 2-thienlymethyl analogue 4b showed a slight decrease in the inhibitory activity (IC50 of 106 ± 28 nM). 4a with a simple benzyl capping group was also found to be a potent inhibitor of drHDAC10 (IC50 of 37 ± 10 nM).
Table 1:
Chemical structures and inhibition of drHDAC10.
| ID | structure | IC50 drHDAC10 [nM] |
|---|---|---|
| 4a |
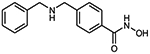
|
37 ± 10 |
| 4b |
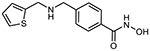
|
106 ± 28 |
| 4c |
|
24 ± 5 |
| 6a |
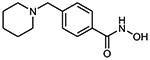
|
4 % @ 1 μM |
| 6b |
|
43 ± 7 |
| 10a |
|
11 ± 1 |
| 10b |
|
29 ± 6 |
| 10c |
|
20 ± 2 |
| 10d |
|
60 ± 5 |
| 10e |
|
530 ± 100 |
| 10f |
|
23 % @ 10 μM |
| 10g |
|
46 % @ 10 μM |
| 13a |
|
62 ± 18 |
| 13b |
|
58 ± 10 |
| 13c |
|
33 ± 3 |
| Tubastatin A |
|
220 ± 20 |
In addition we designed and prepared piperidine-4-acrylhydroxamic acid derivatives bearing different arylethylene or arylmethylene capping groups. These compounds were structurally modified by using docking studies in crystal structure of drHDAC10 and “humanized” drHDAC10. Docking of these derivatives into the crystal structure of HDAC10 showed that they are able to adopt a similar binding orientation as observed for the N8-acetylspermidine analogue inhibitor with an overlap of their basic amino group (Fig. S2 and S3, Supplement). The piperidine-4-acrylhydroxamic acid derivatives were able to undergo extensive interactions in the HDAC10 binding pocket which explain their strong inhibitory activity on this isoform (Fig. 3). The hydroxamate moiety chelates the catalytic zinc ion in a bidentate fashion and shows the typical hydrogen bond interactions with the neighbouring histidine and tyrosine residues. The protonated amine of the piperidine is placed between Asp94 and the gatekeeper residue Glu274 exhibiting a salt bridge interaction. Meanwhile, the capping groups displayed different interactions with the amino acid residues at the edge of the binding pocket due to the use of linkers of distinct lengths.
Figure 3:
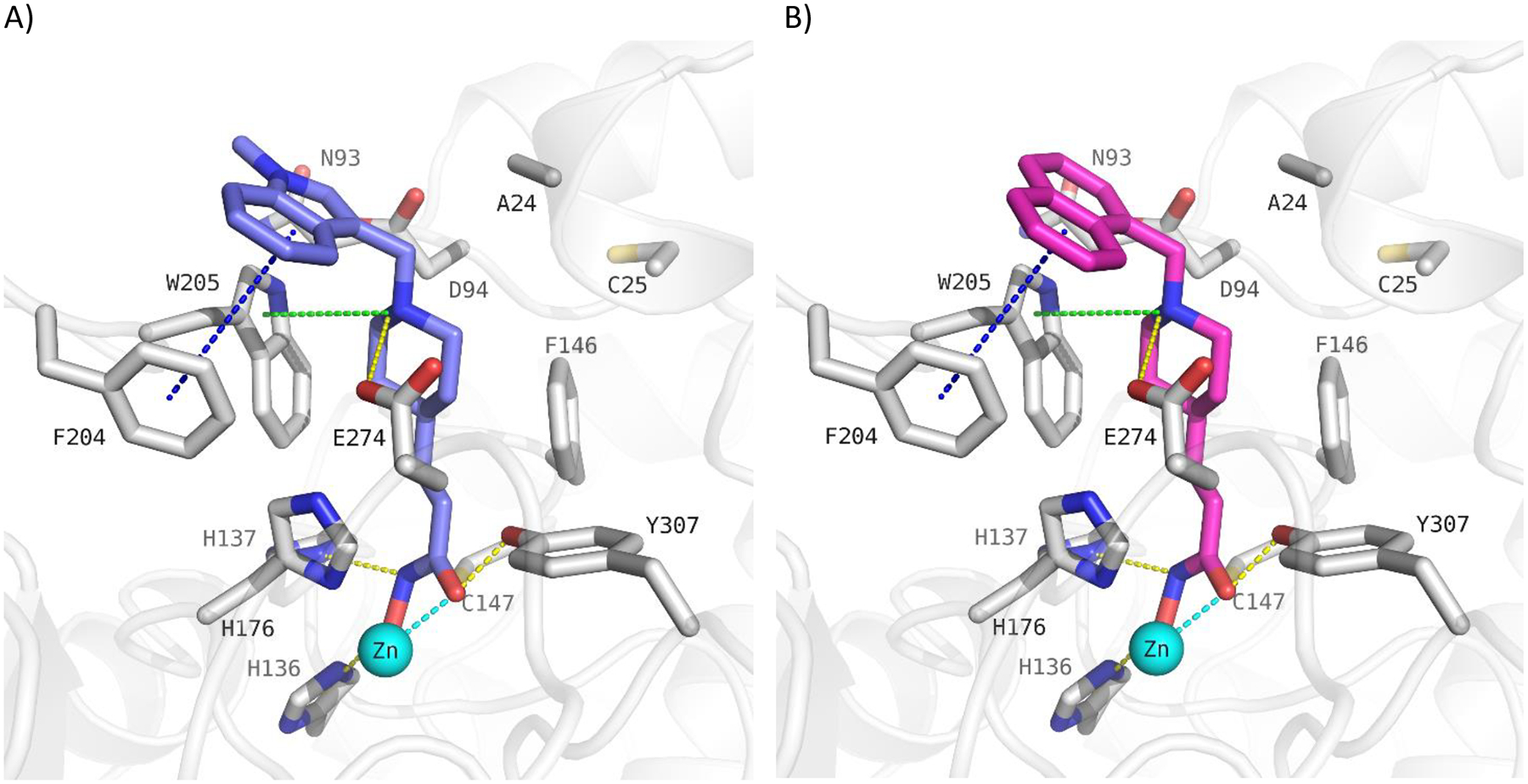
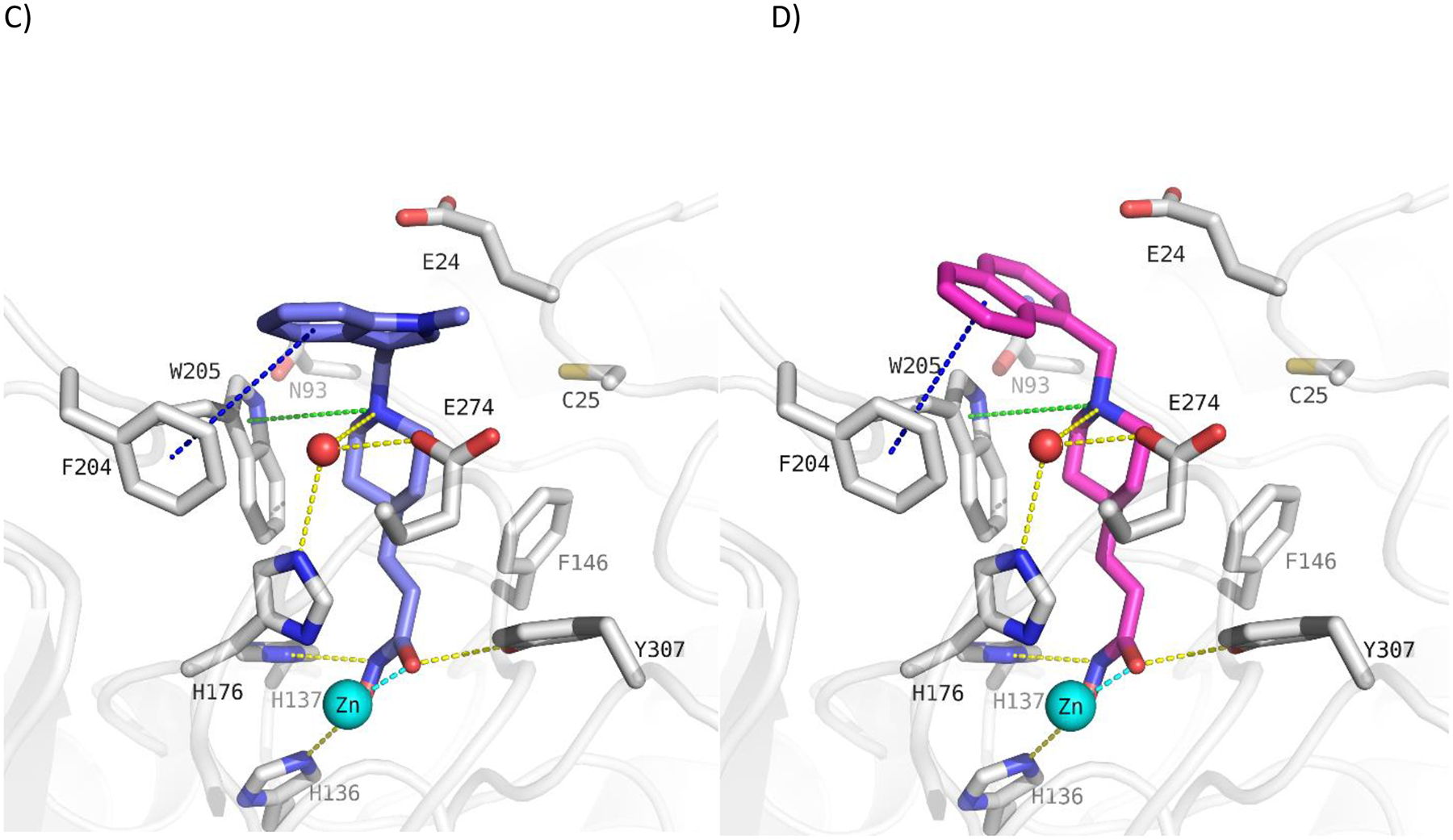
Docking poses calculated for selected HDAC10 inhibitors in the catalytic pocket of drHDAC10 (PDB ID: 5TD7; A and B) and the humanized form of drHDAC10 (PDB ID 6VNQ); C and D)). A) Binding mode of 13b (slate blue sticks) in drHDAC10 (PDB ID: 5TD7); B) Binding mode of 10c (pink sticks) in drHDAC10 (PDB ID: 5TD7); C) Binding of 13b (slate blue sticks) in the humanized form of drHDAC10 (PDB ID 6VNQ); D) Binding of 10c (pink sticks) in the humanized form of drHDAC10 (PDB ID 6VNQ). The Zn ion is shown as cyan sphere and water molecules as red spheres. Yellow dashed lines depict hydrogen bond interactions, cyan dashed lines metal chelation, green dashed lines cation-π interactions and blue-dashed lines π-π interactions.
To validate our approach to model HDAC10-inhibitor complexes and to strengthen our understanding of inhibitor structure-affinity relationships, we determined X-ray crystal structures of selected enzyme-inhibitor complexes. Electron density maps showing the binding of inhibitors 4c, 6b, 10a, and 10b to “humanized” drHDAC10 are presented in Fig. 4. Interestingly, hydroxamate-Zn2+ coordination is monodentate for 6b and bidentate for 4c, 10a, and 10b. The secondary amino group of 4c and the tertiary amino groups of 10a and 10b make a water-mediated hydrogen bond interaction with the active site gatekeeper residue, E274. A similar water-mediated interaction with E274 is observed for the binding of substrate N8-acetylspermidine, so this interaction presumably contributes to inhibitor selectivity for HDAC10.
Figure 4:
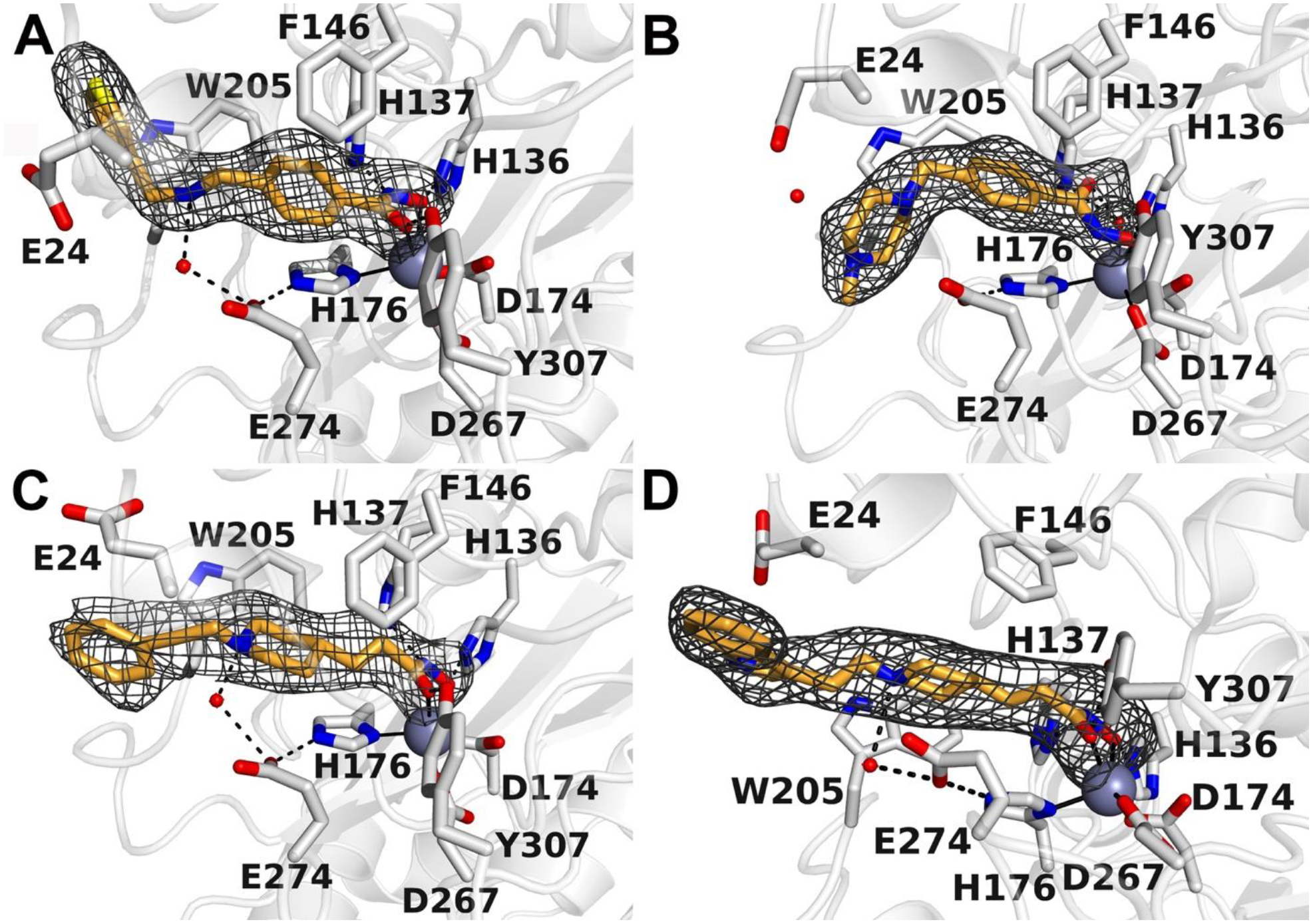
Polder omit electron density maps showing the binding of 4c (A), 6b (B), 10a (C), and 10b (D) in the active site of “humanized” drHDAC10. Atoms are color-coded as follows: C = gray, O = red, N = blue, S = yellow. Water molecules appear as small red spheres and the catalytic zinc ion is a large gray sphere. Hydrogen bond interactions are indicated by dashed black lines.
Except for 10f and 10g, which carry a p-tolylmethylene or biphenyl capping group, all piperidine-4-acrylhydroxamic acid derivatives described herein exhibited submicromolar inhibition of HDAC10, with the phenethyl derivative 10a showing the highest inhibitory potency (IC50 11 ± 1 nM). A consistent impact of the linker length on inhibitory activity could not be observed. While the benzyl and p-tolylmethylene derivatives (10e and 10f, respectively) showed a significant decrease in the HDAC10 inhibitory activity, the m- and p-chlorobenzyl analogues (10d and 13c, respectively) showed strong HDAC10 inhibition (IC50 60 ± 5 nM and 33 ± 3 nM, respectively).
Potent drHDAC10 inhibition was also found for compounds 10b, 10c, 13a and 13b, which all bear a bicyclic aromatic moiety as a capping group with either a methylene or ethylene spacer to the basic amino acid group. 10c has, just as 10e, a methyl linker between the tertiary amine and the capping group, but the aromatic system (naphthyl) is significantly larger so that a π- π stacking with Trp205 may occur. 10b has an ethyl linker and an indole cap group, which shows, that a combination of ethyl linker and bigger cap group can also lead to potent inhibitors. 13b and 13a show a slightly decreased inhibitory activity against drHDAC10 compared to 10b. This shows that the ethyl linker in combination with an indole capping group is favorable compared to a methyl linker in combination with an N-methylindole (13b) or benzothiophene capping group (13a). As exemplified by the predicted binding modes of 10c and 13b (Fig. 3) in drHDAC10 (PDB ID: 5TD7; A and B) and the humanized form of drHDAC10 (PDB ID 6VNQ), it can be observed that the bicyclic capping group is situated above Phe204 where it undergoes π-π stacking interactions. Additional interactions include the essential Zn2+ coordination via the hydroxamate group, as well as the commonly observed hydrogen bond interactions of the hydroxamic acid warhead with the side chains of His136, His137 and Tyr307. The protonated piperidine-NH forms a salt-bridge interaction to the gatekeeper amino acid Glu274 as well as cation-π interaction with Trp205.
The previously obtained docking poses in drHDAC10 (PDB ID:5TD7) and humanized drHDAC10 (PDB ID:6VNQ) were compared with the new crystal structures in order to assess the accuracy of the predicted binding mode.
For compound 4c, RMSD values of 2.8 Å and 1.7 Å were obtained for the ligands’ heavy atoms in drHDAC10 (PDB ID:5TD7) and in humanized drHDAC10 (PDB ID:6VNQ), respectively. The major deviations were observed for the capping group. In humanized drHDAC10 two residues are mutated as compared to drHDAC10, namely Asp94 to Ala94 and Ala24 to Glu24. The orientation of Glu24 in drHDAC10 hinders the capping thiophene group from occupying the same sub-pocket as observed in the resolved crystal structure of humanized drHDAC10 with 4c. Meanwhile, the obtained docking pose of 4c in humanized drHDAC10 (PDB ID:6VNQ) shows a stronger deviation from the experimentally determined binding mode. Whilst the key interactions with the neighboring residues are maintained, some deviations in the linker and capping group are observed. It’s worth mentioning that some difference in the used crystal structure with respect to the determined crystal structure are detected, which mainly include the conformation of the gate-keeper residue Glu274 and the location of the neighboring water molecule. These differences explain the variation in the obtained binding poses (Fig. S4a and S5a, Supplement).
For compound 6b, RMSD values of 1.3 Å in drHDAC10 (PDB ID:5TD7) and 1.5 Å in humanized drHDAC10 (PDB ID:6VNQ) were calculated for the docking poses of the ligand with respect to the experimentally determined binding mode. As previously described, the main difference lies in the coordination fashion of the zinc ion; in the crystal structure the hydroxamate moiety chelates the zinc ion in a monodentate manner (Fig. S4b and S5b, Supplement).
Meanwhile, for compounds 10a and 10b we observed a good overlap of the hydroxamate and linker moieties when comparing the respective crystal structures with the obtained docking poses. However, as observed for compound 4c, the orientation of the solvent-exposed capping groups shows stronger deviations with calculated RMSD values of 1.2 Å and 2.4 Å for 10a in drHDAC10 (PDB ID:5TD7) humanized drHDAC10 (PDB ID: 6VNQ) and 2.7 Å and 3.7 Å for 10b in drHDAC10 and humanized drHDAC10, respectively (Fig. S4c, d and S5c, d Supplement).
In summary, it can be stated that the piperidine-4-acrylhydroxamic acid scaffold is capable of making the desired interactions with the Zn2+ ion and the gatekeeper residue Glu274. The different orientations of the capping groups show that additional interactions can take place in the outer active site which might influence the inhibitory activity.
2.2. In vitro selectivity testing
To evaluate the HDAC selectivity profile, we tested all synthesized compounds against drHDAC10 as well as human HDACs 1, 6, and 8 in vitro. The benzhydroxamic acids show good hHDAC1 and hHDAC8 selectivity. The top hHDAC1 selectivity of this class was observed for 4c with a selectivity index (SI) of 417, the lowest selectivity was observed for 4b with an SI of 85. For the hHDAC8 selectivity, the SI are between 30 for 4b and 94 for 4a. The benzhydroxamic acid derivatives barely show selectivity against hHDAC6, only 4a, 4c and 6b showed a slight selectivity with an SI of 17, 20 and 9; respectively. Since HDAC6 is structurally related to HDAC10, one of the main tasks for the development of HDAC10 inhibitors is to achieve good selectivity against hHDAC6. This has been achieved with the piperidine-4-acrylhydroxamic acid scaffold. 10a shows a strong selectivity with an SI of 395 against hHDAC6 and 1454 against hHDAC1. In addition to the strongest drHDAC10 inhibition, 10a also shows the highest selectivity against hHDAC1 and hHDAC6. In addition, a good hHDAC8 selectivity with an SI of 23 for 10a is observed. Most of the piperidine-4-acrylhydroxamic acid derivatives are weak HDAC6 inhibitors with the exception of 13c (430 ± 50 nM).
2.3. Cellular testing
Next, we tested the specificity of the most promising HDAC10 inhibitors from the in vitro characterization (10c and 13b) in cells. An increased lysosomal compartment has been identified as a marker for HDAC10 inhibition in neuroblastoma cells.11 Compounds with potent drHDAC10 inhibitory activity and good selectivity against hHDAC1, hHDAC6 and hHDAC8, were first tested in SK-N-BE(2)-C neuroblastoma cells. 10c and 13b induced the expansion and acidification of the lysosomal compartment, as measured using the pH-dependent LysoTracker DND-99 fluorescent probe (Fig. S6 Supplement). This increase of the lysosomal compartment is in line with the described role for HDAC10 in lysosomal homeostasis and autophagy in neuroblastoma.7, 11
Since there are no data on a possible role for HDAC10 in autophagy regulation in leukemic cells, we applied 10c and 13b to MV4–11 cells (Fig. 5). These cells are derived from a childhood leukemia and carry an internal tandem duplication in the FMS-like tyrosine kinase (FLT3-ITD). This subtype of acute myeloid leukemia (AML) cells is characterized by poor prognosis and relapse.11, 25 The dye cyto-ID is incorporated into and therefore a marker of autophagic vesicles (pre-autophagosomes, autophagosomes, phagolysosomes),26 which have been shown to accumulate in neuroblastoma cells with inhibited HDAC10.7 Flow cytometry illustrated that 10c and 13b significantly induced the accumulation of cyto-ID positive vesicles in MV4–11 cells after 24 h (Fig. 5A). This encouraged us to further analyze the specificity of 10c and 13b in cells.
Figure 5:
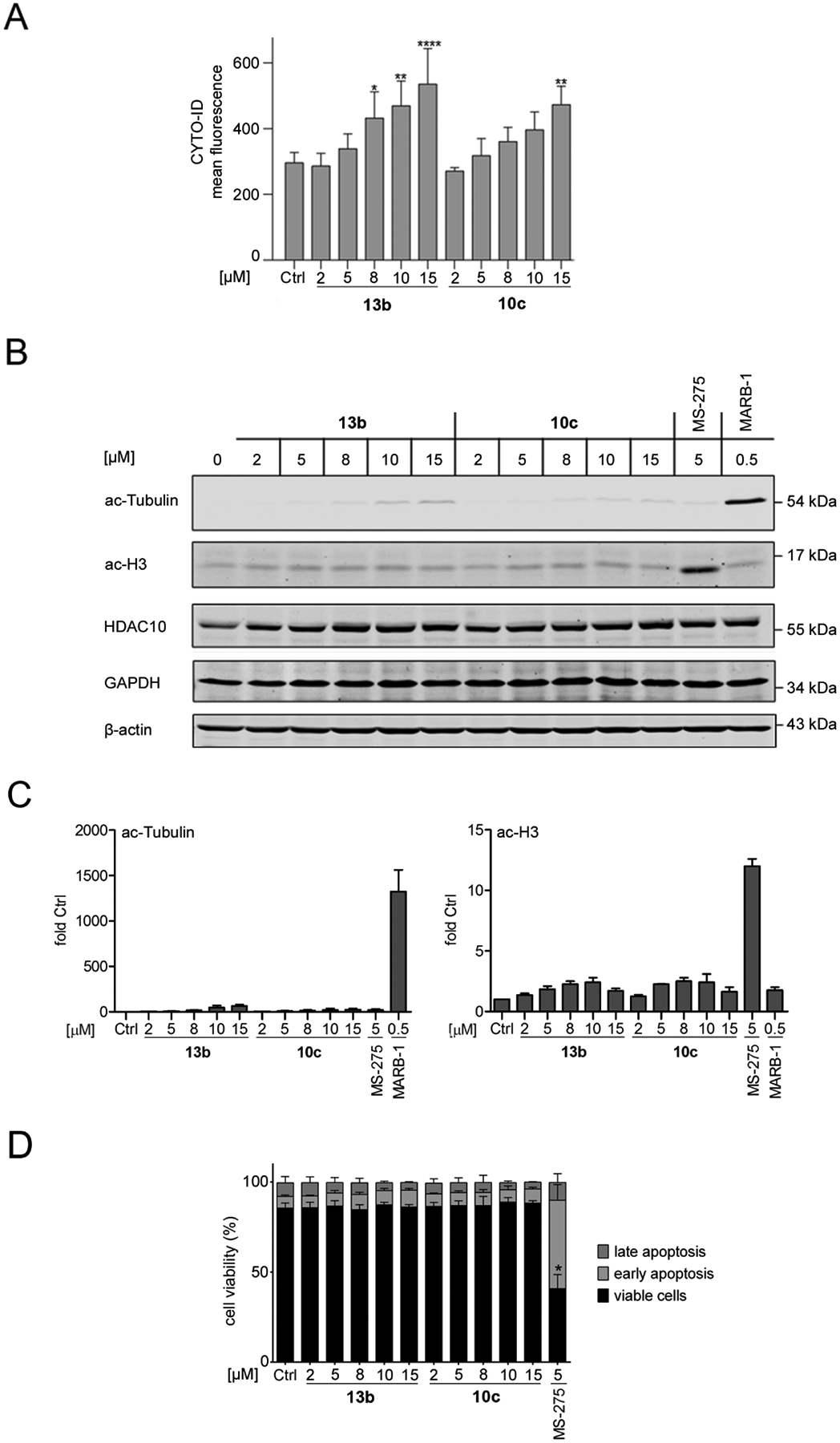
10c and 13b preferentially inhibit HDAC10 in leukemic cells. A: MV4–11 cells were treated with increasing concentrations of 10c and 13b, from 2–15 μM for 24 h; Ctrl, solvent treated samples. 10 μM chloroquine were added to all samples to facilitates the detection of cyto-ID positive vesicles by reduced lysosomal acidification; n = 3. Statistical significance was determined using Two-way ANOVA, *p<0.05, **p<0.01, ****p<0.0001.
B: MV4–11 cells were incubated with 2–15 μM 10c and 13b, 5 μM MS-275, or 0.5 μM marbostat-100 (MARB-1) for 24 h (Ctrl, solvent control). Immunoblot was done as indicated, with β-actin as loading control; n = 2.
C: Quantification of the immunoblots B using Image Studio Lite (LI-COR Biosciences); n = 2.
D: MV4–11 were treated with 2–15 μM 10c and 13b (Ctrl, solvent control), incubated for 24 h and subjected to flow cytometry analyses for annexin-V-FITC and propidium iodide (PI); n = 3. Two-way ANOVA, *p<0.05.
The enzymatic in vitro testing described above measured only weak activity of 10c and 13b against HDAC1. Immunoblots with whole cell lysates from MV4–11 cells coherently showed that 2–15 μM 10c and 13b did not induce a significant accumulation of acetylated histone H3 (Fig. 5A), which is the prototypical target of class I HDACs.27 This indicates that class I HDACs are not substantially affected by 10c and 13b in these cells. 5 μM of the class I HDACi entinostat (MS275, used as positive control, Fig. 2) induced a highly significant accumulation of acetylated histone H3 (Fig. 5D).
The accumulation of acetylated tubulin-α serves as a marker for the inhibition of HDAC6.27, 28 We used the recently identified highly selective inhibitor of HDAC6, marbostat-100, as a positive control for HDAC6 inhibition.28 Compared to 0.5 μM marbostat-100, which enhanced tubulin acetylation 1324-fold on average compared to non-treated cells, 15 μM 10c and 13b induced the acetylation of tubulin 36–80 fold and this effect did not reach significance (Fig. 5B, 5C). These data are in line with the above-described in vitro data (Table 1, 2). The impact of 10c and 13b on the accumulation of cyto-ID positive vesicles was not associated with a loss of HDAC10. We rather noted slightly increased levels of HDAC10 upon its inhibition (Fig. 5B). Moreover, we noted no change of GAPDH levels (Fig. 5B). This finding disfavors that 10c and 13b promote chaperone-mediated autophagy (CMA) in MV4–11 cells. Since CMA was induced by a genetic elimination of HDAC10 in solid tumor-derived cells,12 the unchanged expression of HDAC10 in 10c and 13b treated MV4–11 cells can explain why GAPDH remained unchanged.
Table 2:
In vitro selectivity of the synthesized HDAC10 inhibitors.
| ID | hHDAC1 | hHDAC6 IC50 [nM] | hHDAC8 IC50 [nM] |
|---|---|---|---|
| 4a | 49 % @ 10 μM 19 % @ 1 μM |
210 ± 26 | 3500 ± 520 |
| 4b | 68 % @ 10 μM 15 % @ 1 μM |
158 ± 21 | 3200 ± 410 |
| 4c | 50 % @ 10 μM 16 % @ 1 μM |
177 ± 28 | 1900 ± 340 |
| 6a | n.d. | n.d. | 5 % @ 1 μM |
| 6b | 54 % @ 10 μM 6 % @ 1 μM |
280 ± 25 | 2400 ± 210 |
| 10a | 16 ± 9 μM | 4400 ± 400 | 250 ± 50 |
| 10b | 54 % @ 10 μM 7 % @ 1 μM |
4800 ± 1100 | 420 ± 55 |
| 10c | 3.0 ± 0.2 μM | 3700 ± 450 | 470 ± 70 |
| 10d | 67 % @ 10 μM 24 % @ 1 μM |
83 % @ 10 μM 40 % @ 1 μM |
2500 ± 260 |
| 10e | 62 % @ 10 μM 12 % @ 1 μM |
89 % @ 10 μM 34 % @ 1 μM |
n.d. |
| 10f | n.d. | n.d. | n.d. |
| 10g | 56 % @ 10 μM 22 % @ 1 μM |
72 % @ 10 μM 29 % @ 1 μM |
91 % @ 10 μM 54 % @ 1 μM |
| 13a | 66 % @ 10 μM 29 % @ 1 μM |
1400 ± 140 | 1300 ± 170 |
| 13b | 67 % @ 10 μM 12 % @ 1 μM |
2420 ± 530 | 920 ± 170 |
| 13c | 9 ± 1 μM | 430 ± 50 | 950 ± 260 |
| Entinostat (MS-275) | 0.93 ± 0.1 μM | n.i. | n.i. |
| Tubastatin A | 1.91 ± 0.42 μM | 34 ± 17 | 1440 ± 120 |
n.d. = not determined; n.i. = no inhibition
Next, we tested whether the accumulation of autophagic vesicles upon HDAC10 inhibition is a consequence of apoptosis of MV4–11 cells. Early apoptosis is determined by positive annexin-V-FITC staining caused by the exposure of phosphatidylserine on the cell surface. Late apoptosis and necrosis are indicated by positive annexin-V-FITC staining as well as accumulation of propidium iodide due to the loss of the membrane integrity.29 After a 24-h incubation, 2–15 μM 10c and 13b did not trigger apoptosis (Fig. 5C); unlike class I HDAC inhibition with MS-275 (Fig. 5C). This finding suggests that the inhibition of HDAC10 induce autophagy but not pro-apoptotic mechanisms in MV4–11 cells after 24 h. While these results confirm previous data for pro-survival functions of class I HDACs in such cells, we currently cannot exclude pro-survival functions of HDAC10 in other leukemic cells and upon prolonged HDAC10 inhibition. We further noted that our new HDAC10 inhibitors were not toxic for human embryonic kidney-derived HEK293 cells, even at a high concentration of 50 μM (Table 3).
Table 3:
Cytotoxicity against HEK293 cells at 50 μM inhibitor concentration.
| ID | cell viability [%] |
|---|---|
| 4a | 77.1 ± 5.0 |
| 4b | 71.6 ± 6.1 |
| 4c | 81.5 ± 2.8 |
| 6a | 92.6 ± 13.4 |
| 6b | 86.4 ± 2.3 |
| 10a | 54.4 ± 1.4 |
| 10b | 77.3 ± 4.4 |
| 10c | 51.6 ± 3.9 |
| 10d | 88.6 ± 2.2 |
| 10e | 100.9 ± 6.9 |
| 10f | 104.9 ± 0.8 |
| 10g | 55.0 ± 4.4 |
| 13a | 88.2 ± 1.0 |
| 13b | 68.2 ± 4.8 |
| 13c | 84.6 ± 1.5 |
3. Chemistry
The compounds in the present work were synthesized using the synthetic pathways outlined in Scheme 1 and 2. The synthetic strategy relied primarily on the synthesis of the carboxylic acid intermediates (3a-c, 9a-g and 12a-c) which were subsequently converted to the corresponding hydroxamates through the well-established method by amide coupling with O-(Tetrahydro-2H-pyran-2-yl) hydroxylamine followed by acidic deprotection.23 Scheme 1 illustrates the synthesis of target benzhydroxamate derivatives 4a-c and 6a-b. The respective aldehyde and ethyl 4-aminomethylbenzoate served as starting materials, where reductive amination of primary amine followed by basic hydrolysis afforded the carboxylic acid intermediates 3a-c. As shown in Scheme 2, the N-substituted-piperidineacrylic acid derivatives were obtained either via the alkylation or reductive amination of the unsubstituted piperidine derivative 7 followed by alkaline hydrolysis of the ester function to yield the carboxylic acids 9a-g, 12a-c. These were subsequently converted to the corresponding hydroxamates 10a-g and 13a-c.
Scheme 1.
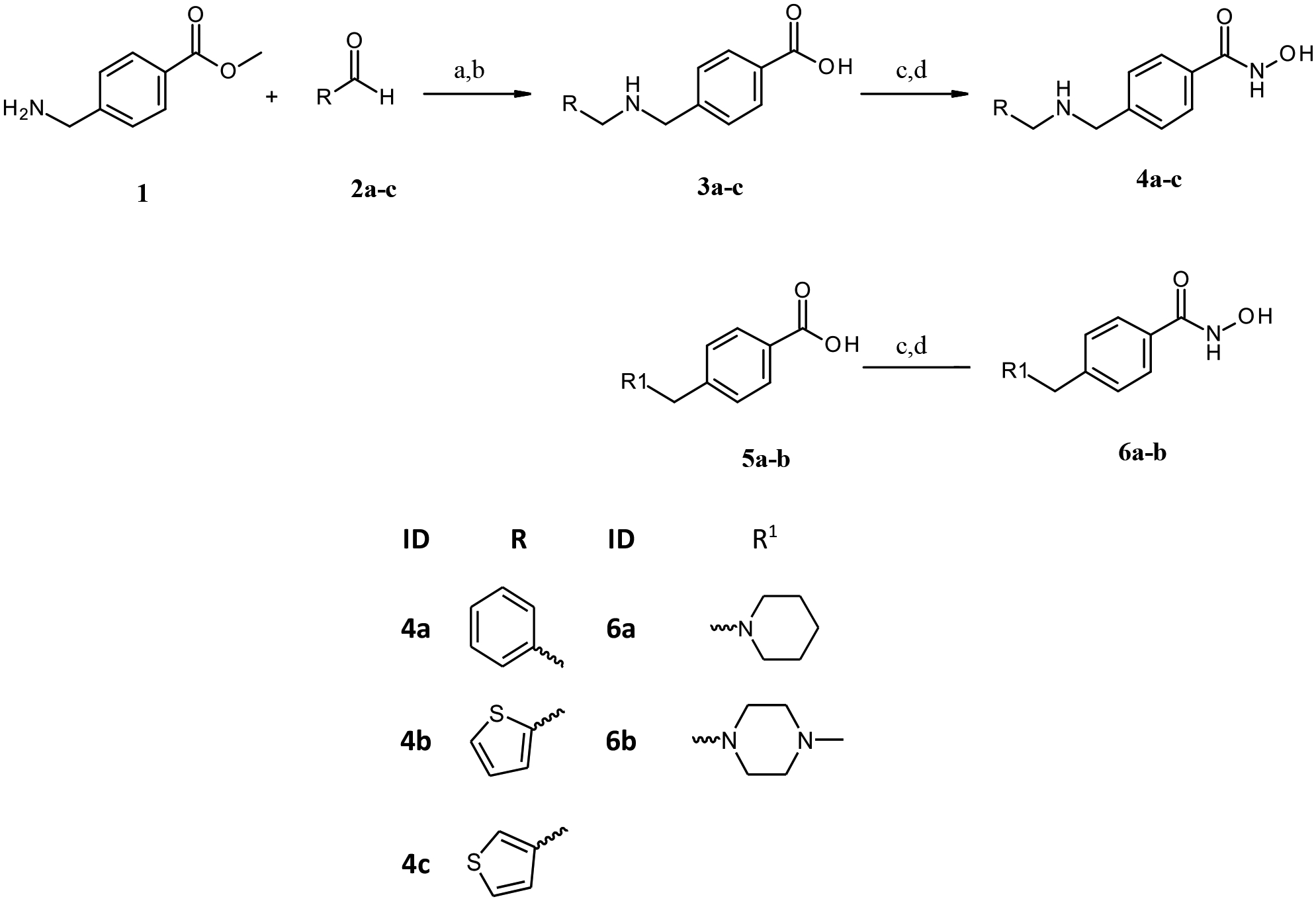
Reagents and conditions: (a) DCM, MeOH, Na(AcO)3BH, r.t, overnight; (b) i)1M. NaOH aq., MeOH, r.t, 48h, ii) dil. HCl; (c) PyBOP, DIPEA, H2NOTHP, THF, r.t, overnight; d) catalytic. HCl, THF, r.t, overnight.
Scheme 2.
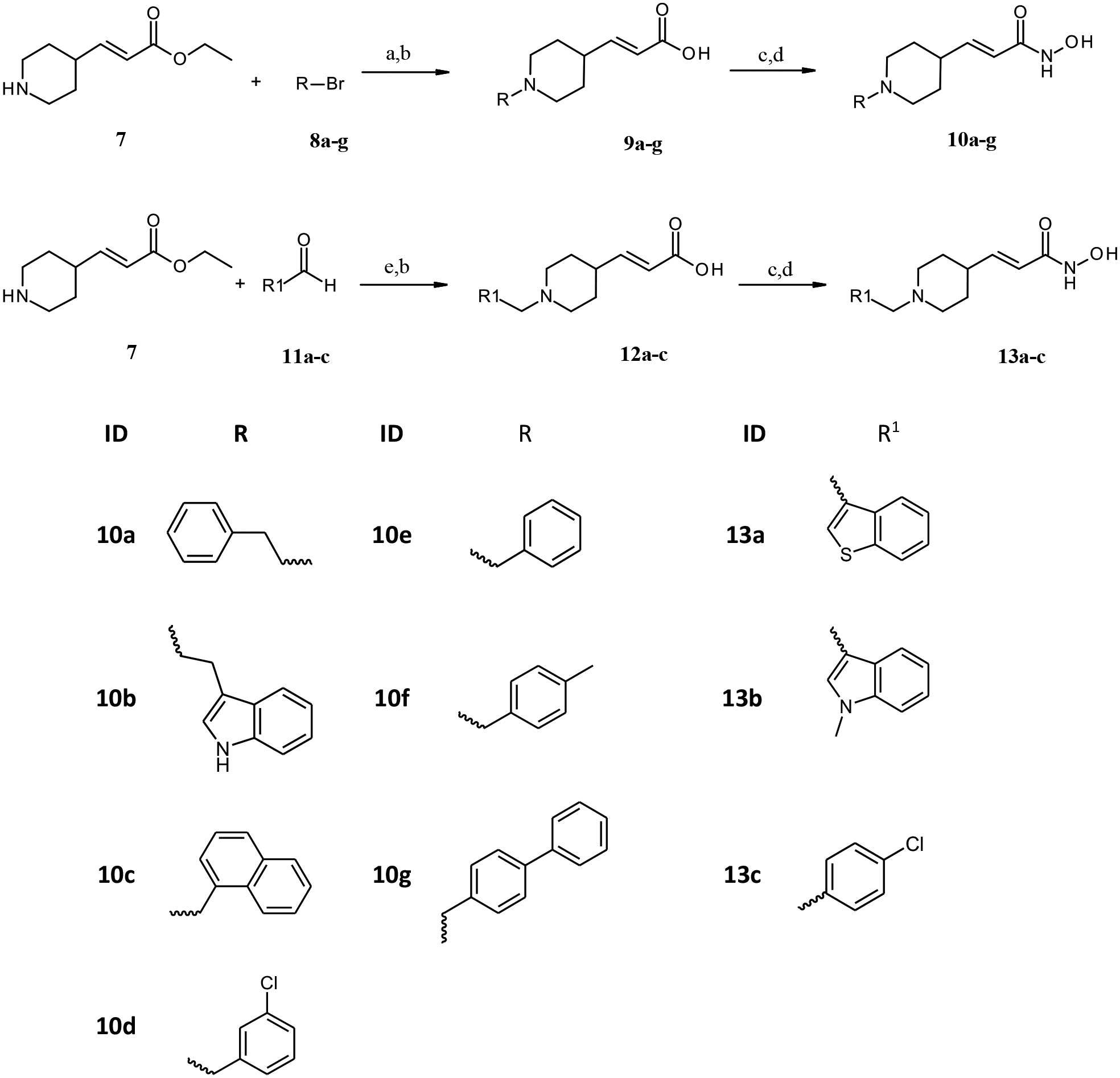
Reagents and conditions: (a) K2CO3, DMF, catalytic KI; r.t.,72h; (b) THF, H2O, LiOH. H2O, r.t., 12h; (c) PyBOP, DIPEA, H2NOTHP, THF, r.t, overnight; d) catalytic. HCl, THF, r.t, overnight; (e) EtOH, Na(AcO)3BH, r.t., 48h.
4. Conclusion
We present here a new chemotype of HDAC10 inhibitor that we obtained by structure-based optimization. We confirmed the interaction of four representative inhibitors with HDAC10 by solving their crystal structures. Our data reveal that piperidine-4-acrylhydroxamates inhibit HDAC10 with good selectivity and activity in vitro and in cultured cells. Consistent with previously published data in neuroblastoma cells,7 we show that pharmacological modulation with hitherto unknown specific HDAC10 inhibitors modulates autophagy in aggressive FLT3-ITD positive AML cells. We can exclude that this is a downstream effect of cell death. Thus, like inhibition of HDAC6 in these and other leukemic cells,4, 30–32 inhibition of HDAC10 modulates biological processes. Highly consistent with these data are results from knock-out mice with a deletion of HDAC6 or HDAC10. In both cases, the animals are viable and show altered responses upon stress.17, 33 Overall, the developed HDAC10 inhibitors 10c and 13b are useful tools to investigate the physiological functions of HDAC10 in future studies.
5. Materials and Methods
5.1. General
All materials and reagents were purchased from Sigma-Aldrich Co. Ltd. and abcr GmbH. All solvents were analytically pure and dried before use. Thin layer chromatography was carried out on aluminum sheets coated with silica gel 60 F254 (Merck, Darmstadt, Germany). For column chromatography under normal pressure silica gel 60 (0.036−0.200 mm) was used.
Final compounds were confirmed to be of >95% purity based on HPLC. Purity was measured by UV absorbance at 254 nm. The HPLC consists of an XTerra RP18 column (3.5 μm, 3.9 mm × 100 mm) from the manufacturer Waters (Milford, MA, USA) and two LC-10AD pumps, a SPD-M10A VP PDA detector, and a SIL-HT autosampler, all from the manufacturer Shimadzu (Kyoto, Japan). For preparative tasks a XTerra RP18 column (7 μm, 19 mm × 150 mm) from the manufacturer Waters (Milford, MA, USA) and two LC-20AD pumps were used. The mobile phase was in all cases a gradient of methanol/ water (starting at 95% water going to 5% water).
Mass spectrometry analyses were performed with a Finnigan MAT710C (Thermo Separation Products, San Jose, CA, USA) for the ESIMS spectra and with a LTQ (linear ion trap) Orbitrap XL hybrid mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) for the HRMS-ESI (high resolution mass spectrometry) spectra. For the HRMS analyses the signal for the isotopes with the highest prevalence was given and calculated (35Cl, 79Br).
1H NMR and 13C NMR spectra were taken on a Varian Inova 500 using deuterated chloroform and deuterated DMSO as solvent. Chemical shifts are referenced to the residual solvent signals.
The following abbreviations and formulas for solvents and reagents were used: dimethylformamide (DMF), dimethylsulfoxide (DMSO), tetrahydrofuran (THF), N,N-diisopropylethylamine (DIPEA) and hydrochloric acid (HCl), methanol (MeOH), water (H2O), sodium hydroxide (NaOH), dichloromehane(DCM), sodium triacetoxyborohydride (Na(AcO)3BH), lithium hydroxide monohydrate (LiOH.H2O), (PyBOP), O-(Tetrahydro-2H-pyran-2-yl)hydroxylamine (H2NOTHP), ethanol (EtOH), benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP), Potassium carbonate (K2CO3),Potassium iodide(KI).
5.2. General synthesis methods
Method A: reductive amination of primary amines
The amine (1 eq) was disolved in dichloromethane (20 ml). The aldehyde (0.95 eq) was added dropwise, and the mixture was stirred at room temperature for 30 min. Afterwards sodium triacetoxyborohydride (2 eq) was added and the mixture was stirred over-night at room temperature. Methanol (5 ml) was added, and the solvents were evaporated under reduced pressure. The products were purified with MPLC using chloroform/methanol as eluent.
Method B: reductive amination of secondary amines
The amine (1 eq) was disolved in ethanol (15 ml). The aldehyde (5 eq) was added and the mixture was stirred at room temperature for 15 min. Afterwards sodium triacetoxyborohydride (5 eq) was added and the mixture was stirred over-night at room temperature. Sodium triacetoxyborohydride (5 eq) was added and the reaction was stirred over night at room temperature. Methanol (5 ml) was added, and the solvents were evaporated under reduced pressure. The products were purified with MPLC using chloroform/methanol as eluent.
Method C: alkylation of secondary amines
The amine (1 eq) was solved in dimethylformamide (6 ml). Potassium carbonate (4 eq) was added, the mixture was stirred for 30 min at room temperature. Afterwards potassium carbonate (4 eq), the alkyl bromide (3 eq) and potassium iodide (1 spatula tip) were added. The mixture was stirred for 72 h at room temperature. The solvent was evaporated under reduced pressure. Further purifications were not performed.
Method D: ester hydrolysis with NaOH
The ester (1 eq) was disolved in methanol (50 ml) and 1M NaOHaq (5 ml) and refluxed for 4 h. The mixture was neutralized with 1M HClaq and evaporated under reduced pressure. The products were purified with MPLC using chloroform/methanol as eluent.
Method E: ester hydrolysis with LiOH
A mixture of the appropriate ester (1 eq) and lithium hydroxide monohydrate (2 eq) in 1:1 mixture of tetrahydrofuran and water (50 ml) was stirred over night at room temperature. The solvents were evaporated under reduced pressure. The products were purified with MPLC using chloroform/methanol as eluent.
Method F: synthesis of THP-protected hydroxamic acids
The appropriate carboxylic acid (1 eq) was disolved in tetrahydrofuran (25 ml). N,N-diisopropyl-ethylamine (12 eq), benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (3 eq) and O-(tetrahydro-2H-pyran-2-yl)hydroxylamine (3 eq) were added. The mixture was stirred at room temperature for 72 h. The solvents were evaporated under reduced pressure. The products were purified with MPLC using chloroform/methanol as eluent.
Method G: THP-deprotection
The THP protected hydroxamic acid (1 eq) was solved in tetrahydrofuran (25 ml), water (12.5 ml) and 1M HClaq (25 drops). The mixture was stirred over night at room temperature. The solvents were evaporated under reduced pressure. The products were purified with MPLC using chloroform/ methanol as eluent.
5.3. Characterization of the final compounds
4a 4-[(benzylamino)methyl]-benzhydroxamic acid
The title compound was synthesized from methyl 4-(aminomethyl)benzoate and benzaldehyde using method A, followed by hydrolysis of the ester product using method D. The hydroxamic acid was synthesized using method F followed by method G.
HRMS m/z: 257.1287 [M+H]+
1H NMR (400 MHz, DMSO-d6) δ = 4.12 (s, 4H), 7.39 (s, 3H), 7.58 (m, 4H), 7.77 (s, 2H), 9.07 (s, 1H), 10.05 (s, 2H), 11.34 (s, 1H).
13C NMR (101 MHz, DMSO) δ 164.11, 133.45, 132.73, 130.56, 129.20, 128.97, 127.99, 127.44, 110.01, 50.36, 49.82.
Purity: 96% (HPLC)
4b 4-{[(thiophen-2-ylmethyl) amino]methyl}benzhydroxamic acid
The title compound was synthesized from methyl 4-(aminomethyl)benzoate and 2-thiophenecarboxaldehyde using method A, followed by hydrolysis of the ester product using method D. The hydroxamic acid was synthesized using method F followed by method G.
HRMS m/z: 263.0850 [M+H]+
1H NMR (400 MHz, DMSO-d6): δ = 4.10 (s, 2H), 4.30 (s, 2H), 7.08 (dd, J1 = 3,6 Hz, J2 = 5,2 Hz, 1H), 7.27 (d, J = 2.4 Hz, 1H), 7.56 (m, 3H), 7.75 (m, 2H), 8.98 (m, 3H), 11.26 (s, 1H).
13C NMR (101 MHz, DMSO) δ 161.49, 137.61, 135.75, 133.00, 132.90, 131.25, 129.91, 127.66, 127.45, 44.69, 50.09.
Purity 97% (HPLC)
4c 4-{[(thiophen-3-ylmethyl)amino]methyl}benzhydroxamic acid
The title compound was synthesized from methyl 4-(aminomethyl)benzoate and 3-thiophenecarboxaldehyde using method A, followed by hydrolysis of the ester product using method D. The hydroxamic acid was synthesized using method F followed by method G.
HRMS m/z: 263.0845 [M+H]+
1H NMR (400 MHz, DMSO-d6) δ = 4.14 (s, 4H), 7.33 (dd, J1 = 1.6 Hz, J2 = 5.2 Hz, 1H), 7.59 (m, 3H), 7.71 (dd, J1 = 1.2 Hz, J2 = 2.8 Hz, 1H), 7.76 (m, 2H), 9.09 (s, 1H), 9.83 (m, 2H), 11.30 (s, 1H).
13C NMR (101 MHz, DMSO) δ 163.77, 135.21, 133.39, 132.88, 130.50, 129.22, 127.56, 116.10, 112.91, 49.39, 46.03.
Purity 98% (HPLC)
6a 4-(piperidin-1-ylmethyl)benzhydroxamic acid
The title compound was synthesized from 4-(piperidin-1-yl) methyl)benzoic acid using method F followed by method G.
HRMS m/z: 235.1442 [M+H]+
1H NMR (400 MHz, DMSO-d6) δ = 1.17 (m, 6H), 1.55 (s, 1H), 1.80 (s, 3H), 3.00 (m, 2H), 7.44 (s, 1H), 7.89 (s, 1H), 8.21 (s, 2H).
13C NMR (101 MHz, DMSO) δ 164.12, 136.61, 132.64, 128.85, 128.08, 61.97, 54.21, 25.6, 24.30 Purity 97% (HPLC)
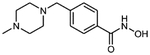
6b 4-[(N-methylpiperazin-1-yl)methyl]benzhydroxamic acid
The title compound was synthesized from 4-[(N-methylpiperazinyl)methyl]benzoic acid using method F followed by method G.
HRMS m/z: 250.1551 [M+H]+
1H NMR (400 MHz, DMSO-d6) δ = 2.77 (s, 3H), 3.41 (m, 8H), 4.32 (s, 2H), 7.69 (m, 2H), 7.79 (m, 2H), 11.30 (s, 1H).
13C NMR (101 MHz, DMSO) δ164.12, 136.61, 132.69, 128.81, 127.89, 61.99, 55.04, 53.57, 45.73 Purity 100% (HPLC)
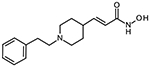
10a (E)-3-(1-phenethylpiperidin-4-yl)acrylhydroxamic acid
The title compound was synthesized from ethyl (E)-3-(piperidin-4-yl)acrylate and (2-bromoethyl)benzene using Method C, followed by hydrolysis of the ester product using method E. The hydroxamic acid was synthesized using method F followed by method G.
HRMS m/z: 275.1755 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ = 1.77 (m, 2H), 1.87 (d, J1 = 13.6 Hz, 2H), 2.69 (m, 1H), 2.97 (m, 2H), 3.09 (m, 2H), 3.20 (m, 2H), 3.56 (d, J1 = 11.6 Hz, 2H), 5.78 (d, J1 = 15.6 Hz, 1H), 6.56 (dd, J1 = 6.0 Hz, J2 = 15.6 Hz, 1H), 7.25 (m, 3H), 7.32 (m, 2H), 8.96 (s, 1H), 10.74 (s, 1H).
13C NMR (101 MHz, DMSO) δ 162.90, 156.08, 143.95, 137.59, 128.82, 127.25, 120.92, 56.84, 51.42, 35.69, 34.28, 29.81.
Purity 96% (HPLC)
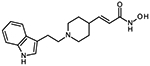
10b (E)-3-{1-[2-(1H-indol-3-yl)ethyl]piperidin-4-yl}acrylhydroxamic acid
The title compound was synthesized from ethyl (E)-3-(piperidin-4-yl)acrylate and 3-(2-bromoethyl)indole using Method C, followed by hydrolysis of the ester product using method E. The hydroxamic acid was synthesized using method F followed by method G.
HRMS m/z: 314.1862 [M+H]+
1H NMR (400 MHz, DMSO-d6) δ = 1.59 (m, 2H), 1.92 (d, J1 = 13.2 Hz, 2H), 2.40 (m, 1H), 3.02 (m, 2H), 3.11 (m, 2H), 3.31 (m, 2H), 3.65 (d, J1 = 11.6 Hz, 2H), 5.77 (d, 1H), 6.59 (dd, J1 = 6.0 Hz, J2 = 15.6 Hz, 1H), 7.00 (t, J1 = 7.2 Hz, 1H), 7.09 (t, J1 = 6.8 Hz, 1H), 7.23 (s, 1H), 7.35 (d, J1 = 8.0 Hz, 1H), 7.57 (d, J1 = 7.6 Hz, 1H), 9.67 (s, 1H), 10.98 (s, 1H).
13C NMR (101 MHz, DMSO) δ 162.89, 144.34, 136.70, 127.09, 123.51, 121.71, 120.88, 118.97, 118.65, 112.02, 109.39, 56.37, 51.43, 34.98, 28.40, 20.10.
Purity 97% (HPLC)
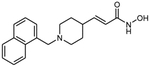
10c (E)-3-[1-(naphthalen-1-ylmethyl)piperidin-4-yl]acrylhydroxamic acid
The title compound was synthesized from ethyl (E)-3-(piperidin-4-yl)acrylate and 1-(bromomethyl)naphthalene using Method C, followed by hydrolysis of the ester product using method E. The hydroxamic acid was synthesized using method F followed by method G.
HRMS m/z: 311.1755 [M+H]+
1H NMR (400 MHz, DMSO-d6) δ = 1.55 (m, 2H), 1.75 (m, 2H), 2.26 (m, 1H), 3.12 (m, 2H), 5.77 (d, J1 = 15.6 Hz, 1H), 6.55 (m, 1H), 7.56 (m, 4H), 7.94 (m, 2H), 8.34 (s, 1H), 8.87 (s, 1H), 10.69 (s, 1H).
13C NMR (101 MHz, DMSO) δ 162.69, 133.85, 132.53, 132.34, 130.06, 129.03, 128.96, 126.46, 126.34, 125.84, 124.82, 124.75, 110.10, 52.97, 51.40, 35.42, 28.59.
Purity 95% (HPLC)
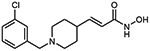
10d (E)-3-[1-(3-chlorobenzyl)piperidin-4-yl]acrylhydroxamic acid
The title compound was synthesized from ethyl (E)-3-(piperidin-4-yl)acrylate and 3-chlorobenzyl bromide using Method C, followed by hydrolysis of the ester product using method E. The hydroxamic acid was synthesized using method F followed by method G.
HRMS m/z: 295.1205 [M+H]+
1H NMR (400 MHz, CD3OD) δ = 1.67 (m, 2H), 1.94 (d, J1 = 10.0 Hz, 2H), 2.43 (m, 2H), 2.84 (m, 2H), 2.98 (s, 1H), 4.12 (s, 2H), 5.83 (d, J1 = 12.4 Hz, 1H), 6.73 (dd, J1 = 4.8 Hz, J2 = 12.0 Hz, 1H), 7.45 (m, 3H), 7.54 (s, 1H), 8.41 (s, 1H).
13C NMR (101 MHz, DMSO) δ 162.70, 156.23, 143.88, 137.34, 128.71, 127.52, 127.16, 123.01, 120.76, 56.65, 51.26, 34.02, 29.60.
Purity 99% (HPLC)
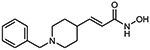
10e (E)-3-(1-benzylpiperidin-4-yl)acrylhydroxamic acid
The title compound was synthesized from ethyl (E)-3-(piperidin-4-yl)acrylate and benzyl bromide using Method C, followed by hydrolysis of the ester product using method E. The hydroxamic acid was synthesized using method F followed by method G.
HRMS m/z: 261.1595 [M+H]+
1H NMR (400 MHz, CD3OD) δ = 2.02 (m, 2H), 2.29 (m, 2H), 2.86 (m, 2H), 3.35 (s, 1H), 3.51 (d, J1 = 10.4 Hz, 2H), 4.42 (s, 2H), 5.85 (d, J1 = 12.4 Hz, 1H), 6.73 (dd, J1 = 4.0 Hz, J2 = 12.4 Hz, 1H), 7.47 (s, 1H), 7.54 (m, 1H), 7.55 (m, 2H), 7.60 (m, 1H).#
13C NMR (101 MHz, DMSO) δ 162.70, 156.33, 143.85, 137.32, 128.11, 127.55, 120.72, 56.55, 51.29, 34.02, 29.65.
Purity 96% (HPLC)
10f (E)-3-[1-(4-methylbenzyl) piperidin-4-yl]acrylhydroxamic acid
The title compound was synthesized from ethyl (E)-3-(piperidin-4-yl)acrylate and 4-methylbenzyl bromide using Method C, followed by hydrolysis of the ester product using method E. The hydroxamic acid was synthesized using method F followed by method G.
HRMS m/z: 275.1751 [M+H]+
1H NMR(400 MHz, CD3OD) δ = 1.65 (m, 2H), 1.96 (d, J1 = 10.4 Hz, 2H), 2.37 (s, 3H), 2.44 (s, 1H), 2.89 (m, 2H), 3.37 (d, J1 = 9.6 Hz, 2H), 4.13 (s, 2H), 5.86 (d, J1 = 12.4 Hz, 1H), 6.71 (dd, J1 = 5.2 Hz, J2 = 12.4 Hz, 1H), 7.32 (dd, J1 = 6.4 Hz, J2 = 26.0 Hz, 4H), 8.42 (s, 1H).
13C NMR (101 MHz, DMSO) δ 164.72, 151.56, 139.87, 139.56, 133.15, 128.89, 123.04, 61.19, 52.35, 42.54, 29.02, 21.31.
Purity 91% (HPLC)
10g (E)-3-{1-[(1,1´-biphenyl)-4-ylmethyl]piperidin-4-yl} acrylhydroxamic acid
The title compound was synthesized from ethyl (E)-3-(piperidin-4-yl)acrylate and 4-brommethylbiphenyl using Method C, followed by hydrolysis of the ester product using method E. The hydroxamic acid was synthesized using method F followed by method G.
HR-MS m/z: 337.1908 [M+H]+
1H NMR (400 MHz, CD3OD) δ = 1.72 (m, 2H), 2.01 (m, 2H), 2.50 (s, 1H), 3.04 (t, J1 = 9.6 Hz, 2H), 3.48 (d, J1 = 10.4 Hz, 2H), 4.31 (s, 2H), 5.86 (d, J1 = 12.4 Hz, 1H), 6.74 (dd, J1 = 5.2 Hz, J2 = 12.4 Hz, 1H), 7.38 (m, 1H), 7.47 (m, 2H), 7.59 (m, 2H), 7.64 (m, 2H), 7.74 (m, 2H), 8.33 (s, 1H).
13C NMR (101 MHz, DMSO) δ 163.66, 162.93, 145.06, 140.15, 131.11, 129.43, 128.09, 127.25, 127.13, 120.39, 110.05, 52.32, 52.14, 31.96, 28.18.
Purity 98% (HPLC)
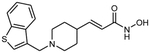
13a (E)-3-{1-[benzothiophen-3-ylmethyl]piperidin-4-yl} acrylhydroxamic acid
The title compound was synthesized from ethyl (E)-3-(piperidin-4-yl)acrylate and 1-Benzothiophen-3-carboxaldehyd using Method B, followed by hydrolysis of the ester product using method E. The hydroxamic acid was synthesized using method F followed by method G.
HRMS m/z: 317.1319 [M+H]+
1H NMR (400 MHz, DMSO-d6) δ = 1.82 (m, 4H), 2.32 (m, 1H), 3.08 (m, 4H), 4.55 (s, 2H), 5.74 (d, J1 = 15.6 Hz, 1H), 6.55 (dd, J1 = 6.0 Hz, J2 = 15.6 Hz, 1H), 7.44 (m, 2H), 8.06 (d, J1 = 8.0 Hz, 1H), 8,18 (d, J1 = 7.6 Hz, 1H), 8.28 (s, 1H), 8.91 (s, 1H), 10.74 (s, 1H).
13C NMR (101 MHz, DMSO) δ 168.73, 138.54, 137.41, 136.88, 128.53, 128.42, 127.36, 123.01, 122.86, 122.51, 121.54, 52.45, 52.23, 42.51, 29.09.
Purity 96% (HPLC)
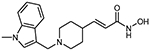
13b (E)-3-{1-[(1-methylindol-3-yl)methyl]piperidin-4-yl} acrylhydroxamic acid
The title compound was synthesized from ethyl (E)-3-(piperidin-4-yl)acrylate and 1-Methylindol-3-carboxaldehyd using Method B, followed by hydrolysis of the ester product using method E. The hydroxamic acid was synthesized using method F followed by method G.
HR-MS m/z: 314.1865 [M+H]+
1H NMR (400 MHz, DMSO-d6) δ = 1.29 (m, 2H), 1.59 (d, J1 = 10.8 Hz, 2H), 1.95 (m, 2H), 2.37 (m, 1H), 2.85 (d, J1 = 11.2 Hz, 2H), 3.59 (s, 2H), 3.73 (s, 3H), 5.73 (d, J1 = 15.6 Hz, 1H), 6.55 (dd, J1 = 6.8 Hz, J2 = 15.6 Hz, 1H), 7.19 (s, 1H), 7.34 (d, J1 = 8.4 Hz, 1H), 7.43 (m, 1H), 7.56 (m, 1H), 7.59 (d, J1 = 76 Hz, 1H), 10.76 (s, 1H).
13C NMR (101 MHz, DMSO) δ 164.00, 146.28, 137.09, 129.87, 128.42, 121.55, 119.91, 119.60, 119.12, 110.04, 109.35, 53.10, 52.60, 37.73, 32.76, 30.90.
Purity 98% (HPLC)
13c (E)-3-[1-(4-chlorobenzyl)piperidin-4-yl]acrylhydroxamic acid
The title compound was synthesized from ethyl (E)-3-(piperidin-4-yl)acrylate and 4-chlorobenzaldehyde using Method B, followed by hydrolysis of the ester product using method E. The hydroxamic acid was synthesized using method F followed by method G.
HRMS m/z: 295.1212 [M+H]+
1H NMR (400 MHz, DMSO-d6) δ = 1.51 (m, 2H), 1.86 (d, J1 = 15.6 Hz, 2H), 2.36 (m, 1H), 2.92 (m, 2H), 3.35 (d, J1 = 12.4 Hz, 2H), 4.28 (s, 1H), 5.74 (d, J1 = 15.6 Hz, 1H), 6.53 (dd, J1 = 6.4 Hz, J2 = 15.6 Hz, 1H), 7.53 (m, 4H), 8.59 (s, 1H), 10.60 (s, 1H).
13C NMR (101 MHz, DMSO) δ 168.75, 136.81, 136.65, 133.73, 128.75, 128.54, 123.04, 61.95, 52.33, 42.54, 29.08.
Purity 99% (HPLC)
5.3. In vitro HDAC inhibitory activity
HDAC1, HDAC6 and HDAC8
The in vitro testing on recombinant HDACs were performed as previously described.34 Recombinant human HDAC1 and −6 were purchased from BPS Biosciences. The enzyme inhibition was determined by using a reported homogenous fluorescence assay.35 The enzymes were incubated for 90 min at 37°C, with the fluorogenic substrate ZMAL (Z-(Ac)Lys-AMC) in a concentration of 10.5 mM and increasing concentrations of inhibitors with subsequent addition of 60 mL of buffer containing trypsin (1 mg/ml) and TSA (2.75 mM) and further incubation for 20 min at 37°C. Fluorescence intensity was measured at an excitation wavelength of 390 nm and an emission wavelength of 460 nm in a microtiter plate reader (BMG Polarstar).
Recombinant hHDAC8 was produced by Romier et al. in Strasbourg.36 The HDAC8 activity assays were performed according to the commercial HDAC8 Fluorometric Drug Discovery Kit [Fluor de Lys(R)-HDAC8, BML-KI178] corresponding to the manufacturer’s instructions. As substrate a tetrapeptide connected to aminomethylcoumarin (AMC) H2N-Arg- His-Lys(Ac)-Lys(Ac)-AMC was synthesized as previously described.34 The enzyme was incubated for 90 min at 37 °C, with a substrate concentration of 50 μM and increasing concentrations of inhibitors. The stop-solution containing inhibitor, to stop the hHDAC8 activity, and Trypsin, to release the AMC, was added. The solution was incubated for 20 min at 37 °C to develop the assay. Fluorescence intensity was measured at an excitation wavelength of 355 nm and an emission wavelength of 460 nm in a microtiter plate reader (BMG Polarstar).
drHDAC10
Cells containing the drHDAC10 plasmid8 were grown in 70 mL of 2x YT medium supplemented with 50 μg/mL kanamycin at 37 °C overnight with shaking (250 rpm).22 The saturated culture (5 mL) was added to 1 L of 2x YT containing 50 μg/mL kanamycin and allowed to grow until OD600 = 0.8. The temperature was then lowered to 16 °C and shaking was stopped. After cooling for 30 min, expression was induced by adding 200 μM IPTG and 250 μM ZnSO4. Cell cultures were grown for 18 h at 16 °C with shaking (250 rpm), after which cells were centrifuged at 5,422 g. Cell pellets were frozen and stored at −80 °C until further use.
Immediately prior to purification (≤ 2 h), pellets were thawed in a water bath at 4 °C. Buffer A [50 mM HEPES (pH 7.5), 300 mM KCl, 10% glycerol, 2 mM TCEP, 10 μM ZnSO4, 30 mM imidazole, 0.4 mg/mL lysozyme (MPbiomedicals), 2.8 units/mL benzonase nuclease (Sigma), and protease inhibitor tablets (Roche Applied Science)] was added to the cell pellet [lysis buffer:cell pellet = 2:1 (v:w)]. The suspension was stirred at 4 °C for 45 min prior to sonication. Lysate was centrifuged at 41,657 g at 4 °C for 1 h, then loaded onto a 5-mL HisTrap (GE) column pre-equilibrated with buffer B [50 mM HEPES (pH 7.5), 300 mM KCl, 10% glycerol, 2 mM TCEP, 10 μM ZnSO4, and 30 mM imidazole]. Protein was eluted with buffer C [50 mM HEPES (pH 7.5), 300 mM KCl, 10% glycerol, 2 mM TCEP, 10 μM ZnSO4, and 500 mM imidazole]. Protein fractions were pooled and digested with TEV protease (overnight) and dialyzed into loading buffer. Finally, the protein solution was loaded onto a tandem MBPTrap (GE)-HisTrap column pre-equilibrated with buffer B. The eluent was collected, pooled, and loaded onto a HiLoad Superdex 200 column equilibrated with buffer D [50 mM HEPES (pH 7.5), 300 mM KCl, 5% glycerol, and 1 mM TCEP]. Protein fractions were pooled, concentrated to ~10 mg/mL, and frozen at −80 °C until further use. All stock solutions were prepared in DMSO; NDA (16 mM) and Ac-spermidine-AMC (10 mM). Compounds for testing were solved and diluted to 12-fold higher than test concentration in DMSO. Ac-spermidine-AMC stock solutions was diluted with assay buffer (20 mM Na2HPO4, pH 7.9, 100 mM NaCl, 0.25 mM EDTA, 10 % (v/v) glycerol, 10 mM Mesna, 0.01 % TWEEN 20) to 126 μM. For assay determination stop solution was prepared, containing 5 μL NDA (16 mM), and 190 μL borat buffer (100 mM boric acid, pH 9.5) per well. Directly before using enzyme solution (0.0054 mg/ml) was prepared in assay buffer.
The assay was performed in black 96-well plates (PerkinElmer, OptiPlateTM-96 F). Assay buffer was presented in the plate, 55 μL for the blank, 45 μL for the blank containing enzyme solution, 50 μL for the negative control and 40 μL for the positive control and test compounds. 5 μL of DMSO were added to the wells of blanks, positive and negative control. Corresponding to the DMSO 5 μL of increasing concentrations of inhibitors in DMSO were added to the relevant wells. After adding 10 μL of enzyme solution (12 nM final assay concentration) to blank containing enzyme, positive control and test compounds, 5 μL Ac-spermidine-AMC solution (10.5 μM final assay concentration) were added to negative control, positive control and test compounds. The plate was incubated for 25 min at 25 °C. Before measuring fluorescence (POLARstar plate reader, λex = 330 nm, λem = 390 nm) each well was filled with 200 μL stop solution.
IC50 calculation: Inhibition was measured at increasing concentration and IC50 was calculated by nonlinear regression with Origin 9.0G software.
5.4. Cytotoxicity assay
To determine the cytotoxicity of the developed compounds, a human epithelial kidney cell line (HEK293) was used. HEK293 cells (DSMZ Braunschweig, ACC305) were incubated at 37 °C in a humidified incubator with 5% CO2 in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal calf serum and 5mM glutamine. Cells were seeded out at 1.5 × 103 cells per well in a 96-well cell culture plate (TPP, Switzerland). The compounds were added immediately to the medium at 50 μM. After 24 h, AlamarBlue reagent (Invitrogen, CA) was added according to the manufacturer’s instructions and incubated for 21 h before samples were analyzed. Detection of viable cells, which convert the resazurine reagent into the highly fluorescent resorufin, was performed by using a FLUOstarOPTIMA microplate reader (BMG Labtec) and the following filter set: Ex 530 nm/Em 590 nm. Measurements were performed in triplicate and data are means with standard deviation < 14%. As a positive control daunorubicin was used and an IC50 value of 12.55 ± 0.07 μM was obtained.
5.5. Molecular docking
The available X-ray structures of drHDAC10 in complex with different inhibitors as listed in Table 4 were downloaded from the Protein Databank (PDB, www.rcsb.org). Protein preparation was done using the protein preparation wizard implemented in Schroedinger version 2019.1 by adding hydrogen atoms, assigning protonation states and minimizing the protein using the OPLS force field implementing the default settings. Ligands structures were generated in MOE [Molecular Operating Environment (MOE), 2020.01; Chemical Computing Group Inc., 1010 Sherbooke St.West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2018]. The ligands were subsequently prepared for docking using the LigPrep tool as implemented in Schroedinger’s software (version 2019.1) and energy minimized using the OPLS3e force field. 25 conformers of all ligands were subsequently generated with ConfGen. Docking of the generated conformers into the prepared protein structures was performed using the program Glide (Schroedinger-release 2019.1) in the Standard Precision mode. To test the ability of the docking approach to correctly reproduce the drHDAC10-inhibitor complexes we docked the cocrystallized inhibitors using the described docking setup. For all complexes the docking setup resulted in docking poses with root mean square deviation (RMSD) values below 2.0 Å (Table 4) for the top-ranked solution (Glidescore SP) with the exception of one structure (PDB ID 6UIM). If considering all docking solutions RMSD values below 0.9 Å were determined for all ligands indicating that the docking setup is able to predict the interaction of the diverse inhibitors correctly. The novel inhibitors were subsequently docked to drHDAC10 (PDB ID: 5TD7) and humanized HDAC10 (PDB ID: 6VNQ) as representative protein structures.
Table 4.
Validation of docking setup for HDAC10-inhibitor complexes from the Protein DataBank
| PDB ID | Cocryst. ligand | Enzyme (Resolution) | RMSD (Å) of top ranked pose | Best RMSD (Å) among top-5 ranked poses | Best RMSD (Å) and docking rank |
|---|---|---|---|---|---|
| 5TD7 | 7-[(3-aminopropyl)amino]-1,1,1-trifluoroheptane-2,2-diol | drHDAC10 (2.65 Å) | 1.91 | 1.78 | 0.89 (119) |
| 6UFN | 7-[(3-aminopropyl)amino]heptan-2-one | drHDAC10 (2.70 Å) | 1.22 | 0.84 | 0.66 (292) |
| 6UFO | 7-[(3-aminopropyl)amino]-1-methoxyheptan-2-one | drHDAC10 (2.68 Å) | 1.08 | 1.07 | 0.51 (283) |
| 6UHV | 6-[(3-aminopropyl)amino]-N-hydroxyhexanamide | drHDAC10 (2.53 Å) | 1.25 | 1.15 | 0.75 (110) |
| 6UHU | 5-[(3-aminopropyl)amino]pentyl boronic acid | drHDAC10 (2.80 Å) | 1.60 | 1.45 | 0.72 (166) |
| 6UII | 5-[(3-aminopropyl)amino]pentane-1-thiol | drHDAC10 (2.65 Å) | 0.81 | 0.59 | 0.59 (3) |
| 6UIJ | S-{5-[(3-aminopropyl)amino]pentyl} thioacetate | drHDAC10 (2.90 Å) | 1.06 | 1.05 | 0.51 (117) |
| 6UIL | 7-[(3-aminopropyl)amino]-1,1,1-trifluoroheptan-2-one | drHDAC10 (2.85 Å) | 0.79 | 0.78 | 0.57 (216) |
| 6UIM | 7-{[(3-aminopropyl)amino]-2-oxoheptyl} thioacetate | drHDAC10 (2.75 Å) | 2.93 | 1.31 | 0.69 (62) |
| 6VNQ | bishydroxamate | humanized drHDAc10 (2.05 Å) | 0.35 | 0.35 | 0.32 (16) |
| 6WBQ | hydroxamate | humanized drHDAc10 (2.00 Å) | 0.54 | 0.54 | 0.54 (1) |
| 6WDV | hydroxamate | humanized drHDAc10 (2.40 Å) | 0.53 | 0.53 | 0.47 (26) |
| 6WDW | hydroxamate | humanized drHDAc10 (2.20 Å) | 0.35 | 0.39 | 0.34 (59) |
| 6WDX | hydroxamate | humanized drHDAc10 (2.65 Å) | 0.62 | 0.52 | 0.36 (30) |
| 6WDY | hydroxamate | humanized drHDAc10 (2.65 Å) | 0.62 | 0.58 | 0.52 (6) |
| 7KUQ | acetyl-spermidine | drHDAC10 Y307F mutant (2.10 Å) | 1.16 | 0.44 | 0.24 (55) |
| 7KUR | acetyl-putrescine | drHDAC10 Y307F mutant (2.10 Å) | 0.73 | 0.73 | 0.18 (64) |
| 7KUS | acetyl-spermidine | drHDAC10 H137A mutant (2.00 Å) | 1.09 | 1.09 | 0.62 (128) |
| 7KUT | acetyl-putrescine | drHDAC10 H137A mutant (2.05 Å) | 0.69 | 0.64 | 0.25 (97) |
5.6. X-ray crystallography
For X-ray crystal structure determinations of “humanized” drHDAC10-inhibitor complexes, inhibitor 4c, 6b, 10a, or 10b was added to a final concentration of 2 mM in a 10 mg/mL solution of protein in size exclusion buffer and equilibrated on ice for 1 h. Trypsin was then added (1:1000 trypsin: “humanized” drHDAC10 molar ratio). The protein solution was filtered (0.22-μm centrifuge filter) after 1 h digestion at room temperature.
All enzyme-inhibitor complexes were crystallized by the sitting drop vapor diffusion method at 4 °C using a Mosquito crystallization robot (TTP Labtech). Typically, a 100-nL drop of protein solution was added to a 100-nL drop of precipitant solution and equilibrated against 80 μL of precipitant solution in a 96-well crystallization plate. Crystallization of the “humanized” drHDAC10 complexes with 4c, 6b, and 10a was achieved using 0.1 M NaH2PO4, 0.1 M Na2HPO4, and 20% PEG 3350 as precipitant. Crystallization of the “humanized” drHDAC10-10b complex was achieved using 0.125 M NaH2PO4, 0.075 M Na2HPO4, and 20% PEG 3350 as precipitant. Micro-seed crystals of the HDAC10–Tubastatin complex were also added to each crystallization drop.
X-ray diffraction data from crystals of each complex was collected on NE-CAT beamline 24-ID-C at the Advanced Photon Source, Argonne National Laboratory (Argonne, IL). Raw intensities were integrated using iMosflm37 and scaled using Aimless in the CCP4 program suite.38,39 All structures were solved by molecular replacement using the crystal structure of the Y307F drHDAC10–FKS complex (5TD7) with ligands and water molecules removed as a search model with the program Phaser.40 Model building was performed using COOT and crystallographic refinement was achieved using Phenix.41,42 The bound inhibitor and water molecules were fit to the electron density map in the final stages of refinement. MolProbity43 was used to validate each refined structure. Data collection and refinement statistics are recorded in Table 5.
Table 5.
Crystallographic data collection and refinement statisticsa
| “Humanized” drHDAC10 complex | 4c | 6b | 10a | 10b |
|---|---|---|---|---|
| Space group | P3121 | P3121 | P3121 | P3121 |
| a,b,c (Å) | 80.75, 80.75, 248.92 | 80.75, 80.75, 248.87 | 80.39, 80.39, 247.51 | 80.50, 80.50, 247.68 |
| α, β, γ (°) | 90, 90, 120 | 90, 90, 120 | 90, 90, 120 | 90, 90, 120 |
| R merge b | 0.093 (1.266) | 0.066 (0.524) | 0.108 (0.726) | 0.163 (1.032) |
| R pim c | 0.046 (0.622) | 0.055 (0.412) | 0.091 (0.584) | 0.096 (0.611) |
| CC 1/2 d | 0.999 (0.690) | 0.992 (0.702) | 0.992 (0.594) | 0.990 (0.713) |
| Redundancy | 9.5 (9.9) | 3.5 (3.4) | 3.4 (3.5) | 7.0 (7.2) |
| Completeness (%) | 100 (100) | 98.0 (98.3) | 97.5 (98.9) | 99.8 (100) |
| I/σ | 13.8 (2.4) | 8.9 (2.0) | 7.2 (2.0) | 7.7 (2.4) |
| Refinement | ||||
| (2.33 – 2.25) | (2.18 – 2.10) | (2.59 – 2.50) | (2.69 – 2.60) | |
| No. reflections | 45730 (4484) | 54740 (5526) | 31956 (3211) | 29495 (2884) |
| (0.2678/0.3128) | (0.2767/0.2914) | (0.2526/0.3298) | (0.2442/0.3029) | |
| Number of Atomsf | ||||
| Protein | 4817 | 4881 | 4772 | 4786 |
| Ligand | 40 | 36 | 38 | 41 |
| Solvent | 157 | 174 | 113 | 70 |
| Average B factor (Å2) | ||||
| Protein | 53 | 46 | 45 | 50 |
| Ligand | 57 | 50 | 54 | 61 |
| Solvent | 50 | 43 | 40 | 42 |
| Root-Mean-Square Deviation (RMSD) | ||||
| Bond lengths (Å) | 0.008 | 0.007 | 0.008 | 0.008 |
| Bond angles (°) | 0.9 | 0.9 | 1.0 | 1.0 |
| Ramachandran Plot (%)g | ||||
| Favored | 96.03 | 95.40 | 95.56 | 95.53 |
| Allowed | 3.66 | 4.13 | 4.29 | 3.99 |
| Outliers | 0.32 | 0.48 | 0.16 | 0.48 |
| PDB Entry | 7U6A | 7U3M | 7U69 | 7U6B |
Values in parentheses refer to the highest-resolution shell of data.
Rmerge = ΣhΣi/Ii,h − 〈I〉h|/ΣhΣiIi,h, where 〈I〉h is the average intensity calculated for reflection h from i replicate measurements.
Rp.i.m. = (Σh(1/(N-1))1/2Σi|Ii,h −〈I〉h|)/ΣhΣi Ii,h, where N is the number of reflections and 〈I〉h is the average intensity calculated for reflection h from replicate measurements.
Pearson correlation coefficient between random half-datasets.
Rwork = Σ||Fo| − |Fc||/Σ|Fo| for reflections contained in the working set. |Fo| and |Fc| are the observed and calculated structure factor amplitudes, respectively. Rfree is calculated using the same expression for reflections contained in the test set held aside during refinement.
Per asymmetric unit.
Calculated with MolProbity.
5.7. PAINS filter
All the herein described compounds were filtered for pan-assay interference compounds (PAINS).44 For this purpose, PAINS1, PAINS2 and PAINS3 filters, as implemented in Schroedinger’s Canvas program, were employed. None of the compounds was flagged as a PAIN.
5.8. Immunoblot
Immunoblots were performed as described.27,45 Antibodies were: GAPDH (#ab128915) from Abcam, Cambridge, U.K.; β-actin (#sc-47778), HDAC10 (#sc-54215), HSP70 (#sc-66048), p62/SQSTM1 (#sc-25575) from Santa Cruz, Heidelberg, Germany; ac-H3 (#06–599) from Millipore-Merck, Darmstadt, Germany; ac-Tubulin (#T7451) from Sigma-Aldrich, Darmstadt, Germany. As protein ladders served the prestained Scientific™ PageRuler™ (#26617) and the PageRuler™ Plus (#26620) from Thermo Fisher, Braunschweig, Germany.
5.9. Detection of modulation of autophagy
Modulation of autophagy was detected with techniques that we described46 using the Cyto-ID® Autophagy Dectection Kit (Enzo Life Science, Lörrach, Germany). In brief, MV4–11 cells were treated with 2 to 15 μM 13b or 10c as well as 10 μM chloroquine and incubated for 24 h at 37°C, 5 % CO2. Cells were then harvested, washed with PBS (phosphate-buffered saline), and stained with Cyto-ID® Green (1:1000 in phenol-red free RPMI, supplemented with 5% FCS, 30 min, 37 °C). After incubation time, cells were washed with PBS twice and measured via flow cytometry using a FACSCanto™ II (BD Biosciences, Heidelberg, Germany). Analysis was done using the FACSDiva software (BD Biosciences).
5.10. Annexin-V-FITC / PI Flow Cytometry measurements
Detection of cell death was done as described,38 MV4–11 cells were treated with 2 – 15 μM 13b or 10c or 5 μM MS-275 and incubated for 24 h at 37°C, 5 % CO2. Cells were then harvested, washed with PBS, and resuspended in 50 μL 1x annexin-V binding buffer containing 2.5 μL annexin V-FITC (Miltenyi Biotec, Bergisch Gladbach, Germany). Samples were incubated for minimum 20 min in the dark at room temperature. Then, PI (50 μg/mL) diluted 1:44 in 440 μL 1x annexin-V binding buffer were added. Samples were then subjected to flow cytometry using a FACSCanto™ II. Analysis was performed using the FACSDiva software (BD Biosciences).
5.11. LysoTracker Assay
Human neuroblastoma SK-N-BE(2)-C cells (European Collection of Authenticated Cell Cultures, ECACC, Salisbury, UK) were seeded into 6-well dishes at a density of 1.5 × 105 cells per well and treated for 24 h with compounds of interest. Cells were stained for 1 h with LysoTracker Red DND-99 (50 nM) in medium under standard cell culture conditions. Cells were washed with ice-cold RPMI without phenol-red and trypsinized for 3 min at 37 °C. Detached cells were centrifuged for 3 min at 8600g and re-suspended in ice-cold RPMI without phenol-red. Mean LysoTracker fluorescence was quantified on a BD FACSCanto II platform using the PE filter setting.
Supplementary Material
Acknowledgement
We thank Prof. S. Mahboobi, University of Regensburg, Germany, for providing marbostat-100. We thank Christina Brachetti, Andrea Piée-Staffa, and Simone Kniesa for excellent technical assistance.
Funding
This work was funded by the Deutsche Forschungsgemeinschaft (DFG) SI868/22–1, project number 46995445 (to W.S.), and KR2291/9–1, project number 427404172; KR2291/12–1, project number 445785155; KR2291/14–1, project number 469954457; KR2291/15–1, project number 495271833; SFB 1361 sub-project 11, project number 393547839 to (O.H.K). The work was further funded by the Brigitte und Dr. Konstanze Wegener Foundation (project number 65), the Wilhelm Sander-Foundation, project number 2019.086.1 (to O.H.K). Work done by D.W.C. and C.J.H.-G. is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165 and GM49758). The Pilatus 6M detector on beamline 24-ID-C is funded by a NIH-ORIP HEI grant (S10 RR029205). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02–06CH11357 (to D.W.C.).
Footnotes
Declaration of Interests
OHK declares the patent WO2016020369A1, which covers substances used in this article.
Data Availability
Atomic coordinates and structure factors for “humanized” drHDAC10 complexes with inhibitors 4c, 6b, 10a, and 10b have been deposited in the Protein Data Bank with accession codes 7U6A, 7U3M, 7U69, and 7U6B, respectively.
References
- 1.San Jose-Eneriz E, Gimenez-Camino N, Agirre X, Prosper F. HDAC Inhibitors in Acute Myeloid Leukemia. Cancers (Basel) 2019, 11 (11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li G, Tian Y, Zhu WG. The Roles of Histone Deacetylases and Their Inhibitors in Cancer Therapy. Front Cell Dev Biol 2020, 8, 576946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krämer OH. HDAC2: a critical factor in health and disease. Trends Pharmacol Sci 2009, 30 (12), 647–55. [DOI] [PubMed] [Google Scholar]
- 4.Beyer M, Romanski A, Mustafa AM, Pons M, Buchler I, Vogel A, et al. HDAC3 Activity is Essential for Human Leukemic Cell Growth and the Expression of beta-catenin, MYC, and WT1. Cancers (Basel) 2019, 11 (10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nebbioso A, Carafa V, Conte M, Tambaro FP, Abbondanza C, Martens J, et al. c-Myc Modulation and Acetylation Is a Key HDAC Inhibitor Target in Cancer. Clin Cancer Res 2017, 23 (10), 2542–55. [DOI] [PubMed] [Google Scholar]
- 6.Hai Y, Shinsky SA, Porter NJ, Christianson DW. Histone deacetylase 10 structure and molecular function as a polyamine deacetylase. Nat Commun 2017, 8, 15368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oehme I, Linke JP, Bock BC, Milde T, Lodrini M, Hartenstein B, et al. Histone deacetylase 10 promotes autophagy-mediated cell survival. Proc Natl Acad Sci U S A 2013, 110 (28), E2592–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shinsky SA, Christianson DW. Polyamine Deacetylase Structure and Catalysis: Prokaryotic Acetylpolyamine Amidohydrolase and Eukaryotic HDAC10. Biochemistry 2018, 57 (22), 3105–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang D, Tang B, Xie X, Xiao Y-F, Yang S-M, Zhang J-W. The interplay between DNA repair and autophagy in cancer therapy. Cancer biology & therapy 2015, 16 (7), 1005–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang W, Chen X, Ji C, Zhang W, Song J, Li J, et al. Key Regulators of Autophagosome Closure. Cells 2021, 10 (11), 2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ridinger J, Koeneke E, Kolbinger FR, Koerholz K, Mahboobi S, Hellweg L, et al. Dual role of HDAC10 in lysosomal exocytosis and DNA repair promotes neuroblastoma chemoresistance. Scientific reports 2018, 8 (1), 10039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Obayashi H, Nagano Y, Takahashi T, Seki T, Tanaka S, Sakai N, et al. Histone deacetylase 10 knockout activates chaperone-mediated autophagy and accelerates the decomposition of its substrate. Biochemical and biophysical research communications 2020, 523 (1), 246–52. [DOI] [PubMed] [Google Scholar]
- 13.Powers J, Lienlaf M, Perez-Villarroel P, Deng S, Knox T, Villagra A, et al. Expression and Function of Histone Deacetylase 10 (HDAC10) in B Cell Malignancies. Methods Mol Biol 2016, 1436, 129–45. [DOI] [PubMed] [Google Scholar]
- 14.Blum KA, Advani A, Fernandez L, Van Der Jagt R, Brandwein J, Kambhampati S, et al. Phase II study of the histone deacetylase inhibitor MGCD0103 in patients with previously treated chronic lymphocytic leukaemia. British journal of haematology 2009, 147 (4), 507–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang JC, Kafeel MI, Avezbakiyev B, Chen C, Sun Y, Rathnasabapathy C, et al. Histone deacetylase in chronic lymphocytic leukemia. Oncology 2011, 81 (5–6), 325–9. [DOI] [PubMed] [Google Scholar]
- 16.Van Damme M, Crompot E, Meuleman N, Mineur P, Bron D, Lagneaux L, et al. HDAC isoenzyme expression is deregulated in chronic lymphocytic leukemia B-cells and has a complex prognostic significance. Epigenetics 2012, 7 (12), 1403–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dahiya S, Beier UH, Wang L, Han R, Jiao J, Akimova T, et al. HDAC10 deletion promotes Foxp3(+) T-regulatory cell function. Scientific reports 2020, 10 (1), 424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herbst-Gervasoni CJ, Steimbach RR, Morgen M, Miller AK, Christianson DW. Structural Basis for the Selective Inhibition of HDAC10, the Cytosolic Polyamine Deacetylase. ACS Chem Biol 2020, 15 (8), 2154–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Geraldy M, Morgen M, Sehr P, Steimbach RR, Moi D, Ridinger J, et al. Selective Inhibition of Histone Deacetylase 10: Hydrogen Bonding to the Gatekeeper Residue is Implicated. J Med Chem 2019, 62 (9), 4426–43. [DOI] [PubMed] [Google Scholar]
- 20.Kolbinger FR, Koeneke E, Ridinger J, Heimburg T, Muller M, Bayer T, et al. The HDAC6/8/10 inhibitor TH34 induces DNA damage-mediated cell death in human high-grade neuroblastoma cell lines. Arch Toxicol 2018, 92 (8), 2649–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Y, Woster PM. Discovery of a new class of histone deacetylase inhibitors with a novel zinc binding group. Medchemcomm 2015, 6 (4), 613–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herbst-Gervasoni CJ, Christianson DW. X-ray Crystallographic Snapshots of Substrate Binding in the Active Site of Histone Deacetylase 10. 2021, 60 (4), 303–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heimburg T, Kolbinger FR, Zeyen P, Ghazy E, Herp D, Schmidtkunz K, et al. Structure-Based Design and Biological Characterization of Selective Histone Deacetylase 8 (HDAC8) Inhibitors with AntiNeuroblastoma Activity. 2017, 60 (24), 10188–204. [DOI] [PubMed] [Google Scholar]
- 24.Herp D, Ridinger J, Robaa D, Shinsky SA, Schmidtkunz K, Yesiloglu TZ, et al. First fluorescent acetylspermidine deacetylation assay for HDAC10 identifies inhibitors of neuroblastoma cell colony growth that increase lysosome accumulation. 2020. [Google Scholar]
- 25.Beyer M, Henninger SJ, Haehnel PS, Mustafa A-HM, Gurdal E, Schubert B, et al. Identification of a highly efficient dual type I/II FMS-like tyrosine kinase inhibitor that disrupts the growth of leukemic cells. Cell Chemical Biology 2021. [DOI] [PubMed] [Google Scholar]
- 26.Silva MC, Nandi GA, Tentarelli S, Gurrell IK, Jamier T, Lucente D, et al. Prolonged tau clearance and stress vulnerability rescue by pharmacological activation of autophagy in tauopathy neurons. Nat Commun 2020, 11 (1), 3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beyer M, Kiweler N, Mahboobi S, Kramer OH. How to Distinguish Between the Activity of HDAC1–3 and HDAC6 with Western Blot. Methods Mol Biol 2017, 1510, 355–64. [DOI] [PubMed] [Google Scholar]
- 28.Sellmer A, Stangl H, Beyer M, Grünstein E, Leonhardt M, Pongratz H, et al. Marbostat-100 Defines a New Class of Potent and Selective Antiinflammatory and Antirheumatic Histone Deacetylase 6 Inhibitors. J Med Chem 2018, 61 (8), 3454–77. [DOI] [PubMed] [Google Scholar]
- 29.Pons M, Reichardt CM, Hennig D, Nathan A, Kiweler N, Stocking C, et al. Loss of Wilms tumor 1 protein is a marker for apoptosis in response to replicative stress in leukemic cells. Archives of Toxicology 2018, 92 (6), 2119–35. [DOI] [PubMed] [Google Scholar]
- 30.Leonhardt M, Sellmer A, Krämer OH, Dove S, Elz S, Kraus B, et al. Design and biological evaluation of tetrahydro-beta-carboline derivatives as highly potent histone deacetylase 6 (HDAC6) inhibitors. European journal of medicinal chemistry 2018, 152, 329–57. [DOI] [PubMed] [Google Scholar]
- 31.Pons M, Nagel G, Zeyn Y, Beyer M, Laguna T, Brachetti T, et al. Human platelet lysate as validated replacement for animal serum to assess chemosensitivity. Altex 2019, 36 (2), 277–88. [DOI] [PubMed] [Google Scholar]
- 32.Toutah K, Nawar N, Timonen S, Sorger H, Raouf YS, Bukhari S, et al. Development of HDAC Inhibitors Exhibiting Therapeutic Potential in T-Cell Prolymphocytic Leukemia. 2021, 64 (12), 8486–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y, Kwon S, Yamaguchi T, Cubizolles F, Rousseaux S, Kneissel M, et al. Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Molecular and cellular biology 2008, 28 (5), 1688–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ghazy E, Zeyen P, Herp D, Hügle M, Schmidtkunz K, Erdmann F, et al. Design, synthesis, and biological evaluation of dual targeting inhibitors of histone deacetylase 6/8 and bromodomain BRPF1. European journal of medicinal chemistry 2020, 200, 112338. [DOI] [PubMed] [Google Scholar]
- 35.Stolfa DA, Stefanachi A, Gajer JM, Nebbioso A, Altucci L, Cellamare S, et al. Design, Synthesis, and Biological Evaluation of 2-Aminobenzanilide Derivatives as Potent and Selective HDAC Inhibitors. ChemMedChem 2012, 7 (7), 1256–66. [DOI] [PubMed] [Google Scholar]
- 36.Marek M, Kannan S, Hauser A-T, Moraes Mourão M, Caby S, Cura V, et al. Structural basis for the inhibition of histone deacetylase 8 (HDAC8), a key epigenetic player in the blood fluke Schistosoma mansoni. PLoS pathogens 2013, 9 (9), e1003645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Battye TGG, Kontogiannis L, Johnson O, Powell HR, and Leslie AGW (2011) iMOSFLM: A new graphical interface for diffraction-image processing with MOSFLM Acta Cryst. D67, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Evans PR, and Murshudov GN (2013) How good are my data and what is the resolution? Acta Cryst. D69, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A & Wilson KS (2011) Overview of the CCP4 suite and current developments. Acta Cryst. D67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007) Phaser crystallographic software J. Appl. Cryst 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010) Features and development of Coot. Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ,Richardson DC, Richardson JS, Terwilliger TC, and Zwart PH (2010) PHENIX: a comprehensive python-based system for macromolecular structure solution Acta Cryst. D66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen VB, Arendal WB III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baell JB. chemistry, GHJOM, 2010.(2010) New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. ACS Publications 53, 2719–40. [DOI] [PubMed] [Google Scholar]
- 45.Pons M, Reichardt CM, Hennig D, Nathan A, Kiweler N, Stocking C, et al. Loss of Wilms tumor 1 protein is a marker for apoptosis in response to replicative stress in leukemic cells. Arch Toxicol 2018, 92 (6), 2119–35. [DOI] [PubMed] [Google Scholar]
- 46.Göder A, Mahendrarajah N, Krämer OH. Detection of Autophagy Induction After HDAC Inhibitor Treatment in Leukemic Cells. Methods Mol Biol 2017, 1510, 3–10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structure factors for “humanized” drHDAC10 complexes with inhibitors 4c, 6b, 10a, and 10b have been deposited in the Protein Data Bank with accession codes 7U6A, 7U3M, 7U69, and 7U6B, respectively.