

Abstract
BACKGROUND:
Ischemic heart disease remains a leading cause of death worldwide. In this study, we test the hypothesis that microRNA-210 protects the heart from myocardial ischemia-reperfusion (IR) injury by controlling mitochondrial bioenergetics and reactive oxygen species (ROS) flux.
METHODS:
Myocardial infarction in an acute setting of IR was examined via comparing loss vs. gain of function experiments in microRNA-210-deficient and wild type mice. Cardiac function was evaluated by echocardiography. Myocardial mitochondria bioenergetics was examined using a Seahorse XF24 Analyzer.
RESULTS:
MicroRNA-210 deficiency significantly exaggerated cardiac dysfunction up to 6 weeks after myocardial IR in male, but not female, mice. Intravenous injection of microRNA-210 mimic blocked the effect and recovered the increased myocardial IR injury and cardiac dysfunction. Analysis of mitochondrial metabolism revealed that microRNA-210 inhibited mitochondrial oxygen consumption, increased glycolytic activity, and reduced mitochondrial ROS flux in the heart during IR injury. Consistently, inhibition of mitochondrial ROS with MitoQ reversed the effect of microRNA-210 deficiency. Mechanistically, we showed that mitochondrial glycerol-3-phosphate dehydrogenase (GPD2) is a novel target of microRNA-210 in the heart, and loss-of-function and gain-of-function experiments revealed that GPD2 played a key role in the microRNA-210-mediated effect on mitochondrial metabolism and ROS flux in the setting of heart IR injury. Knockdown of GPD2 negated microRNA-210 deficiency-induced increases in mitochondrial ROS production and myocardial infarction, and improved left ventricular fractional shortening and ejection fraction after the IR treatment.
CONCLUSIONS:
MicroRNA-210 targeting GPD2 controls mitochondrial bioenergetics and ROS flux and improves cardiac function in a murine model of myocardial infarction in the setting of IR injury. The findings suggest new insights into the mechanisms and therapeutic targets for treatment of ischemic heart disease.
Keywords: myocardial infarction, microRNA-210, glycerol-3-phosphate dehydrogenase, mitochondrial bioenergetics, ROS production
Introduction
Despite recent advances in treatment of acute myocardial infarction (AMI) and improvement of patient survival, ischemic heart disease remains a leading cause of death worldwide and the primary cause of chronic heart failure.1 Novel cardioprotective strategies are still required to attenuate the detrimental effects of AMI, and to improve adverse heart remodeling and cardiac dysfunction in patients with coronary heart disease. Animal studies have suggested microRNA 210 (miR-210) as a potential therapy for treatment of ischemic heart disease to improve cardiac function in a murine model of myocardial infarction.2,3 Human clinical studies have revealed miR-210 as a biomarker in coronary artery disease.4–6 Mature miR-210 of 22 nt is highly homologous across species and is identical among the human, pig and rodent, revealing a translational potential of preclinical study of miR-210 in the rodent. MiR-210 is a master regulator of the cellular hypoxic response by targeting mitochondrial energy metabolism and ROS flux. Mitochondria constitute about 30% of total cardiomyocyte volume. In addition to being the main source of ATP for the contracting cell through oxidative phosphorylation, mitochondria are a key source of cellular ROS production. Reprogramming of mitochondrial metabolism and ROS flux in the setting of ischemia and reperfusion (IR) is a crucial driver of heart injury.7–12 Thus, inhibition of mitochondrial respiratory chain and reduction of mitochondrial oxygen consumption during IR afford protection of ischemic heart disease.9,10
In the present study, we seek to test the hypothesis that miR-210 protects the heart from myocardial IR injury by controlling mitochondrial bioenergetics and ROS flux via comparing gain vs. loss of function experiments in miR-210-deficient and wild type mice. We identify that mitochondrial glycerol-3-phosphate dehydrogenase (GPD2) is a highly conserved target of miR-210. GPD2 is an integral component of the mammalian respiratory chain and glycerophosphate (GP)-shuttle connecting mitochondrial and cytosolic processes, and is a crossroad of glycolysis, fatty acid metabolism and oxidative phosphorylation.13 In addition to ROS production at complex I and succinate dehydrogenase via reverse electron transport,9, 10 GPD2 contributes to overall mitochondrial ROS production.9,10,13–16 Levels of ROS production from GPD2 are high and comparable with maximum rates for complex III when inhibited with antimycin A, and it is an important ROS source in the heart.17,18 The findings of present study provide novel insights into the mechanistic link of miR-210 and GPD2 in controlling mitochondrial bioenergetics and ROS flux during IR in the heart, and present potential therapeutic targets for treatment of ischemic heart disease.
Methods
Supporting data and methods can be found in the Supplementary material online. The authors declare that all supporting data are available within the article and its online supplementary files.
Animals
MiR-210 heterozygous mice on C57BL/6J genetic background were generously provided by Drs. Xin Huang and Yoel Sadovsky.19 STOCK mir210tm1Mtm/Mmjax female mice were first crossed with B6.129S4-Gt(ROSA)26Sortm1(FLP1)Dym/RainJ males to delete the Neo cassette, followed by crossing with B6.C-Tg(CMV-cre)1Cgn/J to delete miR-210.19 Genotypes of wild type (WT) and miR-210 knockout (KO) offspring were confirmed by PCR. Mice were around 8 weeks old at the commencement of experiments. Animal experiments and surgical procedures were performed according to protocols approved by the Institutional Animal Care and Use Committee of Loma Linda University and followed the guidelines by the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Statistical Analysis
Data are expressed as means ± standard deviation or medians and interquartile ranges (IQRs) obtained from the number of experimental animals. Data were analyzed with GraphPad Prism (GraphPad Software, San Diego, CA, USA). Differences were evaluated for statistical significance (P<0.05) by two-way, two-way repeated-measures or three-way analysis of variance (ANOVA) followed by Tukey test, Independent-Samples t test, or non-parametric Kruskal–Wallis test followed by Dunn’s test or Mann-Whitney U test where appropriate.
Results
MiR-210 deficiency exaggerates IR-induced cardiac dysfunction in male mice in a sex-dependent manner
MiR-210 KO and their WT littermate control mice with C57Bl/6 background at 2 months old underwent myocardial infarction (MI) induced by in vivo heart IR treatment via ligation of the mid-left anterior descending (LAD) coronary artery for 30 min followed by reperfusion. Sham-operated animals without ligation of LAD coronary artery served as naïve controls. Hearts were isolated after 4 h or 24 h reperfusion and miR-210 in the heart was measured. As shown in Figure 1, A, miR-210 was significantly increased in the heart at both time points in WT, but not KO, mice. No significant differences between female and male baselines and between female and male IR treatments were found in miR-210 levels. Cardiac function was evaluated by echocardiography prior to and up to 6 weeks after in vivo IR treatment. As shown in Figure 1, B–E and Supplemental Figure I, A–G, there were no significant differences in echocardiography between miR-210 KO and WT mice at baseline and in sham-operated animals. In male WT mice, IR caused a decrease in IVSd, IVSs, LVPWd, LVPWs, EF, FS, and an increase in LVIDd, LVIDs, EDV and ESV. In female WT mice, IR decreased EF and FS, and increased LVIDs and ESV. Of great interest, miR-210 deficiency revealed a striking sex-difference and significantly exaggerated IR-induced cardiac dysfunction in male mice without affecting female animals.
Figure 1. MiR-210 deficiency exaggerates cardiac dysfunction after myocardial infarction in male mice.
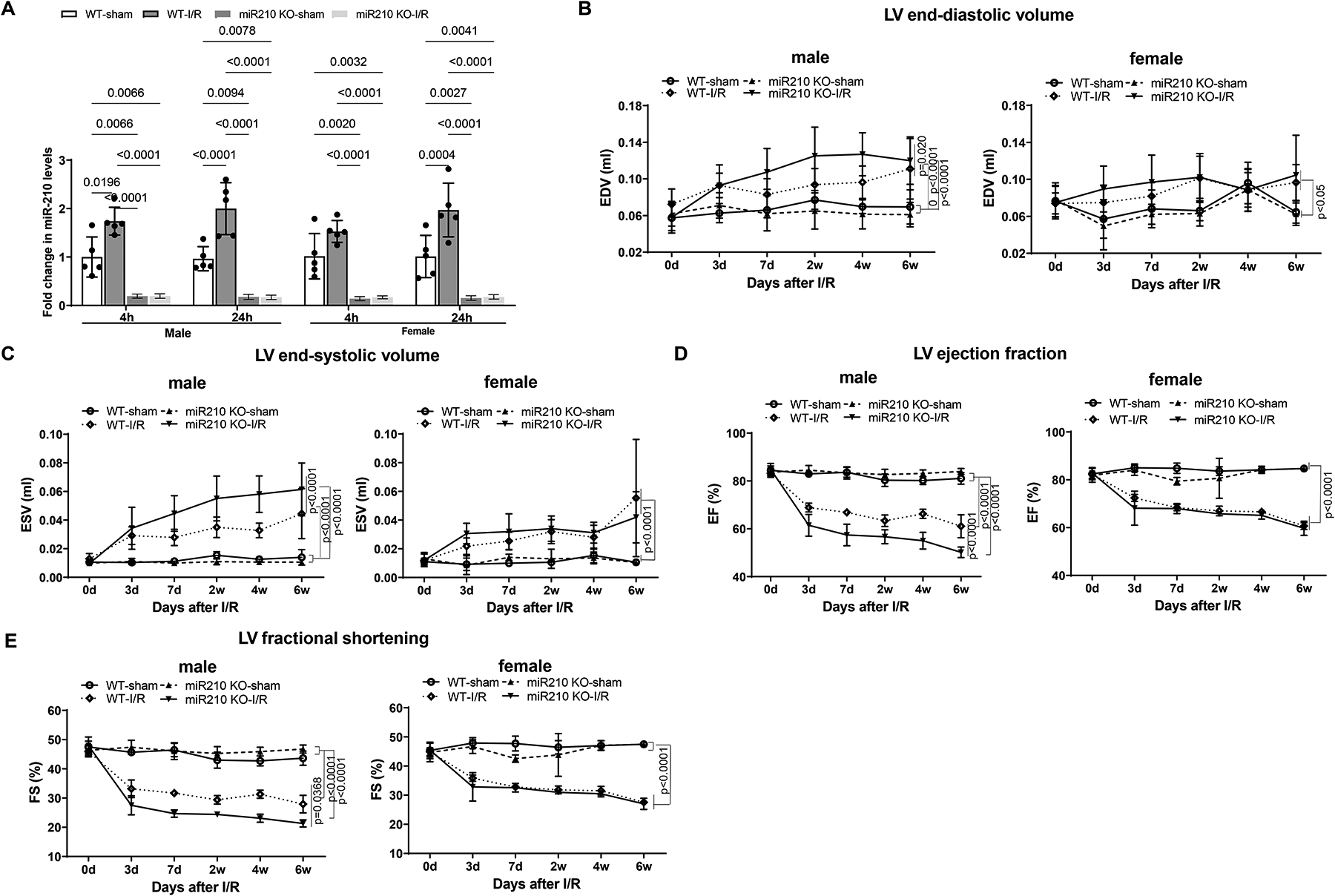
MiR-210 knockout (KO) and their wild-type (WT) littermate control mice with C57Bl/6 background at 2 months old underwent myocardial infarction (MI) induced by in vivo heart ischemia and reperfusion (IR) treatment via ligation of the mid-left anterior descending coronary artery for 30 min followed by reperfusion. Sham-operated animals without ligation of LAD coronary artery served as naïve controls. Hearts were isolated after 4 h or 24 h reperfusion and miR-210 in the heart was measured (A). Cardiac function was evaluated by echocardiography prior to or up to 6 weeks after in vivo IR treatment (B-E). Data are means ± SD, with n=5–6 animals per group, and analyzed by three-way ANOVA (A) or two-way repeated-measures ANOVA (B-E) followed by Tukey’s test. P values are shown in the Figure.
MiR-210 mimic rescues the effect of miR-210 deficiency on IR-induced MI and cardiac dysfunction
Given the finding that miR-210 deficiency affected males only, our following studies were focusing on male animals. We first examined whether and to what extent miR-210 mimic might rescue the effect of miR-210 deficiency on IR-induced MI and cardiac dysfunction. We observed that intravenous administration of miR-210 mimic (10 nmol/kg) increased miR-210 levels in hearts from both miR-210 KO and WT mice 4 h and 24 h after the injection (Supplemental Figure II). Thus, miR-210 mimic (10 nmol/kg) or the negative control was intravenously administered 1 h prior to the IR treatment in miR-210 KO or WT mice, and MI and functional echocardiography were evaluated 72 h after IR. We found that miR-210 deficiency increased MI volume in the setting of acute IR injury compared with WT controls, and miR-210 mimic rescued the effect of miR-210 deficiency on MI (Figure 2, A and B). Likewise, miR-210 deficiency enhanced dysfunction of left ventricular ejection fraction (EF) and fractional shortening (FS), and miR-210 mimic blocked the exaggerated dysfunction in miR-210 KO mice (Figure 2, C and D). Other systolic and diastolic parameters such as IVSd, IVSs, LVPWd, LVPWs, LVIDd, LVIDs, EDV, ESV, and SV were not or slightly altered by miR-210 mimic in WT and miR-210 KO mice 3 days after IR (Supplemental Figure III).
Figure 2. MiR-210 mimic exerts a protective effect on IR-induced MI and cardiac dysfunction.
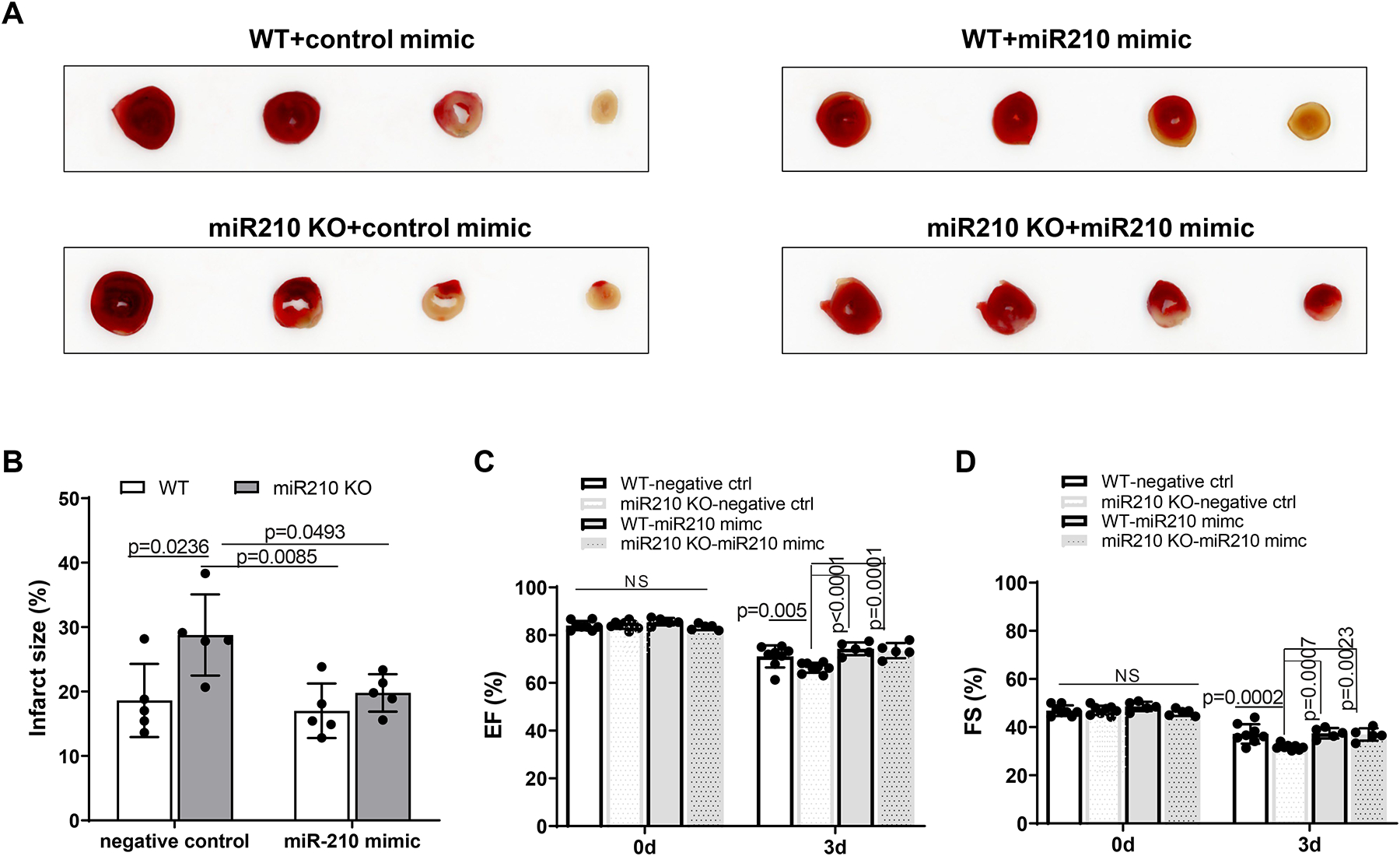
Male miR-210 knockout (KO) and their wild-type (WT) littermate control mice with C57Bl/6 background at 2 months old underwent myocardial infarction (MI) induced by in vivo heart ischemia and reperfusion (IR) treatment via ligation of the mid-left anterior descending coronary artery for 30 min followed by reperfusion. MiR-210 mimic (10 nmol/kg) or negative control was intravenously administered 1 h prior to the IR treatment. MI (A, B) and functional echocardiography (C, D) were evaluated 72 h after IR. Data are means ± SD, with n=5–8 animals per group, and analyzed by two-way ANOVA followed by Tukey’s test. P values are shown in the Figure.
MiR-210 controls mitochondrial bioenergetics in the acute heart IR
The mitochondrion is a major target of miR-210 in cellular hypoxic response. Inhibition of mitochondrial oxygen consumption and bioenergetics during IR affords protection of ischemic heart disease. We thus investigated the role of miR-210 in controlling mitochondrial bioenergetics in the setting of acute heart IR. In concert, we performed Seahorse flux analysis to assess the oxygen consumption rate (OCR, an index of oxidative phosphorylation) and lactate production as assessed by the extracellular acidification rate (ECAR; an index of glycolysis) in isolated cardiac muscle fiber bundles. Supplemental Figure IV illustrates the flow charts for the measurement of OCR and ECAR using a Seahorse XF24 Analyzer. Real-time traces and averaged data of OCR and ECAR measured by Seahorse are shown in Figure 3, A and G. After 30 min of myocardial ischemia and 24 h reperfusion, we observed significant and reproducible increases in basal OCR and maximal respiration capacity in the heart of miR-210 KO mice, as compared to WT animals (Figure 3, B and C). However, oxygen usage devoted to ATP production was significantly decreased (Figure 3, D), companied with a significant increase in OCR linked to proton leak (Figure 3, E), resulting in significantly reduced coupling efficiency of energy production (Figure 3, F). In concert, we observed decreases in basal ECAR and glycolytic capacity in miR-210 KO mice (Figure 3, H and I). These effects of miR-210 deficiency on mitochondrial bioenergetics in the heart were recovered by miR-210 mimic (Figure 3, A–I). Non-mitochondrial respiration, spare respiratory capacity and glycolytic reserve were not significantly affected (Supplemental Figure V). These findings explicitly demonstrate that miR-210 promotes mitochondrial metabolic switch from oxidative phosphorylation to glycolysis in fine-tuning heart response to acute IR and MI development.
Figure 3. MiR-210 controls mitochondrial bioenergetics in the acute heart IR.
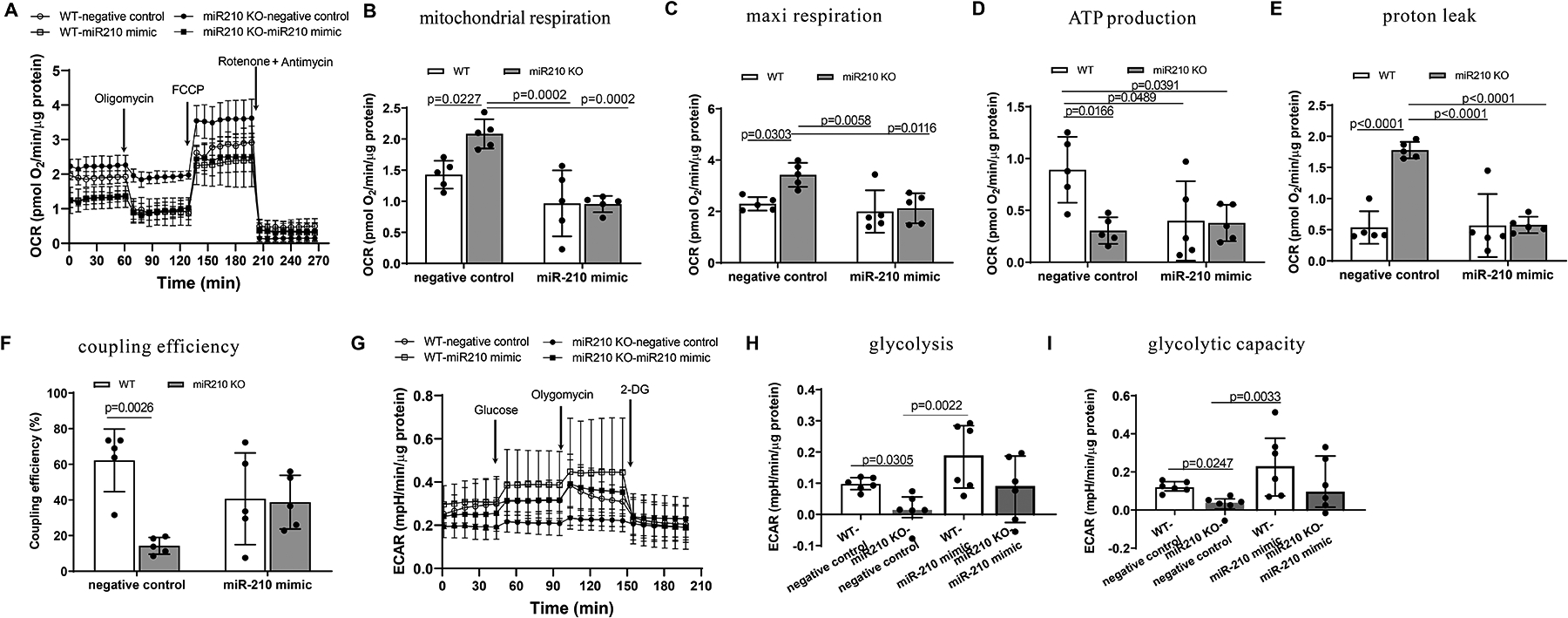
Male miR-210 knockout (KO) and their wild-type (WT) littermate control mice with C57Bl/6 background at 2 months old underwent myocardial infarction (MI) induced by in vivo heart ischemia and reperfusion (IR) treatment via ligation of the mid-left anterior descending coronary artery for 30 min followed by reperfusion. MiR-210 mimic (10 nmol/kg) or negative control was intravenously administered 1 h prior to the IR treatment. The hearts were harvested 24 h after IR and mitochondrial oxygen consumption rate (OCR) (A-F) and extracellular acidification rate (ECAR) (G-I) were analyzed by Seahorse XFe24 Analyzer in isolated cardiac muscle fiber bundles. Seahorse real-time traces and averaged data for OCR (A) and ECAR (G) including the chemical agents that dissect the metabolic responses are shown. Data are means ± SD, and analyzed by two-way ANOVA followed by Tukey’s test (A-E) or median and IQRs with non-parametric Kruskal–Wallis test followed by Dunn’s test (F, G), with n=5–6 animals per group. P values are shown in the Figure.
MiR-210 suppresses mitochondrial ROS in acute heart IR injury and cardiac dysfunction
In the setting of heart IR, it is likely that increased miR-210 acting as a homeostatic mechanism to inhibit mitochondrial bioenergetics and oxygen consumption relieves the mismatch of electron transport and oxygen concentration and decreases mitochondrial ROS (mtROS) flux. We next examined a mechanistic link of miR-210 in the regulation of mtROS production in the heart during IR with the approaches of gain-of-function and loss-of-function using miR-210 mimic and miR-210 KO mice. MiR-210 KO and WT control mice were subjected to 30 min of ischemia followed by 24 h reperfusion, and mitochondria-derived ROS in the heart were measured using a specific mtROS fluorescence probe, MitoSOX.20 We found that mtROS were significantly increased in the heart of KO mice, which was blocked by miR-210 mimic administered intravenously 1 h prior to the IR treatment (Figure 4, A). In agreement, we observed that mitochondria-specific antioxidant, MitoQ (4 mg/kg) given intravenously 15 min prior to the IR treatment decreased mtROS in the heart of WT mice and negated the increased mtROS in miR-210 KO mice (Figure 4, B). We then examined whether and to what extent MitoQ might recover the effect of miR-210 deficiency on IR-induced MI and cardiac dysfunction. We found that MitoQ reduced IR-induced MI in WT mice and negated the effect of miR-210 deficiency on IR-mediated MI (Figure 4, C and D). In concert, we observed that MitoQ rescued the effect of miR-210 deficiency on exaggerated dysfunction of left ventricular EF and FS in miR-210 KO mice 3 days after IR (Figure 4, E and F). The other systolic and diastolic parameters such as IVSd, IVSs, and LVPWd were increased, while LVIDd, LVIDs, EDV, and ESV were decreased. LVPWs and SV were not or slightly altered by mitoQ in WT and miR-210 KO mice exposed to 3 d of IR (Supplemental Figure VI).
Figure 4. MiR-210 suppresses mitochondrial ROS in acute heart IR injury and cardiac dysfunction.
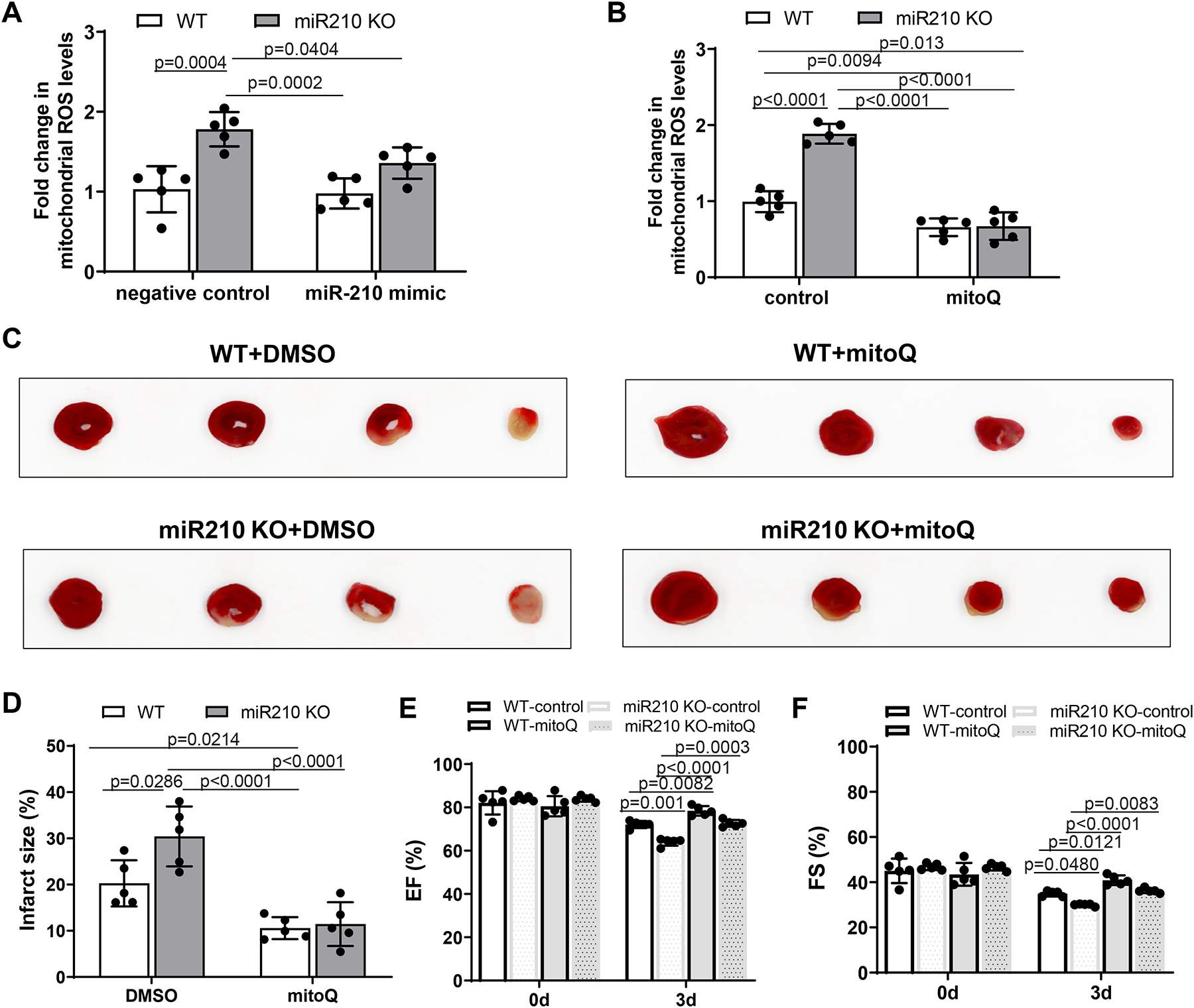
Male miR-210 knockout (KO) and their wild-type (WT) littermate control mice with C57Bl/6 background at 2 months old underwent myocardial infarction (MI) induced by in vivo heart ischemia and reperfusion (IR) treatment via ligation of the mid-left anterior descending coronary artery for 30 min followed by reperfusion. MiR-210 mimic (10 nmol/kg) or negative control was intravenously administered 1 h prior to the IR treatment. MitoQ (4 mg/kg) or vehicle control (DMSO, 1%) was intravenously applied 15 min prior to the IR. Mitochondria were isolated from the hearts 24 h after IR and mitochondria-derived ROS was analyzed with MitoSOX Red (A, B). MI (C, D) and functional echocardiography (E, F) were evaluated 72 h after IR. Data are means ± SD, with n=5 animals per group, and analyzed by two-way ANOVA followed by Tukey’s test. P values are shown in the Figure.
We next examined the effect of inhibition of mtROS production by MitoQ on mitochondrial respiration and glycolysis in the heart of WT and miR-210 KO mice. We observed that MitoQ had no significant effect on basal mitochondrial OCR and maximal respiration capacity in WT mice in the setting of acute IR, but blocked the effect of miR-210 deficiency on mitochondrial OCR (Figure 5, B and C). Of interest, MitoQ significantly increased oxygen usage devoted to ATP production and decreased OCR linked to proton leak in the heart of WT animals, and negated the effect of miR-210 deficiency (Figure 5, D and E). In concert, despite inhibition of mitochondrial respiration, MitoQ significantly increased coupling efficiency of energy production (Figure 5, F). In addition, we found that MitoQ decreased basal ECAR and glycolytic capacity in WT mice and negated the effect of miR-210 deficiency on glycolysis in the heart in the setting of acute IR (Figure 5, H and I). Non-mitochondrial respiration, spare respiratory capacity and glycolytic reserve were not significantly affected (Supplemental Figure VII). Together, these findings suggest that the protective effects of MitoQ are mediated by its effects on mitochondrial respiration and mtROS production and releasing glycolysis stress in the heart in the setting of acute IR.
Figure 5. MitoQ rescues the effect of miR-210 deficiency on mitochondrial bioenergetics.
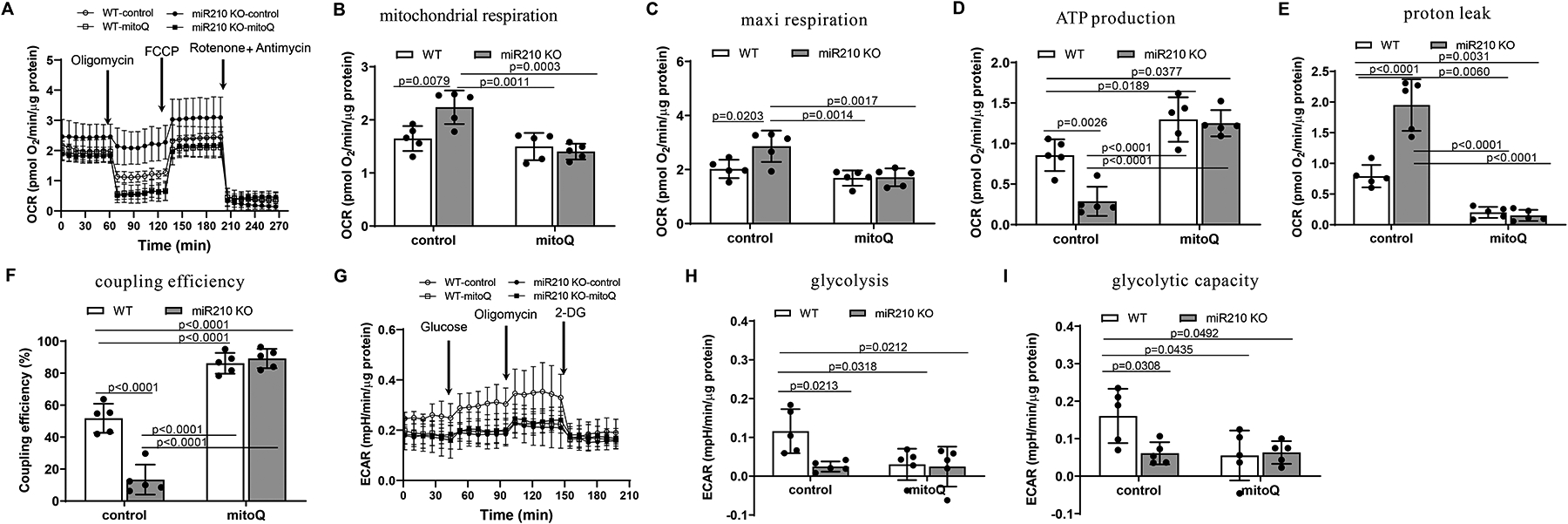
Male miR-210 knockout (KO) and their wild-type (WT) littermate control mice with C57Bl/6 background at 2 months old underwent myocardial infarction (MI) induced by in vivo heart ischemia and reperfusion (IR) treatment via ligation of the mid-left anterior descending coronary artery for 30 min followed by reperfusion. MitoQ (4 mg/kg) or vehicle control (DMSO, 1%) was intravenously applied 15 min prior to IR. The hearts were harvested 24 h after IR and mitochondrial oxygen consumption rate (OCR) (A-F) and extracellular acidification rate (ECAR) (G-I) were analyzed by Seahorse XFe24 Analyzer in isolated cardiac muscle fiber bundles. Seahorse real-time traces and averaged data for OCR (A) and ECAR (G) including the chemical agents that dissect the metabolic responses are shown. Data are means ± SD, with n=5 animals per group, and analyzed by two-way ANOVA followed by Tukey’s test. P values are shown in the Figure.
GPD2 is a novel target of miR-210 controlling mitochondrial ROS in cardiomyocytes
We first examined the role of miR-210 in targeting 3’UTR of GPD2 transcript. Figure 6, A depicts sequences of GPD2 3’UTR with miR-210 targeting sites and a schematic illustration of miR-210 silencing GPD2 mRNA translation. We identified a potential miR-210 complementary binding site in GPD2 3’UTR using the sequence alignment program MultAlin (http://multalin.toulouse.inra.fr/multalin/multalin.html), and demonstrated using a RISC-IP assay that GPD2 3’UTR was enriched by ~5-fold in miR-210 mimic-transfected mouse neonatal cardiomyocytes, compared to negative control-treated cells (Figure 6, B), indicating that GPD2 is a direct target of miR-210. We then evaluated the role of GPD2 in miR-210 mediated cardiomyocyte protection in an oxygen-glucose-deprivation (OGD) model in vitro. We first confirmed that 2 h of OGD with 1% O2 and 24 h of reoxygenation markedly upregulated the level of miR-210 in cardiomyocytes, which was blocked by miR-210 LNA (Figure 6, C). We found that inhibition of miR-210 with miR-210 LNA upregulated GPD2 protein abundance in OGD-treated cardiomyocytes, which was negated by GPD2 siRNA treatment (Figure 6, D). Importantly, miR-210 LNA significantly enhanced OGD/reoxygenation-induced cardiomyocyte injury and LDH release, which was negated by knocking down GPD2 with siRNAs (Figure 6, E), revealing a role of GPD2 downregulation in miR-210-mediated cardiomyocyte protection to hypoxic injury. Next, we observed that compared with the normal controls, the presence of miR-210 LNA increased mtROS in OGD/reoxygenation-treated cardiomyocytes (Figure 6, F). We found that knockdown of GPD2 reduced OGD/reoxygenation-induced mtROS and negated the effect of miR-210 LNA on OGD/reoxygenation-mediated increase in mtROS (Figure 6, F). Since GPD2 knockdown negated the effect of miR-210 inhibition on the increase in mtROS production and injury in cardiomyocytes to OGD/reoxygenation, we then investigated the effect of GPD2 knockdown on mitochondrial respiration and glycolysis in cardiomyocytes. We observed that GPD2 knockdown had no significant effect on non-mitochondrial respiration and spare respiratory capacity in cardiomyocytes exposed to OGD/reoxygenation (data not shown), but blocked the effect of miR-210 inhibition on mitochondrial OCR (Figure 6, G–I). Moreover, GPD2 knockdown significantly increased oxygen usage to ATP production, decreased OCR linked to proton leak, increased coupling efficiency of mitochondrial energy production in OGD/reoxygenation-treated cardiomyocytes and negated the effect induced by miR-210 LNA (Figure 6, J–L). Likewise, we found that GPD2 knockdown reduced glycolysis and glycolytic capacity in cardiomyocytes exposed to OGD/reoxygenation and negated the effect of miR-210 inhibition (Figures 6M–O).
Figure 6. MiR-210 targeting GPD2 inhibits mitochondrial bioenergetics in modulating cardiomyocyte hypoxic injury.
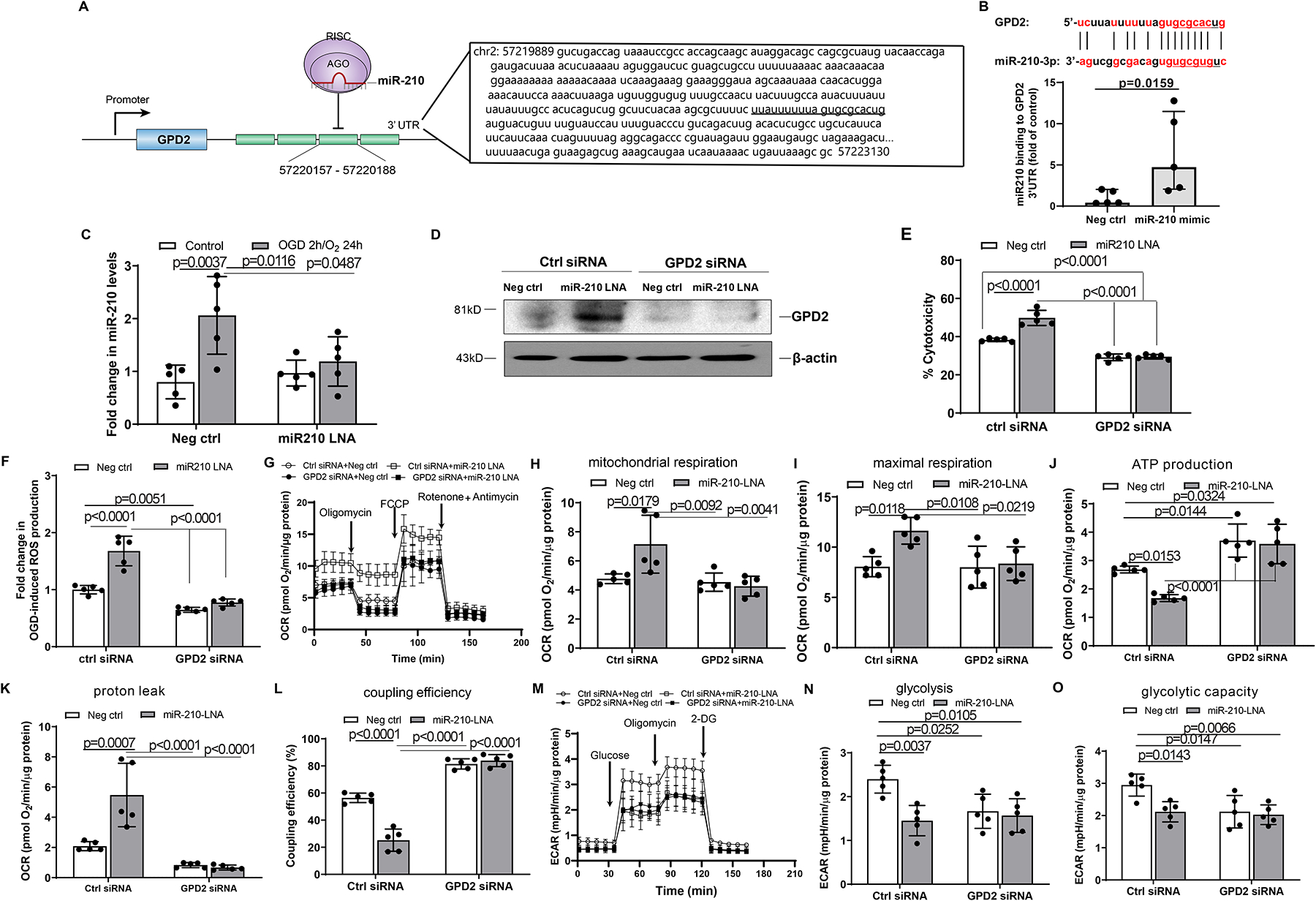
Sequence of GPD2 3’UTR with miR-210 targeting sites and a schematic depiction of miR-210 silencing the GPD2 mRNA translation are shown in panel A. The binding of miR-210 to GPD2 3’UTR was assessed by RISC-IP assay in mouse neonatal cardiomyocytes (B). Mouse neonatal cardiomyocytes were treated with 50 nM miR-210-LNA or scramble LNA (Neg. Ctrl) for 24 h, followed by subjecting to 2 h OGD and 24 h reoxygenation. Control cells were cultured in the normoxic condition for the same durations. The levels of miR-210 were measured 24 h after reoxygenation (C). Mouse neonatal cardiomyocytes were treated with 100 nM GPD2 siRNA and 50 nM miR-210-LNA or scramble LNA (Neg. Ctrl) for 24 h, and then were subjected to OGD followed by reoxygenation (D-O). GPD2 protein abundance was determined with Western blots (D). Cell injury was measured with LDH release assay (E). Mitochondria-derived ROS was analyzed with MitoSOX Red (F). Mitochondrial oxygen consumption rate (OCR) (G-L) and extracellular acidification rate (ECAR) (M-O) were analyzed by Seahorse XFe24 Analyzer. Data are median and IQRs with Mann-Whitney U test (B), or means ± SD and analyzed by two-way ANOVA followed by Tukey’s test (C, E-O), with n=5 independent cultures per group. P values are shown in the Figure.
We next investigated the specific effect of MitoQ on the mitochondrion underlying miR210-GPD2-mtROS signaling in cardiomyocyte hypoxic injury. We found that inhibition of miR-210 with miR-210 LNA significantly increased ROS in the mitochondrion in OGD-treated cardiomyocytes, which was specifically blocked by MitoQ (Figure 7, A and B). In accordance, MitoQ negated the miR-210 LNA-mediated increase in cardiomyocyte injury induced by OGD/reoxygenation (Figure 7, C), revealing a mechanistic link of MitoQ acting on mitochondrial ROS in the miR-210 signaling pathway in cardiomyocytes. Given that miR-210 LNA upregulated GPD2 protein abundance in OGD-treated cardiomyocytes, we then evaluated the specific effect of GPD2 overexpression on mitochondrial ROS in cardiomyocytes. We observed that in the way similar to miR-210 LNA, GPD2 overexpression increased ROS in the mitochondrion and cardiomyocyte injury induced by OGD/reoxygenation, which were blocked by MitoQ (Figure 7, D–F). In addition, we found that GPD2 overexpression enhanced mitochondrial OCR, decreased oxygen usage to ATP production, increased OCR linked to proton leak, decreased coupling efficiency of mitochondrial energy production, and decreased glycolysis and glycolytic capacity in OGD/reoxygenation-treated cardiomyocytes. Of importance, MitoQ negated the effects induced by GPD2 overexpression (Supplemental Figure VIII). Thus, both loss-of-function and gain-of-function findings provide explicit evidence that GPD2 is a key player in the effect of miR-210 on mitochondrial ROS generation in cardiomyocytes.
Figure 7. MitoQ inhibits miR-210 LNA and GPD2 overexpression-induced mtROS generation and cardiomyocyte hypoxic injury.
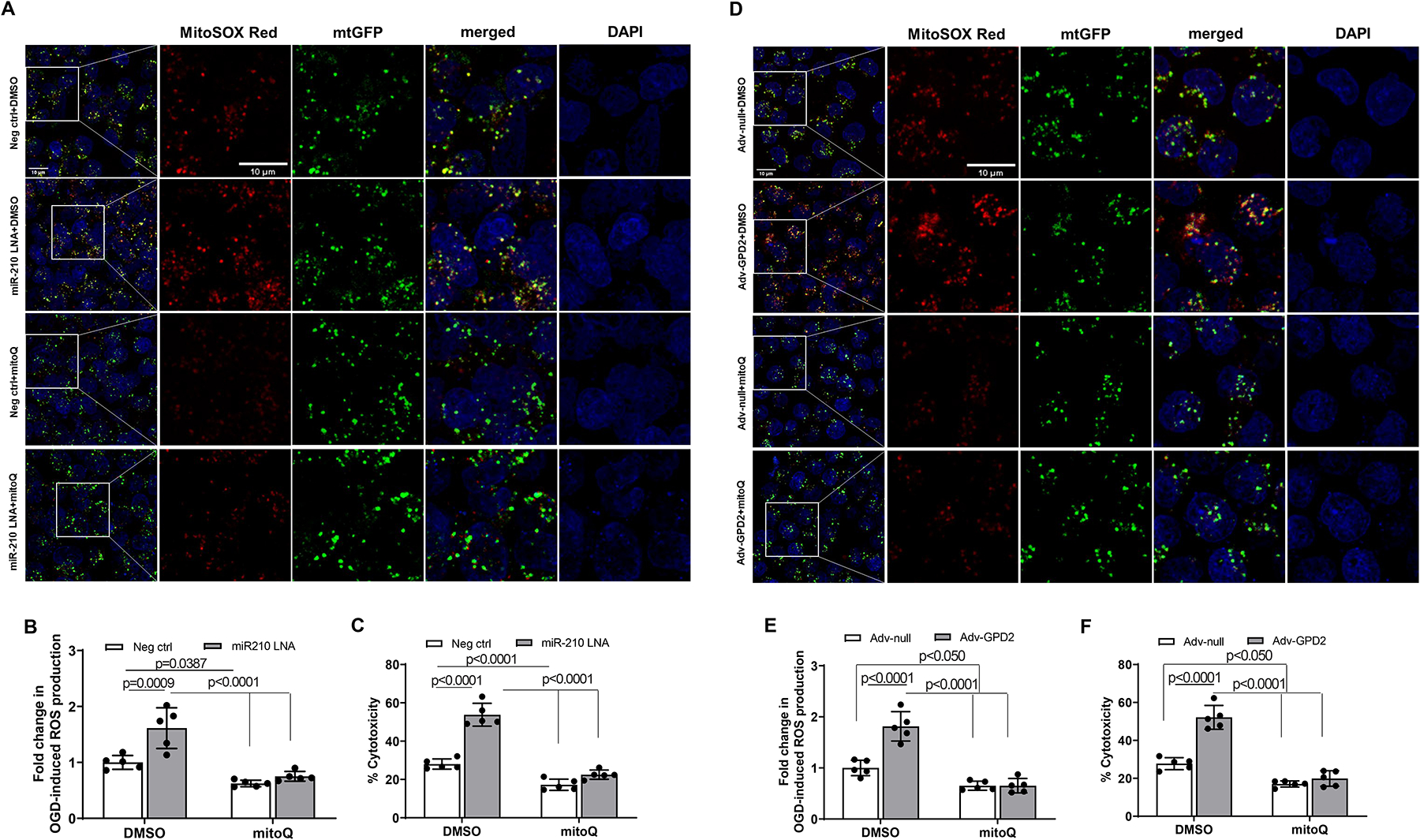
Mouse neonatal cardiomyocytes were treated with miR-210 LNA (50 nM) or scramble LNA (Neg. Ctrl), Adv-GPD2 (1.6 × 107 pfu/ml) or Adv-null in the presence of MitoQ (250 nM) or vehicle control DMSO overnight, followed by subjecting to 2 h OGD and 24 h reoxygenation. Mitochondria-derived ROS was visualized with MitoSOX Red and fluorescence was colocalized with mitochondria, as visualized with Mitochondria-GFP (mtGFP) using fluorescent confocal microscopy (A and D). Panels B and E show the quantitative data of mtROS. Cell injury was measured with LDH release assay (C and F). Data are means ± SD with n=5 independent cultures per group, and analyzed by two-way ANOVA followed by Tukey’s test. P values are shown in the Figure.
Knockdown of GPD2 protects the heart and negates the effect of miR-210 deficiency on IR injury
We then determined whether and to what extent endogenous miR-210 regulates GPD2 in the heart. We found that GPD2 protein abundance was significantly increased in the heart of miR-210 KO mice, as compared to WT mice (Figure 8, A). Increased GPD2 was maintained and enhanced in the heart of miR-210 KO mice after IR at 4 h and 24 h, respectively (Figure 8, B). We verified that GPD2 is a target of miR-210 in MI and demonstrated the binding of endogenous miR-210 to GPD2 3’UTR in vivo in the heart after ischemia for 30 min and reperfusion for 24 h. In agreement with the increase in miR-210 binding to GPD2 3’UTR in miR-210 mimic-transfected cardiomyocytes, IR significantly increased the binding of miR-210 to GPD2 3’UTR in the heart, which was not observed in miR-210 KO mice (Figure 8, C). In concert, intravenous administration of miR-210 mimic prior to the IR treatment decreased GPD2 protein abundance in the heart of WT mice and blocked the effect of miR-210 deficiency on GPD2 protein in the setting of acute heart IR (Figure 8, D).
Figure 8. Knockdown of GPD2 protects the heart and negates the effect of miR-210 deficiency on IR injury.
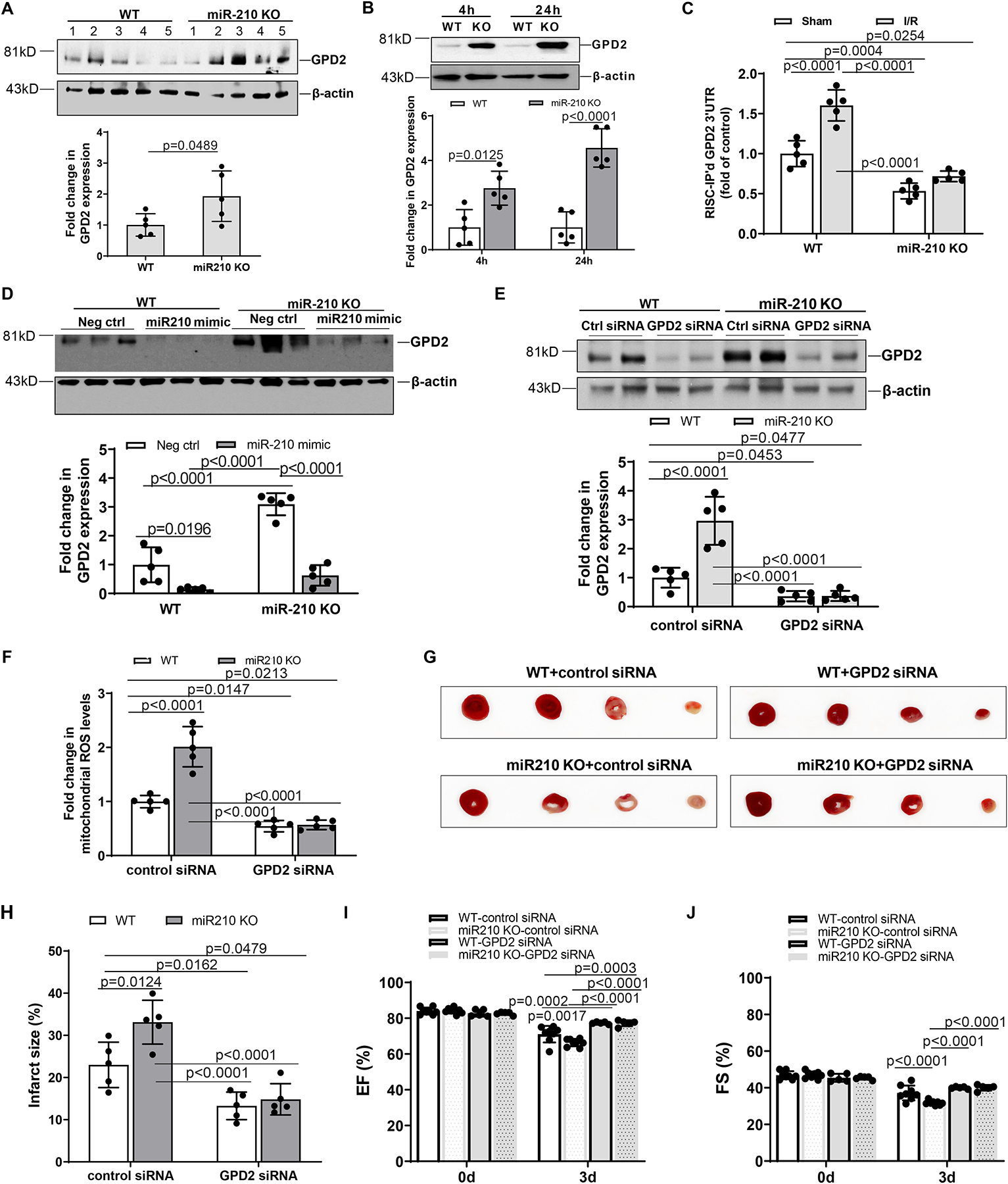
Male miR-210 knockout (KO) and their wild-type (WT) littermate control mice with C57Bl/6 background at 2 months old underwent myocardial infarction (MI) induced by in vivo heart ischemia and reperfusion (IR) treatment via ligation of the mid-left anterior descending coronary artery for 30 min followed by reperfusion. GPD2 protein abundance was measured in the heart before IR (A) and after IR (B) in WT and KO mice. Panel C shows the binding of endogenous miR-210 to GPD2 3’UTR in vivo in the heart after ischemia for 30 min and reperfusion for 24 h in WT and miR-210 KO mice. Panel D shows that intravenous administration of miR-210 mimic 1 h prior to the IR treatment decreases GPD2 protein abundance in the heart of WT mice and blocks the effect of miR-210 deficiency on GPD2 protein in the setting of acute heart IR. GPD2 siRNAs (2 mg/kg) or control siRNAs were intravenously applied 24 h prior to the IR treatment, and hearts were isolated 24 h after IR. GPD2 protein abundance (E) was determined with Western blots and mitochondria-derived ROS (F) was analyzed with MitoSOX Red. MI (G, H) and functional echocardiography (I, J) were evaluated 72 h after IR. Data are means ± SD, with n=5–8 animals per group, and analyzed by t-test (A) and two-way ANOVA followed by Tukey’s test (B-J). P values are shown in the Figure.
We further determined whether and to what extent knockdown of GPD2 inhibits IR-induced mtROS production and MI in WT and miR-210 KO mice. We first validated that GPD2 siRNAs knockdown GPD2 protein expression in the heart of both WT and miR-210 KO mice underwent acute IR treatment (Figure 8, E). We then observed that knockdown of GPD2 protein abundance decreased IR-induced mtROS production and MI in WT and KO mice, and negated the effect of miR-210 deficiency on increased mtROS production and MI (Figure 8, F–H). In concert, knockdown of GPD2 in the heart recovered exaggerated dysfunction of left ventricular EF and FS observed in miR-210 KO mice 3 days after IR (Figure 8, I and J). Other systolic and diastolic parameters were not or slightly altered by GPD2 knockdown in WT and miR-210 KO mice exposed to IR (Supplemental Figure IX).
Discussion
The present finding with loss-of-function and gain-of-function in miR-210 KO mice and using miR-210 mimic provides cause-and-effect evidence that endogenous miR-210 in the heart serves a compensatory mechanism and protects the heart in IR-induced AMI and resultant cardiac dysfunction. The observation of the sex-specific effect in male animals is of great interest, albeit the mechanisms remain elusive. Epidemiological data show that pre-menopausal females have reduced risk and death rates for coronary heart disease. Although female sex hormones may be beneficial in protecting the heart, a large clinical trial failed to show cardioprotection for postmenopausal women on estrogen-progestin replacement,21 highlighting the complexity of gender-based differences in mechanisms of protection in myocardial IR injury. Among other mechanisms, an intricate interplay of sex-biased miRNA networks and intrinsic sex differences in mitochondrial genotype and phenotype may contribute to gender differences in cardiovascular disease.22–24 The discovery of novel regulatory mechanisms of miRNA in the pathogenesis of heart disease has presented new opportunities.5,25–27 Several studies revealed cardioprotective effects of miR-199a, miR-210, miR-499, antimiR-21 and antimiR-92a in treatment of AMI and adverse infarct remodeling in rodent and pig models.2,28–31 Yet, a certain causal role for miRNA in cardioprotection is still controversial. In this study, we demonstrate explicitly in miR-210 KO mice a causative effect of endogenous miR-210 on protecting the heart and improving cardiac function in a murine model of myocardial infarction. Indeed, the role of miR-210 in regulating oxidative stress and hypoxic response is in clinical trials for peripheral artery disease (ClinicalTrials.gov Identifier: NCT04089943) and diabetes (ClinicalTrials.gov Identifier: NCT02629406).
In this study we provide evidence that the stress response of miR-210 in the heart presents a compensatory mechanism of inhibiting mitochondrial oxygen consumption during IR and protects the heart from IR injury. MiR-210 is robustly induced by hypoxia, and is a master regulator of the cellular hypoxic response by targeting mitochondria and inhibiting complex I activity and mitochondrial oxygen consumption.32–34 We found that miR-210 deficiency significantly increases mitochondrial oxygen consumption in the heart after myocardial ischemia and during reperfusion, which is accompanied by an increase in IR-induced mtROS and AMI. Of importance, we observed that miR-210 mimic negates the effect of exaggerated mitochondrial respiration, mtROS production and MI in miR-210 KO mice. These findings of loss-of-function and gain-of-function provide explicit evidence that miR-210 is necessary and sufficient in controlling mitochondrial bioenergetics and ROS generation in the setting of heart IR injury. The observation that despite an increase in mitochondrial oxygen consumption, the coupling efficiency of energy production is significantly decreased with a reduction of ATP generation and an increase in proton leak in miR-210 KO mice, is of great interest. Although proton leak contributes to the reduction of mtROS generation and facilitates exertion of the protective loop that helps to reduce the effects caused by ROS on biological systems, excessive proton leak promotes ROS production, damage of mitochondria and cell viability in the heart.35–37 Mitochondrial dysfunction and energy deficiency have been strongly implicated in the development of heart failure with reduced ejection fraction (HFrEF).38–40 MiR-210 deficiency-mediated decrease in ATP production through oxidative phosphorylation in conjunction with reduced glycolytic activity during IR is likely to contribute to exaggerated dysfunction of left ventricular ejection fraction and fractional shortening observed in miR-210 KO mice and promote the development of HFrEF in ischemic heart disease. Increasing glycolysis contributes to slowing ATP depletion and maintaining higher free energy from ATP hydrolysis, which positively affects systolic and diastolic function during IR.41 The finding that miR-210 mimic rescued the effect of miR-210 deficiency on reduced energy production and ejection fraction/fractional shortening supports a beneficial role of miR-210 in preserving cardiomyocyte energy and fine-tuning heart response to IR and development of heart failure.
Despite the large body of evidence supporting the critical role of ROS production and oxidative stress in ischemic heart disease, large clinical trials with untargeted antioxidants failed to deliver clinically significant benefits.42,43 Inhibition of mtROS generation on reperfusion by antioxidants targeting mitochondria may provide better clinical outcomes. MitoQ is a mitochondria-targeted antioxidant and has been shown to be cardioprotective in a rat model of cardiac ischemia-reperfusion injury.44 In the present study, we observed that MitoQ decreases mtROS in the heart of WT and KO mice and negates the increased mtROS in miR-210 KO mice. Of importance, MitoQ reduces IR-induced AMI and improves left ventricular EF and FS in WT and KO mice, and negates the effect of miR-210 deficiency on IR-mediated AMI and left ventricular dysfunction. These findings support a therapeutic potential of MitoQ in ischemic heart disease and provide evidence of a causal role of mtROS in exaggerated AMI and heart dysfunction observed in miR-210 KO mice in the setting of IR. The mitochondrion is not only the main source of ROS but also a primary target of ROS damage. We found that MitoQ blocks the effect of miR-210 deficiency on mitochondrial respiration and significantly improves the coupling efficiency of energy production in the heart of both WT and miR-210 KO mice, revealing a mechanistic link of inhibition of mtROS and improvement of mitochondrial bioenergetics during heart IR. This is consistent with the observation of improvement of left ventricular EF and FS in both WT and miR-210 KO mice. The intricate effect of MitoQ on increasing coupling efficiency of ATP production through oxidative phosphorylation without significantly increasing mitochondrial oxygen consumption during IR is of great interest and may be of a significant benefit in treatment of heart failure. Indeed, MitoQ improves mitochondrial dysfunction and increases β-oxidation and ATP generation in heart failure induced by pressure overload.45 Of importance, MitoQ has now been in multiple clinical trials including that of heart failure.46
From a therapeutic perspective, blocking the excessive ROS production that occurs on ischemia and reperfusion at its source of mitochondria rather than scavenging ROS after it has been produced presents a more effective strategy, which may alleviate the potential harmful effect of antioxidant treatment on physiological ROS in cellular homeostasis. In addition to succinate dehydrogenase-dependent ROS production in mitochondria during IR9,47, we identified GPD2 as a novel target of miR-210 in the heart undergoing IR-induced MI. GPD2 is a membrane-bound protein to the outer face of mitochondrial inner membrane, and together with cytosolic GPD1 regulates GP-shuttle between cytosolic and mitochondrial energy production and redox balance. GPD2 was the first of mitochondrial dehydrogenases to be characterized as ROS producer.13, 48,49 Indeed, GPD2 produces high amounts of ROS, accounting for ~39%−52% of the total H2O2 generating capacity in mouse heart mitochondria.50 The most likely source of GPD2-dependent ROS production is the Q-binding pocket of GPD2 and superoxide is produced on both sides of the mitochondrial inner membrane in the matrix and intermembrane space.51 ROS production by GPD2 is regulated by the expression level of GPD2, glycerol 3-phosphate (G3P) and calcium, and raised levels of G3P and calcium during ischemia increase superoxide production from GPD2. We demonstrate that GPD2 is a direct target of miR-210 and both loss-of-function and gain-of-function findings provide explicit evidence that downregulation of GPD2 plays a key role in the specific effect of miR-210 on mitochondrial ROS generation in protection of cardiomyocyte viability in response to OGD/reoxygenation. Of importance, we found that miR-210 deficiency markedly upregulates GPD2 protein abundance in the heart in the setting of IR, suggesting that endogenous miR-210 maintains low levels of GPD2 in the heart. Consistent with the findings in cardiomyocytes of OGD/reoxygenation model, we observed that knockdown of GPD2 protein abundance decreases IR-induced mtROS production and AMI in WT and KO mice and blocks the effect of miR-210 deficiency on increased mtROS production and AMI. In concert, knockdown of GPD2 in the heart negates the exaggeration of left ventricular dysfunction in miR-210 KO mice after IR. Besides GPD2, both GPD1 and recently identified phosphoglycolate phosphatase acting as a G3P phosphatase (G3PP)52 are involved in regulation of the GP pathway. However, GPD2 is a rate-limiting step in the GP-shuttle. In addition, G3PP has a significantly higher Km as compared to the G3P dehydrogenase. Rather than affecting the metabolic flux through the glycolytic or lipid biosynthetic pathways under normal cellular levels of G3P, G3PP may protect the heart by removing the excess G3P produced when glucose and lipid levels are high, thereby prevent glucotoxicity and glucolipotoxicity that have been implicated in diabetic cardiomyopathy and heart failure.53 Taken together, these findings reveal a novel mechanistic link of GPD2 in miR-210-mediated regulation of mtROS production in the heart during IR and present a new therapeutic target in treatment of ischemic heart disease and heart failure. Whereas it is out of scope in this study, it remains intriguing for future investigation whether GPD2-dependent ROS production may affect the mitochondrial inner membrane potential that contributes to post-ischemic arrhythmias.54,55
The limitations of this research have revolved around a small animal mode, i.e. the mouse. The findings must be validated in large animal models before further interpretation of clinically meaningful targets and interventions revealed in the study. Another limitation is that the study focuses on male animals based on the initial finding that miR-210 deficiency shows a sex-specific effect on cardiac dysfunction after AMI in male mice. Further study must involve female animals and examine in great detail the sex-difference in mechanisms of protection in myocardial IR injury, particularly the differences in miRNA networks and mitochondrial phenotype. In addition, the effect and mechanistic role of miR-210 in different cardiac cells such as myocardial cells, endothelial cells, and immune cells have not been thoroughly investigated since the study primarily covers the cytopathic metabolism phenotype in the heart tissue.
In conclusion, we demonstrate explicitly in miR-210 KO mice a causative effect of endogenous miR-210 on protecting the heart and improving cardiac function in a murine model of myocardial infarction. The findings of loss-of-function and gain-of-function provide clear evidence that miR-210 is necessary and sufficient in controlling mitochondrial bioenergetics and ROS generation, and support a beneficial role of miR-210 in preserving cardiomyocyte energy in the setting of heart IR injury. We identify a novel mechanistic link of GPD2 in miR-210-mediated regulation of mtROS production in the heart. The study also reveals the intricate effect of MitoQ on inhibiting mtROS and maintaining mitochondrial ATP production, and presents a therapeutic potential of MitoQ in treatment of ischemic heart disease and heart failure.
Supplementary Material
Clinical Perspective.
What Is New?
MiR-210 deficiency induces an ischemic sensitive phenotype and exaggerates AMI and cardiac dysfunction after MI in males in a gender-dependent pattern in a murine model of myocardial infarction.
The study identifies a novel mechanism of miR-210 targeting GPD2 in controlling mitochondrial energy metabolism and ROS flux and improving cardiac function in the setting of acute IR injury.
MiR-210 mimic and MitoQ negate the increase in mtROS and exert a protective effect of AMI and resultant cardiac dysfunction in heart IR.
What Are the Clinical Implications?
Ischemic heart disease remains the most common cardiovascular disease and the leading cause of death globally.
Novel cardioprotective strategies are still required to attenuate the detrimental effects of AMI and to improve adverse heart remodeling and cardiac dysfunction in patients with coronary heart disease.
The present findings reveal new insights into the mechanisms and therapeutic targets for treatment of ischemic heart disease.
Sources of Funding
This work was supported in part by National Institutes of Health Grants P01 HD083132 (L. Zhang).
Non-standard Abbreviations and Acronyms
- IR
ischemia-reperfusion
- ROS
reactive oxygen species
- GPD2
glycerol-3-phosphate dehydrogenase
- AMI
acute myocardial infarction
- miR-210
microRNA 210
- WT
wild type
- KO
knockout
- IQRs
interquartile ranges
- ANOVA
analysis of variance
- LAD
left anterior descending
- PEI
polyethylenimine
- IVSd
interventricular septal end diastole
- IVSs
interventricular septal end systole
- LVIDd
LV end-diastolic dimension
- LVIDs
LV end-systolic dimension
- LVPWD
left ventricular posterior wall thickness at end diastole
- LVPWs
left ventricular posterior wall thickness at end systole
- EDV
end-diastolic volume
- ESV
end-systolic volume
- EF
ejection fraction
- SV
stroke volume
- FS
fractional shortening
- TTC
2,3,5-triphenylte-trazolium chloride
- mtROS
mitochondrial ROS
- PBS
phosphate buffer saline
- OCR
oxygen consumption rate
- ECAR
extracellular acidification rate
- 2-DG
2-deoxyglucose
- OGD
oxygen-glucose-deprivation
- RISC-IP
RNA-induced silencing complex-immunoprecipitation
- LDH
lactate dehydrogenase
- Adv-GPD2
adenoviral vector expressing GPD2
- HFrEF
heart failure with reduced ejection fraction
- GP
glycerophosphate
- G3P
glycerol 3-phosphate
Footnotes
Disclosures
None.
References
- 1.Mortality GBD, Causes of Death C. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;385:117–171. doi: 10.1016/S0140-6736(14)61682-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hu S, Huang M, Li Z, Jia F, Ghosh Z, Lijkwan MA, Fasanaro P, Sun N, Wang X, Martelli F, et al. MicroRNA-210 as a novel therapy for treatment of ischemic heart disease. Circulation. 2010;122:S124–131. doi: 10.1161/CIRCULATIONAHA.109.928424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ong SG, Lee WH, Huang M, Dey D, Kodo K, Sanchez-Freire V, Gold JD, Wu JC. Cross talk of combined gene and cell therapy in ischemic heart disease: role of exosomal microRNA transfer. Circulation. 2014;130:S60–69. doi: 10.1161/CIRCULATIONAHA.113.007917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patterson AJ, Song MA, Choe D, Xiao D, Foster G, Zhang L. Early Detection of Coronary Artery Disease by Micro-RNA Analysis in Asymptomatic Patients Stratified by Coronary CT Angiography. Diagnostics (Basel). 2020;10:875. doi: 10.3390/diagnostics10110875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karakas M, Schulte C, Appelbaum S, Ojeda F, Lackner KJ, Munzel T, Schnabel RB, Blankenberg S, Zeller T. Circulating microRNAs strongly predict cardiovascular death in patients with coronary artery disease-results from the large AtheroGene study. Eur Heart J. 2017;38:516–523. doi: 10.1093/eurheartj/ehw250 [DOI] [PubMed] [Google Scholar]
- 6.Wang Y, Pan X, Fan Y, Hu X, Liu X, Xiang M, Wang J. Dysregulated expression of microRNAs and mRNAs in myocardial infarction. Am J Transl Res. 2015;7:2291–2304. [PMC free article] [PubMed] [Google Scholar]
- 7.Sawyer DB, Colucci WS. Mitochondrial oxidative stress in heart failure: “oxygen wastage” revisited. Circ Res. 2000;86:119–120. doi: 10.1161/01.res.86.2.119 [DOI] [PubMed] [Google Scholar]
- 8.Hausenloy DJ, Yellon DM. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest. 2013;123:92–100. doi: 10.1172/JCI62874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pell VR, Chouchani ET, Murphy MP, Brookes PS, Krieg T. Moving Forwards by Blocking Back-Flow: The Yin and Yang of MI Therapy. Circ Res. 2016;118:898–906. doi: 10.1161/CIRCRESAHA.115.306569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431–435. doi: 10.1038/nature13909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lesnefsky EJ, Chen Q, Moghaddas S, Hassan MO, Tandler B, Hoppel CL. Blockade of electron transport during ischemia protects cardiac mitochondria. J Biol Chem. 2004;279:47961–47967. doi: 10.1074/jbc.M409720200 [DOI] [PubMed] [Google Scholar]
- 13.Mracek T, Drahota Z, Houstek J. The function and the role of the mitochondrial glycerol-3-phosphate dehydrogenase in mammalian tissues. Biochim Biophys Acta. 2013;1827:401–410. doi: 10.1016/j.bbabio.2012.11.014 [DOI] [PubMed] [Google Scholar]
- 14.Kaminski MM, Sauer SW, Kaminski M, Opp S, Ruppert T, Grigaravicius P, Grudnik P, Grone HJ, Krammer PH, Gulow K. T cell activation is driven by an ADP-dependent glucokinase linking enhanced glycolysis with mitochondrial reactive oxygen species generation. Cell Rep. 2012;2:1300–1315. doi: 10.1016/j.celrep.2012.10.009 [DOI] [PubMed] [Google Scholar]
- 15.Langston PK, Nambu A, Jung J, Shibata M, Aksoylar HI, Lei J, Xu P, Doan MT, Jiang H, MacArthur MR, et al. Glycerol phosphate shuttle enzyme GPD2 regulates macrophage inflammatory responses. Nat Immunol. 2019;20:1186–1195. doi: 10.1038/s41590-019-0453-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mracek T, Holzerova E, Drahota Z, Kovarova N, Vrbacky M, Jesina P, Houstek J. ROS generation and multiple forms of mammalian mitochondrial glycerol-3-phosphate dehydrogenase. Biochim Biophys Acta. 2014;1837:98–111. doi: 10.1016/j.bbabio.2013.08.007 [DOI] [PubMed] [Google Scholar]
- 17.Miwa S, Brand MD. Mitochondrial matrix reactive oxygen species production is very sensitive to mild uncoupling. Biochem Soc Trans. 2003;31:1300–1301. doi: 10.1042/bst0311300 [DOI] [PubMed] [Google Scholar]
- 18.Mracek T, Pecinova A, Vrbacky M, Drahota Z, Houstek J. High efficiency of ROS production by glycerophosphate dehydrogenase in mammalian mitochondria. Arch Biochem Biophys. 2009;481:30–36. doi: 10.1016/j.abb.2008.10.011 [DOI] [PubMed] [Google Scholar]
- 19.Krawczynski K, Mishima T, Huang X, Sadovsky Y. Intact feto-placental growth in microRNA-210 deficient mice. Placenta. 2016;47:113–115. doi: 10.1016/j.placenta.2016.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robinson KM, Janes MS, Pehar M, Monette JS, Ross MF, Hagen TM, Murphy MP, Beckman JS. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc Natl Acad Sci U S A. 2006;103:15038–15043. doi: 10.1073/pnas.0601945103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women’s Health Initiative randomized controlled trial. JAMA. 2002;288:321–333. doi: 10.1001/jama.288.3.321 [DOI] [PubMed] [Google Scholar]
- 22.Florijn BW, Bijkerk R, van der Veer EP, van Zonneveld AJ. Gender and cardiovascular disease: are sex-biased microRNA networks a driving force behind heart failure with preserved ejection fraction in women? Cardiovasc Res. 2018;114:210–225. doi: 10.1093/cvr/cvx223 [DOI] [PubMed] [Google Scholar]
- 23.Colom B, Oliver J, Roca P, Garcia-Palmer FJ. Caloric restriction and gender modulate cardiac muscle mitochondrial H2O2 production and oxidative damage. Cardiovasc Res. 2007;74:456–465. doi: 10.1016/j.cardiores.2007.02.001 [DOI] [PubMed] [Google Scholar]
- 24.Lagranha CJ, Deschamps A, Aponte A, Steenbergen C, Murphy E. Sex differences in the phosphorylation of mitochondrial proteins result in reduced production of reactive oxygen species and cardioprotection in females. Circ Res. 2010;106:1681–1691. doi: 10.1161/CIRCRESAHA.109.213645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Basson M. MicroRNAs loom large in the heart. Nat Med. 2007;13:541. doi: 10.1038/nm0507-541 [DOI] [PubMed] [Google Scholar]
- 26.Chien KR. Molecular medicine: microRNAs and the tell-tale heart. Nature. 2007;447:389–390. doi: 10.1038/447389a [DOI] [PubMed] [Google Scholar]
- 27.Quiat D, Olson EN. MicroRNAs in cardiovascular disease: from pathogenesis to prevention and treatment. J Clin Invest. 2013;123:11–18. doi: 10.1172/JCI62876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hinkel R, Penzkofer D, Zuhlke S, Fischer A, Husada W, Xu QF, Baloch E, van Rooij E, Zeiher AM, Kupatt C, et al. Inhibition of microRNA-92a protects against ischemia/reperfusion injury in a large-animal model. Circulation. 2013;128:1066–1075. doi: 10.1161/CIRCULATIONAHA.113.001904 [DOI] [PubMed] [Google Scholar]
- 29.Gabisonia K, Prosdocimo G, Aquaro GD, Carlucci L, Zentilin L, Secco I, Ali H, Braga L, Gorgodze N, Bernini F, et al. MicroRNA therapy stimulates uncontrolled cardiac repair after myocardial infarction in pigs. Nature. 2019;569:418–422. doi: 10.1038/s41586-019-1191-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang JX, Jiao JQ, Li Q, Long B, Wang K, Liu JP, Li YR, Li PF. miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat Med. 2011;17:71–78. doi: 10.1038/nm.2282 [DOI] [PubMed] [Google Scholar]
- 31.Hinkel R, Ramanujam D, Kaczmarek V, Howe A, Klett K, Beck C, Dueck A, Thum T, Laugwitz KL, Maegdefessel L, et al. AntimiR-21 Prevents Myocardial Dysfunction in a Pig Model of Ischemia/Reperfusion Injury. J Am Coll Cardiol. 2020;75:1788–1800. doi: 10.1016/j.jacc.2020.02.041 [DOI] [PubMed] [Google Scholar]
- 32.Chan YC, Banerjee J, Choi SY, Sen CK. miR-210: the master hypoxamir. Microcirculation. 2012;19:215–223. doi: 10.1111/j.1549-8719.2011.00154.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan SY, Zhang YY, Hemann C, Mahoney CE, Zweier JL, Loscalzo J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. 2009;10:273–284. doi: 10.1016/j.cmet.2009.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.White K, Lu Y, Annis S, Hale AE, Chau BN, Dahlman JE, Hemann C, Opotowsky AR, Vargas SO, Rosas I, et al. Genetic and hypoxic alterations of the microRNA-210-ISCU1/2 axis promote iron-sulfur deficiency and pulmonary hypertension. EMBO Mol Med. 2015;7:695–713. doi: 10.15252/emmm.201404511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nanayakkara GK, Wang H, Yang X. Proton leak regulates mitochondrial reactive oxygen species generation in endothelial cell activation and inflammation - A novel concept. Arch Biochem Biophys. 2019;662:68–74. doi: 10.1016/j.abb.2018.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao RZ, Jiang S, Zhang L, Yu ZB. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int J Mol Med. 2019;44:3–15. doi: 10.3892/ijmm.2019.4188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang H, Alder NN, Wang W, Szeto H, Marcinek DJ, Rabinovitch PS. Reduction of elevated proton leak rejuvenates mitochondria in the aged cardiomyocyte. Elife. 2020; 9:e60827. doi: 10.7554/eLife.60827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ventura-Clapier R, Garnier A, Veksler V, Joubert F. Bioenergetics of the failing heart. Biochim Biophys Acta. 2011;1813:1360–1372. doi: 10.1016/j.bbamcr.2010.09.006 [DOI] [PubMed] [Google Scholar]
- 39.Neubauer S. The failing heart--an engine out of fuel. N Engl J Med. 2007;356:1140–1151. doi: 10.1056/NEJMra063052 [DOI] [PubMed] [Google Scholar]
- 40.Zhou B, Tian R. Mitochondrial dysfunction in pathophysiology of heart failure. J Clin Invest. 2018;128:3716–3726. doi: 10.1172/JCI120849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cave AC, Ingwall JS, Friedrich J, Liao R, Saupe KW, Apstein CS, Eberli FR. ATP synthesis during low-flow ischemia: influence of increased glycolytic substrate. Circulation. 2000;101:2090–2096. doi: 10.1161/01.cir.101.17.2090 [DOI] [PubMed] [Google Scholar]
- 42.Heart Outcomes Prevention Evaluation Study I, Yusuf S, Dagenais G, Pogue J, Bosch J, Sleight P. Vitamin E supplementation and cardiovascular events in high-risk patients. N Engl J Med. 2000;342:154–160. doi: 10.1056/NEJM200001203420302 [DOI] [PubMed] [Google Scholar]
- 43.Sugamura K, Keaney JF Jr. Reactive oxygen species in cardiovascular disease. Free Radic Biol Med. 2011;51:978–992. doi: 10.1016/j.freeradbiomed.2011.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RA, Murphy MP, Sammut IA. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005;19:1088–1095. doi: 10.1096/fj.05-3718com [DOI] [PubMed] [Google Scholar]
- 45.Ribeiro Junior RF, Dabkowski ER, Shekar KC, KA OC, Hecker PA, Murphy MP. MitoQ improves mitochondrial dysfunction in heart failure induced by pressure overload. Free Radic Biol Med. 2018;117:18–29. doi: 10.1016/j.freeradbiomed.2018.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zinovkin RA, Zamyatnin AA. Mitochondria-Targeted Drugs. Curr Mol Pharmacol. 2019;12:202–214. doi: 10.2174/1874467212666181127151059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tian R, Colucci WS, Arany Z, Bachschmid MM, Ballinger SW, Boudina S, Bruce JE, Busija DW, Dikalov S, Dorn GW II, et al. Unlocking the Secrets of Mitochondria in the Cardiovascular System: Path to a Cure in Heart Failure-A Report from the 2018 National Heart, Lung, and Blood Institute Workshop. Circulation. 2019;140:1205–1216. doi: 10.1161/CIRCULATIONAHA.119.040551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sekhar BS, Kurup CK, Ramasarma T. Generation of hydrogen peroxide by brown adipose tissue mitochondria. J Bioenerg Biomembr. 1987;19:397–407. doi: 10.1007/BF00768542 [DOI] [PubMed] [Google Scholar]
- 49.Drahota Z, Chowdhury SK, Floryk D, Mracek T, Wilhelm J, Rauchova H, Lenaz G, Houstek J. Glycerophosphate-dependent hydrogen peroxide production by brown adipose tissue mitochondria and its activation by ferricyanide. J Bioenerg Biomembr. 2002;34:105–113. doi: 10.1023/a:1015123908918 [DOI] [PubMed] [Google Scholar]
- 50.Oldford C, Kuksal N, Gill R, Young A, Mailloux RJ. Estimation of the hydrogen peroxide producing capacities of liver and cardiac mitochondria isolated from C57BL/6N and C57BL/6J mice. Free Radic Biol Med. 2019;135:15–27. doi: 10.1016/j.freeradbiomed.2019.02.012 [DOI] [PubMed] [Google Scholar]
- 51.Orr AL, Quinlan CL, Perevoshchikova IV, Brand MD. A refined analysis of superoxide production by mitochondrial sn-glycerol 3-phosphate dehydrogenase. J Biol Chem. 2012;287:42921–42935. doi: 10.1074/jbc.M112.397828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mugabo Y, Zhao S, Seifried A, Gezzar S, Al-Mass A, Zhang D, Lamontagne J, Attane C, Poursharifi P, Iglesias J, et al. Identification of a mammalian glycerol-3-phosphate phosphatase: Role in metabolism and signaling in pancreatic beta-cells and hepatocytes. Proc Natl Acad Sci U S A. 2016;113:E430–439. doi: 10.1073/pnas.1514375113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Possik E, Madiraju SRM, Prentki M. Glycerol-3-phosphate phosphatase/PGP: Role in intermediary metabolism and target for cardiometabolic diseases. Biochimie. 2017;143:18–28. doi: 10.1016/j.biochi.2017.08.001 [DOI] [PubMed] [Google Scholar]
- 54.Akar FG, Aon MA, Tomaselli GF, O’Rourke B. The mitochondrial origin of postischemic arrhythmias. J Clin Invest. 2005;115:3527–3535. doi: 10.1172/JCI25371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou L, Solhjoo S, Millare B, Plank G, Abraham MR, Cortassa S, Trayanova N, O’Rourke B. Effects of regional mitochondrial depolarization on electrical propagation: implications for arrhythmogenesis. Circ Arrhythm Electrophysiol. 2014;7:143–151. doi: 10.1161/CIRCEP.113.000600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ma Q, Dasgupta C, Li Y, Huang L, Zhang L. MicroRNA-210 Downregulates ISCU and Induces Mitochondrial Dysfunction and Neuronal Death in Neonatal Hypoxic-Ischemic Brain Injury. Mol Neurobiol. 2019;56:5608–5625. doi: 10.1007/s12035-019-1491-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xu Z, Alloush J, Beck E, Weisleder N. A murine model of myocardial ischemia-reperfusion injury through ligation of the left anterior descending artery. J Vis Exp. 2014. doi: 10.3791/51329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang X, Zhang X, Ren XP, Chen J, Liu H, Yang J, Medvedovic M, Hu Z, Fan GC. MicroRNA-494 targeting both proapoptotic and antiapoptotic proteins protects against ischemia/reperfusion-induced cardiac injury. Circulation. 2010;122:1308–1318. doi: 10.1161/CIRCULATIONAHA.110.964684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431–435. doi: 10.1038/nature13909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yoshioka J, Chutkow WA, Lee S, Kim JB, Yan J, Tian R, Lindsey ML, Feener EP, Seidman CE, Seidman JG, et al. Deletion of thioredoxin-interacting protein in mice impairs mitochondrial function but protects the myocardium from ischemia-reperfusion injury. J Clin Invest. 2012;122:267–279. doi: 10.1172/JCI44927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oyama J, Blais C Jr., Liu X, Pu M, Kobzik L, Kelly RA, Bourcier T. Reduced myocardial ischemia-reperfusion injury in toll-like receptor 4-deficient mice. Circulation. 2004;109:784–789. doi: 10.1161/01.CIR.0000112575.66565.84 [DOI] [PubMed] [Google Scholar]
- 62.Werth S, Urban-Klein B, Dai L, Hobel S, Grzelinski M, Bakowsky U, Czubayko F, Aigner A. A low molecular weight fraction of polyethylenimine (PEI) displays increased transfection efficiency of DNA and siRNA in fresh or lyophilized complexes. J Control Release. 2006;112:257–270. doi: 10.1016/j.jconrel.2006.02.009 [DOI] [PubMed] [Google Scholar]
- 63.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Song R, Hu XQ, Romero M, Holguin MA, Kagabo W, Xiao D, Wilson SM, Zhang L. Ryanodine receptor subtypes regulate Ca2+ sparks/spontaneous transient outward currents and myogenic tone of uterine arteries in pregnancy. Cardiovasc Res. 2021;117:792–804. doi: 10.1093/cvr/cvaa089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pesta D, Gnaiger E. High-resolution respirometry: OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. Methods Mol Biol. 2012;810:25–58. doi: 10.1007/978-1-61779-382-0_3 [DOI] [PubMed] [Google Scholar]
- 66.Dott W, Mistry P, Wright J, Cain K, Herbert KE. Modulation of mitochondrial bioenergetics in a skeletal muscle cell line model of mitochondrial toxicity. Redox Biol. 2014;2:224–233. doi: 10.1016/j.redox.2013.12.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang P, Lv J, Li Y, Zhang L, Xiao D. Neonatal Lipopolysaccharide Exposure Gender-Dependently Increases Heart Susceptibility to Ischemia/Reperfusion Injury in Male Rats. Int J Med Sci. 2017;14:1163–1172. doi: 10.7150/ijms.20285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Russo I, Micotti E, Fumagalli F, Magnoli M, Ristagno G, Latini R, Staszewsky L. A novel echocardiographic method closely agrees with cardiac magnetic resonance in the assessment of left ventricular function in infarcted mice. Sci Rep. 2019;9:3580. doi: 10.1038/s41598-019-40393-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pfeffer MA, Pfeffer JM, Fishbein MC, Fletcher PJ, Spadaro J, Kloner RA, Braunwald E. Myocardial infarct size and ventricular function in rats. Circ Res. 1979;44:503–512. doi: 10.1161/01.res.44.4.503 [DOI] [PubMed] [Google Scholar]
- 70.Li B, Dasgupta C, Huang L, Meng X, Zhang L. MiRNA-210 induces microglial activation and regulates microglia-mediated neuroinflammation in neonatal hypoxic-ischemic encephalopathy. Cell Mol Immunol. 2020;17:976–991. doi: 10.1038/s41423-019-0257-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Moonira T, Chachra SS, Ford BE, Marin S, Alshawi A, Adam-Primus NS, Arden C, Al-Oanzi ZH, Foretz M, Viollet B, et al. Metformin lowers glucose 6-phosphate in hepatocytes by activation of glycolysis downstream of glucose phosphorylation. J Biol Chem. 2020;295:3330–3346. doi: 10.1074/jbc.RA120.012533 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.