

Abstract
Labeo rohita (Rohu) is one of the most important fish species produced in world aquaculture. Integrative omics research provides a strong platform to understand the basic biology and translate this knowledge into sustainable solutions in tackling disease outbreak, increasing productivity and ensuring food security. Mass spectrometry-based proteomics has provided insights to understand the biology in a new direction. Very little proteomics work has been done on ‘Rohu’ limiting such resources for the aquaculture community. Here, we utilised an extensive mass spectrometry based proteomic profiling data of 17 histologically normal tissues, plasma and embryo of Rohu to develop an open source PeptideAtlas. The current build of “Rohu PeptideAtlas” has mass-spectrometric evidence for 6015 high confidence canonical proteins at 1% false discovery rate, 2.9 million PSMs and ~150 thousand peptides. This is the first open-source proteomics repository for an aquaculture species. The ‘Rohu PeptideAtlas’ would promote basic and applied aquaculture research to address the most critical challenge of ensuring nutritional security for a growing population.
Subject terms: Protein-protein interaction networks, Agriculture
| Measurement(s) | Proteins and Peptides |
| Technology Type(s) | Mass Spectrometry |
| Sample Characteristic - Organism | Labeo rohita |
| Sample Characteristic - Location | India |
Background & Summary
The average annual increase in global consumption of fish has outpaced population growth. Of the global animal protein consumption, 20% is met by fish suggesting the importance of fish in global food security and nutrition. India ranks second in global aquaculture production and Indian major carps (IMCs) contribute to more than 75% of its aquaculture economy1. Labeo rohita (Rohu) is an IMC and among the top eleven finfish species produced in world aquaculture1. With the emergence of genomic information for Rohu, this species has entered the post-genomic era such as transcriptomics, proteomics and metabolomics research to address key issues like safety, quality and health in aquaculture.
Proteomic approaches have been applied in diverse areas to investigate developmental biology, physiology, disease mechanisms, impact of stress inducers2 and effects of dietary supplements on overall physiology of fish3,4. Application of proteomics studies in zebrafish and Xiphophorus sp. has revealed the role of phosphorylated Ezrin in gastrulation5 and peroxiredoxins in human melanoma6. Proteomics can identify and explore sensitive and specific markers for assessing the quality of fish or fishery related products7. The effect of pesticide mixtures and temperature have also been explored in goldfish (Carassius auratus)8. All these findings suggest the importance of proteomic characterization of fish in addressing basic biological to ecological, environmental and food related issues.
Mass spectrometry (MS) based proteomic approaches are progressively used to disentangle complex biological questions, often associated with other omics disciplines (e.g., genomics, transcriptomics, metabolomics)9,10. Proteome reference maps for many organisms such as human and zebrafish have been generated using high resolution mass spectrometry11–13. A recent publication of Rohu genome reported a prediction of 26,400 protein coding genes14. However, proteomics studies in Rohu are rare with most studies focusing on only a particular tissue in isolation15,16.
Data repositories like PeptideAtlas17, PRIDE18 and Global Proteome Machine Database19 enable successful planning of MS-based experiments for biomedical research. The PeptideAtlas project mainly provides a large collection and precise analysis of available MS-based proteomics data. With the exception of the model organism, Zebrafish, no other aquaculture species is well represented so far in any of the publicly available proteomics databases. Towards this goal, an extensive proteomic profiling of 17 histologically normal tissues in Rohu, embryo and plasma was performed using high-resolution high-mass accuracy mass spectrometry. Here, we provided mass spectrometric evidence of more than 150 thousand peptides corresponding to 6015 high confidence canonical proteins with 1% FDR. This dataset has been utilised to develop the PeptideAtlas repository for Rohu. To our knowledge, this is the first such extensive open-source peptide dataset for Rohu.
This work could be considered as a basis for proteomic research on specific genes related to fish health by studying various aspects like improvement in fertility, muscle quality and molecular alterations during stress conditions20. The PeptideAtlas interface is user friendly and very useful in designing targeted proteomic experiments by evaluating the candidate peptides or transitions suited for targeted proteomics based diagnostic assays for fish disease, safety and quality. Using this dataset, spectral libraries can be generated for designing and validating the targeted proteomics data. We believe this extensive proteomic sequence information would complement the genomic information allowing basic and applied research to move faster in fisheries and aquaculture sectors.
Methods
Fish collection and acclimatisation
Three-month old healthy L. rohita fingerlings of around 10 ± 2 g weight, were collected from Powarkheda Regional Centre of ICAR-CIFE, Madhya Pradesh, India. Laboratory conditions used for fingerling acclimatisation included aeration 24 h, daylight 12 h, 10% daily water exchange, water temperature 28–30 °C and feeding twice by 2% of body weight. Following an acclimatisation of seven days, five healthy fishes were placed in an aquarium under starving conditions for one day followed by euthanization for sample collection. Nineteen different types of samples were collected as shown in Table 1 which includes one whole embryonic tissue sample, blood plasma and 17 tissues. Fifteen of the tissues were collected from fingerlings whereas plasma and gonadal tissues from adult fishes. Blood plasma was collected from female fish and embryos were sampled after four days of fertilisation. Collected samples were stored at −80 °C till further use.
Table 1.
Tissue types and sampling details.
| S. no. | Sample | Collection stage |
|---|---|---|
| 1 | AB (Air bladder) | Fingerling (10 ± 2 g) |
| 2 | Brain | |
| 3 | Eye | |
| 4 | Fin | |
| 5 | GB (Gall bladder) | |
| 6 | Gill | |
| 7 | Gut | |
| 8 | Heart | |
| 9 | Kidney | |
| 10 | Liver | |
| 11 | Muscle | |
| 12 | Scale | |
| 13 | Skin | |
| 14 | SC (Spinal cord) | |
| 15 | Spleen | |
| 16 | FG (Female gonad) | Adult female (1000 ± 100 g) |
| 17 | Plasma | |
| 18 | MG (Male gonad) | Adult male (1000 ± 100 g) |
| 19 | Embryo | 4-day post fertilisation |
Protein extraction for in-depth proteomic profiling
For extraction of proteins, organ wise samples collected from individual fish were pooled and taken forward. For lysing the tissue, urea buffer containing 8 M Urea, 50 mM Tris-HCl, 1 mM MgCl2 and 75 mM NaCl was used. For fifteen of the tissues including spleen, spinal cord, skin, scales, muscle, male gonad, liver, kidney, heart, gut, gill, female gonad, eye, brain and air bladder, pH shift solubilisation method20 was used for protein extraction. For these tissues, proteins were extracted using urea buffer in three different pH i.e., pH 2.5, 8 and 13. To around 75–100 mg of tissue sample, 300 µl of lysis buffer was added followed by sonication for 2-3 times (Vibra-Cell™ Ultrasonic Liquid Processors, VCX 130 (Sonics). The sample was bead beated using Zirconium/Silica beads (Cat. No. 11079110z) for 90 s. It was followed by centrifugation at 8000 rpm at 4 °C for 15 min to get a clear supernatant containing proteins. For the embryo sample, whole embryos were processed using Trizol method21 of protein extraction. Plasma sample were directly (without any depletion) taken for downstream analysis.
Protein quantification and quality check on SDS-PAGE
Protein quantification was performed by Bradford protein assay, using Bovine Serum Albumin (BSA) as a standard. Accordingly, absorbance was taken at 595 nm and standard curve was plotted using BSA dilutions and concentration for all the unknown samples was determined. In order to check the quality of the protein extract, 1-dimensional SDS-PAGE was performed for which 15 ug protein was loaded for each sample onto a mini-vertical gel (Bio-Rad Mini PROTEAN® 3 Cell, Bio-Rad Laboratories), in accordance to Laemmli protocol22. As the extracted protein was present in urea containing buffer, no heating step was performed before SDS-PAGE to avoid the risk of Carbamylation. Gel electrophoresis was performed for 1-2 hours followed by staining in Coomassie blue R350 solution in methanol and acetic acid. Gel was destained to visualise the protein bands (Supplementary Fig. S1a).
Fractionation, in-gel digestion and peptide preparation
For in-gel digestion, 30 µg protein from each sample was run on SDS-PAGE as above. Each sample was run in duplicate and at least six slices per lane were excised (Fig. 1a). For plasma sample, 11 gel fractions were processed for in-gel digestion. The electrophoresis was performed for only 30–40 minutes i.e., ~1 cm in the resolving gel. Before performing the digestion of protein, stain was removed followed by protein reduction and alkylation. For removing stain from the gel pieces an alternate treatment with buffer salt ammonium bicarbonate (NH4HCO3) and organic solvent Acetonitrile (ACN) solution was performed. Proteins were reduced using Dithiothreitol (DTT) and alkylated using Iodoacetamide (IAA). For protein digestion, trypsin was used in ~1:30 enzyme to protein (w/w) ratio. Peptides were extracted from the gel pieces after 16–18 hours of digestion using an increasing gradient of ACN solution. Peptides were desalted using C18 Empore™ SPE Disks matrix (Merck). Peptide quantification was done using Scopes method23 and one µg of peptide was subjected to mass spectrometric analysis.
Fig. 1.

An overview of experimental design and analysis workflow. (a) Fishes were dissected to collect the tissue/samples followed by protein extraction and SDS-PAGE. Gel slices were excised and processed for in-gel based tryptic digestion followed by Liquid chromatography tandem mass spectrometry (LC-MS/MS) and analysis in Trans proteomic pipeline (TPP), (b) Raw data obtained from DDA-MS were processed along the pipeline for building PeptideAtlas. Raw files were first converted to mzml followed by comet search and analysis pipeline including peptide prophet, reSpect, iPROphet, protein prophet and final filtering and validation to compile the atlas.
Data-dependant Acquisition by Liquid Chromatography Tandem Mass spectrometry (LC-MS/MS)
An Easy-nLC nano-flow liquid chromatography 1200 system was used for the separation of peptides following in-gel digestion (Fig. 1a). With a flow rate of 5 µl/min, one µg desalted peptides were loaded to pre-analytical column (Thermo Scientific, PN 164564-CMD, Trap column nanoViper C18, 5 µm, 100 Å, Acclaim PepMap 100- 100 µm x 2 cm). The peptides were run over a gradient of 120 min in solvent B which was a solution of 80% ACN with 0.1% Formic acid (FA). The flow rate was kept as 300 nl/min for resolving peptides on the analytical column (Thermo Scientific, PN ES903, C18- 75 μm × 50 cm, 2 μm particle, PepMap RSLC, 100 Å pore size). Mass spectrometric data was acquired using Orbitrap mass analyser in DDA mode in a full scan range of 375–1700 m/z at a mass resolution of 60,000. For dynamic exclusion, the mass tolerance was set as ± 10 for 40 s and for MS2 precursors, the isolation mass window was set to 1.2 Da. High energy Collision Dissociation (HCD) method was used for MS/MS fragmentation. For MS1 and MS2, AGC target was set to be 400000 and 10000, respectively. A lock mass of 445.12003 m/z was used for positive internal calibration.
The mass spectrometric data used in this study for developing PeptideAtlas of Labeo rohita has been utilised for tissue wise profiling of post-translational modifications (PTMs) and comparative protein expression analysis as reported in our recent study24.
Protein identification, TPP analysis and PeptideAtlas assembly
The raw mass spectrometry data (.raw) generated from the Orbitrap Fusion mass spectrometer was converted to .mzML files using MSconvert 3.0.5533 tool25. The converted mzML files were searched using Comet (2019.01 rev.1)26 tool against L. rohita NCBI protein database. This database consisted of protein sequences generated by translation of coding sequences (CDS) through gene predictions after whole genome sequencing of Labeo rohita (Bio project: PRJNA437789). The database had locus tag IDs (prefix Rohu_) and EMBL/Bank/GenBank/DDBJ CSS IDs (prefix RXN). UniProt database for this species (ProteomeID- UP000290572) consists of a UniProt protein identifier for each CD. The NCBI database had 32687 entries and the UniProt database which was downloaded on 16th August, 2019, has 32379 entries and is the subset of the NCBI database. For initial comet search, NCBI database was used whereas all downstream steps including protein identification and PeptideAtlas assembly were performed using combined database of NCBI and UniProt. We utilized the combined database so that the proteins which are not yet included in the UniProt database, can also be covered in PeptideAtlas build.
To the protein database, an equal number of decoy and contaminant sequences were added. Decoy sequences were generated using “randomize sequences and interleave entries” decoy algorithm whereas the contaminant sequences were taken from common Repository of Adventitious Proteins, cRAP, database (http://www.thegpm.org/crap/). The parameters used for the data analysis in Trans-Proteomic Pipeline (TPP) suite include peptide mass tolerance 20 ppm, fragment ions bin tolerance 0.05 m/z and monoisotopic mass offset 0.0 m/z, two allowed missed cleavages, fully tryptic and semi-tryptic peptides, oxidation of tryptophan and methionine (+15.994915 Da) as variable modifications and carbamidomethylation of cysteine (+57.021464 Da) as static modification. Protein identification was performed using TPP V 5.2.0 Flammagenitus27. To score for peptide spectral match (PSM), integrated tools of PeptideProphet and iProphet were used for individual files and the score unique peptides in combined PeptideProphet files. Finally, ProteinProphet tool was used for protein identification based on iProphet input and true identifications were selected at less than 1% FDR28–30. The whole workflow is represented in Fig. 1b.
The chimeric spectra were accessed by reanalysing the iProphet files using reSpect algorithm31. In brief, reSpect search was performed on iProphet files by increasing the precursor mass tolerance to 3.0 Da. TPP analysis was performed as mentioned earlier and the process of reSpect and TPP analysis was repeated once. A minimum iProphet probability ≥ 0.0 was used for the reSpect search. PeptideAtlas processing pipeline was used to build PeptideAtlas by combining the iProphet results from regular TPP and reSpect search results. The spectrum was filtered at variable probability to get constant peptide spectrum match (PSM) FDR of 0.0008% for each experiment. The statistically significant results were organized in the “Rohu PeptideAtlas”, which is built and maintained by ISB at the given link. http://www.peptideatlas.org/builds/rohu/.
Ortholog analysis for the identified proteome
Ortholog analysis for the total canonical proteins was performed in EGGNOG-mapper genome-wide functional annotation tool32 (http://eggnog-mapper.embl.de/). Firstly, the FASTA sequences were acquired from UniProt33 of all the protein IDs and taken as input list (Supplementary Table S1). During this analysis, taxonomic scope was selected as Actinopterygii, orthology restrictions selected as ‘transfer annotation from any ortholog’, seed ortholog detection criteria were set to be 0.001.
Acquisition of selected reaction monitoring (SRM) data for targeted verification
The targeted proteomic data was acquired using a Thermo TSQ Altis Triple Quadrupole Mass Spectrometer linked to a Thermo Vanquish HPLC system. The data was acquired using an SRM/ MRM (Selected/ Multiple reaction monitoring) acquisition mode. A Hypersil GOLD analytical column (Thermo Fisher Scientific, 100 × 2 mm, C18) was used for the reverse phase separation of peptides. Samples were run at a flow rate of 450 µl/ min. One µg of desalted peptide sample was subjected to the column and run for 10 minutes. The liquid chromatography system used, consisted of 0.1% formic acid (FA) in milliQ water as solvent A and 80% Acetonitrile (ACN) and 0.1% FA as solvent B. Throughout the run, the column temperature was set to be 45 οC and cycle time was kept as 2 s. The Skyline daily software34 (version 20.2.1) was utilised for analysing the data.
Data Records
Data record 1
Mass spectrometry data obtained after DDA-MS experiments includes raw files (.raw) for 19 different sample types of fish (Supplementary File S1). This mass spectrometry data along with the protein databases (.fasta) has been deposited to the ProteomeXchange Consortium via the PRIDE partner repository and can be accessed through the identifier PXD026377 using the link https://www.ebi.ac.uk/pride/archive/projects/PXD02637735. The comet search parameter file and MAYU statistical report (.xlsx) is provided in Supplementary File S2 and S3 respectively. Peptides identified are enlisted in Supplementary Table S2. The details of the proteins and peptides identified along with various interactive data and visualizations are available at PeptideAtlas and can be accessed using the given link https://db.systemsbiology.net/sbeams/cgi/PeptideAtlas/buildDetails?atlas_build_id=50036.
Data record 2
The targeted mass spectrometry data includes spectral library files (.blib), the target peptide list selected based on PeptideAtlas data (.xls), instrument raw files (.raw) and the result imported skyline documents (.sky, .view, .skyd, .skyl). The targeted proteomics data including all skyline documents, raw files and spectral library have been deposited to Panorama web server37. Also, the target peptides and transition lists are given in supplementary Tables S3 to S5.
Data record 3
In the EggNOG database32 based ortholog analysis, the canonical proteins were mapped against orthologs corresponding to wide range of cellular processes and metabolic functions. Around 97% of the mapped orthologs belong to Actinopterygii, the class of ray finned fishes and majority of them were linked to signal transduction mechanism. This information is represented in Fig. 2/ Table 2 and Supplementary Table S1).
Fig. 2.
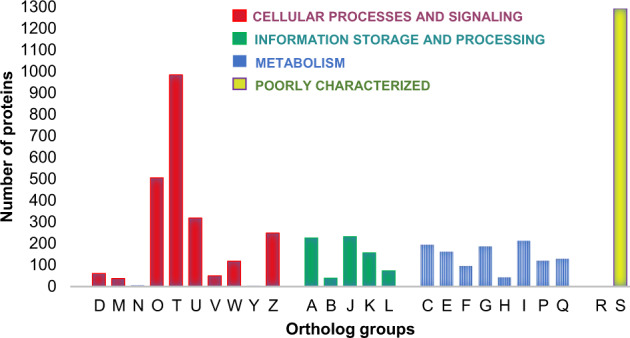
An overview of phylogenetically annotated orthologs for the canonical proteins. The distribution of identified proteins mapped against each ortholog group is presented here (ortholog details in the Table 2).
Table 2.
Distribution of identified canonical proteins across various orthologs*.
| Groups | Description | No. of proteins |
|---|---|---|
| CELLULAR PROCESSES AND SIGNALING | ||
| D | Cell cycle control, cell division, chromosome partitioning | 62 |
| M | Cell wall/membrane/envelope biogenesis | 38 |
| N | Cell motility | 5 |
| O | Post-translational modification, protein turnover, and chaperones | 505 |
| T | Signal transduction mechanisms | 982 |
| U | Intracellular trafficking, secretion, and vesicular transport | 318 |
| V | Defense mechanisms | 50 |
| W | Extracellular structures | 119 |
| Y | Nuclear structure | 4 |
| Z | Cytoskeleton | 249 |
| INFORMATION STORAGE AND PROCESSING | ||
| A | RNA processing and modification | 226 |
| B | Chromatin structure and dynamics | 40 |
| J | Translation, ribosomal structure and biogenesis | 232 |
| K | Transcription | 158 |
| L | Replication, recombination and repair | 74 |
| METABOLISM | ||
| C | Energy production and conversion | 195 |
| E | Amino acid transport and metabolism | 163 |
| F | Nucleotide transport and metabolism | 96 |
| G | Carbohydrate transport and metabolism | 187 |
| H | Coenzyme transport and metabolism | 43 |
| I | Lipid transport and metabolism | 213 |
| P | Inorganic ion transport and metabolism | 120 |
| Q | Secondary metabolites biosynthesis, transport, and catabolism | 129 |
| POORLY CHARACTERIZED | ||
| R | General function prediction only | 0 |
| S | Function unknown | 1289 |
*This data is in continuation of data represented in Fig. 2.
Technical Validation
Building and validation of an extensive PeptideAtlas for Labeo rohita
Targeted proteomics is an emerging approach for acquiring proteome wide qualitative and quantitative information in a targeted manner. Generally, the targeted proteomics involves a hypothesis driven experiment which starts from a list of precise protein/peptide targets to be monitored. PeptideAtlas is a compendium of peptides that can serve as an important resource for designing a targeted experiment or validating the protein/peptide target related to a shotgun experiment. To generate the PeptideAtlas resource for Rohu, the DDA-MS dataset was analysed using a combined non-redundant Uniprot database and NCBI database of Labeo rohita (details in the Methods). To make the data more reliable, accurate and to avoid the identification of false positives, we used MAYU38 tool both at the protein and peptide level. Mayu is a software used to determine false discovery rates (FDRs) for protein identification (protFDR), peptide identification (pepFDR) and peptide-spectrum match (mFDR). All experiments were thresholded at a probability that yields an iProphet model-based PSM-FDR of 0.0008%. The exact probability varies from experiment to experiment depending on how well the modeling can separate correct from incorrect. However, this probability threshold is typically greater than 0.99. For each experiment, the spectra were filtered at variable probability to get constant PSM level FDR of 0.0008%. Throughout the procedure, decoy identifications were retained and then used to compute final decoy-based FDRs. The model-based PSM-FDR was adjusted if the final decoy-based protein FDR is higher than 1%. For protein identification, based on iProphet input, true identifications were selected at less than 1% FDR.
This resulted in the identification of 6015 high confident canonical proteins along with 667 indistinguishable representative proteins, 671 marginally distinguished proteins, 768 representative proteins and 1165 other proteins. The overall summary for Rohu PeptideAtlas is shown in Table 3. Briefly, the current build contains more than 2.96 million identified peptide MS/MS spectra with additional information for a selection of PSMs at FDR level less than or equal to 0.0008% (i.e., 150781 distinct peptides at 0.18% peptide level FDR) (Fig. 3a). This peptide information corresponds to all the identified proteins at less than 1% protein level FDR. All tissues except muscle, fin, scale and plasma have contributed ~15,000-20,000 peptides and ~2000–3000 canonical proteins each to the build (Fig. 3b). Majority of the identified peptides were doubly or triply charged with a length of 10–20 amino acids and most of the identified peptides were without any missed cleavage (Fig. 3c,d, Supplementary Fig. S1b). Each canonical protein has at least 2 unique peptides and ~93% of them had at least ≥3 unique peptides (Fig. 3d, Table S2). As far as the sequence coverage is concerned, observed peptides for ~54% of the canonical proteins spanned >30% of the protein sequence whereas 22% of canonical proteins had >60% coverage (Fig. 3e, Table S2). PeptideAtlas is a user-friendly portal for researchers who can access protein and peptide related information. The Rohu PeptideAtlas hence provides a platform for obtaining detailed information of all identified proteins and peptides that can be helpful for discovery experiments as well as designing targeted assays for L. rohita.
Table 3.
Organ wise numerical summary for the data in Labeo rohita PeptideAtlas.
| Dataset | Experiment Tag | MS Runs | Spectra Searched | Distinct Peptides | Unique Peptides | Cumulative Peptides | Distinct Canonical Proteins | Unique Canonical Proteins | Unique All Proteins | Cumulative Canonical Proteins |
|---|---|---|---|---|---|---|---|---|---|---|
| PrePX245 | Air bladder | 18 | 1073739 | 21148 | 1754 | 21147 | 2428 | 10 | 66 | 2428 |
| PrePX245 | Embryo | 6 | 342968 | 18801 | 1063 | 31710 | 2813 | 14 | 108 | 3360 |
| PrePX245 | Female gonad | 24 | 1299928 | 30777 | 6364 | 49703 | 2828 | 53 | 194 | 4038 |
| PrePX245 | Fin | 6 | 421953 | 9329 | 274 | 51602 | 2074 | 2 | 44 | 4188 |
| PrePX245 | Gallbladder | 9 | 489743 | 16708 | 690 | 55308 | 2953 | 7 | 77 | 4478 |
| PrePX245 | Gill | 18 | 1999739 | 32801 | 2706 | 64160 | 3552 | 17 | 222 | 4808 |
| PrePX245 | Gut | 18 | 1005859 | 28610 | 2781 | 70910 | 3105 | 12 | 147 | 4929 |
| PrePX245 | Female plasma | 8 | 430788 | 6497 | 1743 | 74623 | 562 | 0 | 19 | 4954 |
| PrePX245 | Scales | 15 | 710427 | 1225 | 123 | 74888 | 323 | 0 | 7 | 4956 |
| PrePX245 | Skin | 13 | 776257 | 18838 | 944 | 77876 | 2164 | 1 | 17 | 4970 |
| PrePX245 | Spinal cord | 18 | 1093689 | 43051 | 4862 | 92280 | 3920 | 28 | 169 | 5380 |
| PrePX245 | Brain | 18 | 1113115 | 53736 | 13205 | 109378 | 4343 | 190 | 868 | 5698 |
| PrePX245 | Eye | 18 | 797200 | 29665 | 4328 | 115355 | 2727 | 35 | 116 | 5757 |
| PrePX245 | Kidney | 18 | 1131662 | 34743 | 2527 | 119636 | 3467 | 15 | 107 | 5803 |
| PrePX245 | Liver | 18 | 1203630 | 47487 | 10469 | 132274 | 3544 | 40 | 282 | 5887 |
| PrePX245 | Muscle | 21 | 1034502 | 15167 | 2288 | 134681 | 1697 | 5 | 37 | 5894 |
| PrePX245 | Spleen | 16 | 817942 | 22059 | 1999 | 136880 | 3036 | 11 | 145 | 5916 |
| PrePX245 | Heart | 18 | 1042352 | 36798 | 3751 | 140791 | 3554 | 16 | 160 | 5936 |
| PrePX245 | Male gonad | 18 | 847818 | 40259 | 9990 | 150781 | 3501 | 79 | 277 | 6015 |
Fig. 3.

An overview of Labeo rohita PeptideAtlas build. (a,b), Plots showing cumulative number of peptides and canonical proteins respectively contributed by each experiment. Height of the blue/navy blue bar represents cumulative number of peptides/proteins, height of the orange/red bar represents number of peptides/proteins identified in each experiment and width of the bar (x-axis) represents the number of spectra identified (PSMs) for each experiment, (c) Distribution of peptide spectral matches against the peptide charge, (d) Graph showing the spectral count for the peptides of different lengths and (e). Bar plot representing the number of unique peptides (distinct peptides) per canonical protein where the x-axis shows the bins for number of unique peptides and y-axis show the number of respective canonical proteins, (f) Distribuition of canonical proteins based on percentage sequence coverage [Fig. 3a–e are taken from ‘Experiment Contribution Plots’ section of first page of Labeo rohita PeptideAtlas].
Protein and peptide search in Labeo rohita PeptideAtlas
For any targeted experiment, proteotypic peptides are the ideal targets which can be selected based on several scores assigned to a peptide in PeptideAtlas. For each protein entry, a dynamic page is obtained to provide mass spectral information and peptide modification details about the protein such as total observed peptides and a graphical representation of coverage of protein for each observed peptide. Additionally, all observed peptides are represented in a tabular format and ranked according to their empirical suitability score (ESS) empirical observability score (EOS) (Fig. 4a). ESS is a measure of incidence of observing a protein/peptide in a given sample while EOS represents how much suitable is the observed peptide for the significant proteotypic detection of protein from which it was obtained. Peptides having high value of EOS and map to a unique protein are the most suited candidates to monitor for identifying/quantifying a protein in a given sample. The protein view page also gives the information of all the tissues/sample in which the particular protein was detected.
Fig. 4.

Example of a protein search and peptide search in Rohu PeptideAtlas. (a) Out of several collapsible sections for protein search, three are shown to provide an overview of protein information, observed peptides highlighted in red font and additional information for each observed peptide, respectively. (b) Under peptide view, two sections for one of the observed peptides of the same protein are shown representing general information about peptide and respective annotated MS2 spectrum where x-axis represents the m/z and y-axis shows the intensity.
For any observed peptide, a peptide view page presents all available information of respective peptide including its alignment to particular protein, genome mapping, modification site (if any). It also presents the peptide spectra in each sample where the peptide was observed (Fig. 4b). Spectral quality can be estimated based on the spectral information provided for each peptide in the Lorikeet spectral viewer. Peptide spectra along with the precursor mass and all product ion masses and detected product ions are presented in tabular format.
Utility of PeptideAtlas information in SRM based targeted proteomic experiments
A set of peptides was taken for targeted verification using selected reaction monitoring (SRM) approach. Results were matched with the spectral library for the reliability of the data. This section shows the significance of PeptideAtlas in targeted experiments. We have performed targeted experiment for two proteins in female gonad tissue and similar kind of experiments can be designed and validated using PeptideAtlas information for all studied tissues of Rohu. Following steps were followed for SRM based verification experiment.
Generation of spectral library
PepXML (.pepXML) files obtained after comet search for female gonad sample were used to create a non-redundant spectral library. The spectral library was created using skyline software34 through the ‘build’ option under library tab inside the peptide settings. Finally, a .blib file was created and selected for the experiment.
Peptide and transition selection
Two proteins; Elongation factor 1 alpha (EF1 alpha-A0A498N236) and Zona pellucida sperm binding 3 like protein (zp3- A0A498NTM4) were selected for targeted verification. Only peptides unique to these proteins and without any missed cleavage were considered. Selected peptides were having ESS score greater than or equals to 0.4 and length ranged from 8 to 30 amino acids (Supplementary Table S3). Using skyline software, it was found that 593 transitions corresponding to 30 peptides and 44 precursors of the selected proteins were found in the spectral library of female gonad sample. Hence, two transition lists (TL1-305 transitions and TL2- 288 transitions) were exported for preparing the methods for performing SRM experiment (Supplementary Tables S4, S5).
Performing an SRM based targeted proteomics experiment
Instrument used for SRM experiment was Thermo Altis Triple quadrupole mass spectrometer. Transition lists for selected peptides were used to create respective targeted methods. Peptides obtained from female gonad tissue were run against the prepared methods in replicates (i.e., R1 and R2 for both the transition lists) with a liquid chromatography gradient of 10 minutes (See methods section). Data acquired was imported for further analysis in skyline against the same document from which the transition list was exported.
Validation of data/ spectral information using spectral library
A combination of multiple factors is generally used to correctly identify the peptides in a targeted experiment. The gold standard for this is heavy labelled peptides that co-elute with the peptide of interest. However, when heavy labelled peptides are unavailable as can be the case in most laboratory experiments, fragment ion matching to a spectrum library can be the best method to identify the peptide of interest unambiguously39. In case of spectral library matching, the observed spectra are matched with the existing spectra in the spectral library and a similarity score is calculated called as dot product (dotp). The dotp score is based on the normalised spectral contrast angle, which provides a measure of peak detection confidence. The dotp could range from 0 for lowest similarity to 1 for highest similarity and confident identification40.
In order to determine the promising peptides detected for the selected proteins; we imported the results to skyline. For the precursors, both singly and doubly charged product ions corresponding to y2 through last ion were considered. The spectral information was compared with the spectral library created from PeptideAtlas resource in order to confirm the reliability of the data. This was done based on the dot product metric (dotp) which is a measure of similarity between library spectra and query peaks41. Based on peak shape, peak area and co-elution of fragment ions, many peptides gave consistent results in both the replicate runs with a decent dotp value. Peak area and intensity values were consistent between the replicate runs and no peaks were observed in the blank runs. Table 4 shows respective dotp values for both doubly charged and/triply charged precursor of targeted peptides along with their ESS and EOS scores. For example, the peptide IGGVGTVPVGK and EVAVDFQMR were matched with the spectral library with a dotp value of more than 0.8 and 0.9, respectively in both the replicates (Fig. 5a,b). Similarly, a few more peptides exhibited single peaks for the respective peptide with no ambiguity.
Table 4.
List of peptides selected for SRM based verification along with some details from PeptideAtlas and match score (dotp*) with spectral library.
| Sequence | Accession | ESS | EOS | dotp ( + 2) | dotp ( + 3) |
|---|---|---|---|---|---|
| VFVDSCVATQAPDVNSLPR | PAp07598395 | 0.89 | 1.00 | 0.85 | 0.82 |
| ALWSPMGMASALQSPFGVQEK | PAp07599055 | 0.78 | 0.33 | 0.85 | 0.77 |
| QLLQGPVKPLDWR | PAp07598382 | 0.78 | 0.67 | 0.75 | 0.84 |
| ADGAIVGVQCHYPR | PAp07598197 | 0.76 | 0.67 | 0.79 | 0.81 |
| NMITGTSQADAALLIVSAAK | PAp04190051 | 0.75 | 0.26 | 0.76 | 0.76 |
| YSFIENHGCFVDAK | PAp04184446 | 0.74 | 0.67 | 0.80 | 0.84 |
| QPVTPSSVAVQCSEDR | PAp07601931 | 0.72 | 0.67 | 0.76 | 0.80 |
| FPLVPEVQR | PAp07604998 | 0.69 | 0.83 | 0.88 | NA |
| EVAVDFQMR | PAp07599266 | 0.68 | 0.50 | 0.93 | NA |
| IETGVLKPGMVLTFSPAK | PAp04175036 | 0.62 | 0.32 | 0.79 | 0.85 |
| SIEMHHQGLQTALPGHNVGFNIK | PAp04186345 | 0.60 | 0.26 | NA | 0.65 |
| FMPQTQPEK | PAp07599143 | 0.59 | 0.50 | 0.82 | NA |
| VGYSPVLDCHTTHVSCR | PAp04189673 | 0.55 | 0.37 | 0.56 | 0.70 |
| ATFASVPSDAGR | PAp07604862 | 0.55 | 0.50 | 0.87 | NA |
| TLLEVLDSLLPPVR | PAp07588809 | 0.54 | 0.42 | 0.80 | 0.85 |
| IGGVGTVPVGK | PAp04171392 | 0.54 | 0.42 | 0.85 | NA |
| LVPNKPLCVESFFHYPPLGR | PAp04189456 | 0.53 | 0.26 | 0.65 | 0.77 |
| IHINLVIIGHVDSGK | PAp07601820 | 0.49 | 0.16 | 0.64 | 0.71 |
| YTFTIIDAPGHR | PAp04188238 | 0.49 | 0.32 | 0.70 | 0.87 |
| VYNHVPLR | PAp07604851 | 0.49 | 0.33 | 0.88 | NA |
| MDLTEPPFSQK | PAp04189675 | 0.47 | 0.26 | 0.87 | NA |
| STTTGHLVYK | PAp04190655 | 0.45 | 0.26 | 0.75 | NA |
| GDVAGNAQQDPPSDVSSFIAQIIMLNHPGK | PAp04190359 | 0.44 | 0.16 | NA | 0.52 |
| LEDWPQYLMSGDGATVK | PAp07599462 | 0.44 | 0.11 | 0.69 | 0.46 |
| GEFEAGISR | PAp04185068 | 0.44 | 0.21 | 0.85 | NA |
| IGFEIGAVPFIPVSGWSGENMIAPSQK | PAp07598582 | 0.43 | 0.11 | NA | 0.52 |
| LMLDDWSYERPSNYYFLGNVFNLEASVK | PAp07599621 | 0.41 | 0.17 | NA | 0.47 |
| VQFQLEAFMFQEGQSPSIYITCLLK | PAp07605170 | 0.40 | 0.17 | NA | 0.27 |
| QLMVCVNK | PAp07602504 | 0.40 | 0.11 | 0.88 | NA |
| GITIDISLLK | PAp04189685 | 0.39 | 0.16 | 0.82 | NA |
*dotp represents the measure of similarity between spectral library and experimental data.
Fig. 5.

Targeted proteomic verification using spectral libraries. Left panel shows the peak view for the spectral information obtained for the peptide after performing SRM experiment and right panel shows the peak area view of the replicate runs along with match with the spectral library, (a,b) Spectral information for two peptides showing single, consistent peak with good match with library, (c) Wrongly annotated peak for the given peptide at 5.9 min with a dotp of 0.34 in both the replicate runs (right panel), (d) Correct annotated peak (4.6 min) based on the match with library (0.85/0.84) in both the replicates. [TL1 and TL2 represents the two transition lists, R1 and R2 represents the duplicate runs for the same sample].
However, there were several peptides for which multiple peaks scattered across the LC gradient were observed. These peaks were found to have good shape with co-elution, making it difficult to identify the correct peak in the absence of corresponding heavy labelled peptide. In such cases, spectral libraries play a significant role for determining the best match to obtain reliable and representative fragmentation patterns. For instance, for the peptide GEFEAGISR, two peaks were obtained in both the replicate runs, one at retention time 4.6 min and other at 5.9 min (Fig. 5c,d). Based on the match with spectral library (created using female gonad PepXML files) in both the runs, peak obtained at 4.6 would be the real peak as it has a dotp value of 0.85/0.84 compared to the one at 5.9 with a dotp of 0.34.
Usage Notes
Development and evaluation of a comprehensive PeptideAtlas for Labeo rohita
In the present study, we developed an open resource for fish proteome analysis for the scientific community based on high resolution mass spectrometry data from 19 different sample types of L. rohita (Rohu) using different protein extraction methods and sample fractionation. This is the first and foremost comprehensive fish proteome analysis (along with PTM information that is to be updated soon in PeptideAtlas as a part of another study). The complete building and evaluation process of the Rohu PeptideAtlas is explained elaborately in the Methods section.
A valuable resource for designing targeted proteomics experiments
SRM or MRM based targeted proteomic experiments require unique transitions of the targeted proteins (or peptides) for accurate quantification. PeptideAtlas is the best resource for selecting the unique peptides and respective transitions using several tools in the PeptideAtlas. It also provides the information for best observable or identified tryptic peptides across wide range of sample types and also across different types of mass spectrometry. The interactive interface of PeptideAtlas helps to visualize individual and consensus spectra in PeptideAtlas to select and export either single or multiple targeted peptides/proteins and its respective transitions as.csv/.tsv format which can be imported directly into the mass spectrometry instrument for SRM/MRM experiment.
A valuable resource for spectral library generation and data search
The Rohu PeptideAtlas built is dynamic and can be updated whenever a new proteomics dataset is generated in-house or get uploaded in the public repositories such as PRIDE, MASSIVE42 etc. The data repository in PeptideAtlas, TPP output files (.pepXML) used for generating PeptideAtlas and the results from PeptideAtlas can be used for generating spectral library using SpectraST, an integrated tool in TPP package. Spectral libraries are new generation peptide database with experimentally identified spectra used for the accurate and precise identification/quantification of peptides/proteins in DIA/SWATH analysis or for SRM/MRM data analysis.
Best resource for Proteogenomic analysis and annotation
Accurate annotation of the genome is still a challenging task despite availability of advanced technology and algorithms. Integration of high-resolution mass spectrometry along with genomic data would improve the gene annotations. Rohu genome was sequenced recently and the preliminary annotations are available with no curation and it also contains several hypothetical proteins and pseudogenes14. Currently, in the UniProt database of Labeo rohita, only two proteins are reviewed which have protein evidence (PE) level 2 i.e., experimental evidence at transcript level. However, none of the protein has PE level 1 that represents the protein level evidence. The current dataset can help the UniProt curators, by providing the mass spectrometric based protein level evidence for the existence of Labeo rohita proteome. It has been reported that gene annotation can be improved with the help of mass spectrometric data43,44. Tanner et al. utilised the tandem mass spectra from human peptides and validated 11,000 introns and 39,000 exons at translation level along with identification of novel exons and splicing events45. In a similar manner, the peptide dataset provided in Rohu PeptideAtlas could help to improve the genome annotations and may provide evidence for pseudogenes, alternative splicing events, extended exons and hypothetical proteins.
Supplementary information
Acknowledgements
This work was supported by Department of Biotechnology (BT/PR15285/AAQ/3/753/2015) Govt. of India to S.S and M.G. M.N was supported by University Grants Commission (UGC). R.M would like to acknowledge the US National Institutes for Health, National institute for General Medical Sciences under grant No. GM087221, the Office of the Director 1S10OD026936, the National Institute on Aging grant U19AG023122 and NSF award 1920268. We thank Director General, Indian Council of Agricultural Research; Director, ICAR-Central Institute of Fisheries Education, Mumbai for the support and facility. We acknowledge MASS-FIITB at IIT Bombay supported by the Department of Biotechnology (BT/PR13114/INF/22/206/2015) for mass spectrometric data acquisition. We would like to thank Mr. Saicharan Ghantasala, Deeptarup Biswas and Medha Gayathri J Pai for helpful suggestions and technical support.
Author contributions
Concept and design: M.N., S.S., R.M. and M.G. Maintenance and sampling: N.P., M.N. Method development and Data acquisition: M.N., N.P. Data analysis and Interpretation: M.N., P.J., Z.S., N.P., B.G., R.M. Constructing database: J.R., Z.S., M.N. Writing: Original draft: M.N., N.P., J.R., Z.S., M.G., S.S., R.M. Writing: Review and editing: M.N., N.P., B.G., J.R., Z.S., R.M., M.G., S.S.
Code availability
The authors do not have code specific to this work to disclose.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Panga Jaipal Reddy, Nevil Pinto.
Contributor Information
Mukunda Goswami, Email: mukugoswami@gmail.com.
Sanjeeva Srivastava, Email: sanjeeva@iitb.ac.in.
Supplementary information
The online version contains supplementary material available at 10.1038/s41597-022-01259-9.
References
- 1.FAO. “Sustainability in action.” State of World Fisheries and Aquaculture. Food and Agriculture Organization of the United Nations, Rome, Italy (2020).
- 2.Forne I, Abian J, Cerda J. Fish proteome analysis: model organisms and non-sequenced species. Proteomics. 2010;10:858–872. doi: 10.1002/pmic.200900609. [DOI] [PubMed] [Google Scholar]
- 3.Cerqueira M, et al. How tryptophan levels in plant-based aquafeeds affect fish physiology, metabolism and proteome. Journal of proteomics. 2020;221:103782. doi: 10.1016/j.jprot.2020.103782. [DOI] [PubMed] [Google Scholar]
- 4.Ghaedi G, Keyvanshokooh S, Azarm HM, Akhlaghi M. Proteomic analysis of muscle tissue from rainbow trout (Oncorhynchus mykiss) fed dietary β-glucan. Iranian journal of veterinary research. 2016;17:184. [PMC free article] [PubMed] [Google Scholar]
- 5.Link V, et al. Identification of regulators of germ layer morphogenesis using proteomics in zebrafish. Journal of cell science. 2006;119:2073–2083. doi: 10.1242/jcs.02928. [DOI] [PubMed] [Google Scholar]
- 6.Lokaj K, et al. Quantitative differential proteome analysis in an animal model for human melanoma. J Proteome Res. 2009;8:1818–1827. doi: 10.1021/pr800578a. [DOI] [PubMed] [Google Scholar]
- 7.Pedreschi R, Hertog M, Lilley KS, Nicolai B. Proteomics for the food industry: opportunities and challenges. Critical reviews in food science and nutrition. 2010;50:680–692. doi: 10.1080/10408390903044214. [DOI] [PubMed] [Google Scholar]
- 8.Gandar A, et al. Proteome response of fish under multiple stress exposure: Effects of pesticide mixtures and temperature increase. Aquat Toxicol. 2017;184:61–77. doi: 10.1016/j.aquatox.2017.01.004. [DOI] [PubMed] [Google Scholar]
- 9.Williams, E. G. et al. Systems proteomics of liver mitochondria function. Science352 (2016). [DOI] [PMC free article] [PubMed]
- 10.Chick JM, et al. Defining the consequences of genetic variation on a proteome-wide scale. Nature. 2016;534:500–505. doi: 10.1038/nature18270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim MS, et al. A draft map of the human proteome. Nature. 2014;509:575–581. doi: 10.1038/nature13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilhelm M, et al. Mass-spectrometry-based draft of the human proteome. Nature. 2014;509:582–587. doi: 10.1038/nature13319. [DOI] [PubMed] [Google Scholar]
- 13.Kelkar DS, et al. Annotation of the zebrafish genome through an integrated transcriptomic and proteomic analysis. Molecular & cellular proteomics. 2014;13:3184–3198. doi: 10.1074/mcp.M114.038299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Das P, et al. De novo assembly and genome-wide SNP discovery in Rohu Carp, Labeo rohita. Frontiers in genetics. 2020;11:386. doi: 10.3389/fgene.2020.00386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goswami M, et al. Proteomics Analysis of Liver Tissue of Labeo rohita. Current Proteomics. 2015;12:56–62. doi: 10.2174/157016461201150506202445. [DOI] [Google Scholar]
- 16.Banerjee S, et al. Identification of potential biomarkers of hepatotoxicity by plasma proteome analysis of arsenic-exposed carp Labeo rohita. Journal of hazardous materials. 2017;336:71–80. doi: 10.1016/j.jhazmat.2017.04.054. [DOI] [PubMed] [Google Scholar]
- 17.Deutsch EW, Lam H, Aebersold R. PeptideAtlas: a resource for target selection for emerging targeted proteomics workflows. EMBO reports. 2008;9:429–434. doi: 10.1038/embor.2008.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vizcaíno JA, et al. A guide to the Proteomics Identifications Database proteomics data repository. Proteomics. 2009;9:4276–4283. doi: 10.1002/pmic.200900402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Craig R, Cortens JP, Beavis RC. Open source system for analyzing, validating, and storing protein identification data. Journal of proteome research. 2004;3:1234–1242. doi: 10.1021/pr049882h. [DOI] [PubMed] [Google Scholar]
- 20.Surasani VKR, Tyagi A, Kudre T. Recovery of proteins from rohu processing waste using pH shift method: characterization of isolates. Journal of aquatic food product technology. 2017;26:356–365. doi: 10.1080/10498850.2016.1186130. [DOI] [Google Scholar]
- 21.Jaipal Reddy P, et al. A simple protein extraction method for proteomic analysis of diverse biological specimens. Current proteomics. 2013;10:298–311. doi: 10.2174/15701646113106660004. [DOI] [Google Scholar]
- 22.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 23.Scopes R. Measurement of protein by spectrophotometry at 205 nm. Analytical biochemistry. 1974;59:277–282. doi: 10.1016/0003-2697(74)90034-7. [DOI] [PubMed] [Google Scholar]
- 24.Nissa, M. U. et al. Organ-Based Proteome and Post-Translational Modification Profiling of a Widely Cultivated Tropical Water Fish, Labeo rohita. Journal of proteome research (2021). [DOI] [PubMed]
- 25.Chambers MC, et al. A cross-platform toolkit for mass spectrometry and proteomics. Nature biotechnology. 2012;30:918–920. doi: 10.1038/nbt.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eng JK, Jahan TA, Hoopmann MR. Comet: an open‐source MS/MS sequence database search tool. Proteomics. 2013;13:22–24. doi: 10.1002/pmic.201200439. [DOI] [PubMed] [Google Scholar]
- 27.Deutsch EW, et al. Trans‐Proteomic Pipeline, a standardized data processing pipeline for large‐scale reproducible proteomics informatics. PROTEOMICS–Clinical Applications. 2015;9:745–754. doi: 10.1002/prca.201400164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deutsch EW, et al. State of the human proteome in 2014/2015 as viewed through PeptideAtlas: enhancing accuracy and coverage through the AtlasProphet. Journal of proteome research. 2015;14:3461–3473. doi: 10.1021/acs.jproteome.5b00500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shteynberg D, et al. iProphet: multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates. Molecular & cellular proteomics. 2011;10:M111. 007690. doi: 10.1074/mcp.M111.007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Analytical chemistry. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 31.Shteynberg D, et al. reSpect: software for identification of high and low abundance ion species in chimeric tandem mass spectra. Journal of the American Society for Mass Spectrometry. 2015;26:1837–1847. doi: 10.1007/s13361-015-1252-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huerta-Cepas J, et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic acids research. 2019;47:D309–D314. doi: 10.1093/nar/gky1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Apweiler R, et al. UniProt: the Universal Protein knowledgebase. Nucleic Acids Research. 2004;32:D115–D119. doi: 10.1093/nar/gkh131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.MacLean B, et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nissa MU. 2022. Proteomic profiling of Labeo Rohita; a widely cultivated fish. PRIDE Archive. https://www.ebi.ac.uk/pride/archive/projects/PXD026377
- 36.2022. Labeo rohita PeptideAtlas. PeptideAtlas. https://db.systemsbiology.net/sbeams/cgi/PeptideAtlas/buildDetails?atlas_build_id=500
- 37.Srivastava S. 2022. Multiple reaction monitoring (MRM) based data for targeted validation of proteins in Labeo rohita. Panorama Public. https://panoramaweb.org/rohufemalegonad.url
- 38.Reiter L, et al. Protein identification false discovery rates for very large proteomics data sets generated by tandem mass spectrometry. Molecular & cellular proteomics: MCP. 2009;8:2405–2417. doi: 10.1074/mcp.M900317-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grossegesse M, Nitsche A, Schaade L, Doellinger J. Application of spectral library prediction for parallel reaction monitoring of viral peptides. Proteomics. 2021;21:2000226. doi: 10.1002/pmic.202000226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pino LK, et al. The Skyline ecosystem: Informatics for quantitative mass spectrometry proteomics. Mass spectrometry reviews. 2020;39:229–244. doi: 10.1002/mas.21540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frewen BE, Merrihew GE, Wu CC, Noble WS, MacCoss MJ. Analysis of peptide MS/MS spectra from large-scale proteomics experiments using spectrum libraries. Anal Chem. 2006;78:5678–5684. doi: 10.1021/ac060279n. [DOI] [PubMed] [Google Scholar]
- 42.Choi M, et al. MassIVE. quant: a community resource of quantitative mass spectrometry–based proteomics datasets. Nature methods. 2020;17:981–984. doi: 10.1038/s41592-020-0955-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ignasi F, Joaquin AN, Joan C. Fish proteome analysis: Model organisms and non‐sequenced species. Proteomics. 2010;10:858–872. doi: 10.1002/pmic.200900609. [DOI] [PubMed] [Google Scholar]
- 44.De Souza GA, et al. High accuracy mass spectrometry analysis as a tool to verify and improve gene annotation using Mycobacterium tuberculosis as an example. BMC genomics. 2008;9:1–13. doi: 10.1186/1471-2164-9-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tanner S, et al. Improving gene annotation using peptide mass spectrometry. Genome research. 2007;17:231–239. doi: 10.1101/gr.5646507. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Nissa MU. 2022. Proteomic profiling of Labeo Rohita; a widely cultivated fish. PRIDE Archive. https://www.ebi.ac.uk/pride/archive/projects/PXD026377
- 2022. Labeo rohita PeptideAtlas. PeptideAtlas. https://db.systemsbiology.net/sbeams/cgi/PeptideAtlas/buildDetails?atlas_build_id=500
- Srivastava S. 2022. Multiple reaction monitoring (MRM) based data for targeted validation of proteins in Labeo rohita. Panorama Public. https://panoramaweb.org/rohufemalegonad.url
Supplementary Materials
Data Availability Statement
The authors do not have code specific to this work to disclose.