

Systemic lupus erythematosus (SLE), the most common form of lupus, is a chronic autoimmune disorder characterized by a global loss of self-tolerance and hyper-activation of both innate and adaptive immune systems (Kaul et al., 2016). Neutrophils, the most abundant leukocytes in human blood, have a critical role in maintaining immune surveillance and tissue homeostasis, and its dysregulation is of high relevance to SLE (Ricklin et al., 2010). In patients with SLE, accelerated neutrophil death and the deficiency in clearing dying neutrophils cause nuclear and cytoplasmic antigen exposure, excessive production of type I interferon (IFN), and neutrophil extracellular trap (NET) release, subsequently inducing autoimmune responses (Garcia-Romo et al., 2011). Dysregulated neutrophil death is believed to be a major cause of SLE; however, the underlying mechanism of neutrophil death in SLE is not well-defined.
Ferroptosis is an iron-dependent form of regulated cell death induced by over-accumulation of lipid peroxides on cellular membranes (Dixon et al., 2012). Cells have evolved several ferroptosis defense mechanisms to counteract lipid peroxidation and fight against ferroptosis, prominent among which is glutathione peroxidase 4 (GPX4), which localizes in both cytosol and mitochondria and uses glutathione as its cofactor to convert toxic lipid hydroperoxides into corresponding non-toxic lipid alcohols (Yang et al., 2014; Mao et al., 2021). Ferroptosis is a double-edged sword in cellular life and has been associated with many diseases: while excessive ferroptotic cell death is highly relevant to acute kidney disease, cardiomyopathy, atherosclerosis, and neurodegenerative disorders, ferroptosis impairment can lead to tumor development (Jiang et al., 2021). Although our understanding of the pathological relevance of ferroptosis in human diseases has been significantly advanced in recent years, the potential link between autoimmune diseases and ferroptosis remained unknown. A recent study filled this knowledge gap by revealing that ferroptosis induced by GPX4 transcriptional suppression represents a main form of neutrophil death in lupus (Li et al., 2021). This study further highlights the importance of targeting ferroptosis in SLE treatment in the future.
Through analyzing blood tests and viability detection from patients with autoimmune diseases, Li et al. (2021) confirmed that neutropenia (abnormally low levels of neutrophils) is a common feature in SLE; notably, SLE serum significantly promoted neutrophil death, further suggesting a key role of potential serum factors in regulating neutrophil death and neutropenia. Cytokine array analyses of inflammatory factors identified four cytokines (IFN-α, CXCL11, IL-12p40, and IL-23) with increased levels in SLE serum samples. Additional analyses revealed that blockade of IFN-α, but not other cytokines, restored the viability of neutrophils treated with SLE serum; conversely, the addition of IFN-α or autoantibodies from SLE serum compromised neutrophil viability, suggesting a role of IFN-α and immunoglobulin G (IgG) from SLE serum in mediating neutrophil death (Fig. 1).
Figure 1.
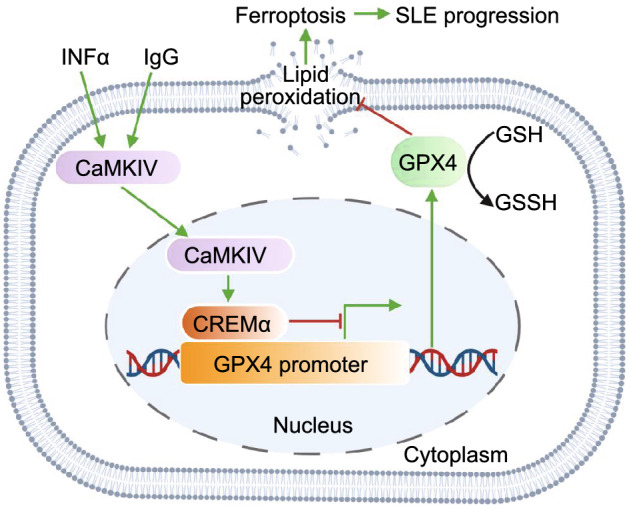
Neutrophil ferroptosis promote SLE pathogenesis. INFα and IgG in SLE serum activate the CaMKIV-CREMα axis, enhance the binding of CREMα to GPX4 promoter, suppress GPX4 expression, followed by increase lipid peroxidation levels. These lead to neutrophil ferroptosis and promote SLE pathogenesis. SLE: systemic lupus erythematosus; IFN-α: interferon-α; IgG: immunoglobulin G; CaMKIV: calcium/calmodulin kinase IV; CREMα: cAMP-responsive element modulator α; GPX4: glutathione peroxidase 4; GSH: glutathione; GSSH: oxidized glutathione
NET release-induced neutrophil death (NETosis, another form of cell death) has been considered as a potential cause of SLE (Kenny et al., 2017). However, Li et al. (2021) showed that NETosis is not dominant in abnormal SLE neutrophil death; instead, this cell death exhibits features of ferroptosis, such as mitochondrial morphological changes and increased lipid peroxidation. Consistently, treatment with SLE serum, IFN-α, or IgG can significantly increase lipid peroxidation levels in neutrophils. Moreover, ferroptosis inhibitors liproxstatin-1 and deferoxamine significantly rescued SLE serum-induced neutrophil death; while other cell death inhibitors could also inhibit this cell death, their rescuing effects were not as statistically significant as those of ferroptosis inhibitors, indicating that ferroptosis inhibitors are the most effective cell death inhibitors in rescuing SLE serum-induced neutrophil death. To understand the underlying mechanisms, Li et al. (2021) performed RNA sequencing analyses and found that GPX4 expression was decreased in neutrophils from SLE patients compared with those from health controls. Further investigations showed that GPX4 expression was significantly reduced in neutrophils cultured with SLE serum, IgG, or IFN-α, suggesting that IFN-α in SLE serum or autoantibodies induce neutrophil ferroptosis potentially through downregulating GPX4 expression.
So how is GPX4 expression controlled in this context? cAMP-responsive element modulator α (CREMα) is a tissue-specific transcriptional repressor in immune cells. CREMα nuclear translocation and its binding ability on promoters are known to be governed by calcium/calmodulin kinase IV (CaMKIV). In SLE, increased nuclear translocation of CaMKIV has been shown to promote CREMα phosphorylation, contributing to aberrant T cell function (Juang et al., 2005). Through promoter sequence analyses, Li et al. (2021) identified a conserved CREMα binding site in the GPX4 promoter, suggesting a potential role of CREMα in regulating GPX4 expression. Consistently, increased nuclear accumulation and enhanced binding of CREMα on the GPX4 promoter were observed in SLE neutrophils or healthy neutrophils cultured with SLE serum, IgG or IFN-α, compared with control neutrophils; further, genetic knockdown of CREMα reversed GPX4 expression suppression by IgG or IFN-α, while CREMα overexpression reduced GPX4 expression. Together, these data indicate that SLE IgG and IFN-α inhibit GPX4 expression through the CaMKIV-CREMα axis.
The authors also examined the relevance of neutrophil ferroptosis to lupus pathophysiology. Li et al. (2021) showed that lupus-prone mice exhibited reduced neutrophil viability and increased lipid peroxidation levels; moreover, compared with the NETosis inhibitor, ferroptosis inhibitors alleviated the disease progression more effectively in lupus-prone mice. To validate the role of GPX4 in neutrophil ferroptosis in vivo, Li et al. (2021) generated myeloid cell-specific Gpx4 haploid-deficient (Gpx4fl/wt) mice. Similar to neutrophils in SLE patients, neutrophils from Gpx4fl/wt mice showed impaired viability with excessive lipid peroxidation levels, and consequently, Gpx4fl/wt mice developed lupus-like disease. Conversely, neutrophils from Camk4 knockout (Camk4−/−) mice exhibited improved cell viability with lower lipid peroxidation levels, which correlated with elevated Gpx4 levels in Camk4−/− mice. Further, combined treatment with pristane and IFN-α adenovirus induced lupus-like disease in wild-type mice, but not in Camk4−/− mice. Collectively, these results suggest that ferroptosis is an important cause of lupus, at least in mouse models.
Altogether, Li et al. (2021) reveal that serum autoantibodies and IFN-α repress GPX4 expression and induce neutrophil ferroptosis through activating the CaMKIV-CREMα axis (Fig. 1). This study likely represents a conceptual breakthrough in the field of SLE research, not only enhancing our mechanistic understanding of lupus pathophysiology, but also indicating ferroptosis inhibitors as potential new therapeutic agents for SLE treatment. Of note, the involvement of IFNs in ferroptosis regulation has been reported previously. For example, cancer immunotherapy has been shown to enhance the effector function of CD8+ T cells, increase IFNγ release to downregulate SLC3A2 and SLC7A11 expression, resulting in reduced cystine uptake and increased ferroptosis in tumor cells (Wang et al., 2019). Furthermore, IFNγ and radiotherapy synergistically repress SLC7A11 expression, further enhancing lipid oxidation and ferroptosis in tumor cells (Lang et al., 2019). Several interesting questions also arise from this study. The underlying mechanisms by which autoantibodies and IFN-α promote CREMα nuclear translocation remains unknown. Further, infections are common in SLE and accounts for 25% to 50% of overall mortality (Petri, 1998). During infection process, host immune cells, such as neutrophils, release large amounts of reactive oxygen species (ROS) at infection sites (Winterbourn et al., 2016). Considering the intimate link between ROS and ferroptosis, the connection of infection and neutrophil ferroptosis remains an interesting topic to be investigated. Since ROS released by NADPH oxidase complex has been demonstrated to induce NETosis (Stoiber et al., 2015), whether there is any crosstalk between ferroptosis and NETosis remains another fascinating question in future studies. Finally, considering that SLE autoantibodies and IFN-α have relatively moderate inhibitory effects on GPX4 expression, it is likely that other unknown mechanisms are involved in inducing neutrophil ferroptosis. Further studies are needed to address these important question.
ACKNOWLEDGEMENTS
We apologize to the colleagues whose relevant work cannot be cited here due to space limitations.
DECLARATIONS
Research in the authors’ lab has been supported by The University of Texas MD Anderson Cancer Center, National Institutes of Health grants R01CA181196, R01CA244144, and R01CA247992 (to BG), and Cancer Center Support (Core) Grant P30 CA016672 from the National Cancer Institute, National Institutes of Health (to The University of Texas MD Anderson Cancer Center).
BG is an inventor on patent applications involving targeting ferroptosis in cancer therapy. Other authors have no conflicts of interest to declare.
This article does not contain any studies with human or animal subjects performed by the any of the authors.
CM drafted the manuscript with additional support from GL for checking relevant literature. CM generated all figures. BG provided revision of the manuscript with additional support from LZ. All authors read and approved the final manuscript.
References
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, Punaro M, Baisch J, Guiducci C, Coffman RL, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. 2011;3:73ra20. doi: 10.1126/scitranslmed.3001201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22:266–282. doi: 10.1038/s41580-020-00324-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juang YT, Wang Y, Solomou EE, Li Y, Mawrin C, Tenbrock K, Kyttaris VC, Tsokos GC. Systemic lupus erythematosus serum IgG increases CREM binding to the IL-2 promoter and suppresses IL-2 production through CaMKIV. J Clin Invest. 2005;115:996–1005. doi: 10.1172/JCI22854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul A, Gordon C, Crow MK, Touma Z, Urowitz MB, van Vollenhoven R, Ruiz-Irastorza G, Hughes G. Systemic lupus erythematosus. Nat Rev Dis Primers. 2016;2:16039. doi: 10.1038/nrdp.2016.39. [DOI] [PubMed] [Google Scholar]
- Kenny EF, Herzig A, Kruger R, Muth A, Mondal S, Thompson PR, Brinkmann V, Bernuth HV, Zychlinsky A. Diverse stimuli engage different neutrophil extracellular trap pathways. Elife. 2017;6:e24437. doi: 10.7554/eLife.24437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang X, Green MD, Wang W, Yu J, Choi JE, Jiang L, Liao P, Zhou J, Zhang Q, Dow A, et al. Radiotherapy and immunotherapy promote tumoral lipid oxidation and ferroptosis via synergistic repression of SLC7A11. Cancer Discov. 2019;9:1673–1685. doi: 10.1158/2159-8290.CD-19-0338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Jiang M, Li K, Li H, Zhou Y, Xiao X, Xu Y, Krishfield S, Lipsky PE, Tsokos GC, et al. Glutathione peroxidase 4-regulated neutrophil ferroptosis induces systemic autoimmunity. Nat Immunol. 2021;22:1107–1117. doi: 10.1038/s41590-021-00993-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, Koppula P, Wu S, Zhuang L, Fang B, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593:586–590. doi: 10.1038/s41586-021-03539-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petri M. Infection in systemic lupus erythematosus. Rheum Dis Clin N Am. 1998;24:423–456. doi: 10.1016/S0889-857X(05)70016-8. [DOI] [PubMed] [Google Scholar]
- Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoiber W, Obermayer A, Steinbacher P, Krautgartner WD. The role of reactive oxygen species (ROS) in the formation of extracellular traps (ETs) in humans. Biomolecules. 2015;5:702–723. doi: 10.3390/biom5020702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, Liao P, Lang X, Kryczek I, Sell A, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569:270–274. doi: 10.1038/s41586-019-1170-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterbourn CC, Kettle AJ, Hampton MB. Reactive oxygen species and neutrophil function. Annu Rev Biochem. 2016;85:765–792. doi: 10.1146/annurev-biochem-060815-014442. [DOI] [PubMed] [Google Scholar]
- Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]