

Abstract
Alpha‐1 antitrypsin deficiency is an autosomal, codominant disorder caused by mutations of the SERPINA1 gene. Several mutations of SERPINA1 have been described associated with the development of pulmonary emphysema and/or chronic liver disease and cirrhosis. Here, we report a very rare PI*Q0parma variant identified for the first time in an Italian family originally from the city of Parma in Northern Italy
Keywords: alpha‐1 antitrypsin deficiency, clinical manifestations, genotype, variant
Here, we report a very rare PI*Q0parma variant identified for the first time in an Italian family originally from the city of Parma in Northern Italy.
INTRODUCTION
Alpha‐1 antitrypsin (AAT) is the main circulating protease inhibitor, its main function is to protect the lung parenchyma from proteolytic degradation. The gene encoding AAT is the highly polymorphic SERPINA1 gene, found at 14q32.1. Mutations in the SERPINA1 gene can lead to AAT deficiency (AATD), a common hereditary disorder but largely under‐recognized despite the recommendations of both the World Health Organization and the medical societies of Europe and the United States. 1 , 2 , 3
Over 150 mutations of SERPINA1 have been described and named by a letter based on the migration speed of the resulting protein on isoelectric focusing (IEF) mutations. 4 AAT variants have been classified into three major categories: normal (characterized by AAT plasma levels within the general population reference ranges), deficient (characterized by decreased, but still detectable, AAT plasma levels) and Null (currently designated Q0, with no detectable AAT plasma levels). 5
In this case series, we report a very rare PI*Q0parma variant identified for the first time in an Italian family originally from the city of Parma in Northern Italy.
METHODS AND RESULTS
Methods
Biochemical and genetic tests to diagnose AATD were performed at the Centre for Diagnosis of Inherited Alpha1‐Antitrypsin Deficiency in Pavia (Italy) with the understanding and written consent of each subject. The samples submitted to the Italian AAT Reference laboratories, because of suspected AATD, underwent a diagnostic algorithm that comprises the determination of AAT and C‐reactive protein serum levels, the phenotyping by IEF analysis, the genotyping for the detection of S and Z variants or A1AT genotyping test and, when needed, SERPINA1 gene sequencing. 6 , 7
The new mutations were identified by sequencing exonic (II–V) and part of the intronic (Ic and IV) portions of the SERPINA1 gene by Sanger methods using the CEQ 8800 genetic analysis System (Beckman Coulter).
This study was approved by the Hospital Ethics Committee of North Emilia Area (approval number: 24474, dated 24 June 2020) in agreement with the Declaration of Helsinki. This research was carried out in accordance with the approved guidelines. Written informed consent was obtained from all participants before inclusion.
Patient 1
A 62‐year‐old male subject, Italian Caucasian, living in Parma, was referred in October 2020 to the Respiratory Disease Unit of the University Hospital of Parma for severe emphysema with respiratory failure. He is a former smoker (about 10 cigarettes a day for 30 years, 15 pack/years) treated with oxygen supplementation therapy, inhaled corticosteroids, long‐acting β2‐agonists and long‐acting muscarinic antagonists. He worked as an HR Director. As the AAT serum concentration was lower than normal (16.6 mg/dl), the patient underwent a genetic investigation in 2016 at San Matteo Hospital of Pavia, Italy, that identified two pathogenic alleles in heterozygosity: Z allele and a new Null variant called Q0parma, from the birthplace of the patient. Q0parma allele consists of the deletion of codon Val413 (rs760849035; 1237_1239del). The final genotype shows a severe AATD with a compound heterozygous, PI*Z/Q0parma.
Upon chest auscultation, the vesicular murmur was significantly reduced. Arterial blood gas analysis showed low oxygen values considering the subject's age: pH 7.43, partial pressure of CO2 36 mmHg, partial oxygen pressure 68 mmHg, HCO3 24 mmol/L and blood oxygen saturation 93%. The 6‐min walk test (6MWT), performed without oxygen supplementation, registered a clinically but not significant desaturation during the test and a slight reduction in the distance walked (445 m measured/469 m predicted).
Due to the presence of obstructive sleep apnoea syndrome with central sleep apnoea and nocturnal hypoxaemia, he was treated with nocturnal continuous positive airway pressure.
Transthoracic cardiac echo colour Doppler shows no indirect signs of pulmonary hypertension even if the test was affected by severe pulmonary emphysema.
Chest high‐resolution computed tomography showed a severe diffuse panlobular emphysema at diagnosis (Figure 1A) and after 4 years of augmentation therapy (Figure 1B).
FIGURE 1.
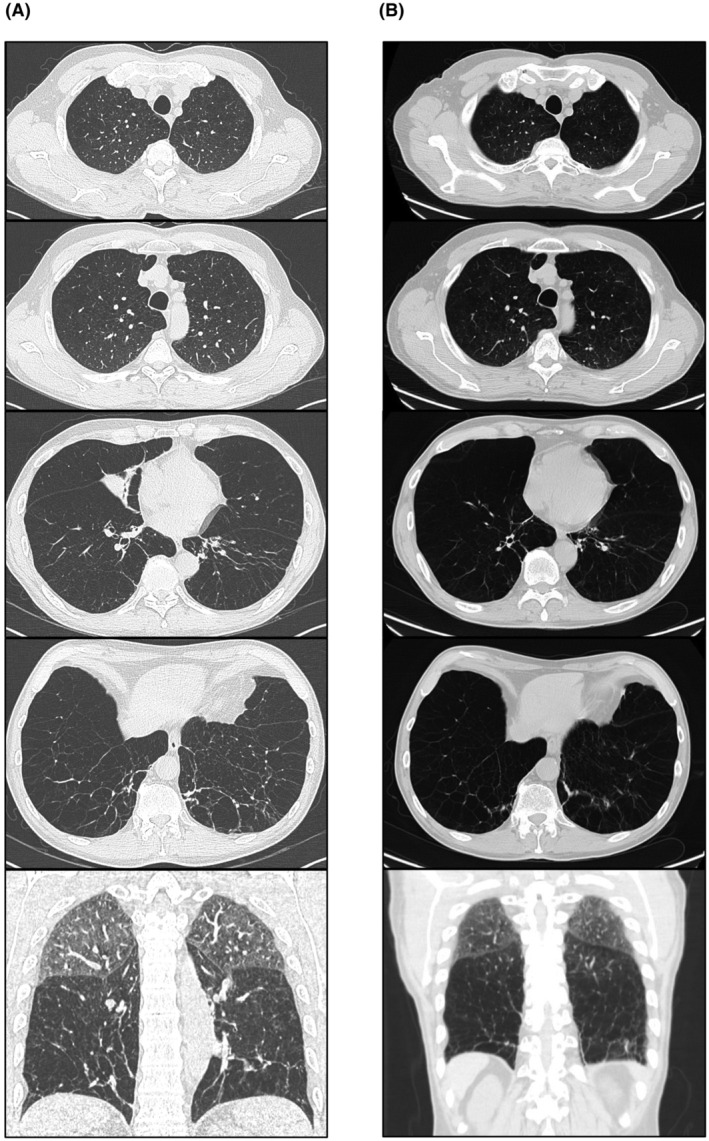
Chest high‐resolution computed tomography of case 1, performed in 2017 (A) and in 2021 (B), showing a severe panlobular emphysema
Cardiopulmonary exercise testing showed a severe reduction in maximal exercise capacity due to ventilatory limitation (maximal oxygen consumption 11.3 ml/kg/min, 38% predicted; workload 79 watt, 34% predicted; ventilatory reserve 0%).
Pulmonary function tests at diagnosis and after 5 years of augmentation therapy revealed an extremely severe airway obstruction, with a negative bronchial reversibility test (Table 1).
TABLE 1.
Spirometric values of patients
| Variables | Case 1 in 2016 | Case 1 in 2021 | Case 2 | Case 3 |
|---|---|---|---|---|
| FEV1, L | 1.53 | 1.02 | 3.73 | 4.90 |
| FEV1, % predicted | 42 | 30 | 112 | 111 |
| FVC, L | 4.55 | 4.29 | 4.15 | 5.65 |
| FVC, % predicted | 91 | 98 | 109 | 109 |
| FEV1/FVC, % | 35 | 23 | 89 | 84 |
| TLC, L | 9.23 | 9 | 6.85 | 8.05 |
| TLC, % predicted | 123 | 125 | 126 | 114 |
| RV, L | 4.68 | 4.60 | 2.56 | 2.41 |
| RV, % predicted | 196 | 187 | 161 | 147 |
| RV/TLC, % | 140 | 135 | 125 | 126 |
| DLCO, % predicted | 41 | 40.8 | 82 | 122 |
| KCO, % predicted | 68 | 56 | 63 | 114 |
Abbreviations: DLCO, diffusing capacity for carbon monoxide; FEV1, forced expiratory volume in 1 s; FVC, forced vital capacity; KCO, transfer coefficient of the lung for carbon monoxide; RV, residual volume; TLC, total lung capacity.
Prick tests were positive for dust mites. Blood chemistry tests (total and fractionated bilirubin, creatine phosphokinase, gamma glutamyl transferase, cholinesterase, aspartate transaminase, alanine transaminase and renal function indices) were in the normal range. Liver and abdominal ultrasound examinations were negative for liver or aneurysmal disease, with normal value of FibroScan equal to 4.7 KPa.
Since 2016, the patient is being treated with AAT augmentation therapy which allow to maintain AAT serum concentration equal to 87.7 mg/dl. The patient was also referred to the lung transplant centre.
Patient 2
A 32‐year‐old female subject, Italian Caucasian, was in good health condition. She lives in Parma, and the diagnosis of AATD was made following genotype analysis performed on the father of the subject (patient 1, with a PI*Z/Q0parma genotype). She was referred to the Respiratory Disease Unit of the University Hospital of Parma. The patient had an AAT serum concentration of 76 mg/dl, with a PI*M Q0parma genotype. She was a former smoker (about 4 cigarettes a day for 5 years, 1 pack/years), and works as a teacher.
She is affected by atopy with prick tests positive for dust mites, grasses, alder, birch and allergic to aspirin. She did not complain of any respiratory symptoms. Arterial oxygen saturation was 99% breathing room air, and the heart rate was 77 bpm. Upon thoracic auscultation, the vesicular murmur was preserved, with no pathological sound. Lung function tests showed an increase of the value of residual volume (RV) and diffusing capacity for carbon monoxide (DLCO) at the lower limits of normal (Table 1). The 6MWT, performed without oxygen supplementation, did not register a clinically significant desaturation but the distance walked was slightly reduced (493 m measured/580 m predicted). Blood count, liver function tests and renal function were in the standard reference range. Chest x‐ray showed no abnormality. Liver and abdominal ultrasound examinations were negative for liver or aneurysmal disease. Echocardiographic parameters were in the normal range.
Patient 3
A 28‐year‐old male subject, Italian Caucasian, was in good health condition. The diagnosis of AATD was made following genotype analysis performed on the father of the subject (patient 1, with a PI*Z/Q0parma genotype). The patient had an AAT serum concentration of 80 mg/dl, with a PI*M Q0parma genotype. He is a former smoker and works as an employee.
He is affected by atopy with prick tests positive for dust mites, grasses and cat. He did not complain of any respiratory symptoms. Lung function tests showed an increase of the value of RV and DLCO (Table 1). He has been living and working abroad for several years and therefore we have little clinical information.
DISCUSSION
AATD predisposes to lung and liver diseases, atopy 8 and rarely to panniculitis, arterial aneurysms and vasculitis, such as granulomatosis with polyangiitis. 9
Generally, the variant Q0 is associated with an increased risk of developing emphysema. The existence of AAT Null alleles was first noted in the early 1970s by several investigators. 7 , 10 The first report of a Null mutation of Italian origin was Q0trastevere, which was detected in an Italian individual with asthma and emphysema. 11
These mutations are extremely rare and can be difficult to diagnose, mainly because IEF is not able to detect Null variants, as they do not produce protein; sequence analysis of SERPINA1 gene is the optimal technique to detect Null. 12
To date, a total of 48 different Null alleles have been detected and characterized. 13
The present study reports for the first time the Q0parma mutation in an Italian family, originally from the city of Parma in Northern Italy, with pulmonary manifestation. Subsequently, Q0parma was reported as Q0montluel in a single case report in French population. 14
The mother of case index (patient 1) was born in 1926 in another city in Northern Italy called Verona, and she had PI* MZ genotype. Her mother and father were also originally from Verona. Her father died in 1944 of respiratory problems. Instead, the father of patient 1 was born in Parma in 1925 and was a smoker, both of his parents were from Parma and were related to each other. It was not possible to carry out the genetic test, but he was certainly a carrier of the Q0parma mutation.
This study underlines the importance of genotyping by whole SERPINA1 gene sequencing, in addition to serum AAT determination, in order to enable detection of AATD in populations and to recommend additional AAT replacement therapy. Augmentation therapy is proved to be effective in improving the quality of life 15 and in slowing down the disease progression, as seen in case 1 chest HRCT scan at diagnosis and after 5 years of augmentation therapy.
In conclusion, our study has expanded the list of Null alleles known to occur within the SERPINA1 gene and underlined the importance of genetic screening, as well as to implement target detection studies, to better understand the rare mutations of AAT, their clinical manifestations and to identify and treat individuals affected by AATD at the earliest possible stage.
CONFLICT OF INTEREST
None declared.
AUTHOR CONTRIBUTION
Alfredo Chetta and Marina Aiello conceived and designed the study, had full access to all of the data and took responsibility for the integrity of the data. Giovanna Pelà, Davide Piloni, Alessandro De Simoni, Annalisa Frizzelli and Lorenzo D'Aloisio carried out clinical and instrumental analysis. Ilaria Ferrarotti, Laura Marchi and Luigino Calzetta contributed to the collection and analysis of data. Marina Aiello, Alfredo Chetta and Ilaria Ferrarotti contributed to the interpretation of the results. Marina Aiello prepared and reviewed the manuscript, in consultation with Alfredo Chetta. All other authors provided critical feedback and approved the final draft.
ETHICS STATEMENT
The authors declare that appropriate written informed consent was obtained for the publication of this manuscript and accompanying images.
Aiello M, Frizzelli A, Marchi L, Ferrarotti I, Piloni D, Pelà G, et al. Clinical manifestations of a new alpha‐1 antitrypsin genetic variant: Q0parma . Respirology Case Reports. 2022;10:e0936. 10.1002/rcr2.936
Associate Editor: Bei He
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- 1. Tsechkovski M, Boulyjenkov V, Heuck C. Alpha 1‐antitrypsin deficiency: memorandum from a WHO meeting. Bull World Health Organ. 1997;75:397–415. [PMC free article] [PubMed] [Google Scholar]
- 2. American Thoracic Society; European Respiratory Society . American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha‐1 antitrypsin deficiency. Am J Respir Crit Care Med. 2003;168:818–900. [DOI] [PubMed] [Google Scholar]
- 3. Miravitlles M, Dirksen A, Ferrarotti I, Koblizek V, Lange P, Mahadeva R, et al. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in A1‐antitrypsin deficiency. Eur Respir J. 2017;50:1700610. [DOI] [PubMed] [Google Scholar]
- 4. Giacopuzzi E, Laffranchi M, Berardelli R, Ravasio V, Ferrarotti I, Gooptu B, et al. Real‐world clinical applicability of pathogenicity predictors assessed on SERPINA1 mutations in alpha‐1‐antitrypsin deficiency. Hum Mutat. 2018;39:1203–13. [DOI] [PubMed] [Google Scholar]
- 5. Ottaviani S, Barzon V, Buxens A, Gorrini M, Larruskain A, El Hamss R, et al. Molecular diagnosis of alpha1‐antitrypsin deficiency: a new method based on Luminex technology. J Clin Lab Anal. 2020;34:e23279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ferrarotti I, Scabini R, Campo I, Ottaviani S, Zorzetto M, Gorrini M, et al. Laboratory diagnosis of alpha1‐antitrypsin deficiency. Transl Res. 2007;150:267–74. [DOI] [PubMed] [Google Scholar]
- 7. Ottaviani S, Gorrini M, Scabini R, Kadija Z, Paracchini E, Mariani F, et al. C reactive protein and alpha1‐antitrypsin: relationship between levels and gene variants. Transl Res. 2011. Jun;157(6):332–8. [DOI] [PubMed] [Google Scholar]
- 8. Aiello M, Frizzelli A, Pisi R, Fantin A, Ghirardini M, Marchi L, et al. Alpha‐1 antitrypsin deficiency is significantly associated to atopy in asthmatic patients. J Asthma. 2020;22:1–12. [DOI] [PubMed] [Google Scholar]
- 9. Ranes J, Stoller JK. A review of alpha1‐antitrypsin deficiency. Am J Respir Crit Care Med. 2012;185(3):246–59. [DOI] [PubMed] [Google Scholar]
- 10. Talamo RC, Langley CE, Reed CE, Makino S. Antitrypsin deficiency: a variant with no detectable α1‐antitrypsin. Science. 1973;181:70–1. [DOI] [PubMed] [Google Scholar]
- 11. Lee J, Novoradovskaya N, Rundquist B, Redwine J, Saltini C, Brantly M. Alpha 1‐antitrypsin nonsense mutation associated with a retained truncated protein and reduced mRNA. Mol Genet Metab. 1998;63:270–80. [DOI] [PubMed] [Google Scholar]
- 12. Blundell G, Cole RB, Nevin NC, Bradley B. Alpha 1‐antitrypsin null gene (pi). Lancet. 1974;2:404. [DOI] [PubMed] [Google Scholar]
- 13. Seixas S, Marques IP. Known mutations at the cause of alpha‐1 antitrypsin deficiency an updated overview of SERPINA1 variation spectrum. Appl Clin Genet. 2021;14:173–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Renoux C, Odou MF, Tosato G, Teoli J, Abbou N, Lombard C, et al. Description of 22 new alpha‐1 antitrypsin genetic variants. Orphanet J Rare Dis. 2018. Sep 17;13(1):161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. López‐Campos JL, Carrasco Hernandez L, Caballero Eraso C. Implications of a change of paradigm in alpha1 antitrypsin deficiency augmentation therapy: from biochemical to clinical efficacy. J Clin Med. 2020. Aug 5;9(8):2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.