

Abstract
This review describes the development of strategies for carbonyl-olefin metathesis reactions relying on stepwise, stoichiometric, or catalytic approaches. A comprehensive overview of currently available methods is provided starting with Paternò-Büchi cycloadditions between carbonyls and alkenes followed by fragmentation of the resulting oxetanes, metal alkylidene-mediated strategies, [3+2]-cycloaddition approaches with strained hydrazines as organocatalysts, Lewis acid-mediated and Lewis acid-catalyzed strategies relying on the formation of intermediate oxetanes, and protocols based on initial carbon-carbon bond formation between carbonyls and alkenes and subsequent Grob-fragmentations. The review concludes with an overview of applications of these currently available methods for carbonyl-olefin metathesis in complex molecule synthesis. Over the past eight years, the field of carbonyl-olefin metathesis has grown significantly and expanded from stoichiometric reaction protocols to efficient catalytic strategies for ring-closing, ring-opening, and cross carbonyl-olefin metathesis. The aim of this review is to capture the status quo of the field and is expected to contribute to further advancements in carbonyl-olefin metathesis in the coming years.
Graphical Abstract

1. INTRODUCTION
The formation of carbon-carbon bonds is of fundamental importance in the field of synthetic chemistry and invaluable for the synthesis of many important biologically active molecules, including current pharmaceuticals and complex natural products. The development of new, sustainable, efficient, and selective catalytic procedures for carbon-carbon bond formation, therefore, represents a key research goal in synthetic chemistry. The metathesis reaction between two alkenes is among the most powerful catalytic carbon-carbon bond forming reactions available and has led to profound synthetic developments in the petroleum, materials, agricultural, and pharmaceutical industries.1–13 The word metathesis, derived from the Greek words for “change” and “position,” directly describes a chemical reaction which redistributes two subunits of two substrates. Olefin-olefin metathesis reactions are classified by a mechanistic profile that relies on olefins 1 and 2 and proceeds through an initial [2+2]-cycloaddition with a metal alkylidene 3 to form a cyclic 4-membered intermediate (Figure 1A). A subsequent [2+2]-cycloreversion forms two new alkene products 4 and 5 that each contain parts of the reactive partners 1 and 2. Since its discovery, olefin-olefin metathesis has spurred important advances in the field of chemistry with broad implications for the synthesis of bioactive compounds and pharmaceuticals. In comparison, the corresponding carbonyl-olefin metathesis reaction between alkenes 1 and carbonyls 6 similarly enables carbon-carbon bond formation to result in new alkenes 4 (Figure 1B); however, this transformation remained, until recently, significantly less developed. Also referred to as a carbonyl-olefin exchange reaction14–20 or oxametathesis21–23 this review provides a comprehensive overview of currently available strategies for this transformation with a particular focus on viable substrate classes and accessible products. Specifically, this review has been organized to discuss the current status of each independent strategy to achieve carbonyl-olefin metathesis.
Figure 1.

Olefin-Olefin Metathesis (A) versus Carbonyl-Olefin Metathesis (B).
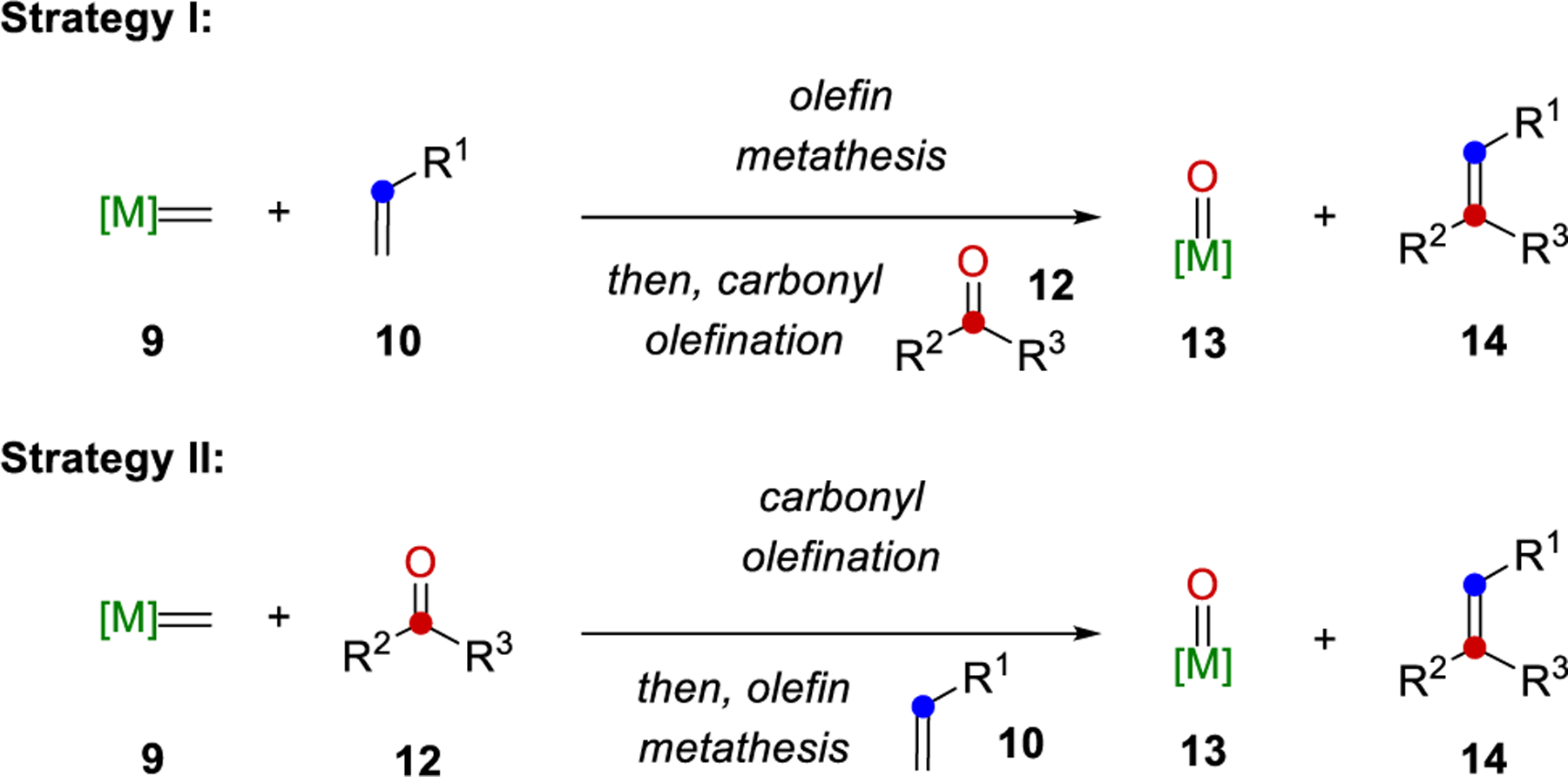
The earliest reported protocols for carbonyl-olefin metathesis rely on a stepwise [2+2]-cycloaddition between alkenes 1 and carbonyls 6 following a Paternò-Büchi reaction protocol under UV-light irradiation to form oxetanes 8 (Figure 2A).28 Fragmentation of 8 under elevated temperatures, acidic conditions, or electron transfer-induction results in alkenes 4 as the desired carbonyl-olefin metathesis products and carbonyls 7 as byproducts. An alternative approach for carbonyl-olefin metathesis is based on metal alkylidenes 9 following one of two strategies (Figure 2B): (1) Upon reaction of alkene 10 with a metal alkylidene 9, complex 11 is formed reminiscent of the initiation step in olefin-olefin metathesis. Subsequent carbonyl olefination with carbonyl 12 results in the desired alkene 14 and metal oxo-species 13. Unfortunately, attempts for the reduction of 13 and its subsequent reintroduction to enable a catalytic process, remain unsuccessful, which renders this strategy stoichiometric in nature.49,50 (2) Alternatively, the steps of carbonyl olefination and olefin metathesis can be reversed to first enable the reaction of carbonyl 12 with metal alkylidene 9 to form a new intermediate alkene 15 together with metal-oxo species 13.64 Olefin metathesis between alkenes 15 and 10 enabled by additional metal alkylidene 9 results in the desired carbonyl-olefin metathesis products 14. Similarly to strategy I, the formation of metal oxo-species 13 renders strategy II stoichiometric in metal alkylidene reagent required. A third and distinct approach for carbonyl-olefin metathesis relies on α,β-unsaturated ketones 16 to provide polyconjugated polymers 18 upon reaction with tungsten-based reagents (Figure 2C).14 The first catalytic strategy for carbonyl-olefin metathesis was based on a [3+2]-cycloaddition reaction paradigm between carbonyls 19 and unsaturated carbocycles 20 with hydrazine 21 as organocatalyst (Figure 2D). Carbonyl-olefin metathesis reactions following this design principle have been reviewed in 2019.24 An alternative catalytic approach for carbonyl-olefin metathesis takes advantage of Lewis acids to activate carbonyls 19 for a [2+2]-cycloaddition with alkenes 1 to result in the formation of intermediate oxetane 23, which upon activation with the Lewis acid undergo retro-[2+2]-cycloaddition to result in alkene 4 and carbonyl 7 as metathesis products (Figure 2E). These Lewis acid-based approaches have been reviewed in 201725 and 2018.26 Importantly, all of these approaches are based on cycloaddition reactions that proceed either directly between carbonyl and alkene substrates, or between one of these starting materials and a suitable catalyst or reagent. A final strategy to achieve formal carbonyl-olefin metathesis reactions is instead centered on the initial formation of a carbon-carbon single bond between carbonyls 24 and alkenes 25 to access diols 26 as reactive intermediates, which can undergo a subsequent Grob fragmentation to form olefins 28 and acetone 27 as products.140,143
Figure 2.
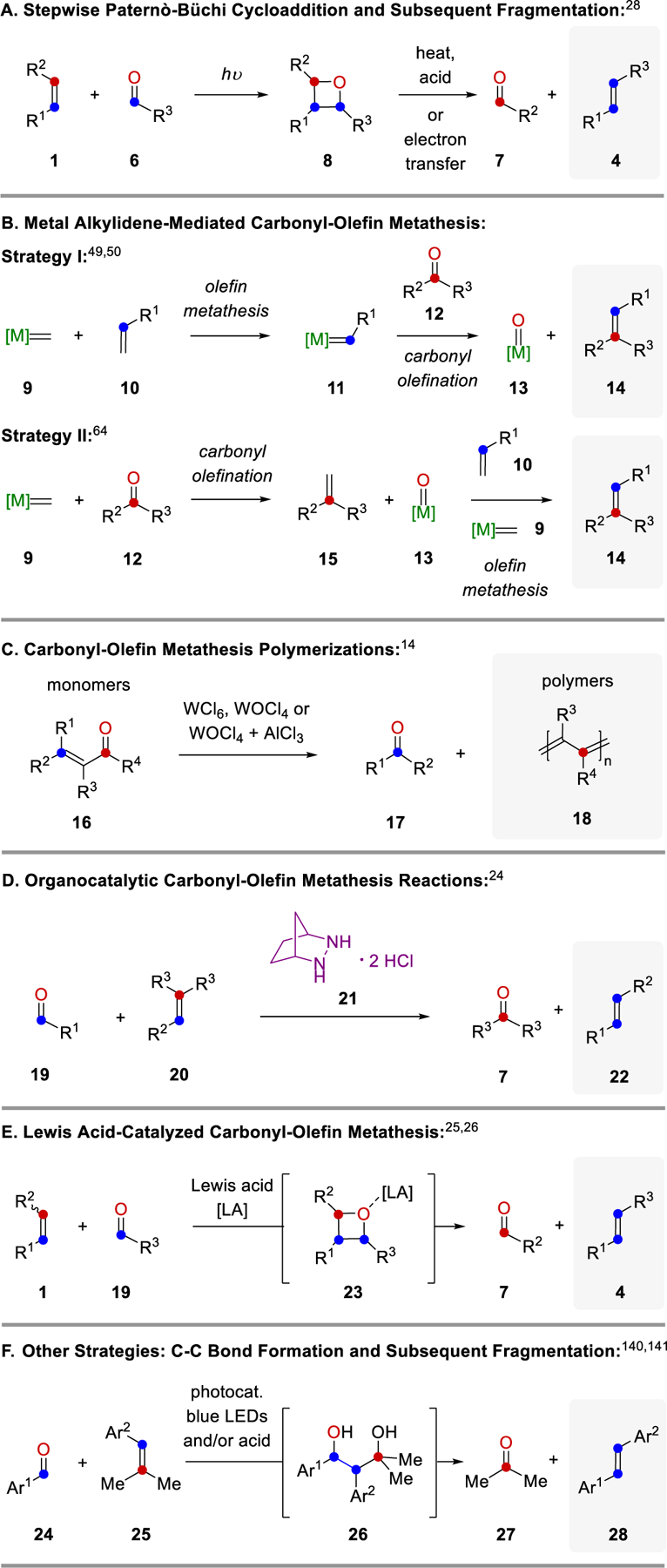
Overview of Currently Available Strategies for Carbonyl-Olefin Metathesis.
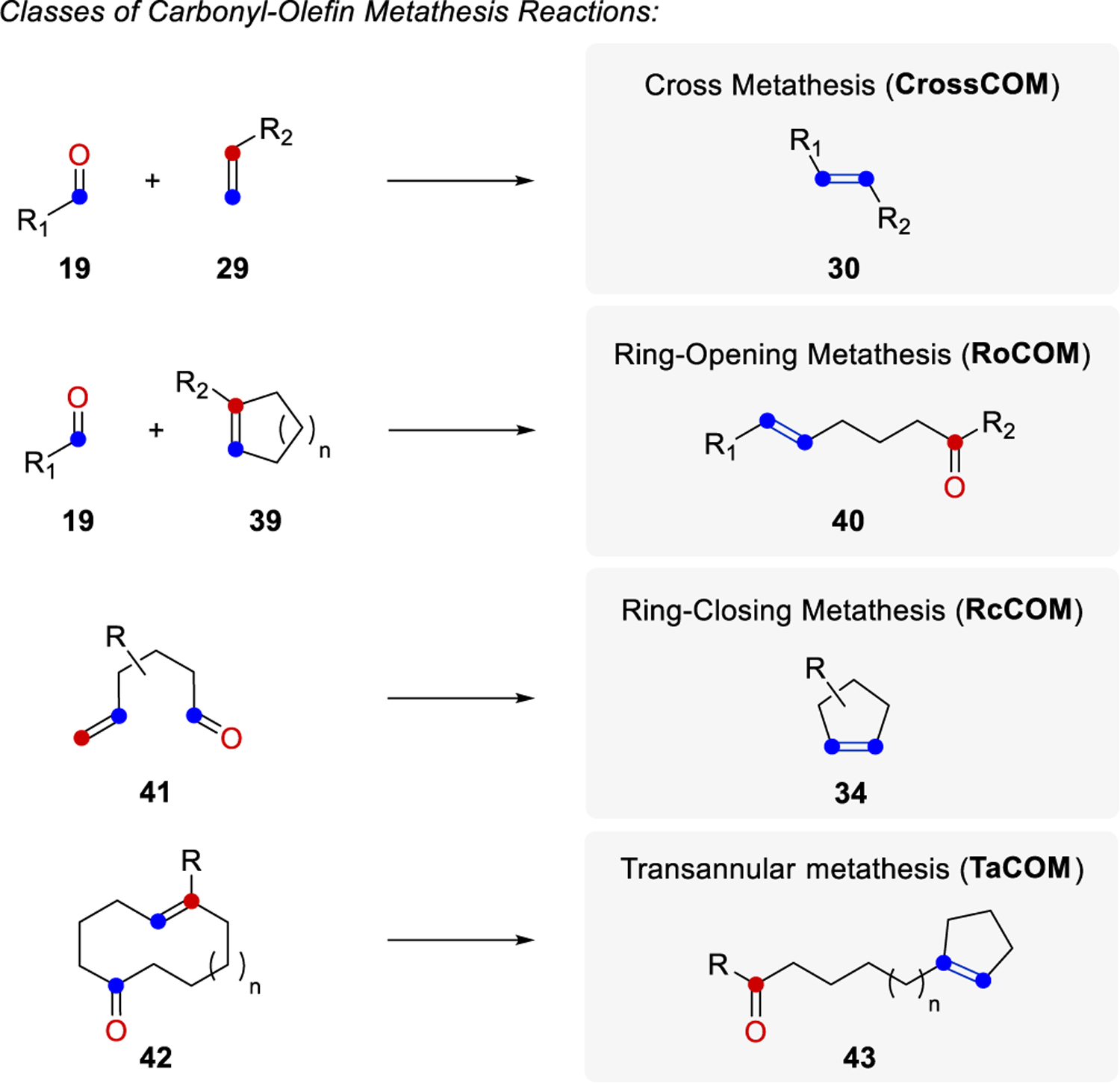
Although less advanced compared to olefin-olefin metathesis, developments in the field of carbonyl-olefin metathesis have grown significantly over the past decade. Importantly, categories of carbonyl-olefin metathesis reactions have been developed that are complementary to those of olefin-olefin metathesis and provide access to similar alkene products (Figure 3). Specifically, olefin-olefin metathesis reactions are classified into three main categories including cross metathesis, ring-opening metathesis, and ring-closing metathesis (Figure 3A). Cross metathesis (CM) reactions between two alkenes 28 and 29 can form more functionalized alkenes 30 upon conversion with a suitable catalyst. Since olefin-olefin metathesis reaction are reversible, the controlled formation of product 30 can be challenging due to its ability to also function as a substrate in subsequent catalytic cycles. Ring-opening metathesis (ROM) reactions between linear alkenes 28 and cyclic alkenes 31 yield acyclic dienes 32 as products, taking advantage of the release of ring-strain as the driving force. The third category represents intramolecular, ring-closing metathesis (RCM) reactions of alkenes 33 to generate unsaturated products 34. Additionally, two different strategies for polymerizations have been developed based on olefin-olefin cross and ring-opening metathesis. Particularly, ring-opening metathesis polymerizations (ROMP) are chain-growth, addition polymerization reactions of cyclic alkenes 35 driven by the release of ring strain while acyclic diene metathesis polymerizations (ADMET) are stepwise-growth, condensation polymerization reactions of terminal dienes 37 that result in polyenes 38 due to the favorable extrusion of ethylene gas as a metathesis byproduct.
Figure 3.

Classes of Olefin-Olefin Metathesis and Carbonyl-Olefin Metathesis Reactions Developed.
The area of carbonyl-olefin metathesis has seen significant advances in recent years, and complementary strategies for cross, ring-opening, and ring-closing carbonyl-olefin metathesis have now been developed (Figure 3B). In comparison to olefin-olefin metathesis, carbonyl-olefin metathesis reactions are often irreversible, which is advantageous with regard to reaction design and achieving high yield and conversation. However, the substrate scope for carbonyl-olefin metathesis currently remains more limited compared to olefin-olefin metathesis. Most examples of cross carbonyl-olefin metathesis reactions (crossCOM) require aryl aldehydes 19 as substrates, which together with alkenes 29 form exclusively (E)-alkenes 30. Similarly, ring-opening carbonyl-olefin metathesis (RoCOM) reactions are dependent on aryl aldehyde substrates 19 and cyclic alkenes 39 to form unsaturated ketones 40. Ring-closing carbonyl-olefin metathesis reactions of unsaturated ketones 41 give access to 5-, 6-, and 7-membered ring systems 34. In addition to these three categories for carbonyl-olefin metathesis, one strategy for polymerization has been reported based on cross metathesis of enones 16 resulting in polyenes 18 similar to ADMET in olefin-olefin metathesis. Finally, transannular carbonyl-olefin metathesis reactions of cyclic, unsaturated ketones 42 were developed following a ring-closing metathesis approach to result in ring-contraction products 43.27
2. CARBONYL-OLEFIN METATHESIS VIA PATERNÒ-BÜCHI CYCLOADDITIONS AND SUBSEQUENT FRAGMENTATIONS
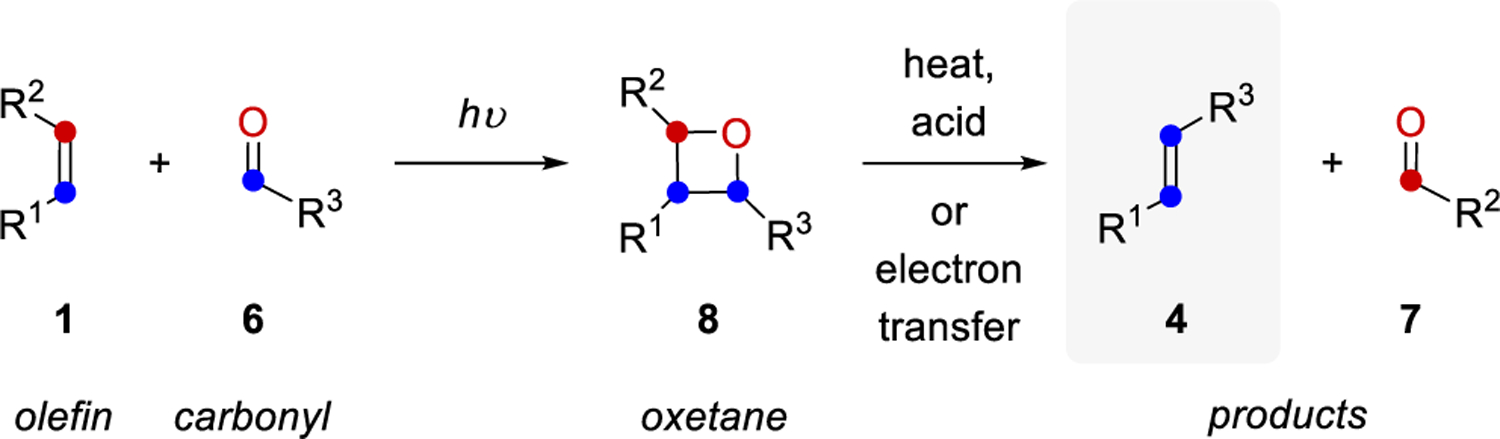
The earliest approaches to realize carbonyl-olefin metathesis take advantage of a two-step process between olefins 1 and carbonyls 6 upon photochemical irradiation following Paternò-Büchi reaction protocols (Scheme 1). Oxetane 8 is formed via a [2+2] photochemical cycloaddition, and subsequent fragmentation can be induced by an acid- or heat-mediated cycloreversion to provide the net carbonyl-olefin metathesis products alkene 4 and aldehyde 7. Although this strategy represents the first approach to facilitate carbonyl-olefin metathesis reactions, its applications remain limited mostly due to a restricted substrate scope and functional group tolerance resulting from the harsh reaction conditions often requiring pyrolysis. This section outlines the developments, advancements, and insights gained regarding carbonyl-olefin metathesis reactions via Paternò-Büchi reactions in combination with fragmentation from the early, simple examples to more recent applications that allow for access to complex polycyclic compounds.
Scheme 1.
Paternò-Büchi Reaction
In 1963, Scharf and Korte28 investigated the photo-induced cyclodimerization of norbornene 44. Following initial unsuccessful attempts to dimerize norbornene in the presence of UV light, aryl ketones were added as photosensitizers. The presence of benzophenone led to a new isolated product that was not the dimer of norbornene, but rather oxetane 45, formed from the Paternò-Büchi reaction between norbornene and benzophenone (Scheme 2). The structure was confirmed by subjecting oxetane 45 to acidic conditions, which provided the fragmentation product 46 that was subsequently oxidized upon cleavage of the alkene fragment to the corresponding dicarboxylic acid. This overall transformation is a net ring-opening carbonyl-olefin metathesis reaction resulting in the formation of 46, albeit serving the purpose of structural elucidation of oxetane 45.
Scheme 2.

Photo-Induced Oxetane Formation of Norbornene and Benzophenone by Scharf and Korte
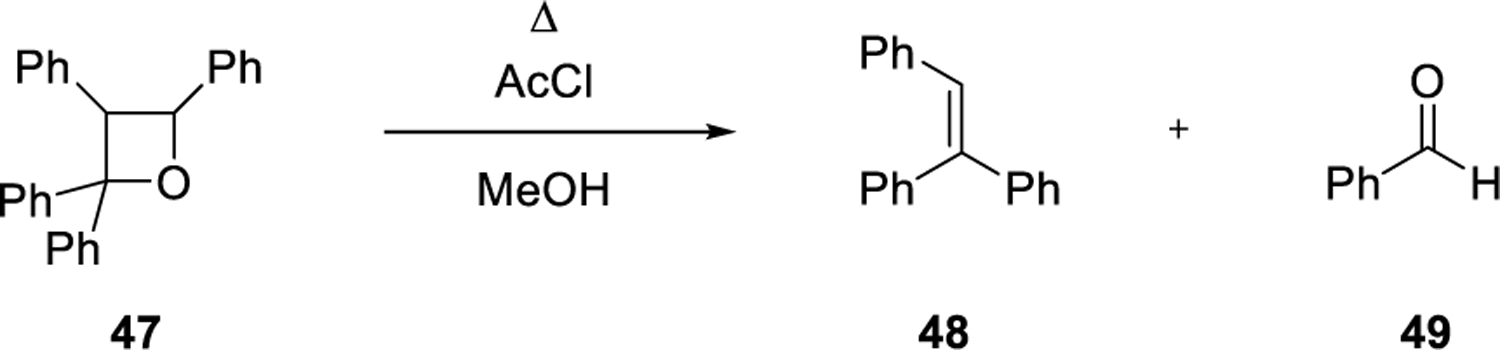
Structural elucidation of oxetane 47 was performed previously by Kohler and Richtmeyer in 1930 (Scheme 3). The reaction of 47 with acetyl chloride in methanol at elevated temperatures resulted in the formation of alkene 48 and benzaldehyde 49, which was confirmed upon conversion into the corresponding phenyl hydrazone.29 Similarly, acid-mediated fragmentations of oxetanes formed upon UV irradiation of 2-methyl-2-butene with benzaldehyde, acetophenone, and n-butyraldehyde were used by Büchi and coworkers for the purpose of structure elucidations and mechanistic investigations of the initial [2+2]-cycloaddition between carbonyls and alkenes.30
Scheme 3.
Structural Elucidation of Oxetanes via Acid-Mediated Fragmentations by Kohler and Richtmeyer
Oxetane fragmentation to carbonyl-olefin metathesis products has also been reported with catalytic amounts of Rh(I) complexes functioning as Lewis acids (Scheme 4).31 Specifically, Grigg and co-workers investigated the rearrangements of epoxides and oxetanes in the presence of [Rh(CO)2Cl]2 and discovered that oxetane 45 fragments to provide 46, quantitatively, incorporating a newly formed aldehyde and tri-substituted olefin. Likewise, oxetane 50 results in furan 51 and benzaldehyde 49 upon treatment under identical reaction conditions, albeit shorter reaction times (Scheme 4). Notably, the authors reported faster transformations when relying on CF3CO2H but a cleaner reaction profile with [Rh(CO)2Cl]2. The direction of the observed oxetane cleavage is predicted by the assumption that the coordination of the rhodium to the ether oxygen atom allows for fragmentation to form the most stable carbonium ion.
Scheme 4.

Rh(I)-Catalyzed Rearrangements of Vinyl Oxetanes by Grigg
In 1973, Jones and co-workers first recognized the synthetic potential of this photolysis-pyrolysis sequence enabling carbonyl-olefin metathesis.32 Their studies were focused on the specifics of oxetane decompositions while the reactive oxetanes were first formed via Paternò-Büchi cycloaddition reactions, which were then heated to 280–300 °C in diphenylmethane to induce fragmentation to the metathesis products (Scheme 5). The authors pointed out that while these transformations may also be performed under mild acid conditions, the pyrolysis route provides a clean product mixture with minimal side reactivity or decomposition. The pyrolysis results are consistent with the regioselective fragmentation of the weakest C-O bond in the rate-determining step, which the authors suggest could proceed via a concerted or stepwise reaction mechanism.
Scheme 5.

Photolysis-Pyrolysis Sequence for Carbonyl–Olefin Metathesis by Jones
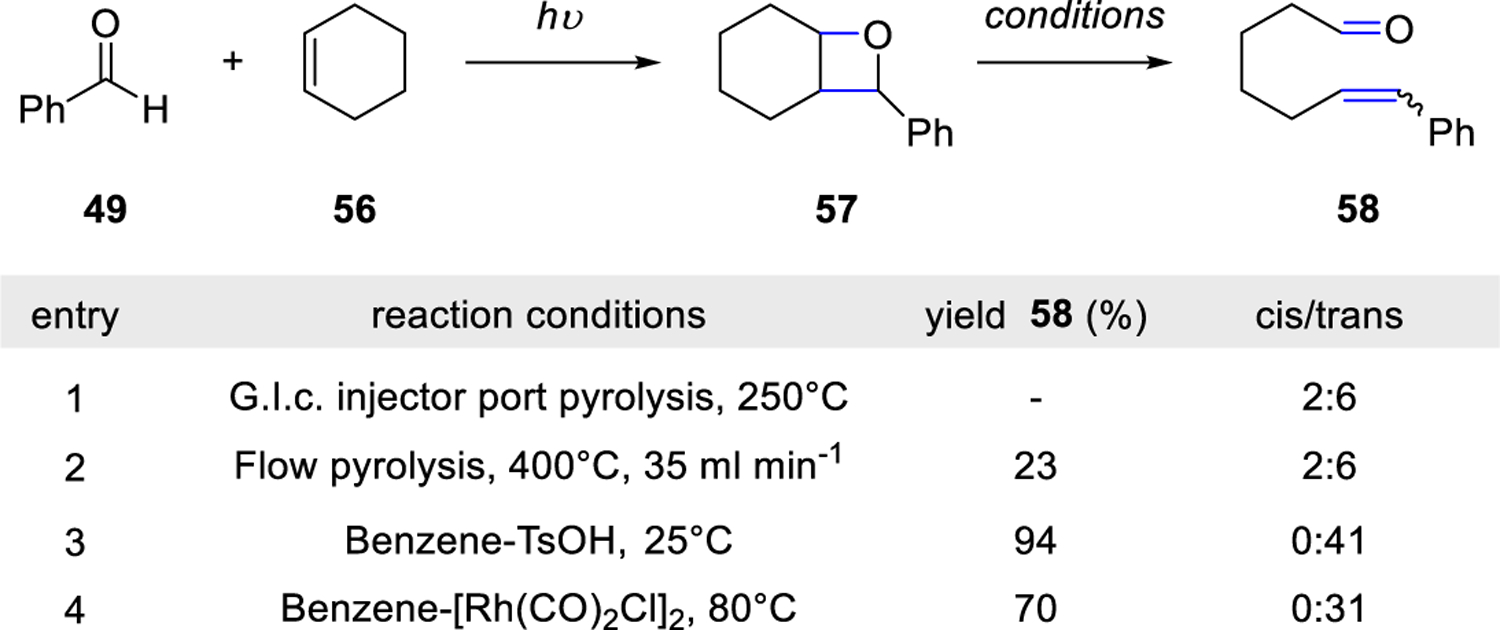
In the following years, Jones and co-workers published the application of the photolysis-pyrolysis sequence for carbonyl-olefin metathesis toward the formation of long-chain enals.33 The photolysis of benzaldehyde 49 and cyclohexene 56 allowed for isolation of oxetane 57 which under various thermolysis conditions provided good yields of cis- and trans-7-phenylhept-6-enal 58, representing an overall ring-opening carbonyl-olefin metathesis reaction (Scheme 6). In the presence of catalytic acid or a Rh(I) complex that has been previously reported17 for the fragmentation of oxetanes to metathesis products, trans-enal 58 was exclusively formed in 94% and 70% yields, respectively, with benzene as the solvent.
Scheme 6.
Formation of Long-Chain Enals via Carbonyl-Olefin Metathesis by Jones
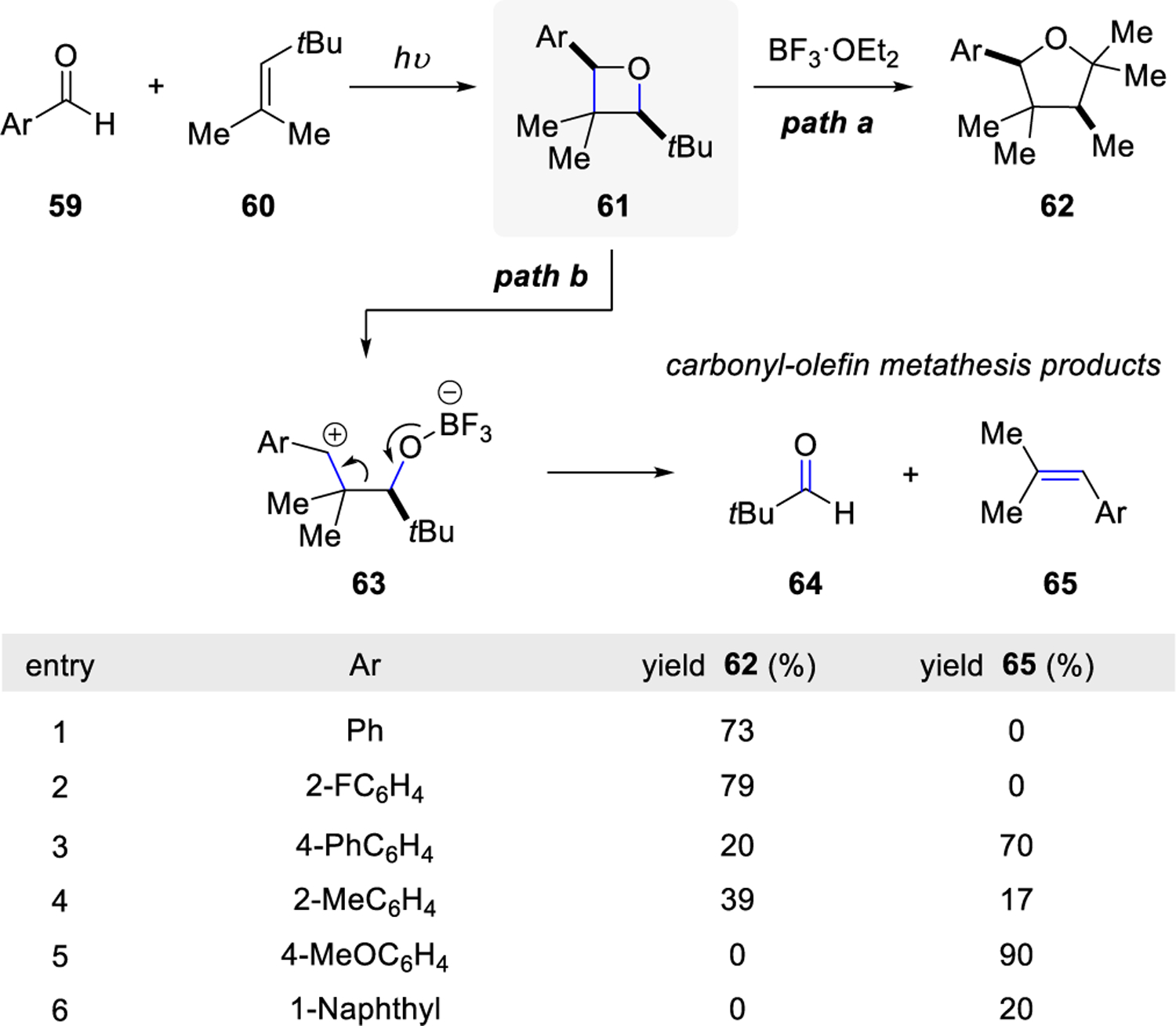
The Lewis acid-promoted fragmentation of oxetanes was reported by Carless and Trivedi in 197934 during their attempts towards the ring-expansion of oxetanes. Oxetane 61, formed from aryl aldehyde 59, provided ring expansion product 62 or the olefin product 65 in the presence of BF3·OEt2, resulting in a two-step metathesis reaction (Scheme 7). The ratio between ring expansion product 62 and the carbonyl-olefin metathesis product 65 was controlled by the electronic nature of the substituents on the aryl group of the aldehyde. Benzaldehyde and aryl aldehydes with electron-withdrawing groups were shown to promote pathway a for ring expansion, whereas electron-donating groups, such as methoxy groups, promoted exclusively pathway b, resulting in carbonyl-olefin metathesis products 64 and 65. The authors suggested that the isolation of ring expansion product 62 supports the formation of intermediate carbocations upon treatment of oxetanes with protic and Lewis acids.
Scheme 7.
Lewis Acid Fragmentation of Oxetanes by Carless
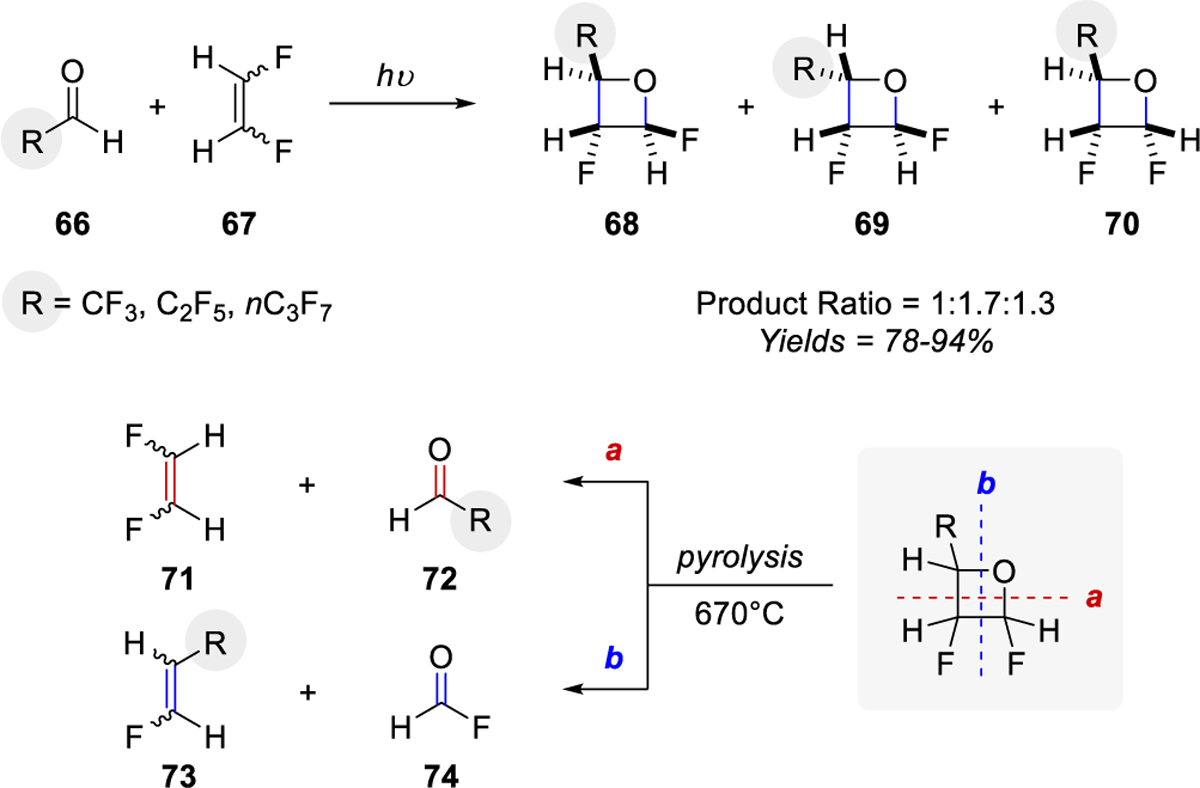
Barlow and co-workers reported the flow pyrolysis of fluorinated oxetanes at 670 °C.35 Following the synthesis of a variety of fluorinated oxetanes 68, 69, and 70 in good yields via a Paternò-Büchi reaction of fluorinated aldehydes 66 and alkene 67, pyrolysis was performed to provide carbonyl-olefin cross-metathesis products via fragmentation pathways a and b (Scheme 8). Interestingly, the flow pyrolysis fragmentation was not selective for the formation of cis or trans olefins but varied with changes in diastereoselectivity and substitution of the oxetane intermediate.
Scheme 8.
Flow Pyrolysis of Fluorinated Oxetanes by Barlow, Coles, and Haszeldine
A qualitative study, which was reported in 198036, aimed to determine the conditions required to favor various fragmentation pathways of oxetanes 75, taking into consideration that previous results obtained based on thermolysis protocols may have been altered by the presence of trace amounts of acid (Scheme 9). In order to determine if the fragmentation was entirely due to thermolysis and not trace acid for acid-catalyzed decomposition, N,N,N’,N’-tetramethylenediamine (TMEDA) was used as the solvent based on previous reports of its beneficial use with extremely acid-sensitive compounds. Nishida and coworkers’ observations are consistent with an earlier report by Jones32 that fragmentation of 75 was indeed sensitive to the reaction conditions. Specifically, base treatment of solvent or the reaction vessel resulted in an increase of fragmentation pathway b leading to alkene 79 and benzophenone (80). Subsequent efforts of Nishida and coworkers centered on mechanistic investigations of oxetane fragmentation to differentiate between a biradical pathway and a possible concerted reaction path. The results obtained suggest that if a concerted fragmentation was feasible, it was not significantly favored over the diradical process.
Scheme 9.

Thermal Fragmentation 2-Aryl Substituted Oxetanes by Nishida
The formation of keto oxetanes 83 was reported in 198137 by Maruyama. Fragmentation of these oxetanes via pyrolysis in TMEDA (to avoid residual acid) provided two different products, olefin 81 and dihydrofuran 87 (Scheme 10). The starting materials, olefin 81 and diketone 82 were regenerated in an overall carbonyl-olefin metathesis reaction following a Paternò-Büchi reaction while the opposite fragmentation provided 87 as the rearranged product in trace amounts. This study also aimed to gain additional support for a radical-based oxetane fragmentation pathway by correlating the bond strengths in the oxetane ring to the structure of the possible intermediate biradicals and the observed results were consistent with the s-character of an intermediate carbon-centered radical.
Scheme 10.

Pyrolysis of Keto Oxetanes by Maruyama
In 1989, Shima and co-workers38 employed the use of photosensitizers for photoinduced ring cleavage in order to develop the regioselective and efficient cleavage of oxetanes. Previous studies of the fragmentation of oxetanes into carbonyl-olefin pairs had not focused on the factors that control regio- and stereochemical reaction outcomes. The ring cleavage was attempted with such electron acceptors as 1-cyanonaphthalene 91, 1,4-dicyanonaphthalene 92, and 9,10-dicyanoanthracene 93 (Scheme 11). Quantum yields for the fragmentation of aryl substituted oxetanes varied depending on the oxetane substitution and aryl groups of the photosensitizer. Notably, the quantum yield was increased with more electron-donating substituents on the oxetane. Yields of the carbonyl-olefin metathesis products, 89 and 90, were improved for oxetanes containing aryl groups with electron-donating substituents such as p-Me or p-OMe (entries 4 and 6, Scheme 11).
Scheme 11.

Photosensitized Ring-Cleavage in the Presence of Electron Acceptors by Shima
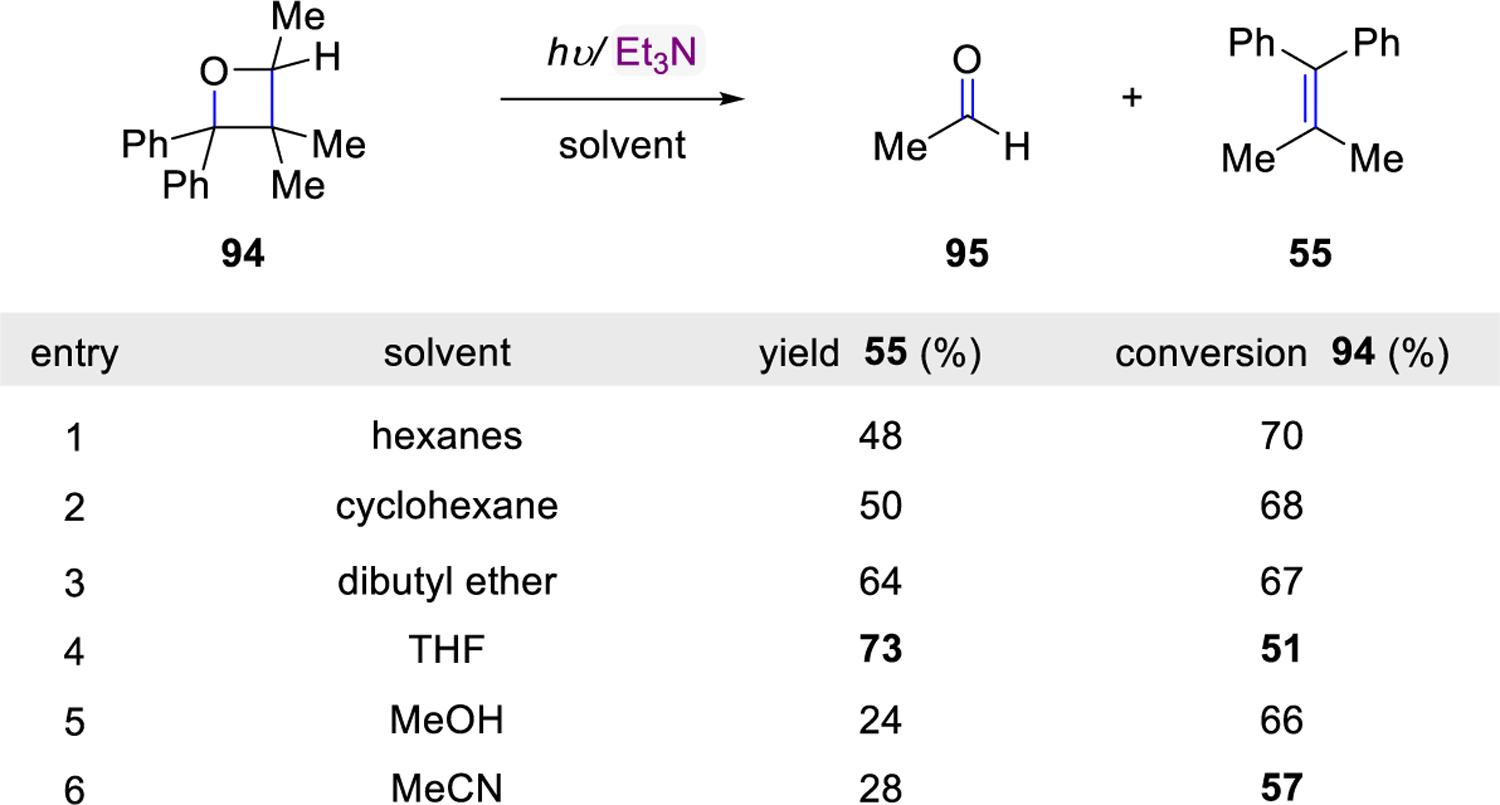
Shima and co-workers followed up on their initial report of regioselective oxetane fragmentation via photochemical electron transfer to aromatic nitriles as electron acceptors with an investigation into related oxetane fragmentations with an electron donor.39 Importantly, the presence of trimethylamine functioning as electron donor resulted in the exclusive formation of 1,1-diarylethene product 55 (Scheme 12) while in contrast, the fragmentation in the presence of electron acceptors38 had provided substituted benzophenones 89 and olefins 90 (Scheme 11). The reaction occurred most efficiently in nonpolar solvents, such as hexanes, but proceeded inefficiently in polar solvents suggesting the formation of an exciplex, as a coordinated complex, between the oxetane and triethylamine in the initiation process.
Scheme 12.
Photosensitized Ring-Cleavage in the Presence of an Electron Donor by Shima
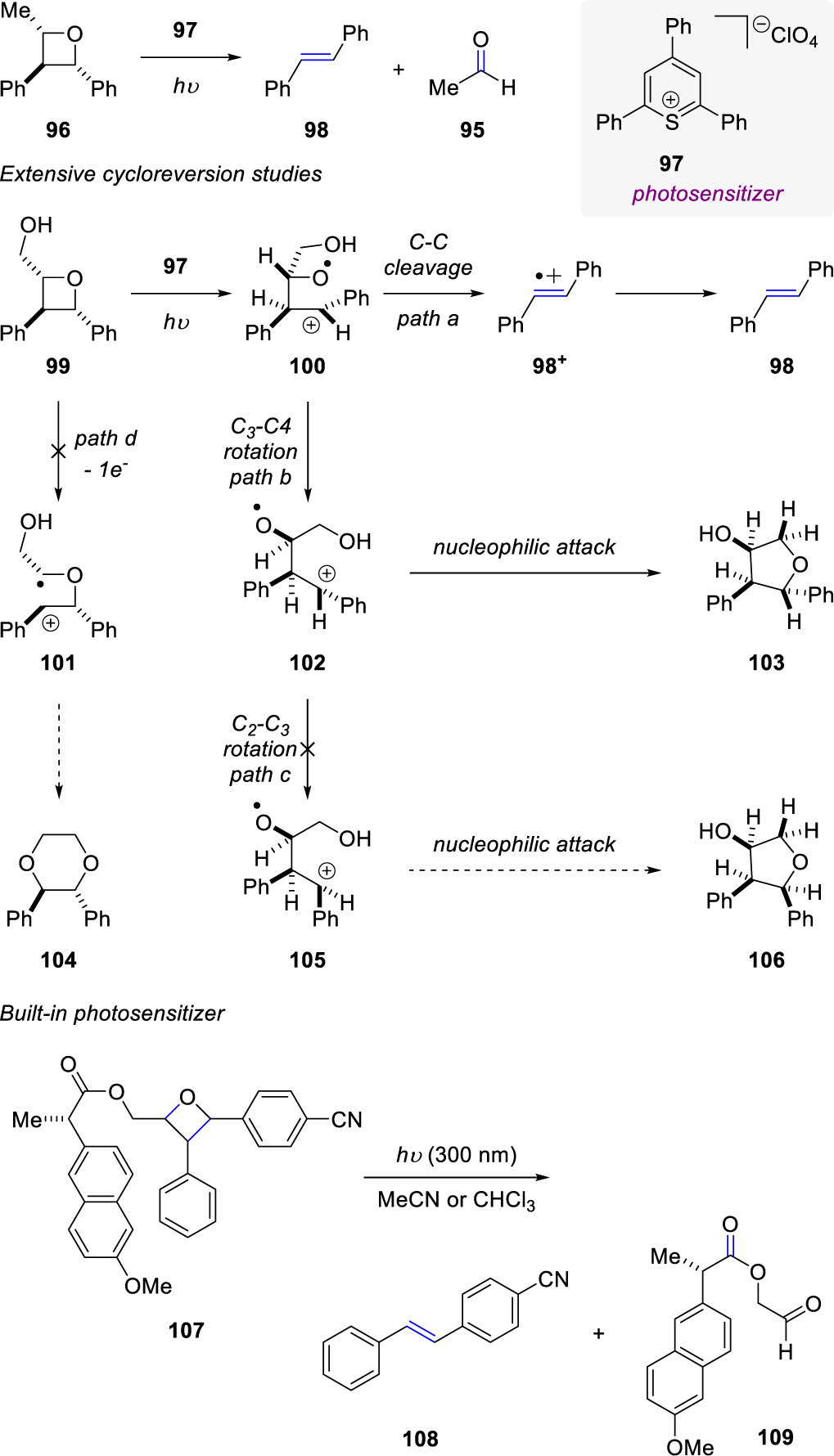
In 2002, Miranda and co-workers utilized photoinduced electron transfer (PET) of triaryl(thia)pyrylium salts (97) to promote cycloreversion of oxetane 96 yielding trans-stilbene 98 and acetaldehyde 95 (Scheme 13).40 They quickly followed up on their initial report with extensive cycloreversion studies employing oxetane 99.41 The predominant electron transfer reaction occurred from the triplet excited state of photosensitizer 97, followed by C-O bond cleavage to form 100. Following path a, an additional C-C bond cleavage formed trans-stilbene radical cation 98+ and ultimately yielded trans-stilbene 98. Miranda and co-workers also observed the formation of 103, which is hypothesized to arise through path b, where rotation of the C3-C4 bond created intermediate 102, followed by intramolecular nucleophilic attack of the pendant alcohol. Potential products 104 and 106 were not observed over the course of their investigations, ruling out paths c and d as active mechanistic pathways. Miranda and co-workers42 aimed to expand their previous work, and in 2005 achieved the cycloreversion of oxetane radical anions generated by PET. The formation of 109 resulted from a Paternò-Büchi reaction of trans-cinnamyl alcohol and p-cyanobenzaldehyde followed by esterification. Fragmentation to olefin 108 and carbonyl 109 was possible due to the “built-in” photosensitizer, 1-methoxynaphthalene, that was introduced via esterification of the oxetane intermediate prior to fragmentation. Interestingly, the choice of solvent, acetonitrile or chloroform, provided distinct results. Acetonitrile as the solvent allowed for more efficient intramolecular fluorescence quenching, whereas stereodifferentiation was increased in chloroform indicating that one diastereomer was more conducive to fragmentation.
Scheme 13.
Photochemical Cycloreversion of Methoxynaphthalene-Oxetane Dyads by Miranda
In 2006, Miranda, Griesbeck, and co-workers43 reported the cleavage of bicyclic oxetane 110 obtained upon photocycloaddition of benzaldehyde and 2,3-dihydrofuran by means of reductive electron-transfer. They described this “metathesis” of oxetanes as an “attractive tool for the synthesis of new carbonyl-ene pairs” (Scheme 14). The cycloreversion of oxetane 110 was photoinduced by the electronically excited reductants in acetonitrile, which led to the formation of the carbonyl-olefin product 111 as a trans/cis mixture of isomers. Mechanistic studies determined that quenching rate constants were dependent on the substitution of the phenyl group and further investigation revealed that the reaction was exergonic in the case of substrates with electron-withdrawing groups. Product analysis established a “photo-photo metathesis” wherein both cycloaddition and cycloreversion were induced by photochemical processes.
Scheme 14.

Carbonyl-Olefin Metathesis of Bicyclic Oxetanes by Miranda and Griesbeck
In 2009, Kutateladze and Valiulin22 recognized the synthetic potential of acid-catalyzed oxetane fragmentations and decided to build on earlier work by Jones and coworkers.33 Specifically, they developed a two-step sequence for carbonyl-olefin metathesis in which strained α-aryl and α-heteroaryl oxetanes 115 were formed and subsequently subjected to mild acid-catalyzed conditions to afford polycyclic aldehydes 117 or hemiacetals 116 (Scheme 15). This reaction design relied on the oxetane moiety itself being part of the strained intermediate formed to ultimately allow for access to a variety of diverse polycyclic systems from readily available starting materials.
Scheme 15.
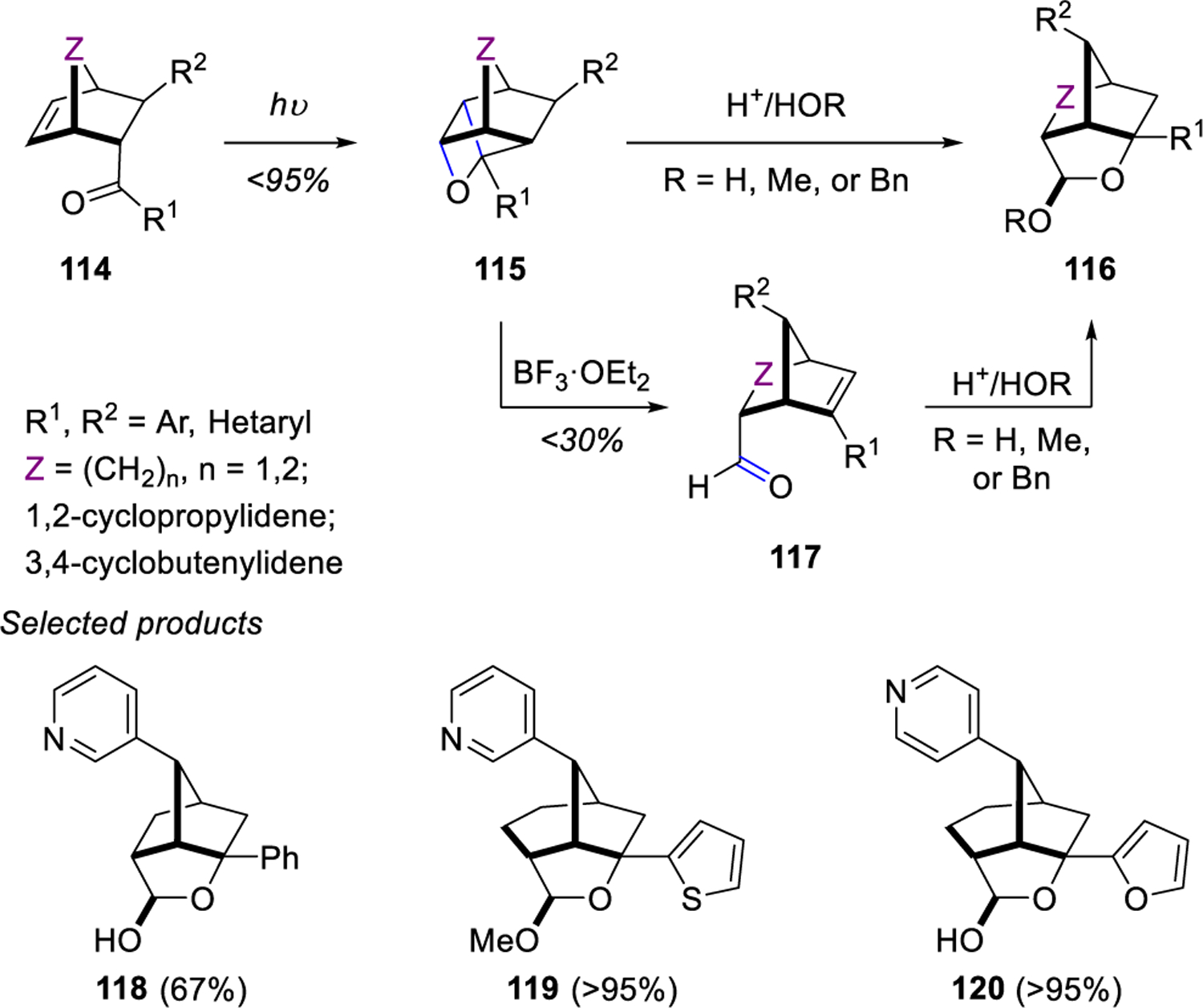
Photoprotolytic Oxametathesis in Polycyclic Systems by Kutateladze
In 2010, D’Auria and co-workers44 reported the metathesis reaction of furans and 2-substituted heterocyclic aldehydes via a proposed intermediate oxetane 122 (Scheme 16). The irradiation of 51 and 121 provided Z,E-olefin 123 as the exclusive product and the Z,E-olefin stereochemistry was observed for all other metathesis products observed (124–127). The selectivity of Z-olefin is set from 51 and the E-olefin arises from the fragmentation of the exo-oxetane intermediates (i.e. exo-122). Interestingly, tri-substituted heterocyclic aldehydes and non-arylated aldehydes provided exclusively oxetane products. The authors propose that the metathesis result of di-substituted heterocyclic aldehydes is due to possible participation of the π-aromatic orbitals in the oxetane C-O bond cleavage.
Scheme 16.
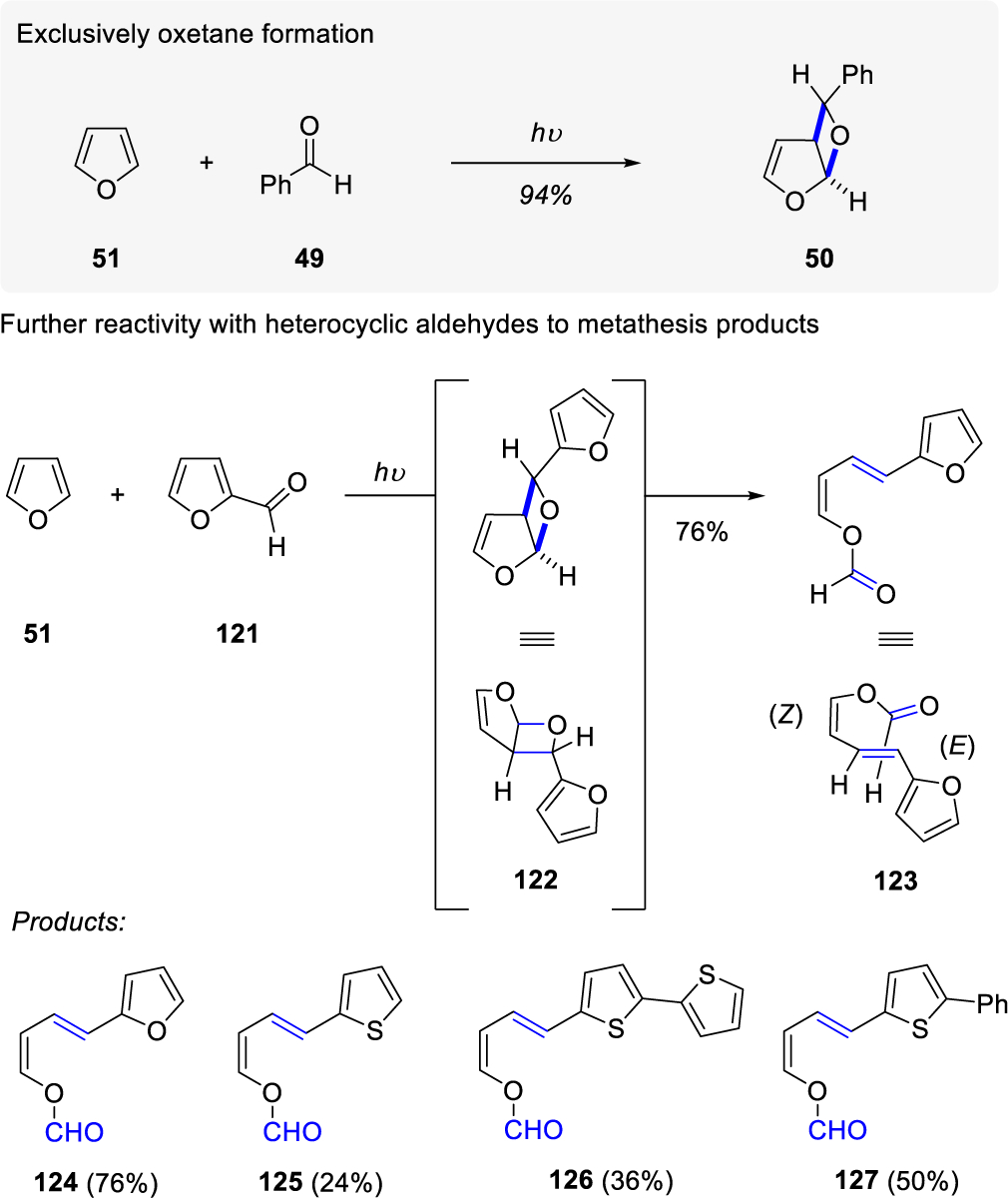
Oxetane Formation vs. Metathesis by D'Auria
In 2011, Kutateladze and co-workers23 reported a photoprotolytic carbonyl-olefin metathesis reaction that expedites the growth of molecular complexity over a few experimentally simple steps (Scheme 17). 128 was subjected to Paternò-Büchi reaction conditions to form oxetane 129 in 85% yield and the subsequent acid-catalyzed cycloreversion produces the alternative carbonyl-olefin pair metathesis product 130. Interestingly, when oxetane intermediate 129 was subjected to acidic conditions, a byproduct also formed in the presence of excess acid (greater than 2.0 equivalents), in which the metathesis product underwent a second electrophilic addition of H+ to the styrene moiety of 130 to generate a benzylic cation that was intercepted by an internal nucleophile, the enol.
Scheme 17.
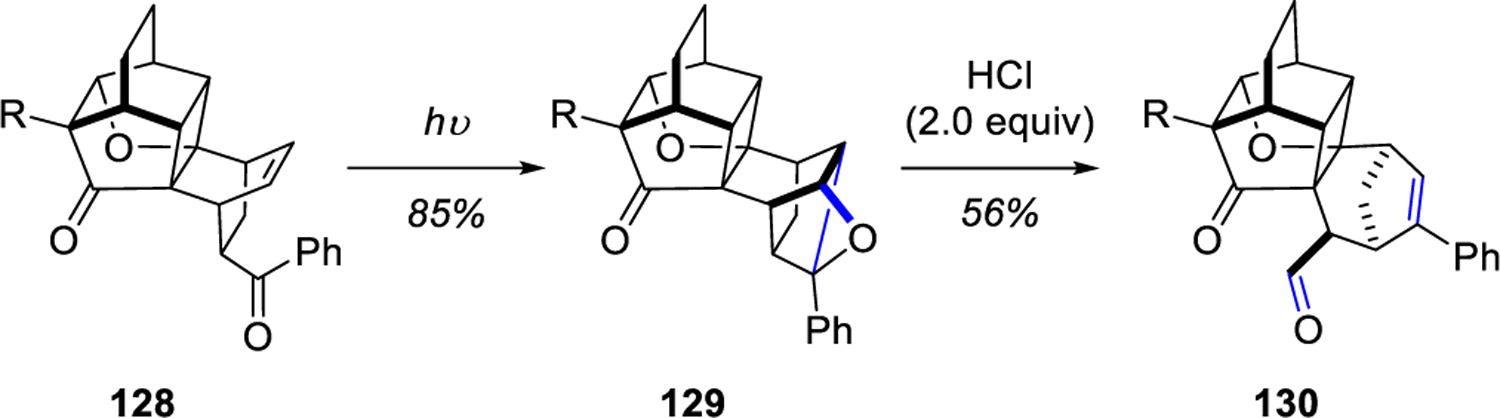
Photoprotolytic Oxametathesis Leading to Molecular Complexity by Kutateladze
A similar method for the construction of polycyclic systems was subsequently reported by Kutateladze in 2013.24 This work expanded the two-step sequence with the photoinduced intramolecular formation of oxetanes and subsequent acid-catalyzed fragmentation to form carbonyl-olefin products (134-136, Scheme 18). Scheme 18 displays the formation of γ-oxetane 132 from acetophenone adduct 131 which subsequently transformed into the metathesis product 133 under acidic conditions. The incorporation of an α-methyl substituent in 137 provided a different reaction pathway that did not result in the carbonyl-olefin product, but rather a mixture of polycyclic compounds 138 and 139 with the stellane core as a result of a radical cyclization in which two C-C bonds were formed.
Scheme 18.
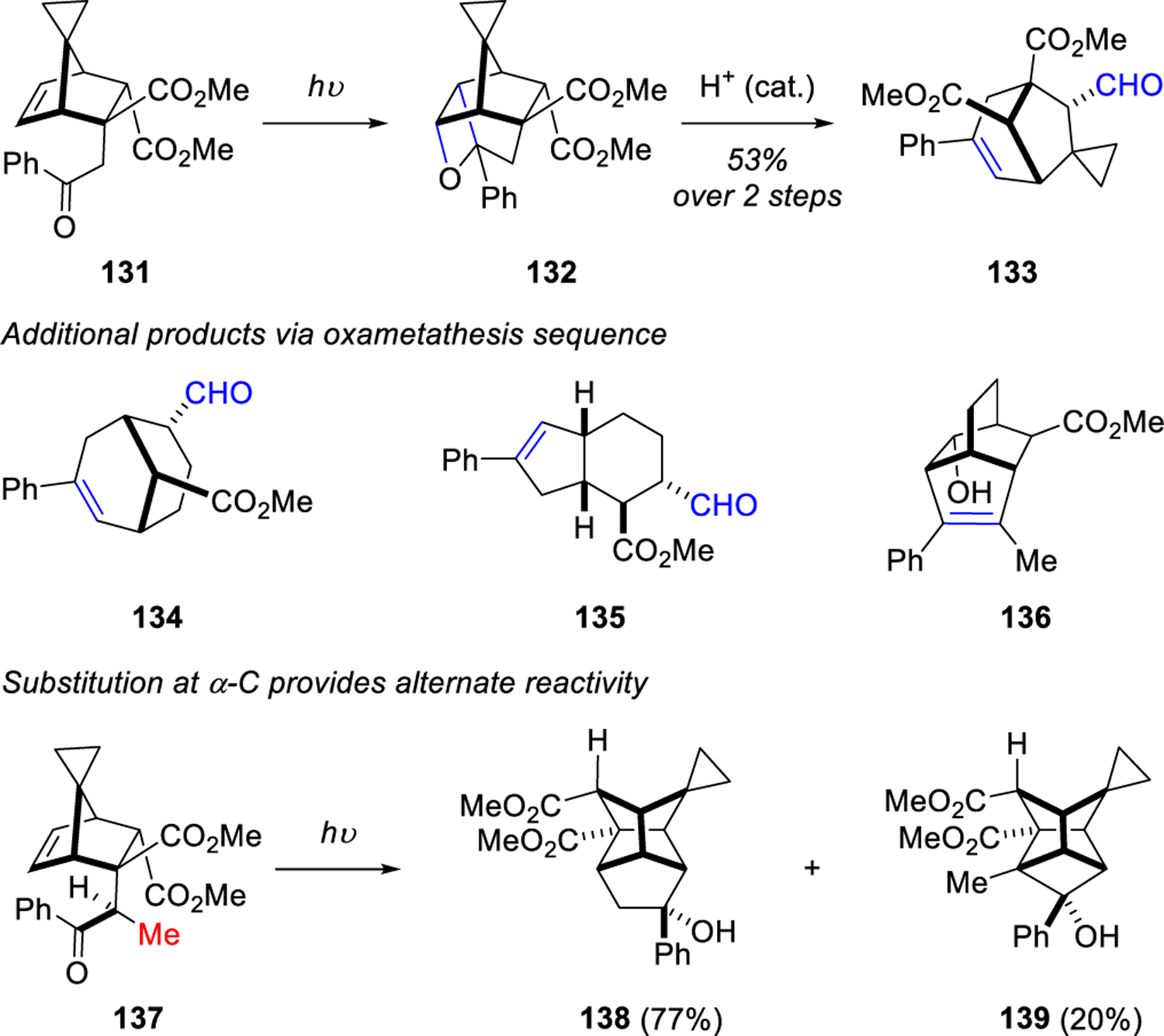
High-Yielding Photoprotolytic Oxametathesis in Polycyclic Systems by Kutateladze
The ability to form and fragment oxetanes has been crucial in the pioneering and understanding of carbonyl-olefin metathesis reactions. Since the discovery of the Paternò-Büchi chemistry, UV light has been utilized to synthesize a variety of oxetanes and their fragmentation patterns have provided valuable insight. Mechanistic elucidations have revealed that from oxetanes, carbonyl-olefin metathesis can be promoted with acid, heat, or UV light to result in the desired alkene products. Limitations of this approach for carbonyl-olefin metathesis are dictated by current shortcomings in Paternò-Büchi reactions to form the required oxetanes, which include the requirement for high energy UV-light and a narrow scope of the carbonyl and alkene substrates. Concomitant to the development of oxetane formation and fragmentation sequences for carbonyl-olefin metathesis, metal-based approaches relying on metal alkylidenes were developed to access distinct alkene products.
3. METAL ALKYLIDENE-MEDIATED CARBONYL-OLEFIN METATHESIS.
Metal alkylidene complexes have been extremely successful in the formation of C-C bonds from the redistribution of two olefin fragments through the scission and regeneration of C-C double bonds, enabling olefin-olefin metathesis. These alkylidene catalysts have made profound impacts in the synthesis of molecules related to the petroleum, materials, agricultural, and pharmaceutical industries.9 The capacity to perform these reactions catalytically is possible due to the ability for turnover of the metal alkylidene active catalyst with one of the olefin fragments following metathesis. The carbonyl-olefin metathesis reaction, however, when utilizing the same metal alkylidene complexes is unable to effect turnover of the metal alkylidene due to the resulting unreactive metal-oxo byproduct (13) formed following the initial metathesis reaction, thereby making these reactions stoichiometric (Scheme 19). For this reaction, these processes are not technically carbonyl-olefin metathesis reactions, because no new carbonyl is formed. Despite this inability to perform the carbonyl-olefin metathesis reaction catalytically with metal alkylidene complexes, the contributions to the field are nevertheless significant. Importantly, carbonyl additives have played an important role in ring-opening metathesis polymerization reactions with W- and Mo-alkylidene catalysts in order to terminate propagation, as the resulting metal-oxo species is unreactive.45 Similarly, Grubbs and coworkers reported the trapping of titanium carbene species with ketones and aldehydes as a method to terminate the living polymerization of norbornene.46 Furthermore, the vast majority of carbonyl-olefin metathesis reactions applied in complex molecule synthesis has thus far relied on metal-mediated approaches, which highlights the synthetic potential and importance that these transformations hold. Although stoichiometric in metal reagent, metal alkylidene-mediated approaches for carbonyl-olefin metathesis have seen important advances since an initial report by Grubbs in 1990. The developments in this area are highlighted in this part of the review with a particular focus on substrate scope and metal reagents used.
Scheme 19.
Strategies for Metathesis with Metal Alkylidenes
Two strategies have been developed for carbonyl-olefin metathesis with metal alkylidene complexes. Strategy I relies on an initial olefin metathesis reaction between the metal alkylidene 9 and alkene 10 through a metallacyclobutane intermediate47,48 (Scheme 19, Strategy I). Carbonyl olefination then occurs with the newly formed metal alkylidene and the carbonyl 12 to provide the desired olefin 14, thereby completing the net carbonyl-olefin metathesis reaction. Strategy II employs the reverse reactivity in which carbonyl olefination occurs initially between metal alkylidene 9 and carbonyl 12 to form a metal-oxo byproduct and alkene intermediate, which then undergoes olefin metathesis with metal alkylidene resulting from 9 and alkene 10 to provide the net carbonyl-olefin metathesis product 14.
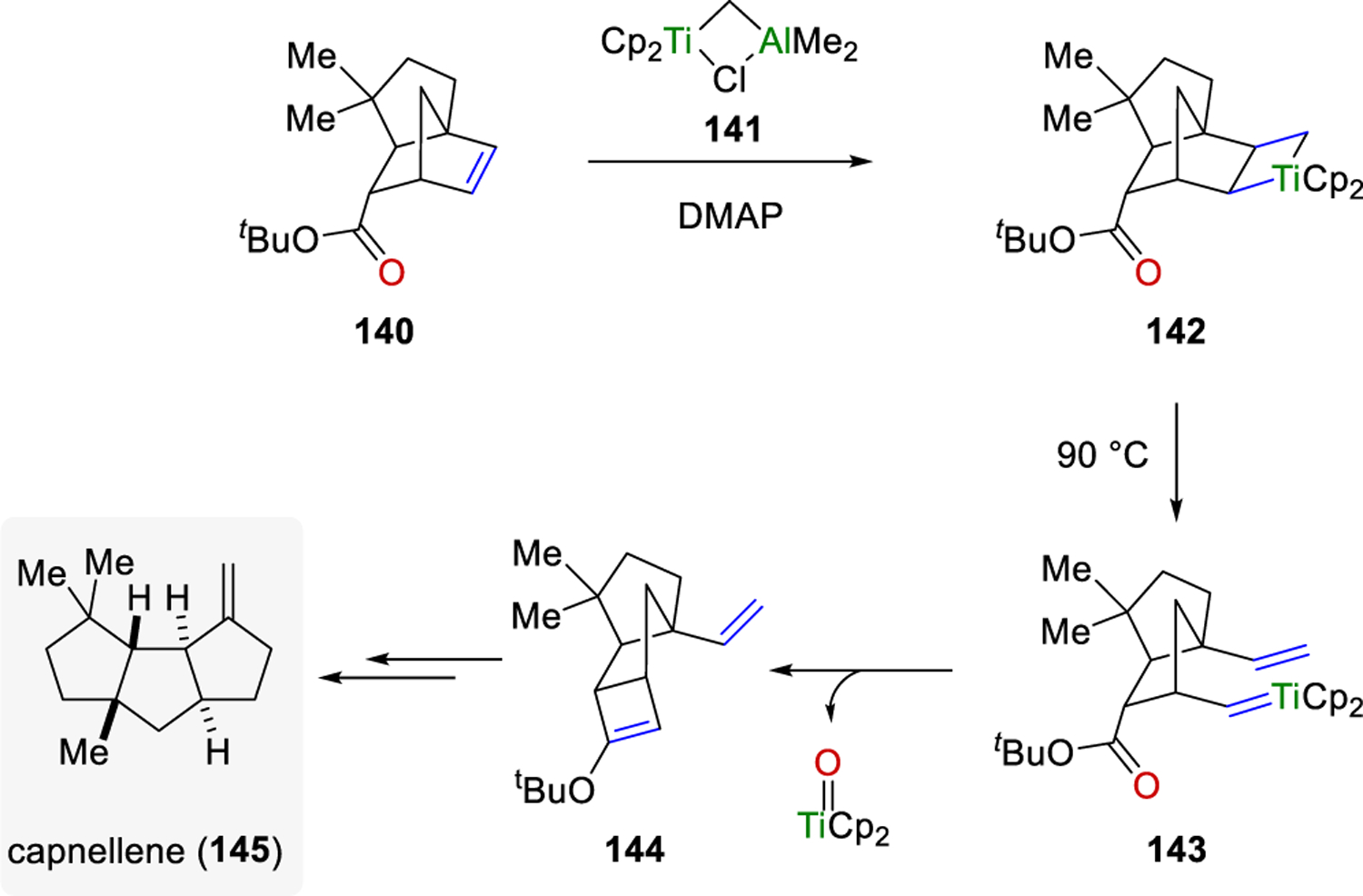
Stille and Grubbs were able to apply strategy I to the synthesis of capnellene (Scheme 20).49,50 Bicyclic intermediate 140 was converted to metallacyclobutane 142 with titanaethylene 141; subsequent heating initiated ring opening to 143 for an initial olefin metathesis reaction. Carbonyl olefination occurred with intramolecular trapping of the alkylidene with the proximal ester species for the net carbonyl-olefin metathesis product 144, which served as a key intermediate towards the total synthesis of (±)-Δ(9,12)-capnellene (145).
Scheme 20.
Application of Strategy I by Grubbs
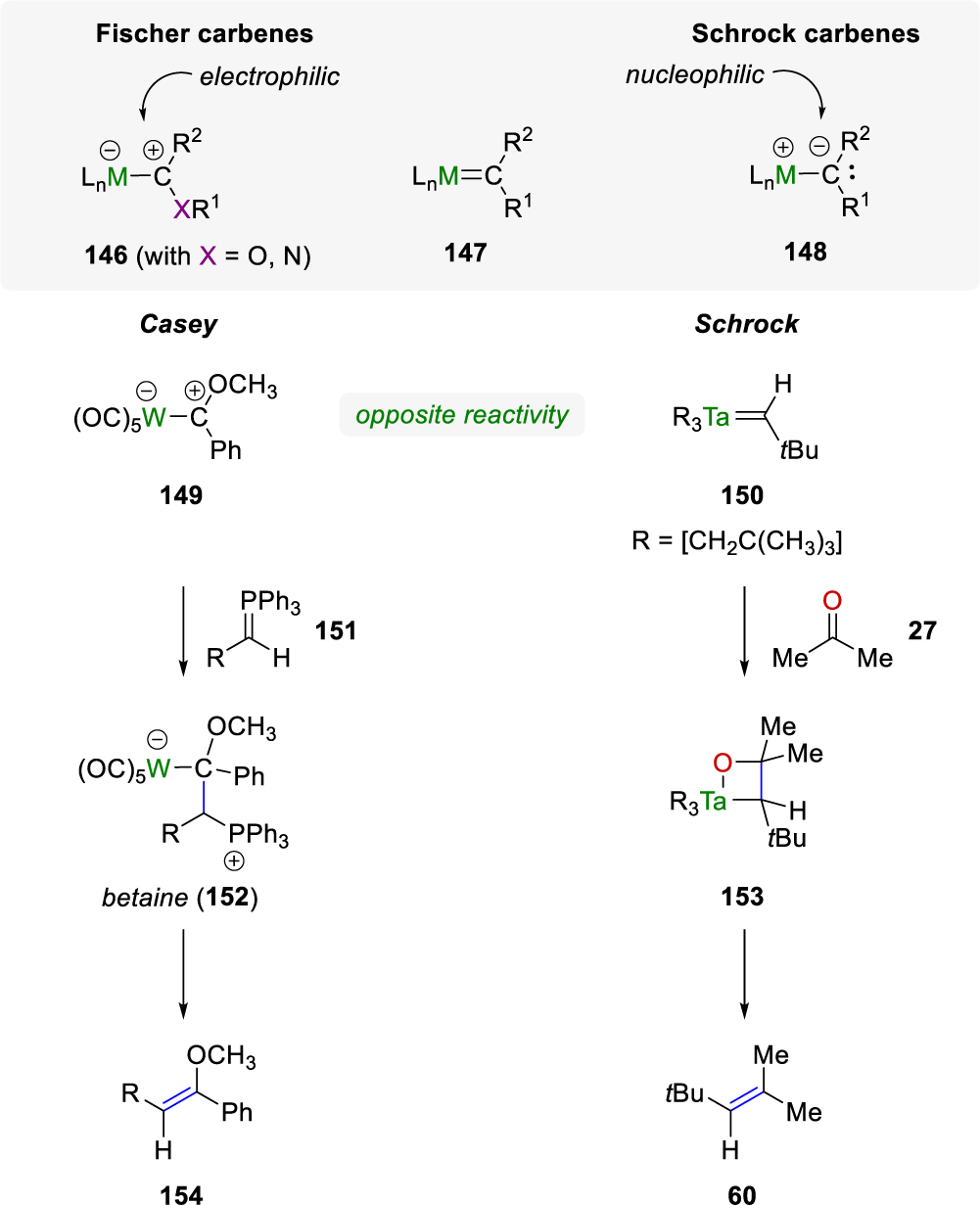
The use of either Fischer51 (146) or Schrock52 (148) metal-carbenes in “Wittig-like” reactions is well-known in the literature. Specifically, in 1972, phenylmethoxycarbenepentacarbonyltungsten(0) 149 was reacted with a carbon nucleophile 151 to form a betaine intermediate in which subsequent fragmentation provided the olefin product 154 (Scheme 21).53 Conversely, Ta[CH2C(CH3)3]3[CHC(CH3)3] (150)54 used by Schrock55 reacted with acetone in a nucleophilic manner to provide the oxametallocycle intermediate 153, which fragmented to the desired olefin product 60. This reactivity was similar to the use of the Tebbe56,57 and Petasis58 reagents whereby a metal-carbenoid reacted with a carbonyl to form a four-membered titanium oxide ring intermediate that fragmented to provide the methylenated product.
Scheme 21.
Fischer and Schrock Metal-Carbenes
The inherent reactivity of titanium carbene species with ketones and aldehydes46,59 has been applied to ring-opening metathesis polymerization reactions (ROMP) for polymer cleavage to enable access to endcapped polymers and metathesis-inactive metal-oxo species 158 (Scheme 22). These living ROMP reactions were therefore quenched deliberately while installing a known moiety in place of the metal. Ethers have been commonly used for Ru-mediated polymerizations while carbonyls, such as benzaldehyde, have been employed for Mo- and W-mediated polymerizations.45
Scheme 22.

Carbonyl-Olefin Metathesis Used for Endcapping Reactions of Polymers by Grubbs
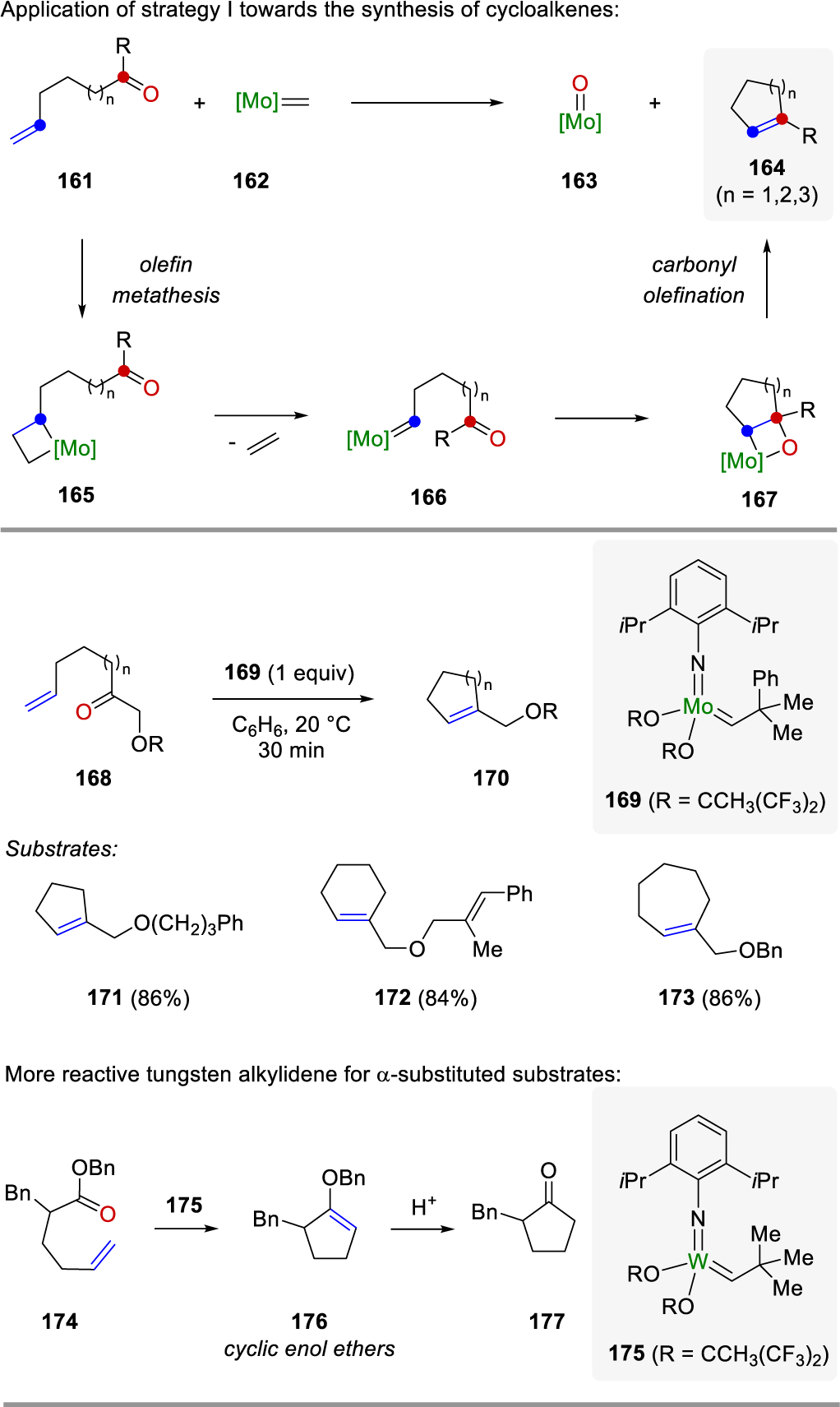
In 1993, Fu and Grubbs60 reported a new method for alkylidene-mediated carbonyl olefination based on the initial discovery by Grubbs during studies toward the synthesis of capnellane49,50 (Scheme 23). Following strategy I, the molybdenum alkylidene complex 162 initially undergoes a [2+2]-cycloaddition with 161 to form a metallacyclobutane intermediate 165, followed by fragmentation to afford a new alkylidene 166. This can then undergo a subsequent intramolecular carbonyl olefination to produce the desired cycloalkene 164. The preference for an initial olefin metathesis reaction over the carbonyl olefination allowed for the ring-closing metathesis to occur instead of the formation of an acyclic diene. Stoichiometric amounts of the metal alkylidene 169 were required to provide a substrate scope with access to 5-, 6-, and 7-membered cycloalkenes (171, 172, and 173, respectively) in high yield. Initial attempts towards ring-closing metathesis employed a specific class of substrates (174) containing esters and α-substitution with the more reactive tungsten alkylidene 175 previously reported by Schrock.61–63 The tungsten alkylidene complex formed cyclic enol ethers 176 that then reacted further to form cyclic ketones 177.
Scheme 23.
Olefin Metathesis and Carbonyl Olefination by Grubbs
A few years later, Nicolaou and co-workers reported a new method that utilized strategy II for the generation of cyclic enol ethers from olefins and esters using excess amounts of the Tebbe and Petasis reagents (Scheme 24).64 The proposed mechanism begins with the initial formation of an acyclic enol ether 180 and oxo-titanium complex 179 through a carbonyl olefination reaction of 178 with the Tebbe reagent. Then, a second equivalent of the Tebbe reagent reacts to form the titanacyclobutane 181. Subsequent fragmentation to afford the titanium alkylidene 182 then allows for an intramolecular cyclization to form a second titanacyclobutane 183, which fragments to the desired cyclic enol ether metathesis product 184 via olefin metathesis. This method is able to generate both 6- and 7-membered cyclic enol ethers (191–193) and is tolerant of a number of functional groups within the substrates (188–190).
Scheme 24.

Application of Strategy II for the Conversion of Olefinic Esters to Cyclic Enol Ethers by Nicolaou
The use of strategy II with metal alkylidenes for the formation of cycloalkenes and cyclic enol ethers was further exploited by Allwein and Rainier in 1998 with an application to the synthesis of a family of compounds associated with neurotoxicity, “red tide” catastrophes, and potent antimicrobial activity.65 They developed a three-step protocol for the synthesis of fused ether ring systems 198 that included an enol ether epoxidation of 194 to provide 195, C-C bond formation to form 196, and finally a ring-closing metathesis of 196 to 197 (Scheme 25). The ring-closing metathesis was performed through pre-functionalization of the carbonyl in olefinic-ester substrate 199 with Takai’s conditions,66 providing primarily the olefinated ester 200 and only trace amounts of ring-closing metathesis product 202. A stoichiometric amount of Schrock’s molybdenum alkylidene catalyst 201 was then employed to perform the ring-closing metathesis in 76% yield.
Scheme 25.

Iterative Pathway Towards Fused Ether Ring Systems by Allwein and Rainier
In the following years Takai-Utimoto reaction conditions were developed to realize similar cyclization strategies. Specifically, Takai-Utimoto conditions for a Wittig-type olefination were reported in 1994 that utilized an alkyl halide, PbX2, TiCl4, and Zn.66 The reaction begins with the formation of a zinc-carbenoid species 204, followed by transmetalation with PbX2 as an accelerant (Scheme 26). Reduction and a second transmetalation lead to geminal dizinc 207. The formation of titanium-containing geminal dimetallic compound 208a occurs prior to olefination of carbonyl compound 17. The Takai and Utimoto group assumed the Tebbe-type complex 208b as another possible key intermediate for the methylenation of carbonyl compounds. These conditions employed strategy II where initial carbonyl olefination is followed by olefin metathesis.
Scheme 26.
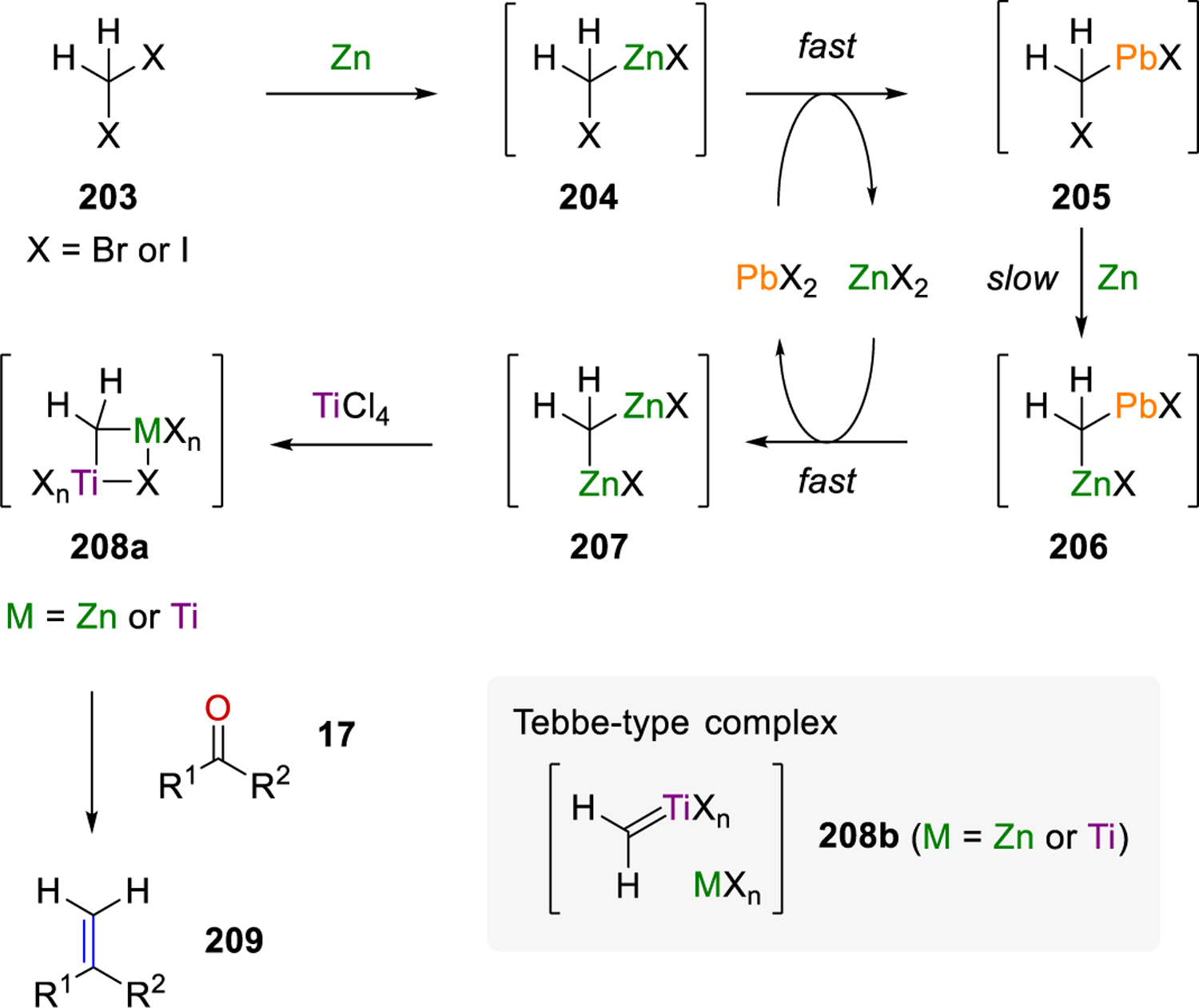
Mechanism of the Takai-Utimoto Conditions
Bennasar and co-workers67 employed strategy II for the synthesis of 1,4-dihydroquinolines using the Petasis reagent.58 Methylenation of N-protected olefinic amides following a ruthenium-catalyzed ring-closing metathesis of the resulting enamides allowed for the formation of 1,4-dihydroquinolines 213a-c in up to 75% yield (Scheme 27). Acetanilide 214 was obtained in only 20% yield after treatment with dimethyl titanocene, presumably as a consequence of its competitive interaction with the amide and carbamate carbonyl groups. Enamides 210a and 210b underwent olefin metathesis reaction with the second-generation Grubbs catalyst 212, also in 75% yield, showing that the steric hindrance of the protecting group does not affect the reactivity of the catalyst. The Bennasar group also transformed the hydroquinolines obtained into quinolines. One year later, the Bennasar group expanded this protocol to the syntheses of indoles 215, 1,2-dihydroisoquinolines 217a, and dihydrobenzoazepines 217b.68 In general, the synthesis of dihydrobenzoazepines resulted in lower yields and mixture of products due to alkene isomerization reactions during the ring-closing metathesis step. An exception was 217a and 217b, where the addition of benzoquinone to the reaction mixture reduced the isomerization process and increased the yield to 50%.
Scheme 27.

Synthetic Route to Benzo-Fused N-heterocycles by Bennasar
Majumder and Rainier were able to further optimize the metathesis reaction69 using the Takai-Utimoto titanium alkylidene66 to achieve the preferential formation of the cyclic enol ether product 219a over the acyclic enol ether product 219b (Scheme 28). Initial investigations using the Takai-Utimoto alkylidene demonstrated the mild nature of the protocol and additionally determined that unlike Nicolaou’s report,64 in which cyclic enol ethers resulted from acyclic enol ether intermediates and a subsequent enol ether ring-closing metathesis, the acyclic enol ethers were not precursors to cyclic enol ethers. Therefore, a Strategy I reaction mechanism was proposed wherein an olefin metathesis reaction occurred followed by a carbonyl olefination reaction forming the cyclic enol ether metathesis product. It was determined that these metathesis reactions utilizing the Takai-Utimoto reagent protocol were dependent on the steric environment of both the ester and olefin moieties.
Scheme 28.
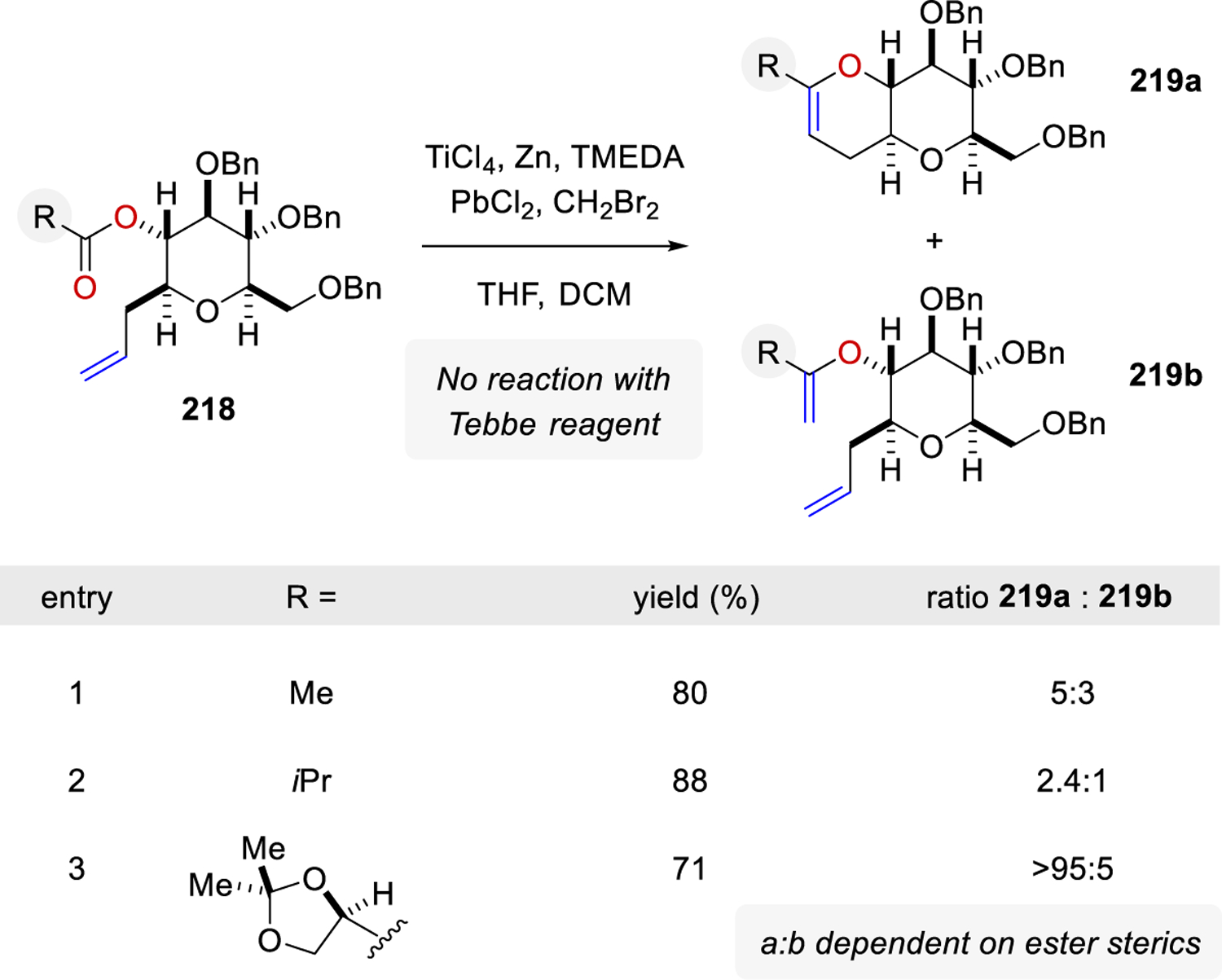
Modified Takai-Utimoto Reduced Titanium Reagent for the Olefinic Ester Cyclization by Rainier
Iyer and Rainier later discovered that the specific type of in situ generated titanium alkylidene reagent was crucial to the ratio of acyclic to cyclic enol ether metathesis products (221 and 222, Scheme 29).70 The titanium methylidene reagent derived from dibromomethane as the alkylidene source favored the formation of the acyclic enol ether product, while the corresponding ethylidene reagent resulting from dibromoethane produced the cyclic enol ether as the sole metathesis product. Further exploration into the substrate scope with this updated protocol determined that the ratio of products greatly favored or provided solely the cyclic enol ether metathesis product 224 in up to 82% yield.
Scheme 29.

Reactivity of Reduced Titanium Alkylidenes for Cyclic Enol Ether Formation by Rainier and Iyer
In 2009, the application of reduced titanium ethylidene reagents was reported for the rapid entry into polycyclic ether skeletons common in the brevetoxin class of compounds.71 This two-directional approach allowed for the formation of tricyclic ether rings via the ring-closing metathesis reaction of substrates containing two olefin ester moieties. The optimized reaction conditions provide 6- and 7-membered cyclic enol ethers and could perform the double metathesis reaction in up to 65% yield (230-233, Scheme 30). The use of this carbonyl-olefin metathesis strategy for the synthesis of cyclic enol ethers allowed for the formation of a heptacyclic compound from C-glycoside precursor in just six steps.
Scheme 30.

Polycyclic Ether Skeletons Synthesis by Rainier
Following their success with the carbonyl-olefin metathesis reaction to form cyclic enol ethers, the Rainier group reported a similar application to the synthesis of macrocycles.72 Macrocyclic motifs, as shown in Scheme 31, are present in a number of biologically active small molecules73 and general synthetic protocols for their formation are desirable. Rainier was able to apply the previously reported conditions70 relying on the use of dibromoethane to access macrocyclic ethers in excellent yields. Specifically, 6- and 7-membered cyclic enol ethers 236-238 were formed within the structure of macrocyclic compounds in up to 87% yield.
Scheme 31.

Olefinic Lactone Cyclization by Rainier
Lastly, Zhou and Rainier were able to further exploit this reduced, in situ generated titanium ethylidene reagent for the formation of cyclic enamides74 from olefinic-amide and olefinic-lactam substrates in good yield and high selectivity towards the cyclic enamide (Scheme 32). 5-, 6- and 7-membered lactams (241–243) provided 75–82% yield of the carbonyl-olefin metathesis products with greater selectivity for the cyclic product with increased ring size of the lactam.
Scheme 32.
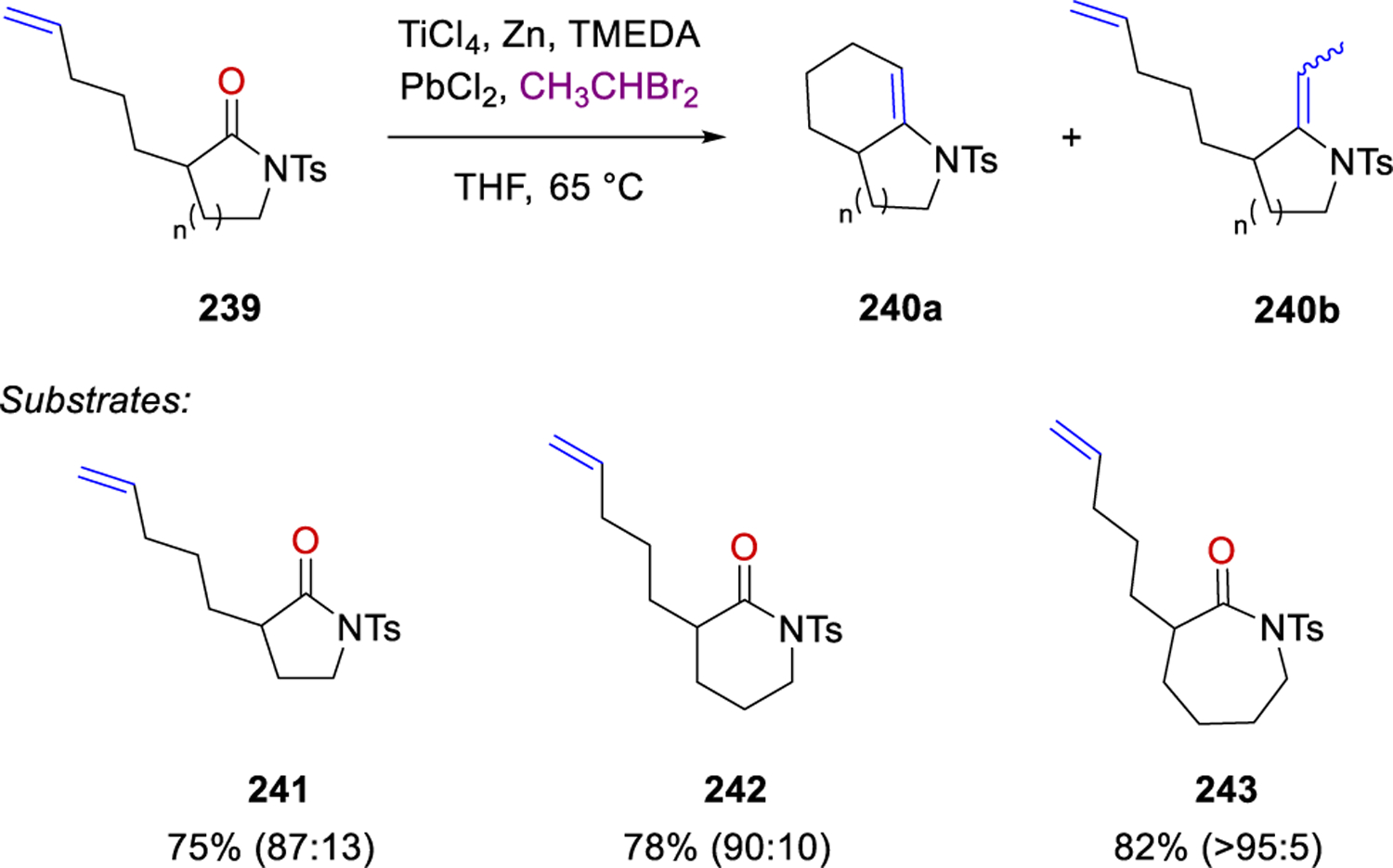
Synthesis of Cyclic Enamides by Rainier
In 2016, Chakraborty and Roy reported a study aimed at the synthesis of cyclopentanes and medium-sized rings relying on a Grubbs II (246) catalyzed ring-closing olefin-olefin metathesis sequence and Grubbs II-mediated carbonyl-olefin metathesis.75 Specifically, terminal alkenes 244 and 245 are converted with 20 mol% Grubbs II (246) to result in the formation of cyclopentene 247 and cyclohexene 248 in 13% and 20% yield, respectively, as the carbonyl-olefin metathesis products. Additionally, the formation of the olefin-olefin metathesis product (not shown) is observed in both reactions in up to 40% yield. The authors state that ring-closing carbonyl-olefin metathesis is known to require stoichiometric amounts of Grubbs II (246) to proceed, however, carbonyl-olefin metathesis has never been observed with ruthenium alkylidenes and reported examples rely exclusively on stoichiometric amounts of molybdenum alkylidenes as reagents. Based on the 1H-NMR and 13C-NMR spectra obtained by Roy and Chakraborty, we postulate that the products obtained are in fact the dimers 249 and 250 resulting upon cross olefin-olefin metathesis. Spectra included in the original report do not extend to the carbonyl region, which consequently do not allow for differentiation between the carbonyl-olefin metathesis products lacking the carbonyl functionality or the dimeric products formed. It thus appears that compounds 247 and 248 were misassigned and ruthenium-alkylidenes such as 246 do not promote carbonyl-olefin metathesis.
The use of metal alkylidenes, specifically Ti-, Ru-, W-, and Mo-alkylidenes, has been a seminal reaction paradigm for carbonyl-olefin metathesis. Two strategies to utilize these alkylidenes for carbonyl-olefin metathesis have been developed, relying on either an initial olefin metathesis, followed by a carbonyl-olefination (Strategy I) or the reversed approach, employing an initial carbonyl-olefination, followed by olefin metathesis (Strategy II) to access the desired olefin. Both of these strategies have been applied to synthesize a myriad of highly functionalized products including cyclic enol ethers, benzo-fused N-heterocycles, and cyclic enamides. Notably, many of these products remain inaccessible with recently developed catalytic protocols for carbonyl-olefin metathesis, which shows the synthetic value of these stoichiometric approaches. Additionally, metal alkylidene-mediated carbonyl-olefin metathesis reactions remain the only strategies currently used in reports of natural product synthesis with the exception of one early example relying on a Paternò-Büchi approach.
4. CARBONYL-OLEFIN METATHESIS POLYMERIZATIONS
Amongst the earliest reported carbonyl-olefin metathesis reactions were the polymerizations of enones 16 with tungsten-based reagents, resulting in the formation of conjugate polymers 18 (Scheme 34). These types of carbonyl-olefin metathesis reactions were initially referred to as “carbonyl-olefin exchange reactions,” and were reported as early as 1983.14 Although the polymers accessible by this approach are important in various areas of materials science, the transformation remained limited in its scope with only three reported monomers undergoing carbonyl-olefin metathesis. Nevertheless, the reaction holds great synthetic potential and raises interesting mechanistic questions.
Scheme 34.
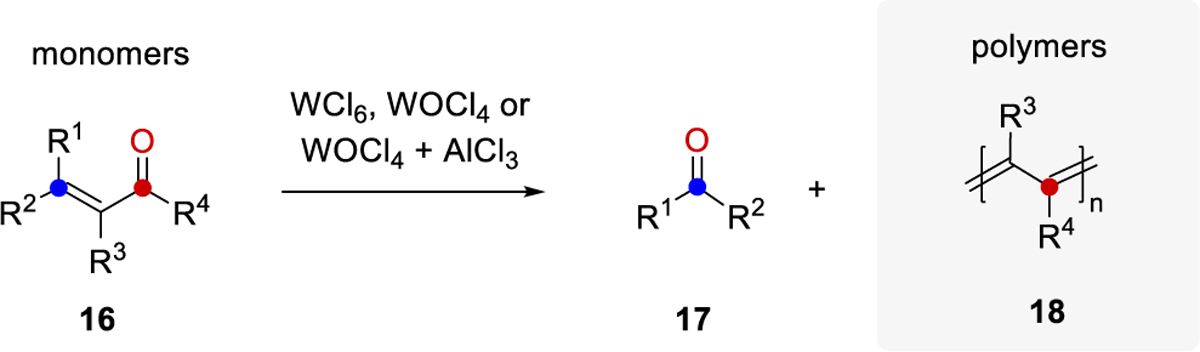
Carbonyl-Olefin Exchange Reactions Enabling Polymerizations
In particular, a consecutive mechanism relying on olefin metathesis, carbonyl-olefin metathesis and final carbonyl metathesis steps was proposed that postulated efficient reactivity of metal-oxo intermediates in subsequent alkene additions. Interestingly, this reactivity has proven elusive in attempts to render metal alkylidene-mediated carbonyl-olefin metathesis reactions catalytic. An additional interesting aspect of carbonyl-olefin metathesis polymerizations is the reliance on tungsten-based reagents to promote the desired transformation, which could exhibit Lewis acid characteristics or form tungsten alkylidenes in situ. This chapter summarizes the developments in the area of carbonyl-olefin metathesis polymerizations and focuses on reaction scope, catalyst investigation, optimization of reaction conditions, and mechanistic insights obtained.
Conjugated polymers are an important part of polymer science, with applications in the fields of bioengineering, materials and biosensors. This class of materials has unique and valuable properties—such as desirable electrical characteristics, conductivity, photoconductivity, paramagnetism, and catalytic activity—that have been extensively investigated.76 The general methods to prepare conjugated polymers include polymerizations, polycondensation, and polymer analogous conversion.77 In 1983, Schopov discovered the carbonyl-olefin exchange reaction (COER) as an additional tool for accessing conjugated polymers.14 Specifically, upon treatment of α,β-unsaturated ketone 251 with WCl6, polyphenylacetylene 252 and benzaldehyde (253, R = H, Scheme 35) were formed. Additionally, polymethylacetylene 255 was also accessible from mesityl oxide 254 and WCl6.14,78 Subsequent investigations revealed that substituting the R-group in 251 for a methyl or phenyl group similarly enabled the formation of 252, and the nature of byproduct 253 affected the yield of the polymer; specifically, benzaldehyde and acetophenone were found to interact with WCl6, while benzophenone did not react or affect the reactivity, leading to higher yields of 252.15 Chain propagation, the first step of COER, is described as double bond cleavage to form a new carbonyl compound 256 and a new C–C double bond in 256, similar to the redistribution of atoms observed in olefin metathesis reactions.16 Newly formed dimer 256 contains a carbonyl and olefin end-groups that are able to undergo COER with a second α,β-unsaturated ketone and continue the process to obtain the conjugated polymer. This polymerization is thus considered a stepwise process and a polycondensation reaction.
Scheme 35.
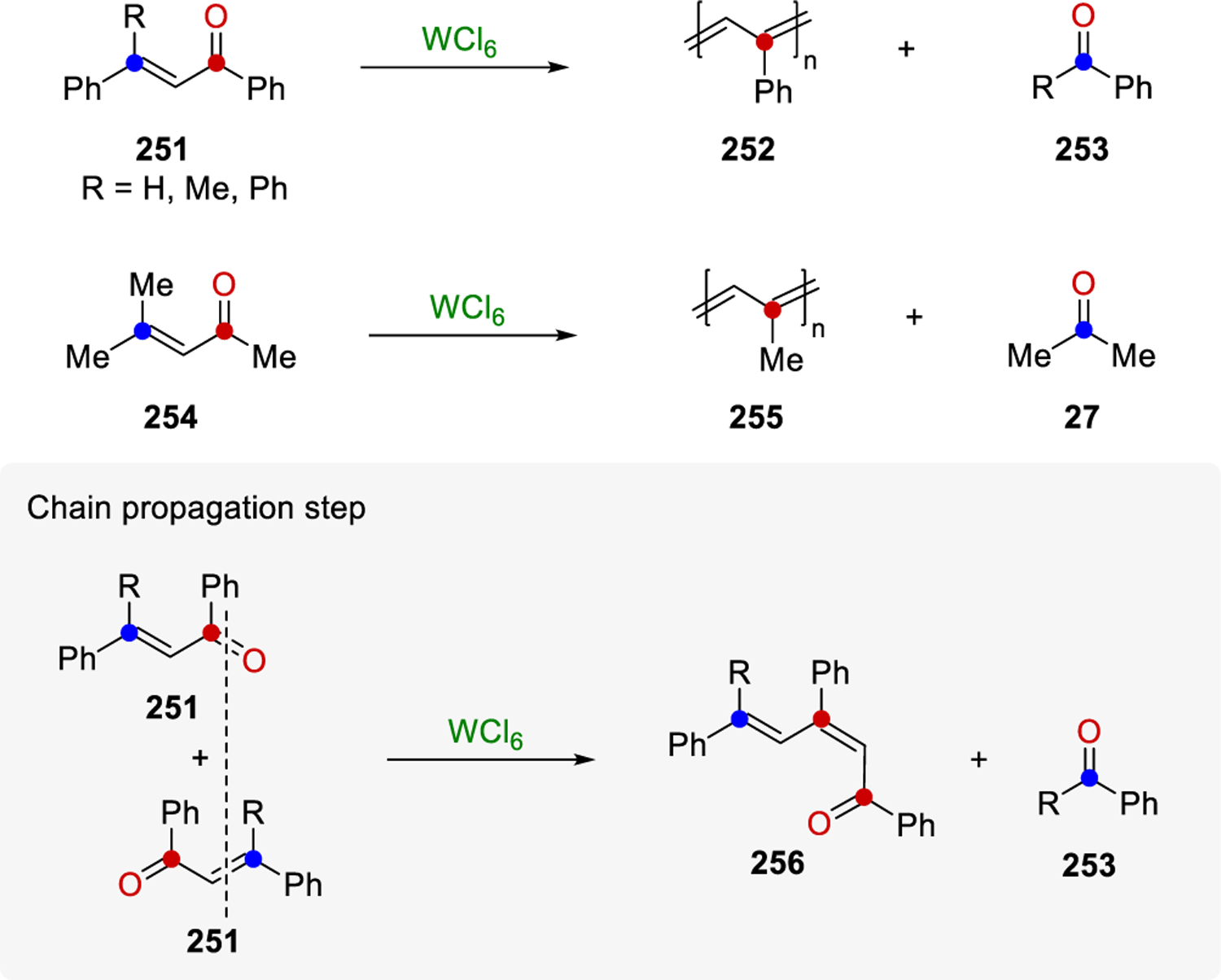
Carbonyl-Olefin Exchange Reaction as Synthetic Route to Polyconjugated Polymers by Schopov
The groups of Schopov and Jossifov carried out several investigations on COER to determine the optimized reaction conditions and important experimental results were obtained: a) the general formula of the monomers is a substituted propenone 1614,15,78 (Scheme 36); more substituted monomers lead to higher yields,18 b) the reaction can be carried out without solvent, or in benzene or chlorobenzene as the optimal solvents,14,15 c) higher yield and molecular weight were observed when increasing the amount of WCl6 (up to equimolar ratios), time, and temperature (120 °C), 15,19 d) during the reaction tungsten changes its degree of oxidation,78 and e) the polymer is obtained as a doped complex with WCl6 while polymer 18 can be obtained after a treatment with concentrated sodium hydroxide solution.18 Additionally, it was found that WOCl4 or WOCl4 with AlCl3 as additives led to higher yields of 18 and, in some cases, quantitative transformation of the monomer.20 Several polyconjugated polymers were obtained under the optimized reaction conditions such as polydiphenylacetylene (257),18 polycamphor (258),79 and poly(1,2-acenaphthenediylidene) (259).14
Scheme 36.
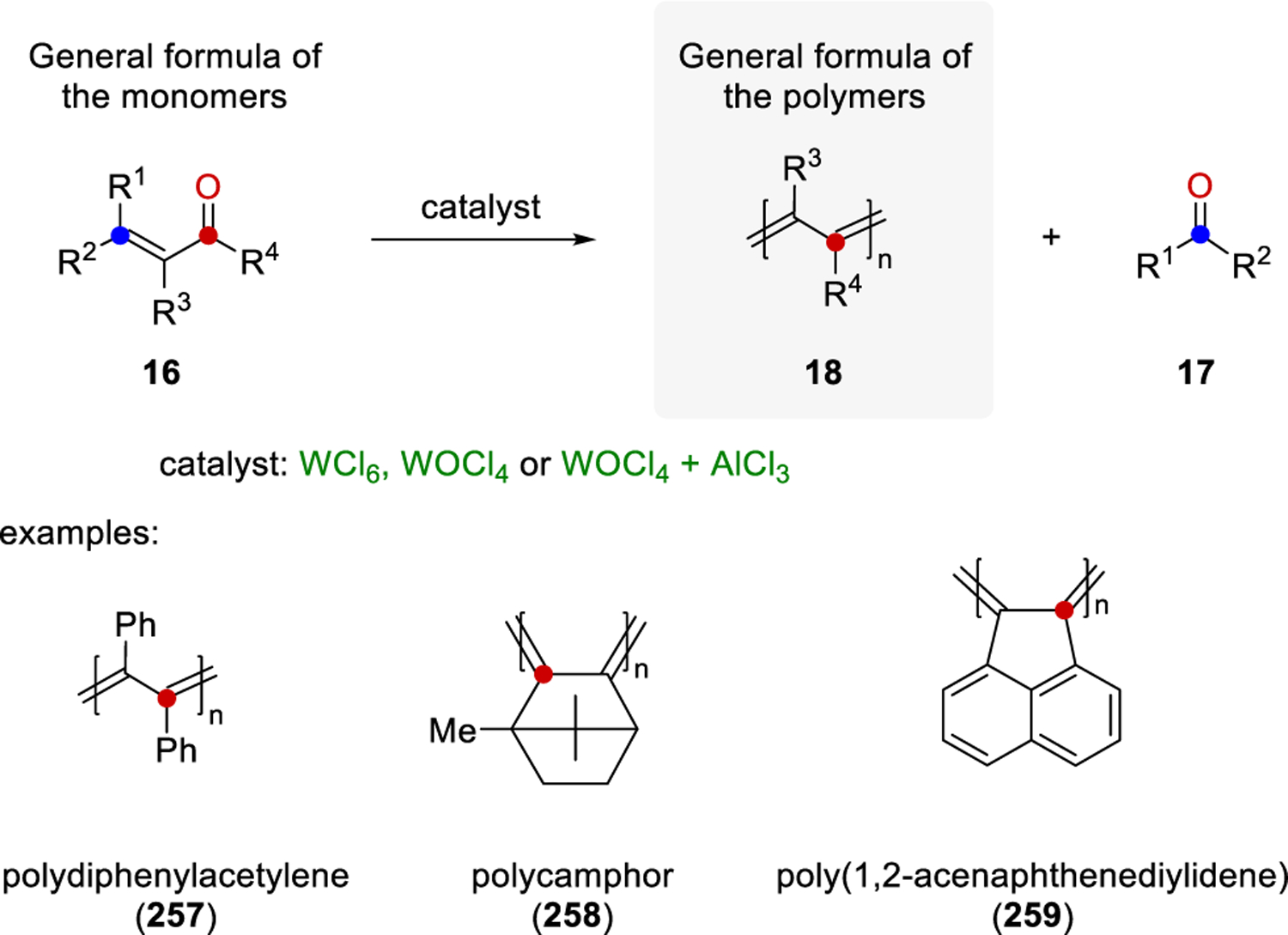
General Formulas and Examples of Conjugated Polymers Obtained with Carbonyl-Olefin Exchange Reaction
Importantly, several compounds including benzophenone 80, tetraphenylethylene 262, traces of molecular oxygen 264, and dibenzyl 265 were identified as byproducts of this polymerization21 (Scheme 37). The presence of these compounds supported the hypothesis that several double bond forming reactions were taking place during the polymerization process. Considering the olefin moiety the “head” and the carbonyl group the “tail,” three types of propagation steps can occur: COER (considered a head to tail propagation), olefin metathesis (OM, considered a head to head propagation), and reductive coupling (RC, considered a tail to tail propagation). Compound 265 was not observed as part of the byproducts reported and the origin of 266 is probably due to an inner double bond metathesis with a carbonyl group.
Scheme 37.

Types of Propagation Steps Proposed by Jossifov
The proposed mechanism for the tungsten-catalyzed polymerization by Schopov21 and Jossifov21,80,81 involves all three types of propagation steps, specifically OM, COM (or COER), and RC82 transformations taking place simultaneously (Scheme 38). The OM involves the formation of metallacyclobutane intermediates from olefins and metal alkylidene complexes via [2+2] cycloadditions and subsequent fragmentations. The COM reaction consists of the redistribution of the metal carbene generated from OM and a carbonyl group to form a new olefin and an oxo-metal complex. Then, the oxo-metal species could either react with a second olefin or with a carbonyl through a [2+2] cycloaddition to start the next cycle of CM. This cycle entailed the formation of a dioxometallacyclobutane and subsequent cleavage to the metal carbene and molecular oxygen. The metal carbene could then react with a second carbonyl group to form an olefin and regenerate the oxo-metal complex to continue the cycle. The hypothesis is based on the existence of similar catalytic systems,83 the established [2+2]-metathesis reactions mechanism relying on transition metal carbene complexes,84 the byproducts observed, and the tungsten oxo-alkylidene complexes reported by Bryan and Mayer.85
Scheme 38.

Possible Mechanism for the Synthesis of Polyconjugated Polymers by Schopov and Jossifov
Notably, Bryan and Mayer reported a series of oxo-alkylidene complexes generated via oxidative addition of ketones to form two divalent ligands with tungsten (Scheme 39). WCl2(PMePh2)4 reacts with two equivalents of ketone forming a metallaoxirane complex 267 via substitution of two phosphine ligands. Some of these complexes decomposed to the oxo-alkylidene complex 268 at room temperature in a few days. However, small changes in the ketones affected the reactivity: tetrasubstitutedethylene compounds, ketones, and other unidentified byproducts were formed together with oxo-tungsten complex (W(O)Cl2L3). Therefore, this proved not to be a general method for preparing oxo-alkylidene complexes. Based on the past evidence and existing literature, Jossifov investigated the polymer formation via reductive coupling of diketone 267 with WCl6·AlCl3,86 which was previously used as a two-component catalyst for COER.20 Polydiphenylacetylene with carbonyl end-groups was obtained, although molecular oxygen was not detected, which means that the polymerization proceeded as a normal RC. This result supports part of the “carbene mechanism” (Scheme 38).
Scheme 39.

Synthesis of Tungsten Metallaoxirane and Tungsten Oxo-Alkylidene Complexes by Mayer
In particular, a consecutive mechanism relying on olefin metathesis, carbonyl-olefin metathesis and final carbonyl metathesis steps was proposed that postulated efficient reactivity of metal-oxo intermediates in subsequent alkene additions. Interestingly, this reactivity has proven elusive in attempts to render metal alkylidene-mediated carbonyl-olefin metathesis reactions catalytic. An additional interesting aspect of carbonyl-olefin metathesis polymerizations is the reliance on tungsten-based reagents to promote the desired transformation, which could exhibit Lewis acid characteristics or form tungsten alkylidenes in situ.
In 2018, Dimova and coworkers reported an FeCl3-catalyzed carbonyl-olefin metathesis polymerization reaction of chalcone 251 to form polyphenylacetylene 252.87 Specifically, when chalcone 251 is converted with 15 mol% FeCl3 at 60 °C in 1,2-dichloroethane, the authors observe the formation of polyphenylacetylene 252 in 76% yield as a predominantly trimeric product. In comparison, when the reaction is conducted in toluene at 90 °C under otherwise identical reaction conditions, 252 is obtained in increased yields of 81% while fractions of molecular masses up to 5000 g·mol-1 are registered.
In summary, these examples highlight the potential of carbonyl-olefin metathesis as a tool for the polymerization of enones. Initial mechanistic studies suggest that all three types of propagation occur simultaneously in these transformations to form the desired polymer products. Despite the fact that the polymers synthesized by these transformations are limited in scope and size, the reactions hold tremendous synthetic potential. While new advances have been reported recently for other categories of carbonyl-olefin metathesis, the most recent developments in tungsten-mediated carbonyl-olefin metathesis polymerizations date back to the 1990s. Only a single, more recent report describes the use of Lewis acids for carbonyl-olefin metathesis polymerization reactions.
5. ORGANOCATALYTIC CARBONYL–OLEFIN METATHESIS REACTIONS
The first catalytic strategy for carbonyl-olefin metathesis was reported by Lambert and coworkers in 2012.88 In contrast to the classic [2+2]-cycloaddition and retro-[2+2]-cycloaddition paradigm usually operative in double bond metathesis reactions, their reaction design principle takes advantage of a [3+2]-cycloaddition and retro-[3+2]-cycloaddition using 1,3-dipoles (Scheme 41). The reliance on this alternative class of pericyclic reactions circumvents some of the challenges presented by [2+2] manifolds; however, 1,3-dipolar cycloadditions present their own set of unique issues. Principle amongst these challenges is the typically high stability of the intermediate, five-membered ring cycloadducts. In contrast to the four-membered ring intermediates of both olefin metathesis and carbonyl-olefin metathesis reactions involving oxetanes, the Lambert design requires some means to destabilize the intermediate cycloadducts, whether it is through the use of strained substrates or, ideally, catalyst design.
Scheme 41.

Reaction Design Principle for the First Catalytic Carbonyl-Olefin Metathesis Reaction.
The specifics of the Lambert blueprint call for the use of hydrazine catalysts, which engage the carbonyl component by condensation to form azomethine imines (or more accurately, their conjugate acid hydrazonium ions). The metathesis then proceeds by reversible 1,3-dipolar cycloadditions with the olefin. The key principle in this design is that, by virtue of the locally symmetric nature of the pyrazolidine intermediate, cycloreversion can occur via two different exit channels: one to reform the starting materials and the other to generate new olefin and hydrazonium intermediates. In precisely the same way as olefin metathesis reactions, the equilibrium point of this inherently reversible process is dictated by elements of ring strain, conjugation, or mass action. It should be stressed that a particular advantage of this platform is that the engagement of the carbonyl and olefin occurs through mechanistic processes—condensation and cycloaddition, respectively—that are compatible with a broad range of functionality. This design thus holds promise for substantial generality, even if much of that promise is as yet unrealized. Nevertheless, this reaction paradigm has been shown to be applicable to catalytic ring-opening and ring-closing carbonyl-olefin metathesis reactions with several distinct substrate architectures.
5.1. Ring-Opening Carbonyl-Olefin Metathesis
In the first implementation of the hydrazine-catalyzed design, Griffith, Vanos, and Lambert88 demonstrated the ring-opening carbonyl-olefin metathesis (ROCOM) of cyclopropenes in 2012 (Scheme 42). The optimal catalyst for this transformation was the symmetric bicyclic hydrazine 21 as its bis-hydrochloride salt. With catalytic 21, aryl or aliphatic aldehydes 19 underwent ROCOM with cyclopropenes 20 to furnish β,γ-unsaturated aldehydes 22. Notably, the carbonyl-olefin metathesis products were formed with complete (E)-selectivity.
Scheme 42.

Catalytic Ring-Opening Carbonyl-Olefin Metathesis via 1,3-Dipolar Cycloadditions.
These reactions were found to proceed in up to 95% yield for a variety of electronically differentiated aryl aldehydes including furan and thiophene derivatives. Aliphatic aldehydes proceed in yields of 35% with the use a slow addition protocol (Scheme 43). It was presumed that the lower efficiency of the aliphatic substrates was due at least in part to side reactions resulting from deprotonation/tautomerization of the intermediate hydrazonium intermediates. The proposed mechanistic hypothesis involves initial condensation between aldehydes 19 and the symmetric hydrazine catalyst to form the corresponding hydrazonium ion 269. Interestingly, of the two possible geometric isomers of this hydrazonium ion (E-269 and Z-269), the authors propose (and calculations support) that cycloaddition with cyclopropane 20 is favored for E-269 via an exo-transition state due to minimized steric interactions, despite the fact that Z-269 is the thermodynamically favored isomer. The resulting pyrazolidine cycloadduct 270 undergoes proton transfer to form 271, which then proceeds through strain-relieving cycloreversion to result in hydrazonium ion 272. Hydrolysis of 272 reveals the carbonyl-olefin metathesis product, enal 22, with concomitant regeneration of the hydrazine catalyst 21.
Scheme 43.

Hydrazine-Catalyzed Ring-Opening Carbonyl–Olefin Metathesis by Lambert.
The Lambert and Houk groups89 followed up this initial report with a detailed mechanistic investigation of this hydrazine-catalyzed carbonyl-olefin metathesis reaction platform.88 Computational analysis fully supported the mechanistic rationale described above and underscored the role of ring strain of the cyclopropane substrates (20). Specifically, the substantial cyclopropane ring strain accelerated the initial [3+2] cycloaddition and led to intermediate fused cyclopropane cycloadducts that also benefited from high ring strain to accelerate the [3+2]-cycloreversion step (Scheme 44). Of particular importance was the finding that other strained olefins such as cyclobutenes and cyclopentenes were likely to have significantly higher activation barriers for the key cycloreversion step using hydrazine catalyst 21. This understanding argued strongly for the design of alternative hydrazine structures that could induce more facile cycloreversions with less reliance on substrate ring strain.
Scheme 44.

Distortion-Accelerated and Strain-Release-Promoted [3+2] and retro-[3+2] in Organocatalytic Carbonyl-Olefin Metathesis by Lambert and Houk.
In 2020, Lambert and coworkers expanded the concept of hydrazine-catalyzed ring-opening carbonyl-olefin metathesis reactions of aldehydes to include norbornenes (Scheme 45), using a combined approach of catalyst design and computational analysis.90 The major challenge confronting a shift to a significantly less-strained substrate like norbornene is that the [3+2]-cycloreversion becomes both endergonic and the rate-determining step of the catalytic cycle. In order to identify a hydrazine that would enable a viable catalytic process with such substrates, a virtual screen of catalysts was conducted. Each step of the catalytic cycle was calculated in order to avoid misleading winners (i.e. structures that minimized cycloreversion but at the expense of an energetically inaccessible other step), and indeed a number of important potential pitfalls were identified. From this expansive study, a second-generation [2.2.2]-bicyclic hydrazine catalyst was identified that promised reasonably accessible cycloaddition and cycloreversion energies with norbornene.
Scheme 45.
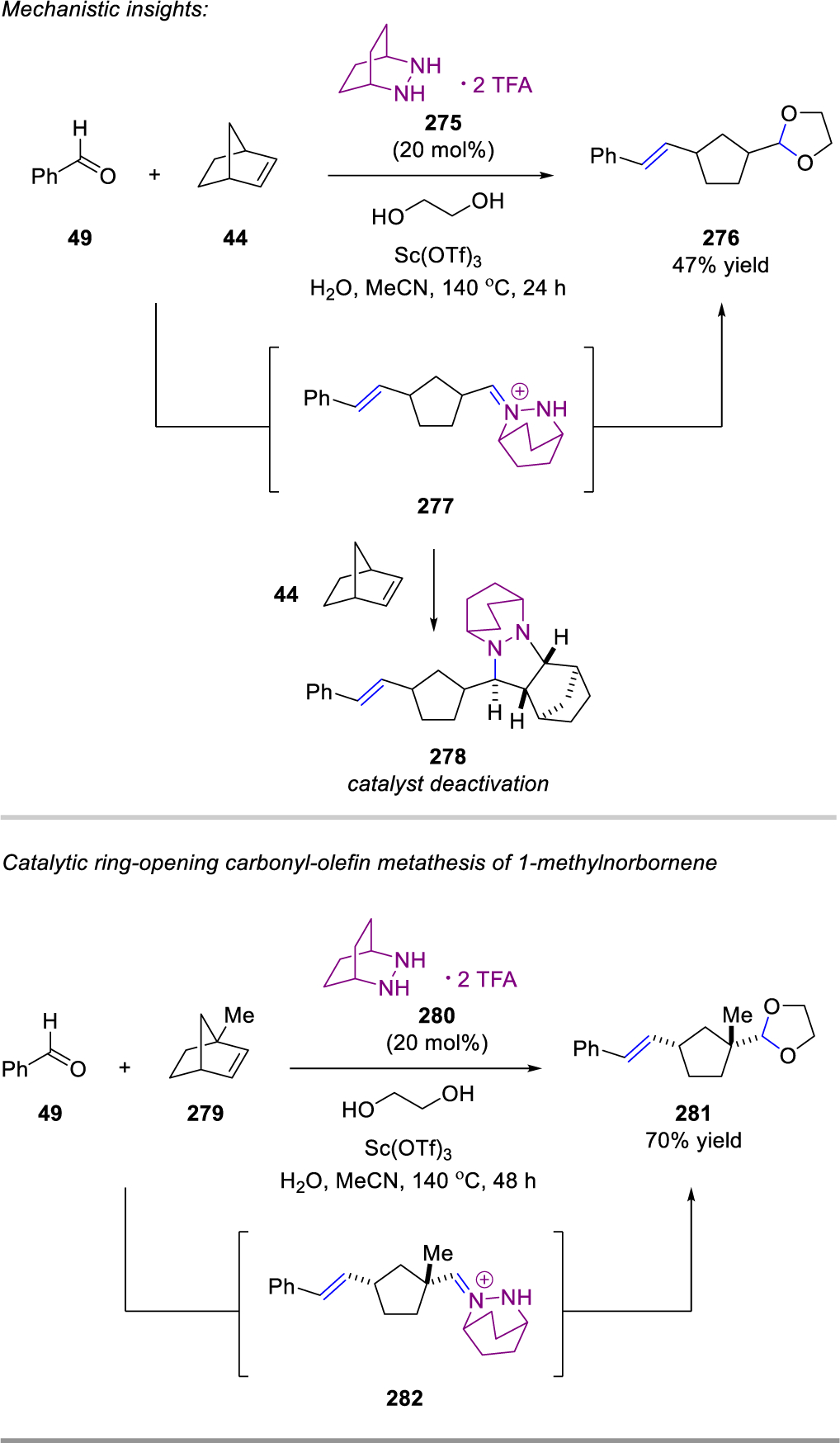
Hydrazine-Catalyzed Ring-Opening Carbonyl-Olefin Metathesis of Norbornenes by Lambert.
Both of the cycloaddition and cycloreversion steps between benzaldehyde (49) and norbornene (44) with the [2.2.2]-bicyclic hydrazine 275 (Scheme 45) could be investigated separately because of the isolable nature of the intermediate cycloadducts. These studies confirmed the computational predictions and suggested a catalytic process should be attainable. Initial results proved encouraging as the use of 20 mol% 275 resulted in the formation of acetal 276 as the ring-opening metathesis product in 47% yield. However, the formation of cycloadduct 278, resulting from cycloaddition of hydrazonium intermediate 277 with a second equivalent of norbornene 44, was also observed. As 278 was resistant to cycloreversion under the reaction conditions, its formation resulted in full catalyst deactivation. Based on these results, it was postulated that hydrolysis of 277 is a key step in the turn-over of the hydrazine catalyst 275. In an attempt to overcome this limitation, methylnorbornene 279 was utilized to destabilize hydrazonium 282 by the introduction of steric strain and hopefully facilitating hydrolysis compared to undesired cycloaddition of a second equivalent of norbornene 279. In fact, the reaction between 49 and 279 provided 74% yield of 281, demonstrating the feasibility of ring-opening carbonyl-olefin metathesis of norbornenes. The authors conclude that competitive cycloaddition of hydrazonium 282 is still observed even with methylnorbornene 279, and thus future efforts should focus on the development of catalysts that promote a more facile hydrolysis step.
5.2. Ring-Closing Carbonyl-Olefin Metathesis
In 2019, Lambert and coworkers extended hydrazine-catalyzed carbonyl-olefin metathesis to the arena of ring-closing metathesis reactions (Scheme 46). Similar to the reaction design for ring-opening metathesis, these transformations rely on the initial condensation between the carbonyl moiety in 283 and the hydrazine catalyst 280 to form a hydrazonium intermediate. This reactive intermediate can then undergo a cycloaddition with the pendant alkene to form the pyrazolidine cycloadduct, which upon retro-[3+2]-cycloaddition and hydrolysis, gives rise to the desired metathesis product 284 and a carbonyl byproduct 285.
Scheme 46.
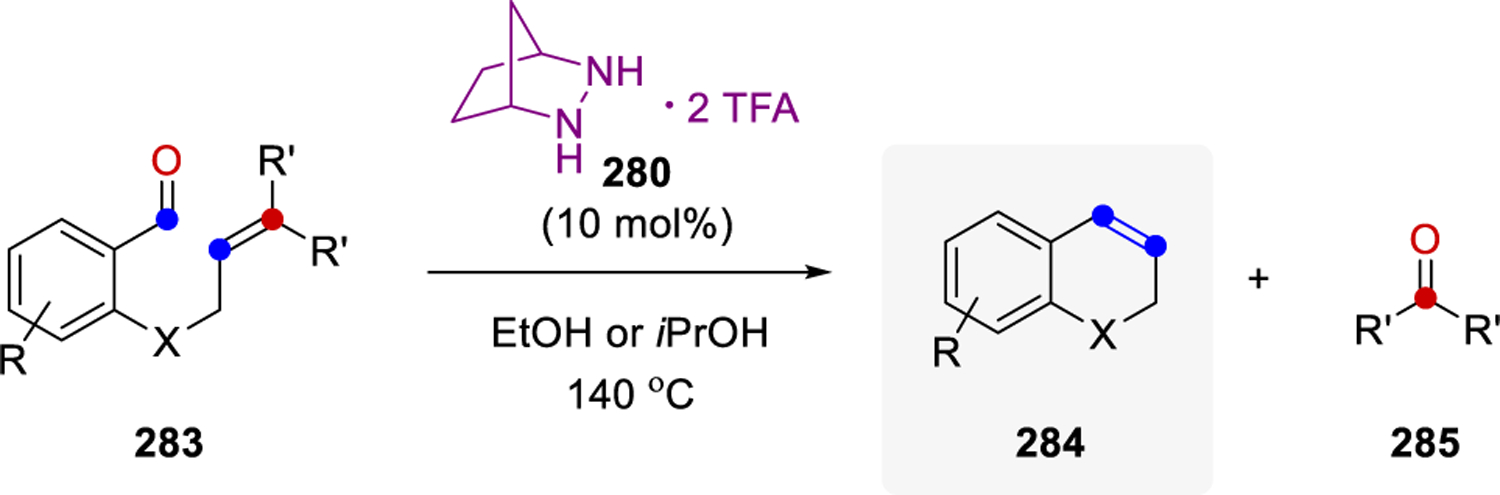
Catalytic Ring-Closing Carbonyl-Olefin Metathesis via 1,3-Dipolar Cycloadditions.
Hydrazine-catalyzed carbonyl-olefin metathesis ring-closing reactions were first established by Lambert and coworkers for salicylaldehyde allylic ethers 286 resulting in the formation of 2H-chromenes 287 (Scheme 47).91 The method utilizes the [2.2.1]-bicyclic hydrazine 280 as the catalyst and showcases a new approach to afford electronically and functionally differentiated 2H-chromenes (287a-287f) in up to 90% yield. Interestingly, the corresponding ketone substrates also prove reactive under the optimal reaction conditions; however, the low yields observed with these substrates point to complications in the condensation and/or cycloaddition steps due to steric strain. Lambert and co-workers were also able to provide additional mechanistic support for the hypothesis of sequential [3+2]- and retro-[3+2]-cycloadditions88–90 with the crystallization of cycloadduct 289. The configuration of 289 was confirmed by X-ray crystallographic analysis. The syn-pentane interaction between the gem-diethyl group was speculated to exert steric pressure on pyrazolidine cycloadduct, potentially accelerating the rate of cycloreversion and suppressing deallylative side reaction. Subsequent exposure of 289 to the optimized reaction conditions resulted in the formation of the desired carbonyl-olefin metathesis product 278a in 73% yield. As opposed to ring-opening metathesis (vide supra), the instability of the ketone hydrazonium byproduct allowed rapid hydrolysis, enabling catalyst turnover and minimizing deactivation.
Scheme 47.

Synthesis of 2H-Chromenes via Hydrazine-Catalyzed Carbonyl–Olefin Metathesis by Lambert
In 2020 Lambert and coworkers further expanded the application of hydrazine-catalyzed carbonyl-olefin ring-closing metathesis reactions to the synthesis of 1,2-dihydroquinolines.92 While the electron donating nitrogen was found to retard the rate of cycloaddition in N-allyl-2-aminobenzaldehyde compared to the salicylaldehyde counterpart, it facilitated a more rapid rate-determining cycloreversion. Such rate enhancement permitted the replacement of the diethylallyl group for a less hindered (and commercially available) prenyl group, while still inhibiting any deallylation side reaction. N-prenylated aminobenzaldehydes 290 underwent the desired transformation to result in a variety of 1,2-dihydroquinolines 291 bearing distinct substitution in up to 93% yield (Scheme 48). Notably, the corresponding N-Boc protected substrates underwent facile in situ deprotection to give rise to quinolones in 61% yield.
Scheme 48.

Synthesis of 1,2-Dihydroquinolines via Hydrazine-Catalyzed Carbonyl–Olefin Metathesis by Lambert
The use of strained hydrazines as catalysts has served as a powerful reaction design to develop catalytic carbonyl-olefin metathesis reactions. The unique [3+2]-cycloaddition strategy relying on the formation of cyclic hydrazonium intermediates enables ring-opening and ring-closing carbonyl-olefin metathesis reactions that allow for the facile synthesis of acyclic unsaturated aldehydes, chromenes, and dihydroquinolines as highly-desired heterocyclic scaffolds. Mechanistic investigations prompted strategic catalyst design to expand the scope of hydrazine-catalyzed carbonyl-olefin metathesis reaction. While the products obtained following ring-closing carbonyl-olefin metathesis protocols are similarly accessible in Lewis acid-catalyzed approaches, the unsaturated aldehydes resulting upon ring-opening carbonyl-olefin metathesis reactions are not, which renders this a particularly powerful and complementary transformation.
6. LEWIS ACID-CATALYZED CARBONYL-OLEFIN METATHESIS REACTIONS
In the early 1970s, the first reports appeared of carbonyl-olefin metathesis reactions between carbonyls and alkenes relying on equimolar amounts of Lewis acids. These transformations also proceed via initial [2+2]-cycloadditions between the alkene 1 and carbonyl 6 moieties to form intermediate oxetanes 8 (Figure 2A). However in comparison to previous stepwise protocols relying on Paternò-Büchi reactions and thus photochemical irradiation, Lewis acid-mediated approaches undergo in situ oxetane formation and fragmentation to provide the desired carbonyl-olefin metathesis products 4 and carbonyl byproducts 7 (Figure 2A). These transformations allow for broader applications and additional substrate complexity while proceeding under overall milder reaction conditions compared to originally reported carbonyl-olefin metathesis conditions based on oxetane pyrolysis.26,27 The Lewis acid takes on a dual role in these reactions to first activate the carbonyl substrate for a [2+2]-cycloaddition and subsequently the oxetane intermediate formed for a [2+2]-cycloreversion. Importantly, this reaction design principle relies on coordinative interactions between catalyst and substrate to forego the formation of metal-oxygen bonds and thus the formation of the corresponding oxidized metal-oxo species compared to metal alkylidene-mediated carbonyl-olefin metathesis approaches. Consequently, this design principle holds great potential as a catalytic strategy for carbonyl-olefin metathesis and has led to important advances in catalytic ring-closing, ring-opening, transannular and cross metathesis of alkenes and carbonyls (Scheme 49). This chapter provides an overview of carbonyl-olefin metathesis reactions initially mediated by Lewis acids, followed by a detailed discussion of currently available Lewis acid-catalyzed variants for each class of carbonyl-olefin metathesis reactions.
Scheme 49.
Lewis Acid-Catalyzed Carbonyl-Olefin Metathesis
6.1. Lewis acid-Mediated Carbonyl-Olefin Metathesis
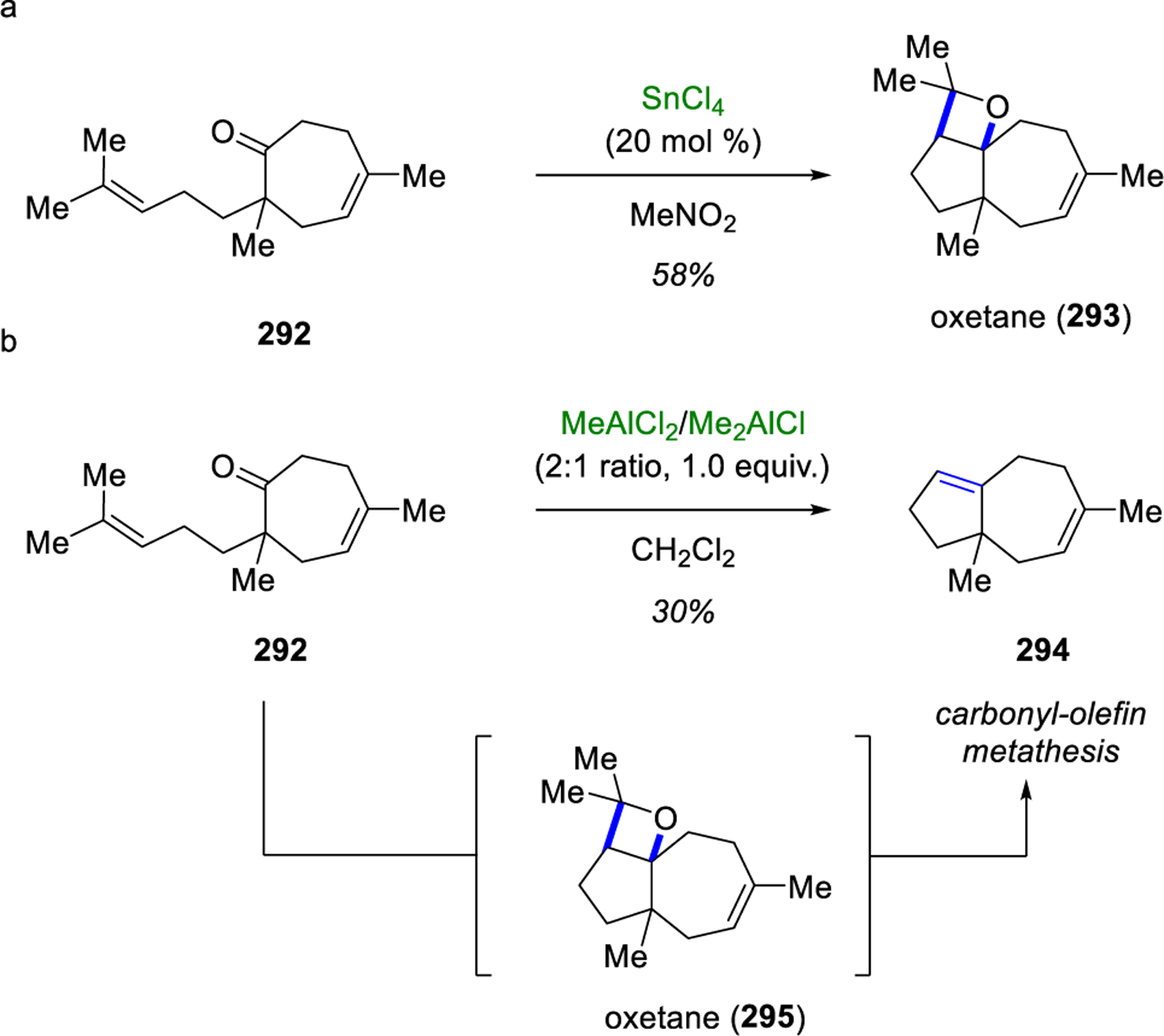
In their studies towards carbon sesquiterpenes, Demole, Enggist and Borer were the first to realize the potential of a one-pot procedure for oxetanes formation relying on Lewis acids. Specifically, cis-oxetane 293 was formed and isolated in 58% yield upon subjection of ketone 292 to 20 mol% SnCl4 via an intramolecular [2+2]-cycloaddition (Scheme 50a).93 Following this report, in 1984 Snider employed the use of the same substrate 292 in his studies of intramolecular ene reactions.94 The treatment of cycloheptanone 292 with stoichiometric amounts of a mixture of MeAlCl2/Me2AlCl in a 2:1 ratio provided the formation of metathesis product 294 in 30% yield (Scheme 50b). Snider proposed that the metathesis reaction proceeds through a stepwise cycloaddition to form oxetane 295 in situ followed by a retro-cycloaddition to provide the metathesis product 294 upon the loss of acetone. Interestingly, the reaction does not occur in the presence of Me1.5AlCl1.5, but a complex mixture is recovered when 292 is treated with stoichiometric amounts of MeAlCl2 indicating the significance of the strength of the Lewis acid in facilitating the carbonyl-olefin metathesis reaction.
Scheme 50.
Lewis acid-mediated metathesis reactions by Demole and Snider.
In 1994, the cross carbonyl-olefin metathesis reaction of benzaldehyde 49 and isobutylene 296 or methyl-2-butene 298 was reported by Bickelhaupt, van Schaik, and Vijn95 to provide yields of 15% and 30%, respectively, of metathesis products 297 and 299 (Scheme 51). A heterogeneous Lewis acid catalyst, EPZ-10,96 consisting of clay-supported ZnCl2 (12% ZnCl2 content) was used to promote metathesis in this reaction. Interestingly, higher temperatures and reactions times increased conversions but with accompanying byproduct formation. A stepwise mechanism is proposed (Scheme 51) that begins with Lewis acid activation of carbonyl 49, followed by nucleophilic addition of olefin 298 to form carbocation 300. An intramolecular addition results in the formation of oxetane 301 which, upon subsequent Lewis acid-assisted fragmentation, yields the carbonyl-olefin metathesis product 299. The reaction is reported to be limited to carbonyl compounds that lack α-hydrogen substituents due to resulting competing aldol condensation reactions with the acetone byproduct of the carbonyl-olefin metathesis reaction.
Scheme 51.

Solid Promoted Lewis Acid-Promoted Carbonyl-Olefin Metathesis by Bickelhaupt and Coworkers
A more complex example of carbonyl-olefin metathesis was reported by Khripach and coworkers in 2006 during their studies towards the synthesis of steroid frameworks.97 Specifically, carbonyl-olefin metathesis product 306 was produced in 60% yield following attempts by Khripach to protect the carbonyl moiety in seco-steroid 304 as a dithioketal upon treatment with excess BF3·OEt2 (Scheme 52). Interestingly, the corresponding Z-olefin isomer 307 failed to undergo the carbonyl-olefin metathesis reaction under identical reaction conditions. The suggested mechanistic hypothesis for this transformation includes an intramolecular, Lewis acid-assisted [2+2]-cycloaddition and cycloreversion of the resulting intermediate oxetane 305.
Scheme 52.

Lewis Acid-Promoted Transannular Carbonyl-Olefin Metathesis by Khripach and Coworkers
In their work towards marine natural product pestalone, Schmalz and coworkers attempted to deprotonate the ether subunits in ortho-prenylated benzophenone and instead detected the formation of an indene-derived metathesis product in 20% yield.98 Subsequent optimization efforts provided 87% yield for the resulting carbonyl-olefin metathesis products with the optimal conditions requiring 1.5 equivalents of BF3·OEt2 in DCM at −40 °C after one hour (Scheme 53). Further application of this protocol was investigated for a series of acetophenone derivatives. Specifically, substrates bearing prenyl (310), geranyl (312) and homoprenyl substituents (313) in the ortho position provided 38–87% yield of the desired carbonyl-olefin metathesis products containing 5- and 6-membered cycloalkenes (311 and 314, Scheme 53). The authors propose a stepwise mechanism for this transformation that relies on initial exo-trig cyclization of 315 following Lewis acid activation of the carbonyl moiety (Scheme 53). Tertiary carbocation 316 is then formed and isomerizes to the more stable benzylic carbocation 317 via an intermediate oxetane. Lastly, fragmentation of carbocation 318 results in the formation of indene 311 as the desired carbonyl-olefin metathesis product with acetone as the byproduct.
Scheme 53.

BF3·OEt2 Induced Metathesis Cyclization of Pestalone Derivatives by Schmalz and Coworkers
6.2. Catalytic Carbonyl-Olefin Ring-Closing Metathesis of Aryl Ketones
Despite the fact that acid-mediated carbonyl-olefin metathesis reactions have been reported in the literature over the last 40 years, acid-catalyzed carbonyl-olefin metathesis reactions have only recently been realized. Lewis acids have now been employed allowing for a catalytic pathway as they activate the carbonyl moiety making it more electrophilic for the nucleophilic attack of the olefin moiety. The coordinating nature of Lewis acids allows for catalyst turnover to occur whereas loss of H+ of Brønsted acids or metal-oxo species in metal alkylidene promoted metathesis reactions, did not allow for this turnover. This section outlines the application of Lewis acids26,27,99–102 towards catalytic carbonyl-olefin ring-closing metathesis reactions relying on aryl ketone substrates.
6.2.1. Formation of 5-Membered Rings
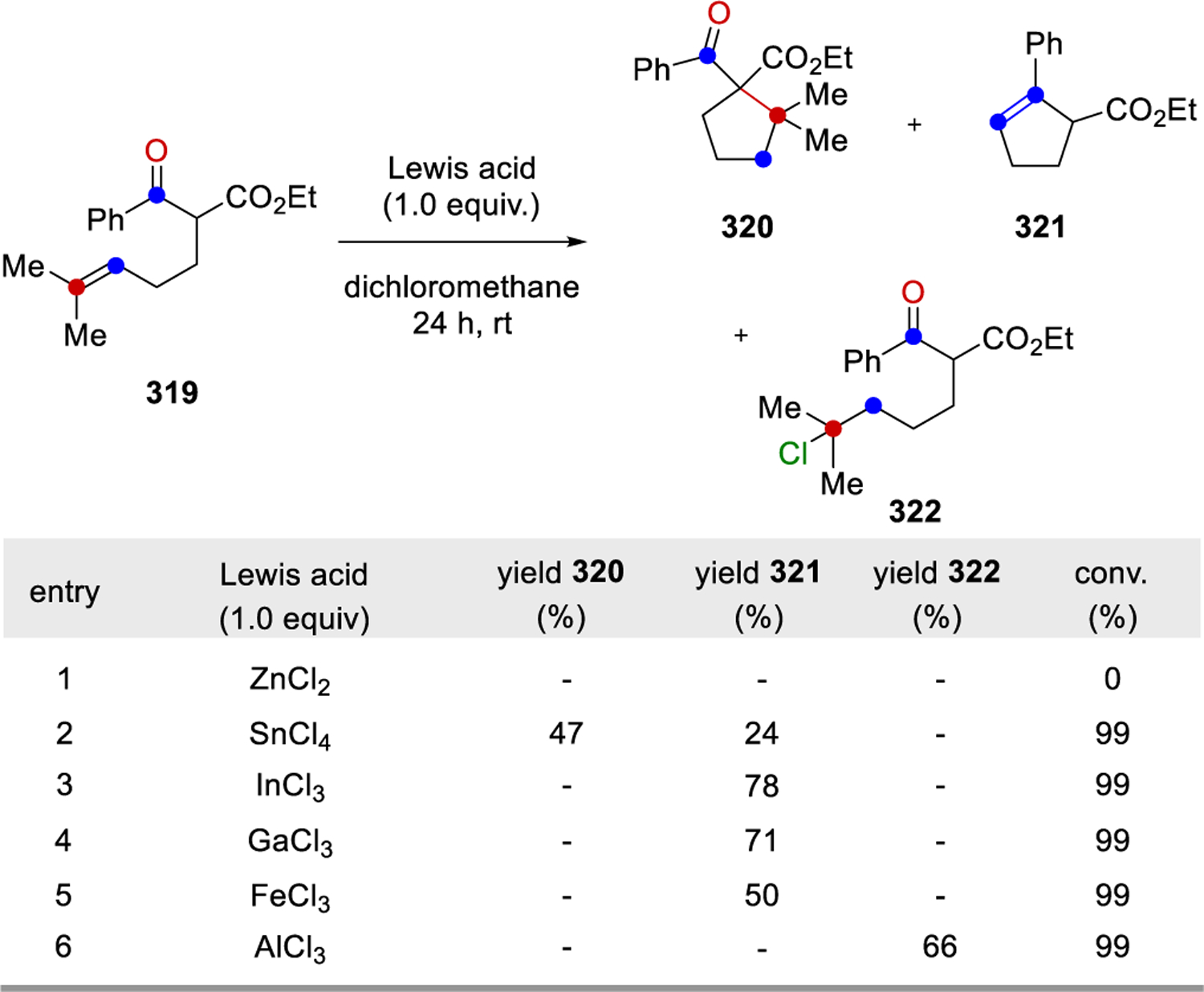
In 2016, Schindler and co-workers reported a carbonyl-olefin ring-closing metathesis reaction relying on FeCl3 as Lewis acid catalyst, which proved uniquely effective for the synthesis of 5-membered rings.99–102,103,104 Initial studies relied on the investigation of β-ketoester 319 with a variety of distinct Lewis acid in stoichiometric reactions (Scheme 54). While weak Lewis acids, such as ZnCl2, resulted in no reactivity, strong Lewis acids including AlCl3 formed the hydrochlorination product 322 exclusively in 66%. Importantly, several Lewis acids, such as SnCl4, InCl3, GaCl3, and FeCl3 proved capable in promoting the desired carbonyl-olefin metathesis reaction while FeCl3 proved superior and resulted in quantitative formation of cyclopentene 321.
Scheme 54.
Initial Studies of Carbonyl-Olefin Ring-Closing Metathesis by Schindler and Coworkers Show Several Lewis Acids Capable of Promoting the Desired Transformation
Reaction conditions relying on catalytic quantities of FeCl3 (5 mol%) in dichloroethane at room temperature were ultimately identified as optimal (Scheme 55). While prenylated alkenes (325) proved superior and formed the desired carbonyl-olefin ring-closing metathesis products quantitatively, styrene derivatives (326a, 327) and their electron-rich (326b) and electron-poor analogs (326c-d) formed the metathesis products in 49 to 70% yield. Importantly, (Z)-styrene (328) was identified as a viable substrate in FeCl3-catalyzed carbonyl-olefin ring-closing metathesis reactions albeit resulting in lower yields than the corresponding (E)-analog 326a. The initial report by the Schindler laboratory included 38 examples of 5-membered rings systems formed in up to 97% yield, while a limited number of successful 6-membered ring formations were shown to proceed in up to 71% yield. Notably, several functional groups, including esters, amides, ethers, lactones, halogens, and sulfones were tolerated well under the optimal reaction conditions resulting in a variety of structurally distinct cyclic products.
Scheme 55.

Scope of the FeCl3-Catalyzed Carbonyl-Olefin Ring-Closing Metathesis Reaction
This initial report suggested two possible mechanistic scenarios for catalytic carbonyl-olefin ring-closing metathesis reactions resulting in the formation of cyclopentenes. Similar to earlier reports relying on stoichiometric quantities of Lewis acids by Bickelhaupt95 and Schmalz,98 a reaction pathway based on intermediate carbocations was considered (Scheme 56). Specifically, upon coordination of the Lewis acid catalyst to β-ketoester 319, initial carbon-carbon bond formation would lead to carbocation 329, which upon intramolecular addition results in the formation of oxetane 331. Subsequent Lewis acid-mediated fragmentation of the carbon-oxygen bond in 331 results in the benzylic carbocation 332 that undergoes carbon-carbon bond rupture to form the carbonyl-olefin metathesis product 321 and acetone 27 as byproduct. However, carbocation trapping experiments conducted by the Schindler group relying on the addition of intramolecular and intermolecular nucleophiles did not give rise to any compounds indicative of the presence of intermediate carbocations and instead gave rise to the desired carbonyl-olefin metathesis products as the exclusive compounds formed. Based on these results, a second mechanistic hypothesis was suggested that similarly relied on the initial Lewis acid-catalyzed activation of β-ketoester 316 albeit in an asynchronous, concerted [2+2]-cycloaddition that does not rely on carbocation intermediates to form oxetane 331. An ensuing asynchronous, concerted retro [2+2]-cycloaddition gives rise to the desired carbonyl-olefin metathesis products 321 and 27. Computational investigations based on DFT studies supported the asynchronous, concerted pathway for oxetane formation and fragmentation via [2+2]-cycloadditions that does not rely on carbocation intermediates.
Scheme 56.
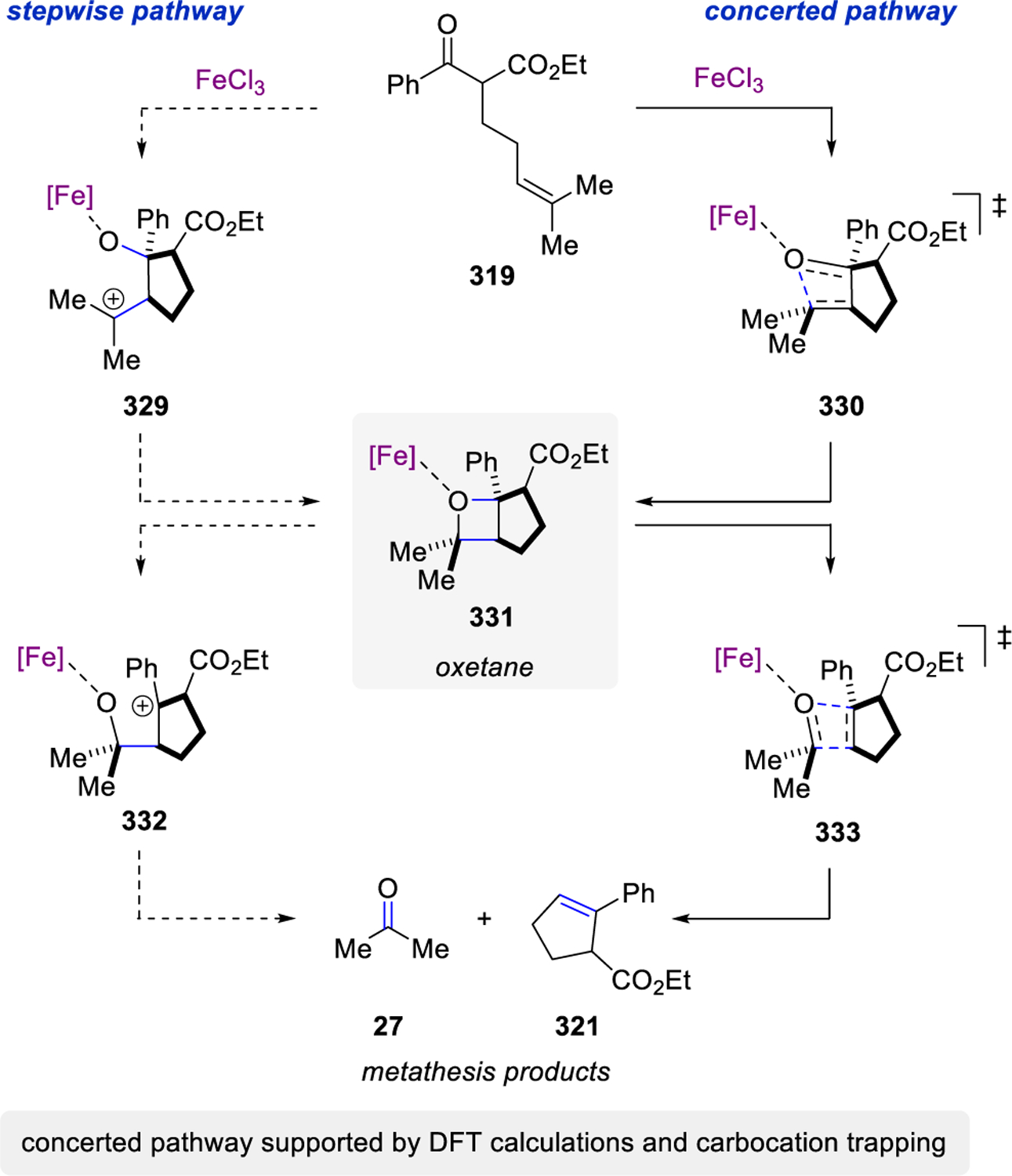
Two Mechanistic Alternatives for FeCl3-Catalyzed Carbonyl-Olefin Ring-Closing Metathesis
Notably, a scalable approach for this FeCl3-catalyzed carbonyl-olefin metathesis was reported by the Schindler group in 2018 as an Organic Synthesis procedure that resulted in 93% yield of the desired metathesis product 337 on a 15 g scale (Scheme 57).105 The revised reaction conditions were modified to be conducted at higher concentration and purification of the product was conducted by distillation to avoid column chromatography.
Scheme 57.

Organic Synthesis Procedure Developed for FeCl3-Catalyzed Carbonyl-Olefin Ring-Closing Metathesis
In 2016, Li and coworkers published an FeCl3-catalyzed carbonyl-olefin ring-closing metathesis reaction that proved efficient for the synthesis of 5-membered rings and a selection of 6-membered ring products in up to 84% yield.106 The initial report relied on two sets of reaction conditions, specifically carbocyclic structures were formed with 10 mol% FeCl3 as the Lewis acid catalyst in dichloroethane at ambient temperatures, while nitrogen-containing 5-membered rings proceeded with catalyst loadings of 20 mol% and the addition of 5.0 equivalents of allyltrimethylsilane (Scheme 58). Interestingly, the approach reported by Li and coworkers relies on styrene derivatives as alkene components for carbonyl-olefin metathesis with the proposed role of allyltrimethylsilane in the reaction mixture being trapping of the benzaldehyde byproduct. Li and coworkers postulate a stepwise reaction mechanism that relies on the initial activation of the substrate 344 with the Lewis acid catalyst to form Lewis acid-base complex 345. Subsequent carbon-carbon bond formation gives rise to diastereomeric carbocations 346a and 346b, of which 346a results in the formation of oxetane 340 and subsequent fragmentation to give rise to the metathesis products 348 and 49. Silylether 349 was isolated in trace quantities from the reaction mixture, which is postulated to arise upon trapping of carbocation 346b with allyltrimethylsilane.
Scheme 58.
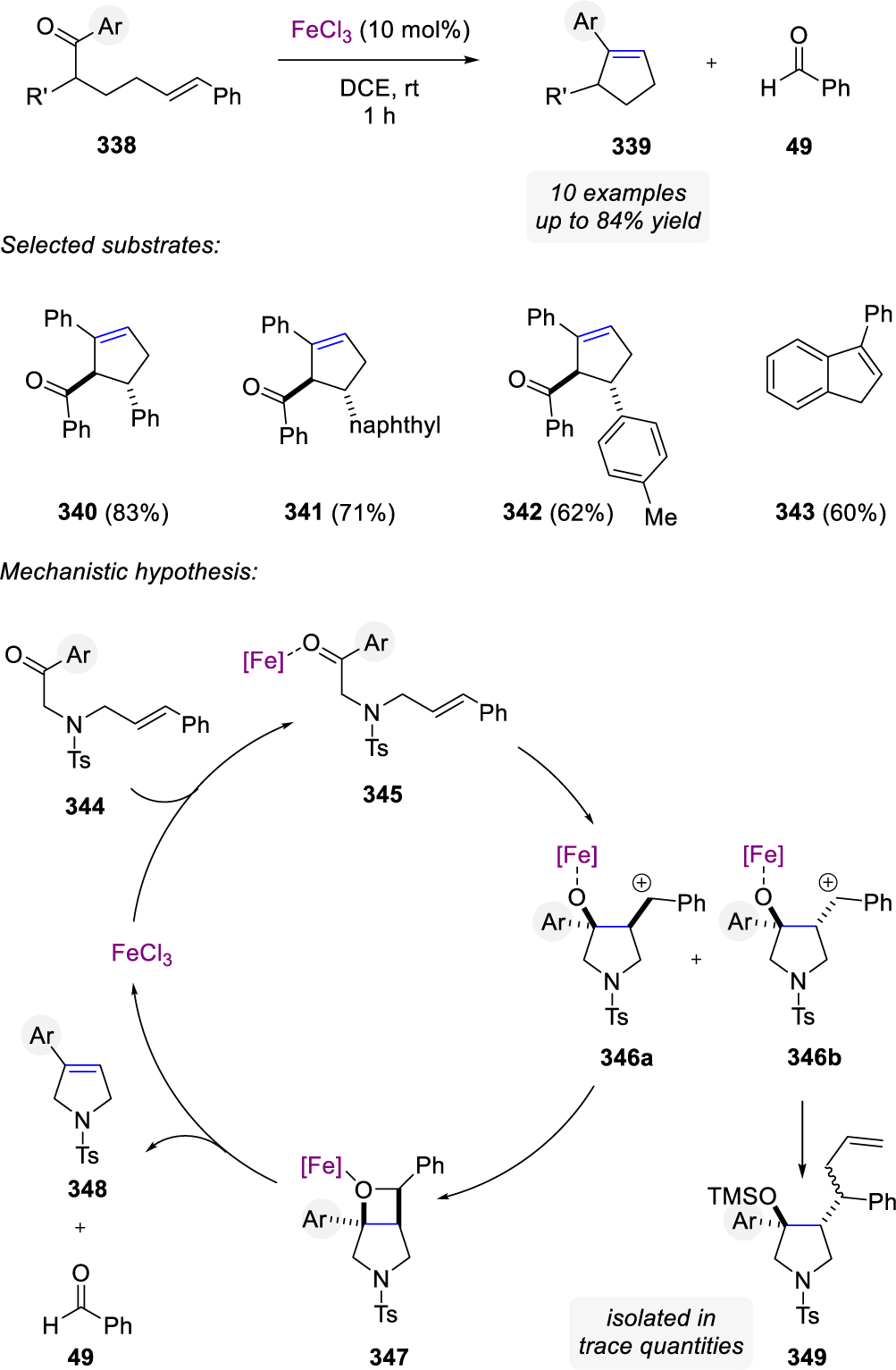
FeCl3-Catalyzed Carbonyl-Olefin Ring-Closing Metathesis by Li and Coworkers
In 2017, Schindler, Devery, Zimmerman, and coworkers conducted detailed mechanistic studies of prenylated alkenes and styrene-derived alkenes as substrates for FeCl3-catalyzed carbonyl-olefin ring-closing metathesis that has led to two distinct proposed catalytic cycles depending on the olefin substitution.107 Results obtained in EPR, kinetic, computational and experimental synthetic studies suggest that the FeCl3 catalyst binds at the carbonyl moiety of the substrate without an electron transfer event for both types of substrates. Additionally, metathesis occurs for a variety of FeCl3 sources regardless of hydration of the metal center while HCl, as a possible hydrolysis product of FeCl3, does not catalyze the transformation. Specifically, prenylated substrates 319 undergo carbonyl-olefin metathesis upon binding of the FeCl3 catalyst to the carbonyl oxygen to form Lewis acid-base complex 350 as the catalyst resting state (Scheme 59). Asynchronous, concerted [2+2]-cycloaddition of 350 via 330 results in the formation of intermediate oxetane 331, which undergoes asynchronous, concerted retro [2+2]-cycloaddition in the product-forming step.
Scheme 59.
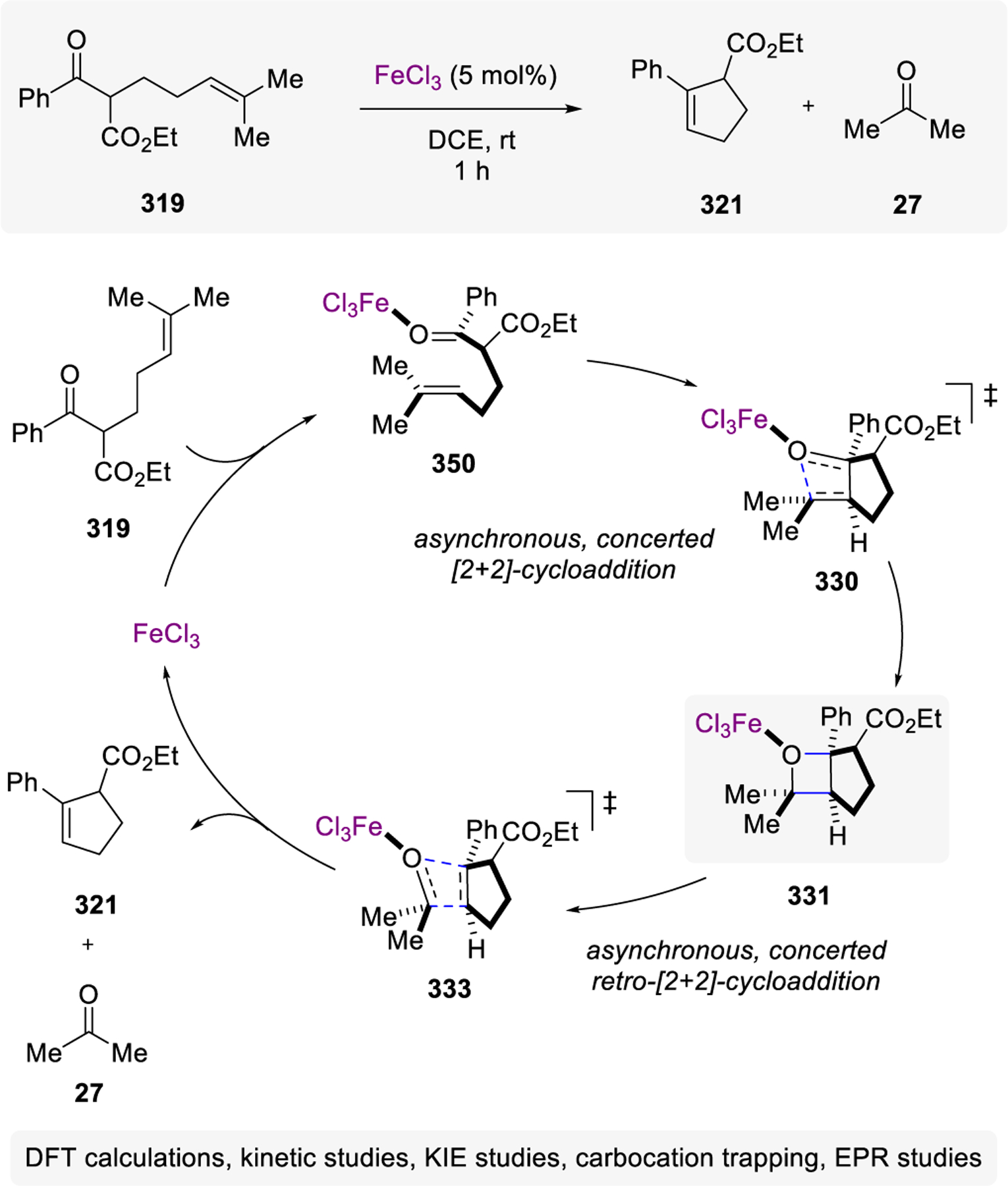
Mechanistic Investigations of Prenylated β-Ketoesters in Fecl3-Catalyzed Carbonyl-Olefin Metathesis by Schindler, Devery, Zimmerman, and Coworkers
In comparison to their prenylated analogs, substrates bearing a styrenyl fragment 351 undergo oxetane fragmentation via a distinct reaction path (Scheme 60). While β-ketoesters 351 undergo activation of the carbonyl oxygen upon binding to the FeCl3 catalyst to form the corresponding Lewis acid-base complex and subsequent asynchronous, concerted [2+2]-cycloaddition to result in oxetane 352, its fragmentation pathway differs. Specifically, heterolysis of oxetane 352 results carbocation 353 as a charged intermediate that subsequently eliminates metathesis product 321 and benzaldehyde 49 as the byproduct.
Scheme 60.
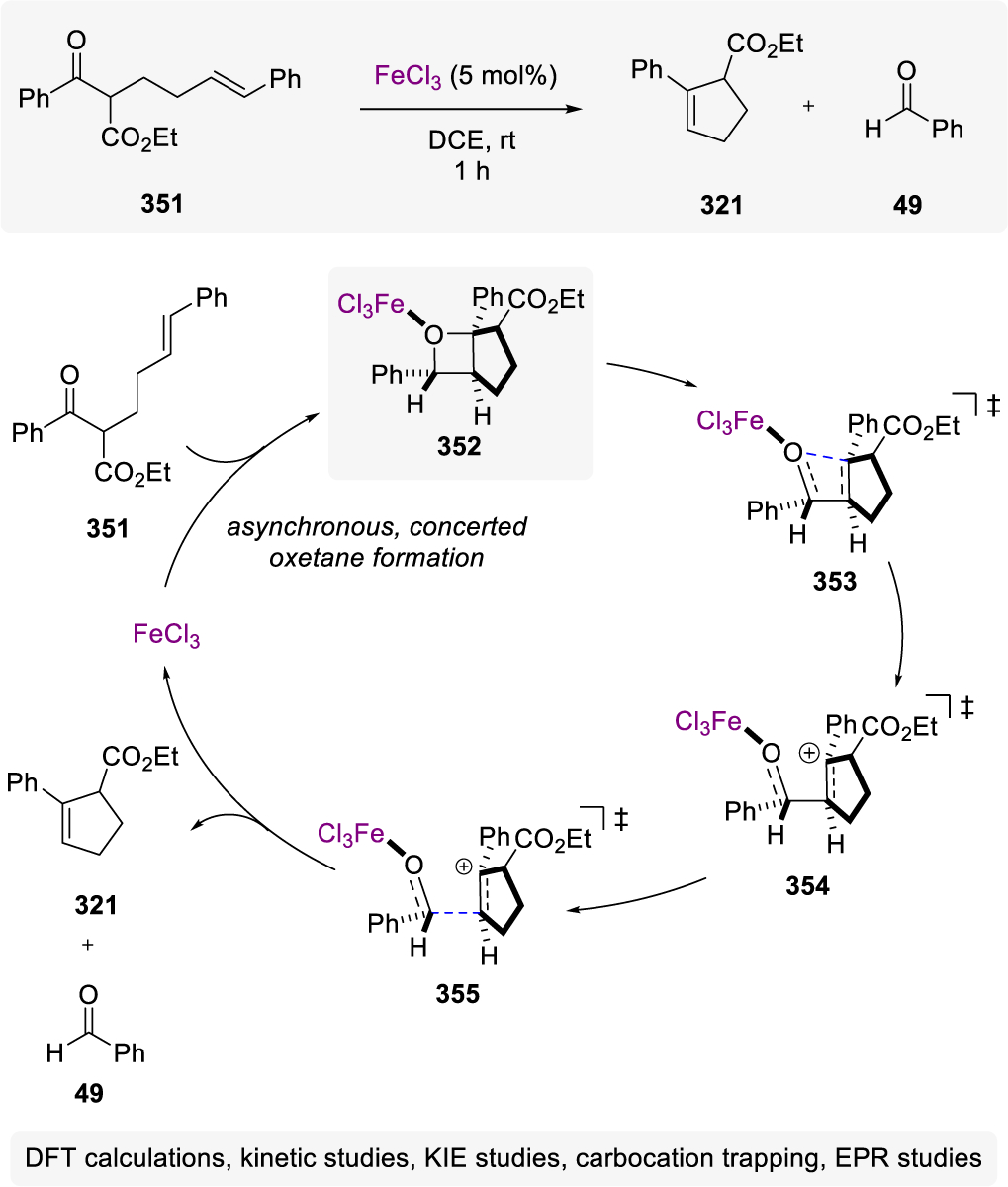
Mechanistic Investigations of Styrenyl-derived β-Ketoesters in FeCl3-Catalyzed Carbonyl-Olefin Metathesis by Schindler, Devery, Zimmerman, and Coworkers
In 2018, the Schindler research group reported an alternative oxetane fragmentation path for prenylated alkenes 356 that interrupts previously established carbonyl-olefin metathesis reactions paths (Scheme 61).108 While aryl ketone 356 undergoes catalytic carbonyl-olefin metathesis relying on FeCl3 as Lewis acid catalyst to give rise to the metathesis product 359 in 71 % yield, conversion of 356 with catalytic amounts of triflic acid results in the exclusive formation of tetrahydrofluorene 361 in 56% yield. Mechanistic studies suggest that both reaction paths proceed via intermediate oxetane 357 although in the Brønsted acid-catalyzed reaction path oxetane fragmentation occurs stepwise to form carbocation 360 that ultimately gives rise to 361 via intramolecular Friedel-Crafts alkylation. These results provide additional support for an asynchronous, concerted oxetane fragmentation of prenylated substrates in carbonyl-olefin metathesis reactions that does not proceed through a carbocation intermediate.
Scheme 61.

Carbonyl-Olefin Metathesis (359) versus Interrupted Carbonyl-Olefin Metathesis Reactions (361).
In 2019, the Devery group investigated the solution structure and catalyst behavior of Lewis acids including FeCl3 and GaCl3 for ring-closing carbonyl-olefin metathesis in kinetic, spectroscopic, colligative, and crystallographic studies.109 While GaCl3 exhibits a classic 1:1 Lewis-acid-Lewis base coordination event, FeCl3 is capable of forming aggregate 363 upon coordination of acetone 27 in solution, which was found to be catalytically active and capable of binding substrate 362 (Scheme 62).
Scheme 62.

Fecl3 Catalyst Behavior and Solution Structure Investigated by Devery and Coworkers
The original report on FeCl3-catalyzed carbonyl-olefin metathesis by Li an coworkers in 2016 also described the formation of 2,5-dihydropyrroles starting from the corresponding styrenyl precursors 367 bearing tosyl-protected secondary amines (Scheme 63).106 The transformation relies on 20 mol% FeCl3 as the Lewis acid catalyst and the addition of 5.0 equivalents allyltrimethylsilane to sequester the benzaldehyde byproduct formed. Prenylated alkenes (369) were identified as viable substrates resulting in 87% yield of the metathesis products together with electron-neutral (370a), -rich (370b), and –poor (370c) styrenes. Notably, the optimal reaction conditions proved efficient for a variety of structurally distinct 2,5-dihydropyrroles resulting in the desired products in up to 91% yield.
Scheme 63.

Formation of 2,5-Dihydropyrroles in Fecl3-Catalyzed Carbonyl-Olefin Metathesis by Li and Coworkers
In 2018, the Schindler group reported a related investigation into the ring-closing carbonyl-olefin metathesis of N-containing substrates to access chiral 3-aryl-2,5-dihydropyrroles relying on commercially available amino acids as chiral pool reagents and FeCl3 as Lewis acid catalyst (Scheme 64).110–111 Initial experimental investigations relying on these substrates had resulted in low yields of the desired products while high loadings of FeCl3 were required. Following their hypothesis that the additional Lewis basic sites present in the amine substrates represent competitive Lewis basic binding sites for the FeCl3 catalyst, experimental and computational investigations were conducted, which ultimately corroborated this hypothesis. The Schindler laboratory postulated that attenuating the electronic properties of the sulfonamide moiety by adding electron-withdrawing substituents to the aromatic ring could disfavor catalyst sequestration and prevent stalling of the metathesis reaction. Evaluation of a variety of electronically distinct sulfonamide-protecting groups based on the hypothesis led to the identification of the 4-trifluoromethylbenzenesulfonyl functionality (FTs in 373, Scheme 64). Interestingly, this electron-deficient sulfonamide renders the protected amine as less Lewis basic and consequently a less competitive binder (374a vs. 374b) for the FeCl3 Lewis acid resulting in yields of up to 99%. Additionally, the enantiomeric excess of the substrates was maintained throughout the course of the transformation resulting in the formation of the desired chiral 2,5-dihydropyrroles in >98% ee. Similar to earlier reports by Li and coworkers, prenyl-derived alkenes 377 proved superior as alkene moieties while styrene derivatives (378a-e) resulted in diminished overall yields. The generality of this approach was demonstrated in 34 examples of electronically and sterically distinct 2,5-dihydropyrroles formed in up to 99% yield and >98% ee. Importantly, the FTs-protecting group was readily cleaved under reductive conditions using SmI2 resulting in the free 2,5-dihydropyrroles while their enantiomeric excess was maintained.
Scheme 64.
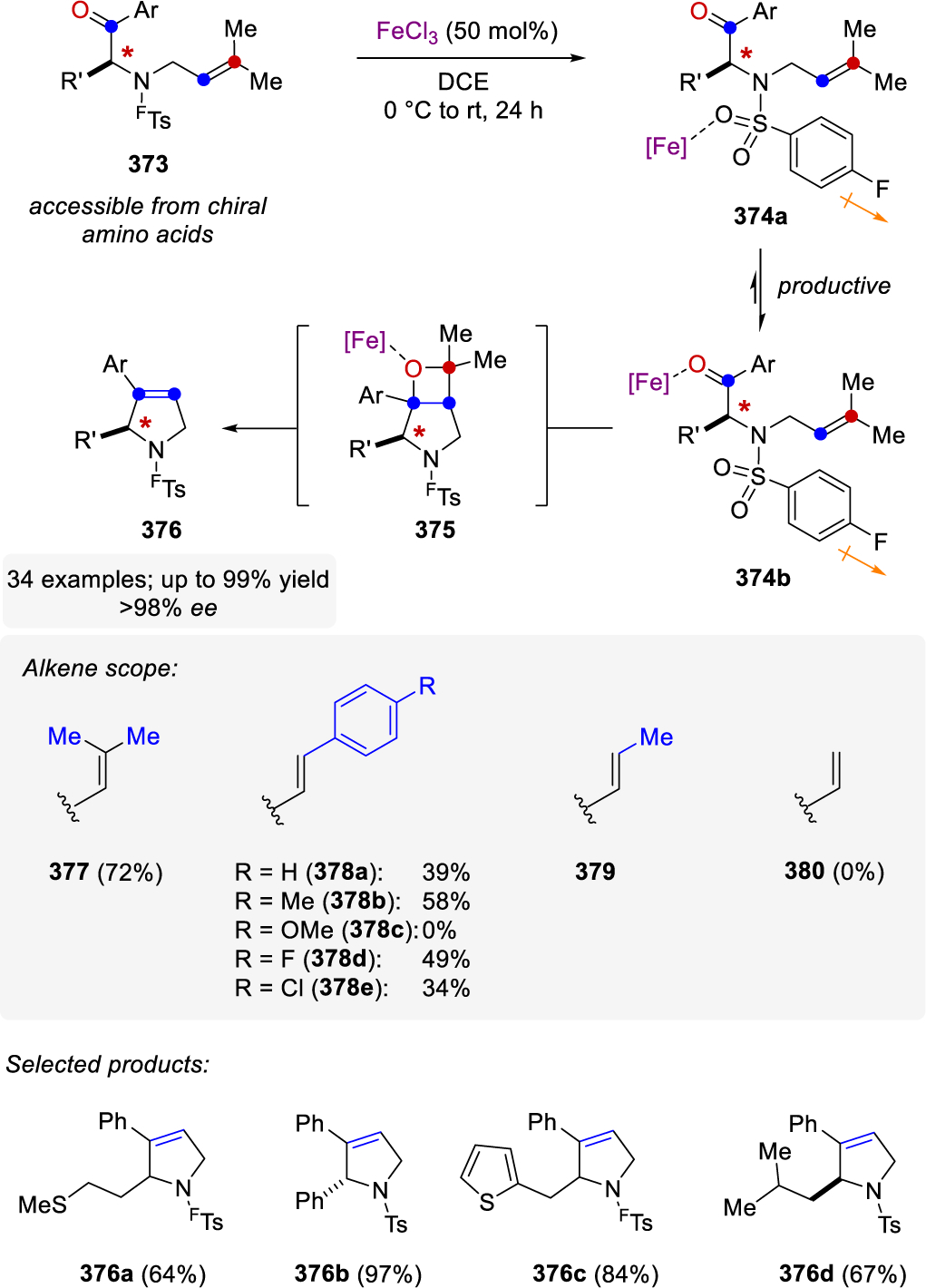
N-Containing Substrates for Carbonyl-Olefin Metathesis by Schindler and Coworkers
In 2018, the Franzén group was able to develop a carbocation-based carbonyl-olefin ring-closing metathesis reaction to access functionalized indenes relying on 4-phenylphenyl-diphenylmethylium tetrafluoroborate (382) as a Lewis acid catalyst (Scheme 65).112 Interestingly, styrene-derived alkenes (386a-c) proved superior as metathesis substrates in this transformation as compared to their prenyl analog (385), which ultimately led to the identification of ortho-methyl styrenes 387 as optimal substrate class. Specifically, Franzén and coworkers identified weakly donating substituents in the ortho-position of the styrene moieties as key structural components to shorten the overall reaction time while reducing competing substrates and product decomposition. The optimized reaction conditions proceeding with 2–5 mol% of 4-phenylphenyl-diphenylmethylium tetrafluoroborate (382) as Lewis acid proved general for 20 examples of indenes 383 bearing steric and electronically distinct substitutions resulting in up to 81% yield of the desired carbonyl-olefin metathesis products.
Scheme 65.

Carbocation-Catalyzed Carbonyl-Olefin Ring-Closing Metathesis by Franzén and Coworkers
In 2018, Nguyen and coworkers showed that tropylium ions are potent Lewis acid catalysts for carbonyl-olefin ring-closing metathesis resulting in the formation of cyclopentene products (Scheme 66).113 Reaction optimization studies conducted by the authors identified substrates incorporating prenyl moieties as alkene components as superior compared to their styrenyl analogs, which resulted in a decrease in yield of the desired carbonyl-olefin metathesis product formed. Optimal conditions rely on 15 mol% of the tropylium catalyst at 90 °C for 6–48 hours neat while the generality of this approach was demonstrated in 18 examples resulting in up to 92% yield (389, Scheme 66).
Scheme 66.

Approaches to Carbonyl-Olefin Ring-Closing Metathesis Relying on Tropylium Cations, Iodine, and AuCl3 as Lewis Acid Catalysts by Nguyen and Lin
In 2019, Nguyen and coworkers built on their prior studies and reported that molecular iodine was similarly capable of catalyzing carbonyl-olefin ring-closing metathesis reactions (Scheme 66).114 Similarly to their earlier report, the optimal reaction conditions proceed neat albeit at ambient temperatures relying on 10 mol% molecular iodine and result in the formation of 24 examples of functionalized cyclopentene products in up to 96% yield. Based on computational investigations conducted of the I2-catalyzed carbonyl-olefin ring-closing metathesis reaction, the authors suggest a reaction pathway that proceeds stepwise via initial carbon-carbon bond formation between the carbonyl and alkene moiety to result in an intermediate carbocation, which subsequently results in an oxetane that then undergoes ensuing stepwise fragmentation via a benzylic carbocation to result in the desired metathesis products.
In 2019, Lin and coworkers reported that catalytic amounts of AuCl3 proved equally efficient as a Lewis acid catalyst for carbonyl-olefin ring-closing metathesis (Scheme 66).115 The optimal reaction conditions reported are based on 5 mol% AuCl3 and proceed in dichloroethane at ambient temperatures for up to 24 hours. Moreover, these conditions prove general for a variety of functionalized cyclopentenes resulting in up to 99% yield of the carbonyl-olefin ring-closing metathesis products. DFT studies performed by the authors support an asynchronous, concerted [2+2]-cycloaddition and retro [2+2]-cycloaddition via intermediate oxetanes while carbocation trapping experiments with MeOH as an exogenous nucleophile did not result in the formation of a new product.
In 2019, Gandon and Bour reported the combination of catalytic carbonyl-olefin ring-closing metathesis and subsequent transfer hydrogenation relying on the Ga-based metal complex 393 and 1,4-cyclohexadiene (1,4-CHD) as a hydrogen donor (Scheme 67).116 Notably, the Ga-complex 393 catalyzes the initial carbonyl-olefin ring-closing metathesis reaction and the ensuing hydrogenation resulting in the formation of functionalized cyclopentane products. The generality of this approach is demonstrated in 24 examples resulting in up to 98% yield. Interestingly, products bearing 1,2-disubstitution are formed in diastereomeric ratios ranging from 4:1 to >20:1 d.r. favoring the cis products. Based on DFT calculations, the authors suggest a reaction pathway that is promoted by a homodimeric Ga-complex formed, which coordinates to the 1,4-cyclohexadiene to activate it for the subsequent reduction of the metathesis products formed.
Scheme 67.
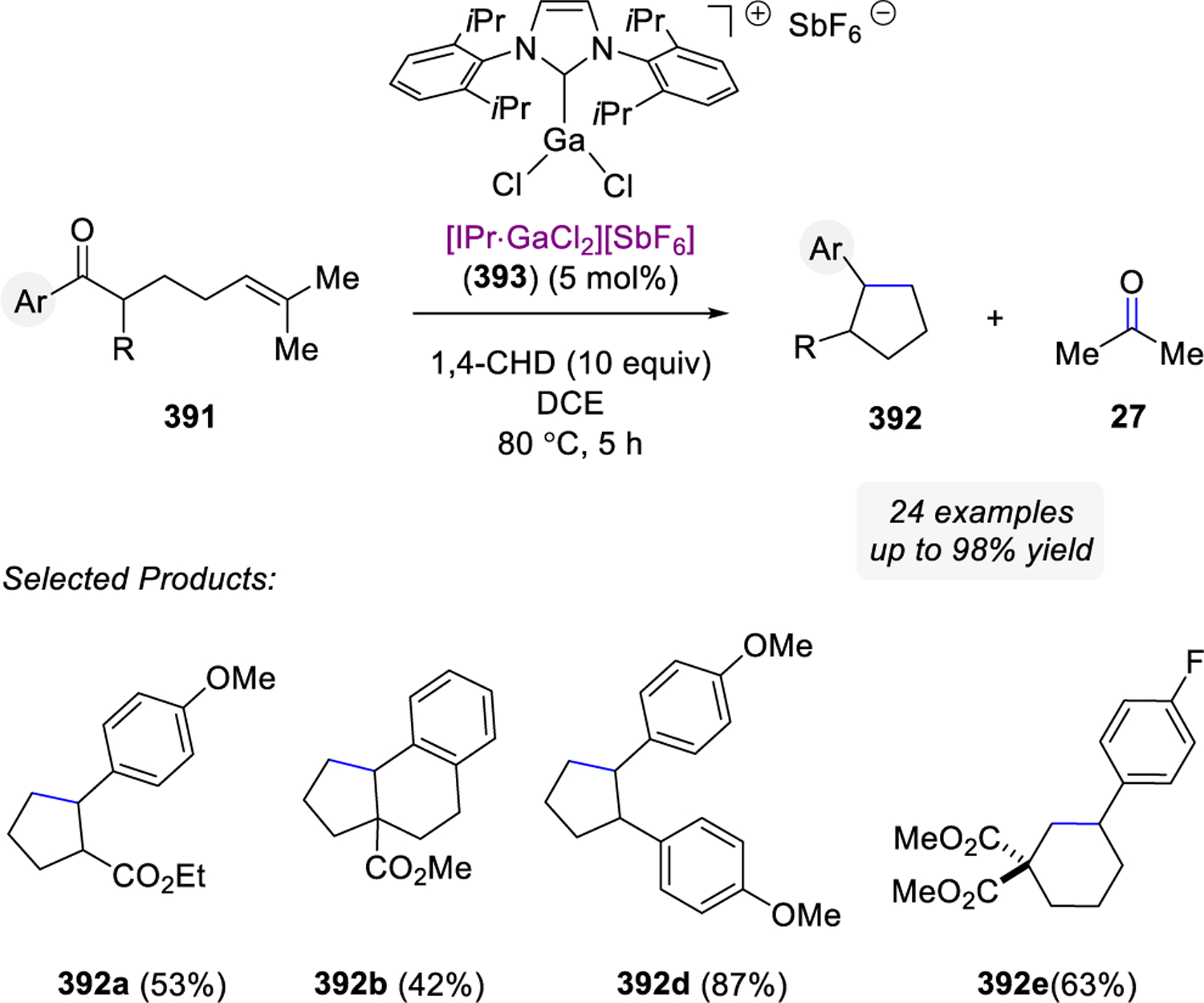
Carbonyl-Olefin Ring-Closing Metathesis and Transfer Hydrogenation by Gandon and Bour
6.2.2. Formation of 6-Membered Rings
In comparison to carbonyl-olefin ring-closing metathesis reactions resulting in 5-membered rings, the analogous transformations forming their higher order homologs in the form of 6- or 7-membered ring products are much less developed. Successful, general reports for Lewis acid-catalyzed carbonyl-olefin ring-closing metathesis reactions currently center on polyaromatic hydrocarbons117 and tetrahydropyridines as products,118 while efficient strategies giving rise to functionalized cyclohexene products have only recently been reported. However, isolated early reports for Lewis acid-catalyzed carbonyl-olefin metathesis reactions for the synthesis of cyclohexenes do exist. For example, studies by Duñach and coworkers in intramolecular carbonyl-ene reactions described the formation of cyclohexene 395 from aryl ketone 394 formed as a byproduct in 36% yield upon conversion with 5 mol% Bi(OTf)3 (Scheme 68).119
Scheme 68.
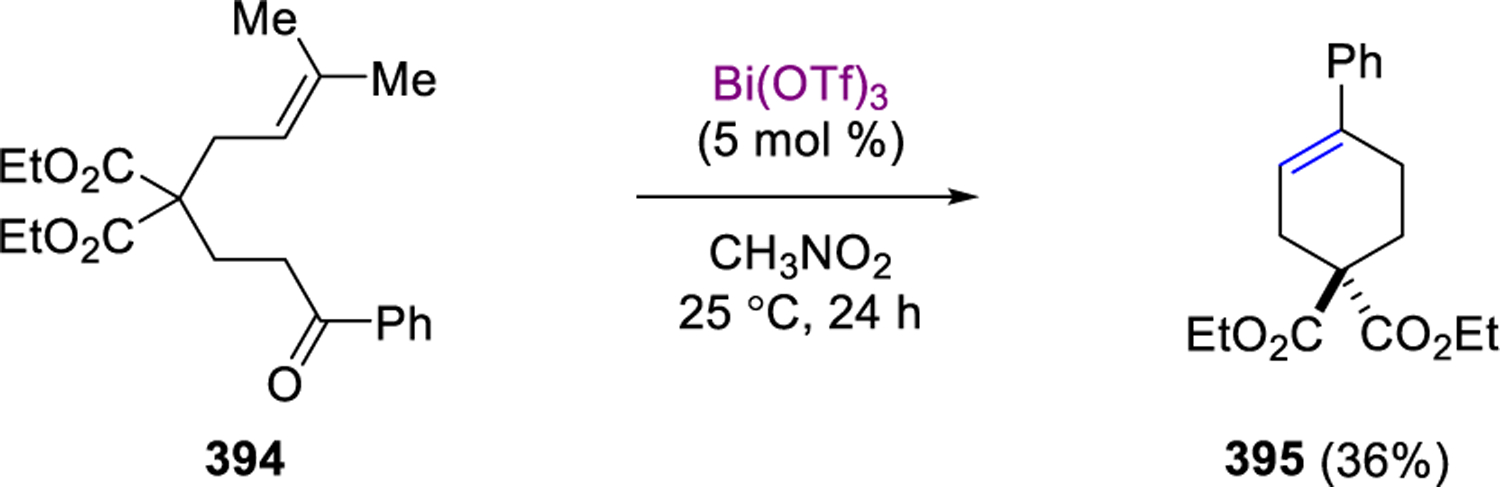
Duñach Observes the Formation of Carbonyl-Olefin Ring-Closing Metathesis Product 395 in Studies of Intramolecular Carbonyl-Ene Reactions
Furthermore, Schindler and coworkers’ early report on FeCl3-catalyzed carbonyl-olefin ring-closing metathesis reactions99 included a limited scope of functionalized cyclohexene products formed (397a-d, Scheme 69). Although these products were formed in up to 70% yield, they remained isolated examples with substrates lacking ortho-substitution on the aryl moiety resulting in significantly diminished yields.
Scheme 69.

Limited Examples for Cyclohexene Products Formed in Catalytic Carbonyl-Olefin Ring-Closing Metathesis by Schindler and Coworkers
Following the development of the ring-closing carbonyl-olefin metathesis reaction of aryl ketones, the Schindler group reported its application for the synthesis of polycyclic aromatic compounds (PACs) in 2017.117 PACs are important structural motifs in natural product synthesis, materials science, and asymmetric catalysis. The reaction conditions originally developed for carbonyl-olefin ring-closing metathesis of cyclopentenes relying on 5 mol% FeCl3 in dichloroethane at ambient temperatures proved efficient in giving rise to a wide variety of electronically and structurally distinct polyaromatic hydrocarbons (Scheme 70). The synthetic generality of this approach was demonstrated in 50 examples proceeding in up to 99% yield. Interestingly, prenylated alkenes (400) resulted in overall lower yields of the metathesis products compared to the corresponding styrenes (401a-e). Subsequent investigations showed that although carbonyl-olefin metathesis is the predominant reaction pathway, prenylated alkenes can undergo competing carbonyl-ene reactions (407) resulting in 21% yield of alkene 408 starting from ketone 404. Subsequent efforts by Schindler and coworkers focused on substrates bearing styrenyl alkenes as these cannot undergo competing carbonyl-ene reactions. Notably, during the course of these optimizations, an oxetane product obtained from a trifluoromethyl ketone substrate was isolated in 45% yield and characterized by X-ray diffraction analysis providing further support for oxetanes as reactive intermediates in catalytic carbonyl-olefin metathesis reactions.
Scheme 70.

Carbonyl-Olefin Metathesis Towards Polycyclic Aromatic Hydrocarbons by Schindler and Coworkers
Additionally, detailed studies focused on the evaluation of the carbonyl moiety were conducted and identified ketones (410), aldehydes (411), as well as ketones bearing sterically constraint substituents as viable substrates (412–413, Scheme 71). Moreover, ketones bearing two aromatic substituents (414 and 415) provided moderate yields of the metathesis products, while enones (416) as well 1,3-diketones (417), and electron deficient ketones (418) resulted in 50%, 72%, and 52% yield, respectively.
Scheme 71.

Investigation of the Carbonyl Scope in Catalytic Carbonyl-Olefin Metathesis Reactions by Schindler and Coworkers
In their 2018 report on carbonyl-olefin ring-closing metathesis reactions relying on tropylium ions as Lewis acid catalysts, the Nguyen group also demonstrated the compatibility of their reaction conditions with the synthesis of polyaromatic systems (Scheme 72).113 Under the reported reaction conditions, prenylated ketone 404 results in the formation of metathesis product 406 in 65% yield together with carbonyl-ene product 408 in 32% yield. Similarly, the 2019 report by Nguyen and coworkers relying on iodine as Lewis acid catalyst included an example resulting in polyaromatic metathesis product 406 in 67% yield from aryl ketone 404 while the carbonyl-ene byproduct 408 was formed in 27% yield (Scheme 72).114
Scheme 72.
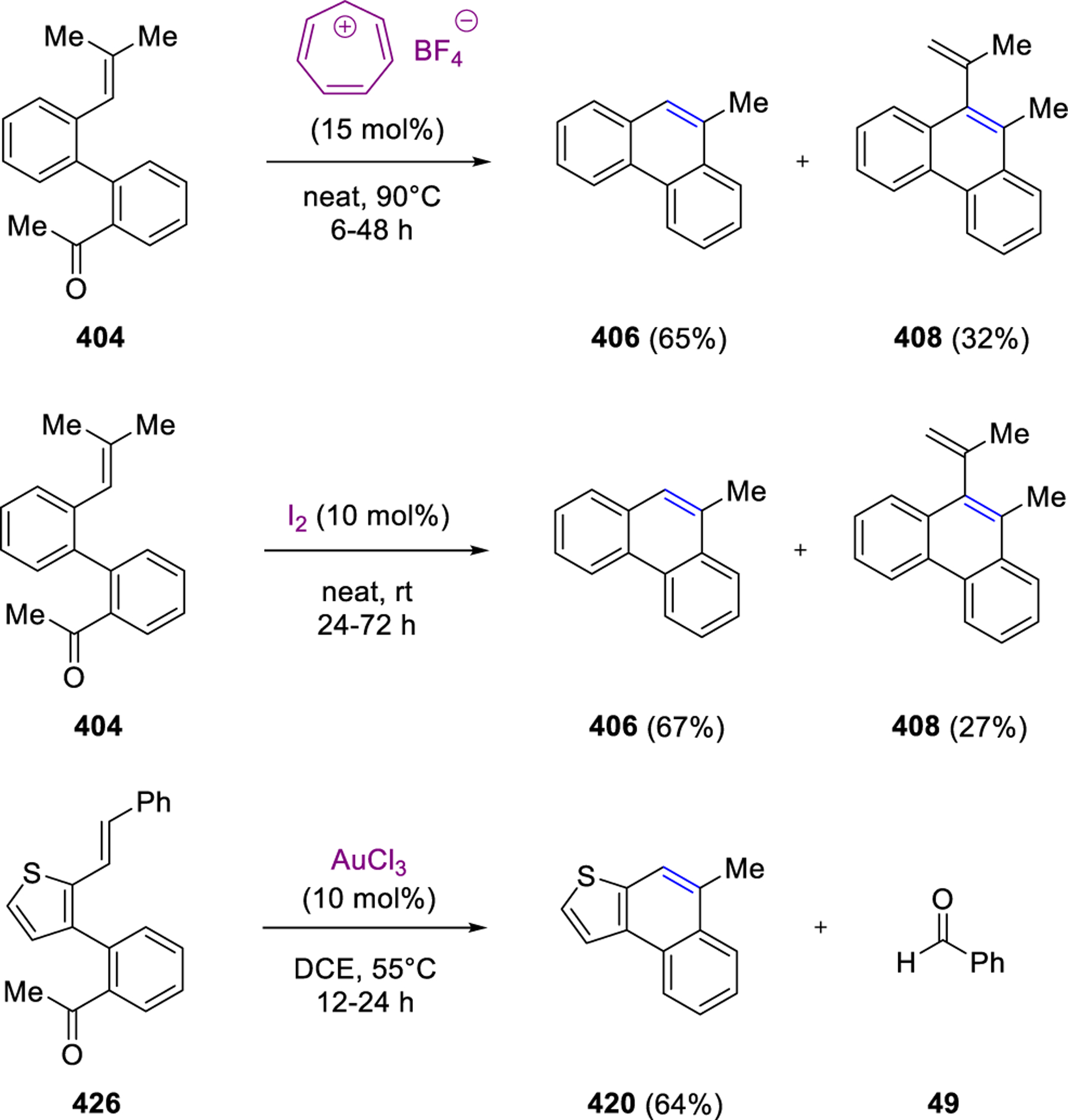
Approaches to Carbonyl-Olefin Ring-Closing Metathesis Relying on Tropylium Cations, Iodine, and AuCl3 as Lewis Acid Catalysts for the Formation of Polyaromatic Hydrocarbons by Nguyen and Lin
The report on AuCl3-catalyzed carbonyl-olefin ring-closing metathesis by Lin and coworkers also included an example resulting in a polyaromatic system demonstrating the compatibility of their optimal conditions with this substrate class (Scheme 72).1145 Specifically, ketone 426 resulted in 64% yield of thiophene when converted with 10 mol% AuCl3 in dichloroethane at 55 °C.
In 2020, Schindler, Reid and coworkers followed up on their initial report on catalytic carbonyl-olefin metathesis for the synthesis of polyaromatic hydrocarbons and focused on the evaluation of additional Lewis acid catalysts for this transformation.120 Importantly, preliminary studies had revealed that the conversion of aldehyde 428a bearing a prenylated alkene moiety with either weaker or stronger Lewis acids compared to FeCl3 favored the carbonyl-ene reaction pathway, while FeCl3 proved unique in promoting predominantly carbonyl-olefin metathesis (Scheme 73). Specifically, catalytic amounts of SnCl4 formed the dehydrated carbonyl-ene product 408 in 95% yield and stoichiometric amounts of Me2AlCl resulted in 94% yield of unsaturated alcohol 428 as the carbonyl-ene product, while 10 mol% FeCl3 provided 33% yield of carbonyl-olefin metathesis product 427 and 19% yield of the dehydrated carbonyl-ene product 408. The fact that even simple Lewis acids demonstrated remarkable selectivity in differentiating between both reaction paths led the authors to investigate the origins of this bias to understand the specific catalyst requirements to favor one reaction path over the other. In the course of their studies, Schindler, Reid and coworkers developed predictive multivariate linear regression models based on kinetic and thermodynamic information obtained in DFT calculations of the complex potential energy surfaces of carbonyl-olefin metathesis and carbonyl-ene reactions as competing reaction paths.
Scheme 73.

Divergent Reactivity Observed in Carbonyl-Olefin Metathesis and Carbonyl-Ene Reactions is not Exclusively Explained by Lewis Acidity as Reported by Schindler and Reid
Importantly, upon binding of the biaryl aldehyde 428a to the Lewis acid, the Lewis acid-base complex 429 is formed, which can undergo a reversible carbonyl-ene reaction with both FeCl3 and Me2AlCl as Lewis acids to form unsaturated alcohol 432 (Scheme 74 ). Importantly, alcohol 432 is isolated as the exclusive product with Me2AlCl as Lewis acid since the competing [2+2]-cycloaddition from 429 is prohibitively high in energy and thus results in the carbonyl-ene reaction as the predominant reaction pathway. In comparison, unsaturated alcohol 432 is formed reversibly when relying on FeCl3 as Lewis acid catalyst in addition to the barrier of competing [2+2]-cycloaddition from 429 being significantly lower compared to the Me2AlCl promoted reaction pathway. Consequently, conversion of biaryl aldehyde 428a with catalytic amounts of FeCl3 results in the formation of carbonyl-olefin metathesis product 431 as the thermodynamic product.
Scheme 74.
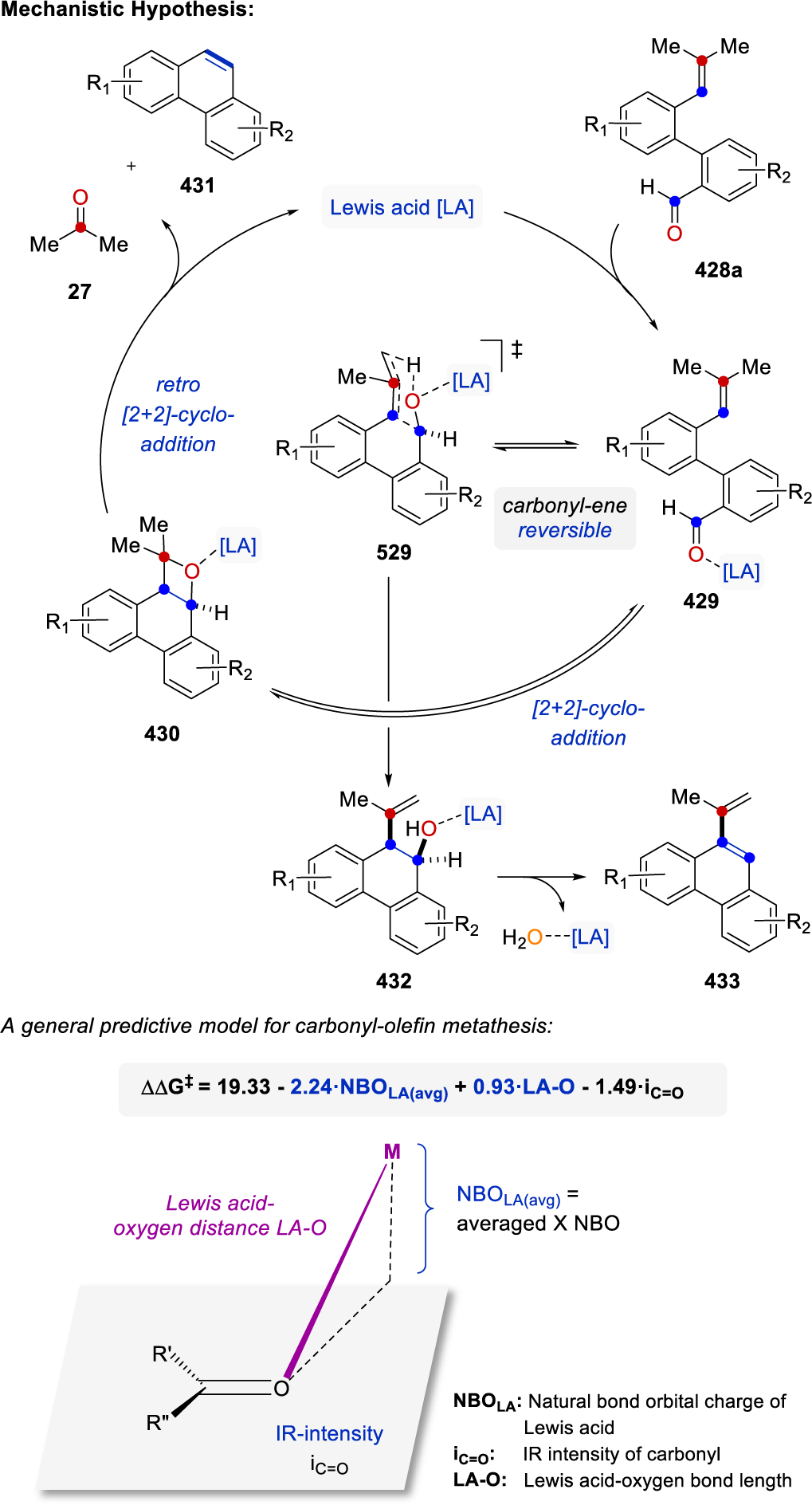
Mechanistic Hypothesis for Competing Carbonyl-Olefin Metathesis and Carbonyl-Ene Reactions of Biaryl Aldehydes by Schindler and Reid
Subsequent efforts centered on the development of multivariate linear regression models to describe the competing reaction pathways. Notably, carbonyl-olefin metathesis and carbonyl-ene reactivity could be correctly predicted based on a set of key parameters for both the substrate and Lewis acid employed, which showed that metathesis reactivity displayed a higher dependency on the choice of Lewis acid employed, whereas carbonyl-ene reactivity was determined to be more substrate dependent. Since carbonyl-olefin metathesis was found to be predominantly determined by the Lewis acid structure, models describing this reactivity mode were expected to be able to adequately predict the reaction barriers for a diverse set of metathesis substrates. Based on descriptors for Lewis acid and substrate from the preceding parameter set, a secondary model for carbonyl-olefin metathesis reaction was developed that relied on three terms centered on the Lewis acid and carbonyl subunit of the substrate (Scheme 74). Specifically, the energy barriers for aryl ketones undergoing Lewis acid-catalyzed carbonyl-olefin metathesis can be predicted based on the IR intensity of the carbonyl (iC=O), the Lewis acid-oxygen bond length (LA-O), and the natural bond orbital charge of the Lewis acid (NBOLA(avg)). Interestingly, the Lewis acid-oxygen distance was identified as an important contributor in the reactivity of distinct Lewis acids during X-ray crystallographic studies of Lewis acid-carbonyl complexes conducted in 1990 by Schreiber and coworkers.121
The Schindler group reported in 2020 the FeCl3-catalyzed formation of functionalized tetrahydropyridines 436.118 Amino acids serve as chiral pool reagents to access N-containing aryl ketones 434 using a variety of electron-poor sulfonamide protecting groups (Scheme 75). Electron-deficient tosylamides functioning as protecting groups for the secondary amines were identified as superior and utilized to promote the desired transformation for a range of 23 substrates proceeding in up to 99% yield. The functionalized metathesis products could be further diversified through a reductive deprotection strategy relying on SmI2 to remove the sulfonamide protecting groups in high yield while maintaining the enantiomeric excess of the products. Notably, prenyl-derived alkenes (437) and terminal alkenes (438) capable of isomerizing in situ were shown to be the only viable alkene substrates for the synthesis of tetrahydropyridines via catalytic carbonyl-olefin metathesis while their stryenyl analogs (439 and 440) resulted in no formation of the desired products.
Scheme 75.
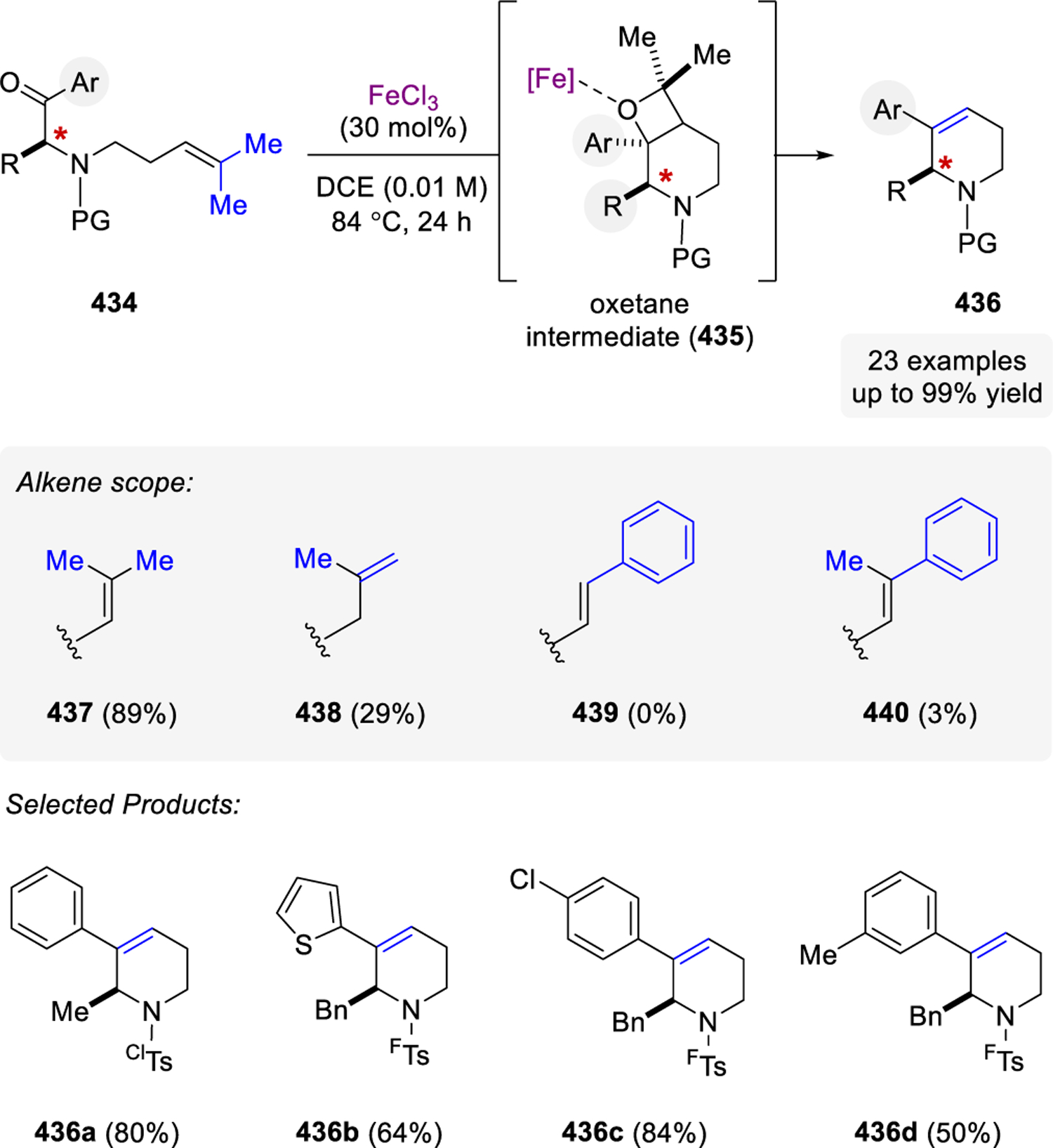
Tetrahydropyridines via FeCl3-Catalyzed Carbonyl-Olefin Metathesis by Schindler and Coworkers
In 2020, Schindler and coworkers were able to extend the scope of 6-membered ring products formed in Lewis acid-catalyzed carbonyl-olefin metathesis reactions from polyaromatic hydrocarbons and tetrahydropyridines to functionalized cyclohexenes accessible from acyclic arylketone substrates.122 Their reaction design is based on Lewis acidic “superelectrophiles”123–125 and observations made during the development of carbonyl-olefin metathesis reactions of less reactive aliphatic ketones.126 Accompanying mechanistic investigations had revealed a second order dependence in FeCl3 and ultimately identified singly bridged dimers (442) formed upon in situ association of FeCl3 monomers (441) as active catalytic species, which together are stronger than the individual FeCl3 monomers (Scheme 76). Schindler and coworkers hypothesized that ion pairs 443 resulting upon heterolytic cleavage of the C-X bond in dimers 442 could function as even stronger Lewis acid catalysts and activate the less reactive acyclic arylketone substrates for carbonyl-olefin metathesis reactions to form cyclohexene products.
Scheme 76.

Lewis Acidic “Superelectrophiles” as Stronger Catalysts for Carbonyl-Olefin Metathesis as Suggested by Schindler and Coworkers
Extensive reaction optimization ultimately revealed that heterobimetallic ion pairs resulting upon chloride abstraction from AlCl3 with Ag(I) additives, such as AgSbF6, are capable of promoting the desired transformation in up to 99% yield as shown for 26 examples while standard Lewis acids including FeCl3 resulted in low yields of less than 20% (Scheme 77). Importantly, the addition of AgSbF6 to AlCl3 leads to the in situ formation of the ion pair [AlCl2][SbF6], which functions as Lewis acidic superelectrophiles. Prenylated alkenes (446) and terminal alkenes (447) capable of isomerizing in situ while resulting in acetone as the metathesis byproduct proved superior forming the desired metathesis products in 80% yield. In comparison, styrene derivatives 448 and 449 resulted in diminished yields of 44% and 27%, respectively. Notably, the reaction conditions also proved viable for a limited number of 7-membered ring products 445g and 445h formed in 43% and 51% yield, respectively.
Scheme 77.
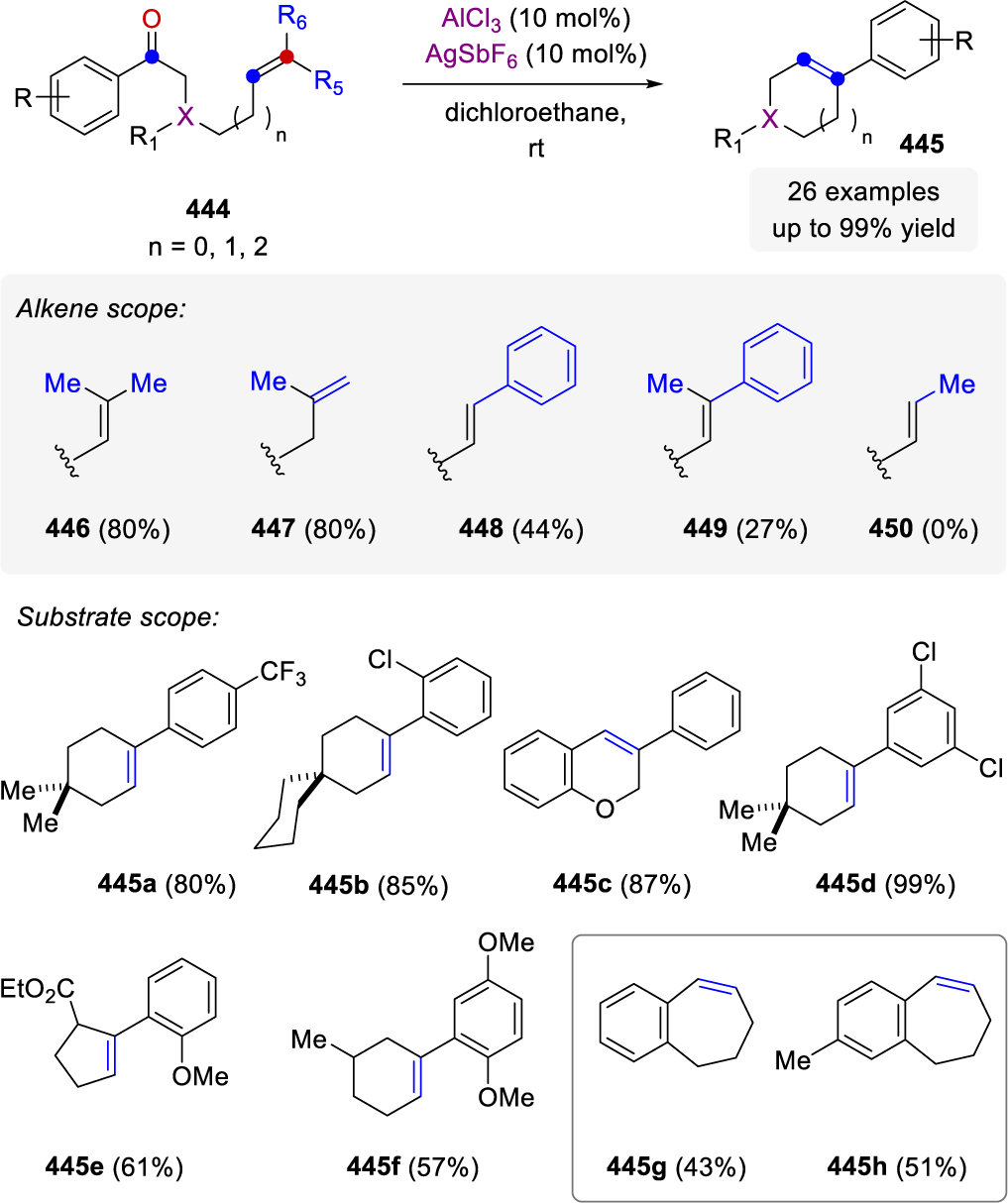
Formation of 6-Membered Rings from Acyclic Arylketones developed by Schindler and coworkers
During their initial reaction optimization of carbonyl-olefin ring-closing metathesis relying on Al(III)-ion pairs, Schindler and coworkers noticed the initial build-up and subsequent depletion of an intermediate. Subsequent efforts to isolate this intermediate identified it as unsaturated alcohol 457 and the product of a carbonyl-ene reaction, which when resubjected to the optimal reaction conditions resulted in the formation of the desired carbonyl-olefin metathesis product 456 (Scheme 78). Ensuing mechanistic investigations relying on 1H-NMR, deuterium-labeling, and computational studies led Schindler and coworkers to postulate a distinct reaction pathway for Al(III)-catalyzed carbonyl-olefin ring-closing metathesis reactions resulting in 6-membered rings based on Lewis acid-activation of aryl ketone 451 to form Lewis acid-base complex 452. A subsequent reversible carbonyl-ene reaction forms unsaturated alcohol 457, which upon Lewis acid-catalyzed hydroalkoxylation gives rise to oxetane 454. A final retro [2+2]-cycloaddition results in the formation of cyclohexene 456 and acetone 27 as the carbonyl-olefin metathesis products. This reaction pathway was found to be 3.3 kcal/mol lower in energy when comparing the reversible carbonyl-ene step to a [2+2]-cycloaddition reaction of Lewis acid-base complex 452 resulting directly in oxetane 454. Nevertheless, substrates incapable of undergoing carbonyl-ene reactions including styrenyl alkene 459 forms cyclohexene 460 as the desired carbonyl-olefin metathesis product under the optimal reaction conditions via [2+2]-cycloaddition and retro [2+2]-cycloaddition albeit in diminished yields of 44%. Notably, mechanistic data obtained in carbonyl-olefin metathesis reactions resulting in 5-membered ring systems supports a distinct reaction pathway that does not rely on a reversible carbonyl-ene reaction. Consequently, a carbonyl-ene step and a subsequent hydroalkoxylation is exclusive to carbonyl-olefin metathesis reactions resulting in 6-membered ring products.
Scheme 78.

Al(III)-Ion Pairs Promote Carbonyl-Olefin Metathesis via a Distinct Reaction Path by Schindler and Coworkers
In 2019, Lin and coworkers reported that their reaction conditions relying on 5 mol% AuCl3 as Lewis acid in dichloroethane at ambient temperatures were equally capable of resulting in the formation of 6-membered 1,2-dihydronapthalenes bearing distinct substitution in up to 96% yield (Scheme 79).115 Furthermore, Lin and coworkers also reported the successful formation of their 7-membered ring analogs as functionalized 6,7-dihydro-5H-benzo[7]annulenes as demonstrated in seven examples and up to 99% yield.
Scheme 79.
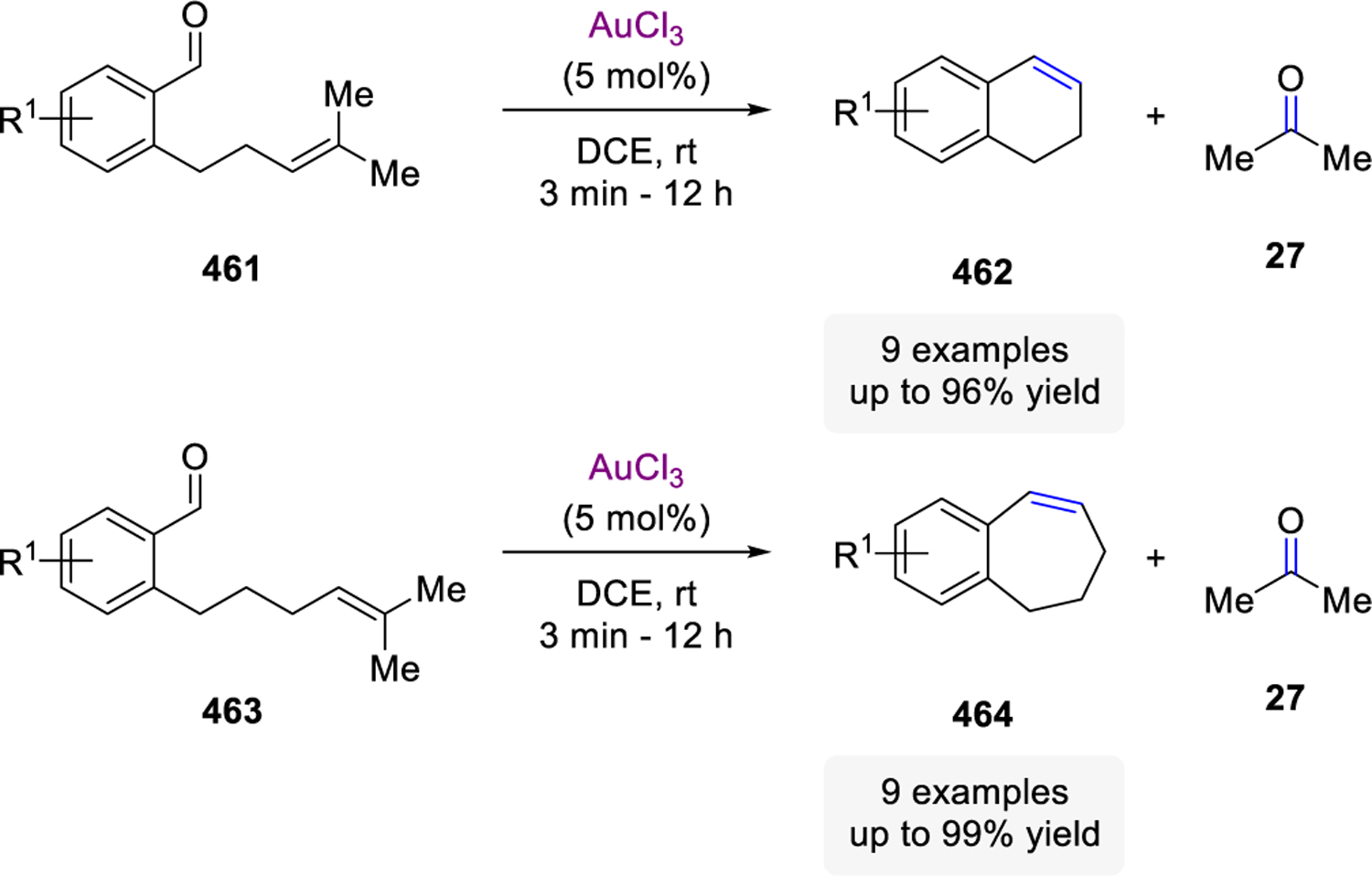
Au(III)-Catalyzed Carbonyl-Olefin Ring-Closing Metathesis for the Formation of 6- and 7-Membered Rings by Lin and Coworkers
6.3. Catalytic Carbonyl-Olefin Ring-Closing Metathesis of Aliphatic Ketones
In comparison to catalytic carbonyl-olefin metathesis reactions of aryl ketones, those efficiently converting aliphatic ketones remain far less advanced. Currently, aliphatic ketone substrates bearing quaternary carbon centers in α-position to the carbonyl moiety undergo efficient carbonyl-olefin metathesis reaction to form cyclopentene products.126 Importantly, aliphatic ketones face several challenges compared to their aromatic analogs that hamper their efficient conversion in carbonyl-olefin metathesis reactions (Scheme 80). Specifically, the aromatic moiety of aryl ketones redistributes electron density and thus aides in transition state stabilization (Scheme 80A). Additionally, alternative oxetane fragmentation pathways exist in which acidic protons in the oxetane intermediate can undergo undesired elimination reactions to result in unsaturated alcohols 466 (Scheme 80B).
Scheme 80.

Challenges in Lewis Acid-Catalyzed Carbonyl-Olefin Metathesis Reactions of Aliphatic Ketones
Due to these challenges, the first protocol for catalytic carbonyl-olefin metathesis of aliphatic ketones was not developed until early 2019 by the Schindler group.126 The majority of substrates undergoing the desired transformation incorporate α-quaternary centers while the generality of this reaction protocol is demonstrated in 41 examples proceeding in up to 91% yield (Scheme 81). Prenylated alkenes prove superior compared to their styrenyl analogs, which require the addition of 5.0 equivalents of allyltrimethylsilane as reported in Li’s protocol for FeCl3-catalyzed carbonyl-olefin metathesis for the formation of 3-aryl-2,5-dihydropyrroles.106 This difference in reactivity is most likely a direct consequence of the metathesis byproducts formed; while acetone can compete with the aliphatic ketone substrates for binding to the FeCl3-catalyst, aromatic aldehyde byproducts are expected to be stronger binders than the corresponding substrates, thus leading to competing catalyst sequestration. Interestingly, the optimal reaction conditions identified require 10 mol% of FeCl3 and prove limited to dichloroethane as reaction solvent while previous examples of catalytic carbonyl-olefin metathesis reactions proceeded efficiently in dichloromethane, toluene, and dichloroethane.99 Additionally, kinetic studies of aliphatic ketone 467 subsequently conducted revealed a second-order dependence in FeCl3 for carbonyl-olefin metathesis reactions of aliphatic ketones resulting in cyclopentene 468 while those relying on aryl ketone substrates show a first-order dependence in FeCl3. The authors followed up their kinetic investigations with spectroscopic and computational investigations, which are consistent with a distinct activation mode for catalytic carbonyl-olefin metathesis reactions of aliphatic ketones resulting forming cyclopentenes (Scheme 81).
Scheme 81.

Carbonyl-Olefin Metathesis of Aliphatic Ketones by Schindler and Coworkers
Specifically, aliphatic ketone 467a coordinates to FeCl3 as the Lewis acid catalyst to form Lewis acid-base complex 480 as the catalyst resting state. In comparison to aryl ketones, this coordination complex is not sufficiently activated for carbonyl-olefin metathesis until a second equivalent of FeCl3 binds to form a Lewis acidic “superelectrophile” in the form of a FeCl3 singly-bridged homo-dimer 481. This homodimer is a stronger Lewis acid compared to the individual Lewis acid monomers and thus capable of activating less reactive substrates. This mechanistic hypothesis aligns with literature reports123–125 from over 60 years ago of superelectrophiles that were proposed to act as stronger Lewis acids in the form of homobimetallic dimers or ion pairs. The “superelectrophilic” Lewis acid-base complex 481 is now sufficiently activated to undergo the desired carbonyl-olefin ring-closing metathesis reaction via [2+2]-cycloaddition to form intermediate oxetane 482 and subsequent retro [2+2]-cycloaddition to ultimately give rise to the metathesis products cyclopentene 468 and acetone 27. Recently, the singly-bridged FeCl3 dimer has been investigated computationally by Zhu and coworkers in their studies of the carbonyl-olefin metathesis reactions to form aromatic species.127
During their studies of intramolecular carbonyl-ene reactions of aliphatic ketone 394, Duñach and coworkers reported evidence of the carbonyl-olefin metathesis product in the investigations of catalytic, intramolecular carbonyl-ene reaction of aliphatic that in addition to the carbonyl-ene product 483 and its dehydrated analog 484, cyclohexene 395 was formed as the carbonyl-olefin metathesis product.119 In the presence of Bi(OTf)3, acting as the Lewis acid catalyst, 394 undergoes the carbonyl-ene reaction to provide cis and trans carbonyl-ene products 483, the elimination product 484 and the metathesis product 395 in a combined yield of 32% and a 41:50:5 ratio (Scheme 83). Interestingly, the use of In(NTf2)3 and Fe(NTf2)2 as the Lewis acid catalyst increased the ratio of the carbonyl-olefin metathesis product. Notably, diene 484 and cyclohexene 395 were formed as the exclusive products in a combined yield of 34% and a 26:74 ratio when relying on catalytic amounts of Fe(NTf2)2 in nitromethane at ambient temperatures for 24 hours. The authors suggest that cyclohexene 395 arises from an intramolecular hydroalkoxylation onto carbonyl-ene product and subsequent fragmentation.
Scheme 83.

Lewis Acid-Catalyzed Intramolecular Carbonyl-Ene Reactions Reported by Duñach and Coworkers
6.4. Catalytic Cross Carbonyl-Olefin Metathesis
In 2015, Franzén and coworkers built on the studies originally published by Bickelhaupt and coworkers95 in 1994 to realize a cross carbonyl-olefin metathesis reaction between benzaldehyde 59 and isobutylene 485 by incorporating important modifications to the procedure. Specifically, the Franzén group had previously identified trityl tetrafluoroborate (486, TrBF4) as a Lewis acid capable of catalyzing a variety of organic transformations, including Diels-Alder and conjugate addition reactions.128 In an initial reaction, an equimolar ratio of benzaldehyde and amylene was converted with 7 mol% TrBF4 (486) to result in 20% yield of the desired cross carbonyl-olefin metathesis product. Subsequent reaction optimization revealed that a 5:1 ratio of aryl aldehyde to alkene with 20 mol% TrBF4 (486) proved optimal and gave rise to the desired cross carbonyl-olefin metathesis products in up to 85% yield (Scheme 84). Additionally, conducting the transformation at lower temperatures albeit for longer reaction times resulted in decreased competing polymerization and catalyst decomposition.
Scheme 84.

E-Selective Cross-Metathesis by Franzén and Coworkers
Following the initial mechanistic hypothesis proposed by Bickelhaupt for a Lewis acid-mediated cross carbonyl-olefin metathesis, Franzén and coworkers suggest a similar proposal that relies on initial LUMO activation of aldehyde 59 by TrBF4 (486) as the Lewis acid catalyst. Nucleophilic addition of alkene 292 onto the Lewis acid-base complex 488 results in carbon-carbon bond formation in carbocation 489 (Scheme 85). Intramolecular addition of the oxygen onto the carbocation forms oxetane 490, which undergoes subsequent C-O bond fragmentation to form benzylic carbocation 491. A final Lewis acid-assisted fragmentation gives rise to alkene 299 and acetone as the cross carbonyl-olefin metathesis products and TrBF4 (486) as the Lewis acid catalyst. Although this mechanistic hypothesis is not supported by experimental investigations, it is based on the known reactivity between carbonyl and alkene functionalities undergoing Prins-cyclizations129 to form 491, which then undergoes a subsequent intramolecular, nucleophilic addition.
Scheme 85.
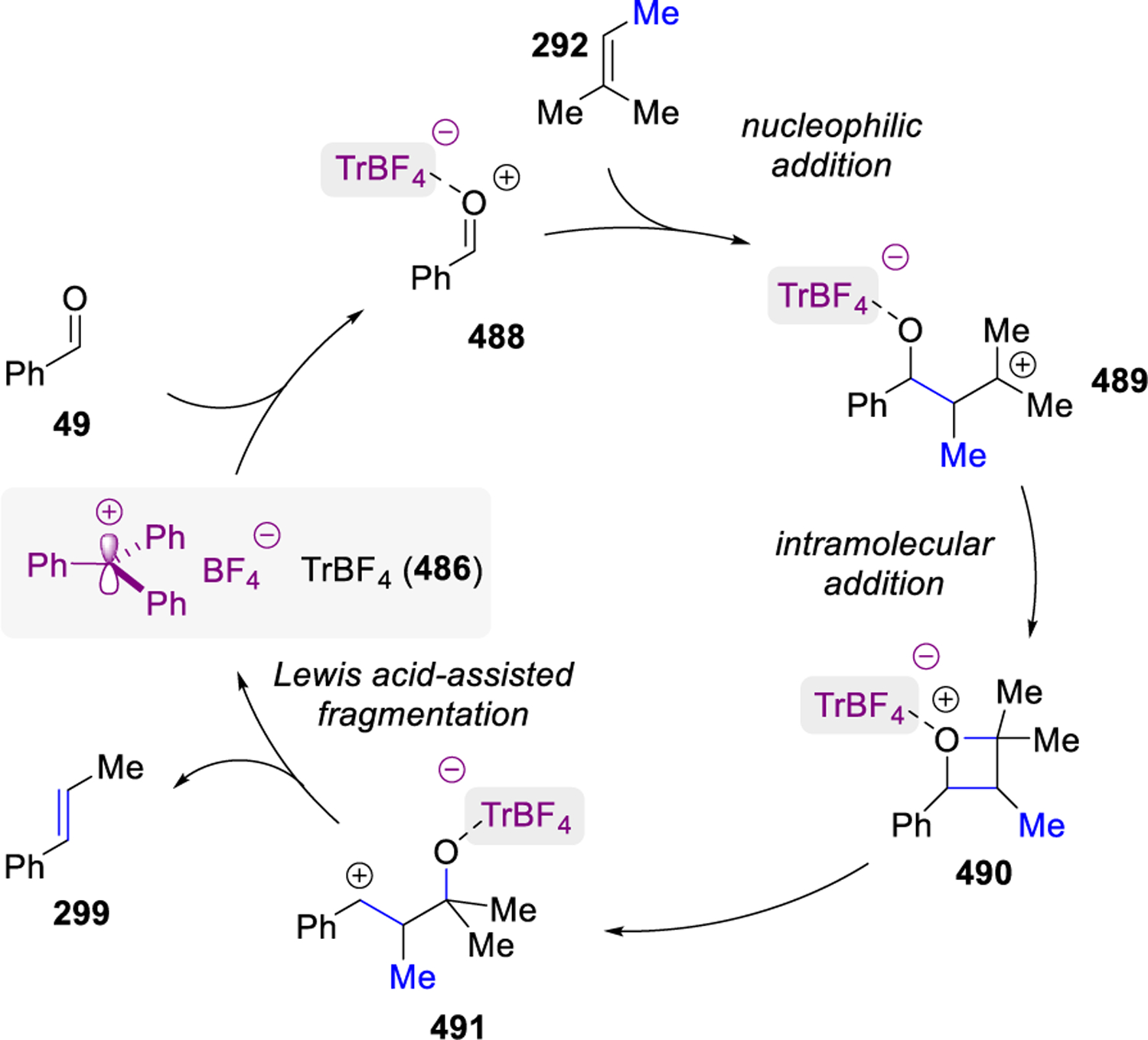
Mechanistic Hypothesis for Catalytic Cross Carbonyl-Olefin Metathesis
In 2018, Nguyen and coworkers showed that catalytic amounts of tropylium tetrafluoroborate (10 mol%) as Lewis acid is similarly capable of promoting the cross carbonyl-olefin metathesis reaction (Scheme 86).113 The optimal reaction conditions also require an excess of aldehyde compared to the alkene substrate (5:1 ratio of aldehyde to alkene) in dichloromethane at 80 °C under microwave irradiation for 30 minutes to form the metathesis products in up to 70% yield. The authors conducted additional mechanistic investigations based on DFT studies, which support a stepwise reaction pathway for intermolecular carbonyl-olefin cross-metathesis reactions similar to that suggested by Franzén and coworkers, in which stepwise oxetane formation and fragmentation are essential components of the reaction pathway. In 2019, Nguyen and coworkers followed up on this report and showed that molecular iodine (10 mol%) also functioned as a suitable Lewis acid to catalyze this transformation.114 Notably, optimal reaction conditions proceed at ambient temperatures with a 2:1 ratio of aldehyde to alkene resulting in the metathesis products in up to 50% yield in reaction times of one to three days (Scheme 86).
Scheme 86.

Tropylium-Ions and Iodine as Lewis Acid Catalysts in Intermolecular Carbonyl-Olefin Cross Metathesis Reactions by Nguyen and Coworkers
The Schindler lab reported a cross carbonyl-olefin metathesis reaction protocol in 2020 relying on Fe(III)-ion pairs as superelectrophilic Lewis acid catalysts.130 Similarly to previous reports by Franzén128 and Nguyen,113,114 this approach is exclusive to aryl aldehydes and relies on an excess of the carbonyl component in a 5:1 ratio compared to the alkene substrate. Nevertheless, the reaction proceeds in dichloromethane as optimal solvent at ambient temperatures within three hours to result in the formation of the desired cross carbonyl-olefin metathesis products in up to 64% yield. An investigation of the alkene scope of this transformation (Scheme 87) revealed that tri-alkylsubstituted alkenes performed best, which is consistent with previous literature reports.
Scheme 87.

E-Selective Intermolecular Carbonyl-Olefin Metathesis by Schindler and Coworkers
As part of these studies, the origin of the high (E)-selectivity of catalytic cross carbonyl-olefin metathesis reactions was investigated (Scheme 88). Importantly, four distinct oxetane stereoisomers could form upon addition of the aldehyde and alkene substrates, which could fragment to form three distinct alkene products. The Schindler group independently synthesized oxetane trans-499 and showed that it results in the selective formation of alkene in 80% while alkene 501, the product obtained in cross carbonyl-olefin metathesis reactions, was not observed. A mixture of trans-502 and trans-499 in a ratio of 1:1.26 resulted in (E)-299 in 47% yield (from trans-594) and 592 in 78% yield (based on trans-593) while no formation of the (Z)-isomer of (E)-299 was observed. A final set of experiments investigated the ability of (Z)-299 to isomerize to (E)-299 under the optimal reaction conditions. While isomerization from (Z)- to (E)-299 is feasible under these conditions, it was determined unlikely to be responsible for the formation of (E)-299 as the exclusive product on the timescale of this transformation. Collectively, these results are consistent with the regioselective formation of a single oxetane intermediate and its subsequent stereospecific fragmentation in the course of cross carbonyl-olefin metathesis reactions to account for the selective formation of (E)-alkene products.
Scheme 88.

E-Selectivity due to Regioselective Oxetane Formation
A further investigation into the mass balance of this reaction determined a competing reaction pathway for the oligomerization of the starting materials to account for the low overall yields observed in catalytic cross carbonyl-olefin metathesis reactions. Similar observations of a polymeric byproduct had previously been reported by Franzén and coworkers.128
Although cross carbonyl-olefin metathesis reactions hold great potential for the catalytic synthesis of new alkene products, they arguably remain among the least advanced classes of this transformations. Importantly, substrates are currently limited to aryl aldehydes and trisubstituted aliphatic alkenes and require a large excess of 5 equivalents of the carbonyl component to proceed. Although progress has been made understanding the exclusive (E)-selectivity of these transformations, the reaction mechanism in addition to the origin of polymeric byproducts remains poorly understood and will require further careful investigations.
6.5. Catalytic Carbonyl-Olefin Ring-Opening Metathesis
In 2018, the first reports of Lewis acid-catalyzed carbonyl-olefin ring-opening metathesis were published by the laboratories of Nguyen and Schindler. The reaction protocol reported by Schindler and coworkers relies on catalytic amounts of GaCl3 as the superior Lewis acid catalyst compared to FeCl3, which also resulted in accompanying decomposition of the metathesis products formed.131 The generality of this approach was demonstrated for a range of 22 examples accessible in up to 47% yield from aryl aldehyde and cyclic alkene substrates (Scheme 89). The initial hypothesis that the ability of cyclic alkenes to undergo carbonyl-olefin ring-opening metathesis was directly related to their inherent ring strain did not prove viable as cyclopentene 503 and cyclohexene 507 underwent the desired transformation in 47% and 18% yield, respectively, while cycloheptene 508 exhibiting a higher ring strain compared to 503 and 507 resulted in no formation of the carbonyl-olefin metathesis products.
Scheme 89.
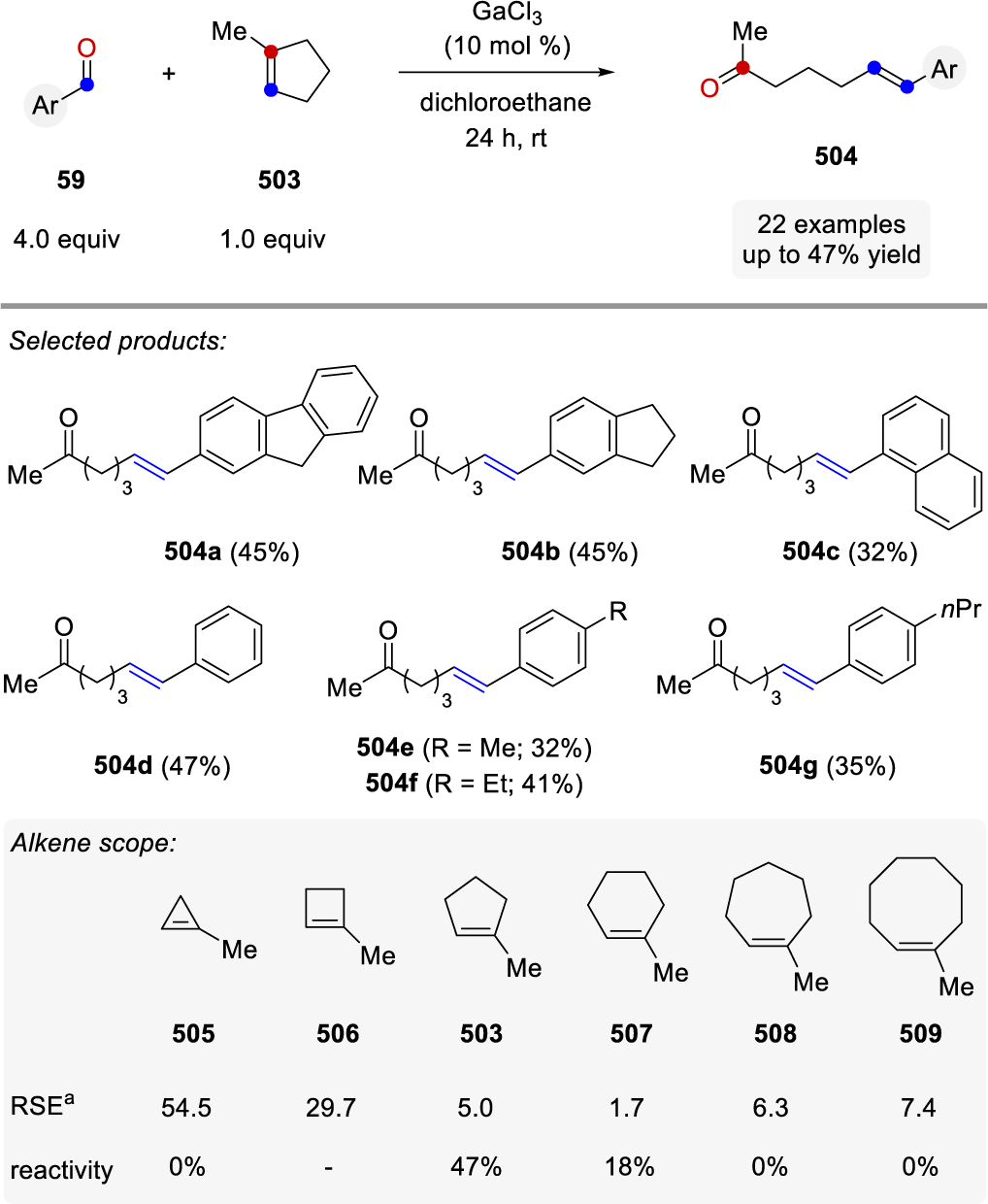
Ring-Opening Carbonyl-Olefin Metathesis by Schindler and Coworkers
Subsequent efforts conducted by Schindler and coworkers focused on investigations related to the overall mass balance of this transformation, which provided at best 47% yield of the desired carbonyl-olefin ring-closing metathesis products (Scheme 90). 1H-NMR studies led to the isolation and subsequent characterization of two additional products formed, specifically cyclic ether 510 formed in 10% yield and bisalkene 514 obtained in 20% yield. The authors hypothesized that both undesired byproducts are formed in a competing carbonyl-ene reaction and subsequent addition of either a second equivalent of aryl aldehyde 59 or cyclopentene 503 to the carbonyl-ene intermediate. Consequently, GaCl3-catalyzed carbonyl-olefin ring-closing metathesis reactions proceed upon coordination of the Lewis acid catalyzed to the aryl aldehyde 59 to form Lewis acid-base complex 511, which can undergo a subsequent [2+2]-cycloaddition with cyclopentene 503 to result in intermediate oxetane 513. A final fragmentation of oxetane 513 via retro [2+2]-cycloaddition gives rise to unsaturated ketone 504 as the desired carbonyl-olefin ring-opening metathesis product. Alternatively, Lewis acid-base complex 511 can undergo a competing carbonyl-ene reaction upon carbon-carbon bond formation with cyclopentene 503 to result in unsaturated alcohol 512. This intermediate can undergo two distinct addition reactions upon dehydration with either aryl aldehyde 59 or cyclopentene 503 to form cyclic ether 510 or bisalkene 514 as competing byproducts responsible for the overall diminished yields observed in carbonyl-olefin ring-closing metathesis.
Scheme 90.

Carbonyl-Olefin Metathesis and Carbonyl-Ene Reaction Paths Compete in Catalytic Ring-Opening Reactions as Proposed by Schindler and Coworkers
Similar observations were reported in 2018 by Nguyen and coworkers in their studies of tropylium tetrafluoroborate-catalyzed carbonyl-olefin ring-opening metathesis reactions.113 The optimized conditions rely on 5 mol% of the Lewis acid catalyst, neat, at 40 °C for up to 24 hours and result in up to 59% yield for a range of 6 examples (Scheme 91). Notably, cyclopentenes were identified as the optimal alkene component while larger ring systems failed to provide the desired metathesis products.
Scheme 91.

Tropylium-Ions and Iodine as Lewis Acid Catalysts in Intermolecular Carbonyl-Olefin Ring-Opening Metathesis Reactions by Nguyen and Coworkers
In 2019, Nguyen and coworkers followed up on this report and showed that molecular iodine (10 mol%) was similarly capable of catalyzing carbonyl-olefin ring-opening metathesis reactions as demonstrated for two examples proceeding in up to 50% yield (Scheme 91).114
Together with carbonyl-olefin cross metathesis reactions, carbonyl-olefin ring-opening metathesis reactions currently remain among the least advanced. 5-Membered cyclic alkenes represent the most viable substrate class with respect to the alkene component, while their 6-membered ring analogs result in significantly diminished yields and higher order ring systems fail to undergo the desired transformation. With regard to the carbonyl substrate, ring-opening metathesis reactions are limited to aryl aldehydes while ketones and aliphatic aldehydes do not undergo carbonyl-olefin ring-opening metathesis. The lower overall yields observed in these transformations of ~50% can be attributed to competing carbonyl-ene reaction pathways that lead to reactive intermediates capable of undergoing subsequent addition reactions with the aryl aldehyde or cyclic alkene substrates.
6.6. Catalytic Transannular Carbonyl-Olefin Metathesis
In 2019, the Schindler laboratory reported a catalytic, transannular carbonyl-olefin metathesis reaction relying on FeCl3 as the Lewis acid catalyst and cyclic, unsaturated ketones 516 as substrates (Scheme 92).27 The development of this transformation was inspired by earlier reports of Khripach and coworkers in 2006 who observed a ring-opening and subsequent ring-contraction during their studies towards the synthesis of steroid frameworks mediated by BF3·OEt2.97 During their reaction optimization, Schindler and coworkers were able to show that catalytic amounts of FeCl3 were capable to catalyze transannular carbonyl-olefin metathesis reactions of 9- and 10-membered ring systems (516) incorporating carbonyl and alkene moieties. The scope of this transformation was demonstrated in 10 examples resulting in up to 84% yield of the desired carbonyl-olefin metathesis products 518–523.
Scheme 92.
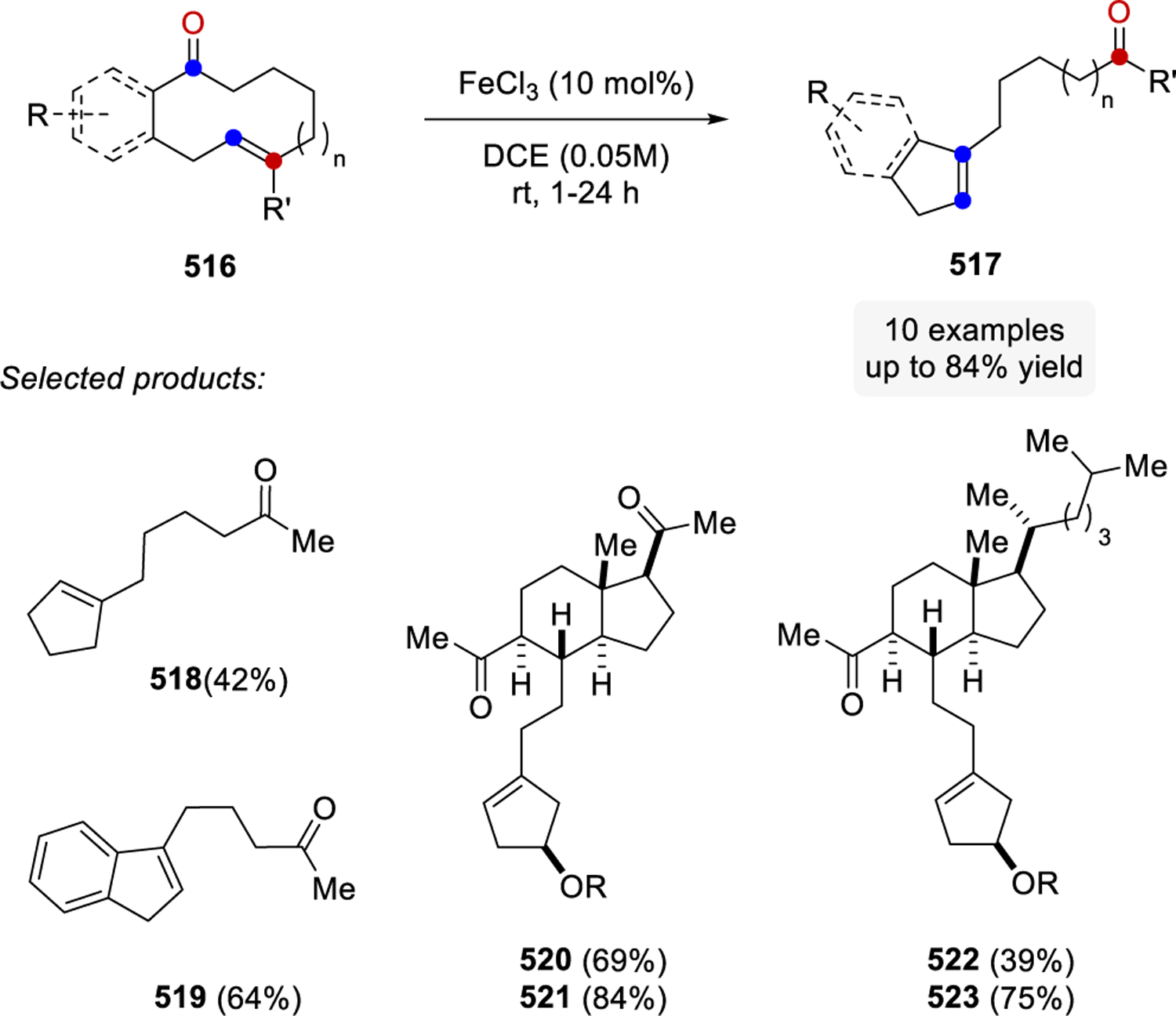
Catalytic, Transannular Carbonyl-Olefin Metathesis by Schindler and Coworkers
Interestingly, initial studies with unsaturated, cyclic ketone 524 aimed at the optimization of this transformation with distinct Lewis acids, including Me2AlCl, FeCl3, and TiCl4 resulted in the formation of three distinct products, specifically carbonyl-ene product 525 when using equimolar amounts of Me2AlCl, carbonyl-olefin metathesis product 526 with catalytic amounts of FeCl3, and tetrahydrofuran 527 with catalytic amounts of TiCl4 (Scheme 93). This divergent reactivity proved general for a variety of 9- and 10-membered ring substrates and prompted additional mechanistic investigations by Schindler and coworkers. Based on these efforts, the authors postulate a mechanistic hypothesis for transannular carbonyl-olefin metathesis in which the substrate 529 binds to FeCl3 as the Lewis acid to form Lewis acid-base complex 530. This complex can undergo either of two reversible transformations, specifically a carbonyl-ene reaction resulting in unsaturated alcohol 532 as the kinetic product or a reversible asynchronous, concerted [2+2]-cycloaddition to form oxetane 531 as the thermodynamic product. Experimental studies support this hypothesis as the kinetic product 532 can be isolated at lower reaction temperatures as a stable product. Oxetane 531 subsequently undergoes FeCl3-catalyzed retro-[2+2]-cycloaddition to result in the formation of 528 as the product of a transannular carbonyl-olefin metathesis reaction (Scheme 94). When cyclodecenone 524 is converted with stoichiometric amounts of Me2AlCl as Lewis acid, the carbonyl-ene reaction path is not reversible as 526 is isolated exclusively as the thermodynamic product. Interestingly, the formation of tetrahydrofuran products 527 has not previously been observed under conditions promoting carbonyl-olefin metathesis reactions. The authors suggest that under TiCl4-catalyzed conditions, oxetane 531 undergoes competing oxetane fragmentation via elimination to form an intermediate unsaturated alcohol capable of undergoing subsequent hydroalkoxylation onto the alkene moiety to result in the formation of tetrahydrofuran 527.
Scheme 93.
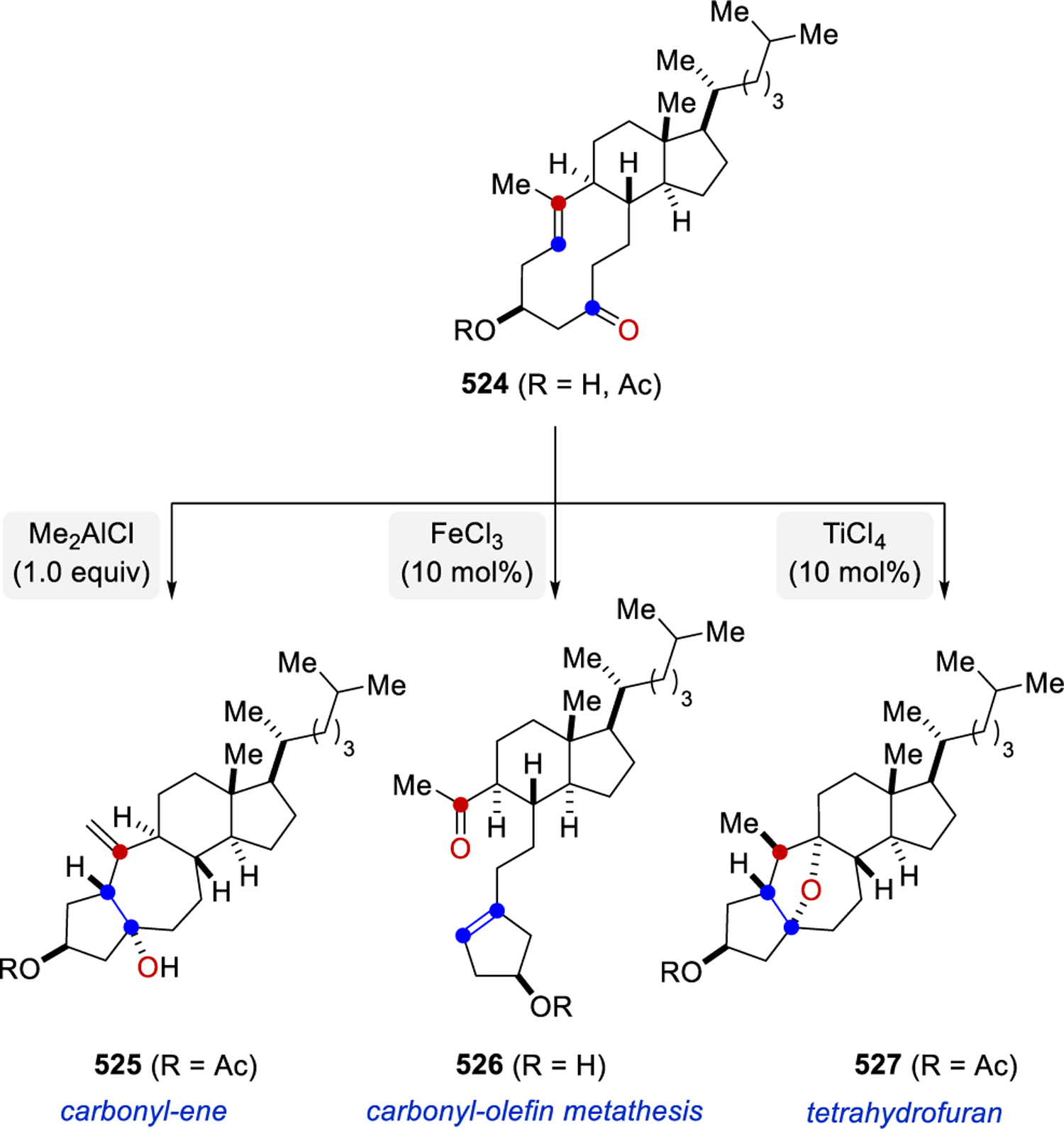
Divergent Reactivity in Transannular Reactions of Unsaturated, Cyclic Ketones 524 with Me2AlC, FeCl3, or TiCl4 as Lewis Acid
Scheme 94.

Mechanistic Hypothesis for Catalytic, Transannular Carbonyl-Olefin Metathesis by Schindler and Coworkers
Lewis acids have proven to be exceptional catalysts for carbonyl-olefin metathesis. Since the first discoveries relying on stoichiometric amounts of a Lewis acid, a diverse array of catalytic systems has been reported to promote ring-closing, ring-opening, cross, and transannular carbonyl-olefin metathesis reactions. Detailed mechanistic studies have been conducted which suggest that the overall reaction pathway is substrate-dependent, relying on either concerted cyclization and fragmentation, carbonyl-ene products as reactive intermediates, or step-wise sequences. Importantly, while the carbonyl-ene pathway was shown to be productive for metathesis in some instances, it can also lead to detrimental byproduct formation in others. Catalytic amounts of FeCl3 has been established as a superior catalytic system for many substrates undergoing ring-closing carbonyl-olefin metathesis. Nevertheless, many other Lewis acids will show reactivity in these transformations albeit often leading to diminished yields of the desired products. Recently, the development of new catalyst systems relying on superelectrophilic Lewis acids significantly broadened to scope of Lewis acid-catalyzed carbonyl-olefin metathesis reactions. However, further advances are necessary to establish carbonyl-olefin metathesis as a general synthetic strategy for alkene synthesis.
7. OTHER CARBONYL–OLEFIN METATHESIS STRATEGIES
7.1. Ring-Closing Carbonyl-Olefin Metathesis
In 2018, Catti and Tiefenbacher132 showed that HCl as a Brønsted acid in combination with a self-assembled host133,134 was able to promote intramolecular ring-closing metathesis of a variety of previously established aryl ketone substrates 319 in moderate to good yields (Scheme 95). Supramolecular hosts have been increasingly investigated for their ability to facilitate reactions that are difficult to perform in bulk solution. One of the most commonly studied host systems, resorcin[4]arene hexamer (534), 135–139 spontaneously self-assembles in apolar solvents like chloroform via hydrogen bonding. Hexamer 534 is proposed to stabilize cationic transition states through cation-π interactions with the aromatic cavity walls.140 Following optimization, the general conditions were determined to be 10 mol% of the self-assembled host and 5–20 mol% of HCl in CHCl3 at 50 °C for 1–8 days. This report suggests that the supramolecular host and the Brønsted acid work synergistically to catalyze the metathesis reaction inside the cavity of the host structure. Initial mechanistic experiments based on trapping studies suggest that the reaction undergoes a stepwise formation of the intermediate oxetane under optimized conditions.
Scheme 95.

Brønsted Acid-Catalyzed Metathesis in a Self-Assembled Supramolecular Host by Tiefenbacher
7.2. Cross Carbonyl-Olefin Metathesis
An alternative catalytic strategy developed for cross carbonyl-olefin metathesis reactions takes advantage of a two-step reaction sequence that can be conducted in situ that relies on initial carbon-carbon bond formation and subsequent Grob fragmentations (Scheme 96). This reaction design principle is currently limited to the formation of styrene derivatives 28 as the Grob Fragmentation requires the formation of stabilized carbocations. Consequently aryl aldehydes have proven superior as substrates following this strategy for catalytic cross carbonyl-olefin metathesis.
Scheme 96.

Cross Carbonyl-Olefin Metathesis Relying on Grob Fragmentations
The Glorius group reported an intermolecular cross carbonyl-olefin metathesis protocol that employed a photocatalysis approach relying on Mes2AcrtBu2BF4 (537) in combination with Brønsted acids (Scheme 97).141 Initial mechanistic investigations conducted by the authors are consistent with a stepwise reaction path that proceeds via carbon-carbon bond formation between aryl aldehyde 24 and olefin 25 to form diol 26 under the aqueous reaction conditions. Subsequent loss of water upon activation of the benzylic alcohol moiety with trifluoroacetic acid results in the formation of carbocation 535, which undergoes Grob fragmentation to result in styrene derivatives 28 and acetone 27 as net carbonyl-olefin metathesis products. Control experiments focused on the conversion of oxetane 536 under the optimal reaction conditions, which failed to provide styrene 538. In comparison, the diol accessible upon carbon-carbon bond formation between benzaldehyde and 2-methyl-1-phenyl-1-propene readily results in the formation of styrene and supports the hypothesis that oxetanes do not function as key intermediates in this transformation.142,143 The reaction design proved general for the synthesis of a variety of styrenes (539-542) in good yields and diastereomeric ratios of >20:1.
Scheme 97.
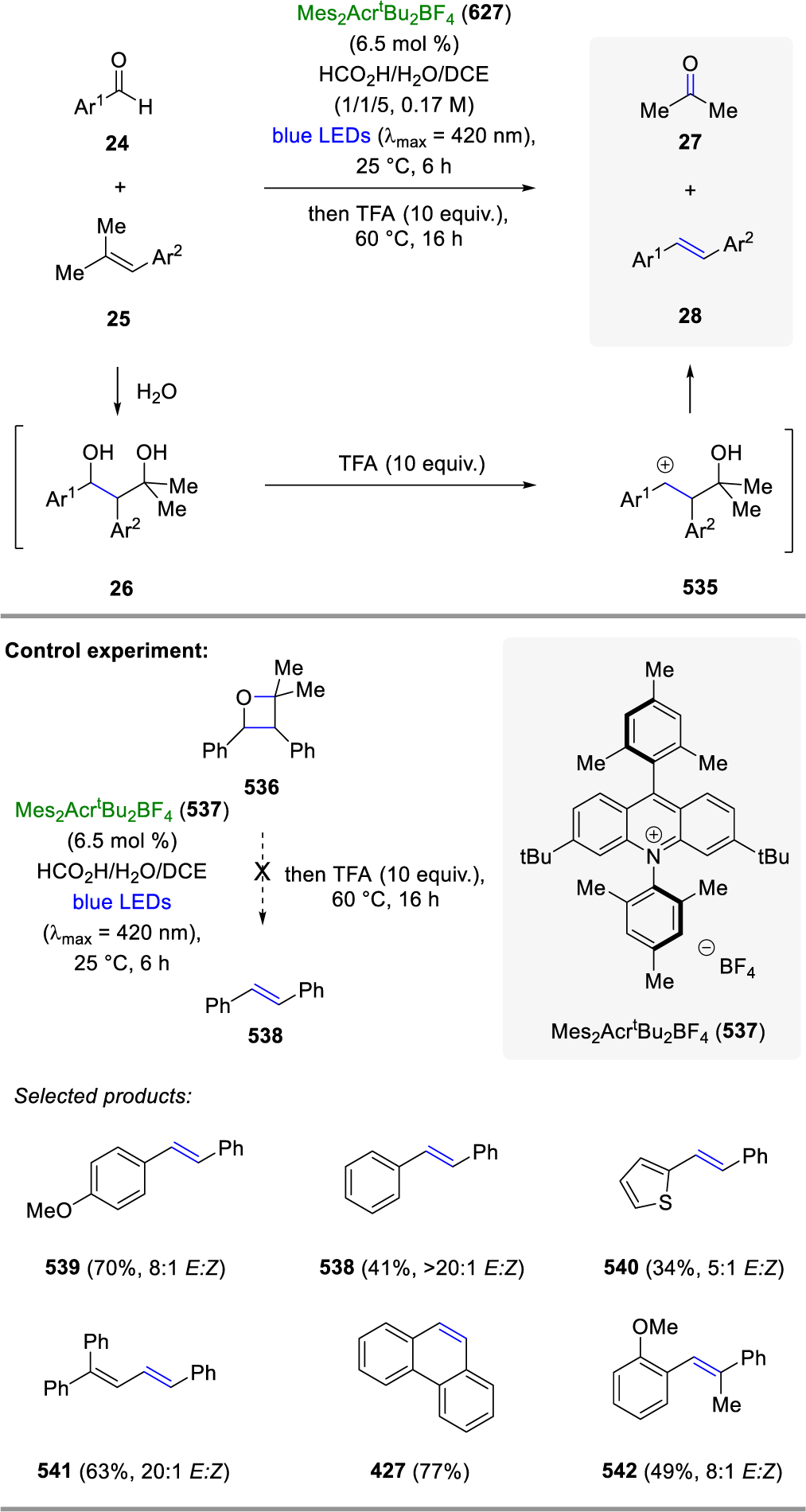
Carbonyl–Olefin Cross-Metathesis via Visible-Light-Induced 1,3-Diol Formation and Fragmentation Sequence by Glorius
Following the report for carbonyl-olefin metathesis by Glorius and coworkers relying on a Grob fragmentation approach, a similar strategy was developed by Leyva-Pérez and coworkers to access styrene derivatives from aryl aldehydes and ketones (543) with vinyl ethers (544).144 This approach utilized an inexpensive, non-toxic aluminosilicate montmorillonite solid-state acid catalyst, montmorillonite K10 (Scheme 98). Solid, fixed-bed acids allowed for the intermolecular carbonyl-olefin metathesis reaction to be conducted in-flow; avoiding unwanted side reactivity of vinyl ether hydrolysis. Computational and experimental results supported an acid-catalyzed mechanism that proceeds through hemiacetal 545 as a key intermediate, which then undergoes a dehydration to form a benzylic carbocation, followed by a subsequent Grob fragmentation to provide ester 547 along with styrene derivative 546 as the desired metathesis product. Notably, only the formation of the E-isomers is observed under the optimized reaction conditions for all substrates investigated.
Scheme 98.

Intermolecular Carbonyl-Olefin Metathesis of Vinyl Ethers Catalyzed by Solid Acids in Flow by Leyva-Pérez
In summary, these examples highlight the ability of Brønsted acids to promote carbonyl-olefin metathesis on a variety of substrate classes. The acidic environment promotes the formation of cationic intermediates which are able to successfully fragment to yield the desired olefin products. Additionally, these conditions were translated to a continuous flow platform resulting in comparable yields of the desired carbonyl-olefin metathesis products. In comparison to Lewis acid-catalyzed approaches, the development of Brønsted acid-catalyzed carbonyl-olefin metathesis reactions is less advanced currently following either the design principle of Lewis acid-catalyzed ring-closing reactions or a Grob fragmentation approach.
8. APPLICATIONS IN NATURAL PRODUCT SYNTHESIS
Carbonyl-olefin metathesis reactions have seen important applications in natural product synthesis, particularly approaches that enable ring-closing transformations. Interestingly, the majority of these applications in total synthesis relies on metal alkylidenes as stoichiometric reagents, which points towards the synthetic potential of carbonyl-olefin metathesis. Since the first strategies for catalytic carbonyl-olefin metathesis have been developed within the past decade, future applications of these reaction protocols in complex settings are to be expected, which will lead to the identification of existing limitations in catalytic carbonyl-olefin metathesis to ultimately spur further important advances in reaction development. This part of the review provides an overview of reported applications for carbonyl-olefin metathesis reactions in the synthesis of natural products since 1975.
In 1975, Jones and coworkers developed a four-step synthetic sequence for the Mediterranean fruit fly pheromone trans-non-6-en-1-ol (553)33 based on a carbonyl-olefin metathesis sequence that relies on initial photochemical [2+2]-cycloaddition and a subsequent Lewis acid-mediated cycloreversion32 (Scheme 99). Paternò-Büchi reaction occurs upon photochemical irradiation with a 450W Hanovia immersion lamp between cyclohexa-1,3-diene 548 and propionaldehyde 549 resulted in the formation of oxetane 550 in 77% yield. Reduction of the internal alkene with PtO2 gave rise to its reduced analog 551, which undergoes a [2+2]-cycloreversion upon treatment with [Rh(CO2)Cl]2 as a Lewis acid under elevated temperatures to form aldehyde 552 in 89% yield. Final reduction of 552 with LiAlH4 results in the quantitative formation of trans-non-6-en-1-ol (553).
Scheme 99.
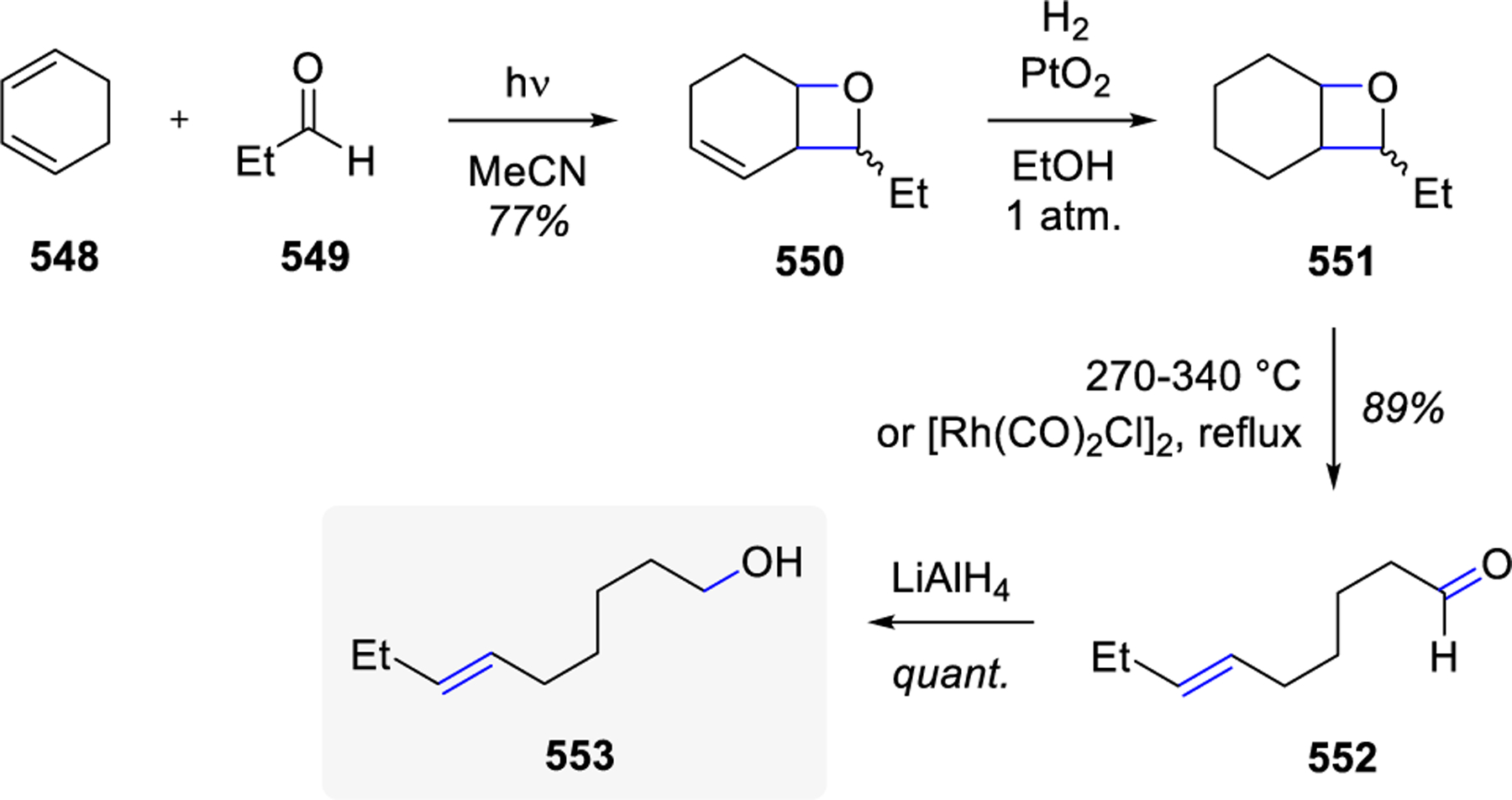
Synthesis of the Mediterranean Fruit Fly Pheromone trans-Non-6-en-1-ol by Jones
Grubbs and Stille published the first application of carbonyl-olefin metathesis towards the synthesis of capnellene in 198649,50 utilizing the Tebbe reagent.56 Capnellene is a proposed precursor to the capnellene family of nonisoprenoid sesquiterpenes and possesses promising antibacterial and antitumor properties.145,146 The approach developed by Grubbs and Stille utilizes a Diels-Alder reaction to form bicyclic 140, which subsequently undergoes an intramolecular carbonyl-olefin ring-opening metathesis relying on stoichiometric amounts of Tebbe reagent to form the cyclobutene 144 in 84% yield (Scheme 100). This tricyclic intermediate was ultimately further advanced to provide the cis-anti-cis tricycle(6.3.0.0)undecane skeletal framework of capnellene 145.
Scheme 100.

Synthesis of (±)-Capnellene by Grubbs and Stille
The structure the potent nonpeptidic toxin, maitotoxin 554, was elucidated in 1996147 and contains 32 ring systems, most of them fused consecutively, and 98 stereocenters (Scheme 101). Nicolaou and coworkers reported the construction of the JKL, OPQ, and UVW ring systems relying on carbonyl-olefin metathesis reactions mediated by the Tebbe reagent. Utilizing their previously reported reaction protocol64 for the formation of cyclic enol ethers from olefin and ester moieties, the P-ring subunit was formed via carbonyl-olefin metathesis in 54% yield starting from ester 555 linking the Q- and O-rings (Scheme 102).148 The V-ring was formed in an analogous manner in 36% yield proving that this this a powerful strategy for the rapid construction of the complex cyclic polyether framework common to maitotoxin and structurally related, smaller natural products.
Scheme 101.
Structure of Maitotoxin, a Natural Toxin Produced by Gambierdiscus toxicus
Scheme 102.

Synthesis of Maitotoxin Subunits Relying on Carbonyl-Olefin Metathesis by Nicolaou and Coworkers
The marine ladder toxins possess highly complex structures and interesting biological properties, including neurotoxicity and antimicrobial properties.149–153 Hemibrevetoxin B (561)154 belongs to the marine ladder toxin family and consists of 4 heterocyclic rings and 10 stereocenters. Rainier and coworkers approached the synthesis of hemibrevetoxin B (561) based on a general strategy to enable the coupling of C-glycosides via carbonyl-olefin metathesis to form the spirocyclic subunit B (560).155 Specifically, they relied on two-step approach previously developed in their laboratory65 to first convert ester 559 in a carbonyl olefination following modified Takai-Utimoto66 conditions and subsequent olefin metathesis reaction of the alkene formed resulting in bicycle 560 in 82% yield over two steps (Scheme 103).
Scheme 103.
Strategy Towards Hemibrevetoxin B by Rainier and Coworkers
The gambieric acids A-D are also members of the marine ladder toxin family and were isolated from the marine dinoflagellate Gambierdiscus toxicus in 1992 by Yasumoto and coworkers.156 This family of toxins incorporates a common skeletal structure consisting of one 9-membered ring, two 7-membered rings, six 6-membered rings and one 5-membered ring along with 27 stereocenters. The construction of the B-ring of the A-E subunit of gambieric acid was achieved via Rainier’s previously reported conditions for metal alkylidene-mediated carbonyl-olefin metathesis65 (Scheme 104). Similar to the previous report, the use of the modified Takai-Utimoto conditions relying on dibromomethane did not result in direct carbonyl-olefin metathesis but rather alkene 563 formed in a carbonyl olefination, which was subsequently subjected to a ruthenium alkylidene catalyst to initiate olefin-olefin metathesis and result in the formation of the desired cyclic enol ether product. Interestingly, when dibromoethane was employed in the reaction protocol to generate the Takai-Utimoto reagent, the cyclic enol ether product 565 corresponding to the direct carbonyl-olefin metathesis product was exclusively formed in 50% yield. Rainier proposed that the carbonyl-olefin metathesis product 565 is formed from an olefin metathesis, carbonyl olefination mechanism suggesting that the more sterically hindered titanium ethylidene preferentially undergoes a reaction with the olefin moiety over the carbonyl in 564.156
Scheme 104.

Strategy Towards Gambieric acids by Rainier and Coworkers
Following their report of the construction of A-E subunits of gambieric acid, Rainier and coworkers reported the total synthesis of brevenal in 2011.158 Brevenal is also part of the ladder toxin family of natural products and displays a wide variety of biological properties including neurotoxicity and antimicrobial activity.159 The presence of cyclic ethers allows for the application of Rainier’s previously reported olefinic-ester cyclization reaction sequence to enable carbonyl-olefin metathesis.65 Importantly, the modified Takai-Utimoto reaction conditions developed relying on dibromoethane as reagent allowed for the successful formation of the A (567)-, E (569)-, C (571)- and D (563)- ring subunits of brevenal 574 in yields of up to 88% (Scheme 105).
Scheme 105.

Strategy Towards Brevenal by Rainier and Coworkers
In 2011, Keck and coworkers160 reported an additional application of carbonyl-olefin metathesis towards the first total synthesis of bryostatin 1, a well-known natural product originally isolated in 1982 by Pettit and coworkers161 that has shown biological activity against a range of cancers162 and exhibited activity similar to those of established oncolytic agents such as Taxol.163 The formation of the C-ring of bryostatin 1 was achieved relying on the modified Takai-Utimoto conditions previously developed by Rainier157 in 2007 to construct glycal 576 in 80% yield (Scheme 106). This high yielding application allowed for the development of the first total synthesis of bryostatin 1 (577) in 30 steps from commercially available (R)-isobutyl lactate.
Scheme 106.

Strategy Towards Bryostatin 1 by Keck and Coworkers
Cocculidine164 is an Erythrina alkaloid with a benz(g)indolizinone scaffold that has been of great interest to the synthetic organic community. Sarpong and coworkers reported the syntheses of (±)-3-demethoxyerythratidinone and (±)-cocculidine 579 in 2013 (Scheme 107).165 Specifically, completion of the synthesis of (±)-cocculidine (579) was achieved in a carbonyl-olefin metathesis reaction mediated by stoichiometric amounts of Schrock’s catalyst166 following reaction conditions originally developed by Grubbs and Fu60 in 84% yield.
Scheme 107.
Synthesis of (±)-Cocculidine by Sarpong and Coworkers
An additional application of Schrock’s metal alkylidene complex for carbonyl-olefin metathesis reactions in natural product synthesis was reported by Lei and coworkers in 2015167 for the total syntheses of (−)-huperzine Q (582) and (+)-lycopladines B (583) and C (584). The Lycopodium alkaloids are a large family of structurally unique natural products with almost 300 isolated members.168,169 (−)-Huperzine Q (582) is a pentacyclic alkaloid that possesses a distinctive aminal moiety and 6 stereogenic centers. The Lei group envisioned a regioselective carbonyl-olefin metathesis reaction of 1,3-diketone 580 to establish the cyclopentene moiety in 581 (Scheme 108). Upon the formation of diketone 580, the direct metathesis reaction between the olefin and carbonyl moieties was explored and after extensive investigation it was determined that in the presence of a stoichiometric amounts of Schrock’s catalyst and ethylene gas (1 atm), carbonyl-olefin metathesis product 581 could be obtained in yields of up to 48%. Control experiments determined that the ethylene gas increased the catalyst’s reactivity by converting the initial metal alkylidene 585 into a more active species 586 that underwent a more facile reaction with 1,3-diketone 580. Furthermore, the ethylene reacted with excess alkylidene 587 regenerating 580 and preventing further byproduct formation.
Scheme 108.

Synthesis of Huperzine Q, Lycoplanadine B and C by Lei and Coworkers
These examples of using carbonyl-olefin ring-closing metathesis for the synthesis of natural products demonstrate the utility and importance of the overall transformation. Currently, the use of metal alkylidenes remains the state-of-the-art strategy for performing such reactions, leaving room for improvement in both sustainability and catalytic turnover. As the field of carbonyl-olefin metathesis continues to expand, it is expected that the translation of current and future catalytic protocols will be utilized in order to overcome challenges in total synthesis.
9. SUMMARY AND FUTURE PERSPECTIVES
In summary, we have provided a comprehensive overview of the currently available methods for carbonyl-olefin metathesis. These include the Paternò-Büchi cycloadditions followed by the fragmentation of the resulting oxetane, metal alkylidene-mediated strategies, [3+2]-cycloaddition approaches with organocatalysts, Lewis acid-mediated and Lewis acid-catalyzed approaches relying on intermediate oxetanes, and lastly, protocols based on initial carbon-carbon bond formation between carbonyls and alkenes and ensuing Grob-fragmentations. While the earliest studies of carbonyl-olefin metathesis reactions date back to the 1960s, many of the developments in the field have occurred within the last eight years. These recent efforts have resulted in efficient protocols for catalytic carbonyl-olefin metathesis reactions, including organocatalytic strategies based on strained hydrazines as catalysts, as well as Lewis acid-catalyzed approaches to enable ring-closing, ring-opening, cross and transannular carbonyl-olefin metathesis.
Despite the recent accomplishments in reaction development, there remain a number of challenges related to the field of carbonyl-olefin metathesis. The employment of metal alkylidenes currently represents the most applied methods for carbonyl-olefin metathesis in organic synthesis despite the requirement for stoichiometric amounts of the metal alkylidene-complex. Consequently, recent catalytic approaches for carbonyl-olefin metathesis need to be further advanced to broaden the substrate scope with regard to functional group tolerance to ultimately enable efficient applications in complex molecule synthesis. Organocatalytic methods for catalytic carbonyl-olefin metathesis provide a promising alternative but are yet limited in substrate scope. However, especially the ring-opening approaches are desirable as they provide access to products that are complementary to those currently accessible with Lewis acid-catalyzed cross carbonyl-olefin metathesis reactions. The ability to transform a wider variety of cyclic olefin substrates in the existing ring-opening carbonyl-olefin metathesis reaction would allow for a significant expansion in methodology. The use of Lewis acids to catalyze carbonyl-olefin metathesis reactions has shown to have a wide range of applications in terms of the categories of metathesis reaction and substrate scope. Notably, strategies for ring-closing, ring-opening, cross, and transannular carbonyl-olefin metathesis have been developed within the past five years for Lewis acid-catalyzed carbonyl-olefin metathesis. However, the ability to tolerate exceedingly Lewis basic sites and the formation of medium- and larger-sized rings has yet to be attained and represents a current frontier in Lewis acid-catalyzed carbonyl-olefin ring-closing metathesis. Cross carbonyl-olefin metathesis reactions are currently low yielding and remain limited in scope despite invested efforts undertaken by multiple research laboratories. A more robust catalytic cross carbonyl-olefin metathesis strategy will have to comprise more variability in the substitution of the olefin partner, which is currently limited to trisubstituted alkenes, and the reduction of the required excess of aryl carbonyl partner. Future catalyst development will have to focus on an enhanced differentiation between distinct reactivity modes of carbonyl and alkene functionalities. Specifically, while carbonyl-ene reactivity was shown to be crucial in ring-closing carbonyl-olefin metathesis for 6-membered ring systems, it can also provide detrimental byproduct(s) in others, such as ring-opening carbonyl-olefin metathesis. Future, improved reaction protocols will have to overcome these synthetic challenges and hamper unwanted side reactivity to further promote the desired transformation. This is particularly the case for the currently low-yielding Lewis acid-catalyzed ring-opening and cross carbonyl-olefin metathesis reactions. Additional future directions for the field include the application of recently reported carbonyl-olefin metathesis strategies towards efficient polymerization reactions. While initial reports dating back to the 1990s relying on the use of WCl6 are promising, mechanistic insights into these carbonyl-olefin metathesis polymerizations are limited and future developments in this area will benefit greatly from a better understanding of the controlling features of these transformations.
Scheme 33.
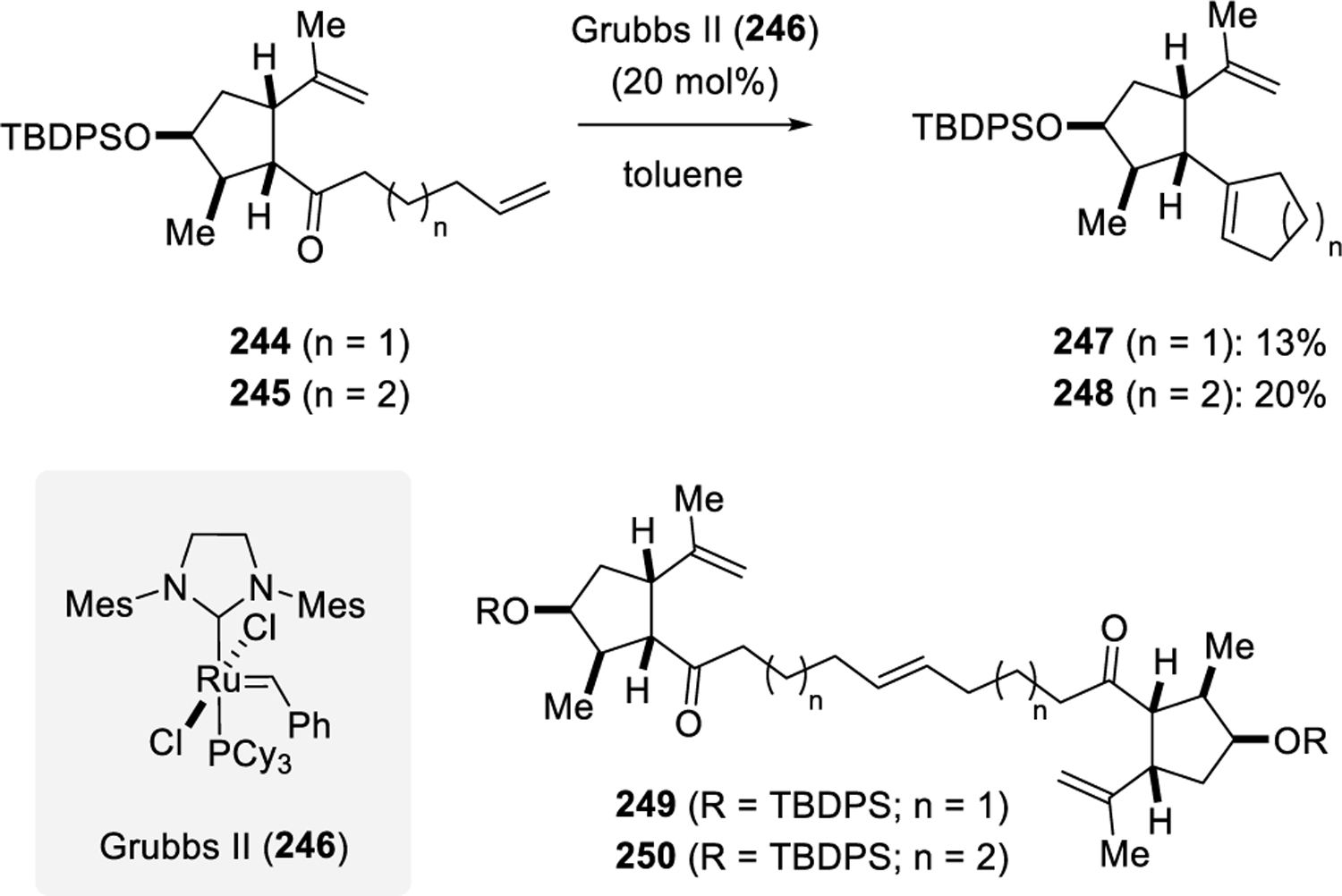
Ruthenium Alkylidene-Mediated Carbonyl-Olefin Metathesis as Reported by Chakraborty and Roy
Scheme 40.
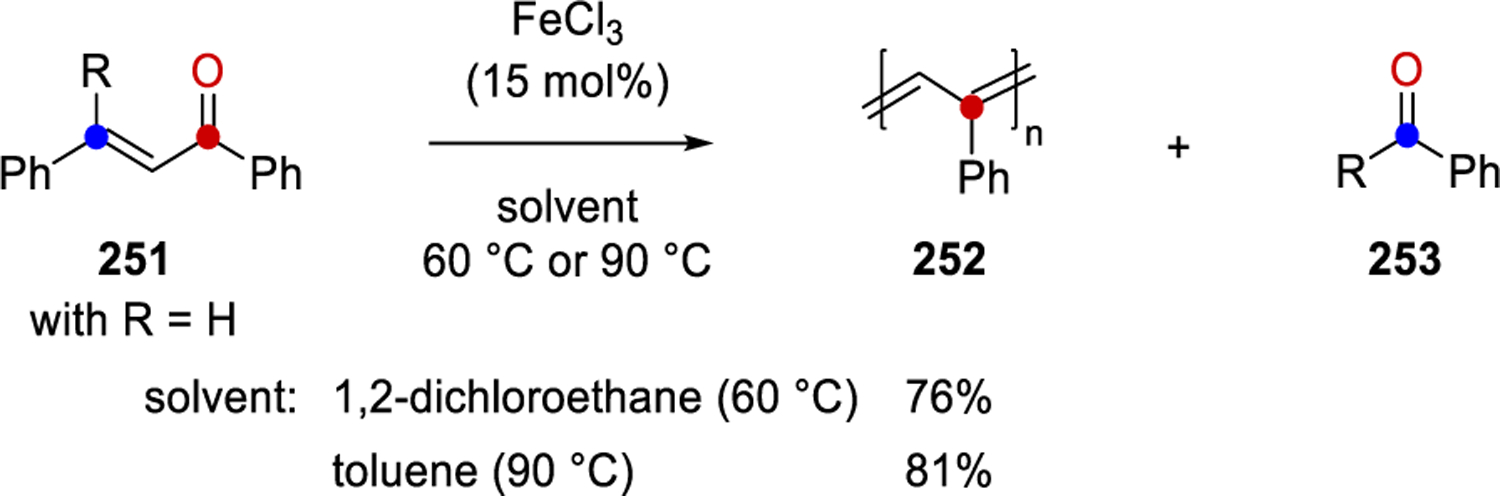
Synthesis of polyphenylacetylene by iron(III) chloride by Dimova
Scheme 82.

FeCl3-Homodimers as Active Catalytic Species in the Carbonyl-Olefin Metathesis of Aliphatic Ketones as Proposed by Schindler and Coworkers
Acknowledgments
This work was supported by the NIH/National Institute of General Medical Sciences (R01-GM118644 to C.S.S.), the NSF (NSF CHE1654223), the Alfred P. Sloan Foundation, the Camille and Henry Dreyfus Foundation, and the David and Lucile Packard Foundation (fellowships to C.S.S.). T.H.L. is grateful for financial support from NIGMS (R35 GM127135).
Author Bios
Haley Albright
Haley Albright received her undergraduate degree from the University of Wisconsin-Madison in 2014 under the supervision of Professor Robert C. West. She then moved to the University of Michigan to pursue her doctoral studies with Professor Corinna S. Schindler where her research focused on the development of a variety of Lewis acid-catalyzed carbonyl-olefin metathesis reaction methodologies. Upon her graduation in 2019, she joined the department at the University of Michigan as a Lecturer before beginning her independent career at Shepherd University in 2021.
Ashlee J. Davis
Ashlee Davis received her undergraduate degree from the University of California, Irvine in 2016 under the supervision of Professor Suzanne A. Blum. In 2017, she moved to the University of Michigan, where she joined the Schindler group to pursue a doctoral degree with a focus on the development and application of Lewis-acid catalyzed carbonyl-olefin metathesis transformations. Her research interests include method development, organometallics, and mechanistic investigation.
Jessica L. Gomez-Lopez
Jessica L. Gomez-Lopez received her M.S. and Ph.D. degree in Chemistry from Technological Institute of Tijuana advised by Miguel P. Parra-Hake in 2013 and Valentín Miranda-Soto in 2016, respectively. She then joined the Schindler group at University of Michigan as a postdoctoral fellow. Her research interests included coordination chemistry, organometallics, and method development
Hannah L. Vonesh
Hannah L. Vonesh received her undergraduate degree from Loyola University Chicago in 2017 under the supervision of Professor James J. Devery, III. She then moved to the University of Michigan to pursue her doctoral studies with Professor Corinna S. Schindler where her research has focused on the elucidation of carbonyl-olefin metathesis reaction mechanisms and the development of Lewis acid-catalyzed methodologies. Her research interests include mechanistic investigation, kinetics, and method development.
Phong K. Quach
Phong K. Quach received his undergraduate degree from Trinity College in 2017 under the supervision of Professor Cheyenne S. Brindle. He then moved to Cornell University to pursue his doctoral studies with Professor Tristan H. Lambert where his research has focused on the development of organocatalytic carbonyl-olefin metathesis methodologies. His research interests include organocatalysis and method development.
Tristan H. Lambert
Tristan H. Lambert graduated from the University of Wisconsin at Platteville with a B.S. in chemistry. He received an M.S. at UC-Berkeley in 2000 and a Ph.D. from Caltech in 2004. After a postdoctoral fellowship at the Memorial Sloan-Kettering Cancer Center, he began his independent career at Columbia University in 2006. He moved to Cornell University in 2018. His research group is interested in catalysis, molecular structure, and method development.
Corinna S. Schindler
Corinna S. Schindler received her undergraduate and M.S. degrees from the Technical University of Munich in Germany, her Ph.D. in chemistry from the ETH Zurich in Switzerland with Prof. Erick M. Carreira in 2010 and completed postdoctoral studies at Harvard University with Prof. Eric N. Jacobsen in 2013. The development and advance of Lewis acid-catalyzed carbonyl-olefin metathesis reactions have been at the forefront of her research efforts since starting her independent career at the University of Michigan in 2013.
REFERENCES
- (1).Fürstner A Olefin Metathesis and Beyond. Angew. Chem., Int. Ed. Engl. 2000, 39, 3012–3043. [PubMed] [Google Scholar]
- (2).Fürstner A Olefinmetathese Und Mehr. Angew. Chem. 2000, 112, 3140–3172. [Google Scholar]
- (3).Chatterjee AK; Choi T-L; Sanders DP; Grubbs RH A General Model for Selectivity in Olefin Cross Metathesis. J. Am. Chem. Soc. 2003, 125, 11360–11370. [DOI] [PubMed] [Google Scholar]
- (4).Schrock RR; Hoveyda AH Molybdenum and Tungsten Imido Alkylidene Complexes as Efficient Olefin-Metathesis Catalysts. Angew. Chem. Int. Ed. 2003, 42, 4592–4633. [DOI] [PubMed] [Google Scholar]
- (5).Connon SJ; Blechert S Recent Developments in Olefin Cross-Metathesis. Angew. Chem., Int. Ed. Engl. 2003, 42, 1900–1923. [DOI] [PubMed] [Google Scholar]
- (6).Connon SJ; Blechert S Recent Advances in Alkene Metathesis. Orgoanomet. Chem. 2004, 93–124. [Google Scholar]
- (7).Grubbs RH Olefin Metathesis. Tetrahedron 2004, 60, 7117–7140. [Google Scholar]
- (8).Nicolaou KC; Bulger PG; Sarlah D Metathesis Reactions in Total Synthesis. Angew. Chem., Int. Ed. Engl. 2005, 44, 4490–4527. [DOI] [PubMed] [Google Scholar]
- (9).Hoveyda AH; Zhugralin AR The Remarkable Metal-Catalysed Olefin Metathesis Reaction. Nature 2007, 450, 243–251. [DOI] [PubMed] [Google Scholar]
- (10).Grela K Olefin Metathesis: Theory and Practice; Wiley-VCH: Weinheim, Germany, 2014. [Google Scholar]
- (11).Handbook of Metathesis, 2nd ed.; Grubbs RH, Wenzel AG, O’Leary DJ, Khosravi E, Eds.; Wiley-VCH: Weinheim, Germany, 2015; Vols. 1–3. [Google Scholar]
- (12).Ogba OM; Warner NC; O’Leary DJ; Grubbs RH Recent Advances in Ruthenium-Based Olefin Metathesis. Chem. Soc. Rev. 2018, 47, 4510–4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Liu Z; Qin C; Koengeter T; Mu Y; Hoveyda AH Impact on Ethylene on Efficiency and Stereochemical in Olefin Metathesis: When to Add It, When to Remove It, and When to Avoid It. Angew. Chem., Int. Ed. Engl. 2020, 59, 22324–22348. [DOI] [PubMed] [Google Scholar]
- (14).Schopov I; Jossifov C A Carbonyl-Olefin Exchange Reaction - New Route to Polyconjugated Polymers, 1. A New Synthesis of Polyphenylacetylene. Makromol. Chem., Rapid Commun 1983, 4, 659–662. [Google Scholar]
- (15).Schopov I; Mladenova L; Kovachev G A Carbonyl-Olefin Exchange-Reaction. New Route to Conjugated Polymers. 3a). Influence of the Reaction Conditions. Makromol. Chem 1988, 189, 1787–1792. [Google Scholar]
- (16).Schopov I; Mladenova L A Carbonyl-Olefin Exchange-Reaction. New Route to Conjugated Polymers. 4a). Chain Growth. Side Reactions. Makromol. Chem 1989, 190, 1483–1488. [Google Scholar]
- (17).Schopov I; Mladenova L A Carbonyl-Olefin Exchange-Reaction - New Route to Conjugated Polymers. 2a). A New Synthesis of Polydiphenylacetylene. Makromol. Chem., Rapid Commun 1985, 6, 659–663. [Google Scholar]
- (18).Schopov I A Carbonyl-Olefin Exchange-Reaction - New Route to Conjugated Polymers. Acta Polym. 1988, 39, 91–94. [Google Scholar]
- (19).Jossifov C; Schopov I Carbonyl-Olefin Exchange-Reaction. A New Route to Conjugated Polymers. 5a). Effect of Different Catalytic Systems. Makromol. Chem 1991, 192, 857–861. [Google Scholar]
- (20).Jossifov C; Schopov I Carbonyl-Olefin Exchange-Reaction. A New Route to Conjugated Polymers. 6a). Low-Molecular-Weight Products. Makromol. Chem. 1991, 192, 863–866. [Google Scholar]
- (21).Valiulin RA; Kutateladze AG Harvesting the Strain Installed by a Paternò-Büchi Step in a Synthetically Useful Way: High-Yielding Photoprotolytic Oxametathesis in Polycyclic Systems. Org. Lett. 2009, 11, 3886–3889. [DOI] [PubMed] [Google Scholar]
- (22).Valiulin RA; Arisco TM; Kutateladze AG Double-Tandem [4π+2π]·[2π+2π]·[4π+2π]·[2π+2π] Synthetic Sequence with Photoprotolytic Oxametathesis and Photoepoxidation in the Chromone Series. J. Org. Chem. 2011, 76, 1319–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Valiulin RA; Arisco TM; Kutateladze AG Photoinduced Intramolecular Cyclopentanation vs Photoprotolytic Oxametathesis in Polycyclic Alkenes Outfitted with Conformationally Constrained Aroylmethyl Chromophores. J. Org. Chem. 2013, 78, 2012–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Lambert TH Development of a Hydrazine-Catalyzed Carbonyl-Olefin Metathesis Reaction. Synlett 2019, 30, 1954–1965. [Google Scholar]
- (25).Ludwig JR; Schindler CS Lewis Acid Catalyzed Carbonyl-Olefin Metathesis. Synlett 2017, 28, 1501–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Ravindar L; Lekkala R; Rakesh KP; Asiri AM; Marwani HM; Qin HL Carbonyl-Olefin Metathesis: a Key Review. Org. Chem. Front. 2018, 5, 1381–1391. [Google Scholar]
- (27).Riehl PS; Nasrallah DJ; Schindler CS Catalytic, Transannular Carbonyl-Olefin Metathesis Reactions. Chem. Sci. 2019, 10, 10267–10274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Scharf D; Korte F Photosensibilisierte Cyclodimerisierung Von Norbornen. Tetrahedron Lett. 1963, 821–823. [Google Scholar]
- (29).Kohler EP; Richtmyer NK Isoxazoline oxides. IX. The reaction between triphenyl isoxazoline oxide and organic magnesium compounds. J. Am. Chem. Soc. 1930, 52, 2038–2046. [Google Scholar]
- (30).Büchi G; Inman CG; Lipinsky ES Light-Catalyzed Organic Reactions. I. The Reaction of Carbonyl Compounds with 2-Methyl-2-Butene in the Presence of UV-Light. J. Am. Chem. Soc. 1954, 76, 4327–4331. [Google Scholar]
- (31).Adames G; Bibby C; Grigg R Rhodium(I) Catalysed Rearrangements of Vinyl Epoxides and Oxetans. J. Chem. Soc., Chem. Commun. 1972, 491–492. [Google Scholar]
- (32).Jones G; Schwartz SB; Marton MT Regiospecific Thermal Cleavage of Some Oxetan Photoadducts: Carbonyl-Olefin Metathesis in Sequential Photochemical and Thermal Steps. J. Chem. Soc., Chem. Commun. 1973, 374–375. [Google Scholar]
- (33).Jones G; Acquadro MA; Carmody MA Long-Chain Enals via Carbonyl-Olefin Metathesis. An Application in Pheromone Synthesis. J. Chem. Soc., Chem. Commun. 1975, 206–207. [Google Scholar]
- (34).Carless HAJ; Trivedi HS New Ring Expansion Reaction of 2-t-Butyloxetans. J. Chem. Soc., Chem. Commun. 1979, 382–383. [Google Scholar]
- (35).Barlow MG; Coles B; Haszeldine RN Heterocyclic Polyfluoro-Compounds. Part 31. Photochemical Oxetan Formation from Fluoroketones and Perfluoroaldehydes and 1,2-Difluoroethylene. J. Chem. Soc., Perkin Trans. 1 1980, 2258–2267. [Google Scholar]
- (36).Imai T; Nishida S Thermal Fragmentation of 3-Alkyl-2-Phenyloxetanes, 3,3-Dimethyl-2-Aryloxetanes, and Related-Compounds. A Case Study of 2-Aryl-Substituted Oxetanes. Can. J. Chem. 1981, 59, 2503–2509. [Google Scholar]
- (37).Maruyama K; Muraoka M; Naruta Y Keto Oxetanes Produced from Photocycloaddition of o-Quinone and Their Thermolysis. Reaction of 9,10-Phenanthrenequinone with Internally Highly Strained Cyclic Olefins. J. Org. Chem. 1981, 46, 983–989. [Google Scholar]
- (38).Nakabayashi K; Kojima J; Tanabe K; Yasuda M; Shima K Organic Photochemical-Reactions. XXXI. Photosensitized Ring-Cleavage Reactions of 2,2-Diaryloxetanes by Aromatic Nitriles. Bull. Chem. Soc. Jpn. 1989, 62, 96–101. [Google Scholar]
- (39).Nakabayashi K; Fujimura S; Yasuda M; Shima K Organic Photochemical-Reactions. XXXII. Photochemical Ring-Cleavage Reactions of 2,2-Diaryloxetanes in the Presence of Electron-Donor. Bull. Chem. Soc. Jpn. 1989, 62, 2733–2735. [Google Scholar]
- (40).Miranda MA; Izquierdo MA; Galindo F Involvement of Triplet Excited States and Olefin Radical Cations in Electron-Transfer Cycloreversion of Four-Membered Ring Compounds Photosensitized by (Thia)Pyrylium Salts. J. Org. Chem. 2002, 67, 4138–4142. [DOI] [PubMed] [Google Scholar]
- (41).Miranda MA; Izquierdo MA Stepwise Cycloreversion of Oxetane Radical Cations with Initial C-O Bond Cleavage. J. Am. Chem. Soc. 2002, 124, 6532–6533. [DOI] [PubMed] [Google Scholar]
- (42).Perez-Ruiz R; Gil S; Miranda MA Stereodifferentiation in the Photochemical Cycloreversion of Diastereomeric Methoxynaphthalene-Oxetane Dyads. J. Org. Chem. 2005, 70, 1376–1381. [DOI] [PubMed] [Google Scholar]
- (43).Perez-Ruiz R; Miranda MA; Alle R; Meerholz K; Griesbeck AG An Efficient Carbonyl-Alkene Metathesis of Bicyclic Oxetanes: Photoinduced Electron Transfer Reduction of the Paternò–Büchi Adducts from 2,3-Dihydrofuran and Aromatic Aldehydes. Photochem. Photobiol. Sci. 2006, 5, 51–55. [DOI] [PubMed] [Google Scholar]
- (44).D'Auria M; Racioppi R; Viggiani L Paternò–Büchi Reaction Between Furan and Heterocyclic Aldehydes: Oxetane Formation vs. Metathesis. Photochem. Photobiol. Sci. 2010, 9, 1134–1138. [DOI] [PubMed] [Google Scholar]
- (45).Bielawski CW; Grubbs RH Living Ring-Opening Metathesis Polymerization. Prog. Polym. Sci. 2007, 32, 1–29. [Google Scholar]
- (46).Gilliom LR; Grubbs RH Titanacyclobutanes Derived from Strained Cyclic Olefins: the Living Polymerization of Norbornene. J. Am. Chem. Soc. 1986, 108, 733–742. [Google Scholar]
- (47).Chauvin YH, Catalyse de Transformation des Oléfines par les Complexes du Tungstène PJ-L II. Télomérisation des Oléfines Cycliques en Présence d'Oléfines Acycliques. Die Makromolekulare Chemie. 1970, 141, 161–176. [Google Scholar]
- (48).Grubbs RH; Carr DD; Hoppin C; Burk PL Consideration of Mechanism of Metal-Catalyzed Olefin Metathesis Reaction. J. Am. Chem. Soc. 1976, 98, 3478–3483. [Google Scholar]
- (49).Stille JR; Grubbs RH Synthesis of (±)-Δ(9,12)-Capnellene Using Titanium Reagents. J. Am. Chem. Soc. 1986, 108, 855–856. [Google Scholar]
- (50).Stille JR; Santarsiero BD; Grubbs RH Rearrangement of Bicyclo[2.2.1]Heptane Ring Systems by Titanocene Alkylidene Complexes to Bicyclo[3.2.0]Heptane Enol Ethers - Total Synthesis of (±)-Δ(9,12)-Capnellene. J. Org. Chem. 1990, 55, 843–862. [Google Scholar]
- (51).Fischer EO; Maasböl A On Existence of Tungsten Carbonyl Carbene Complex. Angewandte Chemie-International Edition; 1964, 3, 580–581. [Google Scholar]
- (52).Schrock RR First Isolable Transition Metal Methylene Complex and Analogs. Characterization, Mode of Decomposition, and Some Simple Reactions. J. Am. Chem. Soc. 1975, 97, 6577–6578. [Google Scholar]
- (53).Casey CP; Burkhard.Tj. Reaction of Metal-Carbene Complexes with Wittig Reagents. New Vinyl Ether Synthesis. J. Am. Chem. Soc. 1972, 94, 6543–6544. [Google Scholar]
- (54).Guggenberger LJ; Schrock RR Structure of Bis(Cyclopentadienyl)Methylmethylenetantalum and Estimated Barrier to Rotation About Tantalum-Methylene Bond. J. Am. Chem. Soc. 1975, 97, 6578–6579. [Google Scholar]
- (55).Schrock RR Multiple Metal-Carbon Bonds. 5. The Reaction of Niobium and Tantalum Neopentylidene Complexes with the Carbonyl Function. J. Am. Chem. Soc. 1976, 98, 5399–5400. [Google Scholar]
- (56).Tebbe FN; Parshall GW; Reddy GS Olefin Homologation with Titanium Methylene Compounds. J. Am. Chem. Soc. 1978, 100, 3611–3613. [Google Scholar]
- (57).Pine SH; Zahler R; Evans DA; Grubbs RH Titanium-Mediated Methylene-Transfer Reactions. Direct Conversion of Esters into Vinyl Ethers. J. Am. Chem. Soc. 1980, 102, 3270–3272. [Google Scholar]
- (58).Petasis NA; Bzowej EI Titanium-Mediated Carbonyl Olefinations. 1. Methylenations of Carbonyl-Compounds with Dimethyltitanocene. J. Am. Chem. Soc. 1990, 112, 6392–6394. [Google Scholar]
- (59).Brown-Wensley KA; Buchwald SL; Cannizzo L; Clawson L; Ho S; Meinhardt D; Stille JR; Straus D; Grubbs RH Cp2TiCH2 Complexes in Synthetic Applications. Pure Appl. Chem. 1983, 55, 1733–1744. [Google Scholar]
- (60).Fu GC; Grubbs RH Synthesis of Cycloalkenes via Alkylidene-Mediated Olefin Metathesis and Carbonyl Olefination. J. Am. Chem. Soc. 1993, 115, 3800–3801. [Google Scholar]
- (61).Murdzek JS; Schrock RR Well-Characterized Olefin Metathesis Catalysts that Contain Molybdenum. Organometallics 1987, 6, 1373–1374. [Google Scholar]
- (62).Schrock RR; Depue RT; Feldman J; Schaverien CJ; Dewan JC; Liu AH Preparation and Reactivity of Several Alkylidene Complexes of the Type W(CHR’)(N-2,6-C6H3-i-Pr2)(OR)2 and Related Tungstacyclobutane Complexes. Controlling Metathesis Activity Through the Choice of Alkoxide Ligand. J. Am. Chem. Soc. 1988, 110, 1423–1435. [Google Scholar]
- (63).Schrock RR; Depue RT; Feldman J; Yap KB; Yang DC; Davis WM; Park L; Dimare M; Schofield M; Anhaus J et al. Further Studies of Imido Alkylidene Complexes of Tungsten, Well-Characterized Olefin Metathesis Catalysts with Controllable Activity. Organometallics 1990, 9, 2262–2275. [Google Scholar]
- (64).Nicolaou KC; Postema MHD; Claiborne CF Olefin Metathesis in Cyclic Ether Formation. Direct Conversion of Olefinic Esters to Cyclic Enol Ethers with Tebbe-type Reagents. J. Am. Chem. Soc. 1996, 118, 1565–1566. [Google Scholar]
- (65).Rainier JD; Allwein SP An Iterative Approach to Fused Ether Ring Systems. J. Org. Chem. 1998, 63, 5310–5311. [Google Scholar]
- (66).Takai K; Kakiuchi T; Kataoka Y; Utimoto K A Novel Catalytic Effect of Lead on the Reduction of a Zinc Carbenoid with Zinc Metal Leading to a Geminal Dizinc Compound. Acceleration of the Wittig-Type Olefination with the RCHX2-TiCl4-Zn Systems by Addition of Lead. J. Org. Chem. 1994, 59, 2668–2670. [Google Scholar]
- (67).Bennasar ML; Roca T; Monerris M; García-Díaz D Sequential N-Acylamide Methylenation-Enamide Ring-Closing Metathesis: a Synthetic Entry to 1,4-Dihydroquinolines. Tetrahedron Lett. 2005, 46, 4035–4038. [Google Scholar]
- (68).Bennasar ML; Roca T; Monerris M; García-Díaz D Sequential N-Acylamide Methylenation-Enamide Ring-Closing Metathesis: Construction of Benzo-Fused Nitrogen Heterocycles. J. Org. Chem. 2006, 71, 7028–7034. [DOI] [PubMed] [Google Scholar]
- (69).Majumder U; Rainier JD Olefinic-Ester Cyclizations Using Takai-Utimoto Reduced Titanium Alkylidenes. Tetrahedron Lett. 2005, 46, 7209–7211. [Google Scholar]
- (70).Iyer K; Rainier JD Olefinic Ester and Diene Ring-Closing Metathesis Using a Reduced Titanium Alkylidene. J. Am. Chem. Soc. 2007, 129, 12604–12605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Zhang Y; Rainier JD Two-Directional Olefinic-Ester Ring-Closing Metathesis using Reduced Ti Alkylidenes. A Rapid Entry into Polycyclic Ether Skeletons. Org. Lett. 2009, 11, 237–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Rohanna JC; Rainier JD Olefinic-Lactone Cyclizations to Macrocycles. Org. Lett. 2009, 11, 493–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Mallinson J; Collins I Macrocycles in New Drug Discovery. Future Med. Chem. 2012, 4, 1409–1438. [DOI] [PubMed] [Google Scholar]
- (74).Zhou J; Rainier JD Olefinic-Amide and Olefinic-Lactam Cyclizations. Org. Lett. 2009, 11, 3774–3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Chakraborty P; Roy SC Study Towards Diversity Oriented Synthesis of Optically Active Substituted Cyclopentane Fused Carbocyclic and Oxacyclic Medium-Sized Rings: Competition Between Grubbs-II Catalyzed Ring Closing Olefin Metathesis and Ring Closing Carbonyl-Olefin Metathesis. J. Chem. Sci. 2016, 128, 1831–1840. [Google Scholar]
- (76).Nezakati T; Seifalian A; Tan A; Seifalian AM Conductive Polymers: Opportunities and Challenges in Biomedical Applications. Chem. Rev. 2018, 118, 6766–6843. [DOI] [PubMed] [Google Scholar]
- (77).Design and Synthesis of Conjugated Polymers; Lecrec M; Morin J-F, Eds.; Wiley-VCH: Weinheim, Germany, 2010. [Google Scholar]
- (78).Jossifov C; Schopov I Synthesis of Polymethylacetylene from Mesityl Oxide. Eur. Polym. J. 1993, 29, 621–623. [Google Scholar]
- (79).Schopov I; Mladenova L Synthesis and Properties of Methylene Bridged Polyenes. Synth. Met. 1992, 48, 249–258. [Google Scholar]
- (80).Jossifov C Speculations on a Possible Mechanism of a Tungsten Catalyzed 1,2,3,3-Tetraphenylprop-2-En-1-One Polymerization. Eur. Polym. J. 1993, 29, 9–13. [Google Scholar]
- (81).Jossifov C Speculations on the Possible Mechanism of the New Routes to Polymer Synthesis by Friedel-Crafts Metathesis Catalytic Systems; Kluwer Academics Publishers: Dordrecht, Netherlands, 2002. [Google Scholar]
- (82).Mcmurry JE Carbonyl-Coupling Reactions Using Low-Valent Titanium. Chem. Rev. 1989, 89, 1513–1524. [Google Scholar]
- (83).Dragutan V; Balaban AT; Dimonie M Olefin Metathesis and Ring-Opening Polymerizations of Cycloolefins; Wiley-Interscience: New York, 1986. [Google Scholar]
- (84).Ivin KJ; Mol JC Olefin Metathesis and Metathesis Polymerization; 2nd ed.; Academic Press: San Diego, USA, 1997. [Google Scholar]
- (85).Bryan JC; Mayer JM Oxidative Addition of Carbon-Oxygen and Carbon-Nitrogen Double Bonds to WCl2(PMePh2)4. Synthesis of Tungsten Metallaoxirane and Tungsten Oxo-and Imido-Alkylidene Complexes. J. Am. Chem. Soc. 1990, 112, 2298–2308. [Google Scholar]
- (86).Jossifov C Polymer Formation via Reductive Coupling of a Diketone by Metathesis Catalytic Systems. Eur. Polym. J. 1998, 34, 883–885. [Google Scholar]
- (87).Penchev H; Dimova SS; Zaharieva KL; Ublekov FS; Novakov Ch; Sinigersky V Synthesis of polyphenylacetylene by iron(III) chloride catalyzed carbonyl olefin metathesis polymerization of chalcone. Bulgarian Chemical Communications, 2018, 50, 169–173. [Google Scholar]
- (88).Griffith AK; Vanos CM; Lambert TH Organocatalytic Carbonyl-Olefin Metathesis. J. Am. Chem. Soc. 2012, 134, 18581–18584. [DOI] [PubMed] [Google Scholar]
- (89).Hong X; Liang Y; Griffith AK; Lambert TH; Houk KN Distortion-Accelerated Cycloadditions and Strain-Release-Promoted Cycloreversions in the Organocatalytic Carbonyl-Olefin Metathesis. Chem. Sci. 2014, 5, 471–475. [Google Scholar]
- (90).Jermaks J; Quach PK; Seibel ZM; Pomarole J; Lambert TH Ring-Opening Carbonyl-Olefin Metathesis of Norbornenes. Chem. Sci. 2020, 11, 7884–7895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Zhang YF; Jermaks J; MacMillan SN; Lambert TH Synthesis of 2H-Chromenes via Hydrazine-Catalyzed Ring-Closing Carbonyl-Olefin Metathesis. ACS Catal. 2019, 9, 9259–9264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Zhang YS, J. H.; MacMillan SN; Lambert TH Synthesis of 1,2-Dihydroquinolines via Hydrazine-Catalyzed Ring-Closing Carbonyl-Olefin MetathesisOr. Org. Lett. 2020, 22, 6026–6030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Demole E Enggist P; Borer MC Applications Synthétiques de la Cyclisaton D’alcools Tertiaries γ-éthyléniques en α-Bromotétrahydrofurannes Sous L’action Du N-Bromosuccinimide. Helv. Chim. Acta 1971, 54, 1845–1864. [Google Scholar]
- (94).Jackson AC; Goldman BE; Snider BB Intramolecular and Intermolecular Lewis Acid-Catalyzed Ene Reactions Using Ketones as Enophiles. J. Org. Chem. 1984, 49, 3988–3994. [Google Scholar]
- (95).van Schaik HP; Vijn RJ; Bickelhaupt F Acid-Catalyzed Olefination of Benzaldehyde. Angew. Chem. Int. Ed. 1994, 33, 1611–1612. [Google Scholar]
- (96).Barlow SJ; Bastock TW; Clark JH; Cullen SR Explanation of an Unusual Substituent Effect in the Benzylation of Anisole and Identification of the Origin of the Active-Sites in Clayzic. Tetrahedron Lett. 1993, 34, 3339–3342. [Google Scholar]
- (97).Khripach VA; Zhabinskii VN; Kuchto AI; Zhiburtovich YY; Gromak VV; Groen MB; van der Louw J; de Groot A Intramolecular Cycloaddition/Cycloreversion of (E)-3β,17β-Diacetoxy-5,10-secoandrost-1(10)-en-5-one. Tetrahedron Lett. 2006, 47, 6715–6718. [Google Scholar]
- (98).Soicke A; Slavov N; Neudorfl JM; Schmalz HG Metal-Free Intramolecular Carbonyl-Olefin Metathesis of ortho Prenylaryl Ketones. Synlett 2011, 2487–2490. [Google Scholar]
- (99).Ludwig JR; Zimmerman PM; Gianino JB; Schindler CS Iron(III)-Catalysed Carbonyl-Olefin Metathesis. Nature 2016, 533, 374–379. [DOI] [PubMed] [Google Scholar]
- (100).Hennessy ET; Jacobsen EN Organometallic Chemistry: A New Metathesis. Nat. Chem. 2016, 8, 741–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Saá C Iron(III)-Catalyzed Ring-Closing Carbonyl-Olefin Metathesis. Angew. Chem. Int. Ed. 2016, 55, 10960–10961. [DOI] [PubMed] [Google Scholar]
- (102).Riehl PS; Schindler CS Lewis Acid-Catalyzed Carbonyl-Olefin Metathesis. Trends Chem 2019, 1, 272–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Halford B Robust Route for Carbonyl-Olefin Metathesis. C&EN 2016, 94, 8. [Google Scholar]
- (104).Deska J Carbonyl-Olefin Metathese. Nachr. Chem. 2016, 64, 604. [Google Scholar]
- (105).Becker MR; Rykaczewski KA; Ludwig JR; Schindler CS Carbonyl-Olefin Metathesis for the Synthesis of Cyclic Olefins. Org. Synth. 2018, 95, 472–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Ma LN; Li WJ; Xi H; Bai XH; Ma EL; Yan XY; Li ZP FeCl3-Catalyzed Ring-Closing Carbonyl-Olefin Metathesis. Angew. Chem. Int. Ed. 2016, 55, 10410–10413. [DOI] [PubMed] [Google Scholar]
- (107).Ludwig JR; Phan S; McAtee CC; Zimmerman PM; Devery JJ; Schindler CS Mechanistic Investigations of the Iron(III)-Catalyzed Carbonyl-Olefin Metathesis Reaction. J. Am. Chem. Soc. 2017, 139, 10832–10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Ludwig JR; Watson RB; Nasrallah DJ; Gianino JB; Zimmerman PM; Wiscons RA; Schindler CS Interrupted Carbonyl-Olefin Metathesis via Oxygen Atom Transfer. Science 2018, 361, 1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Hanson CS; Psaltakis MC; Cortes JJ; Devery JJ Catalyst Behavior in Metal-Catalyzed Carbonyl-Olefin Metathesis. J. Am. Chem. Soc. 2019, 141, 11870–11880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Groso EJ; Golonka AN; Harding RA; Alexander BW; Sodano TM; Schindler CS 3-Aryl-2,5-Dihydropyrroles via Catalytic Carbonyl-Olefin Metathesis. ACS Catal. 2018, 8, 2006–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Groso EJ; Schindler CS Recent Advances in the Application of Ring-Closing Metathesis for the Synthesis of Unsaturated Nitrogen Heterocycles. Synthesis 2019, 51, 1100–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Ni SJ; Franzén J Carbocation Catalysed Ring Closing Aldehyde-Olefin Metathesis. Chem. Commun. 2018, 54, 12982–12985. [DOI] [PubMed] [Google Scholar]
- (113).Tran UPN; Oss G; Pace DP; Ho JM; Nguyen TV Tropylium-Promoted Carbonyl-Olefin Metathesis Reactions. Chem. Sci. 2018, 9, 5145–5151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Tran UPN; Oss G; Breugst M; Detmar E; Pace DP; Liyanto K; Nguyen TV Carbonyl-Olefin Metathesis Catalyzed by Molecular Iodine. ACS Catal. 2019, 9, 912–919. [Google Scholar]
- (115).Wang R; Chen Y; Shu M; Zhao WW; Tao ML; Du C; Fu XY; Li A; Lin ZH AuCl3-Catalyzed Ring-Closing Carbonyl-Olefin Metathesis. Chem. Eur. J. 2019, 26, 1941–1946. [DOI] [PubMed] [Google Scholar]
- (116).Djuroyic A; Vayer M; Li ZL; Guillot R; Baltaze JP; Gandon V; Bour C Synthesis of Medium-Sized Carbocycles by Gallium-Catalyzed Tandem Carbonyl-Olefin Metathesis/Transfer Hydrogenation. Org. Lett. 2019, 21, 8132–8137. [DOI] [PubMed] [Google Scholar]
- (117).McAtee CC; Riehl PS; Schindler CS Polycyclic Aromatic Hydrocarbons via Iron(III)-Catalyzed Carbonyl-Olefin Metathesis. J. Am. Chem. Soc. 2017, 139, 2960–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Rykaczewski KA; Groso EJ; Vonesh HL; Gaviria MA; Richardson AD; Zehnder TE; Schindler CS Tetrahydropyridines via FeCl3-Catalyzed Carbonyl-Olefin Metathesis. Org. Lett. 2020, 22, 2844–2848. [DOI] [PubMed] [Google Scholar]
- (119).Tremel P; Iacobucci C; Massi L; Olivero S; Gal JF; Duñach E Catalytic Intramolecular Carbonyl-Ene Reaction with Ketones: Evidence for a Retro-Ene Process. New J. Chem. 2015, 39, 7453–7458. [Google Scholar]
- (120).Becker MR; Reid JP; Rykaczewski KA; Schindler CS Models for Understanding Divergent Reactivity in Lewis Acid-Catalyzed Transformations of Carbonyls and Olefins. ACS Catal. 2020, 10, 4387–4397. [Google Scholar]
- (121).Shambayati S; Crowe WE; Schreiber SL On the Conformation and Structure of Organometal Complexes in the Solid-State: Two Studies Relevant to Chemical Synthesis. Angew. Chem. Int. Ed. 1990, 29, 256–272. [Google Scholar]
- (122).Davis AJ; Watson RB; Nasrallah DJ; Gomez-Lopez JL; Schindler CS Superelectrophilic Aluminum(III)-Ion Pairs Promote a Distinct Reaction Path for Carbonyl-Olefin Ring Closing Metathesis. Nat. Catal 2020, 3, 787–796. [Google Scholar]
- (123).Negishi E Principle of Activation of Electrophiles by Electrophiles through Dimeric Association - Two are Better than One. Chem. Eur. J. 1999, 5, 411–420. [Google Scholar]
- (124).Olah GA Superelectrophiles. Angew. Chem. Int. Ed. 1993, 32, 767–788. [Google Scholar]
- (125).Olah GA; Klumpp DA Superelectrophiles and their Chemistry; John Wiley & Sons: New Jersey, USA, 2007. [Google Scholar]
- (126).Albright H; Riehl PS; McAtee CC; Reid JP; Ludwig JR; Karp LA; Zimmerman PM; Sigman MS; Schindler CS Catalytic Carbonyl-Olefin Metathesis of Aliphatic Ketones: Iron(III) Homo-Dimers as Lewis Acidic Superelectrophiles. J. Am. Chem. Soc. 2019, 141, 1690–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Chen D; Zhuang D; Zhao Y; Xie Q; Zhu J Reaction Mechanisms of Iron(III) Catalyzed Carbonyl–Olefin Metatheses in 2,5- and 3,5-hexadienals: Significant Substituent and Aromaticity Effects. Org. Chem. Front. 2019, 6, 3917–3924. [Google Scholar]
- (128).Naidu VR; Bah J; Franzen J Direct Organocatalytic Oxo-Metathesis, a trans-Selective Carbocation-Catalyzed Olefination of Aldehydes. Eur. J. Org. Chem. 2015, 2015, 1834–1839. [Google Scholar]
- (129).Prins HJ Condensation of Formaldehyde with Some Unsaturated Compounds. Chem. Weekbl. 1919, 16, 1072–1073. [Google Scholar]
- (130).Albright H; Vonesh HL; Schindler CS Superelectrophilic Fe(III)-Ion Pairs as Stronger Lewis Acid Catalysts for (E)-Selective Intermolecular Carbonyl-Olefin Metathesis. Org. Lett. 2020, 22, 3155–3160. [DOI] [PubMed] [Google Scholar]
- (131).Albright H; Vonesh HL; Becker MR; Alexander BW; Ludwig JR; Wiscons RA; Schindler CS GaCl3-Catalyzed Ring-Opening Carbonyl-Olefin Metathesis. Org. Lett. 2018, 20, 4954–4958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Catti L; Tiefenbacher K Bronsted Acid-Catalyzed Carbonyl-Olefin Metathesis Inside a Self-Assembled Supramolecular Host. Angew. Chem. Int. Ed. 2018, 57, 14589–14592. [DOI] [PubMed] [Google Scholar]
- (133).MacGillivray LR; Atwood JL A Chiral Spherical Molecular Assembly Held Together by 60 Hydrogen Bonds. Nature 1997, 389, 469–472. [Google Scholar]
- (134).Avram L; Cohen Y The Role of Water Molecules in a Resorcinarene Capsule as Probed by NMR Diffusion Measurements. Org. Lett. 2002, 4, 4365–4368. [DOI] [PubMed] [Google Scholar]
- (135).Shivanyuk A; Rebek J Reversible Encapsulation by Self-Assembling Resorcinarene Subunits. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 7662–7665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Yamanaka M; Shivanyuk A; Rebek J Kinetics and Thermodynamics of Hexameric Capsule Formation. J. Am. Chem. Soc. 2004, 126, 2939–2943. [DOI] [PubMed] [Google Scholar]
- (137).Avram L; Cohen Y Self-Recognition, Structure, Stability, and Guest Affinity of Pyrogallol[4]Arene and Resorcin[4]Arene Capsules in Solution. J. Am. Chem. Soc. 2004, 126, 11556–11563. [DOI] [PubMed] [Google Scholar]
- (138).Evan-Salem T; Baruch I; Avram L; Cohen Y; Palmer LC; Rebek J Resorcinarenes are Hexameric Capsules in Solution. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 12296–12300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Barrett ES; Dale TJ; Rebek J Stability, Dynamics, and Selectivity in the Assembly of Hydrogen-Bonded Hexameric Capsules. J. Am. Chem. Soc. 2008, 130, 2344–2350. [DOI] [PubMed] [Google Scholar]
- (140).Zhang Q; Tiefenbacher K Hexameric Resorcinarene Capsule is a Brønsted Acid: Investigation and Application to Synthesis and Catalysis. J. Am. Chem. Soc. 2013, 135, 16213–16219. [DOI] [PubMed] [Google Scholar]
- (141).Pitzer L; Sandfort F; Strieth-Kalthoff F; Glorius F Carbonyl-Olefin Cross-Metathesis through a Visible-Light-Induced 1,3-Diol Formation and Fragmentation Sequence. Angew. Chem. Int. Ed. 2018, 57, 16219–16223. [DOI] [PubMed] [Google Scholar]
- (142).Studer A; Curran DP Catalysis of Radical Reactions: A Radical Chemistry Perspective. Angew. Chem. Int. Ed. 2016, 55, 58–102. [DOI] [PubMed] [Google Scholar]
- (143).Studer A; Curran DP The Electron is a Catalyst. Nat. Chem. 2014, 6, 765–773. [DOI] [PubMed] [Google Scholar]
- (144).Rivero-Crespo MA; Tejeda-Serrano M; Pérez-Sánchez H; Cerón-Carrasco JP; Leyva-Pérez A Intermolecular Carbonyl-Olefin Metathesis with Vinyl Ethers Catalyzed by Homogeneous and Solid Acids in Flow. Angew. Chem. Int. Ed. 2020, 59, 3846–3849. [DOI] [PubMed] [Google Scholar]
- (145).Burkholder PR; Burkholder LM Antimicrobial Activity of Horny Corals. Science 1958, 127, 1174. [DOI] [PubMed] [Google Scholar]
- (146).Ciereszko LS; Karns TKB In Biology and Geology of Coral Reefs; Jones OA;Endean R, Eds.; Academic Press: New York, 1973; Vol. 11. [Google Scholar]
- (147).Nonomura T; Sasaki M; Matsumori N; Murata M; Tachibana K; Yasumoto T The Complete Structure of Maitotoxin. Part II: Configuration of the C135-C142 Side Chain and Absolute Configuration of the Entire Molecule. Angew. Chem. Int. Ed. 1996, 35, 1675–1678. [Google Scholar]
- (148).Nicolaou KC; Postema MHD; Yue EW; Nadin A An Olefin Metathesis Based Strategy for the Construction of the JKL, OPQ, and UVW Ring Systems of Maitotoxin. J. Am. Chem. Soc. 1996, 118, 10335–10336. [Google Scholar]
- (149).Shimizu Y Marine Natural Products; Academic Press: New York, 1978. [Google Scholar]
- (150).Lin YY; Risk M; Ray SM; Vanengen D; Clardy J; Golik J; James JC; Nakanishi K Isolation and Structure of Brevetoxin B from the "Red Tide" Dinoflagellate Ptychodiscus brevis (Gymnodinium breve). J. Am. Chem. Soc. 1981, 103, 6773–6775. [Google Scholar]
- (151).Shimizu Y; Chou HN; Bando H; Vanduyne G; Clardy JC Structure of Brevetoxin A (GB-1 Toxin), the Most Potent Toxin in the Florida Red Tide Organism Gymnodinium breve (Ptychodiscus brevis). J. Am. Chem. Soc. 1986, 108, 514–515. [DOI] [PubMed] [Google Scholar]
- (152).Zheng WJ; DeMattei JA; Wu JP; Duan JJW; Cook LR; Oinuma H; Kishi Y Complete Relative Stereochemistry of Maitotoxin. J. Am. Chem. Soc. 1996, 118, 7946–7968. [Google Scholar]
- (153).Nagai H; Torigoe K; Satake M; Murata M; Yasumoto T; Hirota H Gambieric Acids: Unprecedented Potent Antifungal Substances Isolated from Cultures of a Marine Dinoflagellate Gambierdiscus toxicus. J. Am. Chem. Soc. 1992, 114, 1102–1103. [Google Scholar]
- (154).Prasad AVK; Shimizu Y The Structure of Hemibrevetoxin-B: a New Type of Toxin in the Gulf of Mexico Red Tide Organism. J. Am. Chem. Soc. 1989, 111, 6476–6477. [Google Scholar]
- (155).Rainier JD; Allwein SP; Cox JM C-Glycosides to Fused Polycyclic Ethers. A Formal Synthesis of (±)-Hemibrevetoxin B. J. Org. Chem. 2001, 66, 1380–1386. [DOI] [PubMed] [Google Scholar]
- (156).Nagai H; Murata M; Torigoe K; Satake M; Yasumoto T Gambieric Acids, New Potent Antifungal Substances with Unprecedented Polyether Structures from a Marine Dinoflagellate Gambierdiscus toxicus. J. Org. Chem. 1992, 57, 5448–5453. [Google Scholar]
- (157).Roberts SW; Rainier JD Synthesis of an A-E Gambieric Acid Subunit with Use of a C-Glycoside Centered Strategy. Org. Lett. 2007, 9, 2227–2230. [DOI] [PubMed] [Google Scholar]
- (158).Zhang YA; Rohanna J; Zhou J; Iyer K; Rainier JD Total Synthesis of Brevenal. J. Am. Chem. Soc. 2011, 133, 3208–3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Bourdelais AJ; Jacocks HM; Wright JLC; Bigwarfe PM; Baden DG A New Polyether Ladder Compound Produced by the Dinoflagellate Karenia brevis. J. Nat. Prod. 2005, 68, 2–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (160).Keck GE; Poudel YB; Cummins TJ; Rudra A; Covel JA Total Synthesis of Bryostatin 1. J. Am. Chem. Soc. 2011, 133, 744–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).Pettit GR; Herald CL; Doubek DL; Herald DL; Arnold E; Clardy J Isolation and Structure of Bryostatin 1. J. Am. Chem. Soc. 1982, 104, 6846–6848. [Google Scholar]
- (162).Banerjee S; Wang Z; Mohammad M; Sarkar FH; Mohammad RM Efficacy of Selected Natural Products as Therapeutic Agents Against Cancer. J. Nat. Prod. 2008, 71, 492–496. [DOI] [PubMed] [Google Scholar]
- (163).Alushin GM; Lander GC; Kellogg EH; Zhang R; Baker D; Nogales E High-Resolution Microtubule Structures Reveal the Structural Transitions in αβ-Tubulin upon GTP Hydrolysis. Cell 2014, 157, 1117–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).Yunusov SY; Razakov R The structure of Cocculine and Cocculidine. Chem. Nat. Compd. 1970, 6, 69–73. [Google Scholar]
- (165).Heller ST; Kiho T; Narayan ARH; Sarpong R Protic-Solvent-Mediated Cycloisomerization of Quinoline and Isoquinoline Propargylic Alcohols: Syntheses of (±)-3-Demethoxyerythratidinone and (±)-Cocculidine. Angew. Chem. Int. Ed. 2013, 52, 11129–11133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Schrock RR High-Oxidation-State Molybdenum and Tungsten Alkylidyne Complexes. Acc. Chem. Res. 1986, 19, 342–348. [Google Scholar]
- (167).Hong BK; Li HH; Wu JB; Zhang J; Lei XG Total Syntheses of (−)-Huperzine Q and (+)-Lycopladines B and C. Angew. Chem. Int. Ed. 2015, 54, 1011–1015. [DOI] [PubMed] [Google Scholar]
- (168).Tan CH; Ma XQ; Chen GF; Zhu DY Two Novel Lycopodium Alkaloids from Huperzia Serrata. Helv. Chim. Acta 2002, 85, 1058–1061. [Google Scholar]
- (169).Ma XQ; Gang DR The Lycopodium Alkaloids. Nat. Prod. Rep. 2004, 21, 752–772. [DOI] [PubMed] [Google Scholar]