

Abstract
Organs age differently, causing wide heterogeneity in multimorbidity, but underlying mechanisms are largely elusive. To investigate the basis of organ‐specific ageing, we utilized progeroid repair‐deficient Ercc1Δ /− mouse mutants and systematically compared at the tissue, stem cell and organoid level two organs representing ageing extremes. Ercc1Δ /− intestine shows hardly any accelerated ageing. Nevertheless, we found apoptosis and reduced numbers of intestinal stem cells (ISCs), but cell loss appears compensated by over‐proliferation. ISCs retain their organoid‐forming capacity, but organoids perform poorly in culture, compared with WT. Conversely, liver ages dramatically, even causing early death in Ercc1‐KO mice. Apoptosis, p21, polyploidization and proliferation of various (stem) cells were prominently elevated in Ercc1Δ /− liver and stem cell populations were either largely unaffected (Sox9+), or expanding (Lgr5+), but were functionally exhausted in organoid formation and development in vitro. Paradoxically, while intestine displays less ageing, repair in WT ISCs appears inferior to liver as shown by enhanced sensitivity to various DNA‐damaging agents, and lower lesion removal. Our findings reveal organ‐specific anti‐ageing strategies. Intestine, with short lifespan limiting time for damage accumulation and repair, favours apoptosis of damaged cells relying on ISC plasticity. Liver with low renewal rates depends more on repair pathways specifically protecting the transcribed compartment of the genome to promote sustained functionality and cell preservation. As shown before, the hematopoietic system with intermediate self‐renewal mainly invokes replication‐linked mechanisms, apoptosis and senescence. Hence, organs employ different genome maintenance strategies, explaining heterogeneity in organ ageing and the segmental nature of DNA‐repair‐deficient progerias.
Keywords: adult stem cells, DNA damage response, ERCC1, genome maintenance, liver, nucleotide excision repair, organoids, small intestine
Organs age differently. To investigate the basis of organ‐specific ageing we systematically compared at the tissue, stem cell and organoid level two organs representing ageing extremes, from accelerated aged, repair‐deficient Ercc1 Δ /− mouse mutants. Very short‐lived cells, such as those of intestinal epithelium, have low damage tolerance and opt primarily for apoptosis and cell replacement. On the other extreme, liver (and by inference also neuronal tissue) with long‐lived cells, invest in cell survival by damage sensing and repair primarily linked with the transcribed compartment. They also may have high damage tolerance and consequently higher senescence levels. The differences in genome maintenance strategies we reveal, explain heterogeneity in organ ageing and the segmental nature of DNA‐repair‐deficient progerias.

Abbreviations
- DDR
DNA damage response
- GG‐NER
Global genome nucleotide excision repair
- HR
Homologous recombination
- HSC
Hematopoietic stem cell
- ICS
Intestinal stem cell
- LCS
Liver stem cell
- NHEJ
Non‐homologous end‐joining
- SC
Stem cell
- SI
Small intestine
- TCR
Transcription‐coupled repair
- TLS
Translesion synthesis
- XLR
Cross‐link repair
1. INTRODUCTION
Accumulation of DNA damage is recognized as a principal cause of systemic ageing (Hoeijmakers, 2009; Niedernhofer et al., 2018; Schumacher et al., 2021). DNA lesions interfere with DNA function and activate an intricate DNA damage response (DDR), which triggers repair and decides on cell fate including cell cycle arrest (senescence), mutagenesis, premature differentiation or cell death. Since DNA is at the top of the informational hierarchy, genetic erosion has very diverse, lasting consequences, impairing cell and tissue functioning causing pathology and cancer (Marteijn et al., 2014; Vermeij, Dollé, et al., 2016; Vermeij, Hoeijmakers, & Pothof, 2016). However, how DNA damage shapes ageing and how this relates to organ/tissue‐specific ageing trajectories is largely unknown.
Adult stem cells (SC) gradually lose functionality with age (McNeely, 2020). Mouse models mimicking DNA‐repair‐deficient human syndromes, revealed numerous features of ageing pathology (Marteijn et al., 2014), including functional impairment of hematopoietic stem cells (HSCs) (Cho et al., 2013; Navarro et al., 2006; Nijnik et al., 2007; Prasher et al., 2005; Rossi et al., 2007; Walter et al., 2015). Also, damage likely compromises HSCs and progenitor functionality in wt mice and humans (Beerman et al., 2014; Flach et al., 2014; Rube et al., 2011). In telomerase‐deficient mice, intestinal stem cells (ISCs) and progenitors undergo apoptosis or cell cycle arrest (Begus‐Nahrmann et al., 2009; Sperka et al., 2011). Hair follicle stem cells prematurely differentiate in a mouse model of the human progeroid DNA repair syndrome trichothiodystrophy and upon exogenous damage, similarly to naturally aged mice (Matsumura et al., 2016). Muscle stem cells decline in number and function in Ercc1‐repair‐deficient progeroid mutants, as in aged mice (Alyodawi et al., 2019; Lavasani et al., 2012).
Here, we compared intestine and liver, and their stem cells, organoids and damage responses in the Ercc1‐deficient mouse model of the XFE human progeroid syndrome (Niedernhofer et al., 2006). ERCC1 is in a complex with XPF involved in damage excision in multiple DNA repair systems: global genome nucleotide excision repair (GG‐NER) and transcription‐coupled repair (TCR), which remove helix‐distorting and transcription‐stalling lesions, (deficient in the rare human genetic disorders xeroderma pigmentosum and Cockayne syndrome, respectively (de Laat et al., 1999; Marteijn et al., 2014)), as well as interstrand cross‐link repair (defective in Fanconi's anaemia) and single‐strand annealing repair of persistent double strand breaks (Ahmad et al., 2008; Gillet & Scharer, 2006; Kuraoka et al., 2000; Niedernhofer et al., 2004). Thus, Ercc1Δ /− mice carrying one hypomorphic truncation allele and one null allele, harbour defects in four repair systems and combine multiple human genome instability disorders. They have largely normal embryonal development, but show after birth numerous progressive progeroid symptoms (Dolle et al., 2011), strikingly similar to natural ageing, which limit lifespan to 4–6 months (Vermeij et al., 2016b). Using this model, we analysed at tissue, SCs and organoids levels liver and small intestine, which vastly differ in ageing features in order to comprehend why organs and tissues age differently.
2. RESULTS
2.1. Contrasting ageing physiology and checkpoints of small intestine and liver from Ercc1Δ /− mice
While liver suffers dramatic ageing pathology in Ercc1 Δ /− mutants and in full Ercc1‐KO is lifespan limiting, small intestine (SI) seems unaffected (Dolle et al., 2011; Gregg et al., 2012; Wang et al., 2012). To compare these extremes in ageing, we choose the age of 15 weeks, when Ercc1 Δ /− mice manifest numerous ageing symptoms, without being moribund (Dolle et al., 2011). Histochemical evaluation of Ercc1Δ /− SI revealed no overt abnormalities (Figure 1a), aside from a smaller intestinal tract and perimeter (Figure 1b,c) in line with the reported cachexia of the mutant (Dolle et al., 2011) and smaller overall size of Ercc1Δ /− mice. The number and density of crypts appeared similar to WT (Figure 1d,e) and the ability of goblet cells to secrete mucus seems unchanged, suggesting normal intestinal function (Figure S1A). However, apoptosis—an outcome linked with ageing‐related homeostatic deregulation (Muradian & Schachtschabel, 2001) in naturally aged crypts (Martin et al., 1998) appeared significantly elevated with cells dying at the bottom and higher‐up in both Ercc1Δ /− villi and crypts. Hence, apoptosis is not restricted to a specific cellular compartment or stage of differentiation (Figure 1f–h), consistent with a stochastic origin and congruent with accelerated ageing.
FIGURE 1.

Aging‐related phenotypic features of small intestinal and liver from progeroid Ercc1Δ /− mice. (a) Intestinal tissue from 15‐week‐old Ercc1Δ /− and control mice stained with haematoxylin and eosin. Bars 200 μm. (b, c) Intestinal length (b, n = 2) and perimeter (c, n = 4) from 15‐week‐old mice of indicated genotypes. (d) Jejunal crypt density of 15‐week‐old wt and mutant mice. Crypts were counted on paraffin‐embedded 4μm slices of intestinal tissue. The number of crypts of progeroid Ercc1Δ /− mice is not significantly reduced in spite of the overall cachexia and decreased organ size, p = 0.7328 (n = 3). (e) Cell density in jejunal crypts of 15‐week‐old wt and Ercc1Δ /− mice. Cells were counted on DAPI stained 4μm intestinal tissue slices as in (d), p = 0.5415 (n = 3 mice). (f) Immunofluorescent images of small intestine crypt and villi from sections stained for apoptosis (TUNEL), counterstained with DAPI. Bars 50 μm. (g, h) Apoptosis index in crypts (g) and villi (h), (n = 3 mice). (i) Liver tissue from 15‐week‐old Ercc1Δ /− and wt mice assessed for apoptosis (TUNEL). Red arrows: TUNEL+ cells; black cut‐out depicts a TUNEL+ large hepatocyte. Bars 100 μm. (j–m) Apoptosis index in the liver, parenchymal, non‐parenchymal (l), and biliary cell (m) population of 15‐week‐old wt and mutant mice (n = at least 3 mice for WT, n = 5 mice for mutant groups). Quantification of TUNEL+ nuclei was performed on DAB‐stained liver sections. Data: mean ± SEM. *p < 0.05, **p < 0.01
Contrary to SI, Ercc1Δ /− liver is known to suffer from severe ageing pathology (Dolle et al., 2011; Gregg et al., 2012; Vermeij et al., 2016b; Weeda et al., 1997), confirmed for the Lgr5EGFPErcc1Δ /− model used here in Figure S1B–E. TUNEL immunostaining revealed apoptosis to be clearly enhanced in Ercc1Δ /− liver (Figure 1i–j). Close morphological inspection revealed that nearly all cell types are affected (Figure 1k–m). In fact, the increase of TUNEL + hepatocytes was relatively modest, although, notably, apoptosis included also polykaryons, that is, the equivalent of many diploid hepatocytes. Moreover, hepatocytes expressing cyclin‐dependent kinase inhibitor p21 were ~40‐fold increased (Figure S2A–B) and a ~4‐fold increase in biliary cells was found (Figure S2C), consistent with the systemic nature of the repair defect and wide‐spread premature ageing features.
p21 expression in mice carrying (liver‐specific) DNA repair deficiency is correlated with senescence (Ogrodnik et al., 2017). Recently, Yousefzadeh and coworkers (Yousefzadeh et al., 2020) convincingly demonstrated extensive senescence in 10 organs of Ercc1Δ /− mice, including liver, starting at the age of 12 weeks, progressively increasing with time. We examined liver tissue of 15‐week‐old Ercc1Δ /− mice, but unexpectedly, despite multiple trials and positive controls (see below), did not detect significant loss of LaminB1 and nuclear HMGB1 immunosignals nor significantly increased IL‐6 expression that would suggest overt senescence, at this age in our Ercc1Δ /− mouse line (Figure S3A–F). Further investigation is warranted to find out whether differences in, for example housing conditions, play a role in the age of onset of senescence in liver, including food, for which Ercc1Δ /− mutants are extremely sensitive regarding accelerated ageing (Vermeij, Dollé, et al., 2016) and which likely influences anti‐oxidant buffering and DNA damage load (Milanese et al., 2019).
In conclusion, in this and other studies (Dolle et al., 2011; Weeda et al., 1997; Yousefzadeh et al., 2020), Ercc1Δ /− liver displays numerous accelerated ageing features, in hepatocytes and biliary cholangiocytes, in sharp contrast to SI, although elevated apoptosis in villi and the regenerative and amplifying compartments of crypts suggests altered homeostatic regulation in this organ as well.
2.2. Tissue‐specific regenerative responses parallel homeostatic deregulation and pathology in Ercc1Δ /− small intestine and liver
Previous research provided evidence for limited regenerative proficiency of Ercc1Δ /− liver following partial hepatectomy (Gregg et al., 2012), but which cells are implicated and whether SI is affected as well is unknown. Ki67 immunostaining indicated that the proliferative index of Ercc1Δ /− intestinal crypts did not differ from controls (Figure 2a,b). In contrast, Ercc1Δ /− liver showed a prominent upregulation of Ki67+ cells in nearly all cell populations (Figure 2c–g). Thus, in progeroid Ercc1Δ /− mice SI displays normal regeneration rates, in spite of increased apoptotic events. However, compared with the very high cell turn‐over, cell loss is relatively small and compensatory over‐proliferation would likely go undetected. In contrast, in the liver, which normally has low turn‐over, homeostasis is severely affected, as for other NER‐deficient mouse mutants (Barnhoorn et al., 2014). Hence, DNA repair deficiency triggers cell loss in many cell types in liver, but also enhanced compensatory cell division.
FIGURE 2.
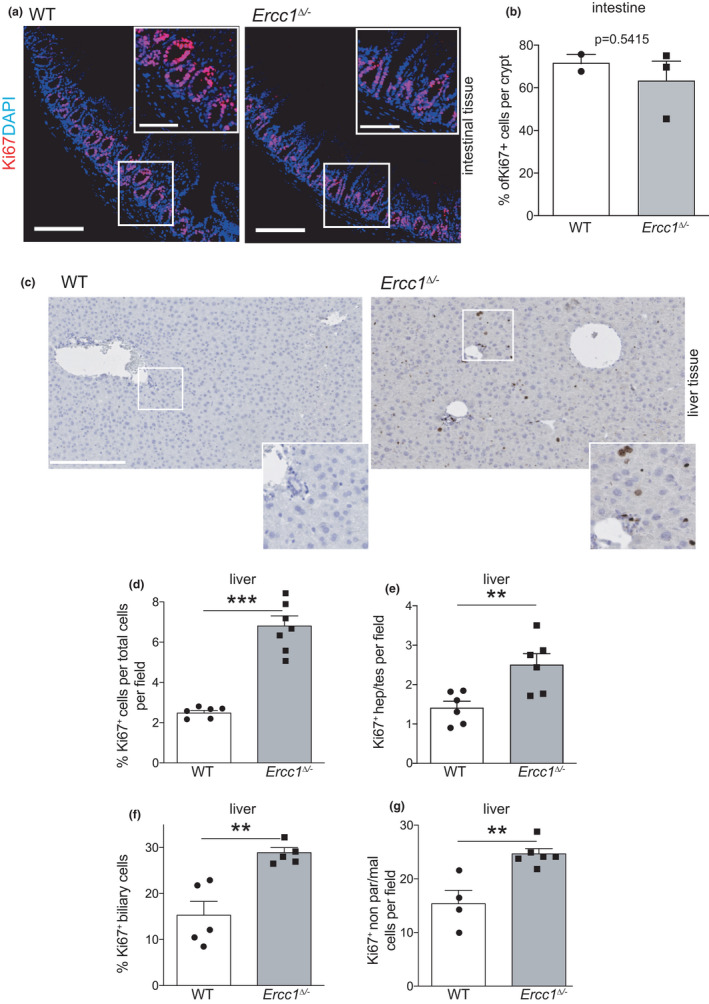
Steady‐state regenerative potential of intestine and liver of progeroid Ercc1Δ /− mice. (a) Immunofluorescence of intestinal tissue sections from 15‐week‐old wt and mutant mice stained for proliferation marker Ki67, nuclei counterstained with DAPI. Insets here and in other panels are digitally magnified images. Bars 10 μm (5 μm for insets). (b) Steady‐state proliferative index of small intestine from 15‐week‐old Ercc1Δ /− (n = 3) and control mice (n = 2). (c) Immunohistochemical staining for proliferation marker Ki67 of liver from 15‐week‐old Lgr5EGFPErcc1Δ /− and wt mice. Nuclei counterstained with haematoxylin. Bars 250 μm. (d) Proliferative index of Lgr5EGFPErcc1Δ /− liver. Quantitation of Ki67+ cells per total cells in a field as represented in (c) from 15‐week‐old mutant and wt mice. More than 5 fields were quantified from n = 7 mice per genotype. (e–g) Proliferative index of various cell populations (identified on morphology and location) in progeroid Lgr5EGFPErcc1Δ /− and wt liver. Note that nearly all cell populations in Ercc1Δ /− liver show increased proliferation. Over 5 fields were quantified from at least 4 mice per genotype. Data: mean ± SEM, **p < 0.01, ***p < 0.001
2.3. Distinct responses of Ercc1Δ /− SC types to unrepaired damage
To further investigate the fate of SCs, we focused on Lgr5 + stem cells responsible for steady‐state intestinal homeostasis but also damage‐induced liver regeneration (Barker et al., 2007; Huch et al., 2013). We crossed Lgr5EGFP − ires − CreERT2 (Barker et al., 2007) and Ercc1Δ /− mice and monitored EGFP‐expressing (EGFP+) cells in 15‐week‐old Lgr5EGFPErcc1Δ /− SI and liver. Flow cytometry revealed a strikingly reduced number of high EGFP‐expressing (EGFPhi) ISCs in progeroid mice (Figure 3a,b). Immunohistology and flow cytometry disclosed also erosion of the total EGFP+ population in mutant intestine (Figure 3c). Unaltered ratios of EGFPlow to EGFPhi cells (Figure 3d) in crypts and seemingly unaffected intestinal functionality (Figure S1A) suggest that ICSs in progeroid mice are eliminated randomly through apoptosis (as shown in Figure 1g,h), rather than premature differentiation. To examine how Ercc1Δ /− intestine maintains homeostasis, given the increased apoptosis and reduced numbers of SCs, we analysed SI sections for co‐occurrence of Ki67 and EGFP expression and found that mutant crypts bear a higher percentage of Ki67+ EGFPhi SCs and progenitors indicating that they are more in cycle than crypts of Lgr5EGFP control mice (Figure 3e,f).
FIGURE 3.

Characterization of small intestinal and liver stem cell populations in progeroid Ercc1Δ /− mice. (a) Enhanced green fluorescent protein (EGFP) immunostaining in crypts of the small intestine from 15‐week‐old mutant and wt mice. Nuclei counterstained with DAPI. Bars 10 μm. (b) Quantitation of intestinal crypt cells expressing high levels of GFP (GFPhi) by flow cytometry of 15‐week‐old wt (n = 4) and Ercc1Δ /− (n = 3) mice. (c) Quantitation of the total EGFP‐expressing (EGFP+) cell population in small intestinal crypts of wt (n = 4) and mutant (n = 3) mice by flow cytometry. (d) Ratio of crypt cells expressing low (EGFPlow, i.e., downstream progenitor population) versus high levels of EGFP (GFPhi, i.e., stem cells) in the intestine of the indicated mice. Each column represents collectively the values for individual crypts for 3 mice per genotype. (e) Quantitation of the GFPhi intestinal population that expresses Ki67 proliferation marker from immunofluorescently stained tissue of 15‐week‐old mice of the indicated genotypes (n = 3 mice per genotype). Note the increase of proliferative stem cells in progeroid Ercc1 mutant mice. (f) Immunofluorescence images of paraffin‐embedded intestinal tissue from 15‐week‐old Lgr5EGFP Ercc1Δ /− and wt mice, stained for EGFP and Ki67. Bars 10 μm. (g) Immunofluorescent staining for Sox9 expression (green) in bile duct cells of liver sections from 15‐week‐old Lgr5EGFP Ercc1Δ /− and wt mice, along with Ki67 (red). Bar 10 μm. (h) Percentage of Sox9+ bile cell population per total cells in liver ducts of 15‐week‐old Ercc1Δ /− and wt mice, quantified from 4μm liver sections stained as in (g) (n = 3 mice). (i) Average number of cells per bile duct in livers of 15‐week‐old Ercc1Δ /− and wt mice, quantified from 4 μm liver sections. Data represent means with SD from quantitation of cells in round bile ducts from 3 mice per genotype and at least 10 bile ducts per mouse. (j) EGFP‐expressing cells in the liver of Lgr5EGFPErcc1Δ /− (n = 3) and control (n = 2) mice. (k) EGFP + Lgr5‐marked stem cells in liver of 15‐week‐old Lgr5EGFPErcc1Δ /− and control mice stained for GFP. Bars 10 μm. Data: mean ± SEM unless otherwise specified. *p < 0.05, ****p < 0.0001
A liver cell type, attributed with SC properties, is the Sox9+ cholangiocyte (Gilgenkrantz & Collin de l'Hortet, 2018). Close examination of bile ducts revealed unaltered Sox9+ cell pools (Figure 3h), but increased bile cell nuclear size (Figure S1c,d) and a more disorganized structure of the ducts in Ercc1Δ /− liver (Figure 3g). In addition, the number of cells per bile duct was significantly reduced (Figure 3i), in line with the evidence for apoptosis of this cell population in mutant liver (Figure 1m). Apparently, cell loss is not adequately compensated by increased cell division leading to aberrant duct morphology and possibly function.
To further investigate the impact of Ercc1 deficiency on liver stem cells (LSCs), we quantified Lgr5EGFP + cells on paraffin‐embedded liver sections. EGFP+ mutant SCs appeared more abundant and display intenser GFP fluorescence than in WT (Figure 3j,k). A prominent characteristic of Ercc1Δ /− liver is the abundance of large polyploid nuclei (McWhir et al., 1993; Weeda et al., 1997). We found that a significant fraction of Ercc1Δ /− Lgr5EGFP + cells has enlarged nuclear size. But despite some EGFP+ cells in mutant mice looking polyploid (Figure S4), overall, nuclei of EGFP+ cells seem smaller than the rest of the (GFP−) population in both genotypes (Figure S4B). We conclude that, in contrast to intestine, the number of Lgr5+ cells in Ercc1Δ /− liver is increased. A fraction of LSCs suffers from polyploidy most likely as a consequence of accumulated DNA damage suggesting limited functional potential.
2.4. Functional exhaustion of liver but not intestinal SC populations of progeroid mice
To assess functional consequences of Ercc1 deficiency on SCs, we examined the ability of mutant and WT ISCs to expand into organoids. After seeding an equivalent number of crypts, organoid‐forming capacity of ISCs seems similar for both genotypes (Figure 4a). However, mutant differentiated organoids, appeared smaller than WT with lower numbers of organoid‐budding crypts (Figure 4b–d). Culturing crypts in 3% O2 did not significantly improve Ercc1Δ /− (or WT) organoid development (Figure S5A) ruling out that it was caused by increased oxidative stress due to culturing in atmospheric oxygen, as previously noted in Ercc1 −/− primary mouse embryonic fibroblasts (Fuhrmann‐Stroissnigg et al., 2017; Niedernhofer et al., 2006; Tilstra et al., 2012). Therefore, the reduced number of ICSs in isolated crypts in Ercc1∆ /− intestine (Figure 3b,c) might explain the difference in organoid size.
FIGURE 4.
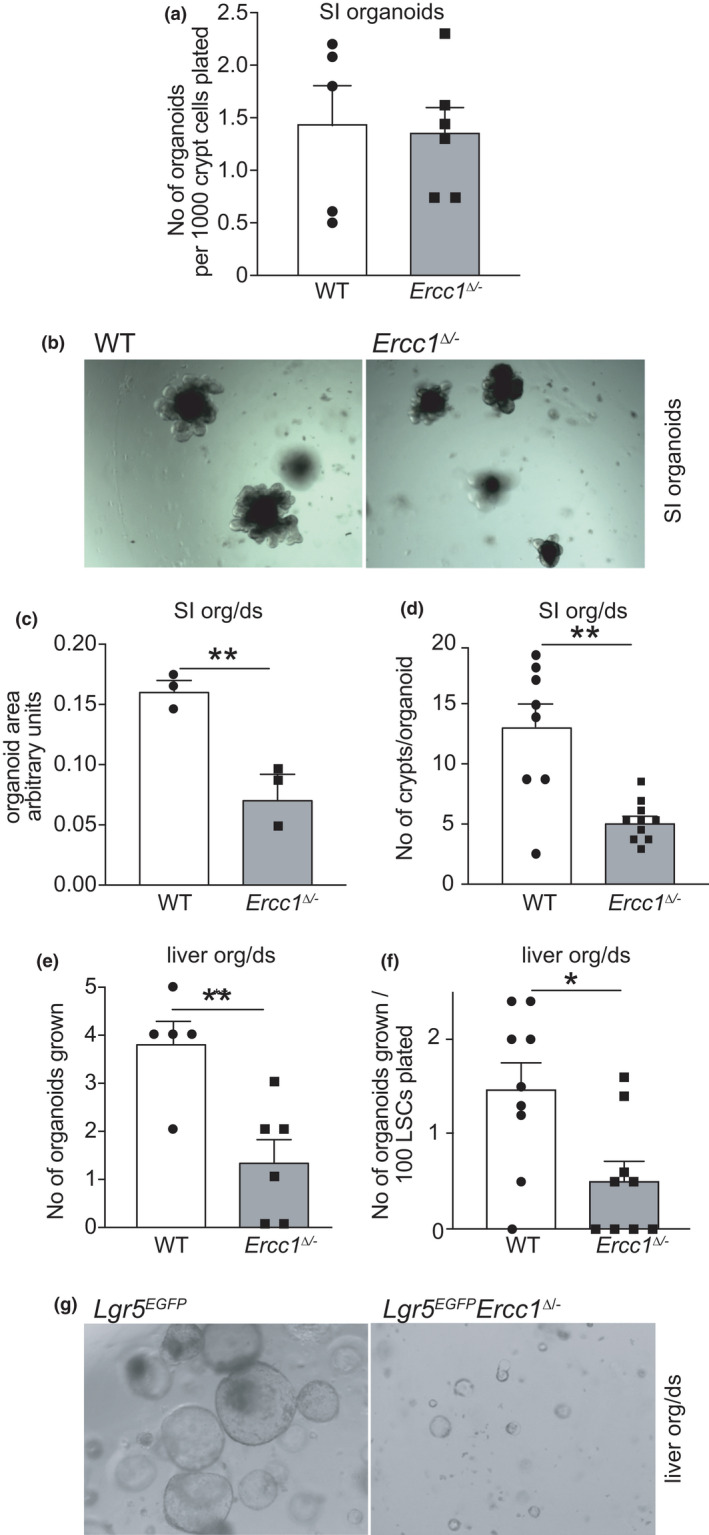
Ex vivo culture of intestinal and liver SCs from Ercc1Δ /− mice. (a) Organoids grown from intestinal crypt cell suspensions, derived from 15‐week‐old Ercc1Δ /− and wt mice, after 9 days in culture, p = not significant (n = 5 mice per genotype). (b) In vitro cultures of freshly isolated crypts from 15‐week‐old mice of the indicated genotypes. Images after 9 days in culture. (c) Average organoid size of the indicated genotypes after 9 days in culture (n = 3 independent cultures derived from different mice per group). (d) Average number of crypts budded in organoids of the indicated genotypes after 9 days in culture (at least two cultures for each mouse and 3 mice per genotype). (e) Number of organoids grown after plating bile cell containing liver cell suspensions, derived from 15‐week‐old Ercc1Δ /− mice and wt controls (at least two cultures for each mouse and 3 mice per genotype). Organoids were counted at Day 7 of culture. (f) Number of liver organoids grown in secondary cultures, after plating single liver stem cells derived from a primary organoid culture (at least three cultures were measured for each mouse and 3 mice per genotype). Organoids were counted at Day 7 of culture. (g) In vitro cultures of liver organoids grown from bile cell suspensions derived from livers of 15‐week‐old mice of the indicated genotypes. Images were taken after 7 days in culture. Data: mean ± SEM. *p < 0.05, **p < 0.01
To probe Ercc1Δ /− LSC self‐renewal potential, we used ex vivo organoid cultures (Huch et al., 2013). After seeding equal numbers of isolated liver cells, bile cells from mutant liver yielded far fewer organoids than controls (Figure 4e). To examine the possible effect of a difference in starting amount of bile cells (Figure 2f), from which organoids spawn, we plated secondary cultures from the organoids. This showed that organoid‐forming ability of Ercc1Δ /− LSCs is impaired (Figure 4f). Moreover, mutant organoids are dramatically smaller (Figure 4g). Both phenotypes were not rescued using low (3%) oxygen culture conditions (Figure S5B).
To conclude, whereas organoid‐forming capacity seems unaltered, Ercc1∆ /− ISCs show reduced growth and formation of budding crypts. Mutant LSCs, however, display strongly diminished ability to form organoids, and drastically compromised development, suggesting SC exhaustion.
2.5. Diverse functional outcomes of DNA damage in Ercc1Δ /− intestinal and liver SCs
Ercc1Δ /− SI organoids show Ki67+ cell content similar to WT (Figure 5a), indicating that proliferative capacity in vitro is largely unaffected, consistent with the in vivo findings. TUNEL and cleaved Caspase‐3 staining revealed that Ercc1Δ /− SI organoid crypts contain cells undergoing apoptosis (Figure 5b), also recapitulating the mouse tissue phenotype. Interestingly, mutant crypts were found positive for senescence‐associated β‐galactosidase (SA‐β‐Gal), indicating the features of senescence (Figure 5c).
FIGURE 5.
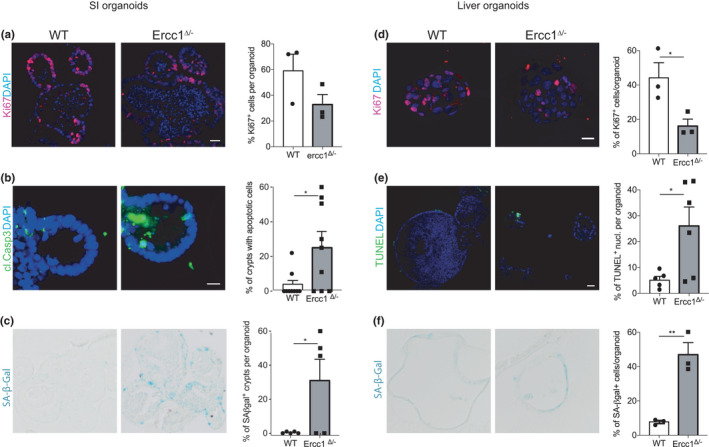
Cell fate phenotypes in Ercc1Δ /− small intestinal and liver organoids. (a) Proliferation index of Ercc1Δ /− intestinal organoids. Immunostaining for Ki67 (red) in Ercc1Δ /− and wt control SI organoids in medium without Wnt3a supplementation. Bar 10 μm. Right, quantitation of proliferative, Ki67+ cells in organoids of the indicated genotypes (n = 3 mice per genotype). (b) DNA damage responses in Ercc1Δ /− SI organoids. Images of crypts of organoids, derived from 15‐week‐old Ercc1Δ /− and control mouse intestines, immunostained for cleaved Caspase‐3. Bar 10 μm. SI organoid crypts containing TUNEL+ cells. Each dot represents the measurement of individual organoid cultures and data from a sample size of 3 mice per genotype. (c) Images of SI organoids from 15‐week‐old mutant and wt mice, stained for senescence‐associated β‐galactosidase (SA‐βGal) activity. Crypts positive for SA‐βGal in organoids of the indicated genotypes. Each dot individual culture of organoids, data from 3 mice per genotype. (d) Immunostaining for Ki67 of liver organoids from 15‐week‐old Ercc1Δ /− and wt mice. Bar 20μm. Proliferation index of organoids (n = 3 mice per genotype). (e) Apoptosis in Ercc1Δ /− and wt liver organoids from 15‐week‐old mice. Bar 50 μm. Percentage TUNEL+ cells per organoid (n = 5 mice per genotype). (f) Histochemical staining for SA‐β‐Gal activity. Percentage of SA‐β‐Gal+ cells per Ercc1Δ /− and wt liver organoid (n = 3 mice per genotype). Data: mean ± SEM. *p < 0.05, **p < 0.01
In contrast, Ercc1Δ /− liver organoids display considerably less Ki67+ cells (Figure 5d) and a significant fraction of SCs was apoptotic or positive for senescence (Figure 5e,f), the latter deviating from our findings in 15‐week‐old liver (Figure S3A–F), suggesting that culture conditions are more stressful than in situ and reach the threshold for senescence. LSCs show moderately compromised proliferation (as assessed by EdU incorporation Figure 5d and S6A,B) and γH2AX foci indicative of DNA breaks (Figure S6C). We found little overlap of γH2AX signal with replicating cells (Figure S6D), consistent with the notion that Ercc1 defects also include repair systems such as TCR (amplified by GG‐NER deficiency) linked with transcription, affecting all cell cycle stages.
Flow cytometry for polyploidy showed a trend for more >4n cells in Ercc1Δ /− organoids, however, increased DNA content did not reach statistical significance (Figure S6E). Therefore, we measured the perimeter of EdU+ nuclei 48 hours after a short pulse, analysing exclusively cells that have undergone replication. Indeed, a significant fraction of Ercc1Δ /− LSC nuclei seems to be larger and some very large, indicating that they further progress in polyploidization in vitro (Figure S6F,G). Staining for albumin, a marker of differentiated hepatocytes, showed that Ercc1Δ /− LSCs in culture do not spontaneously undergo differentiation (data not shown). Collectively, we reveal diverse DNA damage responses in Ercc1Δ /− SI and liver SCs in organoid cultures, affecting stemness and regenerative capacity.
2.6. Comparison of functional DNA repair capacity of intestinal and liver stem cells
Since unrepaired DNA damage is the most logical culprit for the SC phenotypes of Ercc1Δ /− mice, we wished to examine SC responses to different classes of DNA‐damaging agents. We chose UV that causes base‐pair‐disrupting photoproducts mainly repaired by GG‐NER; Illudin S, which induces lesions that block transcription and are repaired by TCR (Jaspers et al., 2002); and interstrand cross‐links inflicted by cisplatin, which require cross‐link repair and mostly affect replication. Ercc1 mutants are deficient in all of these pathways (Marteijn et al., 2014; Niedernhofer et al., 2006). We assessed the ability of cultured repair‐proficient ISCs and LSCs to survive and expand into organoids, following increasing doses of the above genotoxins. Surprisingly, although the intestine compared with liver shows less (premature) ageing in WT and when Ercc1 is mutated, ISCs appeared consistently more sensitive to all genotoxins than LSCs (Figure 6a–d). ISCs were previously shown to be more sensitive to γ‐irradiation (Barker, 2014). Cytochrome C release following treatment with illudin S and cisplatin illustrates the hypersensitivity of ISCs to apoptotic cell death compared to LSCs (Figure 6e,f). Apparently, after equal DNA damage ISCs opt for cell death and LSCs prioritize survival. We wondered whether this differential genotoxin sensitivity correlates with differential repair capacity. Previously, we noted that core NER genes are higher expressed in LSCs than ISCs (Jager et al., 2019). However, expression levels do not always correlate with repair efficiency (Naipal et al., 2015). We examined 6,4‐photoproduct resolution after treatment of SCs with UVB, reflecting mainly GG‐NER and to a lesser extent TCR activities. As shown by the remaining 6,4‐photoproduct, immunosignals LSCs appear superior in repairing these lesions (Figure 6g). Inefficient repair likely enhances the propensity of ISCs to undergo apoptosis. Taken together, despite the virtual absence of accelerated ageing features in intestine, ISCs appear inferior in major DNA repair systems when assayed in parallel with LSCs.
FIGURE 6.

Differential responses of repair‐proficient liver and intestinal stem cells to exogenous damage. (a) Schematic of experimental set‐up for organoid formation assay following DNA damage induction. For each genotoxic agent, cultures from 2 wt mice were used and for each mouse, organoids grown in triplicates were quantified. (b‐d) Clonogenic survival of wt liver and intestinal stem cells upon UVC‐exposure (b); 2 h. treatment with the indicated concentrations of illudin S, which is only removed by transcription‐coupled repair (c) and the DNA‐crosslinking agent cisplatin (d). Organoids from single stem cell containing cultures were counted 5 days after treatment (n = 2 mice per group). (e, f) Number of stem cells that have lost cytochrome c at various time points following treatment with cisplatin (e) or illudin S (f). Data: means and SD of replicate samples from 2 mice in total. (g) Quantitation of 6,4‐photoproducts’ immunosignal in organoid stem cells at the indicated time points following UVB irradiation. Plotted is the percentage of remaining fluorescence relative to the 5 min time point from at least 500 nuclei of separate organoids of each mouse (3 mice per time point). p‐values were calculated using Student's t test. Data: mean ± SEM unless otherwise specified. **p < 0.01, ***p < 0.001, ****p < 0.0001
3. DISCUSSION
At tissue level, we found no overt features of accelerated ageing in Ercc1Δ /− SI. Since Ercc1Δ /− mutants include cross‐link repair and GG‐NER defects, which are highly relevant for replication, this is unexpected as intestine is the most proliferative organ. Cell death affects many tissues of naturally aged mice (Pollack et al., 2002), including SI (Martin et al., 1998; Nalapareddy et al., 2017; Steegenga et al., 2017). Interestingly, Ercc1Δ /− crypts, transient amplifying zone and villi, reveal increased cell death in all stages of intestinal development, consistent with underlying stochastic damage events. Overall regenerative potential of the crypts is maintained, in line with the plasticity reported for ISC populations (Ritsma et al., 2014; Tetteh et al., 2016; Tian et al., 2011) and the presence of slow‐cycling, reserve ISCs (Barriga et al., 2017). Although we cannot exclude alterations in cell cycle rates, elevated spontaneous apoptosis had no detectable effect on tissue proliferation. Possibly, this went undetected amidst the high cell renewal in intestine. In sharp contrast, Ercc1Δ /− liver displays dramatic ageing pathology (Gregg et al., 2012; Vermeij, Dollé, et al., 2016; Weeda et al., 1997). Here, we show that apoptosis is prominent in nearly all liver cell types and p21 elevated in a fraction of cells. The intercellular heterogeneity in p21 levels parallels p53 in Ercc1 −/− liver (McWhir et al., 1993), consistent with stochastic DNA damage. The p21 response likely reflects resistance to apoptosis—explaining the modest increase in hepatocyte death, prior to reaching moribund stages, where senescence and apoptosis are strongly raised (Gregg et al., 2012; Yousefzadeh et al., 2021). The elevated proliferative index, persistent damage and different DDR, likely explain the increased mutations in LSCs compared with ISCs (Jager et al., 2019).
At the SC level, Ercc1Δ /− ISCs are diminished in number, possibly through apoptosis. However, remaining ISCs appear functionally normal. Nevertheless, the number of quiescent Lgr5 + cells is reduced in Ercc1Δ /− crypts, which can be either due to apoptosis and/or cell cycle entry to restore regenerative capacity. In contrast, Ercc1Δ /− liver displays increased levels of Lgr5 + cells, but these cells show evidence of impaired functionality: some are polyploid—indicative of genomic stress. The bile cell population appears compromised. Elevated apoptosis and p21 upregulation point to loss of stemness properties and reduced regenerative potential.
At the organoid level, Ercc1Δ /− ISCs show ~WT ability to form organoids, which are smaller and contain less crypts, suggesting reduced ability to grow and differentiate. Although proliferative potential is marginally decreased, apoptosis and senescence in these crypts are elevated, in line with time‐dependent stochastic DNA damage causing the organoid's limited net growth under culture conditions. In comparison, LSCs appear to be functionally even more compromised in all regards: they form fewer organoids, which are also much smaller and contain fewer proliferative cells, suggesting proliferative exhaustion. Apoptosis, senescence, polyploidy and irregularly shaped nuclei are also increased in liver organoids, accounting for their poor growth. Increased γH2AX foci, mainly in non‐replicating cells provides evidence for DNA damage likely related to transcription (rather replication) stress, a genome‐wide ageing phenomenon, first discovered in Ercc1Δ /− and Xpg −/− liver and subsequently also in natural ageing (Vermeij, Dollé, et al., 2016), which is associated with functional decline, cell cycle arrest, senescence and cell death. This novel ageing feature occurs primarily in organs with low cell renewal (since DNA replication dilutes damage) and affects expression preferentially of large genes, consistent with accumulating random DNA lesions compromising transcription in a gene‐length‐dependent manner (Vermeij, Dollé, et al., 2016). The origin of the endogenous DNA damage is unclear, but in our in vitro studies appears largely independent from O2 levels (see Figure S5b), fitting our observation (Milanese et al., 2019) that TCR‐ defects trigger a potent anti‐oxidant defence (Garinis et al., 2009; Niedernhofer et al., 2006; van der Pluijm et al., 2007).
Directly comparing the DDR and repair properties of WT ISCs and LSCs, we show that ISCs are more sensitive to diverse genotoxins attesting different repair systems. Individual ISC types have distinct damage sensitivities (Shivdasani, 2014). Quiescent Lgr5 + cells appear more resistant, while fast‐dividing ISCs have a low apoptotic threshold, as shown for ionizing radiation (Barker, 2014; Barriga et al., 2017). Consistent with low expression of NER genes (Jager et al., 2019), we find ISCs to be less efficient in repair of UV‐lesions and more sensitive to UV, illudin S and cisplatint damages eliminated by GG‐NER, TCR and cross‐link repair, respectively, supporting the idea that the entire repair machinery is functionally inferior in ISCs.
As liver exhibits much more ageing features than intestine, one might expect genome maintenance in ISCs to be superior. However, in a direct comparison, repair in LSCs appears superior. This explains why Ercc1 (and all combined TCR/GG‐NER) mutants display much less ageing in intestine than liver (and, as evident from human and mouse TCR/GG‐NER mutants, also e.g., neurons and kidney). Apparently, organs utilize different anti‐ageing strategies, involving DDR mechanisms. This raises the question—why do ISCs not invest maximally in DNA repair? One reason may be their tight replicative and differentiation schedule imposed by the rapid renewal of intestinal epithelium, which may not permit spending much time and energy for repair. Intestinal epithelial cells have only a lifespan of only 3–5 days (Gehart & Clevers, 2015) with limited time for DNA damage to accumulate, rendering repair less critical, and hence, prefer they opt for apoptosis. The high proliferation rate of ISCs and the reserve capacity probably can easily compensate for the loss of a relatively small fraction of cells.
The above strategy may be less suitable when cell turnover is slower with more time for lesions to accrue, increasing dependence on repair. This matches with the severe functional and numerical SC exhaustion in Ercc1 mutant mice for the hematopoietic system (Cho et al., 2013; Prasher et al., 2005; Rossi et al., 2007), in which SCs have an estimated average turn‐over of ~2 months. Presumably, exhaustion of HSCs in the Ercc1 mutant is largely caused by its replication‐associated cross‐link repair defect, as in Fanconi's anaemia (FA), in line with the notion that defects in the ERCC1 partner XPF can cause FA (Bogliolo et al., 2013; Kuraoka et al., 2000).
Presumably, the investments in high intestinal tissue turn‐over cannot be afforded by many other organs and cell turn‐over is incompatible with the primary function of, for example most post‐mitotic neurons. Moreover, the intestine resides in a very hostile environment, including the microbiome, with all metabolites passing through its epithelium rendering the organ with the highest exposure to exogenous compounds, explaining its 3–5 day cell turn‐over. Although liver, as main detoxification organ, is also exposed to numerous toxics, we assume that its superior repair and high damage tolerance enable hepatocytes to live longer. Since cells in liver replicate only occasionally, they must rely on replication‐independent DNA repair pathways. Particularly, their transcribed compartment is vital for sustained functionality, explaining why TCR mutants display a segmental progeroid phenotype strongly biased towards post‐mitotic organs and tissues, such as the neuronal system, liver, kidney, fat tissue and skeleton, but less bone marrow and intestine (Lans et al., 2019). A combined TCR/GG‐NER deficiency as in Ercc1 mutants dramatically augments these segmental ageing features, as all GG‐NER lesions also hamper transcription (Andressoo et al., 2006; de Boer et al., 2002).
3.1. A model for the relationship between DNA damage responses and tissue‐specific ageing trajectories
Based on the central role of DNA damage for ageing, we propose a tentative model (Figure 7), in which different DNA repair and damage response systems linked with replication and transcription (cell cycle arrest, senescence, cell death and mutagenesis) are differentially employed as anti‐ageing/cancer strategies. Very frequent replicating cells with a short lifespan, as in intestinal epithelium opt primarily for apoptosis and cell replacement. On the other extreme, liver (and by inference from the phenotypes of mouse and human progeroid mutants with defects in the same repair pathways also neurons, kidney, and skeleton with mostly post‐mitotic cells with a long lifespan invest in cell survival by damage sensing and repair primarily linked with transcription and the transcribed compartment (i.e., the most important functional part), involving TCR with the help of global genome repair systems (GG‐NER and base excision repair). They also may have high damage tolerance and consequent senescence levels. Tissues with an intermediate stem cell turn‐over and lifespan, such as the hematopoietic system, invoke both apoptosis and replication‐linked DDR such as cross‐link and double‐strand break repair by non‐homologous‐endjoing and homologous recombination repair. Global genome and replication‐linked repair and response mechanisms are critical for preventing mutagenesis and cancer. Such a model provides an explanation for the segmental ageing phenotypes of progeroid syndromes in man and mouse mutants in which deficiencies in different DNA repair systems are associated with a different subset of accelerated ageing symptoms. Natural variation in genome maintenance mechanisms, metabolism and exogenous exposure likely contributes to the different ageing trajectories between organs and inter‐individual differences in normal ageing. Finally, we show that organoid cultures from progeroid mice faithfully recapitulate various ageing features and may be useful tools for studying regenerative interventions.
FIGURE 7.

Tentative model for organ‐specific anti‐ageing strategies. Remaining cellular lifespan largely determines which DNA damage response strategy is preferred by organs/tissues to counteract ageing. For intestine, cell death is preferred, as cells have to function only for 3–5 days and cell loss can be compensated by increased (stem) cell proliferation. Obviously, this is very energy‐demanding (daily a human body produces 200 gram intestinal epithelium) and unaffordable for many organs. Other tissues with continuous but slower cell renewal such as the hematopoietic system (average cell turn‐over ~2 months) rely mostly on replication‐related repair (such as NHEJ/HR and XLR) and apoptosis (Hoeijmakers, 2001). Skin, as an organ with high UV exposure and also intermediate cell renewal combines GG‐NER and TCR with apoptosis and premature differentiation of damaged stem cells (Kim et al., 2020). Finally, tissues with slow (e.g., liver, on average ~1 year) or no cell turn‐over (e.g., the central nervous system, life time) depend on constitutive (cell cycle independent) DNA repair systems, most notably TCR to permit long‐term unperturbed use of the transcribed compartment of the genome, needed for sustained proper cellular functioning. Global genome repair systems (base and nucleotide excision repair) are probably important for all organs and tissues for preventing mutagenesis and permit survival. TLS allows replication bypass of lesions to rescue stalled replication and cellular proliferative capacity, however, at the expense of elevated mutagenesis and cancer risk. Cellular (replicative) senescence opposes cell death in most organs. This model explains the segmental nature of repair‐deficient progeroid syndromes, in which inherited deficiencies in different repair systems are associated with a different subset of organs and tissues displaying accelerated ageing.
1Not including mismatch repair, which is a replication error correction system, important for preventing mutations and cancer, particularly in highly proliferative tissues such as intestine. GG‐NER, global genome nucleotide excision repair; NHEJ/HR, Non‐Homologous End‐Joining/Homologous recombination repair two pathways for double strand break repair; TCR, transcription‐coupled repair; TLS, translesion synthesis; XLR, cross‐link repair
4. METHODS
4.1. Mice
All animals were housed in the Erasmus Medical experimental animal center and handled in accordance with the recommendations and regulations of the animal ethical committee (DEC‐consult) and national/EU legislation. Ercc1∆ /− mice were bred in a hybrid F1 genetic background (FVB/N:C57Bl/6J) and genotyped as previously described (Vermeij, Dollé, et al., 2016). The Lgr5EGFP − IRES − CRE2 transgenic mouse line has been previously described (Barker et al., 2007). Lgr5EGFP − IRES − CRE2 Ercc1Δ /− mice were generated by first crossing male Ercc1 +/− mice with female Lgr5EGFP − IRES − CRE2 , all in a pure C57BL/6J background. Female Lgr5EGFP − IRES − CRE2 Ercc1 +/− C57BL/6J progeny were subsequently crossed with male Ercc1∆ /+ mice, in a pure FVB background and the desired Lgr5EGFP − IRES − CRE2 Ercc1Δ /− offspring were obtained in a 50/50 F1 C57BL6J/FVB hybrid background. Ercc1∆ /+ and Ercc1 +/− mice have been previously described (Weeda et al., 1997).
4.2. Immunohistochemistry and immunocytochemistry, microspcopy, FACS and flow cytometric analysis
Immunohistochemistry and immunocytochemistry of intestinal and liver tissues were done according to established procedures and detailed in supplementary materials, including the specific reagents (e.g., antibodies) used. Stainings of paraffin‐embedded SI and liver tissue sections and liver organoids for senescence markers were performed as described (Baar et al., 2017). Procedures for microscopy, FACS and flow cytometric analysis and equipment are specified in Supplementary materials.
4.3. Apoptosis
TUNEL staining of tissue sections and organoid cultures was performed using an in situ cell death kit (Roche) or the Apoptag Plus Peroxidase in situ apoptosis detection kit (Millipore). Apoptosis of ISCs and LSCs after treatment with illudin S and cisplatin was assessed by staining for cytochrome c of single ISC and LSC suspensions—derived from undifferentiated organoid cultures—embedded in Matrigel using standard immunofluorescence protocol for cells (Baar et al., 2017).
4.4. In vitro small intestinal organoid culture
Intestinal organoid formation from crypts was performed as described (Sato et al., 2009). A total of 1000 crypts was mixed with 50 μl of Matrigel and plated in 24‐well plates. Intestinal organoids were grown in ENR medium (Advanced DMEM/F12, B27 and N2 supplement (Invitrogen), 50 ng μl−1 epidermal growth factor, 100 ng ml−1 Noggin (Peprotech), 500 ng ml−1 R‐spondin‐1, 1.25 mM N‐acetylcystein) supplemented with Wnt3a‐conditioned medium34 when appropriate to keep organoids in undifferentiated state. For organoid growth from single cell crypt suspensions, crypts were incubated with TrypLE (Invitrogen) and passed through a 40 μm cell strainer. Resulting cell suspensions were mixed with Matrigel and plated as described with supplementation of growth medium with 10μM Rock inhibitor Y27632 (Sigma). The medium was refreshed every two days. Organoids were passaged every 10 days.
4.5. In vitro liver organoid culture
Growth of organoid cultures from liver bile duct enriched cell suspensions was performed as described (Broutier et al., 2016; Huch et al., 2013, 2015). A total of 100,000 cells was mixed with 50 μl of Matrigel and plated on 24‐well plates. Culture medium was refreshed every two days, and organoids were passaged every 8 to 10 days. Growth of liver organoids from single cells was performed with fluorescence‐activated cell sorting of cell suspensions derived from TrypLE‐digested primary cultures.
4.6. EdU incorporation assay
In intestinal and liver organoid culture medium, EdU was added for 2 h at a final concentration of 10 μM, cultures were washed with PBS, replenished and at the appropriate time points, fixed with 4% paraformaldehyde in PBS for 15 min at room temperature. Edu immunofluorescence was performed using Click‐iT® EdU Alexa Fluor® Imaging Kit (Invitrogen), according to manufacturers’ instructions.
4.7. Genotoxic sensitivity assay
Organoids were dissociated into single cell suspension using TrypLE reagent and passed through a 40 μm cell strainer. Following cell counting, 10,000 single ISCs and 500 LSCs were resuspended in 20 μl Matrigel and plated in a 48‐well plate. Cisplatin and illudin S were diluted in HBSS at appropriate concentrations and added to each well. Two hours after treatment medium was removed and replaced with appropriate stem cell culture medium supplemented with 10μM Rock inhibitor Y27632 (Sigma). For ISC cultures, Gsk3 inhibitor CHIR‐9921 (Axon MedChem) was added to the culture medium at a final concentration of 10 μM.
For UV sensitivity assessment, suspensions of single stem cells were plated in 12‐well plates at different densities and centrifuged at 600 relative centrifugal force for 20 min. at 32°C, medium was carefully removed, and the cells at the well bottom were exposed to the indicated doses of UV (254nm, TUV Lamp Philips). Cells were collected in culture medium, centrifuged at 1500 rpm, resuspended in Matrigel and plated in triplicates in 48‐well plates. Organoids were counted 5 days after plating.
4.8. Assessment of DNA lesion resolution in SCs
Organoids were grown in 8‐well tissue culture slides, exposed to UVB (200J/m2, Philips 40W/12RS UVB lamp) and at selected time points fixed (2% formalin, 20 min. at RT), washed with PBS and 0.1 M glycine, permeabilized with 0,5% Triton‐X‐100 in PBS for 20 min. RT and treated with 2N HCL for 30 min to denature DNA. After extensive washes with PBS and incubation with blocking solution (1% BSA in PBS), 150μl of anti‐6,4‐PP antibody (COSMO‐BIO, Cat#CAC‐NM‐DND‐002) mix (1:500 in blocking solution) was added to each well, and the slides were transferred to a 370C incubator for 3 h. Organoids were washed 3 times with PBST (PBS and 0.05% Triton‐X‐100) and incubated with secondary antibody (Alexa Fluor 555) diluted 1:100 in PBS for 2 h at 37°C. Nuclei were counterstained with Hoechst for 30min. Subsequently, Prolong Diamond mounting medium (Thermo Fisher) was added and slides were incubated overnight in a freezer. Images were captured with a confocal microscope. Fluorescence measurements and analysis were performed using image J software.
4.9. Statistics
Experimental sample size was not strictly chosen based on utilization of statistical methods prior to initiation of the study. Experiments were not standardly performed and analysed in a randomized, blinded fashion. Statistical analysis was performed with GraphPad Prism software. Student's t test and nonparametric Mann‐Whitney test were used to calculate P values. For multiple comparisons, two‐way ANOVA was implemented for evaluation of statistical significance (GraphPad PRISM).
For all other procedures, see Supplementary Materials.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
M.V. designed experimental work, performed experiments, analysed data and wrote the manuscript. J.D. designed, performed and analysed experiments. M.B. performed senescence stainings. S.B. performed flow cytometry, A.M. Ki67, and GFP immunostainings and critically commented on experiments. E.K., R.v.B. and M.J. grown organoid cultures and provided expertise, reagents and protocols. S.M. commented on experiments, data analysis and participated in cell viability experiments. R.M.C.B. helped with the mouse experiments. W.P.V. provided data on p21 and apoptosis and commented on study design and manuscript. J.K. performed immunostainings. R.v.B. and E.C. were involved in the conceptual design. J.P. and J.H.J.H. conceived and supervised the study and wrote the manuscript.
Supporting information
Fig S1
Fig S2
Fig S3
Fig S4
Fig S5
Fig S6
Supplementary Material
ACKNOWLEDGMENTS
M.V., J.H.J.H. and J.P. acknowledge funding from Marie Curie ITN program MARIAGE. Financial support was obtained from NIH/NIA (PO1 AG017242), European Research Council Advanced Grants DamAge, Dam2Age, and PoC Grant Dementia to J.H.J.H., the KWO award and Oncode, supported by the Dutch Cancer Society (5030), Deutsche Forschungsgemeinschaft (DFG) Projekt 73111208 ‐ SFB 829. Additional support was obtained from ‘Memorabel’ (Medical Sciences) and EJP‐RD project TC‐NER. The authors are indebted to Dr. Masaki Akita for 6,4 photoproduct immunostaining, Tsun Wai Kan for cell sorting and Dr. Georgios Gkouridis for comments on the manuscript.
Vougioukalaki, M. , Demmers, J. , Vermeij, W. P. , Baar, M. , Bruens, S. , Magaraki, A. , Kuijk, E. , Jager, M. , Merzouk, S. , Brandt, R. M. C. , Kouwenberg, J. , van Boxtel, R. , Cuppen, E. , Pothof, J. , & Hoeijmakers, J. H. J. (2022). Different responses to DNA damage determine ageing differences between organs. Aging Cell, 21, e13562. 10.1111/acel.13562
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Ahmad, A. , Robinson, A. R. , Duensing, A. , van Drunen, E. , Beverloo, H. B. , Weisberg, D. B. , Hasty, P. , Hoeijmakers, J. H. , & Niedernhofer, L. J. (2008). ERCC1‐XPF endonuclease facilitates DNA double‐strand break repair. Molecular and Cellular Biology, 28, 5082–5092. 10.1128/MCB.00293-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alyodawi, K. , Vermeij, W. P. , Omairi, S. , Kretz, O. , Hopkinson, M. , Solagna, F. , Joch, B. , Brandt, R. M. C. , Barnhoorn, S. , Vliet, N. , Ridwan, Y. , Essers, J. , Mitchell, R. , Morash, T. , Pasternack, A. , Ritvos, O. , Matsakas, A. , Collins‐Hooper, H. , Huber, T. B. , … Patel, K. (2019). Compression of morbidity in a progeroid mouse model through the attenuation of myostatin/activin signalling. Journal of Cachexia, Sarcopenia and Muscle, 10, 662–686. 10.1002/jcsm.12404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andressoo, J.‐O. , Mitchell, J. R. , de Wit, J. , Hoogstraten, D. , Volker, M. , Toussaint, W. , Speksnijder, E. , Beems, R. B. , van Steeg, H. , Jans, J. , de Zeeuw, C. I. , Jaspers, N. G. J. , Raams, A. , Lehmann, A. R. , Vermeulen, W. , Hoeijmakers, J. H. J. , & van der Horst, G. T. J. (2006). An Xpd mouse model for the combined xeroderma pigmentosum/Cockayne syndrome exhibiting both cancer predisposition and segmental progeria. Cancer Cell, 10, 121–132. 10.1016/j.ccr.2006.05.027 [DOI] [PubMed] [Google Scholar]
- Baar, M. P. , Brandt, R. M. C. , Putavet, D. A. , Klein, J. D. D. , Derks, K. W. J. , Bourgeois, B. R. M. , Stryeck, S. , Rijksen, Y. , van Willigenburg, H. , Feijtel, D. A. , van der Pluijm, I. , Essers, J. , van Cappellen, W. A. , van IJcken, W. F. , Houtsmuller, A. B. , Pothof, J. , de Bruin, R. W. F. , Madl, T. , Hoeijmakers, J. H. J. , … de Keizer, P. L. J. (2017). Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell, 169(132–147), e116. 10.1016/j.cell.2017.02.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker, N. (2014). Adult intestinal stem cells: critical drivers of epithelial homeostasis and regeneration. Nature Reviews Molecular Cell Biology, 15, 19–33. 10.1038/nrm3721 [DOI] [PubMed] [Google Scholar]
- Barker, N. , van Es, J. H. , Kuipers, J. , Kujala, P. , van den Born, M. , Cozijnsen, M. , Haegebarth, A. , Korving, J. , Begthel, H. , Peters, P. J. , & Clevers, H. (2007). Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature, 449, 1003–1007. 10.1038/nature06196 [DOI] [PubMed] [Google Scholar]
- Barnhoorn, S. , Uittenboogaard, L. M. , Jaarsma, D. , Vermeij, W. P. , Tresini, M. , Weymaere, M. , Menoni, H. , Brandt, R. M. C. , de Waard, M. C. , Botter, S. M. , Sarker, A. H. , Jaspers, N. G. J. , van der Horst, G. T. J. , Cooper, P. K. , Hoeijmakers, J. H. J. , & van der Pluijm, I. (2014). Cell‐autonomous progeroid changes in conditional mouse models for repair endonuclease XPG deficiency. PLoS Genetics, 10, e1004686. 10.1371/journal.pgen.1004686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barriga, F. M. , Montagni, E. , Mana, M. , Mendez‐Lago, M. , Hernando‐Momblona, X. , Sevillano, M. , Guillaumet‐Adkins, A. , Rodriguez‐Esteban, G. , Buczacki, S. J. A. , Gut, M. , Heyn, H. , Winton, D. J. , Yilmaz, O. H. , Attolini, C.‐O. , Gut, I. , & Batlle, E. (2017). Mex3a marks a slowly dividing subpopulation of Lgr5+ intestinal stem cells. Cell Stem Cell, 20, 801–816.e7. 10.1016/j.stem.2017.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerman, I. , Seita, J. , Inlay, M. A. , Weissman, I. L. , & Rossi, D. J. (2014). Quiescent hematopoietic stem cells accumulate DNA damage during aging that is repaired upon entry into cell cycle. Cell Stem Cell, 15, 37–50. 10.1016/j.stem.2014.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begus‐Nahrmann, Y. , Lechel, A. , Obenauf, A. C. , Nalapareddy, K. , Peit, E. , Hoffmann, E. , Schlaudraff, F. , Liss, B. , Schirmacher, P. , Kestler, H. , Danenberg, E. , Barker, N. , Clevers, H. , Speicher, M. R. , & Rudolph, K. L. (2009). p53 deletion impairs clearance of chromosomal‐instable stem cells in aging telomere‐dysfunctional mice. Nature Genetics, 41, 1138–1143. 10.1038/ng.426 [DOI] [PubMed] [Google Scholar]
- Bogliolo, M. , Schuster, B. , Stoepker, C. , Derkunt, B. , Su, Y. , Raams, A. , Trujillo, J. P. , Minguillón, J. , Ramírez, M. J. , Pujol, R. , Casado, J. A. , Baños, R. , Rio, P. , Knies, K. , Zúñiga, S. , Benítez, J. , Bueren, J. A. , Jaspers, N. G. J. , Schärer, O. D. , … Surrallés, J. (2013). Mutations in ERCC4, encoding the DNA‐repair endonuclease XPF, cause Fanconi anemia. American Journal of Human Genetics, 92, 800–806. 10.1016/j.ajhg.2013.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broutier, L. , Andersson‐Rolf, A. , Hindley, C. J. , Boj, S. F. , Clevers, H. , Koo, B. K. , & Huch, M. (2016). Culture and establishment of self‐renewing human and mouse adult liver and pancreas 3D organoids and their genetic manipulation. Nature Protocols, 11, 1724–1743. 10.1038/nprot.2016.097 [DOI] [PubMed] [Google Scholar]
- Cho, J. S. , Kook, S. H. , Robinson, A. R. , Niedernhofer, L. J. , & Lee, B. C. (2013). Cell autonomous and nonautonomous mechanisms drive hematopoietic stem/progenitor cell loss in the absence of DNA repair. Stem Cells, 31, 511–525. 10.1002/stem.1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer, J. , Andressoo, J. O. , de Wit, J. , Huijmans, J. , Beems, R. B. , van Steeg, H. , Weeda, G. , van der Horst, G. T. J. , van Leeuwen, W. , Themmen, A. P. N. , Meradji, M. , & Hoeijmakers, J. H. J. (2002). Premature aging in mice deficient in DNA repair and transcription. Science, 296, 1276–1279. 10.1126/science.1070174 [DOI] [PubMed] [Google Scholar]
- de Laat, W. L. , Jaspers, N. G. , & Hoeijmakers, J. H. (1999). Molecular mechanism of nucleotide excision repair. Genes & Development, 13, 768–785. 10.1101/gad.13.7.768 [DOI] [PubMed] [Google Scholar]
- Dollé, M. E. T. , Kuiper, R. V. , Roodbergen, M. , Robinson, J. , de Vlugt, S. , Wijnhoven, S. W. P. , Beems, R. B. , de la Fonteyne, L. , de With, P. , van der Pluijm, I. , Niedernhofer, L. J. , Hasty, P. , Vijg, J. , Hoeijmakers, J. H. J. , & van Steeg, H. (2011). Broad segmental progeroid changes in short‐lived Ercc1 −/Δ7 mice. Pathobiology of Aging & Age‐related Diseases, 1(1), 7219. 10.3402/pba.v1i0.7219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flach, J. , Bakker, S. T. , Mohrin, M. , Conroy, P. C. , Pietras, E. M. , Reynaud, D. , Alvarez, S. , Diolaiti, M. E. , Ugarte, F. , Forsberg, E. C. , Le Beau, M. M. , Stohr, B. A. , Méndez, J. , Morrison, C. G. , & Passegué, E. (2014). Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature, 512, 198–202. 10.1038/nature13619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann‐Stroissnigg, H. , Ling, Y. Y. , Zhao, J. , McGowan, S. J. , Zhu, Y. I. , Brooks, R. W. , Grassi, D. , Gregg, S. Q. , Stripay, J. L. , Dorronsoro, A. , Corbo, L. , Tang, P. , Bukata, C. , Ring, N. , Giacca, M. , Li, X. , Tchkonia, T. , Kirkland, J. L. , Niedernhofer, L. J. , & Robbins, P. D. (2017). Identification of HSP90 inhibitors as a novel class of senolytics. Nature Communications, 8, 422. 10.1038/s41467-017-00314-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garinis, G. A. , Uittenboogaard, L. M. , Stachelscheid, H. , Fousteri, M. , van Ijcken, W. , Breit, T. M. , van Steeg, H. , Mullenders, L. H. F. , van der Horst, G. T. J. , Brüning, J. C. , Niessen, C. M. , Hoeijmakers, J. H. J. , & Schumacher, B. (2009). Persistent transcription‐blocking DNA lesions trigger somatic growth attenuation associated with longevity. Nature Cell Biology, 11, 604–615. 10.1038/ncb1866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehart, H. , & Clevers, H. (2015). Repairing organs: lessons from intestine and liver. Trends in Genetics, 31, 344–351. 10.1016/j.tig.2015.04.005 [DOI] [PubMed] [Google Scholar]
- Gilgenkrantz, H. , & Collin de l'Hortet, A. (2018). Understanding Liver Regeneration: From Mechanisms to Regenerative Medicine. The American Journal of Pathology, 188, 1316–1327. 10.1016/j.ajpath.2018.03.008 [DOI] [PubMed] [Google Scholar]
- Gillet, L. C. , & Scharer, O. D. (2006). Molecular mechanisms of mammalian global genome nucleotide excision repair. Chemical Reviews, 106, 253–276. 10.1021/cr040483f [DOI] [PubMed] [Google Scholar]
- Gregg, S. Q. , Gutiérrez, V. , Rasile Robinson, A. , Woodell, T. , Nakao, A. , Ross, M. A. , Michalopoulos, G. K. , Rigatti, L. , Rothermel, C. E. , Kamileri, I. , Garinis, G. A. , Beer Stolz, D. , & Niedernhofer, L. J. (2012). A mouse model of accelerated liver aging caused by a defect in DNA repair. Hepatology, 55, 609–621. 10.1002/hep.24713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeijmakers, J. H. (2001). Genome maintenance mechanisms for preventing cancer. Nature, 411, 366–374. 10.1038/35077232 [DOI] [PubMed] [Google Scholar]
- Hoeijmakers, J. H. (2009). DNA damage, aging, and cancer. The New England Journal of Medicine, 361, 1475–1485. 10.1056/NEJMra0804615 [DOI] [PubMed] [Google Scholar]
- Huch, M. , Dorrell, C. , Boj, S. F. , van Es, J. H. , Li, V. S. W. , van de Wetering, M. , Sato, T. , Hamer, K. , Sasaki, N. , Finegold, M. J. , Haft, A. , Vries, R. G. , Grompe, M. , & Clevers, H. (2013). In vitro expansion of single Lgr5+ liver stem cells induced by Wnt‐driven regeneration. Nature, 494, 247–250. 10.1038/nature11826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huch, M. , Gehart, H. , van Boxtel, R. , Hamer, K. , Blokzijl, F. , Verstegen, M. M. A. , Ellis, E. , van Wenum, M. , Fuchs, S. A. , de Ligt, J. , van de Wetering, M. , Sasaki, N. , Boers, S. J. , Kemperman, H. , de Jonge, J. , Ijzermans, J. N. M. , Nieuwenhuis, E. E. S. , Hoekstra, R. , Strom, S. , … Clevers, H. (2015). Long‐term culture of genome‐stable bipotent stem cells from adult human liver. Cell, 160, 299–312. 10.1016/j.cell.2014.11.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager, M. , Blokzijl, F. , Kuijk, E. , Bertl, J. , Vougioukalaki, M. , Janssen, R. , Besselink, N. , Boymans, S. , de Ligt, J. , Pedersen, J. S. , Hoeijmakers, J. , Pothof, J. , van Boxtel, R. , & Cuppen, E. (2019). Deficiency of nucleotide excision repair is associated with mutational signature observed in cancer. Genome Research, 29, 1067–1077. 10.1101/gr.246223.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspers, N. G. , Raams, A. , Kelner, M. J. , Ng, J. M. , Yamashita, Y. M. , Takeda, S. , McMorris, T. C. , & Hoeijmakers, J. H. (2002). Anti‐tumour compounds illudin S and Irofulven induce DNA lesions ignored by global repair and exclusively processed by transcription‐ and replication‐coupled repair pathways. DNA Repair, 1, 1027–1038. 10.1016/S1568-7864(02)00166-0 [DOI] [PubMed] [Google Scholar]
- Kim, D. E. , Dollé, M. E. T. , Vermeij, W. P. , Gyenis, A. , Vogel, K. , Hoeijmakers, J. H. J. , Wiley, C. D. , Davalos, A. R. , Hasty, P. , Desprez, P.‐Y. , & Campisi, J. (2020). Deficiency in the DNA repair protein ERCC1 triggers a link between senescence and apoptosis in human fibroblasts and mouse skin. Aging Cell, 19, e13072. 10.1111/acel.13072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuraoka, I. , Kobertz, W. R. , Ariza, R. R. , Biggerstaff, M. , Essigmann, J. M. , & Wood, R. D. (2000). Repair of an interstrand DNA cross‐link initiated by ERCC1‐XPF repair/recombination nuclease. The Journal of Biological Chemistry, 275, 26632–26636. 10.1074/jbc.C000337200 [DOI] [PubMed] [Google Scholar]
- Lans, H. , Hoeijmakers, J. H. J. , Vermeulen, W. , & Marteijn, J. A. (2019). The DNA damage response to transcription stress. Nature Reviews Molecular Cell Biology, 20(12), 766–784. 10.1038/s41580-019-0169-4 [DOI] [PubMed] [Google Scholar]
- Lavasani, M. , Robinson, A. R. , Lu, A. , Song, M. , Feduska, J. M. , Ahani, B. , Tilstra, J. S. , Feldman, C. H. , Robbins, P. D. , Niedernhofer, L. J. , & Huard, J. (2012). Muscle‐derived stem/progenitor cell dysfunction limits healthspan and lifespan in a murine progeria model. Nature Communications, 3, 608. 10.1038/ncomms1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marteijn, J. A. , Lans, H. , Vermeulen, W. , & Hoeijmakers, J. H. (2014). Understanding nucleotide excision repair and its roles in cancer and ageing. Nature Reviews Molecular Cell Biology, 15, 465–481. 10.1038/nrm3822 [DOI] [PubMed] [Google Scholar]
- Martin, K. , Kirkwood, T. B. , & Potten, C. S. (1998). Age changes in stem cells of murine small intestinal crypts. Experimental Cell Research, 241, 316–323. 10.1006/excr.1998.4001 [DOI] [PubMed] [Google Scholar]
- Matsumura, H. , Mohri, Y. , Binh, N. T. , Morinaga, H. , Fukuda, M. , Ito, M. , Kurata, S. , Hoeijmakers, J. , & Nishimura, E. K. (2016). Hair follicle aging is driven by transepidermal elimination of stem cells via COL17A1 proteolysis. Science, 351, aad4395. 10.1126/science.aad4395 [DOI] [PubMed] [Google Scholar]
- McNeely, T. , Leone, M. , Yanai, H. , Beerman, I. (2020). DNA damage in aging, the stem cell perspective. Human Genetics, 139(3), 309–331. 10.1007/s00439-019-02047-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWhir, J. , Selfridge, J. , Harrison, D. J. , Squires, S. , & Melton, D. W. (1993). Mice with DNA repair gene (ERCC‐1) deficiency have elevated levels of p53, liver nuclear abnormalities and die before weaning. Nature Genetics, 5, 217–224. 10.1038/ng1193-217 [DOI] [PubMed] [Google Scholar]
- Milanese, C. , Bombardieri, C. R. , Sepe, S. , Barnhoorn, S. , Payán‐Goméz, C. , Caruso, D. , Audano, M. , Pedretti, S. , Vermeij, W. P. , Brandt, R. M. C. , Gyenis, A. , Wamelink, M. M. , de Wit, A. S. , Janssens, R. C. , Leen, R. , van Kuilenburg, A. B. P. , Mitro, N. , Hoeijmakers, J. H. J. , & Mastroberardino, P. G. (2019). DNA damage and transcription stress cause ATP‐mediated redesign of metabolism and potentiation of anti‐oxidant buffering. Nature Communications, 10, 4887. 10.1038/s41467-019-12640-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muradian, K. , & Schachtschabel, D. O. (2001). The role of apoptosis in aging and age‐related disease: update. Zeitschrift Fur Gerontologie Und Geriatrie, 34, 441–446. 10.1007/s003910170015 [DOI] [PubMed] [Google Scholar]
- Naipal, K. A. T. , Raams, A. , Bruens, S. T. , Brandsma, I. , Verkaik, N. S. , Jaspers, N. G. J. , Hoeijmakers, J. H. J. , van Leenders, G. J. L. H. , Pothof, J. , Kanaar, R. , Boormans, J. , & van Gent, D. C. (2015). Attenuated XPC expression is not associated with impaired DNA repair in bladder cancer. PLoS One, 10, e0126029. 10.1371/journal.pone.0126029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalapareddy, K. , Nattamai, K. J. , Kumar, R. S. , Karns, R. , Wikenheiser‐Brokamp, K. A. , Sampson, L. L. , Mahe, M. M. , Sundaram, N. , Yacyshyn, M.‐B. , Yacyshyn, B. , Helmrath, M. A. , Zheng, Y. I. , & Geiger, H. (2017). Canonical Wnt Signaling Ameliorates Aging of Intestinal Stem Cells. Cell Reports, 18, 2608–2621. 10.1016/j.celrep.2017.02.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro, S. , Meza, N. W. , Quintana‐Bustamante, O. , Casado, J. A. , Jacome, A. , McAllister, K. , Puerto, S. , Surralles, J. , Segovia, J. C. , & Bueren, J. A. (2006). Hematopoietic dysfunction in a mouse model for Fanconi anemia group D1. Molecular Therapy: The Journal of the American Society of Gene Therapy, 14, 525–535. 10.1016/j.ymthe.2006.05.018 [DOI] [PubMed] [Google Scholar]
- Niedernhofer, L. J. , Garinis, G. A. , Raams, A. , Lalai, A. S. , Robinson, A. R. , Appeldoorn, E. , Odijk, H. , Oostendorp, R. , Ahmad, A. , van Leeuwen, W. , Theil, A. F. , Vermeulen, W. , van der Horst, G. T. J. , Meinecke, P. , Kleijer, W. J. , Vijg, J. , Jaspers, N. G. J. , & Hoeijmakers, J. H. J. (2006). A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature, 444, 1038–1043. 10.1038/nature05456 [DOI] [PubMed] [Google Scholar]
- Niedernhofer, L. J. , Gurkar, A. U. , Wang, Y. , Vijg, J. , Hoeijmakers, J. H. J. , & Robbins, P. D. (2018). Nuclear Genomic Instability and Aging. Annual Review of Biochemistry, 87, 295–322. 10.1146/annurev-biochem-062917-012239 [DOI] [PubMed] [Google Scholar]
- Niedernhofer, L. J. , Odijk, H. , Budzowska, M. , van Drunen, E. , Maas, A. , Theil, A. F. , de Wit, J. , Jaspers, N. G. J. , Beverloo, H. B. , Hoeijmakers, J. H. J. , & Kanaar, R. (2004). The structure‐specific endonuclease Ercc1‐Xpf is required to resolve DNA interstrand cross‐link‐induced double‐strand breaks. Molecular and Cellular Biology, 24, 5776–5787. 10.1128/MCB.24.13.5776-5787.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijnik, A. , Woodbine, L. , Marchetti, C. , Dawson, S. , Lambe, T. , Liu, C. , Rodrigues, N. P. , Crockford, T. L. , Cabuy, E. , Vindigni, A. , Enver, T. , Bell, J. I. , Slijepcevic, P. , Goodnow, C. C. , Jeggo, P. A. , & Cornall, R. J. (2007). DNA repair is limiting for haematopoietic stem cells during ageing. Nature, 447, 686–690. 10.1038/nature05875 [DOI] [PubMed] [Google Scholar]
- Ogrodnik, M. , Miwa, S. , Tchkonia, T. , Tiniakos, D. , Wilson, C. L. , Lahat, A. , Day, C. P. , Burt, A. , Palmer, A. , Anstee, Q. M. , Grellscheid, S. N. , Hoeijmakers, J. H. J. , Barnhoorn, S. , Mann, D. A. , Bird, T. G. , Vermeij, W. P. , Kirkland, J. L. , Passos, J. F. , von Zglinicki, T. , & Jurk, D. (2017). Cellular senescence drives age‐dependent hepatic steatosis. Nature Communications, 8, 15691. 10.1038/ncomms15691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollack, M. , Phaneuf, S. , Dirks, A. , & Leeuwenburgh, C. (2002). The role of apoptosis in the normal aging brain, skeletal muscle, and heart. Annals of the New York Academy of Sciences, 959, 93–107. 10.1111/j.1749-6632.2002.tb02086.x [DOI] [PubMed] [Google Scholar]
- Prasher, J. M. , Lalai, A. S. , Heijmans‐Antonissen, C. , Ploemacher, R. E. , Hoeijmakers, J. H. , Touw, I. P. , & Niedernhofer, L. J. (2005). Reduced hematopoietic reserves in DNA interstrand crosslink repair‐deficient Ercc1‐/‐ mice. The EMBO Journal, 24, 861–871. 10.1038/sj.emboj.7600542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritsma, L. , Ellenbroek, S. I. J. , Zomer, A. , Snippert, H. J. , de Sauvage, F. J. , Simons, B. D. , Clevers, H. , & van Rheenen, J. (2014). Intestinal crypt homeostasis revealed at single‐stem‐cell level by in vivo live imaging. Nature, 507, 362–365. 10.1038/nature12972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi, D. J. , Bryder, D. , Seita, J. , Nussenzweig, A. , Hoeijmakers, J. , & Weissman, I. L. (2007). Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature, 447, 725–729. 10.1038/nature05862 [DOI] [PubMed] [Google Scholar]
- Rube, C. E. , Fricke, A. , Widmann, T. A. , Furst, T. , Madry, H. , Pfreundschuh, M. , & Rube, C. (2011). Accumulation of DNA damage in hematopoietic stem and progenitor cells during human aging. PLoS One, 6, e17487. 10.1371/journal.pone.0017487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato, T. , Vries, R. G. , Snippert, H. J. , van de Wetering, M. , Barker, N. , Stange, D. E. , van Es, J. H. , Abo, A. , Kujala, P. , Peters, P. J. , & Clevers, H. (2009). Single Lgr5 stem cells build crypt‐villus structures in vitro without a mesenchymal niche. Nature, 459, 262–265. 10.1038/nature07935 [DOI] [PubMed] [Google Scholar]
- Schumacher, B. , Pothof, J. , Vijg, J. , & Hoeijmakers, J. H. J. (2021). The central role of DNA damage in the ageing process. Nature, 592, 695–703. 10.1038/s41586-021-03307-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivdasani, R. A. (2014). Radiation redux: reserve intestinal stem cells miss the call to duty. Cell Stem Cell, 14, 135–136. 10.1016/j.stem.2014.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperka, T. , Song, Z. , Morita, Y. , Nalapareddy, K. , Guachalla, L. M. , Lechel, A. , Begus‐Nahrmann, Y. , Burkhalter, M. D. , Mach, M. , Schlaudraff, F. , Liss, B. , Ju, Z. , Speicher, M. R. , & Rudolph, K. L. (2011). Puma and p21 represent cooperating checkpoints limiting self‐renewal and chromosomal instability of somatic stem cells in response to telomere dysfunction. Nature Cell Biology, 14, 73–79. 10.1038/ncb2388 [DOI] [PubMed] [Google Scholar]
- Steegenga, W. T. , Mischke, M. , Lute, C. , Boekschoten, M. V. , Lendvai, A. , Pruis, M. G. M. , Verkade, H. J. , van de Heijning, B. J. M. , Boekhorst, J. , Timmerman, H. M. , Plösch, T. , Müller, M. , & Hooiveld, G. J. E. J. (2017). Maternal exposure to a Western‐style diet causes differences in intestinal microbiota composition and gene expression of suckling mouse pups. Molecular Nutrition & Food Research, 61(1), 1600141. 10.1002/mnfr.201600141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetteh, P. W. , Basak, O. , Farin, H. F. , Wiebrands, K. , Kretzschmar, K. , Begthel, H. , van den Born, M. , Korving, J. , de Sauvage, F. , van Es, J. H. , van Oudenaarden, A. , & Clevers, H. (2016). Replacement of Lost Lgr5‐Positive Stem Cells through Plasticity of Their Enterocyte‐Lineage Daughters. Cell Stem Cell, 18, 203–213. 10.1016/j.stem.2016.01.001 [DOI] [PubMed] [Google Scholar]
- Tian, H. , Biehs, B. , Warming, S. , Leong, K. G. , Rangell, L. , Klein, O. D. , & de Sauvage, F. J. (2011). A reserve stem cell population in small intestine renders Lgr5‐positive cells dispensable. Nature, 478, 255–259. 10.1038/nature10408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilstra, J. S. , Robinson, A. R. , Wang, J. , Gregg, S. Q. , Clauson, C. L. , Reay, D. P. , Nasto, L. A. , St Croix, C. M. , Usas, A. , Vo, N. et al (2012). NF‐kappaB inhibition delays DNA damage‐induced senescence and aging in mice. The Journal of Clinical Investigation, 122, 2601–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Pluijm, I. , Garinis, G. A. , Brandt, R. M. , Gorgels, T. G. , Wijnhoven, S. W. , Diderich, K. E. , de Wit, J. , Mitchell, J. R. , van Oostrom, C. , Beems, R. , Niedernhofer, L. J. , Velasco, S. , Friedberg, E. C. , Tanaka, K. , van Steeg, H. , Hoeijmakers, J. H. J. , & van der Horst, G. T. J. (2007). Impaired genome maintenance suppresses the growth hormone–insulin‐like growth factor 1 axis in mice with Cockayne syndrome. PLoS Biology, 5, e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeij, W. P. , Dollé, M. E. T. , Reiling, E. , Jaarsma, D. , Payan‐Gomez, C. , Bombardieri, C. R. , Wu, H. , Roks, A. J. M. , Botter, S. M. , van der Eerden, B. C. , Youssef, S. A. , Kuiper, R. V. , Nagarajah, B. , van Oostrom, C. T. , Brandt, R. M. C. , Barnhoorn, S. , Imholz, S. , Pennings, J. L. A. , de Bruin, A. , … Hoeijmakers, J. H. J. (2016a). Restricted diet delays accelerated ageing and genomic stress in DNA‐repair‐deficient mice. Nature, 537, 427–431. 10.1038/nature19329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeij, W. P. , Hoeijmakers, J. H. , & Pothof, J. (2016b). Genome Integrity in Aging: Human Syndromes, Mouse Models, and Therapeutic Options. Annual Review of Pharmacology and Toxicology, 56, 427–445. 10.1146/annurev-pharmtox-010814-124316 [DOI] [PubMed] [Google Scholar]
- Walter, D. , Lier, A. , Geiselhart, A. , Thalheimer, F. B. , Huntscha, S. , Sobotta, M. C. , Moehrle, B. , Brocks, D. , Bayindir, I. , Kaschutnig, P. , Muedder, K. , Klein, C. , Jauch, A. , Schroeder, T. , Geiger, H. , Dick, T. P. , Holland‐Letz, T. , Schmezer, P. , Lane, S. W. , … Milsom, M. D. (2015). Exit from dormancy provokes DNA‐damage‐induced attrition in haematopoietic stem cells. Nature, 520, 549–552. 10.1038/nature14131 [DOI] [PubMed] [Google Scholar]
- Wang, J. , Clauson, C. L. , Robbins, P. D. , Niedernhofer, L. J. , & Wang, Y. (2012). The oxidative DNA lesions 8,5'‐cyclopurines accumulate with aging in a tissue‐specific manner. Aging Cell, 11, 714–716. 10.1111/j.1474-9726.2012.00828.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeda, G. , Donker, I. , de Wit, J. , Morreau, H. , Janssens, R. , Vissers, C. J. , Nigg, A. , van Steeg, H. , Bootsma, D. , & Hoeijmakers, J. H. (1997). Disruption of mouse ERCC1 results in a novel repair syndrome with growth failure, nuclear abnormalities and senescence. Current Biology, 7, 427–439. 10.1016/S0960-9822(06)00190-4 [DOI] [PubMed] [Google Scholar]
- Yousefzadeh, M. , Henpita, C. , Vyas, R. , Soto‐Palma, C. , Robbins, P. , & Niedernhofer, L. (2021). DNA damage—how and why we age? eLife, 10, 10.7554/elife.62852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousefzadeh, M. J. , Zhao, J. , Bukata, C. , Wade, E. A. , McGowan, S. J. , Angelini, L. A. , Bank, M. P. , Gurkar, A. U. , McGuckian, C. A. , Calubag, M. F. , Kato, J. I. , Burd, C. E. , Robbins, P. D. , & Niedernhofer, L. J. (2020). Tissue specificity of senescent cell accumulation during physiologic and accelerated aging of mice. Aging Cell, 19, e13094. 10.1111/acel.13094 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Fig S4
Fig S5
Fig S6
Supplementary Material
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.