

ABSTRACT
We investigated the role of hematopoietically expressed homeobox protein (Hhex) in osteoclast development. Trimethylation of lysine 27 of histone H3 at the cis‐regulatory element of Hhex was maintained and that of lysine 4 was reduced during receptor activator of nuclear factor κB ligand (RANKL)‐induced osteoclastogenesis, which was associated with a reduction of Hhex expression. Overexpression of Hhex in bone marrow–derived macrophages inhibited, whereas Hhex suppression promoted, RANKL‐induced osteoclastogenesis in vitro. Conditional deletion of Hhex in osteoclast‐lineage cells promoted osteoclastogenesis and reduced cancellous bone volume in mice, confirming the negative regulatory role of Hhex in osteoclast differentiation. Expression of cyclin‐dependent kinase inhibitors such as Cdkn2a and Cdkn1b in osteoclast precursors was negatively regulated by Hhex, and Hhex deletion increased the ratio of cells at the G1 phase of the cell cycle. In conclusion, Hhex is an inhibitor of osteoclast differentiation that is regulated in an epigenetic manner and regulates the cell cycle of osteoclast precursors and the skeletal homeostasis. © 2022 The Authors. JBMR Plus published by Wiley Periodicals LLC on behalf of American Society for Bone and Mineral Research.
Keywords: EPIGENETICS, OSTEOCLAST, HHEX, CELL CYCLE, CYCLIN‐DEPENDENT KINASE INHIBITOR
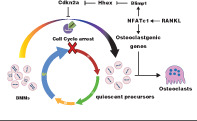
Introduction
Osteoclasts, the primary cells for bone resorption, are involved in both physiologic and pathologic bone resorption.( 1 ) Osteoclast differentiation from monocyte/macrophage‐lineage precursor cells is regulated by two critical cytokines: receptor activator of nuclear factor κB ligand (RANKL) and macrophage colony‐stimulation factor (M‐CSF).( 2 , 3 , 4 ) Transcription factors such as nuclear factor of activated T cell cytoplasmic 1 (NFATc1), nuclear factor κB (NF‐κB), and c‐Fos play critical roles in osteoclast differentiation.( 3 , 5 ) In addition to positive regulators, several negative regulators of osteoclast differentiation have been identified. Expression of interferon regulatory factor 8 (IRF8) is reduced in response to RANKL, and IRF8 negatively regulates osteoclast differentiation. Targeted disruption of Irf8 in mice led to severe osteoporosis due to an increased number of osteoclasts, and enhanced bone destruction induced by lipopolysaccharide administration.( 6 ) We recently reported that a pioneering transcription factor, PU.1, switches its transcription partners from IRF8 to NFATc1, and alters DNA binding regions during osteoclastogenesis; such features were associated with changes in epigenetic profiles and cell‐type–specific gene expression.( 7 , 8 ) In addition to IRF8, transcriptional factors such as B cell lymphoma (Bcl)6( 9 , 10 ) and MAF bZIP transcription factor (Maf)B( 11 ) have been identified as negative regulators of osteoclast differentiation. However, the role of negative regulators and their mechanisms of action have not been fully elucidated compared with positive regulators.
To identify critical negative regulators of osteoclast differentiation, we focused on epigenetic regulation, in particular histone modifications, during RANKL‐induced osteoclastogenesis.( 12 , 13 ) Histone modification plays a crucial role in the regulation of gene expression and cellular differentiation, and trimethylation of lysine 4 of histone H3 (H3K4me3) is associated with transcriptionally active or poised genes. Acetylation of H3K27 (H3K27ac) specifically distinguishes active cis‐regulatory elements and antagonizes polycomb repressive complex 2 (PRC2)‐dependent epigenetic silencing by trimethylation of lysine 27 of histone H3 (H3K27me3). We previously reported that RANKL stimulation reduced the H3K27me3 modification of cis‐regulatory elements of NFATc1 by increasing histone demethylase Jumonji D3.( 13 ) In the present study, we investigated transcription factors whose histone modification status changed during osteoclast differentiation, and identified the hematopoietically expressed homeobox (Hhex, also known as Prh) gene as a negative regulator of osteoclast differentiation. Expression of Hhex was decreased by RANKL stimulation, and Hhex overexpression inhibited and its suppression promoted RANKL‐induced osteoclast differentiation. Interestingly, Hhex regulated the expression of cyclin‐dependent kinase inhibitors (CDKIs) in osteoclast precursors, which play critical roles in the cell cycle progression of osteoclast precursors and their differentiation into mature osteoclasts.
Materials and Methods
Reagents
α‐Minimum Essential Medium (α‐MEM), Dulbecco's Modified Eagle Medium (DMEM), and protease inhibitor cocktail were purchased from Nacalai Tesque (Kyoto, Japan). Fetal bovine serum (FBS), penicillin‐streptomycin, and radioimmunoprecipitation assay (RIPA) buffer were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Human M‐CSF was purchased from PeproTech (Cranbury, NJ, USA). Bacteria‐derived soluble RANKL was purchased from Fujifilm Wako Pure Chemical (Osaka, Japan). A tartrate‐resistant acid phosphatase (TRAP) staining kit was purchased from Cosmo Bio (Tokyo, Japan). Polyinosinic–polycytidylic acid (pIpC) and polybrene transfection reagent were purchased from Sigma Aldrich (St. Louis, MO, USA). Lipofectamine 2000 was purchased from Invitrogen (Carlsbad, CA, USA). Cell Counting Kit‐8 (CCK‐8) and Cell Cycle Assay Solution Blue were purchased from Doujindo (Kumamoto, Japan). The anti‐H3K4me3 antibody (polyclonal antibody, rabbit, 39159, RRID:AB_2615077) was purchased from Active Motif (Carlsbad, CA, USA). Anti‐H3K27me3 (polyclonal antibody, rabbit, 07‐449, RRID:AB_310624) and anti‐H3K27ac antibodies (monoclonal antibody, mouse, 05‐1334, RRID:AB_1977244) were from Millipore (Billerica, MA, USA).
Animal models
Hhex fl/fl (025396) mice on a C57BL/6 background were purchased from Jackson Laboratories (Bar Harbor, ME, USA). Ctsk Cre/+ and Mx‐1 Cre/+ mice were generated as described.( 14 , 15 ) All mice were born and maintained under specific pathogen‐free conditions. Water and food were provided ad libitum. All animal experiments were performed with the approval of the Animal Study Committee of Tokyo University Experimental Animal Ethics Committee (P17‐091) and conformed to relevant guidelines and laws. Hhex fl/fl mice were crossed with CtsK Cre+/− mice to generate CtsK Cre+/−, Hhex fl/fl mice (Hhex ΔOC/−). Hhex fl/fl mice were crossed with Mx‐1 Cre+/− mice to generate Mx‐1 Cre+/−, Hhex fl/fl mice (Hhex MxCre/−). pIpC (12.5 μg/g body weight) was administered to 12‐week‐old mice intraperitoneally dissolved in phosphate‐buffered saline (PBS) once a week at least 3 weeks prior to their use in experiments. Hhex fl/fl mice were used as controls for Hhex ΔOC/− and Hhex ΔMxCre/− mice.
In vitro osteoclastogenesis and osteoclastogenesis assay
To prepare bone marrow macrophages (BMMs), murine bone marrow cells were cultured in α‐MEM with 10% FBS, 1% penicillin‐streptomycin, and 100 ng/mL of M‐CSF for 5 days as described.( 7 ) BMMs were used as osteoclast precursors and further cultured in the presence of 10 ng/mL of M‐CSF and soluble RANKL (sRANKL) (concentrations were tailored to each experiment) for 3 days to generate mature osteoclasts. Cells were fixed with 10% Formalin Neutral Buffer Solution for 5 minutes and stained with a TRAP staining kit. TRAP‐positive cells with more than five nuclei were counted as osteoclasts.
Quantitative reverse‐transcription PCR analysis
Total RNA was extracted with a Direct‐zol RNA Microprep kit (Zymo Research, Irvine, CA, USA) and reverse‐transcribed using a ReverTraAce quantitative reverse‐transcription PCR (RT‐qPCR) Master Mix (Toyobo, Osaka, Japan) to produce single‐stranded cDNA according to the manufacturer's protocol. PCR was performed on Thermal Cycler Dice® Real Time System III (Takara Bio, Shiga, Japan) using THUNDERBIRD SYBR qPCR Mix (Toyobo) according to the manufacturer's instructions. Amplification data were quantified using the standard curve method. Detected signals were confirmed to be specific by a dissociation protocol. All reactions were performed in triplicate.
Primer sequences used for real‐time RT‐PCR analysis were as follows:
Hhex‐F: 5′‐ GTTTCAGAATCGCCGAGCTAAAT‐3′,R: 5′‐ CTGCTCACAGGAAGTGTCCAAA‐3′;Cdkn2a‐F: 5′‐CTGAATCTCCGCGAGGAAAGC‐3′,R: 5′‐GCCCATCATCATCACCTGAATCG‐3′;Nfatc1‐F: 5′‐CAAGTCTCACCACAGGGCTCACTA‐3′,R: 5′‐GCGTGAGAGGTTCATTCTCCAAGT‐3′;Blimp1‐F: 5′‐TTCTTGTGTGGTATTGTCGGGACTT‐3′,R: 5′‐TTGGGGACACTCTTTGGGTAGAGTT‐3′;beta‐Actin‐F: 5′‐CAGCCTTCCTTCTTGGGTATG‐3′,R: 5′‐AGGTCTTTACGGATGTCAACG‐3′;Acp5‐F: 5′‐GACCACAACCTGCAGTATCTTC‐3′R: 5′‐CATAGTGAAACCGCAAGTAGCC‐3′Oscar‐F: 5′‐ATCAGTTTCGAAGGTTCTGGC‐3′R: 5′‐CTGCTGTGCCAATCACAAGTA‐3′Cdkn1b‐F: 5′‐CAGACGTAAACAGCTCCGAATTA‐3′,R: 5′‐TCAGTGCTTATACAGGATGTCCA‐3′;Cdkn1a‐F: 5′‐AGAACGGTGGAACTTTGACT‐3′,R: 5′‐GAGTGCAAGACAGCGACAAG‐3′.Tp53‐F: 5′‐TTCTCCGAAGACTGGATGACTGC‐3′,R:5′‐CGTCCATGCAGTGAGGTGATG‐3′
Immunostaining
After osteoclastogenesis was induced by soluble RANKL and M‐CSF for 2 days, cells were washed with PBS, fixed with 4% (wt/vol) paraformaldehyde for 15 minutes at room temperature, and subsequently permeabilized for 5 minutes with 0.1% (vol/vol) Triton X‐100. Next, cells were rinsed twice with PBS and blocked in 10% (vol/vol) normal goat serum for 1 hour. After blocking, cells were washed with PBS, followed by staining with primary antibody or rhodamine‐conjugated phalloidin (1:200) for 1 hour at room temperature. After washing with PBS, 0.1 μg/mL DAPI (Sigma‐Aldrich) was added for nuclear staining. Fluorescence images were obtained using confocal microscopy (Keyence, Osaka, Japan).
Western blotting
Cells were harvested after washing with ice‐cold PBS and then lysed in RIPA buffer containing protease inhibitor cocktail. Whole‐cell lysates were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE) and transferred onto polyvinylidene fluoride membranes (Millipore). After blocking, the membranes were incubated with the following primary antibodies: anti‐Hhex (ab34222; Abcam, Cambridge, UK), anti‐FLAG (F3165; Sigma‐Aldrich), anti‐GFP (A‐11122; Thermo Fisher Scientific), and horseradish peroxidase (HRP)‐conjugated beta actin (HRP‐60008; Proteintech Technology, Chicago, IL, USA). Antibody detection was accomplished using HRP‐conjugated secondary antibodies (W4021 or W4011; Proteintech) and chemiluminescence signals were developed with Chemi‐Lumi One Super (02230; Nacalai Tesque). Signals were detected and analyzed by a CL1000 Imaging System iBright (Invitrogen).
Plasmid construction and retrovirus transfection
For Hhex overexpression, the full‐length coding sequence of mouse Hhex (NM_008245) was cloned from mature osteoclasts by digestion with EcoRI and XhoI, and subcloned into a retroviral vector (pMX‐IRES‐Puro). The retroviral vector pMX‐Cre has been described.( 10 ) For the chromatin immunoprecipitation sequencing (ChIP) assay, mouse Hhex was cloned into the pMX‐Puro plasmid, whereby the C‐terminal end of the gene contained a 3 × FLAG sequence. To generate retroviral particles, retroviral vectors were transfected into the packaging cell line 293GPG using Lipofectamine 2000 according to the manufacturer's protocol. Supernatants containing retroviruses were collected after 24 hours of transfection. Retroviral particles were used for infection after concentration by centrifugation at 6000g for 16 hours. For retroviral infection, BMMs precultured with M‐CSF were incubated with retrovirus particles in the presence of 10 ng/mL of M‐CSF and 3 μg/mL of polybrene for 6 hours, followed by culture overnight in the presence of 10 ng/mL of M‐CSF. To select transduced BMMs, cells were detached once with trypsin/EDTA (Sigma‐Aldrich) and cultured in α‐MEM containing 10% FBS, 1% penicillin‐streptomycin, 10 ng/mL of M‐CSF, and 3 μg/mL of puromycin for 2 days. After puromycin selection, BMMs were used to generate osteoclasts or for ChIP experiments.
Bone phenotype analysis
Micro–computed tomography (μCT) scanning was performed using a InspeXio SMX‐100CT system (Shimadzu, Kyoto, Japan). Scanning was conducted at 90 kV and 40 μA, and the resolution of a single CT slice was 1024 × 1024 pixels. For bone morphology measurements, a range of 1 mm from the epiphyseal line distal to the femur was analyzed. Three‐dimensional microstructural image data were reconstructed and structural indices were calculated using TRI/3D‐BON software (RATOC Systems, Osaka, Japan).
Histomorphometric analysis
Histomorphometric analysis was performed at magnification ×400 on undecalcified sections from the secondary spongiosa area of the proximal tibia as described.( 16 ) For analysis of bone mineralization, mice were injected intraperitoneally with 8 mg/kg calcein 5 days and 2 days before they were euthanized. The eroded surface (ES), osteoclast surface (Oc.S), and osteoblast surface (Ob.S) were measured. The eroded surface/bone surface ratio (ES/BS), osteoclast surface/bone surface ratio (Oc.S/BS), osteoblast surface/bone surface ratio (Ob.S/BS), and mineral apposition rate (MAR) were calculated. The balance of bone metabolism in the proximal tibia as the % ratios of osteoid surface/bone surface (OS/BS), ES/BS were calculated.
ChIP
ChIP sequencing (ChIP‐seq) was performed as described.( 7 ) In brief, cells were fixed with 1% formaldehyde at room temperature and then neutralized with glycine. Next, cells were harvested, resuspended, sonicated, and then incubated with protein A/G beads that had been preincubated with 4 to 10 μg of the required antibody. Immunoprecipitates were washed and reverse crosslinked. DNA was purified with a PCR purification kit (Qiagen, Hilden, Germany). DNA libraries were prepared for sequencing using the standard Illumina protocol (Illumina, San Diego, CA, USA). Purified DNA was used for cluster generation and sequencing on a cBot Cluster Generation system and Genome Analyzer IIx system (Illumina) according to the manufacturer's instructions. ChIP‐seq reads were mapped onto a mouse reference genome sequence (mm9) using Bowtie software (version 1.1.2; http://www.bowtie‐bio.sourceforge.net/).
Cell proliferation assay and cell cycle analysis
The cell proliferation rate was assessed by CCK‐8 assay. The absorbance of light at 450 nm was measured by a microplate reader to indirectly reflect the number of viable cells. Each group of BMMs was seeded in a 96‐well plate at a density of 2.5 × 104 cells/well in 100 μL medium containing 10 ng/mL M‐CSF. CCK‐8 was added to each well and incubated at 37°C for 2, 24, 48, 72, or 96 hours. The absorbance of the supernatant of each culture was measured at 450 nm using a microplate reader.
Cell‐cycle analysis was performed using Cell Cycle Assay Solution Blue (Doujindo). Briefly, BMMs from Hhex fl/fl and Hhex MxCre/− mice were seeded in six‐well plates. After 24 hours of culture, cells were collected and suspended in 500 μL of PBS. Next, 5 μL of Cell Cycle Assay Solution Blue was added to each cell suspension, and incubated at 37°C for 15 min. The cell‐cycle status of cells was investigated using Cytoflex (Beckman Coulter, Brea, CA, USA); cells at G0/G1, S, and G2/M phases were analyzed using Kaluza analysis software (Beckman Coulter).
Statistical analysis
Statistical tests for each assay were chosen based on their appropriateness for the assay. Each series of experiments was repeated at least three times. All statistical calculations were carried out using Graphpad Prism (GraphPad Software, San Diego, CA, USA). Statistical analyses were performed using unpaired Student's t test. A p value of <0.05 was considered statistically significant. Statistical methods and values including all p values are described in the figure legends.
Results
Hhex gene expression is downregulated in an epigenetic manner during osteoclastogenesis
We first conducted a genomewide screen for transcription factors epigenetically regulated during osteoclast differentiation. To identify negative regulators of osteoclast differentiation, we focused on genes whose histone modifications at the cis‐regulatory element changed from a H3K4me3‐and‐H3K27me3 bivalent status to H3K27me3 monovalent status in response to RANKL stimulation; in addition, a reduction of H3K27ac modification was confirmed. Hhex, a homeodomain transcription factor originally identified as a gene highly expressed in hematopoietic stem cells, is downregulated upon differentiation. H3K27me3 modification at the cis‐regulatory element of Hhex was maintained in osteoclasts, while H3K4me3 and H3K27ac modifications were markedly reduced in mature osteoclasts compared with M‐CSF‐dependent bone marrow‐derived macrophages (BMMs) (Fig. 1A ). Expression of Hhex mRNA in BMMs, as detected by real‐time PCR, was decreased in a time‐dependent manner by RANKL stimulation (Fig. 1B ). Protein levels of Hhex were also markedly decreased after 24 hours of RANKL stimulation, as shown in Fig. 1C by Western blotting. Immunocytochemistry with an anti‐Hhex antibody demonstrated that Hhex is mainly localized in the nuclei of BMMs, and its fluorescence intensity was reduced following RANKL stimulation (Fig. 1D ). These results show that Hhex is down regulated in an epigenetic manner during RANKL‐induced osteoclastogenesis.
Fig. 1.
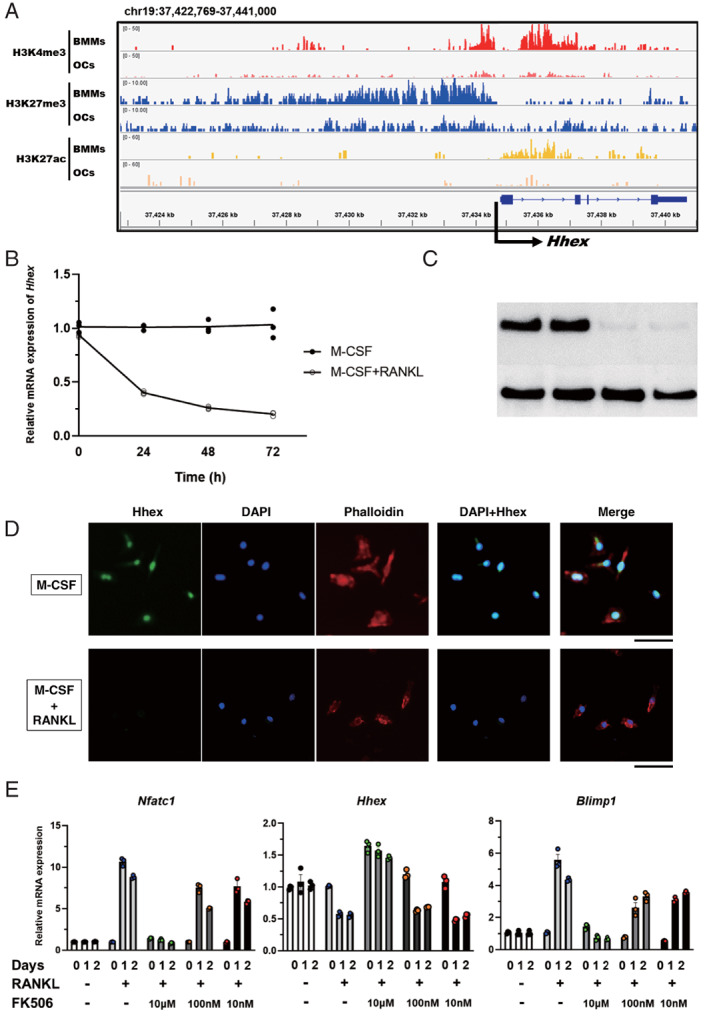
Hhex gene expression is downregulated in an epigenetic manner during osteoclastogenesis. (A) Histone modification of Hhex in BMMs and osteoclasts analyzed by ChIP‐seq of trimethylation of lysine 4 (H3K4me3) and lysine 27 (H3K27me3), as well as acetylation (H3K27ac) of histone H3. (B) Time course of changes in Hhex mRNA expression in BMMs cultured with M‐CSF or M‐CSF and RANKL. (C) Suppression of Hhex protein expression in response to RANKL stimulation as determined by immunoblot analysis. (D) Immunocytochemistry of BMMs cultured with M‐CSF or M‐CSF/RANKL for 2 days. Scale bar = 50 μm. (E) RANKL‐induced suppression of Hhex is mediated by the NFATc1‐Blimp1 axis. Time course of changes in Nfatc1 (left), Hhex (middle), and Blimp1 (right) expression in BMMs treated with M‐CSF alone or M‐CSF and RANKL in the presence of the indicated concentrations of FK506 or DMSO. DMSO = dimethyl sulfoxide.
To determine whether the repression of Hhex by RANKL was dependent on NFATc1, we examined the effect of FK506, a calcineurin inhibitor that blocks the nuclear translocation and autoamplification of NFATc1.( 17 ) As shown in Fig. 1E , FK506 attenuated the RANKL‐induced reduction of Hhex expression in a dose‐dependent manner. Previously, B‐lymphocyte‐induced maturation protein (Blimp)1 (also known as PR domain zinc finger protein 1, PRDM1) was reported to stimulate osteoclast differentiation by downregulating negative regulators such as Irf8, MafB, and Bcl6; notably, Hhex was also negatively regulated by Blimp1.( 9 , 10 ) FK506 dose‐dependently suppressed Blimp1 and Nfatc1 induction by RANKL, which was inversely correlated with Hhex expression (Fig. 1E ). These results suggest that Hhex is negatively regulated by Blimp1 in an NFATc1‐dependent manner.
Hhex is a negative regulator of osteoclastogenesis
To investigate the role of Hhex in osteoclastogenesis, Hhex was overexpressed in BMMs using the retroviral vector pMX‐Hhex‐IRES‐Puro. BMMs infected either by pMX‐Hhex‐IRES‐Puro or a control vector pMX‐IRES‐Puro were treated with RANKL. Overexpression of Hhex in pMX‐Hhex‐IRES‐Puro–infected BMMs was confirmed by quantitative reverse transcription PCR (RT‐qPCR) and immunoblotting (Fig. 2A,B ). As shown in Fig. 2C, Hhex overexpression markedly inhibited RANKL‐induced osteoclast differentiation from BMMs. Negative regulation of osteoclastogenesis by Hhex overexpression was further confirmed using another retroviral vector, pMX‐IRES‐Puro‐Hhex–enhanced green fluorescent protein (EGFP) (data not shown). Conversely, the effect of Hhex suppression on in vitro osteoclastogenesis was examined using Hhex fl/fl mice, which contain loxP sites flanking exon 2 of the Hhex gene. When BMMs obtained from Hhex fl/fl mice (Hhex fl/fl BMMs) were infected with pMx‐Cre retrovirus, expression of Hhex was significantly reduced (Fig. 2D ). RANKL and M‐CSF treatment generated higher number of osteoclasts from Hhex fl/fl BMMs infected with pMx‐Cre virus compared with those infected with control virus (Fig. 2E ). These results suggest that Hhex negatively regulates RANKL‐induced osteoclastogenesis from BMMs. Interestingly, there was no significant difference in the expression of Nfatc1 or osteoclast marker genes when RANKL was stimulated in Hhex‐overexpressing BMMs or Hhex‐depleted BMMs (Fig. 2F,G ).
Fig. 2.
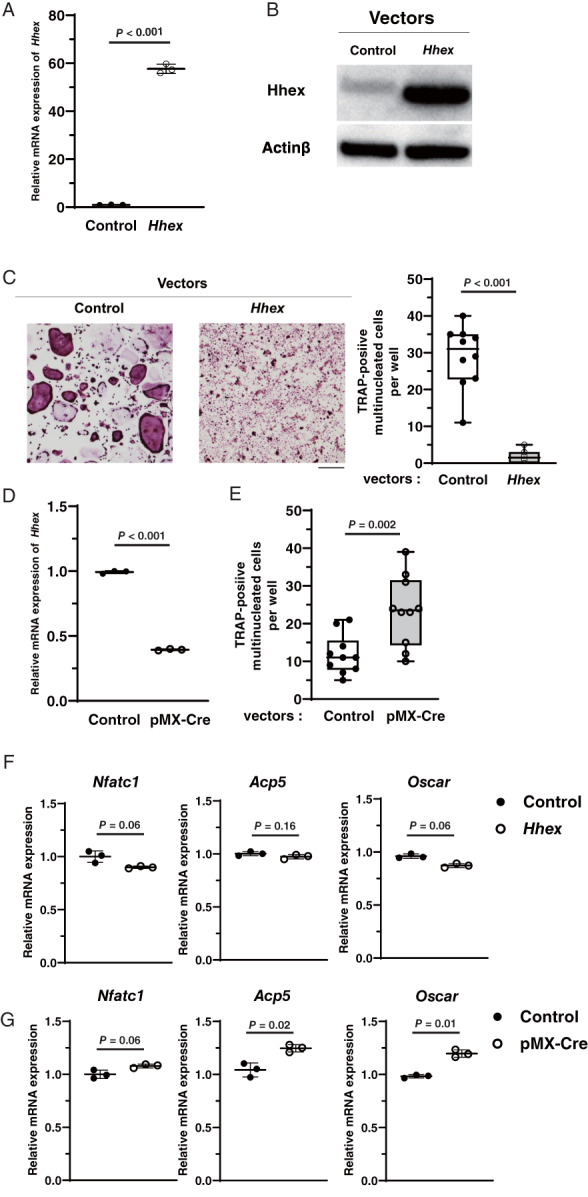
Hhex is a negative regulator of osteoclastogenesis. (A) mRNA expression of Hhex analyzed by real‐time PCR after transfection with pMX‐IRES‐Puro or pMX‐IRES‐Puro‐Hhex. (B) Immunoblot analysis of Hhex protein levels in BMMs after transfection with pMX‐IRES‐Puro and pMX‐IRES‐Puro‐Hhex. (C) Inhibition of RANKL‐induced TRAP‐positive MNC formation by retrovirus‐mediated overexpression of Hhex. (left) TRAP staining. Cultures were performed at least five times and representative pictures are shown. (right) Number of MNCs per well. Data represent the mean ± SD (n = 10). Scale bar = 100 μm. (D) Empty vectors or retrovirus vectors encoding Cre recombinase were infected into BMMs from Hhex flox/flox mice, and expression of Hhex mRNA was analyzed by real‐time PCR. (E) Number of TRAP‐positive MNCs per well after treatment with M‐CSF and RANKL for 3 days (n = 10). (F) mRNA expression levels of Nfatc1, Acp5, and Oscar when RANKL was administered to BMMs transfected with pMX‐IRES‐Puro and pMX‐IRES‐Puro‐Hhex. (G) mRNA expression of Nfatc1, Acp5, and Oscar when RANKL was administered to BMMs from Hhexflox/flox mice transfected with pMX‐IRES‐Puro or pMX‐IRES‐Puro‐Cre. Control = pMX‐IRES‐Puro (Mock); Hhex, pMX‐IRES‐Puro‐Hhex (Hhex overexpression); MNC = multinucleated cell; pMX‐Cre = pMX‐IRES‐Puro‐Cre.
Low‐bone‐mass phenotype induced by Hhex deletion
We next analyzed the effect of Hhex deletion in vivo. Because mice with homozygous disruption of Hhex are embryonic lethal,( 18 , 19 , 20 ) we generated conditional knockout mice using the Cre‐loxP system. We crossed Hhex fl/fl mice with a transgenic line expressing Cre recombinase from a type I interferon inducible promoter (Mx1‐Cre), in which Cre recombinase is induced in response to treatment with pIpC (referred to as Hhex MxCre/− mice). Immunoblotting confirmed that Hhex protein expression was almost completely diminished in BMMs from Hhex MxCre/− mice treated with pIpC (Fig. 3A ). In vitro osteoclast differentiation was increased when Hhex was deleted (Fig. S1 A). To investigate the effect of Hhex deletion in vivo, pIpC (12.5 μg/g of body weight) was injected intraperitoneally into 12‐week‐old male Hhex MxCre/− mice and Hhex fl/fl mice, which were euthanized 8 weeks after the injection to analyze skeletal tissues. μCT analysis of mouse femurs showed that Hhex MxCre/− mice treated with pIpC exhibited a marked reduction in trabecular bone volume, trabecular thickness, trabecular number, and the number of bone nodules compared with Hhex fl/fl mice (Fig. 3B ). The same results were observed in female mice (Fig. S2). TRAP staining revealed an increase in osteoclasts on the bone surface of proximal tibia in Hhex MxCre/− mice as compared with Hhex fl/fl mice (Fig. 3C ). Bone morphometric analysis demonstrated a reduction of bone volume associated with an increase in osteoclast number and an increase in the indicators of bone resorption. Bone formation parameters were slightly decreased in Hhex MxCre/− mice with no significant difference (Fig. 3D ).
Fig. 3.
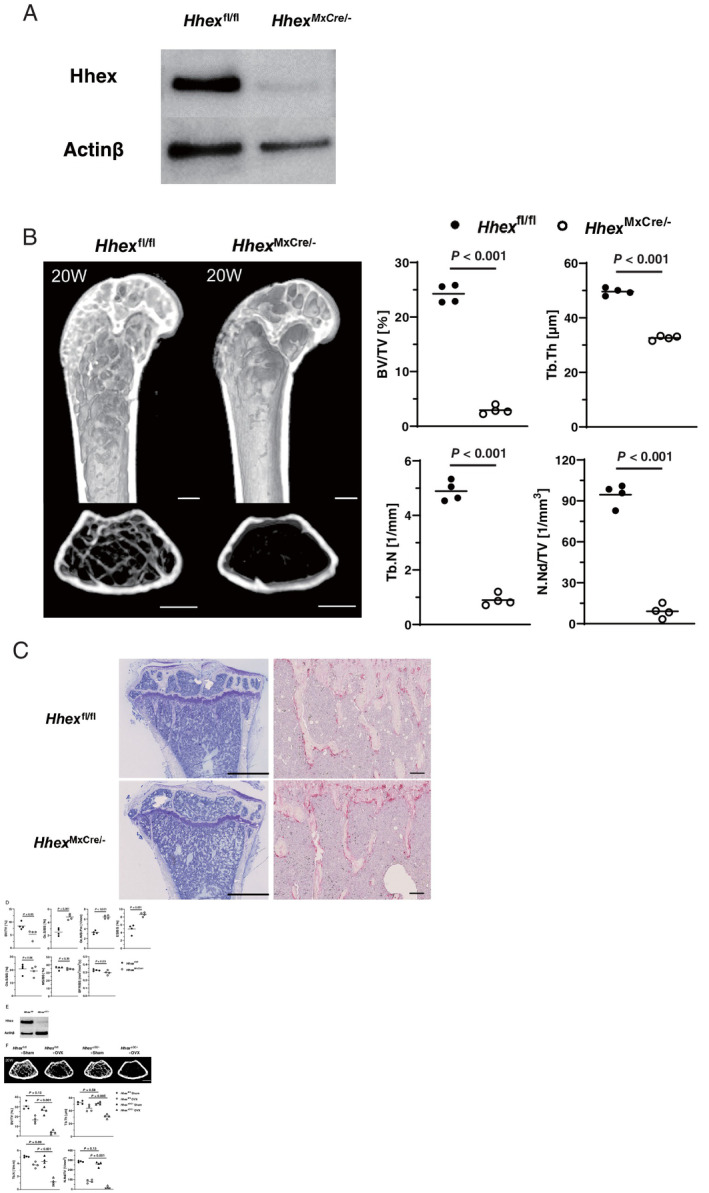
Low‐bone‐mass phenotypes induced by Hhex deletion. (A) Immunoblot analysis of Hhex protein levels in BMMs from Hhex flox/flox and Hhex MxCre/− mice. (B) (left) μCT analysis of femurs of Hhex flox/flox (Hhex fl/fl) and Mx‐1Cre/‐ Hhex flox/flox (Hhex MxCre/−) mice. pIpC injections (12 .5 μg/g of body weight) were administered to 12‐week‐old males in each group, which were euthanized 8 weeks after injection (n = 4 per group). Longitudinal (upper) and axial views (lower) of the metaphyseal region of mice. Scale bars = 500 μm. (right) μCT‐based parameters of the metaphyseal region. (C) Histological analysis of the proximal tibia of Hhex flox/flox and Hhex MxCre/−. pIpC injections (12.5 μg/g of body weight) were administered to 20‐week‐old male mice in each group, which were euthanized after 8 weeks. (left) Toluidine blue staining. Scale bars = 1 mm. (right) TRAP staining. Scale bars = 100 μm. (D) Histomorphometric analysis of tibias from 20‐week‐old mice (n = 4 in each group). Parameters for osteoclastic bone resorption and osteoblastic bone formation in the bone morphometric analysis of Hhex flox/flox and Hhex MxCre/− mice. (E) Immunoblot analysis of Hhex protein levels in BMMs obtained from Hhex flox/flox and CtsK Cre/‐ Hhex flox/flox (Hhex ΔOC/−) mice. (F) μCT analysis of the femurs of Hhex flox/flox (Hhex fl/fl) and CtsK Cre/− Hhex flox/flox (Hhex ΔOC/−) mice. Twelve‐week‐old female mice were subjected to OVX or sham surgery and euthanized 8 weeks later (n = 4 per group). Axial views of the metaphyseal region of mice. Scale bar = 500 μm. μCT‐based parameters of the metaphyseal region. BFR/BS = bone formation rate per bone surface; BV/TV = bone volume per tissue volume; ES/BS = eroded surface per bone surface; MS/BS = mineralized surface per bone surface; N.Nd/TV = number of nodules per tissue volume; Oc.N/B.Pm = osteoclast number per bone perimeter; Ob.S/BS = osteoblast surface per bone surface; Oc.S/BS = osteoclast surface per bone surface; OVX = ovariectomy; Tb.N = trabecular number; Tb.Th = trabecular bone thickness.
Because induction of the Mx1 promoter by pIpC is not restricted to osteoclast‐lineage BMMs, we also generated Hhex conditional knockout mice by crossing Hhex fl/fl mice with cathepsin K‐Cre knock‐in mice, in which the Cre recombinase gene is inserted into the cathepsin K locus and expressed in osteoclast‐lineage cells (referred to as Hhex ΔOC/− mice). A significant reduction of Hhex was observed in BMMs from Hhex ΔOC/− mice after treatment with RANKL and M‐CSF (Fig. 3E ), and osteoclast differentiation was increased when Hhex was deleted (Fig. S1 B). Because we observed only marginal effects by Hhex deletion on bone mass at baseline (Fig. S3 A–C), 12‐week‐old female Hhex ΔOC/− and control Hhex fl/fl mice were subjected to ovariectomy, and euthanized after 8 weeks. μCT analysis showed a significant reduction of trabecular bone volume, trabecular thickness, trabecular number, and the number of bone nodules in ovariectomized Hhex ΔOC/− mice compared with ovariectomized control Hhex fl/fl mice, as shown in Fig. 3F .
These results clearly demonstrate the negative regulatory role of Hhex in osteoclastogenesis both in vitro and in vivo.
Mechanisms of action of Hhex in osteoclast differentiation
We next addressed the mechanism of how Hhex negatively regulates RANKL‐induced osteoclast differentiation. Cell‐cycle regulation is critical for osteoclast differentiation, and previous studies identified cell cycle–arrested quiescent osteoclast precursors as committed osteoclast precursors.( 21 , 22 ) Therefore, we analyzed the effect of Hhex deletion on cell proliferation and cell‐cycle distribution in BMMs. Proliferation of BMMs, as determined by WST‐8, was reduced in Hhex MxCre/− BMMs compared with Hhex fl/fl BMMs (Fig. 4A ). Analyses of cell‐cycle distributions by flow cytometry showed that Hhex deletion in BMMs reduced the proportion of cells at S phase from (mean ± standard deviation [SD]) 14.8% ± 0.7% to 5.1% ± 1.1%, and increased those at G1 phase from 60.7% ± 1.6% to 83.4% ± 1.9%, indicating that Hhex regulated cell cycle status and its deletion induced G1 cell‐cycle arrest in BMMs (Fig. 4B ).
Fig. 4.
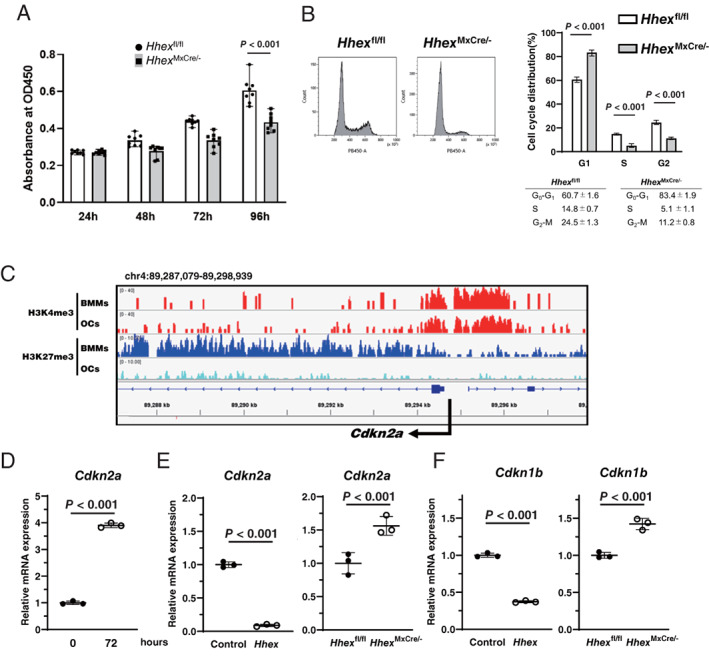
Hhex regulates the cell cycle of BMMs by inhibiting cyclin‐dependent kinase inhibitor expression. (A) Cell proliferation rate was assessed by WST‐8 assay. A significant reduction of cell proliferation was observed in BMMs from Hhex conditional knockout (Hhex MxCre/−) mice compared with Hhex flox/flox (Hhex fl/fl) mice (n = 8). (B) Cell cycle analysis of BMMs from Hhex flox/flox and Hhex MxCre/− mice using flow cytometry (n = 4). (C) Histone modification at the cis‐regulatory element of Cdkn2a, as analyzed by ChIP‐seq in BMMs and osteoclasts. H3K27me3 modification at the cis‐regulatory element of Cdkn2a gene was decreased in mature osteoclasts compared with BMMs, while the H3K4me3 modification did not appear to differ between BMMs and osteoclasts. (D) mRNA expression of Cdkn2a was increased in BMMs by RANKL treatment for 72 hours. (E) Negative regulation of Cdkn2a by Hhex in BMMs. (left) Retrovirus (pMX‐IRES‐Puro‐Hhex)‐induced overexpression of Hhex suppressed Cdkn2a expression in BMMs. (right) Increased expression of Cdkn2a in BMMs from Mx‐1Cre/− Hhex flox/flox mice compared with those from Hhex flox/flox mice. (F) Negative regulation of Cdkn1b by Hhex. (left) Retrovirus‐induced overexpression of Hhex suppressed Cdkn1b expression in BMMs. (right) Increased expression of Cdkn1b in BMMs from Mx‐1Cre/− Hhex flox/flox mice compared with those from Hhex flox/flox mice. Control = pMX‐IRES‐Puro; Hhex = pMX‐IRES‐Puro‐Hhex; Hhex fl/fl = Hhex flox/flox; Hhex ΔMxCre/− = Mx‐1Cre/− Hhex flox/flox.
The mammalian cell cycle is regulated by the balance between cyclin‐dependent kinases and cyclin‐dependent kinase inhibitors (CDKIs), and the terminal differentiation is usually coupled with permanent exit from the cell cycle.( 23 , 24 , 25 ) Hhex reportedly binds to the Cdkn2a locus and directly interacts with PRC2 to enable H3K27me3‐mediated epigenetic repression, and targeted deletion of Hhex stimulates Cdkn2a expression and suppresses self‐renewal of hematopoietic stem cells.( 26 ) Therefore, we next examined the effects of Hhex overexpression and suppression on Cdkn2a expression in BMMs. ChIP‐seq analysis of BMMs and osteoclasts demonstrated that H3K27me3 modification at the cis‐regulatory element of Cdkn2a was decreased in mature osteoclasts compared with BMMs, whereas H3K4me3 modification did not appear to differ between BMMs and osteoclasts (Fig. 4C ), and expression of Cdkn2a was increased after 72 hours of RANKL stimulation (Fig. 4D ). Overexpression of Hhex suppressed RANKL‐induced Cdkn2a expression, which was increased by Hhex deletion (Fig. 4E ). In addition to Cdkn2a, there are at least two distinct families of CDKIs in mammalian cells: the p21/p27 and p16/p18 families. Similar to Cdkn2a, expression of Cdkn1b, which encodes p27, was suppressed by Hhex overexpression and increased by Hhex deletion (Fig. 4F ), whereas expression of Cdkn1a (p21) and Tp53 was not affected by Hhex (Fig. S4).
Discussion
Herein we showed that Hhex, a member of the tinman family of homeodomain‐containing transcription factors, is downregulated by RANKL‐mediated epigenetic modification in BMMs and plays an important role in osteoclast development by regulating cell‐cycle progression. Histone modification of the cis‐regulatory element of Hhex changed from a H3K4me‐and‐H3K27me3 bivalent status to H3K27me3 monovalent status in response to RANKL stimulation, and H3K27ac modification was reduced, which was correlated with a reduction of Hhex expression. These results suggest that Hhex expression was epigenetically silenced during osteoclast differentiation. RANKL‐induced inhibition of Hhex appears to be NFATc1 dependent because suppression of NFATc1 activation by FK506 restored inhibition.
Hhex, a relatively unusual homeodomain protein, exists as a homo‐oligomer( 27 ) and is highly conserved between species.( 28 ) Hhex was first identified in avian and human hematopoietic cells, whereby it was found to regulate cell development and differentiation by both transcriptional and posttranscriptional mechanisms.( 29 , 30 ) Hhex is expressed in early hematopoietic progenitors of all lineages except T‐cell lineages, especially prevalent in hematopoietic stem cells, and downregulated upon terminal differentiation of these cells.( 29 , 31 ) During embryonic development, Hhex is essential for forebrain, liver, and thyroid development; thus, targeted disruption of Hhex in mice leads to embryonic lethality.( 18 , 19 , 20 ) Blastocyst compensation in Hhex −/−; Rag1 −/− chimeric mice or conditional deletion of Hhex in the hematopoietic system showed that Hhex is dispensable for maintenance of hematopoietic stem cells, but critical for early lymphoid specification; in particular, development of B cell lineages.( 32 , 33 , 34 , 35 ) However, the role of Hhex in skeletal homeostasis and osteoclast‐lineage cells has not been clarified.
To analyze the role of Hhex in osteoclast‐lineage cells, we first performed in vitro experiments and found that overexpression of Hhex in BMMs almost completely suppressed RANKL‐induced osteoclast development, whereas its deletion resulted in increased osteoclastogenesis. The role of Hhex in osteoclastogenesis was further confirmed by in vivo experiments using Hhex MxCre/− mice, in which Hhex is inducibly deleted in hematopoietic cells by pIpC administration. Hhex was efficiently deleted in BMMs of Hhex MxCre/− mice, which exhibited reduced bone mass due to increased numbers of osteoclasts. Decreased bone mineral density was also observed in HhexΔOC/− mice. These results demonstrate that Hhex is a negative regulator of osteoclast‐lineage cells both in vitro and in vivo. It should be noted that the skeletal phenotypes of Hhex MxCre/− mice and Hhex ΔOC/− mice are somehow different (Fig. 3A,E ). The exact reason for this difference is unknown, but we speculate that it may be because not all BMMs differentiate into mature osteoclasts in vivo. Furthermore, it is possible that Mx1‐Cre affects other types of hematopoietic cells than monocyte/macrophage‐lineage cells, which indirectly affect osteoclast differentiation.
We next addressed the mechanism how Hhex negatively regulates osteoclastogenesis. It was previously reported that overexpression of Hhex in hematopoietic cells induced murine lymphoid neoplasms,( 36 ) and HHEX is expressed at high levels in human acute myeloid leukemia cells and required for the development of myeloid leukemia driven by the oncogenic fusion protein mixed lineage leukemia‐eleven nineteen leukemia (MLL‐ENL).( 26 ) Moreover, the ability of Hhex to promote myeloid progenitor expansion and acute myeloid leukemogenesis arose from its function of repressing the tumor suppressor gene Cdkn2a, by binding to the Cdkn2a locus and directly interacts with PRC2 to enable H3K27me3‐mediated epigenetic repression.( 26 ) Cdkn2a encodes two CDKIs, p16INK4a and p19ARF, suggesting that Hhex is involved in the regulation of cell cycle progression.
The critical role of cell cycle regulation in osteoclast development has been reported in previous studies. Okahashi and colleagues( 37 ) reported that RANKL induced transient expression of p21 and p27, and suppression of these two CDKIs by antisense oligonucleotides strongly inhibited osteoclast differentiation. Sankar and colleagues( 38 ) reported that mice with double knockout of CDKIs p21 and p27 developed osteopetrosis, with fewer osteoclasts exhibiting lower TRAP activity and abnormal cell morphology present in long bone. Kwon and colleagues( 39 ) reported that M‐CSF deprivation induced cell cycle arrest at the G0/G1 phase and promoted osteoclast differentiation. We previously reported that Cdk6 levels are downregulated in osteoclast precursors by RANKL in a NF‐κB–dependent manner, and overexpression of Cdk6 suppressed osteoclastogenesis.( 40 ) Mizoguchi and colleagues( 21 ) and Muto and colleagues( 22 ) identified cell‐cycle–arrested quiescent osteoclast precursors along bone surfaces that rapidly and directly differentiated into mature osteoclasts in response to RANKL. Collectively, these results suggest that RANKL promotes osteoclast differentiation by inducing cell cycle arrest in BMMs. Therefore, we hypothesized that Hhex regulates osteoclast differentiation by modulating RANKL‐induced cell cycle arrest in BMMs. Analysis of cell‐cycle distribution by flow cytometry showed that Hhex deletion in BMMs reduced the proportion of cells at S phase and increased those at G1 phase. ChIP‐seq analysis of BMMs and osteoclasts demonstrated that the H3K27me3 modification at the cis‐regulatory element of Cdkn2a was decreased in mature osteoclasts as compared with BMMs, whereas the H3K4me3 modification did not appear to differ between BMMs and osteoclasts, and expression of Cdkn2a was increased in osteoclasts (Fig. 4D ). Overexpression of Hhex suppressed RANKL‐induced Cdkn2a expression, which was increased by Hhex deletion (Fig. 4E ). In addition to Cdkn2a, expression of Cdkn1b was negatively regulated by Hhex. These results led us to conclude that Hhex positively regulates the cell‐cycle progression of BMMs by downregulating Cdkn2a and Cdkn1b, and downregulation of Hhex is required for RANKL‐induced cell‐cycle arrest and osteoclastogenesis. Farr and colleagues( 41 ) recently reported that p16 Ink4a‐expressing senescent cells in the bone microenvironment are implicated in age‐related bone loss, which was ameliorated by inducible elimination of p16 Ink4a‐expressing cells. Li and colleagues( 42 ) demonstrated that osteoclast differentiation was suppressed in p16 Ink4a‐deficient mice, which were resistant to ovariectomy‐induced bone loss. In addition, a single nucleotide polymorphism of HHEX, rs2497306, was associated with human aging by regulation of serum dehydroepiandrosterone sulfate levels.( 43 ) These results suggest that the Hhex‐Cdkn2a axis may be involved in age‐related bone loss. However, the role of Hhex in regulation of Cdkn2a and Cdkn1b in bone cells other than osteoclasts remains unknown, and further studies are required to elucidate the role of Hhex in age‐related bone loss.
There are several limitations of this study. First, the mechanism of Hhex‐induced downregulation of Cdk2a and Cdk1b remains unclear. Reportedly, Hhex interacts with members of the Groucho/transducin‐like enhancer of split co‐repressor protein family and may recruit these proteins to a subset of target promoters to bring about transcriptional repression.( 44 ) However, future studies are required to identify the partners of Hhex and global landscape of Hhex‐regulated gene expression in BMMs. Second, it remains unclear how cell‐cycle regulation affects RANKL‐induced osteoclast differentiation. It is widely recognized that there is a temporal coupling between cell‐cycle arrest and terminal differentiation, although cell‐cycle arrest per se is not sufficient for cell differentiation.( 45 ) Muto and colleagues( 22 ) identified cell cycle‐arrested quiescent osteoclast precursors along bone surfaces as committed osteoclast precursors. Motiur Rahman and colleagues( 46 ) reported that RANKL stimulates DNA synthesis and cell proliferation of BMMs during the early phase of osteoclastogenesis, followed by G1 arrest in the latter half of differentiation. Therefore, it is possible that Hhex‐mediated suppression of CDKIs stimulates the proliferation of BMMs at the early stage, whereas subsequent downregulation of Hhex induces cell‐cycle arrest. Further studies are required to identify the mechanism by which cell‐cycle arrest affects osteoclast differentiation. Small sample size in conditional knockout mice is also considered as limitation of this study. Last, the clinical relevance of the present results should be further clarified. It is possible that the Hhex‐Cdkn2a axis is involved in age‐related bone loss by regulating cellular senescence. Interestingly, genomewide association studies demonstrated the association of single nucleotide polymorphisms of HHEX and CDKN2a genes with type 2 diabetes mellitus.( 47 , 48 ) It has been well established that individuals with type 2 diabetes are at an increased risk of osteoporotic fractures, and it is possible that the modulation of HHEX gene expression may play a role in bone fragility in these patients. Accordingly, it is of particular interest to investigate the relationship between HHEX expression levels and bone mineral density in patients with type 2 diabetes in a clinical setting.
In conclusion, we demonstrated that the homeodomain‐containing transcription factor Hhex negatively regulates RANKL‐induced osteoclastogenesis by modulating cell‐cycle progression. This study highlights the regulation of Hhex levels as a potential strategy to inhibit osteoporosis‐induced bone loss and fractures.
Conflicts of Interest
The authors state that they have no conflicts of interest.
Peer Review
The peer review history for this article is available at https://publons.com/publon/10.1002/jbm4.10608.
Supporting information
Fig. S1. (A) Increased osteoclast differentiation in response to M‐CSF and RANKL in BMMs from Hhex MxCre/− mice compared with Hhex flox/flox mice. (Left) TRAP staining (Right) Number of TRAP‐positive multinucleated cell (MNC) per well (n = 10). Bar = 500 μm. Hhex ΔMxCre/−; Mx‐1Cre/− Hhex flox/flox. (B) Increased osteoclast differentiation in response to M‐CSF and RANKL in BMMs from Hhex ΔOC/− mice compared with Hhex flox/flox mice. (Left) TRAP staining. (Right) Number of TRAP‐positive MNC per well (n = 10). Bar = 500 μm. Hhex ΔOC/−; CtsK Cre/− Hhex flox/flox.
Fig. S2. (A) We performed μCT analysis of femurs in females as well as males Hhex flox/flox (Hhex fl/fl) and Mx‐1Cre/− Hhex flox/flox (Hhex MxCre/−) mice. pIpC injections (12.5 μg/g of body weight) were administered to 12‐week‐old females in each group, which were euthanized 8 weeks after injection (n = 4 per group). μCT‐based parameters of the metaphyseal region. BV/TV, bone volume per tissue volume; Tb.Th, trabecular bone thickness; Tb.N, trabecular number; N.Nd/TV, number of nodules per tissue volume.
Fig. S3. (A) Microcomputed tomography (μCT) analysis of the femurs of Hhex flox/flox mice and CtsKCre/− Hhex flox/flox (Hhex ΔOC/−) mice at baseline (10‐week‐old female). Representative μCT images of axial views of the metaphyseal region of Hhex flox/flox mice and Hhex ΔOC/− mice are shown. (B) μCT‐based parameters of the metaphyseal region of 10‐week‐old female. BV/TV, bone volume per tissue volume; Tb.Th, trabecular bone thickness; Tb.N, trabecular number; N.Nd/TV, number of nodules per tissue volume. Hhex fl/fl; Hhex flox/flox, Hhex ΔOC/−; CtsKCre/− Hhex flox/flox. (C) μCT‐based parameters of the metaphyseal region of 10‐week‐old male. BV/TV, bone volume per tissue volume; Tb.Th, trabecular bone thickness; Tb.N, trabecular number; N.Nd/TV, number of nodules per tissue volume. Hhex fl/fl; Hhex flox/flox, Hhex ΔOC/−; CtsKCre/− Hhex flox/flox.
Fig. S4. (A) Effects of Hhex overexpression or depletion on mRNA expression of Cdkn1a in BMMs. (Left) Retrovirus‐induced overexpression of Hhex showed no significant effect on Cdkn1a expression in BMMs. (Right) No significant difference in Cdkn1b or Tp53 expression was observed between BMMs from Hhex flox/flox mice and Hhex MxCre/− mice. Control, pMX‐IRES‐Puro (Mock); Hhex, pMX‐IRES‐Puro‐Hhex (Hhex overexpression). Hhex fl/fl; Hhex flox/flox, Hhex ΔMxCre/−; Mx‐1Cre/‐ Hhex flox/flox. (B) Effects of Hhex overexpression or depletion in BMMs on mRNA expression of Tp53 in BMMs. (Left) Retrovirus‐induced overexpression of Hhex showed no significant effect on Tp53 expression in BMMs. (Right) No significant difference in Tp53 expression was observed between BMMs from Hhex flox/flox mice and Hhex MxCre/− mice. Control, pMX‐IRES‐Puro (Mock); Hhex, pMX‐IRES‐Puro‐Hhex (Hhex overexpression). Hhex fl/fl; Hhex flox/flox, Hhex ΔMxCre/−; Mx‐1Cre/‐ Hhex flox/flox.
Acknowledgments
This work was supported in part by Grants‐in‐Aid from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan to ST (19H05654) and to TM (21 K09316). All the datasets generated in this study are available in the DDBJ Sequence Read Archive (http://trace.ddbj.nig.ac.jp/dra/index_e.html) under the accession numbers DRA006384, DRA007111, and DRA007958. We thank J. Sugita, R. Honma, R. Chijimatsu, (Department of Orthopaedic Surgery, The University of Tokyo), T. Kobayashi, S. Tanigawa, and Y. Sato (Department of Orthopaedic Surgery, The University of Keio) for providing expert technical assistance. We thank Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
Authors' roles: HW, HO, YO, JH, TM, MM, MN, TS, TM, and ST conceived experiments. HW, HO, and YO performed experiments. HO performed biocomputing analysis. HW, HO, JH, YO, TM, MM, MN, TS, TM, and ST analyzed the data. HW and ST wrote the paper. All authors read and approved the final manuscript.
References
- 1. Tanaka S, Tanaka Y, Ishiguro N, Yamanaka H, Takeuchi T. RANKL: a therapeutic target for bone destruction in rheumatoid arthritis. Mod Rheumatol. 2018;28(1):9‐16. [DOI] [PubMed] [Google Scholar]
- 2. Suda T, Takahashi N, Udagawa N, Jimi E, Gillespie MT, Martin TJ. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr Rev. 1999;20(3):345‐357. [DOI] [PubMed] [Google Scholar]
- 3. Tanaka S, Nakamura K, Takahasi N, Suda T. Role of RANKL in physiological and pathological bone resorption and therapeutics targeting the RANKL‐RANK signaling system. Immunol Rev. 2005;208:30‐49. [DOI] [PubMed] [Google Scholar]
- 4. Tanaka S, Takahashi N, Udagawa N, et al. Macrophage colony‐stimulating factor is indispensable for both proliferation and differentiation of osteoclast progenitors. J Clin Invest. 1993;91(1):257‐263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Okamoto K, Nakashima T, Shinohara M, et al. Osteoimmunology: the conceptual framework unifying the immune and skeletal systems. Physiol Rev. 2017;97(4):1295‐1349. [DOI] [PubMed] [Google Scholar]
- 6. Zhao B, Takami M, Yamada A, et al. Interferon regulatory factor‐8 regulates bone metabolism by suppressing osteoclastogenesis. Nat Med. 2009;15(9):1066‐1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Izawa N, Kurotaki D, Nomura S, et al. Cooperation of PU.1 with IRF8 and NFATc1 defines chromatin landscapes during RANKL‐induced osteoclastogenesis. J Bone Miner Res. 2019;34(6):1143‐1154. [DOI] [PubMed] [Google Scholar]
- 8. Kurotaki D, Yoshida H, Tamura T. Epigenetic and transcriptional regulation of osteoclast differentiation. Bone. 2020;138:115471. [DOI] [PubMed] [Google Scholar]
- 9. Miyauchi Y, Ninomiya K, Miyamoto H, et al. The Blimp1‐Bcl6 axis is critical to regulate osteoclast differentiation and bone homeostasis. J Exp Med. 2010;207(4):751‐762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nishikawa K, Nakashima T, Hayashi M, et al. Blimp1‐mediated repression of negative regulators is required for osteoclast differentiation. Proc Natl Acad Sci U S A. 2010;107(7):3117‐3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim K, Kim JH, Lee J, et al. MafB negatively regulates RANKL‐mediated osteoclast differentiation. Blood. 2007;109(8):3253‐3259. [DOI] [PubMed] [Google Scholar]
- 12. Yasui T, Hirose J, Aburatani H, Tanaka S. Epigenetic regulation of osteoclast differentiation. Ann N Y Acad Sci. 2011;1240:7‐13. [DOI] [PubMed] [Google Scholar]
- 13. Yasui T, Hirose J, Tsutsumi S, Nakamura K, Aburatani H, Tanaka S. Epigenetic regulation of osteoclast differentiation: possible involvement of Jmjd3 in the histone demethylation of Nfatc1. J Bone Miner Res. 2011;26(11):2665‐2671. [DOI] [PubMed] [Google Scholar]
- 14. Kühn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269(5229):1427‐1429. [DOI] [PubMed] [Google Scholar]
- 15. Nakamura T, Imai Y, Matsumoto T, et al. Estrogen prevents bone loss via estrogen receptor alpha and induction of Fas ligand in osteoclasts. Cell. 2007;130(5):811‐823. [DOI] [PubMed] [Google Scholar]
- 16. Hirose J, Masuda H, Tokuyama N, et al. Bone resorption is regulated by cell‐autonomous negative feedback loop of Stat5‐Dusp axis in the osteoclast. J Exp Med. 2014;211(1):153‐163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Takayanagi H, Kim S, Koga T, et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3(6):889‐901. [DOI] [PubMed] [Google Scholar]
- 18. Keng VW, Yagi H, Ikawa M, et al. Homeobox gene Hex is essential for onset of mouse embryonic liver development and differentiation of the monocyte lineage. Biochem Biophys Res Commun. 2000;276(3):1155‐1161. [DOI] [PubMed] [Google Scholar]
- 19. Martinez Barbera JP, Clements M, Thomas P, et al. The homeobox gene Hex is required in definitive endodermal tissues for normal forebrain, liver and thyroid formation. Development. 2000;127(11):2433‐2445. [DOI] [PubMed] [Google Scholar]
- 20. Hallaq H, Pinter E, Enciso J, et al. A null mutation of Hhex results in abnormal cardiac development, defective vasculogenesis and elevated Vegfa levels. Development. 2004;131(20):5197‐5209. [DOI] [PubMed] [Google Scholar]
- 21. Mizoguchi T, Muto A, Udagawa N, et al. Identification of cell cycle‐arrested quiescent osteoclast precursors in vivo. J Cell Biol. 2009;184(4):541‐554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Muto A, Mizoguchi T, Udagawa N, et al. Lineage‐committed osteoclast precursors circulate in blood and settle down into bone. J Bone Miner Res. 2011;26(12):2978‐2990. [DOI] [PubMed] [Google Scholar]
- 23. Buttitta LA, Edgar BA. Mechanisms controlling cell cycle exit upon terminal differentiation. Curr Opin Cell Biol. 2007;19(6):697‐704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1‐phase progression. Genes Dev. 1999;13(1512):1501‐1512. [DOI] [PubMed] [Google Scholar]
- 25. Aliprantis AO, Ueki Y, Sulyanto R, et al. NFATc1 in mice represses osteoprotegerin during osteoclastogenesis and dissociates systemic osteopenia from inflammation in cherubism. J Clin Invest. 2008;118(11):3775‐3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shields BJ, Jackson JT, Metcalf D, et al. Acute myeloid leukemia requires Hhex to enable PRC2‐mediated epigenetic repression of Cdkn2a. Genes Dev. 2016;30(1):78‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Soufi A, Jayaraman PS. PRH/Hex: an oligomeric transcription factor and multifunctional regulator of cell fate. Biochem J. 2008;412(3):399‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bedford FK, Ashworth A, Enver T, Wiedemann LM. HEX: a novel homeobox gene expressed during haematopoiesis and conserved between mouse and human. Nucl Acids Res. 1993;21(5):1245‐1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Crompton MR, Bartlett TJ, MacGregor AD, et al. Identification of a novel vertebrate homeobox gene expressed in haematopoietic cells. Nucl Acids Res. 1992;20(21):5661‐5667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Manfioletti G, Gattei V, Buratti E, et al. Differential expression of a novel proline‐rich homeobox gene (Prh) in human hematolymphopoietic cells. Blood. 1995;85(5):1237‐1245. [PubMed] [Google Scholar]
- 31. Jayaraman PS, Frampton J, Goodwin G. The homeodomain protein PRH influences the differentiation of haematopoietic cells. Leuk Res. 2000;24(12):1023‐1031. [DOI] [PubMed] [Google Scholar]
- 32. Jackson JT, Nasa C, Shi W, et al. A crucial role for the homeodomain transcription factor Hhex in lymphopoiesis. Blood. 2015;125(5):803‐814. [DOI] [PubMed] [Google Scholar]
- 33. Goodings C, Smith E, Mathias E, et al. Hhex is required at multiple stages of adult hematopoietic stem and progenitor cell differentiation. Stem Cells. 2015;33(8):2628‐2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jackson JT, Shields BJ, Shi W, et al. Hhex regulates hematopoietic stem cell self‐renewal and stress hematopoiesis via repression of Cdkn2a. Stem Cells. 2017;35(8):1948‐1957. [DOI] [PubMed] [Google Scholar]
- 35. Bogue CW, Zhang PX, McGrath J, Jacobs HC, Fuleihan RL. Impaired B cell development and function in mice with a targeted disruption of the homeobox gene Hex. Proc Natl Acad Sci U S A. 2003;100(2):556‐561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. George A, Morse HC 3rd, Justice MJ. The homeobox gene Hex induces T‐cell‐derived lymphomas when overexpressed in hematopoietic precursor cells. Oncogene. 2003;22(43):6764‐6773. [DOI] [PubMed] [Google Scholar]
- 37. Okahashi N, Murase Y, Koseki T, Sato T, Yamato K, Nishihara T. Osteoclast differentiation is associated with transient upregulation of cyclin‐dependent kinase inhibitors p21(WAF1/CIP1) and p27(KIP1). J Cell Biochem. 2001;80(3):339‐345. [PubMed] [Google Scholar]
- 38. Sankar U, Patel K, Rosol TJ, Ostrowski MC. RANKL coordinates cell cycle withdrawal and differentiation in osteoclasts through the cyclin‐dependent kinase inhibitors p27KIP1 and p21CIP1. J Bone Miner Res. 2004;19(8):1339‐1348. [DOI] [PubMed] [Google Scholar]
- 39. Kwon M, Kim JM, Lee K, et al. Synchronized cell cycle arrest promotes osteoclast differentiation. Int J Mol Sci. 2016;17(8):1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ogasawara T, Katagiri M, Yamamoto A, et al. Osteoclast differentiation by RANKL requires NF‐kappaB‐mediated downregulation of cyclin‐dependent kinase 6 (Cdk6). J Bone Miner Res. 2004;19(7):1128‐1136. [DOI] [PubMed] [Google Scholar]
- 41. Farr JN, Xu M, Weivoda MM, et al. Targeting cellular senescence prevents age‐related bone loss in mice. Nat Med. 2017;23(9):1072‐1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li J, Karim MA, Che H, Geng Q, Miao D. Deletion of p16 prevents estrogen deficiency‐induced osteoporosis by inhibiting oxidative stress and osteocyte senescence. Am J Transl Res. 2020;12(2):672‐683. [PMC free article] [PubMed] [Google Scholar]
- 43. Zhai G, Teumer A, Stolk L, et al. Eight common genetic variants associated with serum DHEAS levels suggest a key role in ageing mechanisms. PLoS Genet. 2011;7(4):e1002025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Swingler TE, Bess KL, Yao J, Stifani S, Jayaraman PS. The proline‐rich homeodomain protein recruits members of the Groucho/Transducin‐like enhancer of split protein family to co‐repress transcription in hematopoietic cells. J Biol Chem. 2004;279(33):34938‐34947. [DOI] [PubMed] [Google Scholar]
- 45. Myster DL, Duronio RJ. To differentiate or not to differentiate? Curr Biol. 2000;10(8):R302‐R304. [DOI] [PubMed] [Google Scholar]
- 46. Motiur Rahman M, Takeshita S, Matsuoka K, et al. Proliferation‐coupled osteoclast differentiation by RANKL: cell density as a determinant of osteoclast formation. Bone. 2015;81:392‐399. [DOI] [PubMed] [Google Scholar]
- 47. Miyake K, Yang W, Hara K, et al. Construction of a prediction model for type 2 diabetes mellitus in the Japanese population based on 11 genes with strong evidence of the association. J Hum Genet. 2009;54(4):236‐241. [DOI] [PubMed] [Google Scholar]
- 48. Saxena R, Voight BF, Lyssenko V, et al. Genome‐wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316(5829):1331‐1336. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. (A) Increased osteoclast differentiation in response to M‐CSF and RANKL in BMMs from Hhex MxCre/− mice compared with Hhex flox/flox mice. (Left) TRAP staining (Right) Number of TRAP‐positive multinucleated cell (MNC) per well (n = 10). Bar = 500 μm. Hhex ΔMxCre/−; Mx‐1Cre/− Hhex flox/flox. (B) Increased osteoclast differentiation in response to M‐CSF and RANKL in BMMs from Hhex ΔOC/− mice compared with Hhex flox/flox mice. (Left) TRAP staining. (Right) Number of TRAP‐positive MNC per well (n = 10). Bar = 500 μm. Hhex ΔOC/−; CtsK Cre/− Hhex flox/flox.
Fig. S2. (A) We performed μCT analysis of femurs in females as well as males Hhex flox/flox (Hhex fl/fl) and Mx‐1Cre/− Hhex flox/flox (Hhex MxCre/−) mice. pIpC injections (12.5 μg/g of body weight) were administered to 12‐week‐old females in each group, which were euthanized 8 weeks after injection (n = 4 per group). μCT‐based parameters of the metaphyseal region. BV/TV, bone volume per tissue volume; Tb.Th, trabecular bone thickness; Tb.N, trabecular number; N.Nd/TV, number of nodules per tissue volume.
Fig. S3. (A) Microcomputed tomography (μCT) analysis of the femurs of Hhex flox/flox mice and CtsKCre/− Hhex flox/flox (Hhex ΔOC/−) mice at baseline (10‐week‐old female). Representative μCT images of axial views of the metaphyseal region of Hhex flox/flox mice and Hhex ΔOC/− mice are shown. (B) μCT‐based parameters of the metaphyseal region of 10‐week‐old female. BV/TV, bone volume per tissue volume; Tb.Th, trabecular bone thickness; Tb.N, trabecular number; N.Nd/TV, number of nodules per tissue volume. Hhex fl/fl; Hhex flox/flox, Hhex ΔOC/−; CtsKCre/− Hhex flox/flox. (C) μCT‐based parameters of the metaphyseal region of 10‐week‐old male. BV/TV, bone volume per tissue volume; Tb.Th, trabecular bone thickness; Tb.N, trabecular number; N.Nd/TV, number of nodules per tissue volume. Hhex fl/fl; Hhex flox/flox, Hhex ΔOC/−; CtsKCre/− Hhex flox/flox.
Fig. S4. (A) Effects of Hhex overexpression or depletion on mRNA expression of Cdkn1a in BMMs. (Left) Retrovirus‐induced overexpression of Hhex showed no significant effect on Cdkn1a expression in BMMs. (Right) No significant difference in Cdkn1b or Tp53 expression was observed between BMMs from Hhex flox/flox mice and Hhex MxCre/− mice. Control, pMX‐IRES‐Puro (Mock); Hhex, pMX‐IRES‐Puro‐Hhex (Hhex overexpression). Hhex fl/fl; Hhex flox/flox, Hhex ΔMxCre/−; Mx‐1Cre/‐ Hhex flox/flox. (B) Effects of Hhex overexpression or depletion in BMMs on mRNA expression of Tp53 in BMMs. (Left) Retrovirus‐induced overexpression of Hhex showed no significant effect on Tp53 expression in BMMs. (Right) No significant difference in Tp53 expression was observed between BMMs from Hhex flox/flox mice and Hhex MxCre/− mice. Control, pMX‐IRES‐Puro (Mock); Hhex, pMX‐IRES‐Puro‐Hhex (Hhex overexpression). Hhex fl/fl; Hhex flox/flox, Hhex ΔMxCre/−; Mx‐1Cre/‐ Hhex flox/flox.