

Tissue and organ failure that results from congenital abnormalities, trauma, disease, or aging contributes to substantial morbidity and mortality. Although the 20th and early 21st centuries have brought dramatic advancements in the use of synthetic and mechanical devices to replace tissues, the restoration of tissue and organ structure and function remains a clinical challenge. Many biologic functions cannot be replicated with such devices, and the unavoidable immune responses that are induced when allografts (see Glossary) of human organs, tissues, or cells are implanted can limit the functionality and longevity of biologic approaches. Regenerative medicine has emerged as a potential alternative approach for tissue and organ restoration in which the engineered tissue is biologically functional. Traditional approaches for regenerative medicine involve biomaterial scaffolds, stem and progenitor cells, and biologic signaling molecules, alone or in combination, to promote new development of healthy tissue (Fig. 1A). A more recent strategy, “regenerative immunology,” promotes tissue healing and regeneration through reprogramming of the host immune system. However, organ transplantation is still the most complete option in regenerative medicine, providing an autologous, allogeneic, or potentially xenogeneic replacement for complete physical and biologic restoration.
Figure 1. Current and Future Strategies for Tissue Replacement.
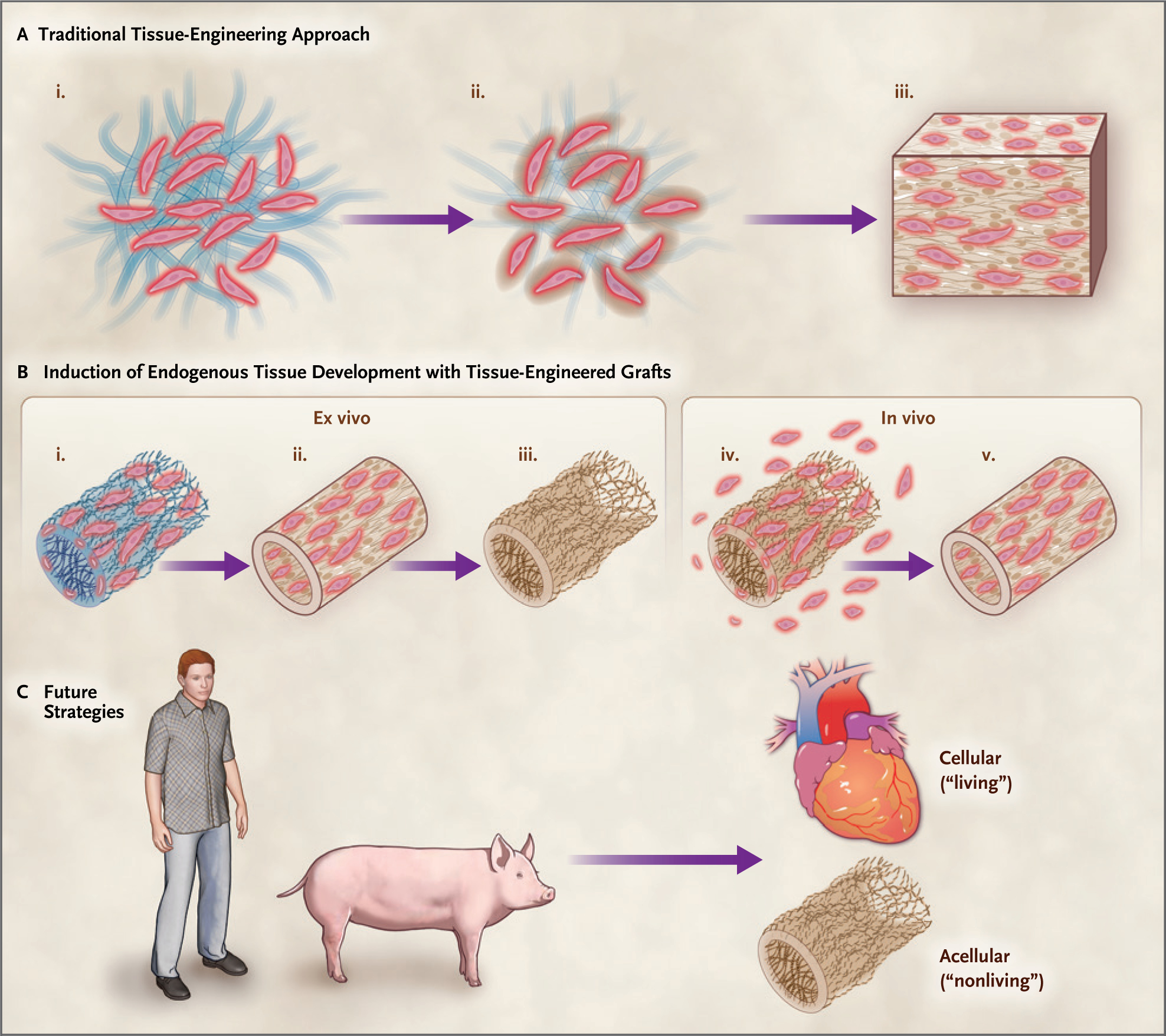
Traditional-tissue engineering approaches (Panel A) seed cells onto a three-dimensional biomaterial scaffold that serves as a framework for new tissue development (i). The scaffold degrades as new extracellular matrix is secreted (ii), resulting in the desired functional tissue (iii). Tissue-engineered grafts (Panel B) can induce endogenous tissue development. Cells from donor tissue are seeded onto a scaffold (i), and mature tissue is produced in bioreactors (ii). Cells are removed (iii), leaving an extracellular matrix that can be readily stored and available when needed. After implantation (iv), cells migrate into the matrix and new tissue develops (v). Future strategies (Panel C) may use either allograft or xenograft tissue sources for transplantable, viable (living) organs or use an acellular tissue extracellular matrix from tissue to mobilize endogenous repair mechanisms.
Advances in immune and genome engineering (or editing) create a foundation for new therapies to accelerate the restoration and replacement of tissues and organs, including those from xenogeneic sources. Regenerative immunology is based on the fact that immune cells such as macrophages and T cells, which are usually considered in their protective role against pathogens or “nonself” cells and as mediators of inflammation, can be made to adopt programs that can promote healing of tissues that have been damaged by the initial inflammatory antimicrobial response.1,2 Such regenerative immune responses can also promote healing after xenogeneic transplantation, provided that the anti-xenogeneic response to nonself tissue can be suppressed. Genome engineering has the capacity to endow xenogeneic tissues with down-modulating, anti-xenogeneic immune responses that can facilitate cross-species transplantation. Therefore, the origins, challenges, innovations, and future of regenerative medicine and transplantation are closely intertwined within the fields of immune and genome engineering. In this review, we summarize some recent developments in this arena.
EXTRACELLULAR MATRIX AS AN INDUCTIVE SCAFFOLD FOR ORGAN ENGINEERING
Xenotransplantation — transplantation across species — marked the earliest example of functional organ transplantation, when a canine kidney was implanted into the neck of a goat in the early 1900s.3 Xenotransplantation has the potential to overcome the barriers of human allograft transplantation, such as donor-tissue availability and preservation. Today, clinical applications of xenotransplantation primarily involve the use of xenogeneic extracellular matrix rather than intact xenogeneic cells or tissues, because intact material induces strong adaptive and innate immune responses as a result of xenogeneic major histocompatibility complex (MHC), other membrane proteins, and glycans. Xenogeneic and allogeneic extracellular matrixes, which are less immunogenic, are currently processed into surgical meshes,4 topical powders, and hydrogels for clinical indications such as hernia and skeletal muscle repair, breast implant surgery, and wound repair.5
In contrast to immune recognition of components of transplanted xenogeneic tissues and organs that result in failure of organ xenotransplantation, extracellular matrixes are now known to have strong immunomodulatory properties that favor constructive tissue and organ remodeling.6,7 These findings render extracellular matrix an attractive component for organ-engineering approaches focused on induction of endogenous tissue repair and reconstruction. In vitro studies indicate that extracellular matrix biomaterials processed for implantation contain cell remnants and other cues present in damaged tissue that are capable of inducing innate repair processes.8 Furthermore, the reservoir of cytokines, chemokines, and matrix-bound nanovesicles released from extracellular matrix bioscaffolds when placed at sites of injury stimulate physiological processes that facilitate the development, maintenance, and repair capabilities of tissues and organs when placed at sites of injury.9,10 These same physiological processes are key to successful organ transplantation.
Tissue-engineering therapies are clinically available — for example, as skin substitutes — but the cost and the challenges of delivering viable cell products limit widespread deployment. Engineered vascular substitutes are poised to become tissue-engineering products with the capacity to reach large-scale clinical delivery, in part through the engineering of inductive extracellular matrix that mobilizes endogenous repair. An off-the-shelf vascular graft is engineered with the use of donor cells, but the functional graft delivered to patients contains no living cells. In this approach, donor cells are seeded onto a tubular biomaterial scaffold (Fig. 1B) that is cultured in bioreactors with flow and pulse rates to stimulate tissue development with physiological properties similar to those of native tissue.11 Cells are then removed from the tissue graft to enable storage and delivery when needed. Similar to allograft and xenograft extracellular matrix products, the final product is composed only of acellular extracellular matrix poised to mobilize endogenous cells once implanted, and these cells will build and remodel the tissue in situ.12
THE SCALING CHALLENGE OF TISSUE ENGINEERING AND STEM CELLS
The somewhat disappointing integration of engineered tissues and organs into clinical practice to date can be attributed at least in part to challenges of requisite scale — manufacturing numbers of cells adequate to build a tissue or organ of clinically relevant size, deliverable to the patient when needed at an acceptable cost. The number of cells required to build an autologous tissue de novo is large and difficult to obtain with patient biopsies. The discovery of adult and induced pluripotent stem cells (iPSCs) as an expandable cell source created excitement as a potential solution for manufacturing adequate numbers of cells capable of forming multiple tissue types. However, the therapeutic regenerative mechanism of transplanted adult stem cells was found to be immunologically based only after clinical testing.13 The early clinical success and regulatory approvals for the use of mesenchymal stem cells (MSCs) for treating graftversus-host disease highlight their immunomodulatory and tolerance-inducing function in recipients of bone marrow transplants. MSCs are currently being tested in combination therapies to promote long-term tolerance of vascularized composite tissue allografts and kidney and liver transplants.14–16 For regenerative-medicine applications, the use of MSCs is more tenuous. After many clinical trials of MSCs for treating myocardial infarction (with mixed results), recent research in a mouse model suggests that the acute wound-healing response to injection, even with dead cells, is the therapeutic mechanism of action.17 Such results again point to the potential of mobilizing endogenous repair mechanisms in tissue repair and transplantation and the importance of the immune system.
A further example of the scaling challenge lies in organoids, derived from iPSCs, which organize into complex tissue structures. The discovery and development of iPSCs raised the possibility of a renewable cell source for engineering autologous stem cells and potentially a universal iPSC that could avoid immunologic rejection.18,19 Organoids of brain, retina, and gastrointestinal tissue replicate the complex cellular composition and organization of native tissue structures in miniature, although, as yet, without vasculature.20,21 These tissue mimics provide realistic models for the study of development and serve as in vitro screening systems, but direct clinical application of organoids for tissue repair is limited by their size (which is on the order of millimeters), their avascular nature, and difficulties in reliably controlling differentiation into specific tissues.
It is now feasible to apply assembly and additive manufacturing techniques to tissue engineering in applications that may be cellular or acellular, in an effort to create organ replacements of appropriate size and relevance to human tissues and organs — and at a scale needed for broader dissemination. A recent example is a three-dimensional–printed, human-size heart (Fig. 1D).22 Large-scale production and practical delivery to patients would need to be achieved if these technologies are to have an effect on clinical practice. Tissue and organ restoration in situ and xenotransplantation through immune and genome engineering remain the alternatives with more potential near-term applications.
THE IMMUNE SYSTEM — WHERE TISSUE REPAIR AND TRANSPLANT TOLERANCE CONVERGE
The immune system is the guardian of tissue and organ integrity. It can be leveraged for repair, immune regulation, and tolerance induction. Immune reactions in tissues guide infection control, tumor surveillance, autoimmunity and tolerance, and tissue repair. The approach of suppressing the immune response in biomaterial implants, tissue repair, and transplantation may shift to actively working with the immune system to exploit its regenerative capacity. The immunologic component of allograft transplant rejection was first observed with tumors and skin grafts more than a century ago, in 1912.23 Intense research efforts led to therapeutic management of the acute rejection induced by the T-cell–dependent allogeneic response that is central to rejection. There remains, however, the problem of chronic allograft rejection, which is often mediated by de novo development of anti-HLA (and other) antibodies. The current prevention of rejection is generally achieved with the use of lifelong immunosuppressive drug regimens. Promoting immune tolerance of allografts and eliminating chronic rejection without sacrificing the integrity of the immune system and its protective effects are central to current research efforts. A recent study involving human transplant biopsy specimens along with murine hearts showed the ability of matrix-bound vesicles to mitigate chronic rejection and fibrosis associated with allograft heart transplantation by stimulating interleukin-33 production that reduced proinflammatory immune activation.24 Cell-based therapies, including stem cells, regulatory T cells (Tregs), and chimeric antigen receptor (CAR) T cells, are undergoing clinical testing for their capacity to regulate the immune response in transplantation. The concept of regulating the immune system instead of suppressing it is a growing direction relevant to both regenerative medicine and transplantation.
In the numerous parallels between transplantation and tissue engineering, the focus on stem cells may have obscured the potential importance of the immune system in tissue repair and regeneration. Endogenous stem cells are critical for tissue homeostasis and renewal, but the therapeutic success of delivering stem cells to promote tissue repair has been limited clinically. In fact, the immune system may be critical for creating a pro-regenerative environment so that stem cells and other tissue-resident cells can be supported, allowing them to repair or rebuild tissues. Local resident and systemically trafficked immune cells, including eosinophils, macrophages, and T cells, contribute to tissue-repair processes. Eosinophils, for example, produce a key cytokine, interleukin-4,25 that is required for muscle repair.26 Macrophages are required for limb regeneration in the axolotl (a neotenic salamander) and contribute to human tissue repair.2 T cells modulate local tissue immune cells and themselves secrete cytokines and growth factors relevant to tissue growth and remodeling.27
A subset of Tregs — for example, Foxp3+ CD4+ Tregs — share relevance to both transplantation and tissue repair (Fig. 2). Tregs secrete immunosuppressive factors such as interleukin-10, transforming growth factor β, and interleukin-35, which mediate tissue tolerance. In addition to immunosuppressive cytokines, Tregs secrete tissue-specific growth factors that mobilize stem cells and promote tissue repair. To mobilize local stem cells and stimulate new tissue development, Tregs secrete amphiregulin in muscle tissue28 and insulin-like growth factor in neural tissue.29 The multinational ONE study (A Unified Approach to Evaluating Cellular Immunotherapy in Solid Organ Transplantation) showed that Treg therapy was safe in combination with standard immunosuppression and that it resulted in a lower incidence of infection and systemic effects of immune suppression after kidney allograft transplantation than drug therapy alone.30 Promoting Treg populations is also attractive for inducing tolerance in autoimmune diseases such as type 1 diabetes, systemic lupus erythematosus, and inflammatory bowel disease.31 Manufacturing techniques continue to be developed to efficiently increase the number of Tregs in culture so that there are adequate numbers for adoptive transfer in people.32 Further refinement of Treg populations for transplantation with antigen specificity and engineered T-cell receptors (e.g., CAR Tregs33,34) is likely to enhance their therapeutic efficacy in the future.
Figure 2. Convergence of Tissue Transplantation and Repair.
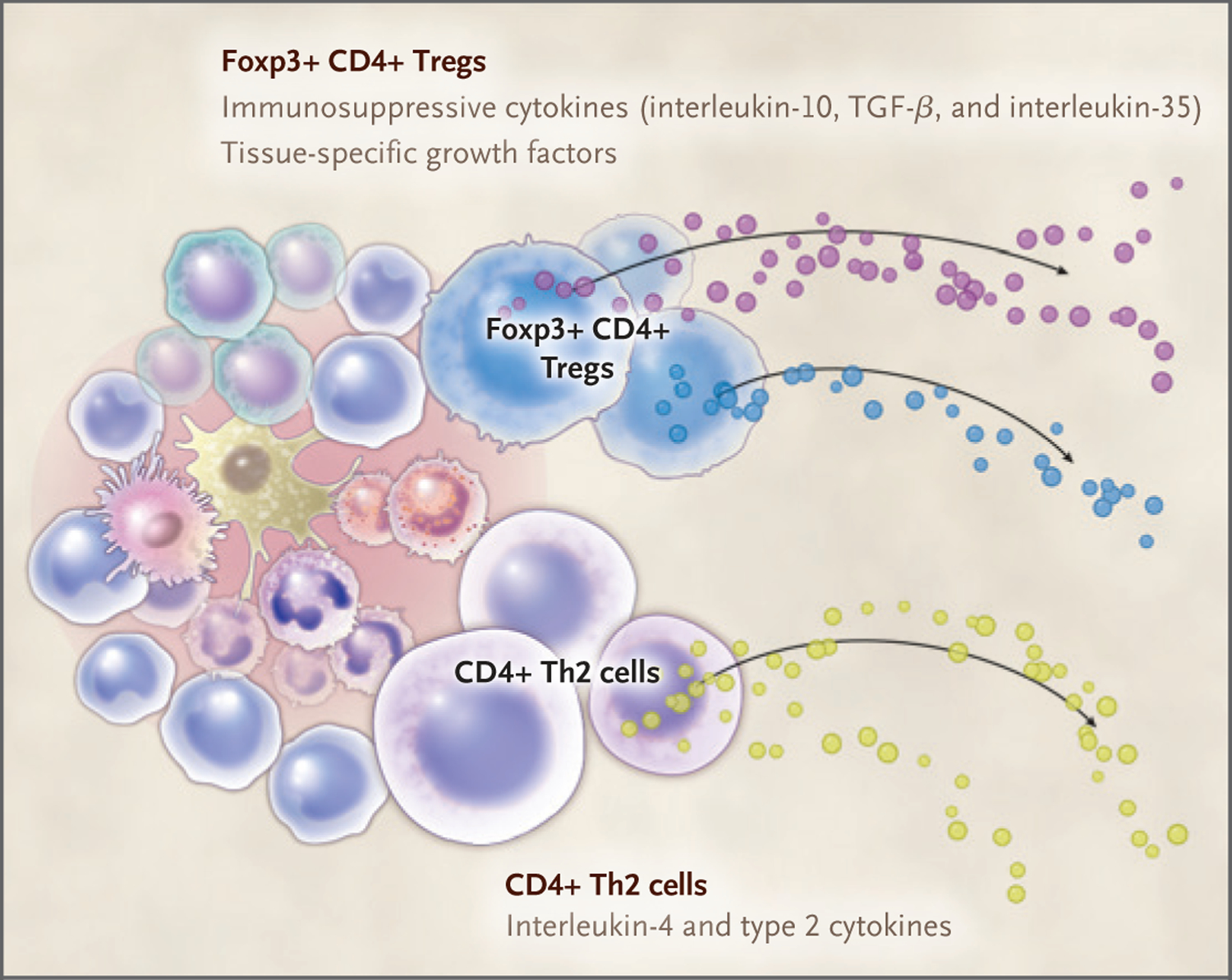
Multiple immune cells contribute to tissue repair and transplant tolerance. T cells, highlighted here, contribute to both tissue repair and transplant tolerance through production of the immunosuppressive cytokines interleukin-10, transforming growth factor β (TGF-β), and interleukin-35 by regulatory T cells (Tregs) and in the context of a wound secrete growth factors specific to the tissue. Other T-cell types, such as type 2 helper T (Th2) cells, promote tissue repair through production of cytokines, including interleukin-4.
XENOTRANSPLANTATION — RIGHT AROUND THE CORNER?
It is hard to imagine factories of beating hearts or expanding and deflating lungs ready for implantation when needed, given both the cost and the necessary logistics. A natural alternative to de novo engineering of organs is to use a live donor of another species as a source. The advantages of this approach are substantial, since a natural living organ is by definition fully functional, but cross-species immune responses create challenges. Engineering to overcome this restriction must be genetic. Specifically, a long-known hyperacute immune response to animal organs is largely eliminated by knocking out genes encoding machinery that synthesizes cell-surface carbohydrate xenoantigens — specifically, the α-1,3-galactosidase encoded by GGTG1. Two other genes (CMAH and B4GALNT2, which encode proteins that produce N-glycolylneuraminic acid) and SD(a) xenoantigens have been studied in knockout models and also reduce cross-reactivity. Other challenges associated with xenotransplants include risks of zoonoses associated with the animal species that supplies the organ and sociocultural reluctance among patients to accept an animal-sourced organ. Pigs are generally considered an ideal potential source of organs for xenotransplantation in humans. Porcine and human organs are similar in size and shape and are readily available, and both inbred and outbred strains are in use for preclinical studies. Moreover, pigs can be cloned by means of somatic-cell nuclear transplantation techniques, and their genomes can now be readily engineered (or “edited”) by designer nucleases such as zinc-finger nucleases, transcription activator–like effector nucleases (TALENs, which include a nonspecific DNA-cleaving nuclease fused to a DNA-binding domain that can target any sequence), and the versatile RNA-targeted CRISPR-Cas9 (clustered regularly interspaced short palindromic repeats and associated Cas9 endonuclease) system. Finally, because pigs are more distantly related to humans than are nonhuman primates, it has been reasoned that they are substantially less likely than nonhuman primates to transmit pathogens through a transplanted organ. Sensitive polymerase-chain-reaction assays are available to detect pathogens, and a robust surveillance program would be required to keep donor animals pathogen-free, but it would be feasible. Furthermore, an in-depth monitoring program for the products would be required to comply with Food and Drug Administration (FDA) requirements. These positives must be balanced against the certainty that the number of “minor antigens” in the form of amino acid sequence variations — and thus the potential for immunologic incompatibility — inexorably increases with phylogenetic distance.
The first whole-organ xenotransplantations were performed in the early 1900s, and the news about them was both good and bad.3,35 The organs were successfully transplanted and functional, but we now recognize that most of them failed rapidly because of hyperacute rejection in response to glycosylated proteins, such as galactose-α-1,3-galactose, which are present in massive amounts in the graft cell membranes of donor animals.36,37 The use of donor pigs with an engineered knockout of GGTG1, which is responsible for primary glycosylation activity, was able to greatly reduce hyperacute rejection experimentally; however, when the tissue was transplanted into nonhuman primates, an imperfect proxy for humans, it was still rejected within weeks.38 In a series of studies, researchers have embarked on a mission to identify human genes, mostly encoding the CD (cluster of differentiation) family of membrane proteins, that can be expressed as transgenic proteins, fusion proteins, or even in the form of antibodies against porcine proteins in the pig to further mitigate human anti-porcine immune responses (Table 1).38 Several such transgenes have been tested, although not systematically, over the past decade, and the proteins they encode fall into three categories: immune cloaking, whereby specific transgenic proteins interfere with killing by natural killer (NK) or T cells, and complement humanization and coagulation humanization, in which specific elements of the human complement or human coagulation pathways are used instead of or in addition to porcine counterparts. The ability of some of these interventions to promote successful xenotransplantation into nonhuman primates has been evaluated, and in some cases substantial improvements in organ rejection have been noted.41,44
Table 1.
Transgenic Proteins and Their Potential Uses in Transplantation.
| Function and Transgenic Protein* | Suspected Mechanism |
|---|---|
| Immune cloaking | |
| CD47 (integrin-associated protein)39,40 | Reduces macrophage-mediated toxicity through signal regulatory protein α (SIRPα); “don’t eat me” signal |
| CD95L (Fas ligand)41 | Prevents apoptosis |
| Anti-pCD152 (anti-pCTLA4 antibody)41 | Minimizes effect of porcine cytotoxic T-cell antigen 4 |
| MGAT3 (GnT-III)41 | Humanizes glycosylation |
| HLA-E–β2 M (fusion protein)42 | Prevents natural killer cell attack in a swine leukocyte antigen knockout |
| SIRPα (signal regulatory protein)40 | Reduces macrophage-mediated toxicity (see CD47) |
| TNF-α (tumor necrosis factor α)43 | Prevents apoptosis |
| TRAIL (TNF-α apoptosis-inducing ligand)41 | Prevents apoptosis |
| Complement humanization | |
| CD46 (membrane cofactor protein)44 | Inhibits complement damage |
| CD55 (decay-accelerating factor)41 | Inhibits complement membrane attack complex |
| CD59 (MAC-inhibitory protein)41 | Inhibits complement membrane attack complex |
| Coagulation humanization | |
| CD39 (ecto-NTP diphosphohydrolase 1)41 | Inhibits platelet aggregation |
| PROCR (endothelial protein C receptor)44 | Enhances anticoagulant serine protease protein C |
| TFPI (tissue factor pathway inhibitor)41 | Inhibits factor Xa and tissue factor, minimizing thrombin formation |
| THBD (thrombomodulin)44 | Activates antithrombotic protein C, minimizing clot formation |
The use of nonhuman primates in preclinical studies may be well intended, but it may add an unnecessary complication to clinical studies. Aside from the expense and ethical challenges surrounding such studies, this approach adds a step to what should be a much simpler question: can we improve the feasibility of transplanting organs from donor species A (pig) into species B (human) by adding a “third party,” species C (nonhuman primate), as a test step? Actually, differences between the immune systems of nonhuman primates and humans, not to mention the differences in size and physiology, would greatly complicate the evaluation of efficacy and safety in going from A to B, the relevant trajectory (Fig. 3). A good example of an “irrelevant” immunologic phenotype is differential glycosylation sensitivity to CMAH-knockout pig cells in nonhuman primates as compared with humans.46 It would be important to develop additional evaluative platforms that avoid the introduction of relevant antigens and organs into nonhuman primates, such as “reversing the polarity” of transplantation by transplanting surplus human tissues or organs into pigs, as well as the more radical notion of using newly dead organ donors to evaluate the human xenoresponse. Such moves might facilitate xenotransplantation implementation in the clinic.47–49
Figure 3. Phylogenetic Relationships among Mammalian Species Germane to Xenotransplantation.
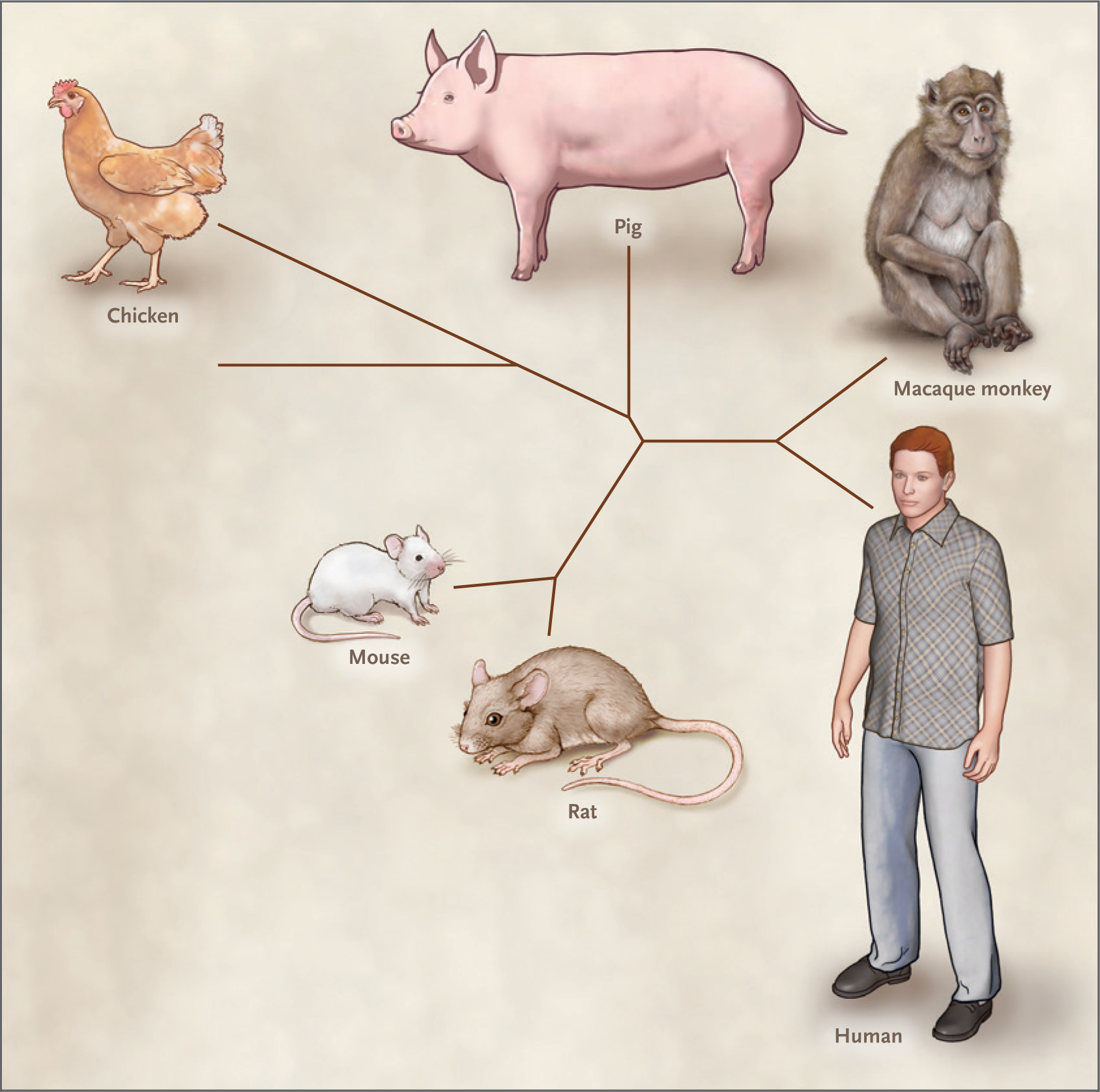
The relationships among the mammalian species cited in this article are shown in this unrooted phylogenetic tree. The branches were formed with the use of an algorithm available at phyloT (https://phylot.biobyte.de) and visualized using iTOL (Interactive Tree of Life; https://itol.embl.de).
TECHNOLOGY DEVELOPMENT IN XENOTRANSPLANTATION
Two noteworthy developments in xenotransplantation have occurred in the past few years. The first relates to the safety concerns (first raised in the 1980s) around transmission of porcine endogenous retrovirus sequences (PERVs) to transformed human tissue culture cells.50,51 To eliminate this risk altogether, CRISPR-Cas9 technology was recently deployed to eliminate all 62 PERV copies that exist (two copies each of 31 loci) from the porcine genome in PK15 porcine kidney cells and fibroblasts.52,53 A second transformative set of experiments with the potential to revolutionize xenotransplantation showed the production of organ-specific interspecies chimeras (sometimes referred to as “exogenesis”), which opened the door to the remarkable possibility of producing organs that contain fully human cells in chimeric animals (Fig. 4). As first shown by Nakauchi and colleagues,54 it is possible to produce chimeras in which the pancreas is entirely replaced by cells of another species. Specifically, in a proof-of-concept experiment, a rat pancreas was produced in a mouse through the introduction of rat stem cells into the inner cell mass of mouse blastocysts carrying a mutation (Pdx1-/-) that specifically blocked murine pancreas development. More recently, similar experiments were performed in pigs, although the pancreas was replaced with that of a genetically distinct line of pig rather than a separate species.55
Figure 4. Xenotransplantation versus Exogenesis — Therapeutic Concepts.
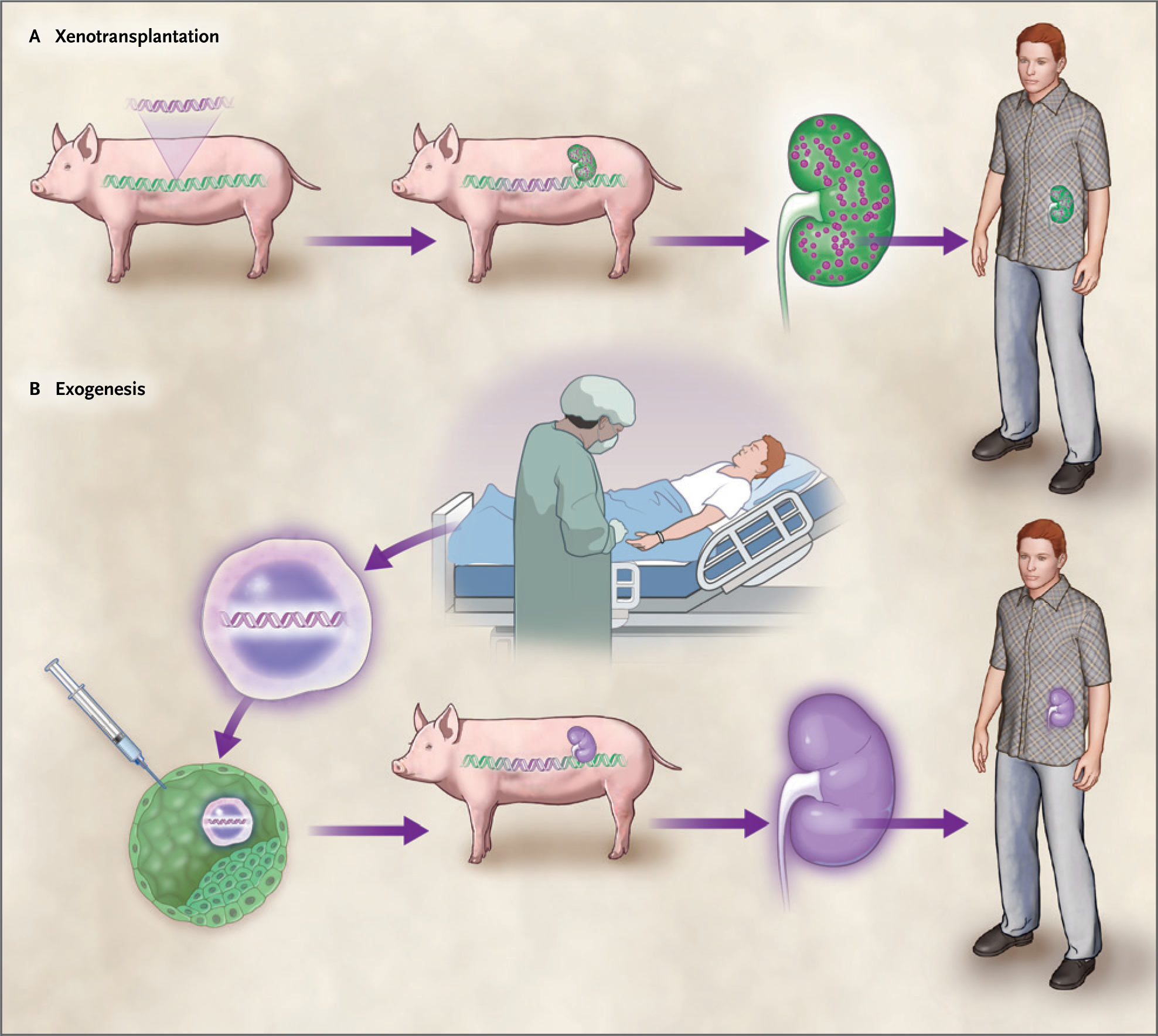
Panel A shows xenotransplantation. (Human components are colored purple, and porcine components are colored green.) The genome of a pig is immunoengineered both by deleting porcine major antigens and by adding specific human transgenes expressing proteins that have been shown or hypothesized to improve the ability of “xeno organs” to persist (e.g., in animal models of xenotransplantation, such as nonhuman primates). Next, the result of the immunoengineering — a donor organism for xenotransplantation — produces an organ modified by immunoengineering. Here, the organ is a kidney, which is green because it has a mostly porcine genome but expresses certain human or humanized proteins (purple circles) intended to improve persistence in humans. Transplantation of the xeno kidney into the recipient restores function. Panel B shows exogenesis, in which a patient is the source of autologous human stem cells (e.g., patient-derived induced pluripotent stem cells [iPSCs]; purple cells). These are injected into a porcine blastocyst derived from a previously immune-engineered pig that has been “humanized” through the deletion of porcine genes and the introduction of human transgenes, as shown in Panel A. The exogenic blastocyst gives rise to a chimera producing a kidney that has a human genome, which can now be transplanted into a recipient.
Nakauchi’s group also led experiments that showed that interspecies organogenesis as described above could generate autologous functional islets through the transplantation of mouse iPSCs into a Pdx1-/- rat. The resulting pancreatic islets were then transplanted into an isogenic mouse in which diabetes had been induced by streptozotocin treatment.56 The transplanted islets had evident function, as indicated by glucose levels that were normalized in the blood of the recipient mice without any immunosuppression administered after the first 5 days after transplantation; these normalized glucose levels persisted for 1 year. This is compelling proof of concept that exogenesis is in our future, under the assumption that a suitable host organism — most likely pig or sheep, in which organ systems are sufficiently similar in size and shape to those of humans — can be engineered to adopt our stem cells as its own. The extent of engineering of both the porcine and human genomes that will be required in order to make exogenesis sufficiently safe and practical and reasonably cost-effective is likely to be substantial, not to mention the extensive safety and efficacy testing that will be required after stable platforms are developed.
Looking ahead, tremendous progress may make “traditional” xenotransplantation not only feasible but pragmatic. Studies involving so-called triple-knockout pigs (i.e., with knockout of GGTA1, CMAH, and B4GALNT2) that lack the carbohydrate xenoantigens on the surface of porcine cells that are normally recognized by normal human serum and peripheral blood mononuclear cells have been performed. The triple-knockout pig is reportedly healthy, and in vitro studies indicate that a substantial proportion of the human population is likely to lack antibody reactivity to renal microvascular epithelial cells derived from these animals.46 Furthermore, the extent of cross-reactivity observed in samples from some patients was similar to cross-reactivity observed in some recipients of human allografts, which suggests that for this group of patients, rejection of xenotransplants from such animals might be controllable through the use of existing immunosuppression regimens. The subset of human cells that remain immunoreactive to triple-knockout cells appeared to recognize swine leukocyte antigen (SLA) class I (the porcine equivalent of HLA [MHC] class I). In a subsequent study,57 the effect that the deletion of porcine SLA genes had on immunoreactivity assays of serum from patients on a transplant wait list was evaluated, and the results suggested that such deletions may increase the proportion of patients who could benefit.
The second independent means of minimizing immunologic damage to porcine organs after transplantation in a human host is the engineering of complement components and pathways. One way to mitigate such damage would be to humanize the porcine surface antigens by expressing one or more human transgenic proteins that down-regulate the activity of human complement and then “cloak” them generally from recognition by the human immune system (Table 1). Because human complement regulatory proteins coevolved with the human complement pathway, human proteins are expected to be more effective at down-regulating the effects of complement than their porcine counterparts. These proteins include CD55 (a complement decay-accelerating factor), CD59 (a membrane attack complex–inhibitory protein), and CD46 (a complement regulatory protein). The gene encoding tumor necrosis factor α has also been proposed as important in controlling acute vascular rejection of porcine xenografts,43 and the CD47 signal protein — which sends a message of “don’t eat me”39 — is considered particularly beneficial for promoting acceptance of porcine tissue. Multiple transgenes can be combined or “stacked” at a single permissive locus, such as Rosa26 in the porcine genome.58 A total of nine “stacked” transgenes were used, in which sets of three protein-coding sequences each were fused and “punctuated” with viral self-cleavage 2A sequences. Just one promoter and terminator are required per set of three protein-coding sequences. These stacked transgenes were combined with a PERV-free, triple-knockout pig genome45; porcine cells with these modifications showed promising in vitro characteristics, including a lack of hyperacute response, appropriate complement responses, and a lack of destruction by human NK cells and macrophages. An ongoing challenge in humanization through the use of transgenes is to identify promoters and insulator sequences (i.e., an enhancer blocker or a barrier) that allow sustained long-term expression for further development.
Hozain et al. recently reported yet another trans-species innovation in transplantation — namely, the rejuvenation of human lungs initially considered too “damaged” for transplantation by connecting them to the circulatory system of a pig.47,48 Such extracorporeal cross-circulation restored these otherwise damaged lungs to a sufficiently healthy state to be potentially suitable for transplantation. Such a strategy could substantially increase the availability of human lungs that would otherwise be discarded rather than transplanted.
CONCLUSIONS
The immune system remains vast, complex, and at times mysterious. But it is beginning to yield mechanistic and practical insights through sustained molecular and cellular probing. With this complexity comes tremendous potential. Immunologic targets have been underexploited clinically in regenerative medicine but are likely to represent the means of defining cell and tissue therapies of the future. Combined with new immunologically based therapeutic strategies, transplantation and regenerative medicine are converging (or reconverging) in the context of immunoengineering and regenerative immunology with the potential to provide transplantable tissues and enhanced tissue-repair strategies.
Acknowledgments
We thank David R. Maestas, Jr., Gerald Brandacher, Drew Pardoll, and Robert Montgomery for their critical review of the manuscript.
Glossary
- 2A sequences
Short amino acid–coding sequences that lead to discontinuous translation, allowing the production of multiple proteins from a single coding sequence
- Allograft
A graft of cells or tissues from one individual member of a species to another
- Chimeric antigen receptor (CAR) T cell
A T cell engineered to express a chimeric antigen receptor
- CD (cluster of differentiation)
Cell-surface molecules that help define cell identity and other properties
- CRISPR-Cas9
Clustered regularly interspaced short palindromic repeats and associated Cas9 endonuclease; a sequence-specific nuclease with sequence specificity conferred by a guide RNA molecule
- Exogenesis
The production of a tissue or organ through transplantation of stem cells from a donor species (e.g., human) into the blastocyst of suitably engineered recipient species (e.g., pig), leading to formation of an organ or tissue from cells with the genome of the donor species in the recipient species
- Immune cloaking
Expression of cell-surface molecules that minimizes immune system damage to a heterologous cell
- Major histocompatibility complex (MHC)
A complex of linked genes encoding cell-surface proteins that display peptides produced by cleavage of intracellular proteins; these proteins help T cells recognize foreign or mutated proteins. Its human form is referred to as HLA, and its porcine counterpart is SLA
- Natural killer (NK) cell
A type of cytotoxic lymphocyte critical to the function of the innate immune system
- Regulatory T cell (Treg)
A specialized type of T cell that dampens the immune response and maintains self-tolerance
- Xenograft
A graft of tissue from a donor to a recipient of a different species
- Xenotransplantation
The transplantation of cells, tissue, or organs to a recipient organism of a different species
Footnotes
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
Contributor Information
Jennifer Elisseeff, Translational Tissue Engineering Center, Wilmer Eye Institute, and the Department of Biomedical Engineering, Johns Hopkins University, Baltimore
Stephen F. Badylak, McGowan Institute of Regenerative Medicine, University of Pittsburgh, Pittsburgh
Jef D. Boeke, Institute for Systems Genetics and the Department of Biochemistry and Molecular Pharmacology, NYU Langone Health, New York Department of Biomedical Engineering, NYU Tandon School of Engineering, New York.
REFERENCES
- 1.Viebahn CS, Benseler V, Holz LE, et al. Invading macrophages play a major role in the liver progenitor cell response to chronic liver injury. J Hepatol 2010;53: 500–7. [DOI] [PubMed] [Google Scholar]
- 2.Godwin JW, Pinto AR, Rosenthal NA. Macrophages are required for adult salamander limb regeneration. Proc Natl Acad Sci U S A 2013;110:9415–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Doyle AM, Lechler RI, Turka LA. Organ transplantation: halfway through the first century. J Am Soc Nephrol 2004;15: 2965–71. [DOI] [PubMed] [Google Scholar]
- 4.Kissane NA, Itani KMF. A decade of ventral incisional hernia repairs with biologic acellular dermal matrix: what have we learned? Plast Reconstr Surg 2012;130: Suppl 2:194S–202S. [DOI] [PubMed] [Google Scholar]
- 5.Dziki J, Badylak S, Yabroudi M, et al. An acellular biologic scaffold treatment for volumetric muscle loss: results of a 13-patient cohort study. NPJ Regen Med 2016;1:16008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dziki JL, Wang DS, Pineda C, Sicari BM, Rausch T, Badylak SF. Solubilized extracellular matrix bioscaffolds derived from diverse source tissues differentially influence macrophage phenotype. J Biomed Mater Res A 2017;105:138–47. [DOI] [PubMed] [Google Scholar]
- 7.Hussey GS, Dziki JL, Lee YC, et al. Matrix bound nanovesicle-associated IL-33 activates a pro-remodeling macrophage phenotype via a non-canonical, ST2-independent pathway. J Immunol Regen Med 2019;3:26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beachley VZ, Wolf MT, Sadtler K, et al. Tissue matrix arrays for high-throughput screening and systems analysis of cell function. Nat Methods 2015;12:1197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blache U, Stevens MM, Gentleman E. Harnessing the secreted extracellular matrix to engineer tissues. Nat Biomed Eng 2020;4:357–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hussey GS, Dziki JL, Badylak SF. Extracellular matrix-based materials for regenerative medicine. Nat Rev Mater 2018; 3:159–73. [Google Scholar]
- 11.Dahl SL, Kypson AP, Lawson JH, et al. Readily available tissue-engineered vascular grafts. Sci Transl Med 2011;3:68ra9. [DOI] [PubMed] [Google Scholar]
- 12.Kirkton RD, Santiago-Maysonet M, Lawson JH, et al. Bioengineered human acellular vessels recellularize and evolve into living blood vessels after human implantation. Sci Transl Med 2019;11(485): eaau6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caplan AI. Why are MSCs therapeutic? New data: new insight. J Pathol 2009; 217:318–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Casiraghi F, Perico N, Remuzzi G. Mesenchymal stromal cells for tolerance induction in organ transplantation. Hum Immunol 2018;79:304–13. [DOI] [PubMed] [Google Scholar]
- 15.Podestà MA, Remuzzi G, Casiraghi F. Mesenchymal stromal cells for transplant tolerance. Front Immunol 2019;10:1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Detry O, Vandermeulen M, Delbouille M-H, et al. Infusion of mesenchymal stromal cells after deceased liver transplantation: a phase I-II, open-label, clinical study. J Hepatol 2017;67:47–55. [DOI] [PubMed] [Google Scholar]
- 17.Vagnozzi RJ, Maillet M, Sargent MA, et al. An acute immune response underlies the benefit of cardiac stem cell therapy. Nature 2020;577:405–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi Y, Inoue H, Wu JC, Yamanaka S. Induced pluripotent stem cell technology: a decade of progress. Nat Rev Drug Discov 2017;16:115–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trounson A, Boyd NR, Boyd RL. Toward a universal solution: editing compatibility into pluripotent stem cells. Cell Stem Cell 2019;24:508–10. [DOI] [PubMed] [Google Scholar]
- 20.Clevers H Modeling development and disease with organoids. Cell 2016;165: 1586–97. [DOI] [PubMed] [Google Scholar]
- 21.Li M, Izpisua Belmonte JC. Organoids — preclinical models of human disease. N Engl J Med 2019;380:569–79. [DOI] [PubMed] [Google Scholar]
- 22.Lee A, Hudson AR, Shiwarski DJ, et al. 3D bioprinting of collagen to rebuild components of the human heart. Science 2019;365:482–7. [DOI] [PubMed] [Google Scholar]
- 23.Barker CF, Markmann JF. Historical overview of transplantation. Cold Spring Harb Perspect Med 2013;3:a014977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li T, Zhang Z, Bartolacci JG, et al. Graft IL-33 regulates infiltrating macrophages to protect against chronic rejection. J Clin Invest 2020;130:5397–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qiu Y, Nguyen KD, Odegaard JI, et al. Eosinophils and type 2 cytokine signaling in macrophages orchestrate development of functional beige fat. Cell 2014;157: 1292–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heredia JE, Mukundan L, Chen FM, et al. Type 2 innate signals stimulate fibro/ adipogenic progenitors to facilitate muscle regeneration. Cell 2013;153:376–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sadtler K, Estrellas K, Allen BW, et al. Developing a pro-regenerative biomaterial scaffold microenvironment requires T helper 2 cells. Science 2016;352:366–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burzyn D, Kuswanto W, Kolodin D, et al. A special population of regulatory T cells potentiates muscle repair. Cell 2013;155:1282–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hui SP, Sheng DZ, Sugimoto K, et al. Zebrafish regulatory T cells mediate organ-specific regenerative programs. Dev Cell 2017;43(6):659–672.e5. [DOI] [PubMed] [Google Scholar]
- 30.Geissler EK. The ONE study compares cell therapy products in organ transplantation: introduction to a review series on suppressive monocyte-derived cells. Transplant Res 2012;1:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raffin C, Vo LT, Bluestone JA. Treg cell-based therapies: challenges and perspectives. Nat Rev Immunol 2020;20:158–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.MacDonald KN, Piret JM, Levings MK. Methods to manufacture regulatory T cells for cell therapy. Clin Exp Immunol 2019;197:52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dawson NA, Lamarche C, Hoeppli RE, et al. Systematic testing and specificity mapping of alloantigen-specific chimeric antigen receptors in regulatory T cells. JCI Insight 2019;4(6):e123672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.MacDonald KG, Hoeppli R, Huang Q, et al. Generation of alloantigen-specific T regulatory cells via a chimeric antigen receptor targeting HLA-A2. J Immunol 2016:196:Suppl 1:140.6. [Google Scholar]
- 35.Reemtsma K Xenotransplantation: a historical perspective. ILAR J 1995;37: 9–12. [DOI] [PubMed] [Google Scholar]
- 36.Galili U Anti-Gal: an abundant human natural antibody of multiple pathogeneses and clinical benefits. Immunology 2013;140:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Joziasse DH, Oriol R. Xenotransplantation: the importance of the Galα1,3Gal epitope in hyperacute vascular rejection. Biochim Biophys Acta 1999;1455:403–18. [DOI] [PubMed] [Google Scholar]
- 38.Cooper DKSV, Satyananda V, Ekser B, et al. Progress in pig-to-non-human primate transplantation models (1998–2013): a comprehensive review of the literature. Xenotransplantation 2014;21:397–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tena AA, Sachs DH, Mallard C, et al. Prolonged survival of pig skin on baboons after administration of pig cells expressing human CD47. Transplantation 2017; 101:316–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ide K, Wang H, Tahara H, et al. Role for CD47-SIRPalpha signaling in xenograft rejection by macrophages. Proc Natl Acad Sci U S A 2007;104:5062–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cooper DK, Ekser B, Ramsoondar J, Phelps C, Ayares D. The role of genetically engineered pigs in xenotransplantation research. J Pathol 2016;238:288–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lilienfeld BG, Crew MD, Forte P, Baumann BC, Seebach JD. Transgenic expression of HLA-E single chain trimer protects porcine endothelial cells against human natural killer cell-mediated cytotoxicity. Xenotransplantation 2007;14:126–34. [DOI] [PubMed] [Google Scholar]
- 43.Oropeza M, Petersen B, Carnwath JW, et al. Transgenic expression of the human A20 gene in cloned pigs provides protection against apoptotic and inflammatory stimuli. Xenotransplantation 2009;16:522–34. [DOI] [PubMed] [Google Scholar]
- 44.Cooper DKC, Hara H, Iwase H, et al. Justification of specific genetic modifications in pigs for clinical organ xenotransplantation. Xenotransplantation 2019;26(4): e12516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yue Y, Xu W, Kan Y, et al. Extensive germline genome engineering in pigs. Nat Biomed Eng 2021;5:134–43. [DOI] [PubMed] [Google Scholar]
- 46.Estrada JL, Martens G, Li P, et al. Evaluation of human and non-human primate antibody binding to pig cells lacking GGTA1/CMAH/β4GalNT2 genes. Xenotransplantation 2015;22:194–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hozain AE, O’Neill JD, Pinezich MR, et al. Xenogeneic cross-circulation for extracorporeal recovery of injured human lungs. Nat Med 2020;26:1102–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hozain AE, Tipograf Y, Pinezich MR, et al. Multiday maintenance of extracorporeal lungs using cross-circulation with conscious swine. J Thorac Cardiovasc Surg 2020;159(4):1640–1653.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parent B, Gelb B, Latham S, et al. The ethics of testing and research of manufactured organs on brain-dead/recently deceased subjects. J Med Ethics 2020;46 199–204. [DOI] [PubMed] [Google Scholar]
- 50.Akiyoshi DE, Denaro M, Zhu H, Greenstein JL, Banerjee P, Fishman JA. Identification of a full-length cDNA for an endogenous retrovirus of miniature swine. J Virol 1998;72:4503–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van der Laan LJ, Lockey C, Griffeth BC, et al. Infection by porcine endogenous retrovirus after islet xenotransplantation in SCID mice. Nature 2000;407:90–4 [DOI] [PubMed] [Google Scholar]
- 52.Yang L, Güell M, Niu D, et al. Genome-wide inactivation of porcine endogenous retroviruses (PERVs). Science 2015; 350:1101–4. [DOI] [PubMed] [Google Scholar]
- 53.Niu D, Wei HJ, Lin L, et al. Inactivation of porcine endogenous retrovirus in pigs using CRISPR-Cas9. Science 2017; 357:1303–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kobayashi T, Yamaguchi T, Hamanaka S, et al. Generation of rat pancreas in mouse by interspecific blastocyst injection of pluripotent stem cells. Cell 2010; 142:787–99. [DOI] [PubMed] [Google Scholar]
- 55.Matsunari H, Nagashima H, Watanabe M, et al. Blastocyst complementation generates exogenic pancreas in vivo in apancreatic cloned pigs. Proc Natl Acad Sci U S A 2013;110:4557–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yamaguchi T, Sato H, Kato-Itoh M, et al. Interspecies organogenesis generates autologous functional islets. Nature:2017;542:191–6. [DOI] [PubMed] [Google Scholar]
- 57.Martens GR, Reyes LM, Li P, et al. Humoral reactivity of renal transplant-wait-listed patients to cells from GGTA1/ CMAH/B4GalNT2, and SLA class I knockout pigs. Transplantation 2017;101(4):e86–e92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fischer K, Kraner-Scheiber S, Petersen B, et al. Efficient production of multi-modified pigs for xenotransplantation by ‘combineering’, gene stacking and gene editing. Sci Rep 2016;6:29081. [DOI] [PMC free article] [PubMed] [Google Scholar]